

Abstract
Treatment with mineralocorticoid receptor (MR) antagonists beginning at the outset of disease, or early thereafter, prevents pulmonary vascular remodeling in preclinical models of pulmonary arterial hypertension (PAH). However, the efficacy of MR blockade in established disease, a more clinically relevant condition, remains unknown. Therefore, we investigated the effectiveness of two MR antagonists, eplerenone (EPL) and spironolactone (SPL), after the development of severe right ventricular (RV) dysfunction in the rat SU5416-hypoxia (SuHx) PAH model. Cardiac magnetic resonance imaging (MRI) in SuHx rats at the end of week 5, before study treatment, confirmed features of established disease including reduced RV ejection fraction and RV hypertrophy, pronounced septal flattening with impaired left ventricular filling and reduced cardiac index. Five weeks of treatment with either EPL or SPL improved left ventricular filling and prevented the further decline in cardiac index compared with placebo. Interventricular septal displacement was reduced by EPL whereas SPL effects were similar, but not significant. Although MR antagonists did not significantly reduce pulmonary artery pressure or vessel remodeling in SuHx rats with established disease, animals with higher drug levels had lower pulmonary pressures. Consistent with effects on cardiac function, EPL treatment tended to suppress MR and proinflammatory gene induction in the RV. In conclusion, MR antagonist treatment led to modest, but consistent beneficial effects on interventricular dependence after the onset of significant RV dysfunction in the SuHx PAH model. These results suggest that measures of RV structure and/or function may be useful endpoints in clinical trials of MR antagonists in patients with PAH.
Keywords: cardiac magnetic resonance imaging, mineralocorticoid receptor antagonist, pulmonary arterial hypertension, right ventricular dysfunction
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a severe and often fatal condition characterized by pathologic narrowing and obliteration of distal pulmonary arteries. Early vasoconstriction, followed by progressive vessel remodeling lead to increased pulmonary vascular resistance, excessive right ventricular (RV) strain and eventually right heart failure (1, 2). Despite the development of more than a dozen drugs for PAH, 50% of patients die within 5–7 yr of diagnosis (3). All currently approved PAH treatments target vasoregulatory pathways to promote pulmonary arterial vasodilation or block vasoconstriction. Although progressive RV failure is the leading cause of death in PAH (4), none of these drugs specifically target RV function.
The prominent role of a dysregulated immune response in PAH pathobiology is well established (5–12). Although less thoroughly investigated, inflammation has been associated with RV dysfunction (13) and may drive maladaptive RV remodeling in PAH (14). Importantly, inflammation has been closely linked to disease pathogenesis in heart failure with reduced ejection fraction (HFrEF) (15–17) and left heart failure following myocardial infarction (18), clinical conditions where mineralocorticoid receptor (MR) antagonist treatment reduces mortality (19, 20). Analogous to HFrEF (21), aldosterone-mediated mineralocorticoid receptor (MR) activation has been identified as a cause of cardiovascular inflammation in PAH (22, 23) where plasma concentrations of aldosterone are elevated and correlate with the severity of hemodynamic derangement (24, 25). These increased levels of aldosterone in PAH have been variably ascribed to renin-angiotensin-aldosterone pathway overactivation (26), and endothelin-1-dependent adrenal and/or local lung aldosterone synthesis (27–29).
Both spironolactone (SPL) and eplerenone (EPL), two MR antagonists used clinically to treat HFrEF, decrease RV systolic pressures and prevent pathologic pulmonary vascular remodeling in the monocrotaline rat and the chronic hypoxia mouse models of pulmonary hypertension (PH) (28–30). In contrast to these PH models, the combination of SU5416, a tyrosine kinase inhibitor, and 3 wk of hypoxia in rats, referred to as the SU5416-hypoxia (SuHx) rat model, more closely mimics the histopathologic findings in patients with PAH (31). In the few preclinical studies that tested MR antagonist therapy in the SuHx model, the majority investigated preventative treatment with EPL (28, 29, 32) whereas one initiated SPL early (day 14), before the establishment of advanced vascular lesions and RV dysfunction (29). Collectively, these investigations have highlighted the beneficial effects of MR antagonists on pulmonary vascular remodeling when treatment is initiated early, before the establishment of advanced disease. Unfortunately, patients with PAH usually present at a later stage of disease than has been simulated in these animal models. Thus, the efficacy of SPL or EPL after the onset of RV dysfunction, a more clinically applicable scenario, remains unknown. In the SuHx PAH rat model, pulmonary pressures peak and cardiac index begins to decline by week 5 (33), providing an opportunity to evaluate the efficacy of MR antagonists on RV dysfunction and pulmonary vascular remodeling at more therapeutically relevant time points. Furthermore, we previously reported that SPL, but not EPL, can suppress endothelial inflammation independent of MR by inducing the proteasomal degradation of xeroderma pigmentosum group B-complementing protein (XPB; official gene symbol ERCC3), a subunit of TFIIH (34). Whether SPL, at clinically achievable dosages, is more effective at treating the inflammatory component of PAH compared with EPL has not been previously examined.
Utilizing serial, high-resolution cardiac cine magnetic resonance imaging (MRI), we show that MR antagonist treatment beginning after week 5 in the SuHx PAH model preserved cardiac index and increased left ventricular (LV) end-diastolic volume index. Improvements in LV filling and ventricular interdependence, as determined by the diastolic and systolic eccentricity index, were more pronounced in EPL-treated animals. Consistent with these beneficial effects on myocardial function, EPL treatment blunted the induction of MR target genes and inflammatory response genes in the RV.
METHODS
SU5416-Hypoxia Pulmonary Arterial Hypertension Model and Mineralocorticoid Receptor Antagonist Treatment
All aspects of animal testing and care were approved by the Animal Care and Use Committee (ACUC) at the National Institutes of Health (NIH) Clinical Center. Animals were housed two per cage, allowed to acclimate for at least 48 h before the study, and kept on a regular 12-h light/dark cycle. Access to water was ad libitum and food was provided to ensure that daily caloric requirements were met.
Male Sprague–Dawley rats [n = 48, arrival weight (mean ± SD) 196 ± 12 g; Charles River Laboratories; Wilmington, MA] were subcutaneously injected with SU5416 (25 mg/kg; Tocris, Minneapolis, MN), exposed to hypoxia (10% ) for 3 wk in a Biospherix Oxycycler (Biospherix Ltd., Parish, NY) and then returned to normoxia for 7 wk (referred to as SuHx rats). SU5416 was suspended in carboxymethylcellulose [0.5% (wt/vol) carboxymethylcellulose sodium, 0.9% (wt/vol) NaCl, 0.4% (vol/vol) polysorbate, 0.9% (vol/vol) benzyl alcohol in deionized water]. The Pfizer Pure Compound Grants Program provided EPL (PF-02845980; USP, micronized powder) and SPL (USP, micronized powder) was purchased from Letco Medical (Wayne, PA).
At the end of week 5 (pretreatment), SuHx rats (n = 43) underwent cardiac MRI followed by blood collection. The rats were then randomly assigned to receive color-coded medicated diets containing either EPL (n = 15, 100 mg/kg/day; Envigo; Fredrick, MD), SPL (n = 15, 40 mg/kg/day; Envigo), or placebo (n = 13; Envigo) from weeks 6–10 (Fig. 1). A total of five SuHx rats died before randomization. Age-matched male Sprague–Dawley rats subcutaneously injected with vehicle alone and maintained in normoxic conditions for 10 wk served as healthy controls. Separate from the SuHx group, normoxic controls underwent cardiac MRI (n = 4; end of week 5), cardiac catheterization and necropsy (n = 4 for both at the end of week 10), and are presented as a visual reference. Tissue was harvested at the end of 10 wk in additional normoxic control rats for RV (n = 9) and lung (n = 6) mRNA expression. Animal weights at the time of SU5416 or vehicle injection, 5, and 10 wk postinjection are provided in Supplemental Table S1.
Figure 1.

Study timeline. A: male Sprague–Dawley rats (N = 48) were subcutaneously injected with SU5416 (25 mg/kg), exposed to hypoxia (10% ) for 3 wk and then returned to normoxia for 7 wk (referred to as SuHx rats). Pretreatment cardiac MRI (n = 43; n = 5 SuHx rats died before MRI) was performed at the end of week 5 followed by blood collection. Animals were then randomly assigned to receive medicated diets containing either eplerenone (EPL; 100 mg/kg/day; n = 15), spironolactone (SPL; 40 mg/kg/day; n = 15), or placebo (n = 13) from weeks 6–10. B: separate from the SuHx group, age-matched male Sprague–Dawley rats subcutaneously injected with vehicle alone and maintained in normoxic conditions underwent cardiac MRI (n = 4; end of week 5), cardiac catheterization, and necropsy (n = 4 for both at the end of week 10). Tissue was obtained in additional normoxic control animals at the end of 10 wk for RV (n = 9) and lung (n = 6) mRNA expression. LHC, left heart catheterization; RHC, right heart catheterization.
Cine Cardiac MRI
MRI was performed in a 7.0 T (Tesla), 16-cm horizontal bore Bruker MR imaging system (Bruker; Billerica, MA) at the end of weeks 5 and 10 using Bruker ParaVision 5.1 software. Rats were anesthetized with 2% isoflurane and imaged with ECG and respiratory gating using a 60 mm Bruker birdcage volume coil or a home built, transmit receive half-saddle surface coil with balanced match. Gradient echo cine images of the heart were acquired in short axis from above the aortic valve to the apex. Short axis cine parameters included repetition time (TR = 15 ms), echo time (TE = 3.4 ms), 30° flip angle, 5.0–6.5 cm isotropic field of view (FOV), 256 × 256 matrix, 2.0-mm slice thickness, 2–3 averages, and 12–15 cine frames depending on heart rate, respiratory rate, and ECG gating. Long axis cine scans were performed for most rats (TR = 15 ms, TE = 5 ms, FOV ∼8.0 × 5.0 cm, 512 × 256 matrix, and 2.0-mm slice thickness, 2.0 mm and 2–4 averages). Magnevist (gadopentate dimeglumine; Bayer HealthCare; Montville, NJ) diluted 1:2 with sterile 0.9% saline was administered subcutaneously at 0.3 mmol Gd/kg. Ejection fraction, ventricular volumes, and associated parameters were calculated from the cine data using CAAS-MRV-FARM software (Pie Medical Imaging, The Netherlands). For analysis, stroke volume and cardiac output were calculated based on LV volumes. Stroke volume and cardiac output calculated based on RV volumes are provided in Supplemental Table S2. Ventricular volumes, stroke volume, and cardiac output were all indexed to body weight (g; see Supplemental Table S3 for unindexed values). The eccentricity index was calculated as previously described (35). Image analysis was performed by investigators who were blinded to treatment assignments (S.A.A and M.L.).
Blood Collection
Blood was collected from the lateral tail vein with a 21-gauge winged infusion set for weeks 5–9. Terminal blood collection at week 10 was performed via cardiac puncture. For lateral tail vein collections, animals were secured in a restraint cone, tails were cleaned with isopropyl alcohol, and a total of 1–2 mL of blood was collected in dipotassium ethylenediaminetetraacetic acid (EDTA) and serum separator tubes (Becton Dickerson; Franklin Lakes, NJ). Whole blood was centrifuged (3,000 rpm for 10 min), serum and plasma were transferred to a new tube, and stored at −80°C until further use.
Serum Drug Concentration Measurements
Liquid chromatography–mass spectrometry (LC-MS) was used to measure serum drug concentrations from weeks 5 to 10. An Agilent 6540 Ultra-High Definition Accurate-Mass Quadrupole Time-of-Flight (Q-TOF) mass spectrometer (Agilent Technologies, Santa Clara, CA) operating at positive jet stream ESI mode was used. The Q-TOF was equipped with an Agilent 1200 HPLC with mobile phases of (A) 0.1% formic acid, 2% acetonitrile, and 98% H2O with 2 mM ammonium fluoride and (B) 0.1% formic acid and 100% methanol. An Agilent Zorbox Eclipse XDB-C18 4.6 × 50 mm, 1.8-µm column was used under isocratic gradient of 10 min 58% mobile phase (B). Chromatograms for each ion were extracted from total ion current (TIC) data with an accuracy window of ±20 ppm and integrated using Agilent MassHunter Qualitative Analysis Software (revision B.05.00). The relative concentrations of canrenone (CAN), one of the two main SPL metabolites, as well as EPL and its major metabolites, a hydrolyzed open-ring form of EPL and 6β hydroxy eplerenone, were calculated based on the ratio of the measured peak intensities and the known concentration of the internal standard, deuterated CAN (1 ng/µL in 50% methanol, 0.1% formic acid). Supplemental Table S4 lists the ions detected, their corresponding mass to charge ratio and retention time.
