

Abstract
Antibody–drug conjugates (ADCs) are an important class of therapeutic agents that harness the highly specific antigen targeting property of antibodies to deliver toxic drugs for targeted cell killing. Site-specific conjugation methods are highly desirable for constructing homogeneous ADCs that possess a well-defined antibody-to-drug ratio, stability, ideal pharmacological profile, and optimal therapeutic index. We report here a facile synthesis of functionalized glycan oxazolines from free sialoglycans that are key donor substrates for enzymatic Fc glycan remodeling and the application of an efficient endoglycosidase mutant (Endo-S2 D184M) for site-specific glycan transfer to construct homogeneous ADCs. We found that by a sequential use of two coupling reagents under optimized conditions, free sialoglycans could be efficiently converted to selectively functionalized glycan oxazolines carrying azide-, cyclopropene-, and norbornene-tags, respectively, in excellent yield and in a simple one-pot manner. We further demonstrated that the recently reported Endo-S2 D184 M mutant was highly efficient for Fc glycan remodeling with the selectively modified glycan oxazolines to introduce tags into an antibody, which required a significantly smaller amount of glycan oxazolines and a much shorter reaction time than that of the Endo-S D233Q-catalyzed reaction, thus minimizing the side reactions. Finally homogeneous ADCs were constructed with three different click reactions. The resulting ADCs showed excellent serum stability, and in vitro cytotoxicity assays indicated that all the three ADCs generated from the distinct click reactions possessed potent and comparable cytotoxicity for targeted cancer cell killing.
Graphical Abstract
INTRODUCTION
Antibody–drug conjugates (ADCs) are a class of therapeutic agents that explore the specificity of antibodies to deliver highly toxic drugs to respective antigen-expressing cells to achieve targeted cell killing.1,2 So far, 11 ADCs have been approved by the US FDA for the treatment of cancers and many more are in the pipeline.3,4 Many factors contribute to the overall in vivo efficacy of an ADC. In addition to the choice of antibody, payload, and linker, the way in which an antibody is conjugated to the drug also plays an important role in dictating the therapeutic outcome of ADCs. The first generation ADCs have been produced through nonspecific conjugations at lysine and/or reduced cysteine residues, which usually result in heterogeneous mixtures of ADC entities that may differ in drug-to-antibody ratios (DARs), sites of attachment, stability, and pharmacokinetic properties.2,5 To overcome the issues of reproducibility, stability, and potential side effects associated with heterogeneous conjugations, the next generation of ADCs has been focused on site-specific conjugations that provide homogeneous conjugates with well-defined pharmacological properties and improved therapeutic index.6-8 Significant progress has been made in recent years in the development of site-specific antibody–drug conjugation strategies.2 Some examples include the following: the introduction of unnatural amino acids or unpaired cysteines for subsequent chemoselective ligation,6,9-15 the selective C-/N-terminal modifications,16,17 the disulfide reduction/rebridging strategy,18,19 and the transglutaminase-mediated chemoenzymatic ligation.20
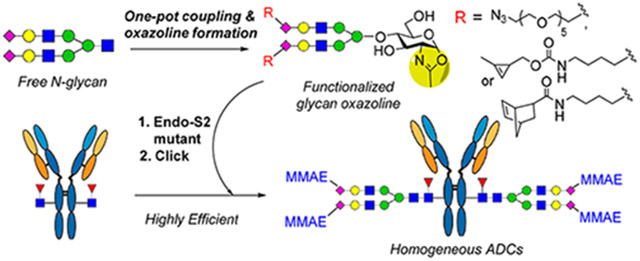
In addition to the site-selective modifications on amino acid residues of the protein domains, another approach is to conjugate drugs at the highly conserved N-glycans located at Asn-297 of the Fc domain.21,22 Since all IgG antibodies carry the conserved Fc N-glycans and they are spatially distant from the antigen-binding region, a unique advantage of the Fc glycan-mediated conjugation is that it does not modify the protein parts, and the attachment of the drug to the Fc glycans usually will not interfere with the Fab-mediated antigen binding. Early attempts to functionalize the Fc glycans through oxidation of adjacent diols of terminal monosaccharides have provided mixtures of conjugates due to the heterogeneity of the glycoforms and the oxidation at different sugar units.23-25 The use of the galactosyltransferase mutants capable of accommodating modified UDP-galactose derivatives as the donor substrates has enabled the incorporation of a selected tag at the Fc glycans for subsequent site-specific conjugation with modified cytotoxic agents.26-29 Nevertheless, this approach usually requires the trimming of the heterogeneous Fc N-glycans to the terminal GlcNAc-glycan forms, and due to the moderate efficiency of the mutant enzyme on the unnatural sugar nucleotide substrate, a large excess of modified sugar nucleotide and enzyme as well as a long incubation time are usually needed to drive the reaction, which often leads to an incomplete reaction and thus heterogeneity of the products. On the other hand, the endoglycosidase-catalyzed glycan remodeling strategy,30,31 enabled by the discovery of the Endo-S and Endo-S2 glycosynthase mutants for efficient glycosylation without product hydrolysis,32-34 has provided a promising method for generating homogeneous glycoforms including ADCs.21,22,35-38 This method includes three key steps: the synthesis of selectively tagged glycan oxazoline as donor substrates, the enzymatic transfer of the tagged glycans to Fc-deglycosylated antibodies, and the click drug conjugation. While this method has demonstrated promise for constructing ADCs, the synthesis of the selectively tagged glycan oxazolines remains to be improved,36,39 and the requirement of a large excess of glycan oxazolines and a relatively long incubation time to drive the reaction, partially due to the moderate activity of the Endo-S D233Q mutant,32 leads to side reactions on the antibody that need significant optimizations.36,38 In this paper, we report a facile one-pot synthesis of functionalized glycan oxazolines carrying azide-, cyclopropene-, and norbornene-tags, respectively, from free natural sialoglycans, and their use for antibody glycan remodeling was catalyzed by the Endo-S2 D184 M mutant with minimized nonenzymatic side reactions.33 The optimized chemoenzymatic method enabled a highly efficient synthesis of selectively tagged antibodies which are readily used for site-specific antibody–drug conjugation. We also performed a comparative study of three click methods for conjugating the drug to make homogeneous antibody–drug conjugates (Figure 1) and evaluated the in vitro cytotoxicity of the resulting ADCs. The one-pot synthesis of functionalized glycan oxazolines, coupled with the efficient Endo-S2 D184M-catalyzed Fc glycan remodeling and click drug conjugation, provides a general and efficient approach to producing structurally well-defined, homogeneous antibody–drug conjugates.
