

Keywords: bone, cancer, hormonal signals, mechanical signals, Sclerostin
Abstract
Osteocytes, former osteoblasts encapsulated by mineralized bone matrix, are far from being passive and metabolically inactive bone cells. Instead, osteocytes are multifunctional and dynamic cells capable of integrating hormonal and mechanical signals and transmitting them to effector cells in bone and in distant tissues. Osteocytes are a major source of molecules that regulate bone homeostasis by integrating both mechanical cues and hormonal signals that coordinate the differentiation and function of osteoclasts and osteoblasts. Osteocyte function is altered in both rare and common bone diseases, suggesting that osteocyte dysfunction is directly involved in the pathophysiology of several disorders affecting the skeleton. Advances in osteocyte biology initiated the development of novel therapeutics interfering with osteocyte-secreted molecules. Moreover, osteocytes are targets and key distributors of biological signals mediating the beneficial effects of several bone therapeutics used in the clinic. Here we review the most recent discoveries in osteocyte biology demonstrating that osteocytes regulate bone homeostasis and bone marrow fat via paracrine signaling, influence body composition and energy metabolism via endocrine signaling, and contribute to the damaging effects of diabetes mellitus and hematologic and metastatic cancers in the skeleton.
CLINICAL HIGHLIGHTS
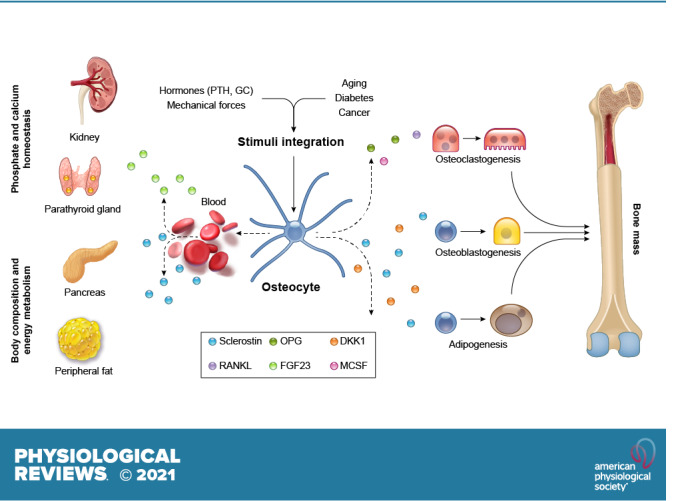
Osteocytes are the most abundant cells in bone and live deep in the mineralized bone matrix. Osteocytes have extensive long cytoplasmic extensions that form a complex network that enables direct communication among osteocytes and with other effector cells in the bone/bone marrow.
Osteocytes respond to mechanical and hormonal stimuli and, in turn, secrete molecules that affect cells of the bone/bone marrow niche by paracrine mechanisms and cells of distant organs by endocrine mechanisms.
Clinical evidence indicates that osteocyte viability and function are altered in common and rare bone diseases, supporting a pathophysiological role of osteocyte-derived molecules in skeletal diseases.
Cancer cells communicate with osteocytes to transform the bone marrow niche into an environment supportive of tumor cell proliferation and bone destruction. Cancer cells have proapoptotic effects on osteocytes and stimulate the production of osteocyte-derived osteoclastogenic cytokines and inhibitors of bone formation.
Therapeutic targeting of osteocytes and their derived factors (Sclerostin, RANKL, and FGF23) has proven to be an effective approach to treat common and rare bone diseases, as well as the deleterious effects of cancer in bone.
1. INTRODUCTION
Scientific advances in the last 25 years demonstrate the profound impact that osteocytes have on bone biology and pathophysiology. The vast advancement in the knowledge about osteocytes and their role in bone homeostasis started in the late 1990s, which was prompted by the discovery of rare skeletal diseases in humans linked to osteocyte dysfunction, the generation of osteocyte-like cell lines, and the development of genetic tools to manipulate in vivo gene expression in osteocytes. Today, it is recognized that osteocytes orchestrate osteoblast and osteoclast function, integrate and transmit hormonal and mechanical cues, regulate the differentiation and activity of bone/bone marrow cells via paracrine signaling, and influence the function of cells in distant tissues by secreting factors that act as endocrine signals. This boom in scientific research prompted the development and commercialization of therapies targeting osteocytes for the treatment of bone diseases, which prevent bone loss and/or increase bone mass and strength.
2. OSTEOCYTOGENESIS AND DEVELOPMENT OF THE OSTEOCYTIC GENE SIGNATURE
2.1. Osteocyte Differentiation from Osteoblasts
Osteocytes are the most fully differentiated cell type of the osteoblastic lineage. Driven by mechanisms still poorly understood, some osteoblasts on the bone surface become surrounded by the matrix proteins they produce and differentiate into osteocytes. Osteoblasts progressively decrease bone matrix production, undergo dramatic changes in morphology, and start expressing particular genes that constitute the osteocyte signature (1–3). Approximately 5–20% of osteoblasts undergo terminal differentiation and become osteocytes. The remainder either die by apoptosis or flatten and turn into cells, known as bone lining cells, that cover the quiescent surfaces of bone. During the process of osteocytogenesis, osteoblast morphology changes to the stellate morphology characteristic of osteocytes. This process is also characterized by an autophagy-mediated (4) reduction in endoplasmic reticulum area and number of mitochondria (5) and the development of cytoplasmic projections. These long cytoplasmic processes run through tunnels carved in the bone mineral known as canaliculi, allowing the osteocytes to physically interact with each other as well as to distribute molecules to adjacent osteocytes and other cells on the bone surface or in the bone marrow compartment (FIGURE 1). During osteoblast-to-osteocyte differentiation, major changes in transcription and translation lead to the expression in osteocytes of molecules that regulate bone homeostasis and phosphate metabolism (2). Different genes have been identified as responsible for the changes in cell morphology and the formation of the complex lacuno-canalicular network. Expression of Podoplanin/E11 is required by osteoid osteocytes (6) to initiate proper dendrite formation (7–9). The osteoblast-to-osteocyte transition is accompanied by increased expression of Dentin matrix protein 1 (DMP1) (10), which is essential for osteocyte maturation (11). The expression of matrix metalloproteinases (MMPs) also increases during osteoblast-osteocyte differentiation, enabling the formation and elongation of dendritic projections (12, 13). Connexin 43 (Cx43) allows communication between adjacent osteocytes and between osteocytes and the bone/bone marrow compartment, preserves osteocyte viability, and mediates osteocyte transduction of mechanical signals (14–16).
FIGURE 1.

Osteocyte morphology and lacuno-canalicular network. A: osteocytes are highly interconnected to each other and to cells on the bone surface. Canaliculi connect osteocytes to the endocortical bone surface and the bone marrow compartment. Electron microscopy imaging of an acid etched bone. B: osteocyte-derived factors circulate through the canalicular system to reach other osteocytes and bone cells on the bone surface or in bone marrow. Immunostaining of Sclerostin in paraffin-embedded bone tissue. C: MLO-Y4 osteocyte-like cells display a stellate morphology with dendritic processes resembling those found in primary osteocytes. White arrows point to individual osteocytes; yellow arrows point to canaliculi.
Osteocytogenesis is also characterized by increased expression of proteins that regulate phosphate metabolism and matrix mineralization (10). One of these regulators is fibroblast growth factor (FGF)23, which binds to FGF receptors and the coreceptor Klotho expressed in tubular cells in the kidney and regulates phosphate metabolism (17). Interestingly, FGF23 expression is in turn regulated by other factors produced by osteocytes, including Phosphate-regulating neutral endopeptidase X-linked (PHEX), DMP1, and Matrix extracellular phosphoglycoprotein (MEPE). In humans, inactivating mutations in PHEX lead to accumulation of FGF23 in the circulation and hypophosphatemia (18). Mice with deletion of PHEX exhibit osteomalacia and an irregular lacuno-canalicular system (19). Similarly, mutations and deletions in DMP1 cause autosomal recessive hypophosphatemic rickets (ARHR), which is associated with high FGF23 production (11, 20). MEPE is another product of osteocytes, and its genetic deletion in mice increases bone mineral density (21). Overall, the dysregulation in osteocytes of any of the genes listed above causes alterations in phosphate metabolism (17, 22–25).
Osteocytes are also a major source of receptor activator of nuclear factor-κΒ ligand (RANKL), the key regulator of osteoclastogenesis, and SOST/Sclerostin, a soluble Wnt signaling antagonist and potent suppressor of bone formation (26–30). The discovery of these molecules identified the long-sought mediators of osteocyte communication with the cells that execute the bone remodeling process. Several cells in the bone/bone marrow niche, such as cells of the osteoblastic lineage, adipocytes, B cells, and T cells, produce RANKL (31–33), yet genetic deletion of RANKL from osteocytes strikingly decreased the number of osteoclasts in bone and increased bone mass. These results contrast with the minimal effects of RANKL deletion in other bone cells, in particular osteoblasts (28–30), and identify osteocytes as a major source of RANKL in the adult skeleton. Regarding SOST/Sclerostin, Sclerostin is not expressed in the early stages of differentiation in the osteoblastic lineage. However, Sclerostin expression increases as osteocytes mature, with maximal levels being reached when the osteocytes become fully surrounded by mineralized bone (34–37). Novel evidence demonstrates that Sclerostin contributes to the regulation of osteocytic RANKL (38, 39), yet the mechanisms regulating osteocytic RANKL and SOST/Sclerostin gene expression in osteocytes are not completely known and likely involve a combination of several transcription factors and epigenetic modifications. Nevertheless, this evidence demonstrates that osteocyte-derived RANKL and SOST/Sclerostin contribute to bone homeostasis by regulating both osteoclastogenesis and osteoblast differentiation and activity (40, 41).
Recent mapping of the transcriptome of murine osteocytes revealed genes and molecular programs that control the intricate osteocytic cell network. The osteocyte signature comprises >1,000 genes differentially expressed in osteocytes compared to other cell types. The skeletal role of ∼80% of these genes is currently unknown. However, many of these genes have been identified as contributors to the formation of neuronal networks, which suggests that these genes may be important for the generation and maintenance of the osteocyte network. By evaluating numerous skeletal indexes in >700 mouse lines with genetic deletions, >26 genes within this osteocyte transcriptome signature were identified as critical regulators of bone structure and function. Furthermore, human homolog genes for some of these 26 genes cause monogenic skeletal dysplasias and are associated with polygenic diseases such as osteoporosis and osteoarthritis (42). These novel findings unravel the genetic identity of the osteocyte network and highlight the critical role of osteocytes in human bone diseases.
2.2. Modulation of Gene Expression Profiles in Osteocytes by Epigenetic Marks
The differentiation of mesenchymal stem cells (MSCs) toward osteoblasts, and ultimately osteocytes, is a process tightly controlled by epigenetic mechanisms (43, 44). Cytosine DNA methylation is a repressive epigenetic mark that negatively regulates gene expression (45). In addition, there are activating (mainly acetylation and phosphorylation) and repressive (methylation, ubiquitination, and sumoylation) histone epigenetic marks that contribute to the regulation of gene expression (46). Furthermore, production of microRNAs (miRNAs) and other small RNAs results in mRNA cleavage or translational repression, depending on the degree of complementarity (47). There are several examples of epigenetic regulation of gene programs leading to the mature phenotype of osteocytes. For instance, coordinated changes in DNA methylation and chromatin modifications determine the expression of several genes, including podoplanin/E11, Alkaline phosphatase (ALP), aromatase, Runt-related transcription factor 2 (RUNX2), RANKL, the osteogenic protein distal-less homeobox 5 (DLX-5), Activating transcription factor 4 (ATF4), Activator protein 1 (AP-1), Osterix, SMADs, SOST, and the estrogen receptor (ER) (43, 44, 48–52). In particular, epigenetic regulation of the SOST gene, whose expression in bone is restricted to osteocytes, is well studied (53). Binding of myocyte enhancer factor-2 (MEF2C) to evolutionarily conserved region 5 (ECR5) plays a critical role in the regulation of SOST expression (54–56). In addition, the loss of epigenetic DNA methyl marks at the SOST proximal promoter enables osteocytes to express SOST (57). Chromatin marks regulated by Sirtuin 1, a histone deacetylase, and HDAC5, a class IIa histone deacetylase, also directly regulate SOST expression in osteocytes (58–62).
In summary, osteoblasts undergo dramatic morphological changes and deep genetic and epigenetic reprogramming to differentiate into mature, matrix-embedded osteocytes. These changes enable osteocytes to become multifunctional cells capable of sensing mechanical loads, regulating bone remodeling, and producing endocrine regulators, yet the signals determining the final fate of osteoblasts (i.e., osteocytes, apoptosis, lining cells) in bone remain unclear. It is anticipated that cell lineage-tracing experiments will provide some light on the fate of osteoblasts. Similarly, the mechanisms regulating the changes in morphology and gene expression during osteoblast to osteocyte transition are yet to be determined. It is likely that epigenetic mechanisms play an important part in these processes, and this area of investigation demands further attention. Finally, it is expected that with the incorporation of single-cell sequencing approaches in the set of tools available to bone research, specific osteocyte gene signatures will be described for both physiological and disease conditions of the skeleton.
3. CHALLENGES OF WORKING WITH OSTEOCYTES IN VITRO AND IN VIVO
Isolating and culturing primary osteocytes, as well as reproducing the complexity of the osteocytic canalicular network in vitro, remain a major challenge for bone biologists. Several protocols to isolate primary osteocytes from bone have been developed, mainly based on controlled digestions of extracellular matrix proteins, followed by the separation of osteocytes from other bone and endothelial cells by virtue of genetic markers expressed by osteocytes (63–72). These approaches result in osteocytes deprived of their native environment, thus markedly changing their molecular signature and cellular phenotype. In addition, once placed in culture, authentic osteocytes frequently lose expression of osteocytic genes and dedifferentiate back to an osteoblastic phenotype (73). An alternative to primary cells is the use of osteocyte-like cell lines of rodent or human origin (60, 74–78) (TABLE 1). However, most of these cell lines do not exhibit all the features of authentic osteocytes, as reviewed recently (79). In this regard, a new osteocyte-like cell line named OmGFP66 has been generated recently. This cell line is capable of differentiating into highly dendritic osteocytes embedded in three-dimensional (3-D) lacunae (78). Yet, like other in vitro two-dimensional (2-D) and 3-D differentiation systems from osteoblast precursor cells, these cultures result in mixed populations of osteocytes, osteoblasts, and less differentiated cells, limiting their use for most purposes.
Table 1.
Osteocyte-like cell lines and their characteristics
| Name | Origin | Characteristics | References |
|---|---|---|---|
| MLO-Y4 | Mouse | Osteocyte | Kato et al. (74) |
| MLO-A5 | Mouse | Late osteoblast-early osteocyte | Kato et al. (75) |
| IDG-SW3 | Mouse | Preosteocyte | Woo et al. (76) |
| Ocy454 | Mouse | Preosteocyte | Spatz et al. (60) |
| OMGFP66 | Mouse | Preosteocyte | Wang et al. (78) |
| HOB-01-C1 | Human | Preosteocyte | Bodine et al. (77) |
The use of ex vivo bone organ cultures has become a convenient alternative to circumvent the limitations of in vitro cultures of isolated osteocytes (80). Osteocytes maintain their natural location, intact lacuna-canalicular network, and 3-D distribution in ex vivo bone organ cultures, making this approach better suited for the investigation of the mechanisms underlying bone growth and bone and cartilage matrix turnover, as well as the effects of cancer in bone (14–20). Ex vivo bone organ cultures also represent low-cost screening models with few ethical considerations and allow the study of both genetic and pharmacological manipulations of osteocytes and other bone cells in their natural environment. Detailed methodological procedures on how to establish ex vivo cultures for the study of osteocytes have been published recently (80).
Several transgenic Cre strains to conditionally manipulate gene expression in osteocytes in vivo are available (81). The design of these transgenic strains has been based on the regulatory regions of two genes primarily expressed by osteocytes. These are the DMP1 promoter, with 10-kb and 8-kb versions (14, 82), and the SOST promoter (28). DMP1 promoters are the most widely used Cre transgenic lines for conditional gene deletion in osteocytes in vivo. However, besides targeting osteocytes, studies with genetic reporters have shown that DMP1-related Cre strains have off-target recombination in mature osteoblasts potentially destined to be osteocytes, muscle, and other soft tissues (81). Similarly, the SOST-Cre strain targets osteocytes, not osteoblasts, but exhibits off-target recombination in hematopoietic stem cells (28). Furthermore, whether all osteocytes are targeted by the SOST-Cre driver has not been confirmed. It is likely that this driver targets a subpopulation of osteocytes more deeply embedded in the mineralized matrix, which, as shown before, express Sclerostin endogenously (36, 83, 84). Tamoxifen-inducible strains for DMP1-10Kb-Cre and SOST-Cre have also been generated recently (85, 86). Limitations with these inducible strains include some degree of “leaky” expression without tamoxifen, the need to carefully optimize tamoxifen doses, and off-target cell recombination (81). Thus many of these osteocyte-targeted Cre approaches are not as specific as intended, yet, despite these limitations, their careful use, inclusion of appropriate controls, performance of comparative studies with Cre lines targeting different cell populations, and confirmation that the deletion of the gene of interest is achieved in osteocytes and no other cells has markedly increased our knowledge about osteocyte functions and will continue to advance our understanding of the role of osteocytes in health and disease. Future research toward the generation of osteocyte-specific promoters without off-target recombination in other bone cells or tissues is warranted.