Hemodynamic Measurements
Anesthesia was induced with 3%–5% isoflurane and maintained at 2% during hemodynamic measurements. Rats were positioned dorsally on a heated platform for right and left heart catheterization. A midline incision was made in the neck and the right external jugular vein was isolated. A curved 2Fr Millar Mikro-Tip solid-state catheter (SPR-513; ADInstruments; Colorado Springs, CO) was introduced into the jugular vein and advanced into the RV. The catheter was removed, and the vessel was tied off for hemostasis. Next, a straight 2Fr Mikro-Tip solid-state catheter (SPR-320; ADInstruments) was introduced into the left carotid artery and advanced into the LV. Pressure tracings from each ventricle were obtained at a sample rate of 1,000 Hz for at least 1 min. Labchart 8 (ADInstruments) was used to measure RV systolic pressure (RVSP), LV end-diastolic pressure (LVEDP), and systemic blood pressure. Rats were euthanized by terminal exsanguination in accordance with NIH ACUC guidelines.
Tissue Collection and Processing
Following terminal exsanguination, the lungs and heart were removed en bloc and carefully dissected away from each other. Pieces of the right cranial and caudal lobes were placed in RNAlater Stabilization Reagent (QIAGEN; Valencia, CA) or snap-frozen in liquid nitrogen, respectively. The left lung was fixed for histology by tracheal instillation of 1% neutral buffered formalin in 0.5% agarose under constant pressure (20 cmH2O). The trachea was ligated after sustained inflation, and the lung was immersed in 10% neutral buffered formalin overnight and then transferred into 70% ethanol. The RV was carefully dissected away from the LV and septum and each weighed to calculate Fulton’s index [RV mass (g)/LV + septum mass (g)]. The RV free wall was divided into separate pieces and placed in RNAlater Stabilization Reagent, snap-frozen in liquid nitrogen or fixed in 10% neutral buffered formalin, and then transferred into 70% ethanol.
Morphometric Analysis for Pulmonary Vascular Remodeling
Lung tissue was cut into 5-mm slices and paraffin embedded. Sections (5 μm) were subsequently stained with fortified hematoxylin and eosin (Histoserv Inc., Germantown, MD) for morphometric analysis. Entire slides were scanned with NanoZoomer digital slide scanner 2.0 RS (Hamamatsu, Bridgewater, NJ). For grading of occlusive lesions, 10 fields at the lung periphery were examined using NDP viewer 2.0 software (Hamamatsu; Supplemental Fig. S1A). A trained pathologist blinded to treatment assignments (Z.X.Y.) examined 100–200 small pulmonary arterioles (diameter < 100 µm) per rat and assigned a grade of 0, 1, or 2 based on the degree of arteriole occlusion (grade 0 = no occlusions; grade 1 < 50% occluded; grade 2 ≥ 50% occluded; Supplemental Fig. S1B) (33). Lung vessel muscularization was assessed in Elastin van Gieson (EVG) stained sections as previously described (36). Briefly, 20 vessels (50–120 µm in external diameter) per rat were examined by an investigator blinded to treatment assignments (M.A.H.S.) using NDP viewer 2.0 software (Hamamatsu). Medial wall thickness of each vessel was calculated as a percentage of the external diameter {[(media thickness × 2)/external diameter] × 100}. Vessel measurements were done using ImageJ software v. 1.53e (National Institutes of Health, Bethesda, MD).
Assessment of Right Ventricular Fibrosis
Paraffin-embedded RV free wall segments were sectioned (5 μm) and stained with Masson Trichrome for assessment of myocardial fibrosis. Slides were scanned with NanoZoomer digital slide scanner 2.0 RS (Hamamatsu) and whole sections were evaluated using NDP viewer 2.0 software (Hamamatsu). A pathologist blinded to treatment assignments (Z.X.Y.) assessed the extent of RV fibrosis. Grade 0 = no fibrotic changes or only perivascular fibrosis; grade 1 = mild, patchy, or focal fibrosis; grade 2 = moderate, with diffuse areas of fibrosis; and grade 3 = severe, with large areas of dense fibrosis or scar formation and myocyte lost.
Right Ventricle and Lung mRNA Expression
Tissue samples from the RV free wall and the cranial lobe of the right lung were cut into 3- to 5-mm pieces and placed into RNAlater Stabilization Reagent (QIAGEN) overnight at 4°C and then stored at −20°C until further processing. RNA was extracted with either the RNeasy Fibrous Tissue Midi Kit (QIAGEN) for the RV samples or the RNeasy Mini Kit (QIAGEN) for the lung samples according to the manufacturer’s instructions and then stored at −80°C. RNA samples were then subjected to reverse transcription using the iScript cDNA Synthesis Kit (Bio-Rad; Hercules, CA). Gene expression assays were performed by quantitative real-time PCR in duplicate using PrimePCR assays and a custom hyperaldosteronism panel (both from Bio-Rad) on the Applied Biosystems ViiATM7 cycler (Life Technologies, Carlsbad, CA) with Sso Advanced Universal SYBR Green Supermix (Bio-Rad). The following PrimePCR assays were used: Ccl2, Ccl3, Ccl5, Cxcl10, Cxcl12, Cxcl1, Cxcl2, Cxcl9, Cx3cl1, Icam1, Vcam1, Xirp1, Ctgf, Cola1, Il1rl1, Il1b, Il33, Lgals3, Plat, Ptgs2, Tgfb, and Tnf. Gene assays included in the hyperaldosteronism panel were Cyp1a1, Ptprd, Edn1, Rgs2, Hk1, Serpine1, Il1r1, Sgk1, Il6, Nampt, Nedd4, and Nrc3c2. Reactions without reverse transcriptase were performed to rule out amplification of residual genomic DNA. Gene expression was calculated based on the 2−ΔΔCt method normalized to Hprt1. ΔΔCt values for placebo-treated SuHx rats were calculated relative to the mean delta Ct of normoxic control rat samples (n = 9 and 10 animals for RV and lung samples, respectively). ΔΔCt values for EPL- and SPL-treated animals were calculated relative to placebo-treated animals within each experimental cycle. When placebo-treated animal samples were not available from within cycle (e.g., due to animal loss), samples from placebo-treated animals in temporally adjacent cycles were used instead.
Cytokine, Chemokine, and Growth Factor Multiplex Immunoassay
Plasma cytokine, chemokine, and growth factor concentrations were determined using Bio-Plex Pro Rat Cytokine 23-Plex Assays (Bio-Rad) on the Bio-Plex 200 system (Bio-Rad) as previously described (34).
Cell Culture and Reagents
Human embryonic kidney (HEK293) cells (ATCC, Manassas, VA) were grown in high-glucose DMEM supplemented with 10% heat-inactivated FBS (Life Technologies, Grand Island, NY). Pulmonary artery endothelial cells (PAECs; Lonza, Walkersville, MD) were cultured for a maximum of six passages in endothelial basal medium-2 (Lonza) supplemented with growth factors containing 2% serum (Lonza) in cell culture flasks coated with type I collagen (50 μg/mL in 0.02 N acetic acid). SPL (Sigma, St. Louis, MO), CAN (Tocris, Minneapolis, MN), and 7α-thiomethyl spironolactone (7α-SPL; Santa Cruz, Dallas, TX) were solubilized in sterile DMSO (Sigma) before dilution in medium. Cells in each experiment were exposed to equivalent volumes of vehicle.
Western Blotting
Tissue samples from the RV free wall and the caudal lobe of the right lung were snap-frozen in liquid nitrogen and stored at −80°C until further processing. For tissue homogenization, samples were thawed on ice and RIPA buffer (5 µL/mg of tissue; Life Technologies) containing Halt Protease and Phosphatase Inhibitor Cocktail (3× final concentration; Life Technologies) was added to each sample. Tissue samples were mechanically disrupted using a TissueLyser (QIAGEN) according to the manufacture’s recommendations. Briefly, the samples were precooled on ice and TissueLyser disruption was performed for 30 s at 30 Hz followed by 10 min incubation on ice. An additional six cycles of disruption were performed (15 s at 30 Hz alternating with 10-min incubation on ice). Following the last cycle, the samples were incubated for 30 min on ice with intermittent gentle agitation. Lysates were cleared by centrifugation (20,000 g for 20 min at 4°C) and the supernatants were transferred to new tubes. HEK293 cells and PAECs were lysed on ice with RIPA buffer (Life Technologies) supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (Life Technologies). Lung and RV homogenates and whole cell lysates were resolved by SDS-PAGE and transferred to nitrocellulose membranes using Trans-Blot Turbo Transfer System (Bio-Rad). Blots were blocked with 5% ECL Primer blocking agent (GE Healthcare, Pittsburgh, PA) and then incubated overnight at 4°C with antibodies for XPB (for RV and lung tissue: 1:5,000, sc293X; Santa Cruz Biotechnology, Inc; Dallas, TX; and for HEK293 cells and PAECs: 1:1,000, AF6349; R&D Systems, Minneapolis, MN) and GAPDH (1:3,000, 2118S; Cell Signaling Technology; Danvers, MA). Blots were washed with 0.1% Tween-20 (Sigma; St. Louis, MO) in PBS and then incubated with HRP-conjugated goat anti-rabbit or donkey anti-goat secondary antibodies (each at 1:5,000, 111-005-003 and 705-005-003, respectively; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) for 1 h at room temperature. Staining for β-actin was done at room temperature for 1 h with an HRP-conjugated anti-β-actin antibody (1:50,000, A3854; Sigma). Blots were developed with an enhanced chemiluminescence substrate (GE Healthcare) using the ChemiDoc XRS+ System (Bio-Rad). Quantification of bands by densitometry analysis was performed using Image Lab software, v. 5.0 (Bio-Rad). Antibody specificity was validated in PAECs transfected with either nontargeting, control siRNA (UGGUUUACAUGUCGACUAA; Dharmacon; Lafayette, CO) or gene-specific siRNA targeting ERCC3 (official gene symbol for XPB) as previously described (34). Two different gene-specific siRNAs targeting ERCC3 were used (GAUCAAGGUUAUAGCUUCA and CCGCGAAGAUGACAA1AAUU; both from Dharmacon). Full-length gels are available in Supplemental Fig. S2. Specificity of one of the XPB antibodies (AF6349; R&D Systems) was previously validated in ERCC3-silenced cells and fibroblasts from patients with known XPB mutations that decrease protein expression levels (34).
Luciferase Reporter Assays
HEK293 cells (1 × 105/well) were transfected with 100 ng of a destabilized luciferase reporter regulated by either NF-κB [pGL 4.32(luc2P/NF-κB-RE/Hygro); Promega, Madison, WI], or activator protein 1 [AP-1; pGL4.44(luc2P/AP-1-RE/Hygro); Promega] along with 50 ng of an internal control vector (pGL4.74/hRluc; Promega) as previously described (37). Empty-vector control plasmid (pCMV6-entry) was added to bring the total amount of plasmid DNA to 300 ng per well. Cell lysates were harvested for luciferase activity using a dual-luciferase assay kit (Promega) according to the manufacturer’s recommendations. Luciferase activity was measured using a VICTOR3 multi-label reader (PerkinElmer; Waltham, MA) and normalized to the activity of the renilla control.
Statistical Analysis
Survival times were plotted using Kaplan–Meier curves and compared using the log-rank test. When there were no repeated measures, we used t tests or ANOVA to compare groups and Pearson’s correlation to correlate continuous variables. Linear mixed models were used to account for the clustering of animals within each cycle and the repeated measures of each animal over time. We first assessed whether there was any evidence that changes from pretreatment values were different across the three groups (i.e., interactions). Post hoc contrasts were tested for comparisons of interest. Data were log-transformed when necessary, to ensure the assumption of normality. Analyses were performed in SAS v. 9.4 and JMP v. 15 (both from SAS Institute; Cary, NC). All P values were two-tailed, unless specifically stated otherwise, and considered significant if P ≤ 0.05.
RESULTS
Severe Right Ventricular Dysfunction in SU5416-Hypoxia Rats before Mineralocorticoid Receptor Antagonist Treatment
The SuHx rat model closely mimics PAH progression in humans, allowing for the assessment of therapeutic interventions at clinically relevant stages of disease (Fig. 1) (31, 33). Contrast-enhanced, cine cardiac MR images of the long and short axis demonstrated both RV dilation, severe septal flattening and RV free wall hypertrophy 5 wk after SU5416 injection (Fig. 2, A–D). As compared with healthy, normoxic control rats, SuHx rats had a reduced RV ejection fraction (EF), whereas LVEF was increased, consistent with a hyperdynamic, underfilled LV in the setting of impaired RV function (Fig. 2, E and F). Consistent with decreased LV filling, the ratio of RV end-diastolic volume (RVEDV) to LVEDV was significantly higher in SuHx rats compared with controls (Fig. 2G), largely due to a reduction in LVEDV (Supplemental Fig. S3). Likewise, cardiac index and stroke volume index were both decreased compared with normoxic control rats by the end of week 5 (Fig. 2, H and I). Due to the circular shape of the LV in the short-axis, the ratio of the LV minor-axis diameter parallel to the septum (D2) to the LV minor-axis diameter perpendicular to the septum (D1), referred to as the eccentricity index, is normally approximately equal to 1 at both end-diastole and end-systole (Fig. 3, A and B) (35). Consistent with both RV volume and pressure overload, the eccentricity indices at end-diastole and end-systole were both significantly increased (Fig. 3, C–F). Collectively, these data demonstrate that SuHx rats have already developed significant RV dysfunction by the end of 5 wk. Importantly, there were no significant differences across the three groups of SuHx rats following randomization but before treatment with placebo, EPL, or SPL (P ≥ 0.20, for each of the post hoc pairwise comparisons of the parameters illustrated in Figs. 2 and 3 and Supplemental Fig. S3).
