

Abstract
Precise pH measurements in the immediate environment of receptors is essential for elucidating the mechanisms through which local pH changes associated with diseased phenotypes manifest into aberrant receptor function. However, current pH sensors lack the ability to localize and target specific receptor molecules required to make these measurements. Herein we present the Litmus-body, our recombinant protein-based pH sensor, which through fusion to an IgG nanobody is capable of piggybacking on IgG antibodies for molecular targeting to specific proteins on the cell surface. By normalizing a pH-dependent green fluorescent protein to a long-Stokes shift red fluorophore or fluorescent protein, we readily report pH independent of sensor concentration using a single 488-nm excitation. Our Litmus-body showed excellent responsiveness in solution, with a greater than 50-fold change across the regime of physiological pH. The sensor was further validated for use on live cells and shown to be specific to the protein of interest. In complex with our Litmus-body, Cetuximab therapeutic antibody retained its functionality in binding and inhibiting ligand interaction of its target epidermal growth factor receptor (EGFR), triggering receptor-mediated endocytosis that allowed tracking of local pH from the cell surface through the endocytic pathway.
Keywords: pH, biosensor, cancer, nanobody, endocytosis, antibody-drug conjugate, glycocalyx
Graphical Abstract
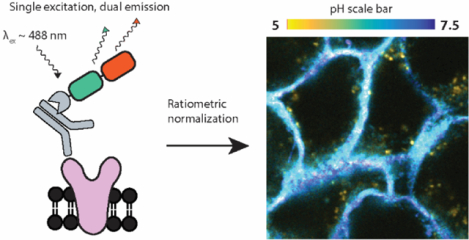
Acidification of the extracellular microenvironment is a hallmark of cancer progression1,2. In response to an increased anabolic demand associated with uncontrolled proliferation, cancer cells upregulate glycolysis and consequently overproduce protons intracellularly3. This excess of protons is then expelled to maintain cellular homeostasis, lowering the pH of the extracellular space. The consequences of extracellular acidosis are diverse and profound: for example, mutant receptors may become permanently activated in a low pH environment4, and acid-adapted cells show a proclivity towards a more aggressive phenotype5. While these effects have been studied in the context of the bulk extracellular pH6,7, work towards elucidating the precise relationship between localized acidity and aberrant cellular function has been limited by the ability to measure pH at precise locations.
Notably, this localized acidity is further exaggerated at the cancer cell surface8. It is suggested that cancer-associated proton secretion creates a concentration gradient of protons that is highest at the cell membrane9,10. In addition, negatively charged residues in the glycocalyx, a carbohydrate-enriched cell coat near the pericellular surface, have also been predicted to accumulate protons that lower the local pH11. In concert with cancer-associated acidosis, the content of anionic moieties in the glycocalyx has been shown to increase12, which could act synergistically with proton secretion to concentrate protons in the vicinity of cell surface receptors. These factors may lead to heterogeneity in the local pH that receptors experience, significantly impacting their functionality. Although cell surface pH can be determined by recently developed pH sensitive fluorescent dyes conjugated to pH-low insertion peptides8 and cell penetrating peptides13, there remains an unmet need in the development of molecularly targeted sensors capable of reporting on the immediate environment of receptors.
Antibody-drug conjugates (ADCs) that bind cell surface receptors would also benefit from targeted pH sensors. ADCs deliver their cytotoxic drugs intracellularly once trafficked into the endocytic pathway, which displays differential compartmental pH that can be crucial for ADC payload delivery14–17. For example, ADCs with acid-labile linkers are designed to release their payload in low pH compartments, such as lysosomes. Furthermore, ADCs with different linker technologies can release payload at different points of the endocytic pathway. Targeted pH sensors capable of specifically binding to ADCs and monitor their endocytic progression could therefore act as an efficient screening tool for the rapidly growing libraries of potential ADC therapeutics18,19.
Recently, nanobodies, or single-domain antibodies, have emerged as attractive option for specific binding and localization to primary antibodies or other molecular epitopes of interest20. For instance, Pleiner et al. have successfully expressed and validated a library for targeting rabbit IgG and all mouse IgG subclasses21. Their small size (~13 kDa), ease of labelling and consistent behaviour in recombinant protein fusions make nanobodies well-suited for fluorescence imaging21–23.
Fluorescent protein-based biosensors are versatile tools that can be fused to nanobodies for specific targeting24. Through extensive protein engineering, derivatives of the green fluorescent protein (GFP) from Aequorea victoria have been selected for pH dependent fluorescence25–28. In particular, pHluorins have been widely used as genetically-encoded sensors for tracking the pH of intracellular compartments25,27. Notably, the bright 488-nm excitable superecliptic pHluorin (SEP) variant is a pH sensor that shows a ~ 50-fold signal change between pH 5.5 and 7.5, making it ideal for applications in physiological conditions. One major drawback is the inability to distinguish pH-dependent fluorescence changes from the local variations in its concentration. Without signal normalization, studies using SEP as a pH sensor have remained qualitative25.
On the other hand, the ratiometric variants of pHluorin exhibit two pH-sensitive excitation peaks that can be normalized against each other25. This feature is an important advantage over the attributes of SEP as it allows for pH quantification independent of sensor concentration. However, the utility of pHluorin and its fluorescence enhanced variant, pHluorin2, suffers from exposing cells to phototoxic UV light and a limited dynamic range of only ~3-fold signal change in physiological conditions25,29. Recently developed ratiometric pH sensors, pHlameleons and pHLemon, have circumvented these shortcomings by tandemly fusing a highly pH sensitive yellow-shifted derivative of GFP to a second fluorescent protein for quantitative pH imaging30,31. However, absolute quantification with fluorescence resonance energy transfer (FRET) sensors typically requires careful correction for spectral bleed-through or sophisticated instrumentation for direct measurement of donor lifetime.
Consequently, we aim to take advantage of the superior pH sensitivity of SEP for pH quantification by normalizing its response against a second fluorophore that displays a large-Stokes shift (LSS). Adopting this strategy could potentially allow the pH response of SEP to be normalized by a single-wavelength co-excitation at 488 nm which, in addition to absolute signal quantification, avoids the photocytotoxicity conferred by UV light excitation. The use of SEP in conjunction with LSS red fluorophores has the added benefit of allowing extra fluorophores to be excited by 594 nm and 633 nm confocal laser lines for multicolor imaging.
Cell surface application of genetically encoded sensors requires their trafficking through cellular secretory pathways. Inevitably, this introduces variabilities in the folding, maturation, oxidation and possible aberrant disulphide bond formation of the sensor proteins that make calibration and precise quantification highly challenging in individual cells and across different cell types32. Recombinant products that can be exogenously applied to the cell surface and be calibrated in the bulk solution would bypass this difficulty.
In this work, we describe the Litmus-body, a tandem protein fusion that incorporates an IgG-specific nanobody and an SEP-based sensor that can normalize its pH response to LSS fluorophores with a single-wavelength excitation. We show here that, as a proof-of-principle, the Litmus-body can be successfully targeted to IgG antibodies and provide localized pH measurements in the vicinity of specific cell surface components, as well as following their transit through the endocytic pathway.
EXPERIMENTAL SECTION
Antibodies and reagents.
The following antibodies were used for immunostaining: mouse anti-human Mucin-1 (Muc1; CD227) monoclonal antibody (555925; BD Biosciences), goat anti-mouse Alexa Fluor 647 (A-21236; Thermo Fisher Scientific) and mouse anti-human epidermal growth factor receptor (EGFR) antibody (225/Cetuximab, MA5–12880; Thermo Fisher Scientific). Biotinylated EGF (EGF-biotin; E3477; Thermo Fisher) and streptavidin Alexa Fluor 647 were used to monitor endocytosis (S21374; Thermo Fisher). α-GFP nanobody (gt-250; Chromotek) was labelled with AFDye 647-NHS Ester (Fluoroprobes) to make 1 mg/mL stocks of AFDye 647-α-GFP nanobody. Doxycycline (sc-204734; Santa Cruz) was used for human cell culture induction, and IPTG (14213-261; IBI Scientific) was used for bacterial culture induction. Kanamycin sulfate (420311; MilliporeSigma) was used for bacterial culture selection. Hoescht 33342 (H1399; Thermo Fisher Scientific) was used for nuclear staining. Normal goat serum (NGS; S-1000; Vector Laboratories) was used as a blocking agent. The following buffers were prepared: 2.5X Ni-NTA binding buffer (375 mM NaCl, 125 mM K2HPO4, 25 mM Tris pH 8.5, 25 mM imidazole), Ni-NTA wash buffer (300 mM NaCl, 50 mM K2HPO4, 20 mM imidazole), Ni-NTA equilibration buffer (300 mM NaCl, 50 mM K2HPO4, 10 mM imidazole), Ni-NTA elution buffer (300 mM NaCl, 50 mM K2HPO4, 250 mM imidazole), and maleimide labelling buffer (MLB; 100 mM K2HPO4, 150 mM NaCl, 1 mM EDTA, 250 mM sucrose). Unless otherwise stated, all chemicals were purchased from MilliporeSigma, cell culture reagents were purchased from Thermo Fisher Scientific and enzymes for molecular cloning were purchased from New England Biolabs (NEB).
Construct generation.
Full cDNA sequences of the sensors used in this study are available in the Supplemental Materials. A dsDNA oligo encoding a cysteine-free SEP (cfSEP) engineered with an additional C-terminal surface cysteine (IDT), using NEB HiFi Assembly, was inserted into a BamHI-HF and NcoI-HF linearized pTP1112 vector (generously provided by Dirk Görlich: Addgene plasmid #104158)21. pTP1112 encodes an N-terminal His14-bdNEDD8 tagged anti-mouse IgG1 Fc specific TP1107 nanobody with an ectopic C-terminal cysteine. This terminal cysteine was removed and replaced with a Gly-Gly-Gly-Gly-Ser flexible linker, to ensure that downstream cysteine-maleimide labelling only occurred at the cfSEP C-terminal cysteine and to improve folding of the fusion protein. These steps generated a His14-bdNEDD8-TP1107-cfSEP construct.
mCyRFP1 was extracted from a custom mMaroon-mCyRFP1 cDNA optimized for bacterial expression (IDT) using the Q5 Hot Start High-Fidelity Master Mix (NEB) with 5’-ATGAACTGTACAAAGGAGGAGGCGGTAGCATGGTTAGTAAAGGCGAAGAAC-3’ (forward) and 5’-CCAAGCTCAGCTAAAGCTTATTTATACAGTTCATCCATGC-3’ (reverse). The His14-bdNEDD8-TP1107-cfSEP construct was linearized with Gibson Assembly compatible ends (forward: 5’-TAAGCTTTAGCTGAGCTTGGAC-3’,and reverse 5’-CATGCTACCGCCTCCTCCTTTGTACAGTTCATCCATG-3’. Afterwards, the linear fragments were combined via NEB HiFi Assembly to form a His14-bdNEDD8-TP1107-cfSEP-mCyRFP1 construct.