Because osteocytes reside deep in mineralized bone, imaging these cells in their environment is another challenging aspect of osteocyte research (87). Until recently, the majority of the imaging work on osteocytes was performed ex vivo, in both undecalcified and decalcified specimens along with conventional sectioning and imaging techniques. The development of confocal microscopy provided the opportunity for osteocyte biologists to image osteocytes in 3-D in their bone microenvironment (88). More recently, the use of micro-computed tomography (μCT) systems, scanning electron microscopy, and transmission microscopy has allowed researchers to image osteocyte lacunae noninvasively and in a 3-D fashion (87). In vivo live imaging is now possible thanks to the generation of transgenic mice with membrane-targeted fluorescent proteins in osteocytes (35, 89, 90). Moving forward, the development of multiphoton microscopy (91), together with tissue clearing techniques (92), is expected to revolutionize the way we image osteocytes and other bone cells.
In conclusion, the study of osteocyte biology is challenging because of the limited accessibility to mineral-embedded osteocytes and the lack of fully representative in vitro models. Researchers are overcoming these limitations with the use of ex vivo models and novel imaging techniques that allow the study of osteocytes in their native 3-D microenvironment. Although the use of the current Cre drivers to study osteocytes in vivo has tremendously advanced our understanding of osteocyte biology, future research is likely to focus on the generation of novel maneuvers to genetically modify osteocytes in a more specific manner.
4. OSTEOCYTE PROGRAMMED CELL DEATH: APOPTOSIS
Osteocyte density in bone is influenced by both the rate of osteoblast differentiation and the rate of apoptosis, as osteocytes are nondividing cells in vivo. Earlier studies of this area demonstrated that osteocyte viability is controlled by a combination of physical and chemical stimuli (93–95). Osteocytes can live for decades; however, these cells undergo programmed cell death by apoptosis like other cells do in bone. The first observations on osteocyte apoptosis were made in the context of estrogen withdrawal, revealing an association between apoptosis and estrogen signaling in osteocytes (96). This work stimulated a number of studies demonstrating that estrogen as well as selective estrogen receptor modulators (SERMs) preserve osteocyte viability and revealing multiple signaling pathways involved (97–106). The bone fragility syndromes ensuing with estrogen or androgen deficiency are characterized by increased osteocyte apoptosis, and similar observations have been made in animal models and human conditions of glucocorticoid (GC) excess, aging, and mechanical unloading (107–109). In contrast, physiological levels of mechanical stimulation (108, 110), estrogen/androgen replacement (100, 101), and treatment with bisphosphonates (111) have been shown to preserve osteocyte viability.
4.1. The Role of Mechanical Signaling in the Regulation of Osteocyte Apoptosis
In vitro studies with osteocytic cells or authentic osteocytes showed that mechanical stimulation prevents apoptosis induced by proapoptotic stimuli, such as GC, tumor necrosis factor-alpha (TNF-α), or reactive oxygen species (ROS) (112, 113). Mechanistic studies revealed that mechanical forces sensed at the osteocyte membrane are transduced into antiapoptotic signals mediated by integrins, the Src and focal adhesion kinases (FAKs), as well as several cytoskeletal proteins. These membrane-associated events lead to the activation of intracellular survival kinases, including extracellular signal-regulated kinase (ERK) (112). Mechanical signals also activate the Wnt/β-catenin signaling pathway (106, 114–117), and there is bidirectional cross talk between survival kinase signaling and the Wnt pathway. For instance, preservation of osteocyte viability by ERK nuclear translocation and antiapoptotic signals induced by mechanical stretching or fluid flow are abrogated by Wnt antagonists, such as the stimulator of β-catenin degradation axis inhibition protein (Axin)2 and Dickkopf Wnt Signaling Pathway Inhibitor 1 (DKK1) (118). Conversely, the phosphorylation and accumulation of β-catenin and glycogen synthase kinase 3β (GSK3β) induced by mechanical cues are prevented by genetic inhibition of ERK or caveolin-1 signaling (118).
Osteocyte life span is also regulated by mechanical signals in vivo. Bones exposed to low or excessive mechanical strains exhibit an increased number of apoptotic osteocytes (93, 108, 119, 120). Under both circumstances, osteoclastic resorption is preceded by osteocyte apoptosis, with bone areas enriched in apoptotic osteocytes being subsequently resorbed by osteoclasts (108). This earlier evidence prompted us to propose that dying osteocytes are initiators of local bone resorption (121) (FIGURE 2). This hypothesis is further supported by the demonstration that genetic induction of osteocyte death stimulates the recruitment of osteoclasts and subsequent bone resorption and induces decreases in bone mass (122). Moreover, the skeleton of mice depleted of osteocytes does not exhibit the expected osteoclastogenic response to unloading (122), demonstrating that osteocytes initiate the cascade of events that lead to bone loss. One of the mechanisms by which dead osteocytes initiate local bone resorption involves increased expression of RANKL in osteocytes surrounding dead osteocytes (123–125). Importantly, an apoptosis inhibitor prevented the increased RANKL and the proresorptive action of either low or excessive mechanical strains (123, 124). Furthermore, mice lacking RANKL in osteocytes do not exhibit the typical increase in osteoclastogenesis and the loss of bone mass induced by unloading (29), demonstrating the requirement of osteocytic RANKL in the bone loss induced by mechanical disuse. Furthermore, treatment with a bisphosphonate that inhibits osteocyte apoptosis without directly affecting osteoclasts prevented the elevation in RANKL expression in osteocytes (and osteoblasts); however, it did not prevent unloading-induced bone loss, suggesting that, under these conditions, other cellular sources of RANKL are responsible for the resorption leading to the loss of bone (125). This suggests that there is a strong cause-effect association between apoptotic osteocytes and the production of RANKL by adjacent osteocytes. Yet, increased osteocytic RANKL does not always result in targeted local bone resorption, suggesting that other osteocyte-derived molecules are involved in osteoclast precursor recruitment to bone areas containing apoptotic osteocytes needing to be resorbed. One potential candidate is osteoprotegerin (OPG), a RANKL-soluble decoy also produced by osteocytes and osteoblasts (126). However, recent evidence showed that OPG deletion in osteocytes (or B lymphocytes) does not affect bone mass, whereas deletion of OPG in osteoblasts increases bone resorption and reduces bone mass, suggesting that under physiological conditions osteoblasts are a major source of OPG (127). Whether osteocytic OPG becomes relevant under pathological situations remains to be determined. Another osteocyte-derived factor involved in osteoclast precursor recruitment is high mobility group box 1 (HMGB1) protein (128), an osteoclast chemotactic cytokine released by apoptotic osteocytes known to regulate the expression of several cytokines controlling osteoclastogenesis, including RANKL, TNF-α, interleukin (IL)-6, and OPG. Moreover, the expression of both RANKL and vascular endothelial growth factor A (VEGF-A) is elevated in living osteocytes surrounding dead osteocytes in overloaded rat bones (129). These findings suggest that apoptotic osteocytes alter in neighboring cells the expression of angiogenesis-inducing molecules that facilitate the recruitment of osteoclast precursors from the circulation to mineralized bone areas that need replacement.
FIGURE 2.

Osteocyte apoptosis: key molecular mechanisms. A: scheme showing that proapoptotic stimuli activate death receptors in osteocytes and initiate caspase-3-mediated programmed cell death. Osteocytes adjacent to apoptotic osteocytes are thought to release proinflammatory and proosteoclastogenic cytokines that attract osteoclasts and promote local bone resorption. B: image showing TUNEL assay to detect degraded DNA in apoptotic osteocytes in paraffin-embedded bones infiltrated with myeloma cancer cells. C: image showing active caspase-3 immunostaining of paraffin-embedded human bone from an osteoporotic patient. Black arrows point to apoptotic osteocytes; red arrows point to healthy osteocytes. AGE, advanced glycation end-products; HMGB1, high mobility group box 1; ROS, reactive oxygen species; TNF, tumor necrosis factor-α; TRAIL, tumor necrosis factor-related apoptosis inducing ligand.
Taken together, these pieces of evidence identify osteocyte apoptosis as the initiating event for bone loss in skeletal disuse scenarios, as occurs with reduced physical activity during aging, bed rest, and motor paralyses (130) or under microgravity conditions during long-term spaceflights (131). Future research efforts are warranted to examine the potential of therapies targeting osteocytes to prevent or treat the bone loss induced by weightlessness.
4.2. Regulation of Osteocyte Apoptosis by Bisphosphonates and Sex Steroids
The loss of sex steroids increases osteocyte apoptosis, whereas estrogens and androgens preserve the viability of both osteocytes and osteoblasts (101, 106). The prosurvival effect of sex steroids is caused by rapid activation of phosphatidylinositol 3-kinase (PI3K) and Src/Shc/ERK, as well as by nongenotropic events downstream of the sex steroid receptors (101, 132). In vitro and in vivo studies show that bisphosphonates promote osteocyte/osteoblast survival via ERK activation and Cx43 signaling (15, 94, 111, 133). Thus, it is possible that the antiapoptotic properties of estrogens and bisphosphonates, and the consequent inhibition of resorption due to the lack of apoptotic osteocytes, contributes to the suppression of bone remodeling seen with these agents.
In conclusion, evidence gathered in the last 20 years demonstrates that untimely premature apoptosis of osteocytes is associated with bone fragility in conditions of dysregulated mechanical and hormonal stimulation. Osteocyte apoptosis triggers signaling mechanisms that drive bone resorption at particular skeletal sites, also known as “targeted bone remodeling.” However, the identity of only some of the potential molecular mediators of the communication between apoptotic osteocytes and osteoclasts and their precursors has been revealed. Future research is warranted to determine the prospective participation of accessory cells as well as additional molecular mechanisms underlying this phenomenon. It is expected that this new knowledge will provide tools to manipulate the apoptotic osteocyte/osteoclast axis toward preserving bone mass and strength.
5. OSTEOCYTE SENESCENCE
The study of osteocyte senescence and the potential of targeting senescent cells in bone as a therapeutic approach to treat bone loss has attracted much attention recently in the musculoskeletal research community. Cellular senescence is a complex process that results in irreversible cell cycle arrest and marked changes in gene expression leading to a senescence-associated secretory phenotype (SASP) (134). In aging cortical bone, ∼10% of osteocytes become senescent, a number that contrasts with <2% in young bones (135, 136). Khosla and colleagues (135, 137, 138) have shown that diabetes and radiation also induce accumulation of senescent osteocytes, which are characterized by high p16Ink4a expression and a development of a SASP. The specific function of senescent osteocytes is yet to be defined. However, emerging data link the accumulation of senescent osteocytes with bone loss and poor bone quality that occurs with aging, radiation, and diabetes (135, 137, 138). Consistent with this notion, SASP accumulation in vitro or in vivo stimulates osteoclastogenesis and suppresses osteoblast differentiation (135). Several pharmacological strategies to either eliminate senescent cells or inhibit the production of SASP have shown prevention of bone loss in preclinical animal models (135, 137, 138). Future studies are warranted to optimize the regimes and timing of therapeutics agents targeting senescent cells in bone.
It is intriguing that accumulation of senescent osteocytes has been described in aging, radiated bones, and diabetes, conditions in which osteocyte apoptosis also is increased (95, 135, 137, 138). To our knowledge, a potential relationship between osteocyte senescence and apoptosis has not been established. Future studies are warranted to determine the timing of senescence versus apoptosis, whether there is a cause-effect relationship between the two phenomena, as well as whether the same or neighboring osteocytes undergo senescence and then apoptosis (or vice versa). Answers to these crucial questions without a doubt will increase the chances of finding targets to regulate these events, establish their potential relationship, and clarify their role in bone pathophysiology.
6. OSTEOCYTE PARACRINE FUNCTIONS
6.1. Regulation of Bone Formation by Osteocytes
Osteocytes sense mechanical forces and transmit bone anabolic signals to other bone effector cells through their canalicular system (139, 140) (FIGURE 3), making the osteocyte the main mechanosensor cell in the skeleton. Ablation of osteocytes by genetic means decreases bone mass and suppresses mechanically induced new bone formation (122). The key role of osteocytes in bone formation is further supported by the identification of genetic alterations in several components of the Wnt signaling pathway, which are accompanied by marked effects on bone mass (141). These clinical discoveries demonstrated that osteocytes are a major source of Wnt signaling antagonists, including DKK1 and Sclerostin (40, 142). The discovery of SOST, the gene encoding Sclerostin, and its impact on bone biology is perhaps one of the major advances in the last decades. Sclerostin was originally described in two rare genetic diseases, sclerosteosis and van Buchem disease, both characterized by high bone mass. Extensive in vitro and in vivo experimentation revealed that Sclerostin negatively regulates the interaction of Wnt ligands with low-density lipoprotein receptor-related proteins (LRPs) 5 and 6 and Frizzled receptors, ultimately preventing beta-catenin translocation to the nucleus (40, 141, 143). Furthermore, SOST knockout (KO) mice exhibit increased bone formation and bone mass, similar to the high bone mass phenotype that inactivating mutations in this gene induced in patients (141, 144) (FIGURE 4). In addition, consistent with the relevant role of osteocytes in mechanotransduction, downregulation of Sclerostin in osteocytes is required to achieve full bone anabolic response to mechanical loads (145, 146). Furthermore, Sclerostin expression is inhibited by parathyroid hormone (PTH), an FDA-approved anabolic agent for osteoporosis in the United States (147–152) (see sect. 8). Although deletion of DKK1 also results in a high-bone mass phenotype, the effects are minor compared with those observed with SOST inhibition. DKK1 inhibition increases SOST/Sclerostin expression in bone, which in turn counteracts the anabolic effects of DKK1 suppression (153). In fact, genetic deletion of both DKK1 and SOST leads to a robust anabolic, additive response greater than those seen when inhibiting each gene alone. Given the prominent role that SOST/Sclerostin and DKK1 have in the regulation of bone formation, several pharmaceutical approaches have been developed to neutralize their actions in the skeleton (discussed in sect. 5).
FIGURE 3.

Osteocyte paracrine signaling in bone. Osteocytes exert paracrine actions on osteoblasts and osteoclasts via cell-to-cell physical communication and/or secretion of molecules that stimulate or inhibit osteoblast or osteoclast differentiation. A: osteocytes produce the proosteoclastogenic cytokines receptor activator of nuclear factor-κΒ ligand (RANKL) and macrophage colony-stimulating factor (M-CSF) in both membrane-bound and soluble forms and the anti-osteoclastogenic factor osteoprotegerin (OPG). B: osteocyte-derived Sclerostin acts on mesenchymal stem cell precursors and promotes bone marrow adipogenesis. C: Wnt antagonists Dkk-1 and Sclerostin produced by osteocytes inhibit osteoblast differentiation and bone forming function. Furthermore, osteocytes express Notch receptors that can communicate physically with osteoblasts and potentially with osteoclasts.
FIGURE 4.

Osteocyte regulation of bone formation via Sclerostin production. A: Sclerostin immunostaining in paraffin-embedded human bone. Black arrow points to Sclerostin-positive osteocytes; red arrow points to Sclerostin-negative osteocytes. B: micro-computed tomography (μCT) image of vertebral cancellous bone from wild-type mice (WT) and mice with global deletion of SOST (SOST KO). C: toluidine blue-tartrate-resistant acid phosphatase (TRAP)-stained histological image of tibial cancellous bone from WT and SOST KO mice. D: dynamic histomorphometry in plastic-embedded bones from WT mice and mice overexpressing human SOST under the control of the DMP1-8kb promoter (Dmp1-8kb-hSOST). Green calcein labels and red alizarin labels are shown. E: toluidine blue-TRAP stained histological image of bones from C57BL/6 mice treated with IgG (control) or neutralizing anti-Sclerostin antibody (anti-Sclerostin-Ab). Yellow arrows point to osteoblasts on the bone surface.
Osteocytes are now considered the bone cells responsible for orchestrating the anabolic actions of the Wnt pathway. Importantly, Wnt signaling activation in osteocytes results in a robust increase in bone mineral density and bone volume (FIGURE 5). These anabolic bone effects are associated with increased osteoblast number and bone matrix production (39). These results contrast with previous work showing that Wnt/β-catenin signaling activation in earlier stages of the osteoblastic lineage (preosteoblasts or osteoblasts) increases OPG and blocks bone resorption with no alteration of physiological bone apposition (154–156).
FIGURE 5.