Figure 1.

A general approach for the glycan-mediated site-specific antibody–drug conjugation via different click reactions.
RESULTS AND DISCUSSION
Improved Synthesis of Azide-Tagged Sialylated N-Glycans from Free Sialoglycans.
Davis and co-workers have previously reported that free sialylated N-glycan could be functionalized by amide coupling between the terminal sialic acids and a tagged amine using 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) as the coupling reagent, but a large excess of an azide-tagged amine (20 equiv) and DMTMM (40 equiv) at an elevated temperature should be applied to give a moderate yield.36 We first repeated the coupling reaction using DMTMM and an azide-PEG-NH2 under similar conditions. Unexpectedly, we found that the amide coupling was accompanied by the simultaneous formation of a glycoside derived from DMTMM, giving the 4,6-dimethoxy-1,3,5-triazin-2-yl α-glycoside (2) instead of the free reducing glycan (3) (Scheme 1). Glycoside 2 was purified by HPLC in excellent yield, and its identity was verified by ESI-MS and NMR analysis. Indeed, Shoda and coworkers have previously reported that reducing sugars can be converted to 4,6-dimethoxy-1,3,5-triazin-2-yl α-glycoside using DMTMM in the presence of a base catalyst such as 2,6-lutidine in an aqueous solution.40,41 We speculated that the large excess of the azido-PEG-NH2 might serve as the base to promote the formation of the glycoside. Interestingly, the α-glycoside (2) could be readily converted to the corresponding free reducing N-glycan by treatment with 0.1% TFA overnight at rt. Recently, Manabe and co-workers have reported that coupling of the N-glycan (1) and amine NH2–(CH2CH2O)3CH2CH2–N3 using DMTMM as the dehydrating agent under the previously described conditions failed to give the expected coupling product, but the use of a phosphonium salt-based reagent, (benzotriazol-1-yloxy)tris(pyrrolidino)phosphonium hexafluorophosphate (PyBOP), provided the desired sialic acid and amine coupling product in 57% yield.39 The resulting azide-tagged antibody has been successfully used for making antibody–drug conjugates.42
Scheme 1.

Synthesis of Functionalized N-Glycans Using DMTMM Coupling
To optimize the amide coupling reaction without the α-glycoside formation, we reasoned that the pH of the reaction, the reaction temperature, and the quantity of the azido-PEG-NH2 and DMTMM might be important factors to modulate the outcome. In search of the reaction conditions, including varying the pH (pH 5–10), the amount of the azido-PEG-NH2 (3–20 equiv), and the temperature (30–60 °C), we found that the pH was critical, and basic conditions led to significant formation of the byproduct, the 4,6-dimethoxy-1,3,5-triazin-2-yl α-glycoside (2). We observed that keeping slightly acidic conditions (pH = 5.5), combined with the use of a significantly reduced amount of azido-PEG-NH2 and DMTMM and an elevated temperature, gave the best coupling yield of the tagged glycan (3) without formation of the α-glycoside (2). Thus, treatment of the sialoglycan (1) with 3 equiv of azido-PEG-NH2 and a total of 10 equiv of DMTMM (added in two portions) in an aqueous buffer (pH 5.5) at 50 °C for 6 h gave the desired amide product (3) in a 92% yield after G15 gel filtration (Scheme 1). The functionalized glycans were readily converted to the corresponding glycan oxazolines in water by using 2-chloro-1,3-dimethylimidazolinium chloride (DMC) as the dehydrating reagent, following the previously reported procedures.40
A “One-Pot” and Optimized Synthesis of the Functionalized N-Glycan Oxazolines.