Single fluorescent proteins were generated via Q5 site-directed deletion of the His14-bdNEDD8-TP1107-cfSEPm-CyRFP1 construct. bdNEDD8, TP1107 nanobody, and the unneeded fluorescent protein were deleted using the following primer pairs: 5’-ATGGTTAGTAAAGGCGAAGAACTGATT-3’ (forward) with 5’-TGATCCGCCGGTATGGTGATGACT-3’ (reverse) to isolate a His14-mCyRFP construct, and 5’-ATGGTGAGTAAAGGAGAAGAACTTTTCACTGG-3’ (forward) with 5’-TGATCCGCCGGTATGGTGATGACT-3’ (reverse) to isolate a His14-cfSEP construct.
His14-SEP was made by a two-fold site-directed mutagenesis of the His14-cfSEP construct in order to generate linear fragments compatible for NEB HiFi Assembly. The S48C mutation was generated using 5’-CTTACCCTTAAATTTATTTGCACTACTGGAAAACTACC-3’ (forward) and 5’-TGATCTGGGTATCTTGAAAAGCATTGAACACCATAAGT-3’, and the M70C mutation was generated with 5’-ACTTATGGTGTTCAATGCTTTTCAAGATACCCAGATCA-3’ (forward) and 5’-GGTAGTTTTCCAGTAGTGCAAATAAATTTAAGGGTAAG-3’ (reverse). The two linear strands were subsequently joined.
The gene encoding human EGFR was a generous gift from Joseph Loturco (Addgene plasmid # 40974)33. It was cloned into a custom tetracycline-inducible, transposon-based PiggyBac expression vector with an internal ribosome entry site (IRES) that expressed cytosolic mNeonGreen and contained a second cassette for bicistronic expression of the reverse tetracycline transactivator, rtTA-M2, and a neomycin resistance cassette. This vector was created by replacing copGFP with mNeonGreen in our previously published vector34.
Cell lines and cultures.
A431 and MCF10A cells were maintained with 100 U/mL Penicillin/Streptomycin at 37°C, 90% relative humidity, and 5% CO2. A previously described MCF10A cell line that stably expressed a doxycycline-inducible rtTA NeoR Mucin-1 (Muc1) deficient of cytoplasmic tail (dCT) was cultured in DMEM/F12 media supplemented with 5% horse serum (16050122; Thermo Fisher), 20 ng/mL EGF (Peprotech), 10 mg/mL insulin, 500 ng/mL hydrocortisone, and 100 ng/mL cholera toxin35. A431 cells (ATCC) were cultured in DMEM supplemented with 10% FBS (10437028; Thermo Fisher), 1 mM pyruvate, and 1X Glutamax.
CHO-S cells were cultured in FreeStyle™ CHO Expression Medium (12541013; ThermoFisher) supplemented with 40 mL of 100X-Glutamax (35050061; Thermo Fisher). Cells were maintained at 37°C and 8% CO2. CHO-S cells were co-transfected with the PiggyBac EGFR expression vector and a hyperactive transposase using polyethyleneimine (PEI) as described in Shurer et al34, and selected with G418 for stable expression.
Recombinant protein production and purification.
All recombinant proteins were expressed in chemically competent NiCo21 (DE3) E. coli (NEB). 5 mL precultures (LB containing 50 μg/ml kanamycin) were grown overnight at 37°C, 220 rpm and then diluted with fresh medium to 0.2 – 1 L, in baffled flasks at no more than a 1:10 media:container volume ratio, and grown to reach OD600 of just below 0.6. The cultures were induced by 0.5 mM IPTG overnight at 24°C, harvested, resuspended in B-PER (Thermo Fisher Scientific) and vortexed for cell lysis. The lysates were cleared by centrifugation at 10,000 g for 20 min at 4°C.
His14-tagged recombinant proteins were purified using immobilized metal affinity chromatography. Supernatant diluted into 1X Ni-NTA binding buffer was bound to equilibrated Ni-NTA resin for 20 min at 4°C, with end-over-end mixing. The resin was added to a spin column, washed thoroughly and incubated with the Ni-NTA elution buffer for 20 min at 4°C, with end-over-end mixing. Recombinant proteins were then eluted and buffer exchanged (fluorescent proteins to pH 7.4 PBS, and bdNEDP1 to 50 mM Tris-HCl, 300 mM NaCl, 250 mM sucrose, 10 mM DTT, pH 7.5) using Zeba 7K MWCO desalting columns or by overnight dialysis with 10K MWCO Snakeskin dialysis tubing. Eluted proteins were then sterile filtered and snap-frozen for long-term storage at −80°C. Nanobody-containing constructs were mixed with 0.1% w/v sodium azide prior to snap-freezing.
NEDD8-removal.
bdNEDP1 protease expressed from the pDG02583 construct (a gift from Dirk Görlich; Addgene plasmid # 104129) was used to remove bdNEDD836. Ni-NTA purified His14-bdNEDD8-tagged proteins were incubated with >500 nM of the protease for 2 h on ice. The protease-protein mixture was incubated with equilibrated Ni-NTA resin for 20 min and spun at 700 g to elute the cleaved protein while leaving the uncleaved protein and the protease bound to the column. The cleaved protein was then buffer exchanged using a Zeba 7K MWCO desalting column to pH 7.4 PBS, prior to filtration and snap-freezing.
ATTO490LS-maleimide labelling.
Cysteine-maleimide labelling was performed as previously described and all steps were kept on ice to protect internal cysteines from the labelling reaction23. Briefly, the engineered cfSEP C-terminal cysteine was reduced by 15 mM TCEP for 10 mins. TCEP was removed by buffer exchange to degassed MLB using a Zeba 7K MWCO desalting column (Thermo Fisher Scientific). ATTO490LS-maleimide (ATTO-TEC Gmbh) was added to the reduced TP1107-cfSEP at a 6:5 molar ratio and the reaction-mixture was brought to pH 7.5 with K2HPO4. The mixture was stirred on ice under nitrogen for 1.5 h. Excess ATTO490LS was removed through buffer exchange to MLB.
SDS-PAGE and western blotting.
Proteins were diluted with 4X NuPAGE LDS Sample Buffer (Thermo Fisher Scientific) and 50 mM DTT and heated at 95°C for 10 min. Proteins were subsequently separated by reducing SDS-PAGE on a 4–12% NuPAGE Bis-Tris pre-cast gradient gel (Thermo Fisher Scientific) at 200 V for 35 min, and transferred to PVDF membranes at 30 V for 1 h. Membranes were blocked with 3% BSA TBST at RT for 1 h, and incubated overnight at 4°C with 1:1,000 of the AFDye 647-α-GFP nanobody. Washed membranes were imaged on a ChemiDoc MP Imaging System (Bio-rad).
Photophysical properties.
Quantum yield was determined at 490 nm in PBS, pH 7.4, by acquiring integrated fluorescence (500 nm – 800 nm) in conjunction with absorbance values in a dilution series from A490 ~ 0.1, to minimize inner filtering effect, using fluorescein as a standard (Quantum yield = 0.925 at 0.1 M NaOH)37. Molar extinction coefficient was determined by measuring mature chromophore concentration under NaOH denaturing conditions. Absorbance at 450 nm was measured immediately after mixing proteins with equal volume of 2 M NaOH38,39. This assumed alkali-denatured chromophore exhibited extinction coefficient 44,000 M−1 cm−1 at 450 nm absorbance. Total protein concentration was determined at 280 nm absorbance. All absorbance was measured on a Cary 300 UV-VIS spectrometer (Agilent Technologies Inc.) and fluorescence spectra were recorded by a QM4 fluorimeter (Horiba Instruments/Photon Technology International). Brightness was calculated as the product between quantum yield and extinction coefficient.
Samples for photobleaching studies were prepared as described in Cranfill et al40. Briefly, 20% polyacrylamide-1 μM fluorescent protein gels buffered with 1X PBS were created by mixing fluorescent protein of interest, 30% acrylamide/bis-acrylamide (1610156; Bio-Rad), 10X PBS at pH 7.4 and ddH2O to a final volume of 250 μL. Gelation chambers were constructed by placing two 22×22mm coverslips approximately 1 cm apart on top of a microscope slide, and mounting a third coverslip atop the empty space between the other two. Polymerization was then initiated and catalyzed with the addition of 3 μL 10% ammonium persulfate and 0.5 μL TEMED. 75 μL of the gel mixture was immediately pipetted into the empty space in the pre-made gelation chambers. This was done via capillary action to ensure a uniform thickness of the samples. Photobleaching data was acquired by imaging the polymerized samples through the glass coverslip with an inverted epifluorescence microscope, using a 60X NA 1.2 water immersion objective (Ti-E, Nikon) and a custom laser excitation system41. A 488 nm excitation laser (Sapphire LP, Coherent) was directed onto the sample by a quad-band dichroic mirror (ZT405/488/561/647rpc, Chroma). The samples were excited continuously for 1,800 s and fluorescence emission was collected through 50 nm bandpass filters centered at 525 nm, in the case of cfSEP, or 595 nm, in the case of mCyRFP (ET525/50m and ET 595/50m, Chroma). Images were acquired at 30 s intervals from the start of excitation on a sCMOS camera (Zyla 4.2, Andor) operated with μManager software (Open Imaging)42. Following acquisition, all pixels in each exposure were averaged to obtain the time-dependent decay data and normalized to the intensity in the first exposure.