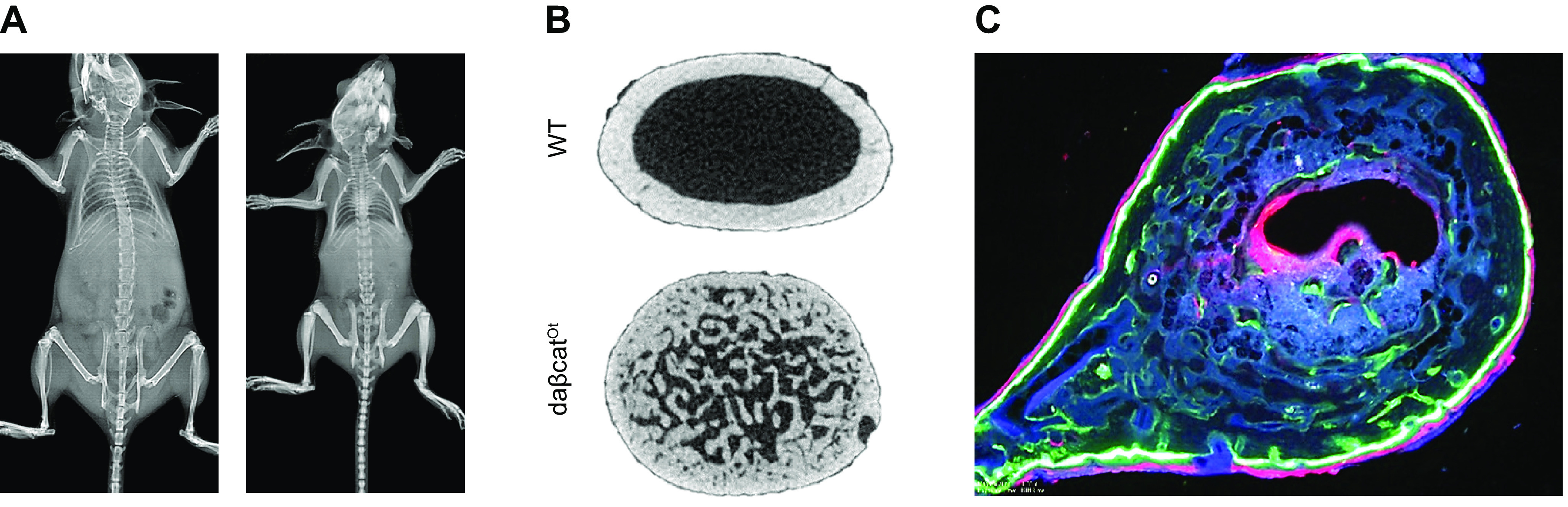
The osteocyte as the anabolic Wnt signaling cell in bone. Mice with genetic constitutive activation of canonical Wnt/β-catenin signaling in osteocytes (daβcatOt) have been used to determine the cell responsible for the bone anabolic effects of canonical Wnt signaling. A: daβcatOt mice are smaller in size compared with wild-type (WT) control littermates and exhibit denser bones. B: daβcatOt mice exhibit exuberant bone formation that expands into the bone marrow cavity of the femoral midshaft. C: dynamic histomorphometry in a plastic-embedded bone from a daβcatOt mouse. Green calcein labels and red alizarin labels are shown.
Osteocytes also can affect osteoblasts through other secreted factors (157) and physical interactions (158). It is possible that the Notch pathway, which regulates direct physical communication between adjacent cells, mediates the physical communication between osteocytes and osteoblasts (159, 160) (see sect. 9).
6.2. Osteocytes as Regulators of Bone Resorption
Osteocytes regulate osteoclast formation and function under both normal and disease conditions (FIGURE 3). Although not the only source, osteocytes are major suppliers of the proosteoclastogenic cytokine RANKL, which regulates the formation and activity of osteoclasts. Genetic deletion of RANKL (driven by Dmp1-Cre) in osteocytes, and some mature osteoblasts, markedly affects bone resorption, and adult mice lacking RANKL in osteocytes exhibit an osteopetrotic phenotype due to lack of mature osteoclasts (29, 30). Supporting the idea that osteocyte-derived RANKL is critical for adult bone remodeling, genetic deletion of RANKL with SOST-Cre, which targets osteocytes but not osteoblasts, resulted in a very similar high-bone mass phenotype (28). More recent studies examined the specific contribution of soluble RANKL resulting from cleavage of the membrane-bound RANKL. Intriguingly, deletion of soluble RANKL results in normal bone mass during growth, whereas adult mice display more cancellous bone due to fewer osteoclasts (161). The similarities between the phenotypes of adult mice with genetic deletion of RANKL/soluble RANKL in osteocytes suggest that soluble RANKL is an important mechanism by which osteocytes modulate bone resorption, yet deletion of soluble RANKL did not mitigate the bone loss induced by estrogen deficiency. In contrast, soluble RANKL appears to accelerate tumor metastasis to bone (162). Nonetheless, the majority of osteocytes are not physically in contact with cells on the bone surface or in bone marrow or near blood vessels. Therefore, how an osteocyte’s membrane-bound RANKL reaches osteoclast progenitors to induce their differentiation to mature, bone-resorbing osteoclasts demands further investigation. Finally, intriguing data presented in a recent report suggest the existence of reverse RANKL signaling in osteoblasts [i.e., receptor activator of NF-κB (RANK) secreted by osteoclasts binds to RANKL in osteoblasts], a mechanism proposed to contribute to the coupling of bone resorption and bone formation in local bone areas (163). Yet, whether reverse RANKL signaling occurs in osteocytes and its consequences remain to be determined.
Osteocytes also secrete OPG, a soluble decoy for RANKL, impeding the binding to its RANK on osteoclasts. OPG production in both osteocytes and osteoblasts is regulated by the Wnt/β-catenin pathway. Supporting this notion, mice lacking β-catenin in osteocytes are osteoporotic because of decreased OPG, increased osteoclast numbers, and bone resorption (126). However, as discussed above, recent evidence suggests that production of OPG by osteoblasts, instead of osteocytes, is responsible for the suppression of osteoclast formation and bone resorption under physiological conditions. Finally, osteocytes also appear to be a source of macrophage colony-stimulating factor (M-CSF), a cytokine essential for osteoclast differentiation (164).
6.3. Osteocytes and Bone Marrow Adipogenesis
Bone marrow adipose tissue (BMAT) is an active and dynamic fat depot confined in the bone marrow cavity that secretes adipokines involved in the regulation of glucose and lipid metabolism and bone homeostasis (165). The exact function and regulation of BMAT are largely unknown. The potential role of osteocytes in BMAT has been reviewed recently (166). Emerging evidence suggests that the Wnt antagonists SOST/Sclerostin and DKK-1 produced by osteocytes could play a role in the regulation of BMAT as well as peripheral fat depots (discussed in sect. 5; FIGURE 3). Supporting this notion, in vitro work shows that recombinant Sclerostin can induce adipocyte differentiation (167, 168). Furthermore, both genetic and pharmacological inhibition of Sclerostin decrease BMAT (168). Mechanistically, this proadipogenic effect is due to inhibition of the Wnt signaling pathway followed by increases in Peroxisome proliferator-activated receptor γ (PPARγ) gene expression (168). Furthermore, pharmacological inhibition of Sclerostin with a neutralizing antibody significantly reduced the increase in BMAT by rosiglitazone, an antidiabetic drug frequently used in the clinic (169).
In summary, the evidence discussed above demonstrates that osteocytes are essential for physiological, normal bone remodeling. Some, but not all, of the mechanisms used by osteocytes to regulate osteoblast and osteoclast function have been discovered during the last decade, leading to the development of new therapeutic approaches to combat low-bone mass conditions. However, it is important to note that bone remodeling is a complex process likely regulated by the coordinated action of several bone cells and factors. The new findings suggesting that osteocytes also influence bone marrow adiposity open the possibility that osteocytes regulate other cell populations in the bone marrow, a topic that demands further investigation.
7. OSTEOCYTE ENDOCRINE FUNCTIONS
7.1. Osteocytes and the Regulation of Whole Body Composition and Metabolism
Osteocyte-derived factors have recently emerged as endocrine signals involved in the cross talk between bone and distant organs regulating peripheral fat and glucose metabolism (170). Genetically induced osteocyte death in mice results in severe lymphocytopenia and peripheral white adipose tissue mass loss (171). Moreover, growing evidence suggests that osteocyte-derived Sclerostin plays a role in the regulation of peripheral fat. In vivo, mice with genetic overproduction of Sclerostin have elevated peripheral fat mass and impaired glucose metabolism. In contrast, mice with global deletion of SOST exhibit low peripheral fat mass compared with control mice (172, 173). Similarly, the elevation in Sclerostin that results from the genetic deletion of LRP4 in osteocytes or the osteocytic activation of β-catenin is accompanied by elevated body fat and peripheral white adipose fat and compromised glucose metabolism (174). Moreover, pharmacological inhibition of Sclerostin with neutralizing antibodies mitigates the high-fat diet-induced body fat accumulation and impairment of glucose metabolism (172). Although clinical data in this area are scarce, circulating Sclerostin is increased in type 2 diabetes patients, and serum Sclerostin levels positively correlate with body and fat mass in healthy adults and diabetic patients (175–177). Similarly, LRP5 mutations blocking the binding of Sclerostin to Wnt coreceptors result in altered fat distribution (178). It is important to note that there are some controversial results. For instance, the increase in Sclerostin that follows the genetic deletion of the stimulatory subunit of G protein Gsα in osteocytes results in loss of gonadal and inguinal white adipose tissue mass (179). This recent body of literature supports the notion that Sclerostin is an endocrine signaling factor that mediates the cross talk between bone and peripheral fat depots and the pancreas. Further investigations are required to determine the clinical relevance of these results, as well as the cellular and molecular mechanisms underlying Sclerostin’s actions on adipocyte biology.
7.2. Regulation of Phosphate Metabolism by Osteocytes: FGF23
Osteocytes express higher levels of genes related to mineralization and phosphate metabolism than osteoblasts, including PHEX, DMP1, and MEPE (2). Osteocytes also produce FGF23, which as discussed above plays a crucial role in the regulation of inorganic phosphate (Pi) homeostasis by inhibiting its renal reabsorption (11, 17, 180–182).
FGF23 secretion is stimulated by high dietary Pi and the active form of vitamin D3, 1,25-dihydroxy-vitamin D3 (1,25-D3) (183–186). FGF23 is a recognized biomarker of altered Pi metabolism as it increases before detectable changes in PTH in early chronic kidney disease (CKD) (187). FGF23 production is also regulated by PTH. Indeed, elevation in both FGF23 and PTH is seen in the hyperphosphatemia exhibited by CKD patients (188, 189), and the increase in FGF23 in a rat model of kidney failure is prevented by parathyroidectomy (190).
FGF23 is also elevated by hyperparathyroidism, as seen in both patients and mouse models (191, 192). Jansen-type metaphyseal chondrodysplasia, caused by missense mutations resulting in constitutive active PTHR1 receptor signaling, is a rare disorder characterized by abnormal bone formation in the metaphysis of long bones. Patients with these mutations have high serum circulating FGF23 levels, regardless of exhibiting normal 1,25-D3 and low Pi levels (193). Moreover, FGF23 expression is increased in osteocytes in mice overexpressing the same mutation in the PTH receptor found in Jansen patients (DMP1-caPTHR1Ot transgenic mice) in DMP1-8kb-expressing cells. Despite circulating FGF23 being elevated in DMP1-8kb-caPTHR1 mice, plasma Pi and renal Pi reabsorption are normal. In addition, the FGF23-FGFR1-KLOTHO signaling axis is active in osteocytes, and osteocytes from DMP1-caPTHR1 mice exhibit increased expression of FGFR1 and Polypeptide N-Acetylgalactosaminyltransferase 3 (GALNT3), downstream targets of FGF23 signaling. Thus, PTH receptor signaling has the ability to integrate the endocrine and paracrine functions of osteocytes by regulating FGF23 through cAMP- and Wnt-dependent mechanisms (194).
The exact mechanisms by which osteocytes sense blood Pi and modulate FGF23 production are still unclear. In a recent article, glycerol-3-phosphate was identified as a potential signaling factor regulating FGF23 in bone (195). These results support the existence of a kidney-to-bone reverse signaling axis by which the kidney, the main target for FGF23 actions, releases glycerol-3-phosphate to regulate osteocyte FGF23 production in bone. Future studies to confirm this hypothesis are warranted.
7.3. Osteocytes and Skeletal Muscle
Growing evidence indicates that there is cross talk between bone and muscle, and recent findings point to osteocytes as potential mediators of this communication, yet the work conducted in this area is very limited. Studies using conditioned media from osteocyte-like cells demonstrated that factors released by osteocytes induce acceleration of myogenesis of C2C12 myoblasts (196, 197). Mechanistic studies identified prostaglandin E2 and Wnt3a released by osteocytes as responsible for the enhanced myogenic differentiation of C2C12 cells in vitro (196, 198). Conditional deletion of Membrane Bound Transcription Factor Peptidase, Site 1 (MBTPS1) in osteocytes leads to an age-related increase in contractile force in adult slow twitch-dominant soleus muscles (197). Osteocytes also express a significant number of cytokines capable of inhibiting myogenesis (199). Conversely, the exercise-induced muscle factor β-aminoisobutyric acid can act as a bone-protective factor and prevents osteocyte cell death induced by ROS in vitro (200). Additional studies are needed to fully understand the role of osteocytes in the cross talk between bone and muscle and to determine the potential of targeting osteocytes to treat bone pathologies accompanied by muscle dysfunction.
In conclusion, emerging data suggest that osteocytes can regulate the function of distant organs through the secretion of endocrine factors. In addition to the well-accepted endocrine role of osteocyte-derived FGF23 in the kidney, evidence from animal models shows that osteocytes can also influence whole body fat, pancreas, and muscle function. However, a causal relationship between these new osteocyte-derived endocrine factors and human metabolic diseases is yet to be established. Future interventional clinical studies are needed to confirm these potential novel endocrine functions of osteocytes.
8. OSTEOCYTES AS MEDIATORS OF HORMONAL SIGNALING
It has long been hypothesized that osteocytes sense changes in mechanical strain and initiate an adaptive skeletal response by coordinating the activity of osteoclasts and osteoblasts. However, that osteocytes are also major target cells in bone and integrators of hormonal signaling was less expected. Evidence accumulated during the last few years confirms that osteocytes are crucial drivers of the skeletal changes induced by systemic hormones with recognized effects on bone.
8.1. Effects of Glucocorticoids on Osteocytes
Osteoblastic cells, and in particular osteocytes, are profoundly affected by GC (FIGURE 6). The role of GCs in different bone cell types has been elucidated by extensive research taking advantage of the enzymatic system of 11β-hydroxysteroid dehydrogenase (11β-HSD) type 1 and type 2, which regulates the local levels of active GC metabolites by a prereceptor mechanism. 11β-HSD2 overexpression in cells early in the osteoblastic differentiation stage revealed that GC signaling is essential for proper bone mass acquisition during growth (201). In addition, elevated prevalence of osteoblast and osteocyte apoptosis is a hallmark of GC excess in the adult skeleton (202, 203). Osteoblast apoptosis explains at least in part the reduced osteoblast number and low bone formation induced by the hormones. In addition, osteocyte apoptosis is likely to disrupt the osteocyte network contributing to GC-induced bone fragility. Indeed, blocking GC action by overexpressing 11β-HSD2 in osteoblasts and osteocytes prevents GC-induced apoptosis, preserving osteoblast function and bone formation (204, 205). GC-induced osteoblast and osteocyte apoptosis depends on the glucocorticoid receptor (GR) and is replicated in vitro, demonstrating that indeed apoptosis is due to direct hormonal effects on osteoblasts and osteocytes (206, 207). Moreover, despite the bone loss seen in osteocalcin gene 2 (OG2)-11β-HSD2 transgenic mice treated with GC, osteocyte apoptosis is prevented and bone strength is preserved, suggesting a direct connection between osteocyte apoptosis and bone fragility (208).
FIGURE 6.

Osteocytes as mediators of mechanical and hormonal signals in bone. A: osteocytes sense mechanical forces in bone and transduce them into biological cues to effector cells. Mechanical forces decrease Sclerostin, increase Wnt signaling, and decrease osteoprotegerin (OPG) expression in osteocytes to enable bone gain. ER, estrogen receptor; PTH1R, parathyroid hormone (PTH) receptor 1. B: glucocorticoid direct actions on osteocytes increase apoptosis (via Pyk2 activation) and Sclerostin expression, leading to decreased Wnt signaling and OPG expression and overall contributing to the bone loss induced by glucocorticoids. C: intermittent administration of PTH (iPTH) requires osteocytic PTH1R signaling to induce full bone anabolism. Mechanistically, PTH decreases SOST expression, activates Wnt signaling, and increases osteoblast number and function. Additionally, iPTH increases receptor activator of nuclear factor-κΒ ligand (RANKL) expression and promotes Notch signaling to regulate PTH-induced bone resorption.
Besides regulating cis- or trans-interactions between the ligand-bound receptor with DNA and the consequent regulation of gene transcription (209, 210), GCs modulate in bone cells the activity of several kinases, including c-Jun NH2-terminal kinase (JNK), Proline-rich tyrosine kinase 2 (PYK2), and ERKs (211–216). In particular, GCs activate in osteoblastic cells PYK2, a kinase related to the focal adhesion kinase FAK. These kinases regulate cell survival by opposing mechanisms. Thus, FAK prevents and PYK2 promotes cell attachment-induced apoptosis (named anoikis) (112, 207, 209, 217–228). Recent evidence showed that genetic deletion or pharmacological inhibition of PYK2 prevents GC-induced bone loss by overriding GC effects on anoikis in osteocytes, osteoblasts, and osteoclasts (207, 229). GC extended osteoclast life span, induced osteoblast/osteocyte apoptosis, increased bone resorption, promoted microarchitectural deterioration, and reduced bone strength in control mice, whereas genetic deletion or pharmacological inhibition of PYK2 prevented all these effects. These findings support the notion that inhibition of PYK2 and anoikis signaling prevents the damaging skeletal effects of GC by promoting osteoblast/osteocyte survival and reducing osteoclast life span and bone resorption activity. Thus, targeting PYK2 is a promising new therapeutic approach for the treatment of GC-induced bone loss.