The chemoenzymatic Fc glycan-specific conjugation requires the synthesis of the functionalized glycan oxazolines as the key donor substrate.21 As the DMTMM-catalyzed amine coupling under the above-described conditions gave an almost quantitative transformation to the azide-tagged N-glycan, we attempted to develop a strategy to directly synthesize the azide-tagged glycan oxazoline from the corresponding sialylated N-glycans in a “one-pot” manner, which are key donor substrates for the endoglycosynthase mutant-catalyzed glycan remodeling and conjugation of antibodies.21,22,32,33 We sought to combine the N-glycan functionalization and oxazoline formation in a one-pot manner by tuning the reaction conditions, including the pH, temperature, and reagents (Scheme 2). The DMTMM-catalyzed amide formation reaction was carried out first under slightly acidic conditions (pH 5.5) at 50 °C, and the reaction was monitored by HPLC until its completion within 6 h. Then, the reaction mixture was cooled on ice, and TEA (70 equiv) and DMC (30 equiv) were added. The formation of the corresponding sugar oxazoline product was complete within 30 min at 0 °C. The oxazoline was purified with P2 size exclusion chromatography to remove all other smaller molecules, affording the pure azide-tagged glycan oxazoline (4a) in 86% isolated yield. Compared with the previous method, this facile one-pot functionalization-oxazoline formation procedure significantly simplified the protocol, resulting in a much-enhanced overall yield (Scheme 2). An attempt to use DMC/TEA under either acidic or basic conditions failed to provide the amine coupling product (data not shown). Thus, DMTMM appeared to be an appropriate dehydrating reagent to enable the “one-pot” conversions. Similarly, the cyclopropene- and norbornene-modified N-glycan oxazolines 4b and 4c were synthesized in a “one-pot” manner in excellent yields, which are two different partners for chemoselective click reactions (Scheme 2).
Scheme 2.

One-Pot Transformation of Free Sialoglycans into Functionalized Glycan Oxazolines
Site-Specific Fc Glycan Remodeling with the Functionalized Glycan Oxazolines Using Endo-S2/Endo-S2 Mutants.
We have previously reported the generation and use of Endo-S mutants for site-specific Fc glycan remodeling of intact antibodies with natural and azide-modified N-glycan oxazolines.32 The Endo-S D233Q mutant has been used for glycan remodeling followed by click reactions to produce antibody–drug conjugates.35-37,42 Since the Endo-S D233Q-catalyzed enzymatic glycan transfer is relatively slow, the reaction conditions should be optimized to minimize nonenzymatic side reactions.36,42 On the other hand, we have reported that the glycosynthase mutant (D184M) derived from Endo-S2, another antibody-specific endoglycosidase from Streptococcus pyogenes of serotype M49, demonstrates significantly enhanced glycosylation efficiency and a much reduced reaction time without detectable side reactions for antibody Fc-glycan remodeling.33,34,43 Thus, we examined the feasibility of the Endo-S2 D184 M mutant to transform the antibody with the functionalized glycan oxazolines, using trastuzumab (Herceptin), an antiepidermal growth factor receptor 2 (HER-2) therapeutic monoclonal antibody, as a model for testing the Fc glycan remodeling and antibody–drug conjugation. Thus, recombinant trastuzumab was treated with an immobilized wild-type Endo-S2 to give the deglycosylated antibody (Fucα1,6GlcNAc-trastuzumab, 6). The wild-type enzyme was readily removed by simple centrifugation after the reaction. It should be mentioned that since Endo-S2 is such an efficient endoglycosidase for Fc deglycosylation, the use of immobilized Endo-S2 for deglycosylation is important, as any trace amount of contamination of Endo-S2, e.g., from trace Fc-associated copurification in the protein A purification steps, could result in slow hydrolysis of the final product during transglycosylation and/or in storage.34 Fucα1,6GlcNAc-trastuzumab (6) was purified by affinity chromatography on a protein A column, the identity of which was verified by LC-ESI-MS analysis (Figure S1, Supporting Information).
The Endo-S2 D184 M-catalyzed transglycosylation of Fucα1,6GlcNAc-trastuzumab (6) with the respective functionalized glycan oxazoline (4a–c) was performed in a Tris buffer (100 mM pH 7.2) at 30 °C, and the reaction was monitored by LC-ESI-MS analysis of intact antibodies. We found that under the above conditions, only 20 mol equiv of the azide-glycan oxazoline (4a) per antibody and less than 0.5% (by weight) of the Endo-S2 mutant would be sufficient for achieving essentially quantitative glycosylation within 20 min to give the azide-tagged antibody (7a) (Scheme 3). The enzymatic reaction was equally efficient for the cyclopropene- and norbornene-modified glycan oxazolines (4b and 4c) to provide the cyclopropene- and norbornene-tagged antibodies (7b and 7c), respectively (Scheme 3). The final products were purified by affinity chromatography on a protein A column in an excellent isolated yield. The identity and homogeneity of the tagged antibodies (7a–7c) were confirmed by LC-ESI-MS analysis of both the intact antibodies and the Fc domains released by IdeS treatment (Figure 2). The observed molecular mass (deconvolution data) of 7a, 7b, and 7c was 151025, 150587, and 150630 Da, which matched well with their calculated value of 151022, 150589, and 150629 Da, respectively. In addition to the intact antibody analysis, LC-ESI-MS analysis of the monomeric Fc domain of 7a–7c released from IdeS treatment further confirmed the site selectivity and homogeneity of the final products (Figure 2). The raw LC-ESI-MS data were shown in Figures S2-S7 (Supporting Information). Notably, previous studies have reported that in the case of the Endo-S D233Q-catalyzed glycosylation of the GlcNAc(α1,6Fuc)-antibody (6), a relatively large excess of the glycan oxazolines (up to 100 equiv in multiple additions) and a large amount of the enzyme (up to 20% by weight) are required to drive the relatively slow reaction to completion, which leads to accumulation of nonenzymatically modified antibody products without optimization of the reaction conditions.36,42,44 To verify the difference in efficiency of the Endo-S D233Q- and Endo-S2 D184 M-catalyzed glycosylations with the selectively modified glycan oxazolines, we performed a side-by-side comparative analysis of the enzymatic glycosylation using the azide-modified glycan oxazoline (4a) as the donor substrate. We found that under optimized conditions (molar ratio of glycan oxazoline 4a to antibody 6, 20:1; antibody concentration 25 mg/mL; Tris buffer, pH 7.2; incubation at 30 °C), the Endo-S2 D184 M-catalyzed reaction gave essentially quantitative conversion to the expected glycosylated antibody product (7c) (M = 151024, Figure S8) within 20 min without any side reactions, while the Endo-S D233Q-catalyzed reaction required 80 min for completion. Interestingly, even in the case of the Endo-S D233Q mutant, we detected only a trace amount of nonenzymatic glycation, which resulted from an addition of an extra glycan moiety (M = 153601, Figure S8). This result was contradictory to the previously reported relatively slow reactions and the side reactions with the Endo-S D233Q mutant when a large amount of glycan oxazolines, longer reaction time, and higher pH were applied.36,42,44 Taken together, these results indicate the high efficiency of the Endo-S2 D184M-catalyzed glycosylation for introducing the functionalized glycans into intact antibodies.