Fluorescence lifetimes were measured by time-correlated single photon counting (TCSPC). Sample solutions were excited by a 445 nm picosecond diode laser (BDL-445-SMC, Becker & Hickl GmbH) pulsed at a 20 MHz repetition rate. Fluorescence decay curves were collected from samples in 1 cm pathlength quartz cuvettes at 90 degrees to the 445 nm excitation using a R3809U-50 microchannel plate photomultiplier tube with a 25 ps transit time spread (Hamamatsu, Bridgewater, NJ). Excitation intensity was attenuated using a Glan-Thompson polarizer to keep the phospholuminescence detection rate less than 0.2% of the repetition rate to avoid photon pile-up. Data was acquired using a TCSPC module (SPC-830, Becker & Hickl GmbH) and fit to a bi-exponential decay using the SPCImage software (Becker & Hickl GmbH).
Solution pH response.
Universal buffer solutions were prepared according to the Carmody buffer series43. This involved mixing a master acid buffer (0.2 M Boric Acid, 0.05 M Citric Acid) and a master base buffer (0.1 M NaHPO4) at previously determined ratios to achieve approximate pH of interest. Fluorescent constructs were diluted to 100 nM in these buffers, and the pH of the buffer-protein solution was recorded using an Orion™ PerpHecT™ ROSS™ Combination pH Micro Electrode (Thermo Fisher Scientific). The buffer-protein solutions had their emission spectra measured using a Tecan 1000M Infinite plate reader at 490 nm excitation. Three sets of triplicates were recorded for each buffer-protein solution.
Curve fitting parameters for pH response.
To describe the sigmoidal response of sensor, the acid dissociation constant, pKa (pH at 50% of max response), and Hill coefficient, nH, were determined by least squares non-linear fitting of the normalized fluorescence response data to a four-parameter logistic model on GraphPad Prism (California USA) using the equation below, as described by Kneen et al44.
Maturation time-course.
Immediately after protease treatment and purification, as above, Litmus-body was buffer exchanged into PBS at pH 7.4 using a Zeba 7K MWCO Desalting Column. At each time point, 200 nM of the protein of interest was analyzed using a Spark 10M (Tecan).
Reversibility of pH sensing.
200 nM of Litmus-body was buffer exchanged between universal buffer at pH 7.35 and pH 9.75, twice over. At each buffer-exchange step, a portion of the solution was withdrawn and fluorescence spectra recorded using a Spark 10M (Tecan). Samples were tested for normality using a Shapiro-Wilk goodness of fit test and equal population variance with Levene’s test to validate the use of a two-tailed t-test on GraphPad Prism (California USA).
Cell binding assay.
Doxycycline-induced MCF10A Muc1dCT cells were fixed with 4% paraformaldehyde for 10 min at RT and blocked in PBS containing 5% NGS for 1 h at RT. All subsequent dilutions were performed in PBS containing 5% NGS. As a positive control, cells were incubated with the primary anti-Muc1 antibody at 1:1,000 dilution overnight at 4°C and detected using the secondary Alexa Fluor 647-labelled goat anti-mouse IgG at 1:1,000 dilution for 1 h at RT. To test Litmus-body as a secondary reagent, cells were incubated with the primary anti-Muc1 antibody at a 1:1,000 dilution overnight at 4°C and then incubated with 30 nM of the Litmus-body for 1 h at RT. For one-step immunostaining, Litmus-body was pre-incubated with the anti-Muc1 antibody at equimolar ratio overnight at 4°C. 30 nM of the pre-incubated IgG-Litmus-body complex was then applied to cells for 1 h at RT. For negative controls, cells were labelled with 30 nM Litmus-body for 1 h at RT in the absence of a primary IgG antibody. All samples were labelled with Hoechst at 1 μg/mL for 15 min. Cells were then imaged on a Zeiss 800 LSM microscope using a 20X air objective (NA 0.8).
CHO-S co-culture study.
EGFR IRES mNeonGreen CHO-S cells and wild-type CHO-S cells were mixed at a 1:1 ratio and incubated on ice in a 5% NGS, 0.1% sodium azide spiked FreeStyle™ CHO Expression Medium for 20 min to inhibit endocytosis. The cell mixture was subsequently incubated with 67 nM pre-coupled Cetuximab-Litmus-body for 20 min on ice, washed and resuspended in fresh, ice-cold media to remove unbound Litmus-body. The live cells were then imaged on a Zeiss 800 LSM microscope using a 20X air objective (NA 0.8).
Muc1 live cell imaging.
Mouse anti-human Muc1 monoclonal antibody and Litmus-body were mixed at equimolar ratio, 4°C, overnight, to form an IgG-Litmus-body complex. Doxycycline-induced MCF10A Muc1dCT cells were incubated at 37°C for 30 min with PBS containing 5% NGS and 0.1% sodium azide to inhibit endocytosis. Cells were then incubated further with 33 nM of the IgG-Litmus-body complex at 37°C for 30 min, washed and imaged in PBS containing 5% NGS and 0.1% sodium azide with pH adjusted to 6, 7.03 and 7.95 by an Orion™ PerpHecT™ ROSS™ Combination pH Micro Electrode (Thermo Fisher Scientific). Spectral imaging was performed using lambda mode, at 488 nm excitation and 9 nm spectral resolution, on a Zeiss LSM880 inverted confocal microscope with a 40X water objective (NA 1.1).
Quantification of lambda mode images.
Thresholding was applied to the 22 channel, 16-bit Lambda-stacked images based on the pixel intensity values of the cfSEP and mCyRFP1 emission channels (cfSEP channel wavelength: 513 nm, mCyRFP1 channel wavelength: 593 nm). Pixels below the threshold (750 AU) in either of the two channels were ignored for the calculation. Mean of the non-thresholded pixels was subsequently used for calculating the average spectra of the selected image subsets, as well as for normalizing the displayed stacks.
Endocytosis studies.
Mouse anti-human EGFR monoclonal antibody (225/Cetuximab) and Litmus-body were mixed at equimolar ratio, 4°C, overnight, to form a Cetuximab-Litmus-body complex. To generate a calibration curve, the Cetuximab-Litmus-body complex was diluted to a final concentration of 200 nM in PBS containing 5% NGS and adjusted to final pH values of 4, 5.04, 5.49, 6.09, 6.56, 7.01, 7.49, 7.91 using an Orion™ PerpHecT™ ROSS™ Combination pH Micro Electrode (Thermo Fisher Scientific). Cetuximab-Litmus-body fluorescence in each pH buffer was then acquired on a Zeiss LSM800 inverted confocal microscope using a 63 X water objective (NA 1.2). The microscope was configured to simultaneously scan for cfSEP and mCyRFP1 signal with 488 nm excitation. To minimize cross-talk, the emission bands were set to collect between 500 – 535 nm for cfSEP and 575 – 700 nm for mCyRFP1.
To quantify EGF binding and uptake, A431 were non-enzymatically detached using PBS with 10 mM EDTA and suspended in serum-free media supplemented with 25 mM HEPES and 0.5% BSA at 37°C for 30 min. Cells were spun down at 4°C, resuspended in ice cold serum-free media containing 0.5% BSA and either kept on ice (for the EGF-biotin or EGF only controls) or incubated with 67 nM of Cetuximab or Cetuximab-Litmus-body on ice for 15 min. Cells were then spiked with 200 ng/mL of EGF-biotin or EGF on ice for 30 min, spun down, resuspended in pre-warmed (37°C) serum-supplemented media and allowed to undergo endocytosis for 0, 3, 5, 10, or 15 min. Endocytosis was arrested by immediately transferring the cells to an ice-bath in addition to supplementing the media with 0.1% sodium azide. Cells were incubated with 1:1,000 streptavidin conjugated with Alexa Fluor 647 for 20 min, washed, and fluorescence analysed on a BD Accuri C6 flow cytometer. Flow cytometry data was analysed using FlowJo software. Median fluorescence was reported for the time course.
For intracellular trafficking experiments, A431 plated overnight at 10,000 cells/cm2 were chilled on ice for 1 h to slow down endocytosis, washed and incubated with 67 nM of the Cetuximab-Litmus-body complex in PBS containing 5% NGS at 37°C for 30 min. Cells were washed and allowed to undergo endocytosis for 1 h at 37°C. They were further washed, placed in a pH 7.5 PBS buffer containing 5% NGS, and imaged. Cells were then placed in a pH 5.2 PBS buffer containing 5% NGS and imaged a second time. Cell imaging was done on a Zeiss LSM800 inverted confocal microscope using a 63X water objective (NA 1.2) and an identical optical configuration was used for generating the calibration curve above.
RESULTS AND DISCUSSION
Litmus-body and expression.
We set out to create Litmus-body, a nanobody-sensor fusion, that can be specifically targeted to IgG antibodies to report on the local pH of cell surface components. Sensor fusion proteins were produced in E. coli for ease of culture and scaling. We took advantage of a highly soluble protease-cleavable tag previously reported by Frey and Görlich (bdNEDD8) to optimize expression of the nanobody component in our sensor36. Removable by its associated protease bdNEDP1, the bdNEDD8 tag has been shown to improve the cytoplasmic yield of nanobodies in E. coli (Supplemental Figure 1). To generate an IgG specific Litmus-body, we fused fluorescent sensor proteins to bdNEDD8-TP1107, an anti-mouse IgG Fc fragment nanobody, that allowed the pH sensor to be applied in a manner analogous to secondary antibodies upon cleavage of NEDD8 (Figure 1A).
Figure 1.