Another mechanism by which GC impacts bone homeostasis is through the accumulation of ROS in bone (230), associated with endoplasmic reticulum (ER) stress that is alleviated by phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) (231, 232). Supporting the relevant role of ROS/ER stress as a mediator of the effects of GC on bone, the eIF2α dephosphorylation inhibitors salubrinal and guanabenz are capable of blocking ROS-induced ER stress (233, 234), as well as blocking the increase in osteoblast and osteocyte apoptosis in vitro and the inhibition of osteoblast differentiation induced by GC (235). In addition, salubrinal inhibited osteoblast and osteocyte apoptosis in vivo and blunted the GC-induced decrease in bone mass and bone formation. These findings provide an additional pathway to interfere with the deleterious actions of GC on bone.
GCs also have profound effects on the gene expression profile of osteocytes. SOST/Sclerostin expression is increased by GC, as well as the expression of several Wnt target genes linked to anabolism/survival (i.e., Cyclin D1) and anticatabolism (i.e., OPG) (236). Consistent with a driving role of SOST/Sclerostin in GC’s skeletal actions, mice lacking the gene SOST are protected from the loss of bone mass, microarchitectural deterioration, and decrease in structural and material strength induced by GC. Remarkably, the preservation of bone integrity in SOST KO mice is due to prevention of the increase in bone resorption induced by GC but not to the restoration of bone formation or the inhibition of osteoblast/osteocyte apoptosis (236). These studies indicate that, in the frame of GC excess, inhibition of SOST/Sclerostin and activation of Wnt/β-catenin signaling opposes bone catabolism and maintains bone integrity. In addition to SOST, GCs decrease the expression of several proteases required for perilacunar remodeling [MMP13, MMP14, tartrate-resistant acid phosphatase (TRAP)] (237). This suppression of perilacunar remodeling causes disruption of the osteocyte lacuno-canalicular network and contributes to the subchondral bone degeneration seen in GC-induced osteonecrosis.
8.2. Osteocytes as Mediators of PTH Signaling in Bone
PTH is an important mediator of extracellular calcium and phosphate levels and has profound effects on the skeleton. Chronic PTH elevation, as occurs in hyperparathyroidism, results in bone loss due to stimulation of bone resorption. In contrast, intermittent PTH elevation, as occurs with daily injections of PTH 1–34, stimulates bone formation more than resorption, an approach used in the clinic to treat osteoporosis (238–240). Mechanistically, the bone anabolic effects of PTH have been linked to its ability to stimulate proliferation of preosteoblasts, to promote osteoblast survival, to transform lining cells into matrix-synthesizing osteoblasts, or to a combination of these effects, downstream of the PTH receptor 1 (PTH1R) (238, 241). Basic and clinical work over the last three decades demonstrates that osteocytes are critical target cells for the anabolic actions of PTH in the skeleton (242) (FIGURE 6). Genetic activation of the PTH1R in osteocytes in mice is sufficient to elicit the effects of PTH on the skeleton (240, 243–245), and, conversely, genetic deletion of the PTH1R in osteocytes leads to a defective bone anabolic response to PTH in mice (85, 194, 246–249).
Parfitt postulated in the 1970s (250) that osteocytes must control the rapid release of calcium, whereas osteoclasts and resorption determine the later phase of calcium release in response to PTH. That osteocytes are involved in the regulation of calcium homeostasis by the hormone is supported by more recent findings showing that mice lacking the PTH1R in osteocytes (tamoxifen-induced DMP1-10kb-Cre) are unable to maintain normal circulating levels of calcium when fed a low-calcium diet (85). During lactation, PTH-related peptide (PTHrP) is produced to stimulate the release of calcium from the skeleton to the blood and enable milk production by the mammary glands. This phenomenon is accompanied by increased osteocyte lacunar size as a result of perilacunar bone remodeling providing the additional source of calcium and contributes to the overall bone loss exhibited during lactation (251–254). Perilacunar remodeling, or “osteocytic osteolysis,” is mediated by the induction of an “osteoclast-like” gene expression profile in osteocytes, including genes that promote acidification and degrade and/or dissolve the extracellular matrix and mineral (251–253). Importantly, conditional deletion of PTH1R in osteocytes mitigates the increase in osteocyte lacunar size and the bone loss seen with lactation in mice, demonstrating the involvement of direct actions of PTH1R signaling in these cells (252).
In contrast to the requirement of the PTH receptor in osteocytes for bone anabolism induced by PTH injections, chronic elevation of PTH in mice lacking the PTH1R in osteocytes fed a low-calcium diet still increased bone resorption and induced the expected bone loss (246). However, T cell depletion or conditional deletion of PTH1R in T cells in mice prevented the bone loss induced by chronic elevation of PTH (255–257). Thus, complementary cellular mechanisms facilitate the skeletal actions of PTH.
The molecular mechanisms downstream from PTH1R signaling involved in the bone effects of PTH remain unclear. It is well documented that salt-inducible kinases (59, 62, 258) mediate PTH decreases in SOST/Sclerostin expression, an effect lost in mice with conditional deletion of PTH1R in osteocytes (147, 148, 246). However, DMP1-8kb-SOST mice, in which SOST is overexpressed in osteocytes and thus cannot be downregulated by PTH, displayed the expected increase in bone formation and Wnt/β-catenin signaling induced by daily PTH injections, demonstrating that SOST downregulation is dispensable for bone anabolism induced by PTH (246). In addition, PTH also increased bone mass and induced new bone formation in global SOST KO mice (259). Furthermore, direct effects of PTH on osteocytes regulate the production of RANKL and promote bone resorption (260). In fact, genetic deletion of the PTH-responsive distal control region (DCR) in the RANKL gene (DCR−/− mice) or pharmacological inhibition of MMP14, a metalloproteinase involved in the generation of soluble RANKL from membrane-bound RANKL, attenuated the exacerbated bone resorption displayed by DMP1-caPTH1ROt mice (245, 260). Altogether, these studies suggest that osteocyte RANKL, and soluble RANKL in particular, are important components of the bone catabolic response to PTH signaling.
In conclusion, today it is recognized that osteocytes are critical target cells for the action of hormones that affect the skeleton, in particular GC and PTH. Extensive evidence from several research laboratories demonstrates that GCs regulate osteocyte survival and induce critical changes in osteocytic gene expression that impact bone remodeling. Regarding PTH, expression of the PTH1R in osteocytes is required for full bone anabolism by the hormone, and PTH controls bone formation and resorption by regulating the expression of several osteocytic genes, including RANKL, SOST/Sclerostin, DKK1, and MMP14. Future research is warranted to reveal the whole spectrum of mechanisms and molecular mediators downstream of signaling in osteocytes induced by GC and PTH, with the ultimate goal of identifying new targets and developing improved bone therapeutics.
9. OSTEOCYTE PATHOPHYSIOLOGY
9.1. The Effects of Aging on Osteocytes
One of the functions of osteocytes is to sense bone microdamage and initiate repair (1, 261). With aging, there is a decline in osteocyte density and an increase in number of empty osteocyte lacunae, an index of premature osteocyte death (262, 263). These changes in osteocytes are associated with accumulation of microdamage in old bones. It is possible that the reduction in osteocyte number is a direct consequence of apoptosis of osteoblasts, which is also increased. Furthermore, osteocyte apoptosis might be due to reduced skeletal loading, elevated levels of ROS in the bone/bone marrow microenvironment (264), and/or increased endogenous GC signaling with age (265). This loss of osteocytes could partially explain the discrepancy between bone quantity and quality that occurs during aging.
Another potential cause of osteocyte death is Cx43 expression, which is reduced with age (266). Cx43 is required to preserve osteocyte viability and regulates the expression of pro- and antiosteoclastogenic cytokines in osteocytes (14, 267). In vivo and in vitro observations show that osteocytes lacking Cx43 are more prone to undergo apoptosis and exhibit an increased RANKL-to-OPG ratio (14, 268). Furthermore, long bones from old rodents and humans, as well as from mice lacking Cx43 in osteocytes, display altered cortical bone geometry resulting from concomitant increased periosteal bone formation and exacerbated endocortical bone resorption (14, 109, 268–270).
9.2. Diabetes Mellitus and Osteocytes
Diabetes mellitus (DM) is associated with elevated bone fracture risk and impaired fracture healing, resulting from alterations in bone structure and decreased strength that compromises bone quality (271–273). Type 1 DM (T1D) patients have reduced bone mass, whereas type 2 DM patients display normal or even high bone mass. In both conditions, however, the bone disease is characterized by decreased bone formation and increased resorption. Yet, the mechanisms leading to DM-induced bone disease are not clear. Using a preclinical mouse model of DM, recent work demonstrated that osteocyte functions are altered in DM. Mice with T1D induced by streptozotocin display low bone mass, decreased structural and material properties, higher osteoclast number and bone resorption, and reduced osteoblast number and bone formation. In addition, T1D mice have increased osteocyte apoptosis and upregulation of Sclerostin, DKK1, and RANKL/OPG expression in bone (274, 275). The changes in formation and resorption rates induced by DM, as well as the bone loss, were rectified by peptides derived from PTH-related peptide (PTHrP)(1–37) and (107–111), which activate PTH1R signaling (274). Comparable effects in DM models have been shown with similar doses of PTH(1–34) (275), and more recent studies demonstrated that abaloparatide, a PTHrP analog that, like PTH, binds to the PTH1R and is an FDA-approved bone anabolic agent, was superior to PTH in increasing or restoring bone mass in control or DM mice, respectively (276, 277). However, both agents corrected the impaired bone structural and material properties, citrate content, and increased Wnt antagonist expression in mice with T1D. Whether the actions of abaloparatide and PTH are due to effects mediated by osteocytes remains to be established.
Osteocytes might be involved in the metabolic changes induced by DM. Regulation of body composition and glucose metabolism in mice fed a high-fat diet have been associated with osteocytic expression of peroxisome proliferator-activated receptor γ (PPARγ) (278). Conditional deletion of osteocytic PPARγ results in mice with less fat mass and increased insulin sensitivity and energy expenditure. In addition, mice lacking PPARγ in osteocytes retain glycemic control when fed with a high-fat diet, supporting the notion that osteocytes regulate glucose homeostasis via a PPARγ-dependent mechanism. Furthermore, PPARγ signaling in osteocytes positively regulates Sclerostin expression (279). These results support the hypothesis that osteocyte signaling mediates the bone-deleterious effects of DM and open the possibility of targeting osteocytes to treat the deleterious effects of DM on the bone.
9.3. Role of Osteocytes in Cancer That Grows in Bone
Bone is a preferred site for the metastasis of cancer cells, and cancer cells communicate with host bone cells to generate a niche conducive for the progression of cancer in bone (280). Advances in the last decade revealed some of the mechanistic underpinnings by which osteocytes contribute to tumor growth, skeletal destruction, and bone pain in cancer (281, 282) (FIGURE 7). Increased osteocyte apoptosis is found in bones from multiple myeloma (MM) patients (283), particularly in bone areas colonized by MM cells (284). Osteocyte apoptosis is triggered by MM-induced activation of Notch signaling in osteocytes, which along with TNF-α secreted by MM cells, induces caspase-3-mediated apoptosis (284). Autophagy might be also involved in the stimulation of osteocyte apoptosis by MM cells (285). The increase in osteocyte apoptosis is accompanied by accumulation of osteoclasts in bone areas adjacent to dead osteocytes (283), supporting the notion that local bone resorption and the development of lytic lesions can be initiated by apoptotic osteocytes. Supporting this hypothesis, MM cells also increase RANKL and decrease OPG expression in osteocytes and enhance the ability of osteocytes to attract osteoclast precursors in vitro (283, 284).
FIGURE 7.
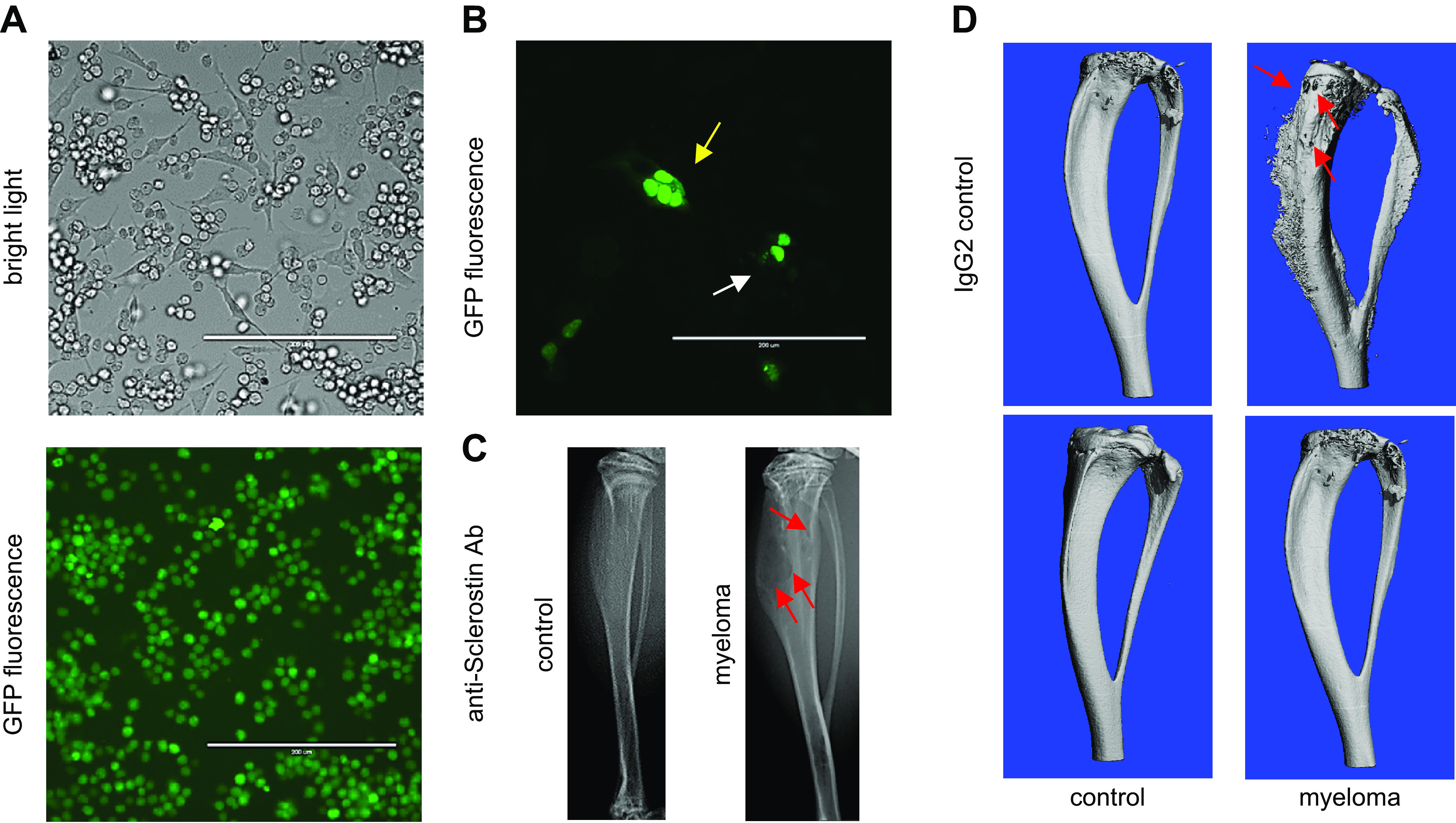
Osteocytes generate a microenvironment supportive for the growth of cancer cells and exacerbated bone resorption. A: cocultures of osteocytes and myeloma-green fluorescent protein (GFP) cancer cells enable the study of communication by cell-to-cell direct cell contact and by the exchange of soluble factors. Under these conditions, myeloma cells establish physical contact with the body and dendritic processes of osteocytes. B: osteocytes transfected with nuclear GFP cocultured with myeloma cancer cells. Fragmented (yellow arrow) and normal (white arrow) nuclei in MLO-Y4 osteocytes cocultured with MM1.S myeloma cells are displayed. C: murine intratibial injection of myeloma cells engrafts and produces multiple myeloma (MM) tumors in the bone marrow tumors that induce osteolytic lesions (red arrows) similar to those seen in myeloma patients. D: pharmacological blockade of the osteocyte-derived factor Sclerostin with a neutralizing antibody (anti-Sclerostin Ab) prevents the progression of the myeloma-induced bone disease in mice bearing myeloma tumors. Three-dimensional micro-computed tomography (μCT) reconstructions of C57BL/KaLwRijHsd bones bearing 5TGM1 myeloma cells are shown.
Osteocytes, through the production of Sclerostin, play a prominent role in the suppression of osteoblast differentiation and new bone formation in MM (284, 286). Sclerostin production by osteocytes is increased in MM-colonized bones (284). In addition, the production of Sclerostin in bones in breast cancer models is associated with increased breast cancer cell migration and invasion, as well as with the development of bone osteolysis (287). Blockade of SOST/Sclerostin’s actions in MM promotes osteoblast formation and stimulates new bone formation without affecting tumor growth, and thus could become an important therapeutic target to combat the bone disease that accompanies MM (287–290) (see sect. 10). Of note, Sclerostin inhibition reduced the proliferation of metastatic breast cancer tumor cells (291). These studies suggest that Sclerostin’s actions on tumor cells are cancer type dependent, and thus use of anti-Sclerostin antibodies requires individual evaluation. Finally, MM cells also increase the expression of FGF23 and VEGFA in osteocytes (292, 293), which in turn elevate heparanase expression and contribute to angiogenesis, respectively.