Scheme 3.

Chemoenzymatic Glycan Remodeling Using the Endo-S2/Endo-S2 D184M Enzyme Pair for Site-Specific Introduction of Functional Tags in the Antibody
Figure 2.
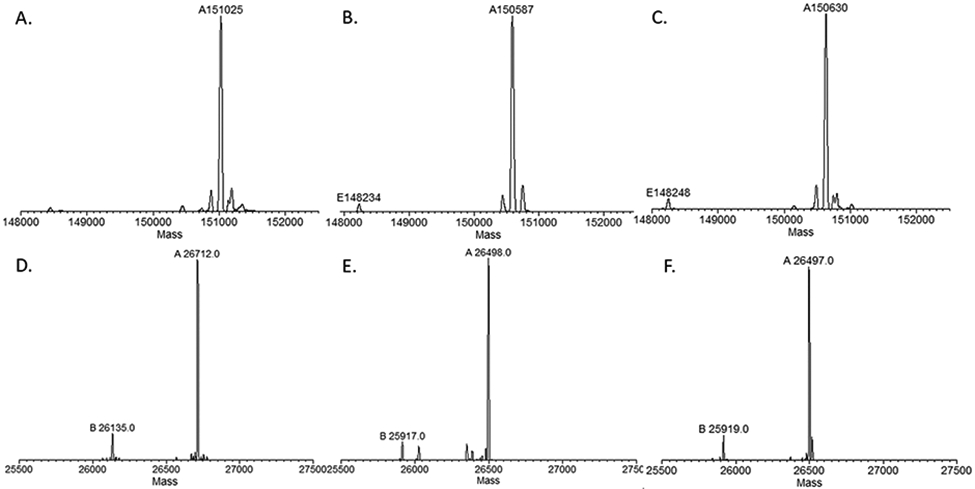
LC-ESI-MS analysis of the functionalized intact antibody (7a–7c) and the Fc domains released by IdeS treatment: A) the deconvoluted mass of intact antibody 7a; B) the deconvoluted mass of intact anibody 7b; C) the deconvoluted mass of intact antibody 7c; D) the deconvoluted mass of the Fc domain of antibody 7a; E) the deconvoluted mass of the Fc domain of antibody 7b; and F) the deconvoluted mass of the Fc domain of antibody 7c.
Synthesis of Homogeneous Antibody–Drug Conjugates via Click Reactions.
In this study, we selected monomethyl auristatin E (MMAE) as the payload for making the antibody–drug conjugates (ADCs). MMAE, coupled with the valine-citrulline cleavable linker, has been used as the payload in four FDA-approved ADCs.2,3 Also, ADCs based on auristatin and trastuzumab have been tested as being effective against T-DM1 resistant cell lines.45 The click reactions between the azide-tagged antibody and dibenzylcyclooctyne (DBCO) have been widely used to generate ADCs.28,37,42,46 Recently, the inverse electron demand Diels–Alder (iEDDA) reaction has also been applied to synthesize site-specific ADCs.47,48 Despite the impressive progress, a side-by-side comparison of these different click reactions in ADCs has not been performed. It is of great interest to see if the distinct click linkages will make a difference in the cancer cell killing performance of the resulting ADCs. To construct the antibody–drug conjugates with the three distinct click conjugations, we synthesized two functionalized MMAE derivatives, the DBCO-modified MMAE (8) and the tetrazine-modified MMAE (9), as distinct click partners for the strain-promoted alkyne–azide cycloaddition reaction and the inverse electron demand Diels–Alder reaction, respectively. A cathepsin B cleavable valine-citrulline linker was introduced in the construct with a self-immolation spacer.25,49 Then the DBCO or the tetrazine moiety was linked to MMAE to give the DBCO- and tetrazine-modified MMAE (8 and 9), respectively (see the Supporting Information).