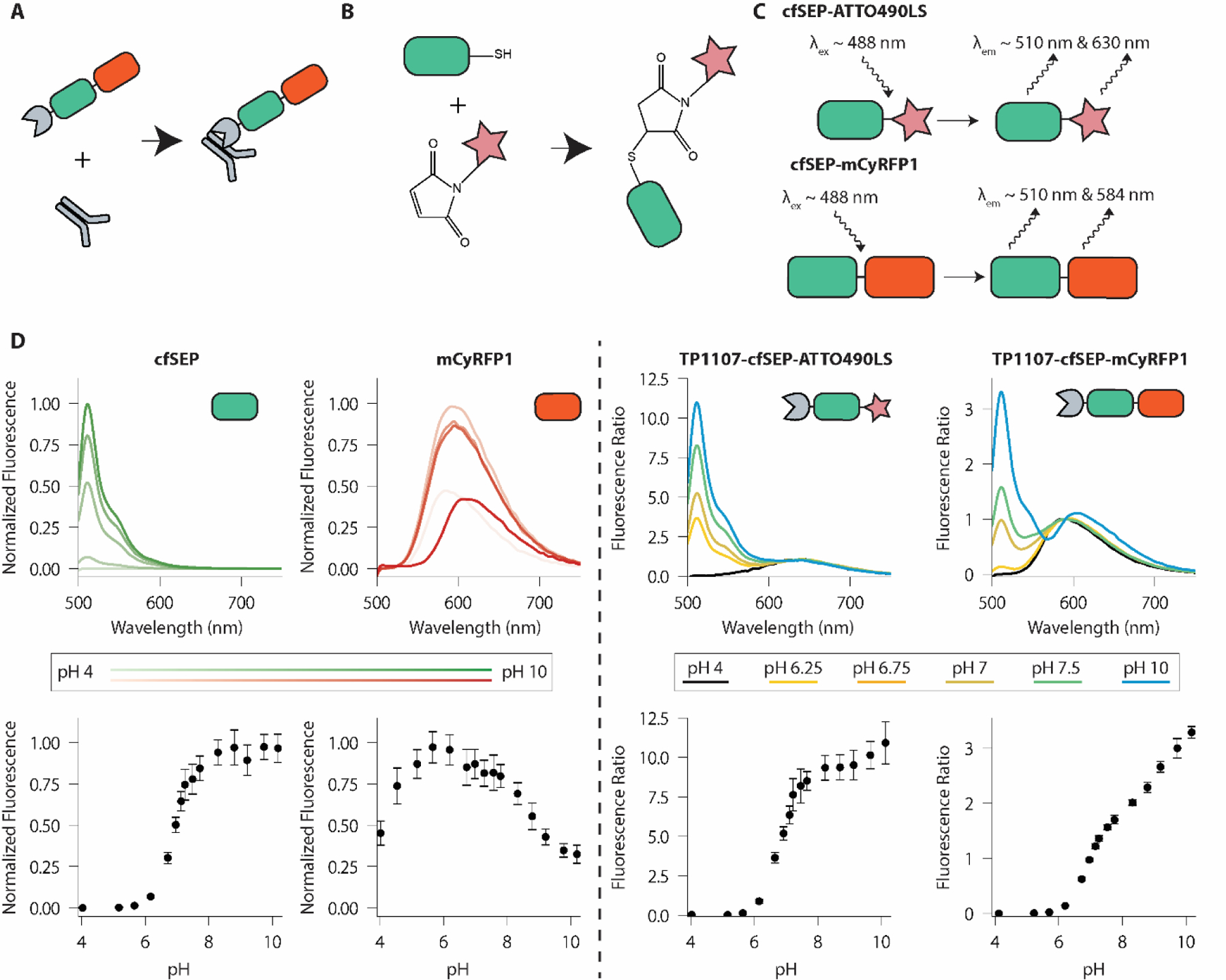
Litmus-body design and solution properties. A) Overview of the Litmus-body. A pH-sensitive GFP derivative (green) and red fluorophore (red) are fused to an anti-IgG nanobody (grey), allowing for targeting of primary antibodies. B) Reaction scheme for the conjugation of cfSEP to a maleimide-modified fluorophore. C) Scheme of the dual emission nature of the Litmus-body. ATTO490LS (top) or mCyRFP1 (bottom) is used as the long Stokes-shift red fluorescence emitter for ratiometric normalization of the green sensor emission. D) Fluorescence spectra and pH responsiveness of TP1107-cfSEP-mCyRFP1, TP1107-cfSEP-ATTO490LS, and their constituent fluorescent proteins acquired on a fluorescence microplate reader. Darker shading in cfSEP and mCyRFP1 spectra corresponds to an increase in pH. The fluorescence ratio for TP1107-cfSEP-mCyRFP1 was calculated as (I510 nm/I590 nm), while the fluorescence ratio for TP1107-cfSEP-ATTO490LS was calculated as (I510 nm/I630 nm). Error bars represent standard deviation of three independent experiments, each with triplicate.
Design and validation of a cysteine-free SEP engineered for maleimide chemistry.
To explore the potential use of a synthetic dye for pH signal normalization, we created a cysteine-free SEP (cfSEP) engineered with an ectopic C-terminal cysteine for site-specific maleimide-dye labelling (Figure 1B). Unlike conventional non-selective modifications, such as via N-hydroxysuccinimide ester, maleimide reaction on C-terminal cysteines has been demonstrated to be an effective protein labelling strategy that produces homogenous protein-dye-ratio conjugates, reduces batch-to-batch variation and does not alter binding properties of the modified protein23,45.
We introduced C48S/C70M mutations to remove the native cysteines in SEP and generated cfSEP. This was to avoid off-target cysteine-maleimide reactions and to ensure that only the engineered C-terminal cysteine would be available for dye-conjugation. C48S/C70M mutations have previously been validated and shown to maintain fluorescence in GFP derivatives46. cfSEP with the engineered C-terminal cysteine was tested for its pH response. It retained similar ideal pKa and responsiveness to SEP in the physiological range, while also displaying consistent photophysical properties to SEP (Table 1 and Supplemental Figure 2). These results pointed to the suitability of cfSEP as a substitute for SEP.
Table 1.
Photophysical properties of superecliptic pHluorin (SEP) proteins.
| Construct | Emission Maxima (nm) | Quantum Yield | Extinction Coefficient (mM−1cm−1) | Brightness (mM−1cm−1) | pKa | Hill Coefficient |
|---|---|---|---|---|---|---|
| SEP | 512 | 0.53 | 31 | 16.50 | 7.3 | 1.3 |
| cfSEP | 510 | 0.48 | 29 | 13.99 | 6.9 | 1.4 |
Expression and pH response of TP1107-cfSEP-ATTO490LS.
ATTO490LS-maleimide was conjugated to TP1107-cfSEP on the engineered C-terminal cysteine to generate a TP1107-cfSEP-ATTO490LS construct (Figure 1B). ATTO490LS is a long-stoke shift synthetic dye with peak excitation at 496 nm. These properties make ATTO490LS an attractive normalization partner for cfSEP as both fluorophores can be simultaneously excited and individually resolved (Figure 1C). We recorded the fluorescence response to solution pH of the TP1107-cfSEP-ATTO490LS construct (Figure 1D). The response of the conjugate at 510 nm emission (Supplemental Figure 3C) showed a similar pKa and responsiveness to the unconjugated cfSEP, indicating that the fusion and labelling of cfSEP did not have a negative impact on its functionality. pH response of TP1107-cfSEP-ATTO490LS was determined by normalizing cfSEP signal against ATTO490LS (Figure 1D). The construct readily detected pH changes in the physiological regime, exhibiting >50-fold signal enhancement from pH 5.5 to 7.5. We noted that the 630 nm emission of the construct increased at a rate greater than both the cfSEP and ATTO490LS individually (Supplemental Figure 3B, 3C), potentially indicating the occurrence of fluorescence resonance energy transfer (FRET) between cfSEP and ATTO490LS. We subsequently found that the fluorescence lifetime of the combined construct (~1.5 ns) was indeed lower than that of cfSEP (~2 ns). However, the occurrence of FRET between the fluorophores should not impede practical application of the sensor.
Expression and pH response of TP1107-cfSEP-mCyRFP1.
To avoid the need for chemical conjugation with a synthetic dye, we looked to monomeric cyan-excitable red fluorescent protein (mCyRFP1) instead as a normalization partner for cfSEP. mCyRFP1 is a long-stokes shift TagRFP derivative with broad-excitation around 500 nm38. Like ATTO490LS, mCyRFP1 can be co-excited with cfSEP while displaying an easily separable emission. The C-terminal cysteine on cfSEP was removed and replaced by a flexible GGGGS peptide linker to fuse to mCyRFP1. The 510 nm emission of the construct showed a pKa and responsiveness similar to the unconjugated cfSEP (Supplemental Figure 3D), suggesting that fusion to a red fluorophore did not impact cfSEP performance. mCyRFP1 showed a distinctive and consistent pH response, which was retained in the 590 nm emission of the fusion construct (Figure 1D, Supplemental Figure 3D).
The pH response of TP1107-cfSEP-mCyRFP1 was determined by normalizing cfSEP signal against mCyRFP1’s contribution (Figure 1D). TP1107-cfSEP-mCyRFP1 performed similarly to TP1107-cfSEP-ATTO490LS, also exhibiting >50-fold signal increase from pH 5.5 to 7.5. We found that the fluorescence ratio between cfSEP and mCyRFP1 in TP1107-cfSEP-mCyRFP1 was stable after the 6 h required to extract the product from E. coli, purify and carry out the protease processing. This indicated that the recombinant product can be applied immediately as a pH measuring tool after processing (Supplemental Figure 4). Moreover, the mCyRFP1 fused construct benefited from the biphasic pH responsiveness of mCyRFP1, allowing for additional ratiometric changes in sensor response above pH 8. This supplemented the overall responsiveness of the sensor upon the saturation of the cfSEP signal and extended the range of pH sensitivity in more basic environments (Figure 1D). Note that TP1107-cfSEP-mCyRFP1 displayed a minor yet irreversible altered pH response after exposure to highly basic environments (pH 9.75; Supplemental Figure 5). This is primarily attributed to the irreversible decrease in mCyRFP fluorescence at basic pH, likely indicating damages to the fluorescent protein or chromophore. Thus, some caution is required when using the cfSEP-mCyRFP sensor at highly basic pH, although these conditions are rarely encountered in cellular applications and tissue imaging.
Molecular targeting and response to environmental pH on live cells.