Figure 2.

SU5416-hypoxia (SuHx) rats develop significant right ventricular (RV) dysfunction before mineralocorticoid receptor antagonist treatment as determined by cardiac MRI. Representative long (A) and short (B) axis images of a normoxic control (CTRL) rat. In contrast, a long axis view of a SuHx rat reveals severe RV dilation and hypertrophy (C), whereas a short axis view demonstrates severe septal flattening (D). All images were taken at end-diastole. Quantification of ventricular function by cardiac MRI demonstrated a significant reduction in the RV ejection fraction (EF; E), whereas the left-ventricular (LV) EF (F) was significantly increased in all three groups of SuHx rats compared with CTRLs without a significant difference between the study groups prior to treatment (P ≥ 0.54 for each of the post hoc pairwise comparisons). Consistent with reduced RV function and impaired LV filling, the RV/LV end-diastolic volume (EDV; G) ratio was increased, whereas both the cardiac index (H) and stroke volume index (I) were reduced. There were no significant differences in RVEDV/LVEDV ratio, cardiac index, or stroke volume index between the three groups prior to treatment (P ≥ 0.20 for each of the post hoc pairwise comparisons). Data are presented as means ± SD of 4 healthy CTRLs and 13–15 animals per treatment group. One-way ANOVA was used to compare the four groups followed by post hoc Tukey’s test for pairwise contrasts. **P < 0.01, ***P < 0.001, ****P < 0.0001. EPL, eplerenone; PL, placebo; SPL, spironolactone.
Figure 3.

SU5416-hypoxia (SuHx) rats develop right ventricular (RV) pressure and volume overload. Representative short axis images at end-diastole (ED; A) and end-systole (ES; B) demonstrate the normal circular appearance of the left-ventricle (LV) in a healthy, normoxic control (CTRL) rat. In contrast, volume and pressure overload of the RV in SuHx rats results in flattening of the intraventricular septum at ED (C) and ES (D). SuHx rats had significant increases in the mean ED (E) and ES (F) eccentricity index compared with CTRL rats. However, there were no significant differences in the ED or ES eccentricity indices between the three groups prior treatment (P ≥ 0.52 for each of the post hoc pairwise comparisons). The eccentricity index refers to the ratio of the LV minor-axis diameter parallel to the septum (D2) divided by the LV minor-axis diameter perpendicular to the septum (D1). Data are presented as means ± SD of 4 normoxic CTRLs and 13–15 animals per treatment group. One-way ANOVA was used to compare the four groups followed by post hoc Tukey’s test for pairwise contrasts. ***P < 0.001, ****P < 0.0001. EPL, eplerenone; PL, placebo; SPL, spironolactone.
Serial Assessment of Serum Concentrations of Eplerenone, Spironolactone and/or Their Major Metabolites in SU5416-Hypoxia Rats
The study drug dose attained (mg/kg/day) was calculated in a subset of EPL- and SPL-treated animals based on the measured amount of feed consumed per cage per week and the known concentrations of EPL (2 mg/g) and SPL (0.8 mg/g) per gram of feed (Supplemental Table S5). Although there were periods during the study when goal doses were not met, the mean calculated daily dose over the 5-wk treatment was at least 85% of the target dose in more than 70% of the EPL- and SPL-treated animals monitored. To further assess study drug consumption over time in the SuHx PAH model, serum concentrations of CAN, one of the two main active metabolites of SPL, EPL, and two major metabolites of EPL were determined by LC-MS. Similar to levels reported in healthy rats administered a comparable dose of oral SPL (50 mg/kg) (38, 39), mean (±SD) CAN concentrations in SPL-treated animals were 181.8 ng/mL (±68.9), 188.7 ng/mL (±126.9), and 180.4 ng/mL (±121.9) in weeks 7, 8, and 9 respectively (Fig. 4A). Although relatively consistent drug levels were achieved from weeks 7 to 9 in SPL-treated animals (P = 0.86, one-way ANOVA), the mean CAN concentration at the end of week 10 was only 43.22 ng/mL (±16.8), significantly lower compared with each of the preceding weeks (P < 0.0001 for comparison across weeks 7–10, one-way ANOVA; P < 0.0001 for each post hoc pairwise comparison of weeks 7, 8, and 9 vs. week 10). Total mean (±SD) concentrations of EPL and its metabolites were 490.1 ng/mL (±20.6), 569.2 ng/mL (±236.0), 514.2 ng/mL (±287.2), 415.9 ng/mL (±192.7), and 298.2 ng/mL (±205.2) in weeks 6, 7, 8, 9, and 10, respectively (Fig. 4B), closely approximating levels in healthy male rats dosed 100 mg/kg orally for 13 wk (40). Although EPL levels were lower at the end of week 10 compared with week 7 (P = 0.03 for the post hoc pairwise comparison), there were no other significant differences in concentrations between weeks.
Figure 4.

Serum drug concentrations in the eplerenone (EPL) and spironolactone (SPL) treatment groups. A: serum concentrations of canrenone, a major SPL metabolite, were determined by liquid chromatography-mass spectrometry at week 5 (n = 15), week 6 (n = 0), week 7 (n = 11), week 8 (n = 15), week 9 (n = 11), and week 10 (n = 11). B: serum concentrations of EPL and its two major metabolites were determined by liquid chromatography-mass spectrometry at week 5 (n = 15), week 6 (n = 2), week 7 (n = 12), week 8 (n = 14), week 9 (n = 10), and week 10 (n = 13). As expected, neither EPL, EPL metabolites nor canrenone were detected in the placebo group (data not shown). Likewise, canrenone was not detected in serum samples from the EPL treatment group and EPL or its metabolites were not detected in serum samples from animals treated with SPL.
Mineralocorticoid Receptor Antagonists Prevented Worsening Right Ventricular Dysfunction
Following treatment randomization, a total of 8 SuHx rats died between weeks 6 and 10 (n = 2 in placebo group; n = 3 in both the EPL and SPL groups); however, there were no significant differences in mortality rates between groups (log-rank test, P = 0.94; Supplemental Fig. S4). After 5 wk of treatment, repeat cardiac MRI was performed in survivors (n = 11 in placebo group; n = 12 in both the EPL and SPL group) and changes from pretreatment were analyzed. Despite ongoing RV dysfunction, as first documented at the end of week 5, cardiac index was stable following treatment with either EPL or SPL (P = 0.04 and 0.03, respectively, for the difference between the effect of each MR antagonist vs. placebo; Fig. 5A). Mean stroke volume index was also maintained following treatment with EPL or SPL (P = 0.048 and 0.06, respectively, for each vs. placebo; Fig. 5B). In contrast, both mean cardiac index and stroke volume index continued to decline in placebo-treated SuHx rats. Importantly, there was no significant difference in mean heart rate used for determining cardiac index across the groups (P = 0.38; Supplemental Fig. S5A), further supporting that MR antagonist treatment effects on cardiac output reflect preservation of stroke volume. Cardiac index was calculated using the LV-derived stroke volume index and a single heart rate value taken during imaging of the mid-left ventricle where the greatest proportion of volume was measured. Notably, this single value was strongly correlated with mean and median heart rates recorded throughout the imaging sessions (r = 0.89, P < 0.0001 for both; Supplemental Fig. S5B).
Figure 5.

Treatment with mineralocorticoid receptor antagonists after the onset of right ventricular (RV) dysfunction blocks further decline in RV performance in SU5416-hypoxia (SuHx) rats. A: treatment of SuHx rats with either eplerenone (EPL; 100 mg/kg/day) or spironolactone (SPL; 40 mg/kg/day) prevented the further decline in cardiac index observed in placebo (PL) treated animals. B: the effect of MR antagonists on cardiac index was due to maintenance of the stroke volume (SV) index in EPL- and SPL-treated animals (P = 0.048 and 0.06, respectively, for each compared with placebo treatment) without a significant difference in heart rate among the three groups (Supplemental Fig. S5A). C: LV end-diastolic volume (LVEDV) index, which represents the LV filling volume normalized by body weight, improved with both SPL and EPL treatment. D: the RVEDV index did not change from pre- to posttreatment in any of three groups (P = 0.97). E: therefore, the improvement in the RVEDV/LVEDV ratio with EPL treatment was primarily due to the increase in LVEDV. Data are presented as means ± SD. Male, Sprague–Dawley rats were treated with SU-5416 (25 mg/kg) and exposed to hypoxia (10% ) for 3 wk, followed by normoxia for 7 wk. The effects of EPL and SPL vs. placebo (n = 11–12 per treatment group) on cardiac function were assessed by cardiac MRI at the end of weeks 5 (pretreatment) and 10 (posttreatment). Linear mixed models with random batch and animal effects were used to compare the changes between the three treatment groups. The horizontal dotted line intersecting the y-axis at 0 highlights no change in the given MRI parameter between weeks 5 and 10. *P < 0.05, **P ≤ 0.01. MR, mineralocorticoid receptor.
Eplerenone Reduced Ventricular Septal Flattening and Increased Left Ventricular Volume
Both MR antagonists increased the mean LVEDV index (P = 0.005 and 0.04 for EPL and SPL vs. placebo, respectively), whereas it declined further in placebo-treated animals, suggesting worsening LV filling over time in SuHx PAH rats (Fig. 5C). The change in mean RVEDV index was similar across the three groups (P = 0.97; Fig. 5D); however, the mean RVEDV/LVEDV ratio was reduced in EPL-treated rats (P = 0.02 for the difference between the effect of EPL treatment vs. placebo; Fig. 5E). Likewise, EPL significantly improved interventricular septal deviation at end-diastole and end-systole (P = 0.03 and 0.02, respectively; Fig. 6, A and B). Importantly, weight gain in all three groups was similar from week 5 to week 10 (P = 0.23; Supplemental Fig. S6A), arguing against a pronounced diuresis due to MR antagonist treatment. Despite effects on cardiac index, stroke volume index and LV filling, neither MR antagonist had a significant effect on RVEF compared with placebo (P = 0.70; Supplemental Fig. S6B). Changes in LVEF (P = 0.34; Supplemental Fig. S6C) and the Fulton index were also similar across all three groups (P = 0.38; Supplemental Fig. S6D).
Figure 6.

Eplerenone (EPL) ameliorated ventricular septal displacement on cardiac MRI. The eccentricity index (EI) at end-diastole (ED; A) and end-systole (ES; B) was lower in EPL-treated animals, indicating less displacement of the interventricular septum due to right ventricular (RV) volume and pressure overload, respectively. Mean eccentricity index at both ED and ES did not significantly improve in the spironolactone (SPL) treatment group (P ≥ 0.25 for each compared with the placebo group). Eccentricity index was calculated as the ratio of the diameter of the left ventricular (LV) minor axis parallel to the interventricular septum divided by the diameter of the LV minor axis perpendicular to the septum. Representative pre- and posttreatment, mid-ventricular images taken at ED (A) and ES (B) are shown to the left, and the corresponding animals are depicted as black dots in each plot. Data are presented as means ± SD of 11–12 animals per group. Linear mixed models with random batch and animal effects were used to compare the changes between the three treatment groups. The horizontal dotted line intersecting the y-axis at 0 highlights no change in the eccentricity index between weeks 5 and 10. *P < 0.05. PL, placebo.