The conjugation between the azide-tagged antibody (7a) and the DBCO-modified MMAE (8) was carried out in 30% DMSO at rt with a final concentration of the antibody at 2 mg/mL and 20 mol equiv of the payload per click handle being used (Scheme 4). The reaction was monitored by LC-ESI-MS, which indicated the completion of conjugation with 8 h to give ADC 10a. The conjugation between the cyclopropene- or norbornene-tagged antibody (7b or 7c) and the tetrazine-modified MMAE (9) was performed under the same conditions as described for the preparation of ADC 10a. We found that the reaction between the cyclopropene-antibody (7b) and the tetrazine-modified MMAE (9) went very fast, which took less than 4 h for completion to give the ADC (10b). This result was consistent with previous observations.47 On the other hand, the click reaction between the norbornene-tagged antibody (7c) and the tetrazine-modified MMAE (9) was relatively slow, which required 16 h to go to completion to give the conjugate (10c) (Scheme 4). The final products (10a, 10b, and 10c) were purified by protein A affinity chromatography, and their identity was confirmed by LC-ESI-MS analysis (Figure 3). The ESI-MS analysis of the intact ADCs (Figure 3A-C) showed that the observed deconvolution data matched well with the expected mass of the intact ADC and that each ADC carried 4 payloads, giving a drug-to-antibody ratio (DAR) of 4. The ESI-MS analysis of the monomeric Fc domain released by IdeS treatment of the intact ADCs (Figure 3D-F) further confirmed that the MMAE was site-specifically attached to the Fc domain of the antibody. The original data on the ESI-MS analysis of the intact antibodies and the Fc domains of ADCs 10a–10c were shown in Figures S9-S14 (Supporting Information). It should be mentioned that a minor peak corresponding to a loss of 762 Da from the intact antibody or the Fc domain was observed in the ESI-MS spectra (Figure 3). This species was presumably generated by fragmentation at the aminobenzyl carbamate moiety in the linker of the ADCs. Similar fragments have been observed in the ESI-MS analysis of ADCs with the same moiety in the linkers in a previous report.14 Finally, ESI-MS analysis of the protein backbone of the intact antibody and the Fc domain after PNGase F treatment to remove the modified Fc N-glycans further confirmed that there were no additional modifications of the protein backbone except the MMAE attachment to the Fc glycans (Figures S15-S17, Supporting Information). To verify if there was any aggregation of the synthetic antibody–drug conjugates, we performed size exclusion chromatography of the three ADCs (10a, 10b, and 10c). We found that ADC 10a did not have any aggregation, while ADCs 10b and 10c generated by the tetrazine-based click reaction showed about 8% aggregation product (Figure S18), indicating some difference in the stability of the respective conjugates. We also examined the serum stability of the three ADCs (10a, 10b, and 10c) using rat serum as a model system. We found that after incubation of the synthetic ADCs with the rat serum at 37 °C for 3 days, there was no payload coming off from the antibody conjugates as indicated by the LC-ESI-MS analysis of the antibodies and the conjugates (Figure S19). These results suggested that the antibody–drug conjugates constructed by the present method had a reasonable serum stability.
Scheme 4.

Synthesis of Homogeneous ADCs through Click Reactions
Figure 3.
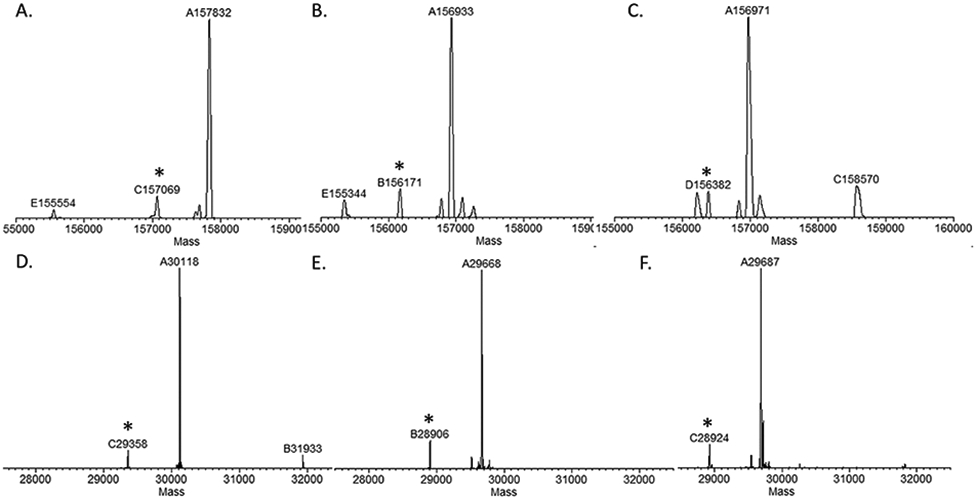
LC-ESI-MS analysis of the intact antibody–drug conjugates (10a–10c) and the Fc domains released by IdeS treatment: A) the deconvoluted mass of intact ADC 10a; B) the deconvoluted mass of intact ADC 10b; C) the deconvoluted mass of intact ADC 10c. D) the deconvoluted mass of the Fc domain of 10a; E) the deconvoluted mass of the Fc domain of 10b; and F) the deconvoluted mass of the Fc domain of 10c. Asterisked peaks indicate the ion fragments derived from the intact antibody or its Fc domain, which corresponds to a loss of 762 Da.
Evaluation of the in Vitro Cytotoxicity of the ADCs.
To compare the potency of the ADCs synthesized with different click reactions, we studied the in vitro cytotoxicity of ADCs with SK-BR-3 (high HER-2 expressing) and T47D (low HER-2 expressing) cells. For the SK-BR-3 cell line, our ADCs demonstrated a dose-dependent killing of the antigen-positive cells (Figure 4A). Meanwhile, the low HER-2 expressing T47D cells were insensitive to the ADCs up to 1 μg/mL (Figure 4B). These results suggest the antibody retains its high specificity on HER2 after all the modifications. The IC50 of the antibody–drug conjugates (10a, 10b, and 10c) against SK-BR-3 cells was measured as 13.9 ng/mL, 15.4 ng/mL, and 21.8 ng/mL, respectively (corresponding to 88 pM, 162 pM, and 138 pM, respectively). The data indicated that the azide–alkyne MMAE conjugate (10a) was a slightly better than the cyclopropene- or norbornene-tetrazine MMAE conjugates (10b and 10c) for cell killing. However, the IC50 data (100–200 pM) are quite comparable to those MMAE-based ADCs with similar DARs.25,42,45,47 The results from the present side-by-side comparison study suggest that the ADCs generated by the two different types of click reactions (SPAAC vs iEDDA reactions) are equally efficient for target cell killing. Taken together, the results suggest that the one-pot synthesis of functionalized glycan oxazolines coupled with the efficient Endo-S2 D184M-catalyzed Fc glycan remodeling and click drug conjugation provides a general and efficient approach to producing structurally well-defined, homogeneous antibody–drug conjugates with high potency.