We moved forward with the TP1107-cfSEP-mCyRFP1 reagent for cellular testing and hereinafter referred to it as the Litmus-body. Litmus-body was targeted to specific cancer cell surface components by its ability to piggyback on IgG antibodies. Mucin-1 (Muc1) was selected as an epitope of interest given its key role in forming the glycocalyx on polarized epithelial cells and cancer cells35,47,48. Fixed Muc1-overexpressing cells stained with a primary anti-Muc1 IgG antibody could be similarly detected either by a secondary antibody or by using the IgG-specific Litmus-body as a secondary reagent (Figure 2A). We also allowed the Litmus-body to react to the primary anti-Muc1 IgG antibody, at an equimolar ratio, before cell treatment to optimize the incubation protocol for live cell applications. The IgG-Litmus-body complex exhibited a similarly high specific binding to the surface of Muc1-overexpressing cells (Figure 2B). In addition, Litmus-body in complex with an antibody that targeted epidermal growth factor receptor (EGFR), showed selective binding to EGFR-overexpressing cells but not EGFR-negative control cells in a co-culture model system (Supplementary Figure 6). Together these results indicated that the Litmus-body did not alter the specificity of IgGs and verified that the specificity of TP1107 binding to IgG antibodies was unaffected in the sensor construct. Litmus-body could therefore be used as a simple, one-step targeting reagent by pre-complexing with an IgG antibody.
Figure 2.
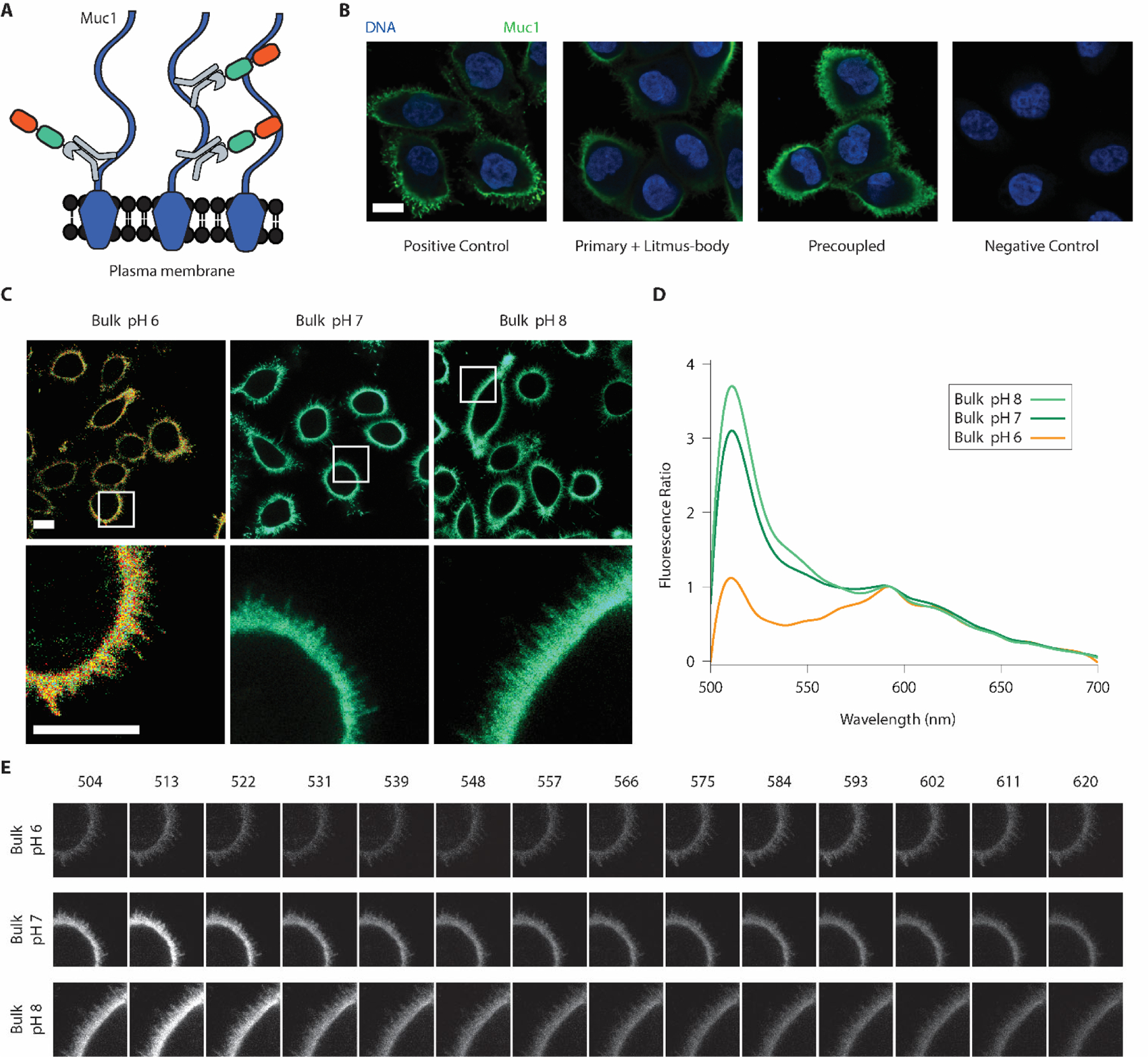
Litmus-bodies bind specifically to their target of interest and respond to bulk pH change on live cells. A) Scheme depicting the nature of binding between Muc1, and pre-coupled anti-Muc1 IgG-Litmus-body on Muc1 overexpressing MCF10A cells. Numerous epitopes for anti-Muc1 IgG are present on the cell surface, allowing for dense labelling. B) Representative immunofluorescence images from three independent experiments depicting the specificity of the Litmus-body as compared against conventional secondary antibody staining (Blue: DNA, and Green: 488 nm excitation) on paraformaldehyde-fixed Muc1-overexpressing MCF10A cells. Positive control depicts treatment with an anti-Muc1 primary IgG and Alexa Fluor 488 anti-IgG secondary antibody. Cells were treated with an anti-Muc1 primary antibody and the Litmus-body was used as a secondary reagent (Primary + Litmus-body). To test for a simple, one-step staining procedure, anti-Muc1 IgG antibody was pre-reacted with the Litmus-body to form an anti-Muc1 IgG-Litmus-body complex before applying to cells (Precoupled). C) Representative live cell images from three independent experiments of pre-coupled anti-Muc1 IgG-Litmus-body binding to Muc1 overexpressing MCF10A cells treated with 0.1% (w/v) sodium azide in various bulk pHs. The bottom row depicts select regions of interest (white boxes). D) Fluorescence spectra normalized against the mCyRFP1 emission (calculated as (intensity/intensity at 593 nm)), from the region of interest presented in C). E) Spectral stacks of the three regions of interests depicted in C), with the wavelengths listed in nm. Panels for each sample were normalized against their mean intensity at 593 nm. Scale bars: 10 μm.
The IgG-Litmus-body complex was targeted to the surface of live Muc1-overexpressing cells and tested for its response to environmental pH. We treated these cells with sodium azide to inhibit endocytosis and minimize the internalization of the pH sensor. cfSEP and mCyRFP1 were simultaneously excited with a 488 nm laser and spectrally imaged. Emission peaks were observed in normalized spectra around the expected 510 nm and 583 nm for cfSEP and mCyRFP1, respectively. Increasing bulk solution pH from 6 to 8 brought about a concurrent signal increase of cfSEP relative to mCyRFP1 on the cell surface (Figure 2C, 2D, 2E). These results suggested that the IgG specific Litmus-body could be readily targeted to the cell surface and may act as a suitable agent to report local pH perturbations on the surface of live cells.
pH imaging of therapeutic antibody Cetuximab and its cell surface receptor following endocytosis.
To demonstrate the practical relevance of the Litmus-body, we chose to follow the intracellular trafficking of a clinically important antibody, Cetuximab, and its target cell surface receptor, EGFR, an overexpressed drug target on multiple cancer types49. Cetuximab is a monoclonal anti-EGFR IgG antibody that blocks ligand binding to EGFR and induces receptor mediated endocytosis50. Thus, trafficking of Cetuximab provides an ideal testbed for validating our Litmus-body, given the diverse range of pH values that are expected in cellular compartments along the endocytic pathway51. Besides its use in treating multiple cancer types including skin, colorectal, head and neck52,53, Cetuximab is under active investigation as an ADC for cancer therapies54, although the pH values it encounters on the cell-surface and following endocytosis have not been reported to our knowledge.
To provide this information, we complexed the Litmus-body with Cetuximab and followed the intracellular trafficking of the Cetuximab-Litmus-body complex after its binding to EGFR (Figure 3A). Confocal imaging experiments were configured to simultaneously excite and collect the emission of both cfSEP and mCyRFP1 to avoid excessive photobleaching. As above, for simple one-step targeting, Litmus-body was first reacted to Cetuximab to form a Cetuximab-Litmus-body complex and a calibration curve was obtained on the confocal microscope by curve fitting of cfSEP/mCyRFP1 fluorescence ratio in solution (Figure 3B). Note this calibration curve yielded cfSEP/mCyRFP fluorescence ratios different to that obtained by a fluorescence plate reader, which confirmed a similar trend of Litmus-body response to pH (Figure 1D). This highlighted the need of calibrating the Litmus-body response against the specific instruments in use for accurate pH measurements.
Figure 3.