Higher Serum Concentrations of Mineralocorticoid Receptor Antagonists Were Associated with Lower Right Ventricular Systolic Pressures and Reduced Total Pulmonary Vascular Resistance but Not Reversal of Pulmonary Vessel Remodeling
Neither mean RVSP, total pulmonary vascular resistance, nor the proportion of remodeled pulmonary vessels were significantly different among the three groups (P = 0.15, P = 0.25 and 0.19, respectively; Fig. 7, A–C) despite evidence of reduced pressure overload based on the end-systolic eccentricity index in EPL-treated SuHx PAH rats. Therefore, we performed an exploratory analysis based on serum drug concentrations over the course of the study. Animals were divided into two groups based on whether mean serum drug concentrations before catheterization (weeks 6–9) were above or below the group means. Mean (±SD) metabolite concentrations were 184.2 (±108.3) and 504.8 (±240.2) ng/mL in SPL- and EPL-treated animals, respectively, before catheterization. Both EPL- and SPL-treated SuHx rats with high mean drug levels had significantly lower RVSP compared to those with low mean drug levels (unpaired t test, P = 0.02 and 0.04, respectively; Fig. 7A). Total pulmonary resistance index (TPRI) was similarly lower in EPL-treated animals with high mean drug levels (unpaired t test, P = 0.05; Fig. 7B) whereas TPRI showed a nonsignificant trend in SPL animals with higher drug concentrations (unpaired t test, P = 0.09; Fig. 7B). Consistent with finding lower RVSP and TPRI in animals with high EPL concentrations, EPL-treated animals that gained more weight (i.e., consumed more EPL medicated diet) tended to have lower RVSPs (r = −0.68, P = 0.07; Supplemental Fig. S7). The percentage of grade 1 and grade 2 pulmonary arteriolar occlusive lesions (see Supplemental Fig. S1B for examples) in animals with high EPL levels tended to be less than those with low drug levels (unpaired t test P = 0.09; Fig. 7C), though the percentage of lesions was similar in SPL-treated animals with high and low drug levels (unpaired t test P = 0.47; Fig. 7C). Pulmonary arteriolar muscularization was similarly unaltered by MR antagonist treatment (P = 0.36; Supplemental Fig. S8). As expected, the mean LVEDP of each treatment group was within the normal range, and there were no significant differences across the three groups (P = 0.97; Supplemental Fig. S9). Mean systemic, systolic, and diastolic blood pressures were also similar among the three groups (P ≥ 0.26 for all; Supplemental Fig. S9). Collectively, these results suggest that the benefit of MR antagonists on stroke volume, LVEDV, and septal displacement in the SuHx PAH model may be due to direct, salutary effects on the RV in addition to any modest effects on pulmonary pressures.
Figure 7.
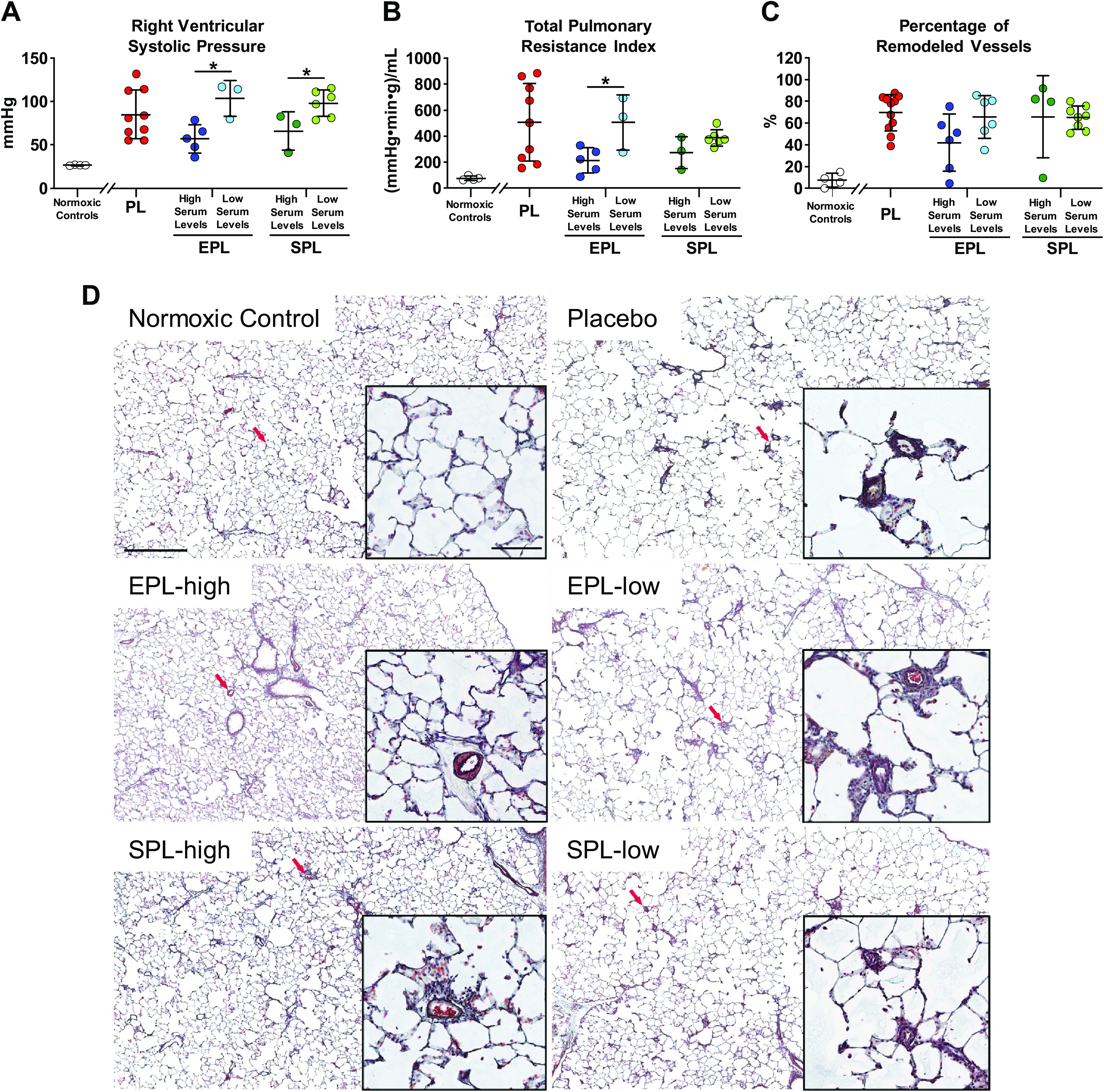
Treatment with mineralocorticoid receptor antagonists had a concentration-dependent effect on right ventricular systolic pressure (RVSP) and total pulmonary resistance index (TPRI) but not pulmonary vascular remodeling. A: RVSP was not significantly different between the three treatment groups (P = 0.15). However, in eplerenone (EPL)- and spironolactone (SPL)-treated animals, those with serum drug concentrations above the mean (high serum levels) had lower RVSPs compared to animals with drug concentrations below the mean (low serum levels; P = 0.02 and 0.04, respectively). RVSP was not successfully measured in some placebo- (n = 2), EPL- (n = 4), and SPL-treated (n = 3) animals. B: TPRI was also not significantly different between the three treatment groups (P = 0.24); however, EPL-treated animals with serum drug concentrations above the mean had lower TPRI compared with those with drug concentrations below the mean (P = 0.05). In contrast, there was a nonsignificant trend of lower TPRI in animals with high compared with low serum levels in the SPL treatment group (P = 0.09). C: pulmonary artery remodeling as defined as the percentage of grade 1 + 2 occlusion of small pulmonary arterioles (diameter < 100 µm; see methods and Supplemental Fig. S1B for details) was also not significantly different across the three treatment groups (P = 0.19). The percentage of remodeled vessels tended to be lower in animals with high versus low serum EPL concentrations (P = 0.09), whereas it was similar in SPL-treated animals with high versus low concentrations (P = 0.47). Data are presented as means ± SD of 8–9 animals per group for RVSP and TPRI, and 11–12 animals per group for histologic grading of vessel occlusion. One-way ANOVA was used to compare the three treatment groups and pooled t tests were used to compare the differences between the high and low serum level groups. Serum was collected between week 5 (pretreatment) and week 10. The mean concentration in each group was calculated based on measured concentrations after treatment initiation until prior to cardiac catheterization (weeks 6–9). See Fig. 4 for all measured concentrations. Normoxic controls (n = 4) are shown for comparison. *P ≤ 0.05. D: representative images of lung vessel histomorphology in a normoxic control (top left) and SuHx animals treated with either placebo (top right), EPL (high serum level: middle row, left; low serum level: middle row, right) or SPL (high serum level: bottom left; low serum level: bottom right). Red arrows indicate location of higher magnification image in inset. Scale bar: 500 and 100 µm (inset). PL, placebo.
Effects of Eplerenone on Mineralocorticoid Receptor Target Gene Induction Mirror Its Impact on Right Ventricular Inflammatory Gene Transcription
Pathological activation of MR target gene expression is closely associated with proinflammatory and profibrotic pathways (22). Therefore, we investigated whether induction of MR and inflammatory target gene expression in the RV was blocked by EPL or SPL treatment. Of the 12 MR target genes examined, 10 were induced in placebo-treated Su/Hx rat RV tissue samples compared with normoxic controls (Cyp1a1, Edn1, Rgs2, Hk1, Serpine1, Il1r1, Sgk1, Nampt, Nedd4, and Nr3c2; one-tailed signed-rank test P ≤ 0.1 for all; Supplemental Fig. S10A). Congruent with its effects on RV function, EPL reduced the expression of all 10 MR target genes induced in the RV of placebo-treated SuHx rats; yet only the effect on Cy1a1 reached statistical significance (mean fold-change difference between EPL- and placebo-treated animals: Cyp1a1 0.42, Edn1 0.80, Hk1 0.73, Il1r1 0.65, Nampt 0.59, Nedd4 0.83, Nr3c2 0.72, Rgs2 0.77, Serpine1 0.61, Sgk1 0.63; Fig. 8A). In contrast, possibly due to the fall in drug levels after week 10, SPL treatment had less impact on a smaller number of MR target genes (mean fold-change difference between SPL- and placebo-treated animals: Il1r1 0.91, Nampt 0.76, Nr3c2 0.93, Rgs2 0.98, Serpine1 0.84, Sgk1 0.73; Fig. 8A).
Figure 8.

Right ventricular (RV) mineralocorticoid receptor (MR) and inflammatory target gene transcription was suppressed by eplerenone (EPL) treatment. A: EPL consistently suppressed RV MR target gene induction, although only the reduction of Cyp1a1 reached statistical significance (P = 0.04). Although directionally similar to changes in EPL-treated animals, none of the changes in RV MR target gene expression were significantly different in the spironolactone (SPL) group compared with placebo-treated SuHx rats. Cyp1a1, Edn1, Rgs2, Hk1, Serpine1, Il1r1, Sgk1, Nampt, Nedd4, and Nr3c2 were induced in placebo-treated Su/Hx RV free wall tissue samples relative to normoxic control rat RVs (one-sample Wilcoxon signed-rank test P ≤ 0.1 for all; see Supplemental Fig. S9A for the relative expression of all MR target genes in the placebo group). B: EPL treatment demonstrated a modest and largely consistent overall suppressive effect on RV inflammatory target gene expression compared with placebo-treated SuHx rats, but none of the changes reached statistical significance. In contrast, SPL treatment had more variable effects on RV inflammatory target gene expression. Ccl2, Cola1, Ctgf, Cxcl10, Cxcl12, Cxcl9, Cx3cl1, Icam1, Lgals3, Plat, Ptgs2, Tgfb, Vcam1, and Xirp1 were induced in placebo-treated Su/Hx RV free wall tissue samples relative to normoxic controls (one-sample Wilcoxon signed-rank test P ≤ 0.1 for all; see Supplemental Fig. S9B. for the relative expression of all 22 inflammatory genes in the placebo group). Gene expression determined by quantitative real-time PCR is presented as geometric mean fold-change ± geometric SE of 11–12 animals per group. ΔΔCt values for EPL- and SPL-treated animals were calculated relative to placebo-treated animals within each experimental cycle. When placebo-treated animal samples were not available from within cycle samples (e.g., due to animal loss), placebo-treated animals in temporally adjacent cycles were used instead. Linear mixed models with random batch and animal effects were used to compare gene expression changes in EPL- and SPL-treated animals relative to the placebo group. The horizontal dashed line intersecting the y-axis at 1 highlights no change in mRNA expression relative to placebo-treated animals. *One-tailed P < 0.05 compared with placebo-treated animals.