Finally, osteocytes also alter the biology of malignant cells, enhancing cancer cell invasiveness and migration capacity and stimulating tumor growth (284, 294–297). Also, novel intriguing findings suggest the existence of cross talk between osteocytes and bone sensory nerves, which might contribute to the pain induced by cancer cell growth in bone (298).
9.4. Osteocytes and Rare Diseases
Analysis of osteocytes and their signaling factors in rare skeletal diseases has unequivocally facilitated the identification of new targets and the development of new therapeutic approaches to combat both rare and common skeletal diseases. Various genetic skeletal rare bone diseases are associated with dysfunctional osteocytes, as recently reviewed (299). As discussed in sect. 2, sclerosteosis and van Buchem disease are characterized by mutations in the osteocyte-derived factor Sclerostin, leading to low or lack of SOST expression and the development of a skeletal dysplasia characterized by high bone mass (300–305). These findings led to the generation of antibodies neutralizing Sclerostin skeletal actions and successful FDA approval for its use in the treatment of postmenopausal bone loss. Mutations in the PHEX and DMP1 genes cause hypophosphatemia and ARHR, respectively, diseases characterized by high levels of osteocyte-derived FGF23 and decreased bone mass (11, 20, 306). Antibodies against FGF23 were generated and recently approved for the treatment of children with X-linked hypophosphatemia (see sect. 10) (307).
Osteogenesis imperfecta is another rare skeletal disease with low bone mass and frequent fractures linked to mutations in COL1A1 and COL1A2 genes (308). Morello and colleagues reported that the osteocyte transcriptome is dysregulated in this disease (309) and that loss of osteocytic RANKL is sufficient to increase cancellous bone mass in murine models of osteogenesis imperfecta (310). Future studies are needed to fully understand the specific role of these mutations in osteocytes versus the effects that environmental clues sent by a diseased bone microenvironment may have on them.
In summary, the function and viability of osteocytes are altered in several disorders that compromise the integrity of bone, yet whether the changes in osteocyte behavior are the origin or just a consequence of the skeletal complications demands further investigation. Given the potential role of osteocytes as regulators of distant organs, the contribution of these cells to metabolic diseases and other nonbone disorders (i.e., sarcopenia) should also be investigated.
10. OSTEOCYTES AS TARGETS AND MEDIATORS OF THERAPEUTIC INTERVENTIONS
10.1. Bisphosphonates
The potent antiresorptive bisphosphonates have been widely used to treat bone diseases for decades and to preserve bone mass by inhibiting osteoclast activity and bone resorption. However, the antifracture properties of bisphosphonates cannot be fully accounted for by the effect on bone mass. Studies in the late 1990s provided an explanation for this apparent discrepancy by demonstrating that bisphosphonates preserve osteocyte (and osteoblast) viability (94) (FIGURE 8). All bisphosphonates inhibit apoptosis of osteoblastic cells, even analogs that are inactive on osteoclasts, supporting that these drugs act through different mechanisms in osteoclasts versus osteocytes/osteoblasts (311). Moreover, an analog that does not inhibit resorption still was able to prevent apoptosis of osteocytes and osteoblasts induced by GC or by unloading (125, 312). Furthermore, this analog protected from GC-induced loss of bone mass and strength. Preservation of osteoblast function and the osteocyte network by promoting cell viability in the absence of antiresorptive effects might be beneficial in the treatment of bone fragility diseases in which decreased bone resorption is not desired.
FIGURE 8.
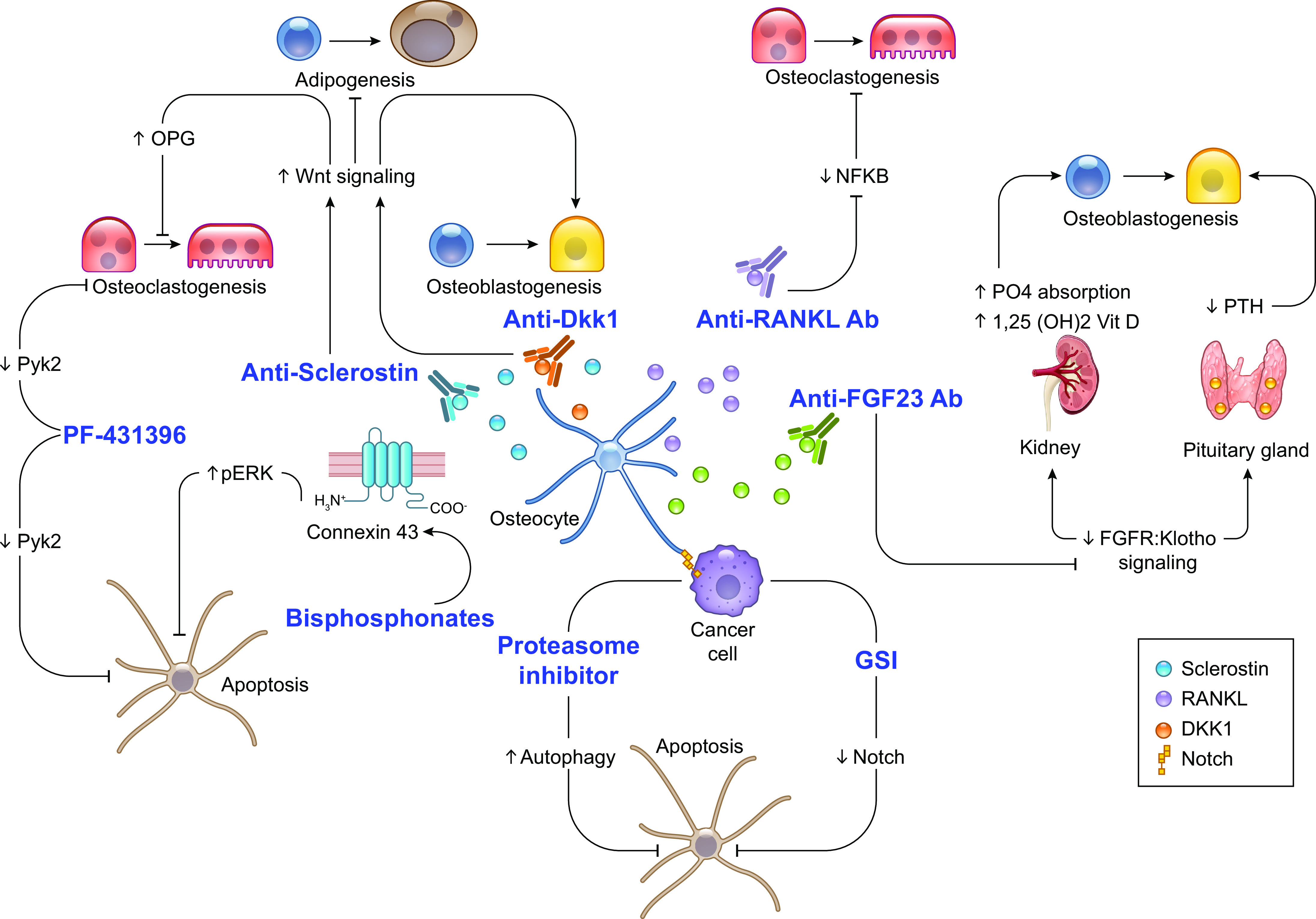
Osteocytes and osteocyte-derived factors as therapeutic targets. A better understanding of osteocyte biology prompted the generation of therapeutics targeting osteocytes and osteocyte-secreted signaling molecules. Antibodies designed to neutralize Sclerostin (clinically tested and FDA approved) and Dkk1 (clinically tested but not FDA approved) function were developed to boost bone formation in osteoporotic patients. Similarly, a neutralizing antibody against receptor activator of nuclear factor-κΒ ligand (RANKL) (clinically tested and FDA approved) was generated to stop bone loss in osteoporotic and cancer patients. More recently, a neutralizing antibody against fibroblast growth factor (FGF)23 (clinically tested and FDA approved) has been approved for the treatment of X-linked hypophosphatemia and tumor-induced osteomalacia. Furthermore, we know now that bisphosphonates (clinically tested and FDA approved), proteasome inhibitors (clinically tested and FDA approved), and pan-inhibitors of the Notch pathway (clinically tested but not FDA approved; bone-targeting molecule (BT)-gamma secretase inhibitor (GSI) experimental] preserve osteocyte viability. Finally, pharmacological inhibition of Pyk2 signaling with PF-431396 (experimental, not FDA approved) decreases glucocorticoid induced osteocyte apoptosis and promotes osteoclast cell death. NFKB, nuclear factor-κΒ; OPG, osteoprotegerin; PTH, parathyroid hormone.
The antiapoptotic action of bisphosphonates is strictly dependent on the expression of Cx43, the most abundant member of this gap junction family of proteins in bone cells (267). Remarkably, the survival effect of bisphosphonates does not require the formation of gap junction channels (15, 111). Instead, bisphosphonates open Cx43 hemichannels, which in turn activate a pathway that inhibits apoptosis by phosphorylating p90RSK kinase and its substrates, C/EBPβ and BAD.
10.2. Neutralizing Antibodies Against Sclerostin
Osteocyte-derived Sclerostin has rapidly become an important therapeutic target for the treatment of bone diseases characterized by low bone mass (313–316) (FIGURE 8). The neutralizing anti-Sclerostin antibody is considered a “dual” therapeutic agent, as it increases osteoblast number and stimulates bone formation but also inhibits osteoclast differentiation and suppresses bone resorption. In contrast to other therapeutic strategies in which bone formation and resorption are decreased (bisphosphonates) or increased (PTH1R ligands) at the same time, the bone gain induced by the anti-Sclerostin antibody results from opposing effects on bone formation (increased) and bone resorption (decreased). However, anti-Sclerostin discontinuation results in BMD loss, which eventually returns to pretreatment levels. In addition, the increased risk of cardiovascular events when the anti-Sclerostin antibody was compared with a bisphosphonate in clinical trials has raised safety issues (317, 318). Despite this observation, anti-Sclerostin was approved by the FDA in 2019 for the treatment of postmenopausal women at high risk of fracture.
Additionally, as discussed in sect. 9, preclinical animal studies showed that anti-Sclerostin antibody induces new bone formation in bones infiltrated with cancer cells without affecting tumor growth (287–290). Future studies are warranted to explore in clinical trials the potential benefits of anti-Sclerostin therapy in patients with cancer-related skeletal complications.
10.3. Neutralizing Antibodies Against RANKL
Taking into consideration that osteocytes are one of the major producers of RANKL in adult bone, it is possible that the antiresorptive properties of neutralizing antibodies against RANKL on bone are due to blockade of RANKL produced by osteocytes (FIGURE 8). Denosumab is a fully human monoclonal antibody against RANKL developed as a therapeutic agent to pharmacologically block bone resorption (319, 320). Clinical and preclinical work demonstrated that denosumab blocks RANKL signaling downstream of RANK in osteoclasts, resulting in inhibition of osteoclast formation and activity and decreased bone resorption. Denosumab is approved by the FDA for the treatment of postmenopausal osteoporosis, GC-induced osteoporosis, and the skeletal complications of metastatic prostate and breast cancer patients (321). A clinical issue that has recently attracted much attention is the bone loss that occurs after denosumab discontinuation, which can be mitigated by bisphosphonates (322). Croucher and McDonald have shown via intravital imaging of osteoclasts in vivo how removal of OPG-Fc, which mimics the effects of denosumab, increases fusion and fission of surviving osteoclasts, leading to the formation of active bone-resorbing osteoclasts potentially responsible for the initial rapid bone loss that ensues after denosumab withdrawal (323). In addition to the current patient populations approved to receive denosumab, a recent clinical study showed that denosumab prevents skeletal-related events in MM patients (324). Although it is clear that RANKL from osteocytes contributes to adult bone remodeling, whether the membrane-bound form, the soluble form, or both derived from osteocytes contribute to cancer-induced bone disease remains unclear and investigation is warranted.
10.4. Neutralizing Antibodies Against FGF23
Osteocytes produce FGF23 and regulate phosphate reabsorption in the kidney in an endocrine manner (24). Earlier this year, the FDA approved the use of burosumab, a neutralizing antibody against FGF23, for the treatment of X-linked hypophosphatemia (XLH) as well as tumor-induced osteomalacia (307) (FIGURE 8). Interestingly, the levels of FGF23, coming from either osteocytes or cancer cells, are elevated in prostate cancer and MM (293, 325–327). Whether targeting FGF23 in cancer would lead to anticancer effects and improvement of bone health is yet to be determined.
10.5. Proteasome Inhibitors and Osteocytes
Proteasome inhibitors (PIs) are drugs capable of blocking the action of the proteasome, a key cellular complex that degrades unneeded or damaged proteins. PIs are considered the backbone of MM treatment because of their ability to arrest tumor growth (328). Although preferentially used to stop the growth of tumors, both preclinical and clinical data demonstrate that PIs (329) also have effects on bone cells (330). Treatment with PIs inhibits RANKL expression in osteoblasts and NF-κB-dependent osteoclast differentiation, thus decreasing bone resorption (331). Furthermore, PIs have transient effects on osteoblasts that increase bone formation. (331). Treatment of MM patients with PIs maintained osteocyte viability (285) and decreased the serum levels of Sclerostin by 50% compared to control subjects (332) (FIGURE 8). However, whether these effects are due to direct actions on osteocytes remains to be determined. In the frame of GC excess, PIs might represent a therapeutic opportunity, as the hormones upregulate in bone (and muscle) the expression of atrogenes, E3 ubiquitin ligases that prime protein for proteasomal degradation (333).
10.6. Targeting Notch Signaling in Osteocytes
The effects of Notch signaling in osteocytes are complex. A series of studies by Canalis and collaborators showed that activation of Notch signaling in osteocytes by genetic overexpression of the Notch receptor intracellular domain (NICD)1 results in increased cancellous bone volume and fewer osteoclasts (160). Surprisingly, deletion of Notch receptors 1 and 2, which should inhibit Notch signaling, also increases trabecular bone mass by decreasing osteoclast number (334). The cause of some of these unexpected skeletal phenotypes might be interference with Notch signaling during bone development. Activation of Notch signaling in osteocytes in adult mice, which avoids potential developmental issues, demonstrated that Notch activation is sufficient to stimulate new bone formation and prevent the bone loss induced by age and ovariectomy (335). Additionally, activation of Notch in osteocytes in vitro, either by culturing osteocytes on Delta-like canonical Notch ligand 1 (DLL1)-coated plates or by coculturing them with MM cells, induces osteocyte apoptosis (284) (FIGURE 8). Moreover, pharmacological inhibition of the Notch pathway with gamma secretase inhibitors (GSIs), which block the cleavage of the NICD, decreases bone resorption (336, 337). However, this approach is clinically limited by severe side effects in the gut (338). To bypass gut toxicity, we generated a novel bone-targeted Notch inhibitor and showed that bone-targeted inhibition of Notch decreases tumor growth and bone destruction, without inducing gut toxicity (336). Importantly, bone-targeted inhibition of Notch does not affect physiological bone formation and seems to preserve the bone gain induced by daily PTH injections (339). An alternative strategy to avoid toxicity is the use of pharmacological targeting of individual Notch components. In particular, neutralizing antibodies against specific Notch receptors or ligands Delta‐1 and Jagged-1 are currently being studied in preclinical models (340, 341). It also has been shown that pharmacological inhibition of Jagged-1 blocks osteoblast mineralization (335). Additional efforts are needed to identify the role of specific components of the Notch pathway in osteocytes in bone homeostasis.
In summary, meticulous studies of osteocyte biology during the last two decades guided the development of new therapeutic approaches targeting osteocytic signaling pathways and derived factors to improve skeletal health. Although not discussed in this review, the role of osteocytes, both as contributors and/or as potential targets, in craniofacial disease and periodontal disease is an important and emerging area of research (342–344). Full characterization of osteocyte dysfunction in common and rare skeletal diseases could provide clues for the discovery of new targets to treat bone-related diseases.
11. CONCLUSIONS AND FUTURE DIRECTIONS
Advances during the last two decades demonstrated that osteocytes are multifunctional signaling cells that regulate osteoblasts and osteoclasts in a paracrine manner to maintain skeletal homeostasis. The knowledge gained about the underlying mechanisms provided the rationale for the development of new approaches to treat skeletal diseases targeting osteocytes. It is also now recognized that osteocytes are affected by mechanical and hormonal signals and by bone therapeutic agents. In addition, osteocytes contribute to the skeletal complications of diabetes and cancer as well as rare bone diseases. More recently, much attention has been attracted to the potential role of osteocytes as endocrine signaling cells in the regulation of body fat and glucose metabolism, beyond the well-recognized role of these cells in the regulation of phosphate metabolism through actions in the kidney. Future studies are needed to establish the contribution of osteocyte-derived molecules to whole body fat and their potential as therapeutic target cells for metabolic diseases.
Despite the tremendous advances in our knowledge of osteocyte biology, key areas of osteocyte biology remain underexplored. For instance, how osteocyte life span and gene expression signature are regulated is not completely understood. Unbiased genomewide approaches such as single-cell RNA sequencing (RNA-seq), proteomics, and epigenomics should advance our knowledge of the molecular changes preceding osteocyte dysfunction and thus guide the development of novel therapeutic targets. Furthermore, a better understanding of osteocyte energetic demands and utilization of glucose, fatty acids, and amino acids is needed. Moreover, although several paracrine and endocrine signaling mechanisms driven by osteocytes have been elucidated, autocrine signaling in osteocytes is underexplored.