Figure 4.

Cytotoxicity assays of the antibody–drug conjugates with the SK-BR-3 (HER2 overexpression) and the T47D (HER2 low expression) cancer cell lines. All assays were performed in triplicate.
CONCLUSION
An efficient, Fc glycan-mediated chemoenzymatic method for site-specific antibody–drug conjugation is described. This improved approach is enabled by the optimized synthesis of selectively modified glycan oxazolines from free sialoglycans in a one-pot manner and the use of the highly efficient endoglycosidase mutant (Endo-S2 D184M) for transferring the tagged glycans to provide the selectively tagged antibodies ready for click drug conjugation. The enhanced enzymatic activity of the Endo-S2 D184 M mutant over the previously used Endo-S D233Q mutant for transferring the selectively modified glycans permits the use of a significantly smaller amount of glycan oxazolines with a much shorter reaction time to complete the enzymatic reaction, thus minimizing the potential nonenzymatic side reactions. The homogeneous ADCs constructed by the present method showed excellent serum stability and demonstrated potent and selective cytotoxicity against Her2-overexpressing cancer cells. This improved method is also quite flexible for introducing different tags into an antibody, allowing site-specific payload conjugation with different click reactions to construct homogeneous antibody–drug conjugates.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH grants R01AI155716 and R01GM096973 to L.X.W.)
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.1c00314.
Detailed experimental procedures; synthesis of functionalized glycan oxazolines; synthesis of DBCO- and tetrazine-tagged MMAE; chemoenzymatic synthesis of antibody–drug conjugates; and copies of ESI-MS data and NMR spectra (PDF)
The authors declare the following competing financial interest(s): L.X.W. is the founder of GlycoT Therapeutics. All other authors declare no conflict of interest.
Contributor Information
Chong Ou, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Chao Li, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Roushu Zhang, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Qiang Yang, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Guanghui Zong, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Yuanwei Dai, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Rebecca L. Francis, Laboratory of Molecular Genetics and Immunology, The Rockefeller University, New York, New York 10065, United States
Stylianos Bournazos, Laboratory of Molecular Genetics and Immunology, The Rockefeller University, New York, New York 10065, United States.
Jeffrey V. Ravetch, Laboratory of Molecular Genetics and Immunology, The Rockefeller University, New York, New York 10065, United States
Lai-Xi Wang, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
REFERENCES
- (1).Chari RV; Miller ML; Widdison WC Antibody-drug conjugates: an emerging concept in cancer therapy. Angew. Chem., Int. Ed 2014, 53 (15), 3796–827. [DOI] [PubMed] [Google Scholar]
- (2).Walsh SJ; Bargh JD; Dannheim FM; Hanby AR; Seki H; Counsell AJ; Ou X; Fowler E; Ashman N; Takada Y; et al. Site-selective modification strategies in antibody-drug conjugates. Chem. Soc. Rev 2021, 50 (2), 1305–1353. [DOI] [PubMed] [Google Scholar]
- (3).do Pazo C; Nawaz K; Webster RM The oncology market for antibody-drug conjugates. Nat. Rev. Drug Discovery 2021, 20, 583. [DOI] [PubMed] [Google Scholar]
- (4).Drago JZ; Modi S; Chandarlapaty S Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol 2021, 18 (6), 327–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Beck A; Goetsch L; Dumontet C; Corvaia N Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discovery 2017, 16 (5), 315–337. [DOI] [PubMed] [Google Scholar]
- (6).Junutula JR; Raab H; Clark S; Bhakta S; Leipold DD; Weir S; Chen Y; Simpson M; Tsai SP; Dennis MS; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol 2008, 26 (8), 925–32. [DOI] [PubMed] [Google Scholar]
- (7).Shen BQ; Xu K; Liu L; Raab H; Bhakta S; Kenrick M; Parsons-Reponte KL; Tien J; Yu SF; Mai E; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol 2012, 30 (2), 184–9. [DOI] [PubMed] [Google Scholar]
- (8).Strop P; Delaria K; Foletti D; Witt JM; Hasa-Moreno A; Poulsen K; Casas MG; Dorywalska M; Farias S; Pios A; et al. Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat. Biotechnol 2015, 33 (7), 694–6. [DOI] [PubMed] [Google Scholar]
- (9).Axup JY; Bajjuri KM; Ritland M; Hutchins BM; Kim CH; Kazane SA; Halder R; Forsyth JS; Santidrian AF; Stafin K; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (40), 16101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).VanBrunt MP; Shanebeck K; Caldwell Z; Johnson J; Thompson P; Martin T; Dong H; Li G; Xu H; D’Hooge F; et al. Genetically Encoded Azide Containing Amino Acid in Mammalian Cells Enables Site-Specific Antibody-Drug Conjugates Using Click Cycloaddition Chemistry. Bioconjugate Chem. 2015, 26 (11), 2249–60. [DOI] [PubMed] [Google Scholar]