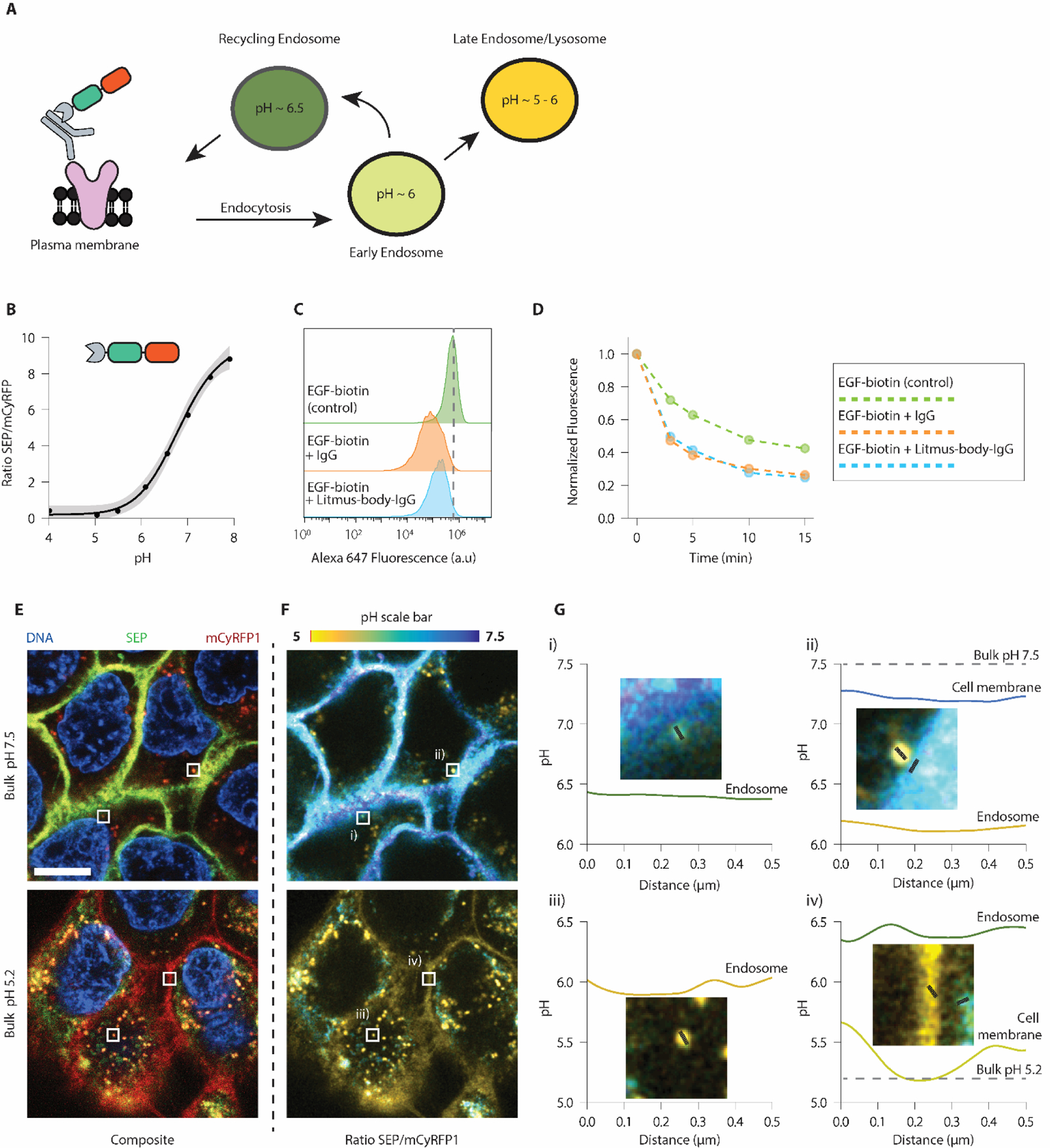
Litmus-body readily detects pH changes along the endocytic pathway. A) Scheme depicting the nature of binding between epidermal growth factor receptor (EGFR) and pre-coupled anti-EGFR IgG-Litmus-body on EGFR overexpressing A431s. Upon binding to EGFR, the IgG-Litmus-body complex is endocytosed, allowing it to monitor changes in pH along the endocytic pathway. B) Insolution calibration of the Litmus-body based on (ISEP/ImCyRFP1) at various bulk pH, acquired on a confocal microscope. Grey-shaded regions represented a 95% CI. C) Flow cytometry histograms depicting the presence of A431 cell surface-bound EGF-biotin under various labelling conditions. >10,000 cells shown for each condition. Dashed line represents the mode of the EGF-biotin only control. D) Time course demonstrating the endocytosis of EGF-biotin under various labelling conditions of A431s. Fluorescence was normalized to values at t = 0. E) Representative live cell fluorescence images of the binding and internalization of pre-coupled anti-EGFR IgG-Litmus-body by EGFR-overexpressing A431s. Top: bulk solution at pH 7.5. Bottom: bulk solution at pH 5.2. F) Ratiometric (ISEP/ImCyRFP1) variants of the images presented in Figure E). Using the calibration curve from A), ratios were converted to pH values. G) Line traces were taken from regions of interest depicted as the corresponding white boxes in E/F. Scale bar: 10 μm.
To ensure that the Litmus-body did not interfere with the ability of Cetuximab to bind EGFR and block ligand-EGFR interaction, we looked at the cell surface binding and internalization of biotinylated epidermal growth factor (EGF) by A431 epidermoid cancer cells, which overexpressed EGFR by gene amplification55. EGF-biotin binding to EGFR on the cell surface was comparably blocked by Cetuximab and the Cetuximab-Litmus-body complex (Figure 3C). Cells treated with Cetuximab or Cetuximab-Litmus-body also showed near identical EGF-biotin uptake over time (Figure 3D, Supplemental Figure 7). We thus concluded that the Litmus-body had minimal effect on the physiological function of Cetuximab or its effect on Cetuximab-EGFR internalization.
Subsequently, we applied the Cetuximab-Litmus-body complex to A431s and monitored its internalization by receptor-mediated endocytosis at pH 7.5 via live-cell imaging. We then switched the bulk solution to pH ~5 to quench cfSEP signal on the cell surface. cfSEP and mCyRFP1 signals were observed in the composite images (Figure 3E) and were processed to represent their fluorescence ratio (Figure 3F). These ratios were converted to pH values based on the calibration curve (Figure 3B, 3F, 3G). Interestingly when the bulk solution was adjusted to pH 7.5, regions on the membrane were observed at a slightly lower pH of ~7.2 (ii), Figure 3G). This is consistent with previous reports suggesting that cancer cell surfaces have a lower pH than the bulk extracellular environment8.
In contrast, Litmus-body sequestered inside endocytic vesicles reported pH ranges from ~5 to 6.5 (Figure 3F, 3G). Note that these Litmus-body-loaded intracellular vesicles were similar in size to endosomes and reported the expected pH values for these compartments (Figure 3A). At a bulk solution of pH 5.2, the cell surface pH reported by Litmus-body closely matched the bulk solution pH (Figure 3G iv)) though its membrane signal became less defined, possibly due to its dissociation from EGFR at low pH56. In contrast, the juxtaposed intracellular compartment reported pH ~6.5 against the low cell surface pH. Altogether, these results suggested that our Litmus-body could be a useful tool for reporting the microenvironmental pH that its molecular targets may experience.
CONCLUSION
Cell surface acidification is a hallmark of aggressive diseases such as cancer1,2. Here, we presented the Litmus-body, an IgG-specific pH sensor in which we fused together an anti-mouse IgG TP1107 nanobody, a pH responsive cysteine-free super-ecliptic pHluorin (cfSEP) and a large-stoke shift monomeric cyan-excitable red fluorescent protein (mCyRFP1), and demonstrated its ability to quantitatively monitor the local pH surrounding cell surface targets. Co-excitation and separable emission of cfSEP and mCyRFP1 made it possible to normalize the pH response of the Litmus-body by a single-wavelength excitation. By engineering cfSEP for maleimide labelling, we also described a synthetic dye-conjugate variant with a large-stoke shift ATTO490LS that replaced mCyRFP1 for signal normalization. The dye-conjugate would benefit from the reduced size of a synthetic dye (ATTO490LS; <1 kDa) compared to a fluorescent protein (mCyRFP1; 26.4 kDa), though the appreciable fluorescence resonance energy transfer (FRET) between cfSEP and ATTO490LS decreases its signal to some extent. We did not further characterize the cfSEP-ATTO490LS version of the Litmus-body and it may potentially be a useful alternative, especially when the size of the reagent is an important factor. Swapping the cfSEP-based sensor module for other pH sensors, including reported lifetime-based sensors30,31 , should be trivial. This highlights the utility of the modular design of the Litmus-body.
The modular design of the Litmus-body would allow the nanobody domain to be swapped for other targeting variants. While we focused our proof-of-principle studies on monoclonal anti-mouse IgG antibodies as their derivatives play important roles in therapeutic antibodies in the clinic57,58, we envisage that the anti-mouse IgG nanobody domain can be easily replaced by anti-IgG nanobodies that are currently available from other species including rabbit21. Furthermore, while unexplored in our work, a diverse palette of nanobodies have been developed for commonly expressed molecular targets including EGFR59. Litmus-body variants that can be directly localized to targets of interest would remove the need for a primary antibody, and further benefit from the small size of the Litmus-body to maximize cell and tissue sample penetration and perfusion.
Preformed IgG-Litmus-body complexes derived from reacting IgG antibodies to the Litmus-body provided a simple, time-effective one-step targeting strategy for live cell applications. This approach benefited from the monomeric and monovalent nature of anti-IgG nanobodies, as they do not cross-link primary antibodies to form large multimeric complexes that impede IgG binding21. Using this strategy, we targeted the Litmus-body to two oncogenic cell surface proteins that are overexpressed in multiple cancer types: Muc1 and EGFR49,60, and demonstrated that the Litmus-body responded well to pH changes surrounding these proteins. Importantly, the Litmus-body reported a decreased pH surrounding EGFR on the cancer cell surface compared to the bulk solution. This observation is consistent with recent reports in the literature that suggest the pH on cancer cell surfaces is lower than the bulk microenvironment8. The ability to target cell surface proteins is a major advantage of the Litmus-body over current strategies in the literature that lack molecular specificity, such as using low pH insertion peptides8, and paves the way to elucidating the mechanisms that give rise to their aberrant function in the cancerous phenotype.
The ability of the Litmus-body to piggyback on the vast diversity and high specificity of IgG antibodies may prove to be a powerful approach for broad classes of molecular targets. It bears noting that the binding of nanobodies to antibodies may interfere with antibody-antigen interactions and potentially alter the binding of antibodies to target, as well as potentially altering target protein functionality, through steric inhibition of the binding sites and allosteric regulation. However, in complex with our Litmus-body, the antibodies tested here retained their high specificity to target, did not show off-target labelling and retained their function in blocking receptor-ligand interactions.
Antibody trafficking into acidic intracellular compartments may be important for payload release in the design of antibody-drug conjugates (ADCs) used in cancer therapies61. In our proof-of-principle studies, Litmus-body reported on the pH surrounding the internalized Cetuximab, an EGFR specific antibody used in cancer treatment that is undergoing active research for ADC based therapies54. This intracellular tracking was enabled by the single-wavelength co-excitable and dual-emission nature of the Litmus-body that permitted the accurate co-localization of cfSEP and mCyRFP1. Simultaneous excitation of fluorophores with a single-wavelength light can minimize the effect of sample movement, uneven sensor distribution and sample thickness variations62. It also reduced image acquisition time, phototoxicity and unnecessary photobleaching. These factors were particularly important when the Litmus-body complexes were sequestered inside fast-moving endosomal vesicles in living cells63.