In conclusion, osteocytes contribute to the physiological and pathophysiological regulation of bone metabolism. A complete comprehension of how osteocytes regulate the formation/function of osteoblast, osteoclasts, bone marrow adipocytes, and potentially other cells in remote organs is key to improving current therapeutic approaches and identifying novel approaches to target osteocytes, with the ultimate goal of finding superior and safer treatments to combat bone diseases.
GRANTS
This work was supported by the National Institutes of Health (R01-AR059357, R01-CA209882, R01-AT008754, and O2S1-AT008754 to T.B. and R37-CA251763 to J.D.-C.); an Arkansas Research Alliance Scholar award to T.B.; the Arkansas COBRE program NIGMS P20GM125503, a Scholar Award by the American Society of Hematology Scholar Award, and a Brian D. Novis Award by the International Myeloma Foundation (to J.D.-C.); the Winthrop P. Rockefeller Cancer Institute at UAMS (to T.B. and J.D.-C.); and the Department of Veterans Affairs (I01 BX002104 and IK6BX004596 to T.B.).
DISCLAIMERS
The contents do not represent the views of the US Department of Veterans Affairs or the United States Government.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.D.-C. prepared figures; J.D.-C. and T.B. drafted manuscript; J.D.-C. and T.B. edited and revised manuscript; J.D.-C. and T.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Drs. David Burr and Amy Y. Sato (University of Arkansas for Medical Sciences) for critical review and editing of the manuscript.
REFERENCES
- 1.Parfitt AM. Life history of osteocytes: relationship to bone age, bone remodeling, and bone fragility. J Musculoskelet Neuronal Interact 2: 499–500, 2002. [PubMed] [Google Scholar]
- 2.Plotkin LI, Bellido T. Osteocytic signalling pathways as therapeutic targets for bone fragility. Nat Rev Endocrinol 12: 593–605, 2016. doi: 10.1038/nrendo.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paic F, Igwe JC, Nori R, Kronenberg MS, Franceschetti T, Harrington P, Kuo L, Shin DG, Rowe DW, Harris SE, Kalajzic I. Identification of differentially expressed genes between osteoblasts and osteocytes. Bone 45: 682–692, 2009. doi: 10.1016/j.bone.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piemontese M, Onal M, Xiong J, Han L, Thostenson JD, Almeida M, O’Brien CA. Low bone mass and changes in the osteocyte network in mice lacking autophagy in the osteoblast lineage. Sci Rep 6: 24262, 2016. doi: 10.1038/srep24262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palumbo C, Palazzini S, Zaffe D, Marotti G. Osteocyte differentiation in the tibia of newborn rabbit: an ultrastructural study of the formation of cytoplasmic processes. Acta Anat (Basel) 137: 350–358, 1990. doi: 10.1159/000146907. [DOI] [PubMed] [Google Scholar]
- 6.Zhang K, Barragan-Adjemian C, Ye L, Kotha S, Dallas M, Lu Y, Zhao S, Harris M, Harris SE, Feng JQ, Bonewald LF. E11/gp38 selective expression in osteocytes: regulation by mechanical strain and role in dendrite elongation. Mol Cell Biol 26: 4539–4552, 2006. doi: 10.1128/MCB.02120-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skupien A, Konopka A, Trzaskoma P, Labus J, Gorlewicz A, Swiech L, Babraj M, Dolezyczek H, Figiel I, Ponimaskin E, Wlodarczyk J, Jaworski J, Wilczynski GM, Dzwonek J. CD44 regulates dendrite morphogenesis through Src tyrosine kinase-dependent positioning of the Golgi. J Cell Sci 127: 5038–5051, 2014. doi: 10.1242/jcs.154542. [DOI] [PubMed] [Google Scholar]
- 8.Hughes DE, Salter DM, Simpson R. CD44 expression in human bone: a novel marker of osteocytic differentiation. J Bone Miner Res 9: 39–44, 1994. doi: 10.1002/jbmr.5650090106. [DOI] [PubMed] [Google Scholar]
- 9.Ohizumi I, Harada N, Taniguchi K, Tsutsumi Y, Nakagawa S, Kaiho S, Mayumi T. Association of CD44 with OTS-8 in tumor vascular endothelial cells. Biochim Biophys Acta 1497: 197–203, 2000. doi: 10.1016/S0167-4889(00)00063-X. [DOI] [PubMed] [Google Scholar]
- 10.Bellido T, Plotkin LI, Bruzzaniti A. Bone cells. In: Basic and Applied Bone Biology, edited by Burr D, Allen MR.. Oxford, UK: Elsevier, 2014, p. 27–45. [Google Scholar]
- 11.Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38: 1310–1315, 2006. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holmbeck K, Bianco P, Pidoux I, Inoue S, Billinghurst RC, Wu W, Chrysovergis K, Yamada S, Birkedal-Hansen H, Poole AR. The metalloproteinase MT1-MMP is required for normal development and maintenance of osteocyte processes in bone. J Cell Sci 118: 147–156, 2005. doi: 10.1242/jcs.01581. [DOI] [PubMed] [Google Scholar]
- 13.Zhao W, Byrne MH, Wang Y, Krane SM. Osteocyte and osteoblast apoptosis and excessive bone deposition accompany failure of collagenase cleavage of collagen. J Clin Invest 106: 941–949, 2000. doi: 10.1172/JCI10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bivi N, Condon KW, Allen MR, Farlow N, Passeri G, Brun LR, Rhee Y, Bellido T, Plotkin LI. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J Bone Miner Res 27: 374–389, 2012. doi: 10.1002/jbmr.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plotkin LI, Manolagas SC, Bellido T. Transduction of cell survival signals by connexin-43 hemichannels. J Biol Chem 277: 8648–8657, 2002. doi: 10.1074/jbc.M108625200. [DOI] [PubMed] [Google Scholar]
- 16.Bivi N, Lezcano V, Romanello M, Bellido T, Plotkin LI. Connexin43 interacts with βarrestin: a pre-requisite for osteoblast survival induced by parathyroid hormone. J Cell Biochem 112: 2920–2930, 2011. doi: 10.1002/jcb.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA 98: 6500–6505, 2001. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng JQ, Clinkenbeard EL, Yuan B, White KE, Drezner MK. Osteocyte regulation of phosphate homeostasis and bone mineralization underlies the pathophysiology of the heritable disorders of rickets and osteomalacia. Bone 54: 213–221, 2013. doi: 10.1016/j.bone.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan B, Takaiwa M, Clemens TL, Feng JQ, Kumar R, Rowe PS, Xie Y, Drezner MK. Aberrant Phex function in osteoblasts and osteocytes alone underlies murine X-linked hypophosphatemia. J Clin Invest 118: 722–734, 2008. doi: 10.1172/JCI32702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farrow EG, Davis SI, Ward LM, Summers LJ, Bubbear JS, Keen R, Stamp TC, Baker LR, Bonewald LF, White KE. Molecular analysis of DMP1 mutants causing autosomal recessive hypophosphatemic rickets. Bone 44: 287–294, 2009. doi: 10.1016/j.bone.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gowen LC, Petersen DN, Mansolf AL, Qi H, Stock JL, Tkalcevic GT, Simmons HA, Crawford DT, Chidsey-Frink KL, Ke HZ, McNeish JD, Brown TA. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem 278: 1998–2007, 2003. doi: 10.1074/jbc.M203250200. [DOI] [PubMed] [Google Scholar]
- 22.Aono Y, Yamazaki Y, Yasutake J, Kawata T, Hasegawa H, Urakawa I, Fujita T, Wada M, Yamashita T, Fukumoto S, Shimada T. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Res 24: 1879–1888, 2009. doi: 10.1359/jbmr.090509. [DOI] [PubMed] [Google Scholar]
- 23.Rowe PS. The wrickkened pathways of FGF23, MEPE and PHEX. Crit Rev Oral Biol Med 15: 264–281, 2004. doi: 10.1177/154411130401500503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quarles LD. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab 285: E1–E9, 2003. doi: 10.1152/ajpendo.00016.2003. [DOI] [PubMed] [Google Scholar]
- 25.White KE, Evans WE, O’Riordan JL, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Strom TM. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26: 345–348, 2000. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 26.van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, Löwik CW. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med 199: 805–814, 2004. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robling AG, Castillo AB, Turner CH. Biomechanical and molecular regulation of bone remodeling. Annu Rev Biomed Eng 8: 455–498, 2006. doi: 10.1146/annurev.bioeng.8.061505.095721. [DOI] [PubMed] [Google Scholar]
- 28.Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, Bonewald L, Manolagas SC, O’Brien CA. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS ONE 10: e0138189, 2015. doi: 10.1371/journal.pone.0138189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med 17: 1235–1241, 2011. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17: 1231–1234, 2011. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 31.O’Brien CA, Nakashima T, Takayanagi H. Osteocyte control of osteoclastogenesis. Bone 54: 258–263, 2013. doi: 10.1016/j.bone.2012.08.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujiwara Y, Piemontese M, Liu Y, Thostenson JD, Xiong J, O’Brien CA. RANKL (Receptor Activator of NFκB Ligand) produced by osteocytes is required for the increase in B cells and bone loss caused by estrogen deficiency in mice. J Biol Chem 291: 24838–24850, 2016. doi: 10.1074/jbc.M116.742452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu W, Zhong L, Yao L, Wei Y, Gui T, Li Z, Kim H, Holdreith N, Jiang X, Tong W, Dyment N, Liu XS, Yang S, Choi Y, Ahn J, Qin L. Bone marrow adipogenic lineage precursors promote osteoclastogenesis in bone remodeling and pathologic bone loss. J Clin Invest 131: e140214, 2021. doi: 10.1172/JCI140214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delgado-Calle J, Arozamena J, García-Renedo R, García-Ibarbia C, Pascual-Carra MA, González-Macias J, Riancho JA. Osteocyte deficiency in hip fractures. Calcif Tissue Int 89: 327–334, 2011. doi: 10.1007/s00223-011-9522-0. [DOI] [PubMed] [Google Scholar]
- 35.Dallas SL, Bonewald LF. Dynamics of the transition from osteoblast to osteocyte. Ann NY Acad Sci 1192: 437–443, 2010. doi: 10.1111/j.1749-6632.2009.05246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Löwik CW, Reeve J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J 19: 1842–1844, 2005. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- 37.Irie K, Ejiri S, Sakakura Y, Shibui T, Yajima T. Matrix mineralization as a trigger for osteocyte maturation. J Histochem Cytochem 56: 561–567, 2008. doi: 10.1369/jhc.2008.950527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE 6: e25900, 2011. doi: 10.1371/journal.pone.0025900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tu X, Delgado-Calle J, Condon KW, Maycas M, Zhang H, Carlesso N, Taketo MM, Burr DB, Plotkin LI, Bellido T. Osteocytes mediate the anabolic actions of canonical Wnt/β-catenin signaling in bone. Proc Natl Acad Sci USA 112: E478–E486, 2015. doi: 10.1073/pnas.1409857112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delgado-Calle J, Sato AY, Bellido T. Role and mechanism of action of sclerostin in bone. Bone 96: 29–37, 2017. doi: 10.1016/j.bone.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bellido T. Osteocyte-driven bone remodeling. Calcif Tissue Int 94: 25–34, 2014. doi: 10.1007/s00223-013-9774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Youlten SE, Kemp JP, Logan JG, Ghirardello EJ, Sergio CM, Dack MR, Guilfoyle SE, et al. Osteocyte transcriptome mapping identifies a molecular landscape controlling skeletal homeostasis and susceptibility to skeletal disease. Nat Commun 12: 2444, 2021. doi: 10.1038/s41467-021-22517-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delgado-Calle J, Riancho JA. The role of DNA methylation in common skeletal disorders. Biology (Basel) 1: 698–713, 2012. doi: 10.3390/biology1030698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delgado-Calle J, Garmilla P, Riancho JA. Do epigenetic marks govern bone mass and homeostasis? Curr Genomics 13: 252–263, 2012. doi: 10.2174/138920212800543129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turek-Plewa J, Jagodziński PP. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol Biol Lett 10: 631–647, 2005. [PubMed] [Google Scholar]
- 46.Santos-Rosa H, Caldas C. Chromatin modifier enzymes, the histone code and cancer. Eur J Cancer 41: 2381–2402, 2005. doi: 10.1016/j.ejca.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 47.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297, 2004. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 48.Jensen ED, Gopalakrishnan R, Westendorf JJ. Regulation of gene expression in osteoblasts. BioFactors 36: 25–32, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hantusch B, Kalt R, Krieger S, Puri C, Kerjaschki D. Sp1/Sp3 and DNA-methylation contribute to basal transcriptional activation of human podoplanin in MG63 versus Saos-2 osteoblastic cells. BMC Mol Biol 8: 20, 2007. doi: 10.1186/1471-2199-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miao D, He B, Karaplis AC, Goltzman D. Parathyroid hormone is essential for normal fetal bone formation. J Clin Invest 109: 1173–1182, 2002. doi: 10.1172/JCI0214817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delgado-Calle J, Sañudo C, Sánchez-Verde L, García-Renedo RJ, Arozamena J, Riancho JA. Epigenetic regulation of alkaline phosphatase in human cells of the osteoblastic lineage. Bone 49: 830–838, 2011. doi: 10.1016/j.bone.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 52.Delgado-Calle J, Sañudo C, Fernández AF, García-Renedo R, Fraga MF, Riancho JA. Role of DNA methylation in the regulation of the RANKL-OPG system in human bone. Epigenetics 7: 83–91, 2012. doi: 10.4161/epi.7.1.18753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.del Real A, Riancho JA, Delgado-Calle J. Epigenetic regulation of Sost/sclerostin expression. Curr Mol Biol Rep 3: 85–93, 2017. doi: 10.1007/s40610-017-0063-9. [DOI] [Google Scholar]
- 54.Leupin O, Kramer I, Collette NM, Loots GG, Natt F, Kneissel M, Keller H. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res 22: 1957–1967, 2007. doi: 10.1359/jbmr.070804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Collette NM, Genetos DC, Economides AN, Xie L, Shahnazari M, Yao W, Lane NE, Harland RM, Loots GG. Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc Natl Acad Sci USA 109: 14092–14097, 2012. doi: 10.1073/pnas.1207188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sebastian A, Loots GG. Transcriptional control of Sost in bone. Bone 96: 76–84, 2017. doi: 10.1016/j.bone.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 57.Delgado-Calle J, Sañudo C, Bolado A, Fernández AF, Arozamena J, Pascual-Carra MA, Rodriguez-Rey JC, Fraga MF, Bonewald L, Riancho JA. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J Bone Miner Res 27: 926–937, 2012. doi: 10.1002/jbmr.1491. [DOI] [PubMed] [Google Scholar]
- 58.Cohen-Kfir E, Artsi H, Levin A, Abramowitz E, Bajayo A, Gurt I, Zhong L, D’Urso A, Toiber D, Mostoslavsky R, Dresner-Pollak R. Sirt1 Is a regulator of bone mass and a repressor of Sost encoding for Sclerostin: a bone formation inhibitor. Endocrinology 152: 4514–4524, 2011. doi: 10.1210/en.2011-1128. [DOI] [PubMed] [Google Scholar]
- 59.Wein MN, Liang Y, Goransson O, Sundberg TB, Wang J, Williams EA, O’Meara MJ, Govea N, Beqo B, Nishimori S, Nagano K, Brooks DJ, Martins JS, Corbin B, Anselmo A, Sadreyev R, Wu JY, Sakamoto K, Foretz M, Xavier RJ, Baron R, Bouxsein ML, Gardella TJ, Divieti-Pajevic P, Gray NS, Kronenberg HM. SIKs control osteocyte responses to parathyroid hormone. Nat Commun 7: 13176, 2016. doi: 10.1038/ncomms13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spatz JM, Wein MN, Gooi JH, Qu Y, Garr JL, Liu S, Barry KJ, Uda Y, Lai F, Dedic C, Balcells-Camps M, Kronenberg HM, Babij P, Pajevic PD. The Wnt-inhibitor Sclerostin is up-regulated by mechanical unloading in osteocytes in-vitro. J Biol Chem 290: 16744–16758, 2015. doi: 10.1074/jbc.M114.628313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sato T, Verma S, Andrade CDC, Omeara M, Campbell N, Wang JS, Cetinbas M, Lang A, Ausk BJ, Brooks DJ, Sadreyev RI, Kronenberg HM, Lagares D, Uda Y, Pajevic PD, Bouxsein ML, Gross TS, Wein MN. A FAK/HDAC5 signaling axis controls osteocyte mechanotransduction. Nat Commun 11: 3282, 2020. doi: 10.1038/s41467-020-17099-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishimori S, O’Meara MJ, Castro CD, Noda H, Cetinbas M, da Silva Martins J, Ayturk U, Brooks DJ, Bruce M, Nagata M, Ono W, Janton CJ, Bouxsein ML, Foretz M, Berdeaux R, Sadreyev RI, Gardella TJ, Jüppner H, Kronenberg HM, Wein MN. Salt-inducible kinases dictate parathyroid hormone receptor action in bone development and remodeling. J Clin Invest 129: 5187–5203, 2019. doi: 10.1172/JCI130126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stern AR, Bonewald LF. Isolation of osteocytes from mature and aged murine bone. Methods Mol Biol 1226: 3–10, 2015. doi: 10.1007/978-1-4939-1619-1_1. [DOI] [PubMed] [Google Scholar]