Of the 22 inflammatory target genes profiled, 14 were induced in placebo-treated Su/Hx rat RV tissue samples compared with normoxic controls (Ccl2, Cola1, Ctgf, Cxcl10, Cxcl12, Cxcl9, Cx3cl1, Icam1, Lgals3, Plat, Ptgs2, Tgfb, Vcam1, and Xirp1; one-tailed signed-rank test P ≤ 0.1 for all; Supplemental Fig. S10B). Similar to its consistent suppression of RV MR gene induction, EPL treatment reduced expression of more than 85% of the inflammatory target genes induced in the RV of placebo-treated SuHx rats. However, no individual gene reached statistical significance (mean fold-change difference between EPL and placebo treated animals: Cola1 0.58, Ctgf 0.64, Cx3cl1 0.79, Cxcl9 0.87, Cxcl10 0.85, Cxcl12 0.84, Icam1 0.88, Lgals3 0.81, Plat 0.69, Tgfb 0.84, Vcam1 0.72, Xirp1 0.83; Fig. 8B). In contrast to EPL, SPL had no discernible effect on inflammatory genes induced in the RV (mean fold-change difference between SPL and placebo-treated animals: Cola1 0.98, Cx3cl1 0.96, Cxcl9 0.92, Cxcl12 0.95, Plat 0.95; Fig. 8B). Interestingly, in SPL-treated animals where drug levels were more variable across the group, suppression of MR and inflammatory target gene expression relative to placebo-treated animals was consistently greater in the animals with high serum levels compared with those with low serum levels (Supplemental Fig. S11). Although these post hoc analyses should be interpreted with caution given the small number of animals in the high and low groups, it is notable that the relative changes in gene expression compared with placebo in SPL-treated animals with high serum levels are consistent with the overall effect of EPL on gene expression observed in the main analysis. Despite the trend toward favorable gene expression changes with EPL, the degree of RV fibrosis was similar among the three groups (Supplemental Fig. S12).
Next, we determined whether inflammatory gene expression in Su/Hx PAH lung tissue was altered by MR antagonist treatment. Although more than a third of the 22 inflammatory target genes profiled in lung tissue homogenates were induced in placebo-treated Su/Hx rats compared with normoxic controls (Ccl2, Ccl3, Cxcl1, Cxcl2, Cxcl9, Lgals3, Tnf, and Xirp1; signed-rank test P ≤ 0.1 for all; Supplemental Fig. S13A), neither EPL nor SPL demonstrated a consistent effect on lung inflammatory gene expression (Supplemental Fig. S13B). Circulating cytokines and chemokines were also examined in plasma samples serially over time using a multiplex assay. In placebo-treated animals, there were no significant changes in the concentrations of the 23 inflammatory mediators between week 5 and weeks 8–10 (Supplemental Table S6). Moreover, neither EPL nor SPL treatment significantly altered plasma concentrations of any of the circulating cytokines and chemokines (Supplemental Table S6).
Absence of Spironolactone-Mediated Effects on In Vivo XPB Protein Expression
Previously, we demonstrated that SPL-induced proteasomal degradation of XPB suppressed proinflammatory cytokine production in cultured, primary human PAECs (34). Consistent with the lack of SPL-mediated suppression of inflammatory gene transcription in the RV and lung of Su/Hx PAH rats, XPB protein expression in these tissues was similar across the three treatment groups (P = 0.51 and 0.53, respectively; Fig. 9, A and B). Unlike SPL, its two major active metabolites, 7α-thiomethylspironolactone (7α-SPL) and CAN, did not suppress canonical NF-κB or AP-1 inflammatory pathway activation (P = 1.0 for the effect of 7α-SPL and CAN on reporter activity compared with vehicle control; Supplemental Fig. S14A) or induce XPB degradation in vitro (P ≥ 0.99 for the effect of 7α-SPL and CAN on XPB protein levels compared with vehicle control; Supplemental Fig. S14B). Furthermore, in cultured primary human PAECs, significant reduction in XPB protein levels only occurred at SPL concentrations ≥ 3 µM (Fig. 9C), several fold higher than the average measured serum concentrations of CAN achieved in SPL-treated Su/Hx rats [mean (±SD) 440 (±326) nM across all time points]. Lastly, XPB levels return to ∼50% of baseline 18 h after SPL treatment in vitro (Fig. 9D). Collectively, the inability of SPL metabolites to induce XPB-degradation, the relatively lower serum drug concentrations detected in vivo, and the potential recovery of XPB protein levels during the intervals between drug consumption by Su/Hx rats may explain preserved RV and lung XPB protein expression and the lack of anti-inflammatory effects through this mechanism in SPL-treated animals.
Figure 9.

Right ventricular (RV) and lung XPB protein expression is preserved in spironolactone (SPL)-treated SU5416-hypoxia (SuHx) rats. Protein expression of XPB in the RV (A) and lung (B) was not significantly different between the three treatment groups (P = 0.51 and P = 0.53). Tissue homogenates from the RV free wall and the caudal lobe of the right lung were resolved by SDS-PAGE and immunoblotted for XPB and GAPDH. In A and B, one-way ANOVA was used to compare the three treatment groups. C: XPB protein levels in primary human pulmonary artery endothelial cells (PAECs) were dose dependently reduced by SPL (P < 0.0001 for the slope of the dose-response curve). *P < 0.05, ****P ≤ 0.0001 for the pairwise contrasts of SPL treatment compared with vehicle control (CTRL). Whole cell lysates from PAECs were collected after a 2-h incubation with vehicle CTRL (DMSO) or increasing doses of SPL, resolved by SDS-PAGE and immunoblotted for XPB and β-actin. D: XPB levels in PAECs return to approximately 50% of baseline 18 h after SPL treatment in vitro. ****P < 0.0001 for the difference in XPB protein levels 18 h after SPL removal compared with 0 h. PAECs were treated with SPL (10 µM) for 2 h followed by replacement of the cell culture media with fresh media without SPL. Whole cell lysates were then collected at 0, 2, 4, 6, and 18 h after the media was changed, resolved by SDS-PAGE and immunoblotted for XPB and β-actin. Representative Western blots are shown below each of the densitometry plots. In C and D, dose- and time-dependent effects of SPL were analyzed by fitting a linear regression line. One-way ANOVA followed by Dunnett’s test was used to examine the effect of each dose and time point.
DISCUSSION
Using serial cine cardiac MRI, we demonstrated that MR antagonist treatment after the onset of significant RV dysfunction in the SuHx PAH model led to modest, but consistent beneficial effects on measures of interventricular dependence. Five weeks after model induction and before treatment initiation, cardiac imaging revealed a reduced RVEF, RV hypertrophy, pronounced diastolic and systolic septal flattening with impairment of LV filling, and reduced cardiac index in SuHx animals. In this setting, treatment with EPL or SPL improved LV filling and prevented the further decline in cardiac index. Likewise, cardiac MRI demonstrated an improvement in interventricular septal displacement in EPL-treated animals whereas concordant but less pronounced effects on the eccentricity index were seen in SPL-treated animals. Yet, despite these seemingly beneficial effects, there was no difference in survival between EPL- or SPL-treated animals and placebo controls. Drug and drug metabolite levels were determined by LC-MS over the course of the study to monitor treatment administration. Although MRI detected improvement in measures of load-dependent RV performance, MR antagonist treatment initiated after established disease in SuHx animals did not result in statistically significant reductions in either pulmonary artery pressure or vessel remodeling. Nonetheless, in exploratory analyses, animals with higher drug levels tended to have lower RVSPs and pulmonary resistance. Lastly, similar to its salutary effects on cardiac function, EPL treatment tended to suppress MR and proinflammatory target gene induction in the RV, although these transcript changes did not reach statistical significance. In contrast, inflammatory gene expression in whole lung tissue and circulating cytokine levels were not reduced by either MR antagonist. The results of our study provide evidence that MR antagonists may be beneficial in PAH even after the onset of RV dysfunction.
Clinically, cardiac MRI is recognized as the gold standard imaging modality for the quantitative evaluation of RV structure and function as well as determination of ventricular volumes (41). In addition, cardiac MRI-derived volumes of both the RV and LV have been previously demonstrated to predict mortality in patients with PAH (42–49). The feasibility and value of longitudinal cardiac MRI were recently demonstrated in the rat SuHx model (50). Although sequential imaging in the same animals was not performed in their study, the authors acknowledged that such studies would have advantages over traditional outcome measures of disease progression or therapeutic response (e.g., invasive hemodynamics, histology). Accordingly, application of serial cardiac MRI in our study offered high-resolution, quantitative determination of structural and functional changes in each individual animal over time in the rat SuHx PAH model. Specifically, cine cardiac MRI detected modest beneficial effects of MR antagonists on measures of interventricular dependence including cardiac index, the RVEDV/LVEDV ratio, and the degree of septal displacement.
In the current study, cardiac MRI-derived stroke volume index (and therefore cardiac index) was determined based on LV volumes. Indeed, prior studies have demonstrated that stroke volume and cardiac output measurements based on MRI-derived LV volumes correlate better with the invasive Fick method in patients with PAH compared with MRI-derived pulmonary artery flow or RV volumes (44, 51). However, due to the lack of comparable data in PAH/PH animal models, it is unknown whether LV volume-based calculation of stroke volume and cardiac output is superior to RV volumetric analysis in rodents. Decreases in LV stroke volume can result from reduced RV stroke volume and/or impairment of early LV diastolic filling due to significant septal flattening (52). Septal flattening, due to delayed peak RV free wall shortening or contraction, impairs LV filling and reduces stroke volume (53), thereby linking the observed effects of EPL on the eccentricity index, LVEDVI, and cardiac index demonstrated here. Although LV contractility was not specifically measured, increases in LVEDV can also increase myocardial stretch and thereby raise cardiac output due to improved contractility by the Frank–Starling mechanism (54). Despite increases in LVEDVI with EPL and SPL, we did not see concurrent decreases in RVEDVI. Similarly, previous clinical studies have demonstrated a lack of improvement in RVEDVI despite 12 mo of PAH-specific therapy (45, 55). This may be due to the greater reproducibility of LV volume measurements compared with the RV where endomyocardial border delineation and trabeculations may introduce more variability (44, 45). Alternatively, previous studies suggest that LVEDVI rather than RVEDVI may be a more informative measure of overall cardiac function (45, 52), highlighting the importance of biventricular assessment by cardiac MRI. Treatment-induced diuresis is unlikely to have contributed significantly to improvements in ventricular interdependence since mean weight gain was similar across the three groups over the duration of the study. In fact, RVSP was negatively correlated with weight gain in EPL-treated animals.
Prior preclinical studies have demonstrated that MR antagonist therapy initiated either at the outset of disease induction, or early thereafter, prevented the development of PAH (28–30, 32). Across these previous studies, early MR antagonist treatment consistently lowered pulmonary artery pressures and attenuated lung vessel remodeling. Yet patients with PAH usually present at a later stage of disease than has been simulated in these studies. Therefore, we examined the efficacy of MR antagonist therapy after the onset of RV dysfunction, as is the case for most patients with PAH. In contrast to these previous studies, later treatment with either EPL or SPL failed to significantly reduce mean RVSP, total pulmonary vascular resistance, or the proportion of remodeled pulmonary vessels. Whereas plexiform lesions are rare until the very late stage of the SuHx model (weeks 13–14) (31), the proportion of severe occlusive lesions in the distal pulmonary arterioles (<100 µm) peaks at week 5 (33). Therefore, even by week 5, vessel remodeling is advanced and likely more difficult to reverse compared with models where therapy was initiated earlier, before establishment of advanced disease. Indeed, compared with strategies aimed at disease reversal, experimental PAH/PH models are generally recognized as being more susceptible to prevention (56). For example, when started before monocrotaline-induced PAH, SPL significantly inhibited pulmonary vessel muscularization, but when SPL treatment was initiated 21 days later in the same model, vessel remodeling was unaffected (30). Notably, RVSP and pulmonary vascular remodeling were similarly unabated in SuHx mice despite either smooth muscle or endothelial cell-specific MR deletion (57). In contrast, systemic MR inhibition with SPL, initiated before SuHx-induced PAH, reduced both lung vascular remodeling and RVSP. Thus, both the timing of treatment and the cells or tissues targeted appear to impact the efficacy of MR antagonism in animal models of PAH (57).
In contrast to preclinical models of PH/PAH, MR antagonists failed to improve cardiac function in the pulmonary artery banding model of RV dysfunction where afterload is mechanically increased due to proximal pulmonary artery constriction without distal pulmonary vascular remodeling (32, 58). The authors of these studies concluded that in the setting of PH/PAH, any benefits of MR antagonists on the RV are largely indirect due to their salutary effects on the pulmonary vasculature. Notably in each of these studies, systemic blood pressure was significantly lower in the treatment group, and therefore, it is possible that RV ischemia may have confounded any direct therapeutic benefits on RV function. In contrast to treatment-induced hypotension in the pulmonary artery banding models, systemic BP was similar across the three treatment groups (placebo, EPL, or SPL) in the current study. Furthermore, the use of cine cardiac MRI in the current study enabled precise measurements of ventricular volumes, stroke volume, and interventricular septal deformation. Previous studies either did not assess these parameters or primarily relied on echocardiography, and therefore, subtle improvements may have gone undetected (32, 58). Notably, many of the measures of improved cardiac performance observed in our study are correlated with pulmonary pressure. Therefore, although not statistically significant, favorable effects of MR antagonists on the pulmonary circulation may in fact have contributed to their benefits on ventricular interdependence.