These properties may be valuable in tracking ADCs transiting through intracellular compartments. ADCs typically exhibit specificity for molecules on the cancer cell surface and deliver cytotoxic payload once internalized and reach acidic lysosomes. For example, drug conjugation strategies that take advantages of acid-labile linkers can release payload in low pH compartments. Traditionally, ADCs are designed to have high affinity for their target at acidic pH. More recently, “acid-switched” ADCs that instead show high affinity for their target at neutral pH can have improved lysosomal trafficking with enhanced payload delivery and cytotoxicity14. In either case, Litmus-body may potentially be useful in monitoring the pH-sensitive payload delivery of ADCs, as well as to screen for ADC variants that are better trafficked into compartments of the desired pH for payload delivery16,17.
The use of recombinant protein sensors for targeted cell-surface applications bypasses the challenges associated with validating the expression, maturation, folding and aggregation of genetically encoded sensors in mammalian cells. Defects and variations in these processes are likely to lead to inconsistent calibration and quantification of cell surface pH by genetically encoded sensors. The exogenous application of recombinant sensors also allows the quantity of the sensor to be precisely controlled to minimize perturbation to cells. This mitigates the risk of cell stress that is often underappreciated with overexpressing genetically encoded fluorescent proteins in mammalian cells64,65 Unlike the Litmus-body, endogenously expressed sensors further suffer from the inability to distinguish between signals from their intracellular biosynthetic pool and those that are internalized from the cell surface. Finally, applying the Litmus-body to a novel cell-target requires simply changing the targeting IgG, potentially improving the throughput of new ADC discovery. The Litmus-body accurately recapitulates the endocytic environment observed by the ADCs, bypassing the potential pitfalls that may manifest with the overexpression of fluorescent proteins in highly confined environments seen in more conventional genetically encoded constructs.
Supplementary Material
ACKNOWLEDGMENT
This investigation was supported by National Institute of Health New Innovator DP2 GM229133 (M.J.P.), National Cancer Institute U54 CA210184-01 (M.J.P. and W.R.Z.), National Institute of General Medical Sciences Ruth L. Kirschstein National Research Service Award 2T32GM008267 (M.J.C.), and National Science Foundation Graduate Research Fellowship DGE-1650441 (M.J.C.). Imaging data was acquired through the Cornell University Biotechnology Resource Center, with NYSTEM C029155 and NIH S10OD018516 funding for the shared Zeiss LSM880 microscope.
Footnotes
Supporting Information. Additional figures and cDNA sequences for the sensors are available as a PDF. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- (1).Corbet C; Feron O Tumour Acidosis: From the Passenger to the Driver’s Seat. Nat. Rev. Cancer 2017, 17 (10), 577–593. 10.1038/nrc.2017.77. [DOI] [PubMed] [Google Scholar]
- (2).Webb BA; Chimenti M; Jacobson MP; Barber DL Dysregulated PH: A Perfect Storm for Cancer Progression. Nat. Rev. Cancer 2011, 11 (9), 671–677. 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- (3).Pavlova NN; Thompson CB The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23 (1), 27–47. 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Szpiech ZA; Strauli NB; White KA; Ruiz DG; Jacobson MP; Barber DL; Hernandez RD Prominent Features of the Amino Acid Mutation Landscape in Cancer. PLOS ONE 2017, 12 (8), e0183273. 10.1371/journal.pone.0183273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Damaghi M; Gillies R Phenotypic Changes of Acid-Adapted Cancer Cells Push Them toward Aggressiveness in Their Evolution in the Tumor Microenvironment. Cell Cycle 2017, 16 (19), 1739–1743. 10.1080/15384101.2016.1231284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hundshammer C; Düwel S; Schilling F Imaging of Extracellular PH Using Hyperpolarized Molecules. Isr. J. Chem 2017, 57 (9), 788–799. 10.1002/ijch.201700017. [DOI] [Google Scholar]
- (7).Hao G; Xu ZP; Li L Manipulating Extracellular Tumour PH: An Effective Target for Cancer Therapy. RSC Adv. 2018, 8 (39), 22182–22192. 10.1039/C8RA02095G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Anderson M; Moshnikova A; Engelman DM; Reshetnyak YK; Andreev OA Probe for the Measurement of Cell Surface PH in Vivo and Ex Vivo. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (29), 8177–8181. 10.1073/pnas.1608247113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chiche J; Brahimi-Horn MC; Pouysségur J Tumour Hypoxia Induces a Metabolic Shift Causing Acidosis: A Common Feature in Cancer. J. Cell. Mol. Med 2010, 14 (4), 771–794. 10.1111/j.15824934.2009.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Parks SK; Chiche J; Pouysségur J Disrupting Proton Dynamics and Energy Metabolism for Cancer Therapy. Nat. Rev. Cancer 2013, 13 (9), 611–623. 10.1038/nrc3579. [DOI] [PubMed] [Google Scholar]
- (11).Schnitzer JE Glycocalyx Electrostatic Potential Profile Analysis: Ion, PH, Steric, and Charge Effects. Yale J. Biol. Med 1988, 61 (5), 427–446. [PMC free article] [PubMed] [Google Scholar]
- (12).Pearce OMT; Läubli H Sialic Acids in Cancer Biology and Immunity. Glycobiology 2016, 26 (2), 111–128. 10.1093/glycob/cwv097. [DOI] [PubMed] [Google Scholar]
- (13).Yang Y; Xia M; Zhao H; Zhang S; Zhang X A Cell-Surface-Specific Ratiometric Fluorescent Probe for Extracellular PH Sensing with Solid-State Fluorophore. ACS Sens. 2018, 3 (11), 2278–2285. 10.1021/acssensors.8b00514. [DOI] [PubMed] [Google Scholar]
- (14).Kang JC; Sun W; Khare P; Karimi M; Wang X; Shen Y; Ober RJ; Ward ES Engineering a HER2-Specific Antibody–Drug Conjugate to Increase Lysosomal Delivery and Therapeutic Efficacy. Nat. Biotechnol 2019, 37 (5), 523–526. 10.1038/s41587-019-0073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Van Epps HA; Senter PD Breaking up Is Hard to Do. Nat. Chem. Biol 2019, 15 (10), 934–935. 10.1038/s41589-019-0351-1. [DOI] [PubMed] [Google Scholar]
- (16).Kalim M; Chen J; Wang S; Lin C; Ullah S; Liang K; Ding Q; Chen S; Zhan J Intracellular Trafficking of New Anticancer Therapeutics: Antibody-Drug Conjugates. Drug Des. Devel. Ther 2017, 11, 2265–2276. 10.2147/DDDT.S135571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tsui CK; Barfield RM; Fischer CR; Morgens DW; Li A; Smith BAH; Gray MA; Bertozzi CR; Rabuka D; Bassik MC CRISPR-Cas9 Screens Identify Regulators of Antibody–Drug Conjugate Toxicity. Nat. Chem. Biol 2019, 15 (10), 949–958. 10.1038/s41589-019-0342-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Birrer MJ; Moore KN; Betella I; Bates RC Antibody-Drug Conjugate-Based Therapeutics: State of the Science. JNCI J. Natl. Cancer Inst 2019, 111 (6), 538–549. 10.1093/jnci/djz035. [DOI] [PubMed] [Google Scholar]
- (19).Khongorzul P; Ling CJ; Khan FU; Ihsan AU; Zhang J Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res 2020, 18 (1), 3–19. 10.1158/1541-7786.MCR-19-0582. [DOI] [PubMed] [Google Scholar]
- (20).Muyldermans S Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem 2013, 82, 775–797. 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- (21).Pleiner T; Bates M; Görlich D A Toolbox of Anti–Mouse and Anti–Rabbit IgG Secondary Nanobodies. J Cell Biol 2017, jcb.201709115. 10.1083/jcb.201709115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Virant D; Traenkle B; Maier J; Kaiser PD; Bodenhöfer M; Schmees C; Vojnovic I; Pisak-Lukáts B; Endesfelder U; Rothbauer U A Peptide Tag-Specific Nanobody Enables High-Quality Labeling for DSTORM Imaging. Nat. Commun 2018, 9 (1), 930. 10.1038/s41467-018-03191-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pleiner T; Bates M; Trakhanov S; Lee C-T; Schliep JE; Chug H; Böhning M; Stark H; Urlaub H; Görlich D Nanobodies: Site-Specific Labeling for Super-Resolution Imaging, Rapid Epitope-Mapping and Native Protein Complex Isolation. eLife 2015, 4, e11349. 10.7554/eLife.11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Rothbauer U; Zolghadr K; Tillib S; Nowak D; Schermelleh L; Gahl A; Backmann N; Conrath K; Muyldermans S; Cardoso MC; Leonhardt H Targeting and Tracing Antigens in Live Cells with Fluorescent Nanobodies. Nat. Methods 2006, 3 (11), 887–889. 10.1038/nmeth953. [DOI] [PubMed] [Google Scholar]
- (25).Miesenböck G; Angelis DAD; Rothman JE Visualizing Secretion and Synaptic Transmission with PH-Sensitive Green Fluorescent Proteins. Nature 1998, 394 (6689), 192–195. 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- (26).Tsien RY The Green Fluorescent Protein. Annu. Rev. Biochem 1998, 67 (1), 509–544. 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- (27).Sankaranarayanan S; De Angelis D; Rothman JE; Ryan TA The Use of PHluorins for Optical Measurements of Presynaptic Activity. Biophys. J 2000, 79 (4), 2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rodriguez EA; Campbell RE; Lin JY; Lin MZ; Miyawaki A; Palmer AE; Shu X; Zhang J; Tsien RY The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends Biochem. Sci 2017, 42 (2), 111–129. 10.1016/j.tibs.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mahon MJ PHluorin2: An Enhanced, Ratiometric, PH-Sensitive Green Florescent Protein. Adv. Biosci. Biotechnol. Print 2011, 2 (3), 132–137. 10.4236/abb.2011.23021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Esposito A; Gralle M; Dani MAC; Lange D; Wouters FS PHlameleons: A Family of FRET-Based Protein Sensors for Quantitative PH Imaging. Biochemistry 2008, 47 (49), 13115–13126. 10.1021/bi8009482. [DOI] [PubMed] [Google Scholar]