- 64.Kalajzic I, Matthews BG, Torreggiani E, Harris MA, Divieti Pajevic P, Harris SE. In vitro and in vivo approaches to study osteocyte biology. Bone 54: 296–306, 2013. doi: 10.1016/j.bone.2012.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Halleux C, Kramer I, Allard C, Kneissel M. Isolation of mouse osteocytes using cell fractionation for gene expression analysis. Methods Mol Biol 816: 55–66, 2012. doi: 10.1007/978-1-61779-415-5_5. [DOI] [PubMed] [Google Scholar]
- 66.Semeins CM, Bakker AD, Klein-Nulend J. Isolation of primary avian osteocytes. Methods Mol Biol 816: 43–53, 2012. doi: 10.1007/978-1-61779-415-5_4. [DOI] [PubMed] [Google Scholar]
- 67.Stern AR, Stern MM, Van Dyke ME, Jähn K, Prideaux M, Bonewald LF. Isolation and culture of primary osteocytes from the long bones of skeletally mature and aged mice. Biotechniques 52: 361–373, 2012. doi: 10.2144/0000113876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stern A, Stern M, Van Dyke M, Bonewald L. The isolation and culture of primary osteocytes from the long bones of skeletally mature and aged mice (Abstract). J Bone Miner Res 26: S471, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gu G, Nars M, Hentunen TA, Metsikkö K, Väänanen HK. Isolated primary osteocytes express functional gap junctions in vitro. Cell Tissue Res 323: 263–271, 2006. doi: 10.1007/s00441-005-0066-3. [DOI] [PubMed] [Google Scholar]
- 70.Nijweide PJ, Van Der Plas A, Alblas MJ, Klein-Nulend J. Osteocyte isolation and culture. Methods Mol Med 80: 41–50, 2003. doi: 10.1385/1-59259-366-6:41. [DOI] [PubMed] [Google Scholar]
- 71.de Rooij KE, de Wilt E, Deckers MM, de Ruiter MC, Que I, Papapoulos SE, Lowik CW. An animal model for the isolation of mammalian osteocytes using a membrane-bound enzyme, which in bone is specifically expressed in osteocytes (Abstract). J Bone Miner Res 18: S82, 2002. [Google Scholar]
- 72.Van Der Plas A, Nijweide PJ. Isolation and purification of osteocytes. J Bone Miner Res 7: 389–396, 1992. doi: 10.1002/jbmr.5650070406. [DOI] [PubMed] [Google Scholar]
- 73.Torreggiani E, Matthews BG, Pejda S, Matic I, Horowitz MC, Grcevic D, Kalajzic I. Preosteocytes/osteocytes have the potential to dedifferentiate becoming a source of osteoblasts. PLoS One 8: e75204, 2013. doi: 10.1371/journal.pone.0075204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kato Y, Windle JJ, Koop BA, Mundy GR, Bonewald LF. Establishment of an osteocyte-like cell line, MLO-Y4. J Bone Miner Res 12: 2014–2023, 1997. doi: 10.1359/jbmr.1997.12.12.2014. [DOI] [PubMed] [Google Scholar]
- 75.Kato Y, Boskey A, Spevak L, Dallas M, Hori M, Bonewald LF. Establishment of an osteoid preosteocyte-like cell MLO-A5 that spontaneously mineralizes in culture. J Bone Miner Res 16: 1622–1633, 2001. doi: 10.1359/jbmr.2001.16.9.1622. [DOI] [PubMed] [Google Scholar]
- 76.Woo SM, Rosser J, Dusevich V, Kalajzic I, Bonewald LF. Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J Bone Miner Res 26: 2634–2646, 2011. doi: 10.1002/jbmr.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bodine PV, Vernon SK, Komm BS. Establishment and hormonal regulation of a conditionally transformed preosteocytic cell line from adult human bone. Endocrinology 137: 4592–4604, 1996. doi: 10.1210/endo.137.11.8895322. [DOI] [PubMed] [Google Scholar]
- 78.Wang K, Le L, Chun BM, Tiede-Lewis LM, Shiflett LA, Prideaux M, Campos RS, Veno PA, Xie Y, Dusevich V, Bonewald LF, Dallas SL. A novel osteogenic cell line that differentiates into GFP-tagged osteocytes and forms mineral with a bone-like lacunocanalicular structure. J Bone Miner Res 34: 979–995, 2019. doi: 10.1002/jbmr.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Divieti Pajevic P. New and old osteocytic cell lines and 3D models. Curr Osteoporos Rep 18: 551–558, 2020. doi: 10.1007/s11914-020-00613-3. [DOI] [PubMed] [Google Scholar]
- 80.Bellido T, Delgado-Calle J. Ex vivo organ cultures as models to study bone biology. JBMR Plus 4: 10345, 2020. doi: 10.1002/jbm4.10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dallas SL, Xie Y, Shiflett LA, Ueki Y. Mouse Cre models for the study of bone diseases. Curr Osteoporos Rep 16: 466–477, 2018. doi: 10.1007/s11914-018-0455-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lu Y, Xie Y, Zhang S, Dusevich V, Bonewald LF, Feng JQ. DMP1-targeted Cre expression in odontoblasts and osteocytes. J Dent Res 86: 320–325, 2007. doi: 10.1177/154405910708600404. [DOI] [PubMed] [Google Scholar]
- 83.Delgado-Calle J, Arozamena J, García-Renedo R, García-Ibarbia C, Pascual-Carra MA, Gonzalez-Macías J, Riancho JA. Osteocyte deficiency in hip fractures. Calcif Tissue Int 89: 327–334, 2011. doi: 10.1007/s00223-011-9522-0. [DOI] [PubMed] [Google Scholar]
- 84.Power J, Poole KE, van Bezooijen R, Doube M, Caballero-Alías AM, Lowik C, Papapoulos S, Reeve J, Loveridge N. Sclerostin and the regulation of bone formation: effects in hip osteoarthritis and femoral neck fracture. J Bone Miner Res 25: 1867–1876, 2010. doi: 10.1002/jbmr.70. [DOI] [PubMed] [Google Scholar]
- 85.Powell WF, Barry KJ, Tulum I, Kobayashi T, Harris SE, Bringhurst F, Divieti Pajevic P. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J Endocrinol 209: 21–32, 2011. doi: 10.1530/JOE-10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maurel DB, Matsumoto T, Vallejo JA, Johnson ML, Dallas SL, Kitase Y, Brotto M, Wacker MJ, Harris MA, Harris SE, Bonewald LF. Characterization of a novel murine Sost ER(T2) Cre model targeting osteocytes. Bone Res 7: 6, 2019. doi: 10.1038/s41413-018-0037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Webster DJ, Schneider P, Dallas SL, Müller R. Studying osteocytes within their environment. Bone 54: 285–295, 2013. doi: 10.1016/j.bone.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Himeno-Ando A, Izumi Y, Yamaguchi A, Iimura T. Structural differences in the osteocyte network between the calvaria and long bone revealed by three-dimensional fluorescence morphometry, possibly reflecting distinct mechano-adaptations and sensitivities. Biochem Biophys Res Commun 417: 765–770, 2012. doi: 10.1016/j.bbrc.2011.12.031. [DOI] [PubMed] [Google Scholar]
- 89.Dallas SL, Veno PA. Live imaging of bone cell and organ cultures. Methods Mol Biol 816: 425–457, 2012. doi: 10.1007/978-1-61779-415-5_26. [DOI] [PubMed] [Google Scholar]
- 90.Kalajzic I, Braut A, Guo D, Jiang X, Kronenberg MS, Mina M, Harris MA, Harris SE, Rowe DW. Dentin matrix protein 1 expression during osteoblastic differentiation, generation of an osteocyte GFP-transgene. Bone 35: 74–82, 2004. doi: 10.1016/j.bone.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 91.Dunn KW, Sutton TA, Sandoval RM. Live-animal imaging of renal function by multiphoton microscopy. Curr Protoc Cytom 83: 12, 2018. doi: 10.1002/0471142956.cy1209s41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Greenbaum A, Chan KY, Dobreva T, Brown D, Balani DH, Boyce R, Kronenberg HM, McBride HJ, Gradinaru V. Bone CLARITY: Clearing, imaging, and computational analysis of osteoprogenitors within intact bone marrow. Sci Transl Med 9: eaah6518, 2017. doi: 10.1126/scitranslmed.aah6518. [DOI] [PubMed] [Google Scholar]
- 93.Noble BS, Peet N, Stevens HY, Brabbs A, Mosley JR, Reilly GC, Reeve J, Skerry TM, Lanyon LE. Mechanical loading: biphasic osteocyte survival and the targeting of osteoclasts for bone destruction in rat cortical bone. Am J Physiol Cell Physiol 284: C934–C943, 2003. doi: 10.1152/ajpcell.00234.2002. [DOI] [PubMed] [Google Scholar]
- 94.Bellido T, Plotkin LI. Novel actions of bisphosphonates in bone: preservation of osteoblast and osteocyte viability. Bone 49: 50–55, 2011. doi: 10.1016/j.bone.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jilka RL, Bellido T, Almeida M, Plotkin LI, O’Brien CA, Weinstein RS, Manolagas SC. Apoptosis in bone cells. In: Principles of Bone Biology edited by Bilezikian JP, Raisz LG, Martin TJ.. San Diego, CA: Academic Press, 2008, p. 237–261. [Google Scholar]
- 96.Tomkinson A, Reeve J, Shaw RW, Noble BS. The death of osteocytes via apoptosis accompanies estrogen withdrawal in human bone. J Clin Endocrinol Metab 82: 3128–3135, 1997. doi: 10.1210/jcem.82.9.4200. [DOI] [PubMed] [Google Scholar]
- 97.Tomkinson A, Gevers EF, Wit JM, Reeve J, Noble BS. The role of estrogen in the control of rat osteocyte apoptosis. J Bone Miner Res 13: 1243–1250, 1998. doi: 10.1359/jbmr.1998.13.8.1243. [DOI] [PubMed] [Google Scholar]
- 98.Huber C, Collishaw S, Mosley JR, Reeve J, Noble BS. Selective estrogen receptor modulator inhibits osteocyte apoptosis during abrupt estrogen withdrawal: implications for bone quality maintenance. Calcif Tissue Int 81: 139–144, 2007. doi: 10.1007/s00223-007-9049-6. [DOI] [PubMed] [Google Scholar]
- 99.Mann V, Huber C, Kogianni G, Collins F, Noble B. The antioxidant effect of estrogen and selective estrogen receptor modulators in the inhibition of osteocyte apoptosis in vitro. Bone 40: 674–684, 2007. doi: 10.1016/j.bone.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 100.Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O’Brien CA, Plotkin LI, Fu Q, Mancino AT, Wen Y, Vertino AM, Powers CC, Stewart SA, Ebert R, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science 298: 843–846, 2002. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- 101.Kousteni S, Bellido T, Plotkin LI, O’Brien CA, Bodenner DL, Han L, Han K, DiGregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, Roberson PK, Weinstein RS, Jilka RL, Manolagas SC. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell 104: 719–730, 2001. doi: 10.1016/S0092-8674(01)00268-9. [DOI] [PubMed] [Google Scholar]
- 102.Plotkin LI, Aguirre JI, Kousteni S, Manolagas SC, Bellido T. Bisphosphonates and estrogens inhibit osteocyte apoptosis via distinct molecular mechanisms downstream of ERK activation. J Biol Chem 280: 7317–7325, 2005. doi: 10.1074/jbc.M412817200. [DOI] [PubMed] [Google Scholar]
- 103.Plotkin LI, Han L, Manolagas SC, Bellido T. An ERK-Mediated anti-apoptotic effect of bisphosphonates, but not estrogen, on osteocytes in vitro, depends on the integrity of gap junctions: evidence for distinct signaling pathways upstream from ERKs (Abstract). J Bone Miner Res 14: S155, 1999. [Google Scholar]
- 104.Jilka RL, Parfitt AM, Manolagas SC, Bellido T, Smith C. Cleavage of collagen by osteoblasts in vitro generates anti-apoptotic signals: a mechanism for the regulation of their functional lifespan and fate during bone formation (Abstract). J Bone Miner Res 14: S343, 1999. [Google Scholar]
- 105.Jilka RL, Takahashi K, Munshi M, Williams DC, Roberson PK, Manolagas SC. Loss of estrogen upregulates osteoblastogenesis in the murine bone marrow. Evidence for autonomy from factors released during bone resorption. J Clin Invest 101: 1942–1950, 1998. doi: 10.1172/JCI1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Boyce BF, Xing L, Jilka RL, Bellido T, Weinstein RS, Parfitt AM, Manolagas SC. Apoptosis in bone cells. In: Principles of Bone Biology, edited by Bilezikian JP, Raisz LG, Rodan GA.. San Diego, CA: Academic Press, 2002, p. 151–168. [Google Scholar]
- 107.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: potential mechanisms of their deleterious effects on bone. J Clin Invest 102: 274–282, 1998. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, Bellido T. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res 21: 605–615, 2006. doi: 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- 109.Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, Kousteni S, O’Brien CA, Bellido T, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem 282: 27285–27297, 2007. doi: 10.1074/jbc.M702810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bellido T. Antagonistic interplay between mechanical forces and glucocorticoids in bone: a tale of kinases. J Cell Biochem 111: 1–6, 2010. doi: 10.1002/jcb.22660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Plotkin LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest 104: 1363–1374, 1999. doi: 10.1172/JCI6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Plotkin LI, Mathov I, Aguirre JI, Parfitt AM, Manolagas SC, Bellido T. Mechanical stimulation prevents osteocyte apoptosis: requirement of integrins, Src kinases and ERKs. Am J Physiol Cell Physiol 289: C633–C643, 2005. doi: 10.1152/ajpcell.00278.2004. [DOI] [PubMed] [Google Scholar]
- 113.Bakker A, Klein-Nulend J, Burger E. Shear stress inhibits while disuse promotes osteocyte apoptosis. Biochem Biophys Res Commun 320: 1163–1168, 2004. doi: 10.1016/j.bbrc.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 114.Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone 42: 606–615, 2008. doi: 10.1016/j.bone.2007.12.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Armstrong VJ, Muzylak M, Sunters A, Zaman G, Saxon LK, Price JS, Lanyon LE. Wnt/β-catenin signaling Is a component of osteoblastic bone cell early responses to load-bearing and requires estrogen receptor α. J Biol Chem 282: 20715–20727, 2007. doi: 10.1074/jbc.M703224200. [DOI] [PubMed] [Google Scholar]
- 116.Sunters A, Armstrong VJ, Zaman G, Kypta RM, Kawano Y, Lanyon LE, Price JS. Mechano-transduction in osteoblastic cells involves strain-regulated, estrogen receptor alpha-mediated, control of IGF-IR sensitivity to ambient IGF, leading to PI3-K/AKT dependent, Wnt/LRP5 receptor-independent activation of beta-catenin signaling. J Biol Chem 285: 8743–8758, 2010. doi: 10.1074/jbc.M109.027086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Almeida M, Han L, Bellido T, Manolagas SC, Kousteni S. Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by beta-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J Biol Chem 280: 41342–41351, 2005. doi: 10.1074/jbc.M502168200. [DOI] [PubMed] [Google Scholar]
- 118.Gortazar AR, Martin-Millan M, Bravo B, Plotkin LI, Bellido T. Crosstalk between caveolin-1/ERKs and ß-catenin survival pathways in osteocyte mechanotransduction. J Biol Chem 288: 8168–8175, 2013. doi: 10.1074/jbc.M112.437921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Verborgt O, Gibson GJ, Schaffler MB. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res 15: 60–67, 2000. doi: 10.1359/jbmr.2000.15.1.60. [DOI] [PubMed] [Google Scholar]
- 120.Verborgt O, Tatton NA, Majeska RJ, Schaffler MB. Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: complementary roles in bone remodeling regulation? J Bone Miner Res 17: 907–914, 2002. doi: 10.1359/jbmr.2002.17.5.907. [DOI] [PubMed] [Google Scholar]
- 121.Bellido T. Osteocyte apoptosis induces bone resorption and impairs the skeletal response to weightlessness. Bonekey-osteovision 4: 252–256, 2007. doi: 10.1138/20070272. [DOI] [Google Scholar]
- 122.Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, Ito M, Takeshita S, Ikeda K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab 5: 464–475, 2007. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 123.Cabahug-Zuckerman P, Frikha-Benayed D, Majeska RJ, Tuthill A, Yakar S, Judex S, Schaffler MB. osteocyte apoptosis caused by hindlimb unloading is required to trigger osteocyte RANKL production and subsequent resorption of cortical and trabecular bone in mice femurs. J Bone Miner Res 31: 1356–1365, 2016. doi: 10.1002/jbmr.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kennedy OD, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Osteocyte apoptosis is required for production of osteoclastogenic signals following bone fatigue in vivo. Bone 64: 132–137, 2014. doi: 10.1016/j.bone.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Plotkin LI, Gortazar AR, Davis HM, Condon KW, Gabilondo H, Maycas M, Allen MR, Bellido T. Inhibition of osteocyte apoptosis prevents the increase in osteocytic RANKL but it does not stop bone resorption or the loss of bone induced by unloading. J Biol Chem 290: 18934–18942, 2015. doi: 10.1074/jbc.M115.642090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, Kneissel M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol 30: 3071–3085, 2010. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cawley KM, Bustamante-Gomez NC, Guha AG, MacLeod RS, Xiong J, Gubrij I, Liu Y, Mulkey R, Palmieri M, Thostenson JD, Goellner JJ, O’Brien CA. Local production of osteoprotegerin by osteoblasts suppresses bone resorption. Cell Rep 32: 108052, 2020. doi: 10.1016/j.celrep.2020.108052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang J, Shah R, Robling AG, Templeton E, Yang H, Tracey KJ, Bidwell JP. HMGB1 is a bone-active cytokine. J Cell Physiol 214: 730–739, 2008. doi: 10.1002/jcp.21268. [DOI] [PubMed] [Google Scholar]