- (11).Nilchan N; Li X; Pedzisa L; Nanna AR; Roush WR; Rader C Dual-mechanistic antibody-drug conjugate via site-specific selenocysteine/cysteine conjugation. Antib Ther 2019, 2 (4), 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shinmi D; Taguchi E; Iwano J; Yamaguchi T; Masuda K; Enokizono J; Shiraishi Y One-Step Conjugation Method for Site-Specific Antibody-Drug Conjugates through Reactive Cysteine-Engineered Antibodies. Bioconjugate Chem. 2016, 27 (5), 1324–31. [DOI] [PubMed] [Google Scholar]
- (13).Dennler P; Chiotellis A; Fischer E; Bregeon D; Belmant C; Gauthier L; Lhospice F; Romagne F; Schibli R Trans-glutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconjugate Chem. 2014, 25 (3), 569–78. [DOI] [PubMed] [Google Scholar]
- (14).Anami Y; Xiong W; Gui X; Deng M; Zhang CC; Zhang N; An Z; Tsuchikama K Enzymatic conjugation using branched linkers for constructing homogeneous antibody-drug conjugates with high potency. Org. Biomol. Chem 2017, 15 (26), 5635–5642. [DOI] [PubMed] [Google Scholar]
- (15).Lin S; Yang X; Jia S; Weeks AM; Hornsby M; Lee PS; Nichiporuk RV; Iavarone AT; Wells JA; Toste FD; Chang CJ Redox-based reagents for chemoselective methionine bio-conjugation. Science 2017, 355 (6325), 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Casi G; Huguenin-Dezot N; Zuberbühler K; Scheuermann J; Neri D Site-Specific Traceless Coupling of Potent Cytotoxic Drugs to Recombinant Antibodies for Pharmacodelivery. J. Am. Chem. Soc 2012, 134 (13), 5887–5892. [DOI] [PubMed] [Google Scholar]
- (17).Zhang C; Dai P; Vinogradov AA; Gates ZP; Pentelute BL Site-Selective Cysteine-Cyclooctyne Conjugation. Angew. Chem., Int. Ed 2018, 57 (22), 6459–6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Forte N; Chudasama V; Baker JR Homogeneous antibody-drug conjugates via site-selective disulfide bridging. Drug Discovery Today: Technol. 2018, 30, 11–20. [DOI] [PubMed] [Google Scholar]
- (19).Badescu G; Bryant P; Bird M; Henseleit K; Swierkosz J; Parekh V; Tommasi R; Pawlisz E; Jurlewicz K; Farys M; et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjugate Chem. 2014, 25 (6), 1124–36. [DOI] [PubMed] [Google Scholar]
- (20).Schneider H; Deweid L; Avrutina O; Kolmar H Recent progress in transglutaminase-mediated assembly of antibody-drug conjugates. Anal. Biochem 2020, 595, 113615. [DOI] [PubMed] [Google Scholar]
- (21).Manabe S; Yamaguchi Y Antibody Glycoengineering and Homogeneous Antibody-Drug Conjugate Preparation. Chem. Rec 2021, 21, 1–11. [DOI] [PubMed] [Google Scholar]
- (22).Wang LX; Tong X; Li C; Giddens JP; Li T Glycoengineering of Antibodies for Modulating Functions. Annu. Rev. Biochem 2019, 88, 433–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Stan AC; Radu DL; Casares S; Bona CA; Brumeanu T-D Antineoplastic Efficacy of Doxorubicin Enzymatically Assembled on Galactose Residues of a Monoclonal Antibody Specific for the Carcinoembryonic Antigen. Cancer Res. 1999, 59 (1), 115–121. [PubMed] [Google Scholar]
- (24).Zhou Q; Stefano JE; Manning C; Kyazike J; Chen B; Gianolio DA; Park A; Busch M; Bird J; Zheng X; et al. Site-specific antibody-drug conjugation through glycoengineering. Bioconjugate Chem. 2014, 25 (3), 510–20. [DOI] [PubMed] [Google Scholar]
- (25).Faridoon; Shi W; Qin K; Tang Y; Li M; Guan D; Tian X; Jiang B; Dong J; Tang F; Huang W New linker structures applied in glycosite-specific antibody drug conjugates. Org. Chem. Front 2019, 6 (17), 3144–3149. [Google Scholar]
- (26).Zhu Z; Ramakrishnan B; Li J; Wang Y; Feng Y; Prabakaran P; Colantonio S; Dyba MA; Qasba PK; Dimitrov DS Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar. MAbs 2014, 6 (5), 1190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).van Geel R; Wijdeven MA; Heesbeen R; Verkade JM; Wasiel AA; van Berkel SS; van Delft FL Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody-Drug Conjugates. Bioconjugate Chem. 2015, 26 (11), 2233–42. [DOI] [PubMed] [Google Scholar]
- (28).Li X; Fang T; Boons GJ Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew. Chem., Int. Ed 2014, 53 (28), 7179–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Okeley NM; Toki BE; Zhang X; Jeffrey SC; Burke PJ; Alley SC; Senter PD Metabolic engineering of monoclonal antibody carbohydrates for antibody-drug conjugation. Bioconjugate Chem. 2013, 24 (10), 1650–5. [DOI] [PubMed] [Google Scholar]
- (30).Li C; Wang LX Chemoenzymatic Methods for the Synthesis of Glycoproteins. Chem. Rev 2018, 118 (17), 8359–8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Fairbanks AJ The ENGases: versatile biocatalysts for the production of homogeneous N-linked glycopeptides and glycoproteins. Chem. Soc. Rev 2017, 46 (16), 5128–5146. [DOI] [PubMed] [Google Scholar]