- (31).Burgstaller S; Bischof H; Gensch T; Stryeck S; Gottschalk B; Ramadani-Muja J; Eroglu E; Rost R; Balfanz S; Baumann A; Waldeck-Weiermair M; Hay JC; Madl T; Graier WF; Malli R PH-Lemon, a Fluorescent Protein-Based PH Reporter for Acidic Compartments. ACS Sens. 2019, 4 (4), 883–891. 10.1021/acssensors.8b01599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Costantini LM; Baloban M; Markwardt ML; Rizzo M; Guo F; Verkhusha VV; Snapp EL A Palette of Fluorescent Proteins Optimized for Diverse Cellular Environments. Nat. Commun 2015, 6 (1), 1–13. 10.1038/ncomms8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chen F; LoTurco J A Method for Stable Transgenesis of Radial Glia Lineage in Rat Neocortex by PiggyBac Mediated Transposition. J. Neurosci. Methods 2012, 207 (2), 172–180. 10.1016/j.jneumeth.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Shurer CR; Head SE; Goudge MC; Paszek MJ Mucin-Coating Technologies for Protection and Reduced Aggregation of Cellular Production Systems. Biotechnol. Bioeng 2019, 116 (5), 994–1005. 10.1002/bit.26916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Shurer CR; Colville MJ; Gupta VK; Head SE; Kai F; Lakins JN; Paszek MJ Genetically Encoded Toolbox for Glycocalyx Engineering: Tunable Control of Cell Adhesion, Survival, and Cancer Cell Behaviors. ACS Biomater. Sci. Eng 2017. 10.1021/acsbiomaterials.7b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Frey S; Görlich D A New Set of Highly Efficient, Tag-Cleaving Proteases for Purifying Recombinant Proteins. J. Chromatogr. A 2014, 1337, 95–105. 10.1016/j.chroma.2014.02.029. [DOI] [PubMed] [Google Scholar]
- (37).Magde D; Wong R; Seybold PG Fluorescence Quantum Yields and Their Relation to Lifetimes of Rhodamine 6G and Fluorescein in Nine Solvents: Improved Absolute Standards for Quantum Yields¶. Photochem. Photobiol 2002, 75 (4), 327–334. 10.1562/0031-8655(2002)0750327FQYATR2.0.CO2. [DOI] [PubMed] [Google Scholar]
- (38).Laviv T; Kim BB; Chu J; Lam AJ; Lin MZ; Yasuda R Simultaneous Dual-Color Fluorescence Lifetime Imaging with Novel Red-Shifted Fluorescent Proteins. Nat. Methods 2016, 13 (12), 989–992. 10.1038/nmeth.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ward WW Biochemical and Physical Properties of Green Fluorescent Protein. In Green Fluorescent Protein; John Wiley & Sons, Ltd, 2005; pp 39–65. 10.1002/0471739499.ch3. [DOI] [PubMed] [Google Scholar]
- (40).Cranfill PJ; Sell BR; Baird MA; Allen JR; Lavagnino Z; de Gruiter HM; Kremers G-J; Davidson MW; Ustione A; Piston DW Quantitative Assessment of Fluorescent Proteins. Nat. Methods 2016, 13 (7), 557–562. 10.1038/nmeth.3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Colville MJ; Park S; Zipfel WR; Paszek MJ High-Speed Device Synchronization in Optical Microscopy with an Open-Source Hardware Control Platform. Sci. Rep 2019, 9 (1), 1–13. 10.1038/s41598-019-48455-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Edelstein AD; Tsuchida MA; Amodaj N; Pinkard H; Vale RD; Stuurman N Advanced Methods of Microscope Control Using MManager Software. J. Biol. Methods 2014, 1 (2). 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Carmody WR Easily Prepared Wide Range Buffer Series. J. Chem. Educ 1961, 38 (11), 559. 10.1021/ed038p559. [DOI] [Google Scholar]
- (44).Kneen M; Farinas J; Li Y; Verkman AS Green Fluorescent Protein as a Noninvasive Intracellular PH Indicator. Biophys. J 1998, 74 (3), 1591–1599. 10.1016/S0006-3495(98)77870-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Cal PMSD; Bernardes GJL; Gois PMP Cysteine-Selective Reactions for Antibody Conjugation. Angew. Chem. Int. Ed 2014, 53 (40), 10585–10587. 10.1002/anie.201405702. [DOI] [PubMed] [Google Scholar]
- (46).Suzuki T; Arai S; Takeuchi M; Sakurai C; Ebana H; Higashi T; Hashimoto H; Hatsuzawa K; Wada I Development of Cysteine-Free Fluorescent Proteins for the Oxidative Environment. PLoS ONE 2012, 7 (5). 10.1371/journal.pone.0037551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kufe DW Mucins in Cancer: Function, Prognosis and Therapy. Nat. Rev. Cancer 2009, 9 (12), 874–885. 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Paszek MJ; DuFort CC; Rossier O; Bainer R; Mouw JK; Godula K; Hudak JE; Lakins JN; Wijekoon AC; Cassereau L; Rubashkin MG; Magbanua MJ; Thorn KS; Davidson MW; Rugo HS; Park JW; Hammer DA; Giannone G; Bertozzi CR; Weaver VM The Cancer Glycocalyx Mechanically Primes Integrin-Mediated Growth and Survival. Nature 2014, 511 (7509), 319–325. 10.1038/nature13535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Arteaga CL; Engelman JA ERBB Receptors: From Oncogene Discovery to Basic Science to Mechanism-Based Cancer Therapeutics. Cancer Cell 2014, 25 (3), 282–303. 10.1016/j.ccr.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Liao H-J; Carpenter G Cetuximab/C225-Induced Intracellular Trafficking of Epidermal Growth Factor Receptor. Cancer Res. 2009, 69 (15), 6179–6183. 10.1158/0008-5472.CAN-09-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Huotari J; Helenius A Endosome Maturation. EMBO J. 2011, 30 (17), 3481–3500. 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Frampton JE Cetuximab: A Review of Its Use in Squamous Cell Carcinoma of the Head and Neck. Drugs 2010, 70 (15), 1987–2010. 10.2165/11205010-000000000-00000. [DOI] [PubMed] [Google Scholar]
- (53).Galizia G; Lieto E; De Vita F; Orditura M; Castellano P; Troiani T; Imperatore V; Ciardiello F Cetuximab, a Chimeric Human Mouse Anti-Epidermal Growth Factor Receptor Monoclonal Antibody, in the Treatment of Human Colorectal Cancer. Oncogene 2007, 26 (25), 3654–3660. 10.1038/sj.onc.1210381. [DOI] [PubMed] [Google Scholar]
- (54).Glatt DM; Beckford Vera DR; Prabhu SS; Mumper RJ; Luft JC; Benhabbour SR; Parrott MC Synthesis and Characterization of Cetuximab–Docetaxel and Panitumumab–Docetaxel Antibody–Drug Conjugates for EGFR-Overexpressing Cancer Therapy. Mol. Pharm 2018, 15 (11), 5089–5102. 10.1021/acs.molpharmaceut.8b00672. [DOI] [PubMed] [Google Scholar]
- (55).Merlino GT; Xu YH; Ishii S; Clark AJ; Semba K; Toyoshima K; Yamamoto T; Pastan I Amplification and Enhanced Expression of the Epidermal Growth Factor Receptor Gene in A431 Human Carcinoma Cells. Science 1984, 224 (4647), 417–419. 10.1126/science.6200934. [DOI] [PubMed] [Google Scholar]
- (56).Theofilopoulos AN; Dixon FJ The Biology and Detection of Immune Complexes. Adv. Immunol 1979, 28, 89–220. [DOI] [PubMed] [Google Scholar]
- (57).Almagro JC; Daniels-Wells TR; Perez-Tapia SM; Penichet ML Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front. Immunol 2017, 8, 1751. 10.3389/fimmu.2017.01751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Chames P; Van Regenmortel M; Weiss E; Baty D Therapeutic Antibodies: Successes, Limitations and Hopes for the Future. Br. J. Pharmacol 2009, 157 (2), 220–233. 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Tintelnot J; Baum N; Schultheiß C; Braig F; Trentmann M; Finter J; Fumey W; Bannas P; Fehse B; Riecken K; Schuetze K; Bokemeyer C; Rösner T; Valerius T; Peipp M; Koch-Nolte F; Binder M Nanobody Targeting of Epidermal Growth Factor Receptor (EGFR) Ectodomain Variants Overcomes Resistance to Therapeutic EGFR Antibodies. Mol. Cancer Ther 2019, 18 (4), 823–833. 10.1158/1535-7163.MCT-18-0849. [DOI] [PubMed] [Google Scholar]
- (60).Nath S; Mukherjee P MUC1: A Multifaceted Oncoprotein with a Key Role in Cancer Progression. Trends Mol. Med 2014, 20 (6), 332–342. 10.1016/j.molmed.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Dan N; Setua S; Kashyap VK; Khan S; Jaggi M; Yallapu MM; Chauhan SC Antibody-Drug Conjugates for Cancer Therapy: Chemistry to Clinical Implications. Pharm. Basel Switz 2018, 11 (2). 10.3390/ph11020032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Woehler A Simultaneous Quantitative Live Cell Imaging of Multiple FRET-Based Biosensors. PLoS ONE 2013, 8 (4). 10.1371/journal.pone.0061096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Condon KH; Ehlers MD CHAPTER 8 - Postsynaptic Machinery for Receptor Trafficking. In Protein Trafficking in Neurons; Bean AJ, Ed.; Academic Press: Burlington, 2007; pp 143–174. 10.1016/B978-012369437-9/50013-X. [DOI] [Google Scholar]
- (64).Mori Y; Yoshida Y; Satoh A; Moriya H Development of an Experimental Method of Systematically Estimating Protein Expression Limits in HEK293 Cells. Sci. Rep 2020, 10 (1), 1–11. 10.1038/s41598-020-61646-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Ganini D; Leinisch F; Kumar A; Jiang J; Tokar EJ; Malone CC; Petrovich RM; Mason RP Fluorescent Proteins Such as EGFP Lead to Catalytic Oxidative Stress in Cells. Redox Biol. 2017, 12, 462–468. 10.1016/j.redox.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.