Doses of SPL (40 mg/kg/day) and EPL (100 mg/kg/day) used in our study were selected because of their minimal effect on systemic blood pressure (21, 59–63). These doses were either the same (30) or higher (28, 29) than those used in prior preclinical rat PAH studies that demonstrated the efficacy of MR antagonists when given early (28–30). Based on in vitro inhibition of aldosterone-mediated MR activation, SPL is 20- to 40-fold more potent than EPL; yet the epoxy modification of EPL’s steroid backbone results in significantly greater MR specificity (64, 65). In vivo, SPL is highly protein bound (>90%) but its major metabolites (CAN and 7α-SPL) are also active as MR antagonists (66). In contrast, EPL is only 50% protein bound and has no active metabolites (66). Despite significantly lower in vitro binding affinity, higher EPL bioavailability may explain its more effective blockade of renal [H3]-aldosterone binding and production of a higher urinary Na+/K+ ratio (a biomarker of anti-MR activity) in rats compared with SPL (64). Although these findings may not indicate their relative potency in extra-renal tissues such as the heart or vasculature, they are in agreement with the more pronounced EPL effects seen in our study. Conversely, in clinical trials directly comparing these two MR antagonists in patients with hypertension, SPL treatment produced significantly greater reductions in blood pressure when compared with equivalent doses of EPL (67–69). Nevertheless, the directionally similar effects of SPL on physiologic measures and MR target genes suggest that the more pronounced effects of EPL could merely be dose related.
Similar to our study, prior preclinical PAH studies administered EPL mixed with feed (28, 29, 32). In contrast, SPL was given to animals differently in the current study as compared with prior studies. In two studies, SPL (25 mg/kg/day) was given in the drinking water (28, 29), and a third study used subcutaneous, continuous release pellets (40 mg/kg/day for rats and 15 mg/kg/day for mice) (30). Although continuous release pellets may sustain stable drug levels, we selected a route of SPL administration that could be replicated with EPL. At the time of our study, there was no precedent for compounding EPL into subcutaneous release pellets and EPL cannot be dissolved in drinking water. Utilizing the same route of administration for both SPL and EPL enabled a proper comparison across the treatments, but perhaps introduced some uncertainty in dosing. To address this issue, relative concentrations of CAN, a SPL metabolite, EPL, and two major EPL metabolites were measured serially over time by LC-MS, and, in a subset of animals, study drug dose was calculated based on the amount of medicated feed consumed. Although blood sample collection could not be harmonized with drug consumption in each animal, the concentrations of each of the analytes measured are expected to have reached steady state based on their half-lives (66). Concentrations of EPL and major EPL metabolites consistently remained within the expected range (40) throughout the study. On the contrary, we detected a significant fall in CAN concentrations at the end of week 10. Despite this decline, both mean cardiac index and LV diastolic filling were maintained in SPL-treated animals, and in all but one animal, measured CAN concentrations at the end of the study were above those achievable in patients treated with clinically relevant doses of SPL (70). In addition, in both EPL- and SPL- treated animals, the mean daily dose was at least 85% of the target dose in more than 70% of the monitored animals.
Previously, we demonstrated that whole lung XPB protein levels were decreased by SPL treatment (40 mg/kg/day delivered by subcutaneous pellet) in a rat model of monocrotaline-induced PH (34). However, in the current study, protein levels of XPB were not decreased in either RV or lung tissue homogenates of SPL-treated animals where concentrations of CAN, a surrogate for SPL, fell significantly at week 10. In addition to the fall in serum drug levels, the half-life of SPL in vivo when delivered orally is fairly short (<2 h) (34). In accordance with prior studies, here we demonstrated that unlike SPL, CAN and 7α-SPL fail to promote XPB degradation (71, 72) and XPB protein levels recover within hours in the absence of SPL in vitro (73). Therefore, it is possible that the lack of XPB degradation observed in the current study might be explained by the inability of chow-based dosing to maintain consistent SPL concentrations as would be expected with continuous release pellets (30). Nevertheless, the absence of SPL-mediated suppression of proinflammatory genes in the RV and lung is in agreement with preserved XPB protein levels in these tissues.
In placebo-treated animals, plasma concentrations of 23 cytokines and chemokines measured at the end of week 10 were similar to week 5 levels. Likewise, treatment with EPL or SPL did not impact circulating inflammatory mediator levels. These findings are in line with a recent study, which demonstrated the relative paucity of systemic inflammation in both the monocrotaline and SuHx rat models of PAH as evidenced by low plasma levels of TNFα, IL-6, and MCP-1 (74). In contrast, we found increased inflammatory target gene expression in both the RV (Ccl2, Cola1, Ctgf, Cxcl10, Cxcl12, Cxcl9, Cx3cl1, Icam1, Lgals3, Plat, Ptgs2, Tgfb, Vcam1, and Xirp1) and lung tissue (Ccl2, Ccl3, Cxcl1, Cxcl2, Cxcl9, Lgals3, Tnf, and Xirp1) of SuHx rats. Although neither EPL or SPL consistently suppressed inflammatory gene induction in SuHx rat lung tissue, MR and inflammatory target genes were reduced by EPL in the RV, although only one gene met the threshold for statistical significance. In the context of improved cardiac performance with MR blockade, our finding of only modest RV gene expression changes without improvement in RV fibrosis is in line with a recent study that found delayed reversal of RV fibrosis even though measures of RV function, including stroke volume, septal flattening, and exercise duration, had significantly recovered (75). Nevertheless, the observed improvements in ventricular interdependence did not translate to improved survival in SuHx rats treated with MR antagonists after the onset of severe RV dysfunction.
There are some notable limitations to the current investigation. Right heart catheterization was successful in only a subset of animals (i.e., sample sizes for cardiac MRI were 18%–34% larger); therefore, it is possible that we were underpowered to detect differences in RVSP across treatment groups. Nonetheless, improvements in pulmonary vascular remodeling in EPL-treated animals did not reach significance even though lung histopathology was assessed in all animals that underwent cardiac MRI. In our exploratory analysis based on drug levels, the findings of lower mean RVSP (EPL and SPL groups) and total pulmonary resistance (EPL group) in animals with higher drug levels do not necessarily imply causality. Rather, these animals may simply have had less severe disease overall, consumed more medicated diet, and therefore had higher drug levels. Though, based on serial cardiac MRI in all animals, we observed effects on load-dependent parameters including cardiac index, stroke volume index and the end-systolic eccentricity index, which would be predicted to correlate with lower pulmonary artery pressures. Since experiments were conducted in male Sprague–Dawley rats to facilitate comparisons with prior preclinical studies, determination of sex-specific responses to MR antagonists is precluded. This is particularly relevant in the context of recent preclinical studies and data from patients that demonstrate increased aldosterone production in response to physiologic and pathophysiological stimuli, increased endothelial MR expression and heightened susceptibility to aldosterone-induced endothelial dysfunction in females compared with males (76–78). In addition, there is evidence to suggest that therapeutic responses to MR antagonists may be greater in females (79–81). Therefore, as PAH disproportionally affects women, future preclinical PAH studies of MR antagonists would benefit from including animals of both sexes. Considering the complexity of PAH in humans, a single preclinical model would not be expected to capture the nuances of disease heterogeneity in patients. The SuHx model was chosen for our study because disease progression and the underlying lung histopathology mirrors that seen in patients with PAH. Although serial cine cardiac MRI is likely to be informative in other preclinical PH/PAH models, it has limited availability and is more costly than other methods of investigation. Nonetheless, the reproducibility of the measures and the capability of performing serial exams in individual animals are of particular value when assessing both the efficacy and safety of experimental PH therapies.
A recent placebo-controlled crossover study demonstrated that SPL was safe and well-tolerated in PAH patients without any occurrences of hyperkalemia (82). Although treatment with SPL for 8 wk did not alter biomarkers of collagen metabolism, the primary end point of the study, it may be that a longer duration of treatment is necessary. Thus, in addition to prior preclinical studies (28–30, 32), the findings reported here support ongoing clinical trials evaluating the benefit of MR antagonists in patients with PAH (83) (NCT01712620, NCT03344159) and suggest that serial measures of RV structure and/or function (e.g., cardiac MRI, echocardiography, NT-proBNP) may be useful for determining efficacy.
DATA AVAILABILITY
Data will be made available on reasonable request. Please see the link to supplemental material in supplemental data.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.14673282;
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.14673291;
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.14673348;
Supplemental Fig. S4: https://doi.org/10.6084/m9.figshare.14673366;
Supplemental Fig. S5: https://doi.org/10.6084/m9.figshare.14673375;
Supplemental Fig. S6: https://doi.org/10.6084/m9.figshare.14673372;
Supplemental Fig. S7: https://doi.org/10.6084/m9.figshare.14673381;
Supplemental Fig. S8: https://doi.org/10.6084/m9.figshare.14673387;
Supplemental Fig. S9: https://doi.org/10.6084/m9.figshare.17014793;
Supplemental Fig. S10: https://doi.org/10.6084/m9.figshare.17014766;
Supplemental Fig. S11: https://doi.org/10.6084/m9.figshare.17014784;
Supplemental Fig. S12: https://doi.org/10.6084/m9.figshare.17014787;
Supplemental Fig. S13: https://doi.org/10.6084/m9.figshare.14673393;
Supplemental Fig. S14: https://doi.org/10.6084/m9.figshare.17014790;
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.14673399;
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.14673405;
Supplemental Table S3: https://doi.org/10.6084/m9.figshare.17000884;
Supplemental Table S4: https://doi.org/10.6084/m9.figshare.17000887;
Supplemental Table S5: https://doi.org/10.6084/m9.figshare.17000896;
Supplemental Table S6: https://doi.org/10.6084/m9.figshare.17000899.
GRANTS
This work was supported by intramural funds from the National Institutes of Health Clinical Center.
DISCLAIMERS
The opinions expressed in this article are those of the authors and do not represent any position or policy of the National Institutes of Health, the US Department of Health and Human Services, or the US government.
DISCLOSURES
The NIH Clinical Center PAH Program previously received support from Aadi Bioscience through a Cooperative Research and Development Agreement. The authors have no personal financial relationship with Aadi Bioscience or any other entity. Eplerenone (PF-02845980) was provided by the Pfizer Pure Compound Grants Program.
AUTHOR CONTRIBUTIONS
R.L.D. and J.M.E. conceived and designed research; M.L., L.-Y.C., S.G., A.J.M., S.A.A., J.N.H.N., A.N., M.A.H.S., E.J.D., Y.Z., K.A.J., Z.-X.Y., H.W., S.B.S. and J.M.E. performed experiments; M.L., S.A.A., S.W., and J.S. analyzed data; M.L., S.W., S.B.S., R.R.V., M.A.S., R.L.D. and J.M.E. interpreted results of experiments; M.L. and J.M.E prepared figures; M.L., R.L.D, and J.M.E. drafted manuscript; M.L., M.A.S., R.L.D, and J.M.E. edited and revised manuscript; M.L., L.-Y.C., S.G., A.J.M., S.A.A., J.N.H.N., A.N., M.A.H.S., E.J.D., Y.Z., K.A.J., Z.-X.Y., H.W., S.W., J.S., S.B.S., R.R.V., M.A.S., R.L.D. and J.M.E approved final version of manuscript.
ACKNOWLEDGMENTS
This research was made possible through the National Institutes of Health (NIH) Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation, Genentech, the American Association for Dental Research, the Colgate-Palmolive Company, and other private donors. The authors thank Kelly Byrne for editorial assistance and Dr. Sachindra Joshi for expert advice on cardiac catheterization in rats.