- (32).Huang W; Giddens J; Fan SQ; Toonstra C; Wang LX Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J. Am. Chem. Soc 2012, 134 (29), 12308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Li T; Tong X; Yang Q; Giddens JP; Wang LX Glycosynthase Mutants of Endoglycosidase S2 Show Potent Trans-glycosylation Activity and Remarkably Relaxed Substrate Specificity for Antibody Glycosylation Remodeling. J. Biol. Chem 2016, 291 (32), 16508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Li T; Li C; Quan DN; Bentley WE; Wang LX Site-specific immobilization of endoglycosidases for streamlined chemoenzymatic glycan remodeling of antibodies. Carbohydr. Res 2018, 458–459, 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tang F; Wang LX; Huang W Chemoenzymatic synthesis of glycoengineered IgG antibodies and glycosite-specific antibody-drug conjugates. Nat. Protoc 2017, 12 (8), 1702–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Parsons TB; Struwe WB; Gault J; Yamamoto K; Taylor TA; Raj R; Wals K; Mohammed S; Robinson CV; Benesch JL; Davis BG Optimal Synthetic Glycosylation of a Therapeutic Antibody. Angew. Chem., Int. Ed 2016, 55 (7), 2361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tang F; Yang Y; Tang Y; Tang S; Yang L; Sun B; Jiang B; Dong J; Liu H; Huang M; et al. One-pot N-glycosylation remodeling of IgG with non-natural sialylglycopeptides enables glycosite-specific and dual-payload antibody-drug conjugates. Org. Biomol. Chem 2016, 14 (40), 9501–9518. [DOI] [PubMed] [Google Scholar]
- (38).Sianturi J; Manabe Y; Li HS; Chiu LT; Chang TC; Tokunaga K; Kabayama K; Tanemura M; Takamatsu S; Miyoshi E; et al. Development of alpha-Gal-Antibody Conjugates to Increase Immune Response by Recruiting Natural Antibodies. Angew. Chem., Int. Ed 2019, 58 (14), 4526–4530. [DOI] [PubMed] [Google Scholar]
- (39).Manabe S; Abe J; Ito Y Amide bond formation of sialic acid in oligosaccharide without protecting group. Heterocycles 2018, 97, 1203–1209. [Google Scholar]
- (40).Noguchi M; Tanaka T; Gyakushi H; Kobayashi A; Shoda S Efficient synthesis of sugar oxazolines from unprotected N-acetyl-2-amino sugars by using chloroformamidinium reagent in water. J. Org. Chem 2009, 74 (5), 2210–2. [DOI] [PubMed] [Google Scholar]
- (41).Noguchi M; Nakamura M; Ohno A; Tanaka T; Kobayashi A; Ishihara M; Fujita M; Tsuchida A; Mizuno M; Shoda S A dimethoxytriazine type glycosyl donor enables a facile chemoenzymatic route toward alpha-linked N-acetylglucosaminyl-galactose disaccharide unit from gastric mucin. Chem. Commun. (Cambridge, U. K.) 2012, 48 (45), 5560–2. [DOI] [PubMed] [Google Scholar]
- (42).Manabe S; Yamaguchi Y; Matsumoto K; Fuchigami H; Kawase T; Hirose K; Mitani A; Sumiyoshi W; Kinoshita T; Abe J; et al. Characterization of Antibody Products Obtained through Enzymatic and Nonenzymatic Glycosylation Reactions with a Glycan Oxazoline and Preparation of a Homogeneous Antibody-Drug Conjugate via Fc N-Glycan. Bioconjugate Chem. 2019, 30 (5), 1343–1355. [DOI] [PubMed] [Google Scholar]
- (43).Li T; DiLillo DJ; Bournazos S; Giddens JP; Ravetch JV; Wang LX Modulating IgG effector function by Fc glycan engineering. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (13), 3485–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Iwamoto M; Sekiguchi Y; Nakamura K; Kawaguchi Y; Honda T; Hasegawa J Generation of efficient mutants of endoglycosidase from Streptococcus pyogenes and their application in a novel one-pot transglycosylation reaction for antibody modification. PLoS One 2018, 13 (2), No. e0193534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Loganzo F; Tan X; Sung M; Jin G; Myers JS; Melamud E; Wang F; Diesl V; Follettie MT; Musto S; et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol. Cancer Ther 2015, 14 (4), 952–63. [DOI] [PubMed] [Google Scholar]
- (46).Chio TI; Gu H; Mukherjee K; Tumey LN; Bane SL Site-Specific Bioconjugation and Multi-Bioorthogonal Labeling via Rapid Formation of a Boron-Nitrogen Heterocycle. Bioconjugate Chem. 2019, 30 (5), 1554–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Oller-Salvia B; Kym G; Chin JW Rapid and Efficient Generation of Stable Antibody-Drug Conjugates via an Encoded Cyclopropene and an Inverse-Electron-Demand Diels-Alder Reaction. Angew. Chem., Int. Ed 2018, 57 (11), 2831–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Walker JA; Bohn JJ; Ledesma F; Sorkin MR; Kabaria SR; Thornlow DN; Alabi CA Substrate Design Enables Heterobifunctional, Dual "Click" Antibody Modification via Microbial Transglutaminase. Bioconjugate Chem. 2019, 30 (9), 2452–2457. [DOI] [PubMed] [Google Scholar]
- (49).Doronina SO; Toki BE; Torgov MY; Mendelsohn BA; Cerveny CG; Chace DF; DeBlanc RL; Gearing RP; Bovee TD; Siegall CB; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol 2003, 21 (7), 778–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.