REFERENCES
- 1.Vonk Noordegraaf A, Chin KM, Haddad F, Hassoun PM, Hemnes AR, Hopkins SR, Kawut SM, Langleben D, Lumens J, Naeije R. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J 53: 1801900, 2019. doi: 10.1183/13993003.01900-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 53: 1801887, 2019. doi: 10.1183/13993003.01887-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 142: 448–456, 2012. doi: 10.1378/chest.11-1460. [DOI] [PubMed] [Google Scholar]
- 4.D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Kernis JT, Levy PS, Pietra GG, Reid LM, Reeves JT, Rich S, Vreim CE, Williams GW, Wu M. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 115: 343–349, 1991. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 5.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 151: 1628–1631, 1995. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 6.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, Walker C, Budd DC, Pepke-Zaba J, Morrell NW. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 122: 920–927, 2010. doi: 10.1161/CIRCULATIONAHA.109.933762. [DOI] [PubMed] [Google Scholar]
- 7.Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, Long CS, Voelkel NF, Nicolls MR. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 109: 867–879, 2011. doi: 10.1161/CIRCRESAHA.110.236927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, Tuder RM. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 261–272, 2012. doi: 10.1164/rccm.201201-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, Scheed A, Ritter C, Dahal BK, Vater A, Klussmann S, Ghofrani HA, Weissmann N, Klepetko W, Banat GA, Seeger W, Grimminger F, Schermuly RT. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 897–908, 2012. doi: 10.1164/rccm.201202-0335OC. [DOI] [PubMed] [Google Scholar]
- 10.Sweatt AJ, Hedlin HK, Balasubramanian V, Hsi A, Blum LK, Robinson WH, Haddad F, Hickey PM, Condliffe R, Lawrie A, Nicolls MR, Rabinovitch M, Khatri P, Zamanian RT. Discovery of distinct immune phenotypes using machine learning in pulmonary arterial hypertension. Circ Res 124: 904–919, 2019. doi: 10.1161/CIRCRESAHA.118.313911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tian W, Jiang X, Sung YK, Shuffle E, Wu TH, Kao PN, Tu AB, Dorfmüller P, Cao A, Wang L, Peng G, Kim Y, Zhang P, Chappell J, Pasupneti S, Dahms P, Maguire P, Chaib H, Zamanian R, Peters-Golden M, Snyder MP, Voelkel NF, Humbert M, Rabinovitch M, Nicolls MR. Phenotypically silent bone morphogenetic protein receptor 2 mutations predispose rats to inflammation-induced pulmonary arterial hypertension by enhancing the risk for neointimal transformation. Circulation 140: 1409–1425, 2019. doi: 10.1161/CIRCULATIONAHA.119.040629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elinoff JM, Mazer AJ, Cai R, Lu M, Graninger G, Harper B, Ferreyra GA, Sun J, Solomon MA, Danner RL. Meta-analysis of blood genome-wide expression profiling studies in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 318: L98–L111, 2020. doi: 10.1152/ajplung.00252.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prins KW, Archer SL, Pritzker M, Rose L, Weir EK, Sharma A, Thenappan T. Interleukin-6 is independently associated with right ventricular function in pulmonary arterial hypertension. J Heart Lung Transplant 37: 376–384, 2018. doi: 10.1016/j.healun.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paulin R, Sutendra G, Gurtu V, Dromparis P, Haromy A, Provencher S, Bonnet S, Michelakis ED. A miR-208-Mef2 axis drives the decompensation of right ventricular function in pulmonary hypertension. Circ Res 116: 56–69, 2015. doi: 10.1161/CIRCRESAHA.115.303910. [DOI] [PubMed] [Google Scholar]
- 15.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med 323: 236–241, 1990. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 16.Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 103: 2055–2059, 2001. doi: 10.1161/01.cir.103.16.2055. [DOI] [PubMed] [Google Scholar]
- 17.Briasoulis A, Androulakis E, Christophides T, Tousoulis D. The role of inflammation and cell death in the pathogenesis, progression and treatment of heart failure. Heart Fail Rev 21: 169–176, 2016. doi: 10.1007/s10741-016-9533-z. [DOI] [PubMed] [Google Scholar]
- 18.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res 110: 159–173, 2012. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 341: 709–717, 1999. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 20.Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleiman J, Gatlin M.; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators . Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 348: 1309–1321, 2003. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 21.Rocha R, Rudolph AE, Frierdich GE, Nachowiak DA, Kekec BK, Blomme EA, McMahon EG, Delyani JA. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol 283: H1802–H1810, 2002. doi: 10.1152/ajpheart.01096.2001. [DOI] [PubMed] [Google Scholar]
- 22.DuPont JJ, Jaffe IZ. 30 years of the mineralocorticoid receptor: the role of the mineralocorticoid receptor in the vasculature. J Endocrinol 234: T67–T82, 2017. doi: 10.1530/JOE-17-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maron BA, Leopold JA. Emerging concepts in the molecular basis of pulmonary arterial hypertension: part II: neurohormonal signaling contributes to the pulmonary vascular and right ventricular pathophenotype of pulmonary arterial hypertension. Circulation 131: 2079–2091, 2015. doi: 10.1161/CIRCULATIONAHA.114.006980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maron BA, Opotowsky AR, Landzberg MJ, Loscalzo J, Waxman AB, Leopold JA. Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur J Heart Fail 15: 277–283, 2013. doi: 10.1093/eurjhf/hfs173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Safdar Z, Thakur A, Singh S, Ji Y, Guffey D, Minard CG, Entman ML. Circulating aldosterone levels and disease severity in pulmonary arterial hypertension. J Pulm Respir Med 5: 295, 2015. doi: 10.4172/2161-105X.1000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, François C, Schalij I, Dorfmüller P, Simonneau G, Fadel E, Perros F, Boonstra A, Postmus PE, van der Velden J, Vonk-Noordegraaf A, Humbert M, Eddahibi S, Guignabert C. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 780–789, 2012. doi: 10.1164/rccm.201203-0411OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossi GP, Albertin G, Neri G, Andreis PG, Hofmann S, Pessina AC, Nussdorfer GG. Endothelin-1 stimulates steroid secretion of human adrenocortical cells ex vivo via both ETA and ETB receptor subtypes. J Clin Endocrinol Metab 82: 3445–3449, 1997. doi: 10.1210/jcem.82.10.4279. [DOI] [PubMed] [Google Scholar]
- 28.Maron BA, Zhang YY, White K, Chan SY, Handy DE, Mahoney CE, Loscalzo J, Leopold JA. Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation 126: 963–974, 2012. doi: 10.1161/CIRCULATIONAHA.112.094722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maron BA, Oldham WM, Chan SY, Vargas SO, Arons E, Zhang YY, Loscalzo J, Leopold JA. Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation 130: 168–179, 2014. doi: 10.1161/CIRCULATIONAHA.113.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Preston IR, Sagliani KD, Warburton RR, Hill NS, Fanburg BL, Jaffe IZ. Mineralocorticoid receptor antagonism attenuates experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 304: L678–L688, 2013. doi: 10.1152/ajplung.00300.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121: 2747–2754, 2010. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 32.Boehm M, Arnold N, Braithwaite A, Pickworth J, Lu C, Novoyatleva T, Kiely DG, Grimminger F, Ghofrani HA, Weissmann N, Seeger W, Lawrie A, Schermuly RT, Kojonazarov B. Eplerenone attenuates pathological pulmonary vascular rather than right ventricular remodeling in pulmonary arterial hypertension. BMC Pulm Med 18: 41, 2018. doi: 10.1186/s12890-018-0604-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toba M, Alzoubi A, O'Neill KD, Gairhe S, Matsumoto Y, Oshima K, Abe K, Oka M, McMurtry IF. Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmonary arterial hypertensive rats. Am J Physiol Heart Circ Physiol 306: H243–H250, 2014. doi: 10.1152/ajpheart.00728.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elinoff JM, Chen LY, Dougherty EJ, Awad KS, Wang S, Biancotto A, Siddiqui AH, Weir NA, Cai R, Sun J, Preston IR, Solomon MA, Danner RL. Spironolactone-induced degradation of the TFIIH core complex XPB subunit suppresses NF-κB and AP-1 signalling. Cardiovasc Res 114: 65–76, 2018. doi: 10.1093/cvr/cvx198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan T, Petrovic O, Dillon JC, Feigenbaum H, Conley MJ, Armstrong WF. An echocardiographic index for separation of right ventricular volume and pressure overload. J Am Coll Cardiol 5: 918–927, 1985. doi: 10.1016/s0735-1097(85)80433-2. [DOI] [PubMed] [Google Scholar]
- 36.Siddique MAH, Satoh K, Kurosawa R, Kikuchi N, Elias-Al-Mamun M, Omura J, Satoh T, Nogi M, Sunamura S, Miyata S, Ueda H, Tokuyama H, Shimokawa H. Identification of emetine as a therapeutic agent for pulmonary arterial hypertension: novel effects of an old drug. Arterioscler Thromb Vasc Biol 39: 2367–2385, 2019. doi: 10.1161/ATVBAHA.119.313309. [DOI] [PubMed] [Google Scholar]
- 37.Dougherty EJ, Elinoff JM, Ferreyra GA, Hou A, Cai R, Sun J, Blaine KP, Wang S, Danner RL. Mineralocorticoid receptor (MR) trans-activation of inflammatory AP-1 signaling: dependence on DNA sequence, MR conformation, and AP-1 family member expression. J Biol Chem 291: 23628–23644, 2016. doi: 10.1074/jbc.M116.732248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erbler HC. Effect of spironolactone and its main metabolite canrenone on the renin-angiotensin-aldosterone-system during long-term treatment in rats. Acta Endocrinol (Copenh) 90: 147–156, 1979. doi: 10.1530/acta.0.0900147. [DOI] [PubMed] [Google Scholar]
- 39.Kaukonen AM, Vuorela P, Vuorela H, Mannermaa JP. High-performance liquid chromatography methods for the separation and quantitation of spironolactone and its degradation products in aqueous formulations and of its metabolites in rat serum. J Chromatogr A 797: 271–281, 1998. doi: 10.1016/S0021-9673(97)01167-9. [DOI] [PubMed] [Google Scholar]
- 40.Cook CS, Zhang L, Ames GB, Fischer J, Zhang J, Levin S. Single- and repeated-dose pharmacokinetics of eplerenone, a selective aldosterone receptor blocker, in rats. Xenobiotica 33: 305–321, 2003. doi: 10.1080/0049825021000049277. [DOI] [PubMed] [Google Scholar]
- 41.Kiely DG, Levin D, Hassoun P, Ivy DD, Jone PN, Bwika J, Kawut SM, Lordan J, Lungu A, Mazurek J, Moledina S, Olschewski H, Peacock A, Puri GD, Rahaghi F, Schafer M, Schiebler M, Screaton N, Tawhai M, Van Beek EJ, Vonk-Noordegraaf A, Vanderpool RR, Wort J, Zhao L, Wild J, Vogel-Claussen J, Swift AJ. EXPRESS: statement on imaging and pulmonary hypertension from the Pulmonary Vascular Research Institute (PVRI). Pulm Circ 9: 1–32, 2019. doi: 10.1177/2045894019841990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Wolferen SA, Marcus JT, Boonstra A, Marques KM, Bronzwaer JG, Spreeuwenberg MD, Postmus PE, Vonk-Noordegraaf A. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J 28: 1250–1257, 2007. doi: 10.1093/eurheartj/ehl477. [DOI] [PubMed] [Google Scholar]
- 43.van de Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, Boonstra A, Marques KM, Westerhof N, Vonk-Noordegraaf A. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 58: 2511–2519, 2011. doi: 10.1016/j.jacc.2011.06.068. [DOI] [PubMed] [Google Scholar]
- 44.Swift AJ, Rajaram S, Campbell MJ, Hurdman J, Thomas S, Capener D, Elliot C, Condliffe R, Wild JM, Kiely DG. Prognostic value of cardiovascular magnetic resonance imaging measurements corrected for age and sex in idiopathic pulmonary arterial hypertension. Circ Cardiovasc Imaging 7: 100–106, 2014. doi: 10.1161/CIRCIMAGING.113.000338. [DOI] [PubMed] [Google Scholar]
- 45.Peacock AJ, Crawley S, McLure L, Blyth KG, Vizza CD, Poscia R, Francone M, Iacucci I, Olschewski H, Kovacs G, Vonk Noordegraaf A, Marcus JT, van de Veerdonk MC, Oosterveer FP. Changes in right ventricular function measured by cardiac magnetic resonance imaging in patients receiving pulmonary arterial hypertension-targeted therapy: the EURO-MR study. Circ Cardiovasc Imaging 7: 107–114, 2014. doi: 10.1161/CIRCIMAGING.113.000629. [DOI] [PubMed] [Google Scholar]
- 46.Swift AJ, Capener D, Johns C, Hamilton N, Rothman A, Elliot C, Condliffe R, Charalampopoulos A, Rajaram S, Lawrie A, Campbell MJ, Wild JM, Kiely DG. Magnetic resonance imaging in the prognostic evaluation of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 196: 228–239, 2017. doi: 10.1164/rccm.201611-2365OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewis RA, Johns CS, Cogliano M, Capener D, Tubman E, Elliot CA, Charalampopoulos A, Sabroe I, Thompson AAR, Billings CG, Hamilton N, Baster K, Laud PJ, Hickey PM, Middleton J, Armstrong IJ, Hurdman JA, Lawrie A, Rothman AMK, Wild JM, Condliffe R, Swift AJ, Kiely DG. Identification of cardiac magnetic resonance imaging thresholds for risk stratification in pulmonary arterial hypertension. Am J Respir Crit Care Med 201: 458–468, 2020. doi: 10.1164/rccm.201909-1771OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baggen VJ, Leiner T, Post MC, van Dijk AP, Roos-Hesselink JW, Boersma E, Habets J, Sieswerda GT. Cardiac magnetic resonance findings predicting mortality in patients with pulmonary arterial hypertension: a systematic review and meta-analysis. Eur Radiol 26: 3771–3780, 2016. doi: 10.1007/s00330-016-4217-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alabed S, Shahin Y, Garg P, Alandejani F, Johns CS, Lewis RA, Condliffe R, Wild JM, Kiely DG, Swift AJ. Cardiac-MRI predicts clinical worsening and mortality in pulmonary arterial hypertension: a systematic review and meta-analysis. JACC Cardiovasc Imaging 14: 931–942, 2021. doi: 10.1016/j.jcmg.2020.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jayasekera G, Wilson KS, Buist H, Woodward R, Uckan A, Hughes C, Nilsen M, Church AC, Johnson MK, Gallagher L, Mullin J, MacLean MR, Holmes WM, Peacock AJ, Welsh DJ. Understanding longitudinal biventricular structural and functional changes in a pulmonary hypertension Sugen-hypoxia rat model by cardiac magnetic resonance imaging. Pulm Circ 10: 2045894019897513, 2020. doi: 10.1177/2045894019897513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mauritz GJ, Marcus JT, Boonstra A, Postmus PE, Westerhof N, Vonk-Noordegraaf A. Non-invasive stroke volume assessment in patients with pulmonary arterial hypertension: left-sided data mandatory. J Cardiovasc Magn Reson 10: 51, 2008. doi: 10.1186/1532-429X-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gan C, Lankhaar JW, Marcus JT, Westerhof N, Marques KM, Bronzwaer JG, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Impaired left ventricular filling due to right-to-left ventricular interaction in patients with pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 290: H1528–H1533, 2006. doi: 10.1152/ajpheart.01031.2005. [DOI] [PubMed] [Google Scholar]
- 53.Marcus JT, Gan CT, Zwanenburg JJ, Boonstra A, Allaart CP, Gotte MJ, Vonk-Noordegraaf A. Interventricular mechanical asynchrony in pulmonary arterial hypertension: left-to-right delay in peak shortening is related to right ventricular overload and left ventricular underfilling. J Am Coll Cardiol 51: 750–757, 2008. doi: 10.1016/j.jacc.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 54.Marcus JT, Vonk Noordegraaf A, Roeleveld RJ, Postmus PE, Heethaar RM, Van Rossum AC, Boonstra A. Impaired left ventricular filling due to right ventricular pressure overload in primary pulmonary hypertension: noninvasive monitoring using MRI. Chest 119: 1761–1765, 2001. doi: 10.1378/chest.119.6.1761. [DOI] [PubMed] [Google Scholar]
- 55.van de Veerdonk MC, Huis In T Veld AE, Marcus JT, Westerhof N, Heymans MW, Bogaard H-J, Vonk-Noordegraaf A. Upfront combination therapy reduces right ventricular volumes in pulmonary arterial hypertension. Eur Respir J 49: 1700007, 2017. doi: 10.1183/13993003.00007-2017. [DOI] [PubMed] [Google Scholar]
- 56.Provencher S, Archer SL, Ramirez FD, Hibbert B, Paulin R, Boucherat O, Lacasse Y, Bonnet S. Standards and methodological rigor in pulmonary arterial hypertension preclinical and translational research. Circ Res 122: 1021–1032, 2018. doi: 10.1161/CIRCRESAHA.117.312579. [DOI] [PubMed] [Google Scholar]
- 57.Menon DP, Qi G, Kim SK, Moss ME, Penumatsa KC, Warburton RR, Toksoz D, Wilson J, Hill NS, Jaffe IZ, Preston IR. Vascular cell-specific roles of mineralocorticoid receptors in pulmonary hypertension. Pulm Circ 11: 20458940211025240, 2021. doi: 10.1177/20458940211025240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Borgdorff MA, Bartelds B, Dickinson MG, Steendijk P, Berger RA. cornerstone of heart failure treatment is not effective in experimental right ventricular failure. Int J Cardiol 169: 183–189, 2013. doi: 10.1016/j.ijcard.2013.08.102. [DOI] [PubMed] [Google Scholar]
- 59.Benetos A, Lacolley P, Safar ME. Prevention of aortic fibrosis by spironolactone in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol 17: 1152–1156, 1997. doi: 10.1161/01.atv.17.6.1152. [DOI] [PubMed] [Google Scholar]
- 60.Lacolley P, Safar ME, Lucet B, Ledudal K, Labat C, Benetos A. Prevention of aortic and cardiac fibrosis by spironolactone in old normotensive rats. J Am Coll Cardiol 37: 662–667, 2001. doi: 10.1016/s0735-1097(00)01129-3. [DOI] [PubMed] [Google Scholar]
- 61.Wahed MI, Watanabe K, Ma M, Yamaguchi K, Takahashi T, Tachikawa H, Kodama M, Aizawa Y. Effects of eplerenone, a selective aldosterone blocker, on the progression of left ventricular dysfunction and remodeling in rats with dilated cardiomyopathy. Pharmacology 73: 81–88, 2005. doi: 10.1159/000081267. [DOI] [PubMed] [Google Scholar]
- 62.Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 67: 97–105, 2005. doi: 10.1016/j.cardiores.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 63.Sakurabayashi-Kitade S, Aoka Y, Nagashima H, Kasanuki H, Hagiwara N, Kawana M. Aldosterone blockade by Spironolactone improves the hypertensive vascular hypertrophy and remodeling in angiotensin II overproducing transgenic mice. Atherosclerosis 206: 54–60, 2009. doi: 10.1016/j.atherosclerosis.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 64.de Gasparo M, Joss U, Ramjoué HP, Whitebread SE, Haenni H, Schenkel L, Kraehenbuehl C, Biollaz M, Grob J, Schmidlin J. and Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 240: 650–656, 1987. [PubMed] [Google Scholar]
- 65.Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 217: 27–31, 2004. doi: 10.1016/j.mce.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 66.Kolkhof P, Bärfacker L. 30 years of the mineralocorticoid receptor: mineralocorticoid receptor antagonists: 60 years of research and development. J Endocrinol 234: T125–T140, 2017. doi: 10.1530/JOE-16-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 15: 709–716, 2002. doi: 10.1016/S0895-7061(02)02957-6. [DOI] [PubMed] [Google Scholar]
- 68.Parthasarathy HK, Ménard J, White WB, Young WF Jr, Williams GH, Williams B, Ruilope LM, McInnes GT, Connell JM, MacDonald TM. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 29: 980–990, 2011. doi: 10.1097/HJH.0b013e3283455ca5. [DOI] [PubMed] [Google Scholar]
- 69.Roush GC, Ernst ME, Kostis JB, Yeasmin S, Sica DA. Dose doubling, relative potency, and dose equivalence of potassium-sparing diuretics affecting blood pressure and serum potassium: systematic review and meta-analyses. J Hypertens 34: 11–19, 2016. doi: 10.1097/HJH.0000000000000762. [DOI] [PubMed] [Google Scholar]
- 70.de Denus S, O'Meara E, Desai AS, Claggett B, Lewis EF, Leclair G, Jutras M, Lavoie J, Solomon SD, Pitt B, Pfeffer MA, Rouleau JL. Spironolactone metabolites in TOPCAT—new insights into regional variation. N Engl J Med 376: 1690–1692, 2017. doi: 10.1056/NEJMc1612601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kemp MG, Krishnamurthy S, Kent MN, Schumacher DL, Sharma P, Excoffon K, Travers JB. Spironolactone depletes the XPB protein and inhibits DNA damage responses in UVB-irradiated human skin. J Invest Dermatol 139: 448–454, 2019. doi: 10.1016/j.jid.2018.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ueda M, Matsuura K, Kawai H, Wakasugi M, Matsunaga T. Spironolactone-induced XPB degradation depends on CDK7 kinase and SCF(FBXL18) E3 ligase. Genes Cells 24: 284–296, 2019. doi: 10.1111/gtc.12674. [DOI] [PubMed] [Google Scholar]
- 73.Alekseev S, Ayadi M, Brino L, Egly JM, Larsen AK, Coin F. A small molecule screen identifies an inhibitor of DNA repair inducing the degradation of TFIIH and the chemosensitization of tumor cells to platinum. Chem Biol 21: 398–407, 2014. doi: 10.1016/j.chembiol.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 74.Schlosser K, Taha M, Deng Y, Jiang B, McIntyre LA, Mei SH, Stewart DJ. Lack of elevation in plasma levels of pro-inflammatory cytokines in common rodent models of pulmonary arterial hypertension: questions of construct validity for human patients. Pulm Circ 7: 476–485, 2017. doi: 10.1177/2045893217705878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boehm M, Tian X, Mao Y, Ichimura K, Dufva MJ, Ali K, Dannewitz Prosseda S, Shi Y, Kuramoto K, Reddy S, Kheyfets VO, Metzger RJ, Spiekerkoetter E. Delineating the molecular and histological events that govern right ventricular recovery using a novel mouse model of pulmonary artery de-banding. Cardiovasc Res 116: 1700–1709, 2020. doi: 10.1093/cvr/cvz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shukri MZ, Tan JW, Manosroi W, Pojoga LH, Rivera A, Williams JS, Seely EW, Adler GK, Jaffe IZ, Karas RH, Williams GH, Romero JR. Biological sex modulates the adrenal and blood pressure responses to angiotensin II. Hypertension 71: 1083–1090, 2018. doi: 10.1161/HYPERTENSIONAHA.117.11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Faulkner JL, Kennard S, Huby AC, Antonova G, Lu Q, Jaffe IZ, Patel VS, Fulton DJR, Belin de Chantemele EJ. Progesterone predisposes females to obesity-associated leptin-mediated endothelial dysfunction via upregulating endothelial MR (mineralocorticoid receptor) expression. Hypertension 74: 678–686, 2019. doi: 10.1161/HYPERTENSIONAHA.119.12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Faulkner JL, Lluch E, Kennard S, Antonova G, Jaffe IZ, Belin de Chantemèle EJ. Selective deletion of endothelial mineralocorticoid receptor protects from vascular dysfunction in sodium-restricted female mice. Biol Sex Differ 11: 64, 2020. doi: 10.1186/s13293-020-00340-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kanashiro-Takeuchi RM, Heidecker B, Lamirault G, Dharamsi JW, Hare JM. Sex-specific impact of aldosterone receptor antagonism on ventricular remodeling and gene expression after myocardial infarction. Clin Transl Sci 2: 134–142, 2009. doi: 10.1111/j.1752-8062.2009.00094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khosla N, Kalaitzidis R, Bakris GL. Predictors of hyperkalemia risk following hypertension control with aldosterone blockade. Am J Nephrol 30: 418–424, 2009. doi: 10.1159/000237742. [DOI] [PubMed] [Google Scholar]
- 81.Merrill M, Sweitzer NK, Lindenfeld J, Kao DP. Sex differences in outcomes and responses to spironolactone in heart failure with preserved ejection fraction: a secondary analysis of TOPCAT trial. JACC Heart Fail 7: 228–238, 2019. doi: 10.1016/j.jchf.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Safdar Z, Frost A, Basant A, Deswal A, O'Brian Smith E, Entman M. Spironolactone in pulmonary arterial hypertension: results of a cross-over study. Pulm Circ 10: 2045894019898030, 2020. doi: 10.1177/2045894019898030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Elinoff JM, Rame JE, Forfia PR, Hall MK, Sun J, Gharib AM, Abd-Elmoniem K, Graninger G, Harper B, Danner RL, Solomon MA. A pilot study of the effect of spironolactone therapy on exercise capacity and endothelial dysfunction in pulmonary arterial hypertension: study protocol for a randomized controlled trial. Trials 14: 91, 2013. doi: 10.1186/1745-6215-14-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.14673282;
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.14673291;
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.14673348;
Supplemental Fig. S4: https://doi.org/10.6084/m9.figshare.14673366;
Supplemental Fig. S5: https://doi.org/10.6084/m9.figshare.14673375;
Supplemental Fig. S6: https://doi.org/10.6084/m9.figshare.14673372;
Supplemental Fig. S7: https://doi.org/10.6084/m9.figshare.14673381;
Supplemental Fig. S8: https://doi.org/10.6084/m9.figshare.14673387;
Supplemental Fig. S9: https://doi.org/10.6084/m9.figshare.17014793;
Supplemental Fig. S10: https://doi.org/10.6084/m9.figshare.17014766;
Supplemental Fig. S11: https://doi.org/10.6084/m9.figshare.17014784;
Supplemental Fig. S12: https://doi.org/10.6084/m9.figshare.17014787;
Supplemental Fig. S13: https://doi.org/10.6084/m9.figshare.14673393;
Supplemental Fig. S14: https://doi.org/10.6084/m9.figshare.17014790;
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.14673399;
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.14673405;
Supplemental Table S3: https://doi.org/10.6084/m9.figshare.17000884;
Supplemental Table S4: https://doi.org/10.6084/m9.figshare.17000887;
Supplemental Table S5: https://doi.org/10.6084/m9.figshare.17000896;
Supplemental Table S6: https://doi.org/10.6084/m9.figshare.17000899.
Data Availability Statement
Data will be made available on reasonable request. Please see the link to supplemental material in supplemental data.