- 129.Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone 50: 1115–1122, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Marcus R. Mechanisms of exercise effects on bone. In: Principles of Bone Biology, edited by Bilezikian JP, Raisz LG, Rodan GA.. San Diego, CA: Academic Press, 2002, p. 1477–1488. [Google Scholar]
- 131.Bikle DD, Halloran BP, Morey-Holton E. Spaceflight and the skeleton: lessons for the earthbound. Gravit Space Biol Bull 10: 119–135, 1997. [PubMed] [Google Scholar]
- 132.Kousteni S, Han L, Chen JR, Almeida M, Plotkin LI, Bellido T, Manolagas SC. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest 111: 1651–1664, 2003. doi: 10.1172/JCI200317261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Plotkin LI, Bellido T. Bisphosphonate-induced, hemichannel-mediated, anti-apoptosis through the Src/ERK pathway: a gap junction-independent action of connexin43. Cell Commun Adhes 8: 377–382, 2001. doi: 10.3109/15419060109080757. [DOI] [PubMed] [Google Scholar]
- 134.Farr JN, Kaur J, Doolittle ML, Khosla S. Osteocyte cellular senescence. Curr Osteoporos Rep 18: 559–567, 2020. doi: 10.1007/s11914-020-00619-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, LeBrasseur NK, Drake MT, Pignolo RJ, Pirtskhalava T, Tchkonia T, Oursler MJ, Kirkland JL, Khosla S. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 23: 1072–1079, 2017. doi: 10.1038/nm.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, Drake MT, Tchkonia T, LeBrasseur NK, Kirkland JL, Bonewald LF, Pignolo RJ, Monroe DG, Khosla S. Identification of senescent cells in the bone microenvironment. J Bone Miner Res 31: 1920–1929, 2016. doi: 10.1002/jbmr.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Eckhardt BA, Rowsey JL, Thicke BS, Fraser DG, O’Grady KL, Bondar OP, Hines JM, Singh RJ, Thoreson AR, Rakshit K, Lagnado AB, Passos JF, Vella A, Matveyenko AV, Khosla S, Monroe DG, Farr JN. Accelerated osteocyte senescence and skeletal fragility in mice with type 2 diabetes. JCI Insight 5: e135236, 2020. doi: 10.1172/jci.insight.135236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chandra A, Lagnado AB, Farr JN, Monroe DG, Park S, Hachfeld C, Tchkonia T, Kirkland JL, Khosla S, Passos JF, Pignolo RJ. Targeted reduction of senescent cell burden alleviates focal radiotherapy-related bone loss. J Bone Miner Res 35: 1119–1131, 2020. doi: 10.1002/jbmr.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Duncan RL, Turner CH. Mechanotransduction and the functional response of bone to mechanical strain. Calcif Tissue Int 57: 344–358, 1995. doi: 10.1007/BF00302070. [DOI] [PubMed] [Google Scholar]
- 140.Burger EH, Klein-Nulend J, van Der Plas A, Nijweide PJ. Function of osteocytes in bone—their role in mechanotransduction. J Nutr 125: 2020S–2023S, 1995. doi: 10.1093/jn/125.suppl_7.2020S. [DOI] [PubMed] [Google Scholar]
- 141.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med 19: 179–192, 2013. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 142.Delgado-Calle J, Bellido T. Osteocytes and skeletal pathophysiology. Curr Mol Biol Rep 1: 157–167, 2015. doi: 10.1007/s40610-015-0026-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chang MK, Kramer I, Huber T, Kinzel B, Guth-Gundel S, Leupin O, Kneissel M. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proc Natl Acad Sci USA 111: E5187–E5195, 2014. doi: 10.1073/pnas.1413828111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res 23: 860–869, 2008. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 145.Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, Stolina M, Turner CH, Robling AG, Plotkin LI, Bellido T. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 50: 209–217, 2012. doi: 10.1016/j.bone.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Robling AG, Turner CH. Mechanical signaling for bone modeling and remodeling. Crit Rev Eukaryot Gene Expr 19: 319–338, 2009. doi: 10.1615/CritRevEukarGeneExpr.v19.i4.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, Jilka RL. Chronic elevation of PTH in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146: 4577–4583, 2005. doi: 10.1210/en.2005-0239. [DOI] [PubMed] [Google Scholar]
- 148.Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone 37: 148–158, 2005. doi: 10.1016/j.bone.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 149.van Lierop AH, Witteveen JE, Hamdy NA, Papapoulos SE. Patients with primary hyperparathyroidism have lower circulating sclerostin levels than euparathyroid controls. Eur J Endocrinol 163: 833–837, 2010. doi: 10.1530/EJE-10-0699. [DOI] [PubMed] [Google Scholar]
- 150.Drake MT, Srinivasan B, Mödder UI, Peterson JM, McCready LK, Riggs BL, Dwyer D, Stolina M, Kostenuik P, Khosla S. Effects of parathyroid hormone treatment on circulating sclerostin levels in postmenopausal women. J Clin Endocrinol Metab 95: 5056–5062, 2010. doi: 10.1210/jc.2010-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mirza FS, Padhi ID, Raisz LG, Lorenzo JA. Serum sclerostin levels negatively correlate with parathyroid hormone levels and free estrogen index in postmenopausal women. J Clin Endocrinol Metab 95: 1991–1997, 2010. doi: 10.1210/jc.2009-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yu EW, Kumbhani R, Siwila-Sackman E, Leder BZ. Acute decline in serum sclerostin in response to PTH infusion in healthy men. J Clin Endocrinol Metab 96: E1848–E1851, 2011. doi: 10.1210/jc.2011-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Witcher PC, Miner SE, Horan DJ, Bullock WA, Lim KE, Kang KS, Adaniya AL, Ross RD, Loots GG, Robling AG. Sclerostin neutralization unleashes the osteoanabolic effects of Dkk1 inhibition. JCI Insight 3: e98673, 2018. doi: 10.1172/jci.insight.98673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Glass DA 2nd, Karsenty G. In vivo analysis of Wnt signaling in bone. Endocrinology 148: 2630–2634, 2007. doi: 10.1210/en.2006-1372. [DOI] [PubMed] [Google Scholar]
- 155.Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R, Brum A, Park D, Galili N, Mukherjee S, Teruya-Feldstein J, Raza A, Rabadan R, Berman E, Kousteni S. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 506: 240–244, 2014. doi: 10.1038/nature12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 8: 751–764, 2005. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 157.Igwe JC, Jiang X, Paic F, Ma L, Adams DJ, Baldock PA, Pilbeam CC, Kalajzic I. Neuropeptide Y is expressed by osteocytes and can inhibit osteoblastic activity. J Cell Biochem 108: 621–630, 2009. doi: 10.1002/jcb.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fujita K, Xing Q, Khosla S, Monroe DG. Mutual enhancement of differentiation of osteoblasts and osteocytes occurs through direct cell-cell contact. J Cell Biochem 115: 2039–2044, 2014. doi: 10.1002/jcb.24880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zanotti S, Canalis E. Notch signaling in skeletal health and disease. Eur J Endocrinol 168: R95–R103, 2013. doi: 10.1530/EJE-13-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Canalis E, Parker K, Feng JQ, Zanotti S. Osteoblast lineage-specific effects of notch activation in the skeleton. Endocrinology 154: 623–634, 2013. doi: 10.1210/en.2012-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Xiong J, Cawley K, Piemontese M, Fujiwara Y, Zhao H, Goellner JJ, O’Brien CA. Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat Commun 9: 2909, 2018. doi: 10.1038/s41467-018-05244-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Asano T, Okamoto K, Nakai Y, Tsutsumi M, Muro R, Suematsu A, Hashimoto K, Okamura T, Ehata S, Nitta T, Takayanagi H. Soluble RANKL is physiologically dispensable but accelerates tumour metastasis to bone. Nat Metab 1: 868–875, 2019. doi: 10.1038/s42255-019-0104-1. [DOI] [PubMed] [Google Scholar]
- 163.Ikebuchi Y, Aoki S, Honma M, Hayashi M, Sugamori Y, Khan M, Kariya Y, Kato G, Tabata Y, Penninger JM, Udagawa N, Aoki K, Suzuki H. Coupling of bone resorption and formation by RANKL reverse signalling. Nature 561: 195–200, 2018. doi: 10.1038/s41586-018-0482-7. [DOI] [PubMed] [Google Scholar]
- 164.Harris SE, MacDougall M, Horn D, Woodruff K, Zimmer SN, Rebel VI, Fajardo R, Feng JQ, Heinrich-Gluhak J, Harris MA, Abboud-Werner SL. Meox2Cre-mediated disruption of CSF-1 leads to osteopetrosis and osteocyte defects. Bone 50: 42–53, 2012. doi: 10.1016/j.bone.2011.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Singh L, Tyagi S, Myers D, Duque G. Good, bad, or ugly: the biological roles of bone marrow fat. Curr Osteoporos Rep 16: 130–137, 2018. doi: 10.1007/s11914-018-0427-y. [DOI] [PubMed] [Google Scholar]
- 166.Fairfield H, Rosen CJ, Reagan MR. Connecting bone and fat: the potential role for Sclerostin. Curr Mol Biol Rep 3: 114–121, 2017. doi: 10.1007/s40610-017-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ukita M, Yamaguchi T, Ohata N, Tamura M. Sclerostin enhances adipocyte differentiation in 3T3-L1 cells. J Cell Biochem 117: 1419–1428, 2016. doi: 10.1002/jcb.25432. [DOI] [PubMed] [Google Scholar]
- 168.Fairfield H, Falank C, Harris E, Demambro V, McDonald M, Pettitt JA, Mohanty ST, Croucher P, Kramer I, Kneissel M, Rosen CJ, Reagan MR. the skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis. J Cell Physiol 233: 1156–1167, 2018. doi: 10.1002/jcp.25976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Farrell M, Fairfield H, Costa S, D’Amico A, Falank C, Brooks DJ, Reagan MR. Sclerostin-neutralizing antibody treatment rescues negative effects of rosiglitazone on mouse bone parameters. J Bone Miner Res 36: 158–169, 2021. doi: 10.1002/jbmr.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Karsenty G, Olson EN. Bone and muscle endocrine functions: unexpected paradigms of inter-organ communication. Cell 164: 1248–1256, 2016. doi: 10.1016/j.cell.2016.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sato M, Asada N, Kawano Y, Wakahashi K, Minagawa K, Kawano H, Sada A, Ikeda K, Matsui T, Katayama Y. Osteocytes regulate primary lymphoid organs and fat metabolism. Cell Metab 18: 749–758, 2013. doi: 10.1016/j.cmet.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 172.Kim SP, Frey JL, Li Z, Kushwaha P, Zoch ML, Tomlinson RE, Da H, Aja S, Noh HL, Kim JK, Hussain MA, Thorek DL, Wolfgang MJ, Riddle RC. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc Natl Acad Sci USA 114: E11238–E11247, 2017. doi: 10.1073/pnas.1707876115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kim SP, Da H, Li Z, Kushwaha P, Beil C, Mei L, Xiong WC, Wolfgang MJ, Clemens TL, Riddle RC. Lrp4 expression by adipocytes and osteoblasts differentially impacts sclerostin's endocrine effects on body composition and glucose metabolism. J Biol Chem 294: 6899–6911, 2019. doi: 10.1074/jbc.RA118.006769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Nelson JH, Davis HM, McAndrews K, Cregor MD, Thompson WR, Plotkin LI, Robling AG, Bellido T, Delgado-Calle J. Sclerostin regulates adipocyte fate and mediates paracrine and endocrine signaling between osteocytes and fat (Abstract). J Bone Miner Res 32, Suppl 1: Abstract SUN-0665, 2018.
- 175.García-Martín A, Rozas-Moreno P, Reyes-García R, Morales-Santana S, García-Fontana B, García-Salcedo JA, Muñoz-Torres M. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab 97: 234–241, 2012. doi: 10.1210/jc.2011-2186. [DOI] [PubMed] [Google Scholar]
- 176.Urano T, Shiraki M, Ouchi Y, Inoue S. Association of circulating sclerostin levels with fat mass and metabolic disease-related markers in Japanese postmenopausal women. J Clin Endocrinol Metab 97: E1473–E1477, 2012. doi: 10.1210/jc.2012-1218. [DOI] [PubMed] [Google Scholar]
- 177.Amrein K, Amrein S, Drexler C, Dimai HP, Dobnig H, Pfeifer K, Tomaschitz A, Pieber TR, Fahrleitner-Pammer A. Sclerostin and its association with physical activity, age, gender, body composition, and bone mineral content in healthy adults. J Clin Endocrinol Metab 97: 148–154, 2012. doi: 10.1210/jc.2011-2152. [DOI] [PubMed] [Google Scholar]
- 178.Loh NY, Neville MJ, Marinou K, Hardcastle SA, Fielding BA, Duncan EL, McCarthy MI, Tobias JH, Gregson CL, Karpe F, Christodoulides C. LRP5 regulates human body fat distribution by modulating adipose progenitor biology in a dose- and depot-specific fashion. Cell Metab 21: 262–273, 2015. doi: 10.1016/j.cmet.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fulzele K, Lai F, Dedic C, Saini V, Uda Y, Shi C, Tuck P, Aronson JL, Liu X, Spatz JM, Wein MN, Divieti Pajevic P. Osteocyte-secreted Wnt signaling inhibitor sclerostin contributes to beige adipogenesis in peripheral fat depots. J Bone Miner Res 32: 373–384, 2017. doi: 10.1002/jbmr.3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yoshiko Y, Wang H, Minamizaki T, Ijuin C, Yamamoto R, Suemune S, Kozai K, Tanne K, Aubin JE, Maeda N. Mineralized tissue cells are a principal source of FGF23. Bone 40: 1565–1573, 2007. doi: 10.1016/j.bone.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 181.Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem 278: 37419–37426, 2003. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- 182.Clinkenbeard EL, Cass TA, Ni P, Hum JM, Bellido T, Allen MR, White KE. Conditional deletion of murine Fgf23: interruption of the normal skeletal responses to phosphate challenge and rescue of genetic hypophosphatemia. J Bone Miner Res 31: 1247–1257, 2016. doi: 10.1002/jbmr.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ito M, Sakai Y, Furumoto M, Segawa H, Haito S, Yamanaka S, Nakamura R, Kuwahata M, Miyamoto K. Vitamin D and phosphate regulate fibroblast growth factor-23 in K-562 cells. Am J Physiol Endocrinol Metab 288: E1101–E1109, 2005. doi: 10.1152/ajpendo.00502.2004. [DOI] [PubMed] [Google Scholar]
- 184.Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology 146: 5358–5364, 2005. doi: 10.1210/en.2005-0777. [DOI] [PubMed] [Google Scholar]
- 185.Burnett SM, Gunawardene SC, Bringhurst FR, Jüppner H, Lee H, Finkelstein JS. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res 21: 1187–1196, 2006. doi: 10.1359/jbmr.060507. [DOI] [PubMed] [Google Scholar]
- 186.Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, Ghishan FK. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol 289: G1036–G1042, 2005. doi: 10.1152/ajpgi.00243.2005. [DOI] [PubMed] [Google Scholar]
- 187.Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 79: 1370–1378, 2011. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Silver J, Naveh-Many T. FGF23 and the parathyroid glands. Pediatr Nephrol 25: 2241–2245, 2010. doi: 10.1007/s00467-010-1565-3. [DOI] [PubMed] [Google Scholar]
- 189.White KE, Larsson TE, Econs MJ. The roles of specific genes implicated as circulating factors involved in normal and disordered phosphate homeostasis: frizzled related protein-4, matrix extracellular phosphoglycoprotein, and fibroblast growth factor 23. Endocr Rev 27: 221–241, 2006. doi: 10.1210/er.2005-0019. [DOI] [PubMed] [Google Scholar]
- 190.Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol 299: F882–F889, 2010. doi: 10.1152/ajprenal.00360.2010. [DOI] [PubMed] [Google Scholar]
- 191.Kobayashi K, Imanishi Y, Miyauchi A, Onoda N, Kawata T, Tahara H, Goto H, Miki T, Ishimura E, Sugimoto T, Ishikawa T, Inaba M, Nishizawa Y. Regulation of plasma fibroblast growth factor 23 by calcium in primary hyperparathyroidism. Eur J Endocrinol 154: 93–99, 2006. doi: 10.1530/eje.1.02053. [DOI] [PubMed] [Google Scholar]
- 192.Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, Nishizawa Y. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol 18: 2683–2688, 2007. doi: 10.1681/ASN.2006070783. [DOI] [PubMed] [Google Scholar]
- 193.Brown WW, Jüppner H, Langman CB, Price H, Farrow EG, White KE, McCormick KL. Hypophosphatemia with elevations in serum fibroblast growth factor 23 in a child with Jansen's metaphyseal chondrodysplasia. J Clin Endocrinol Metab 94: 17–20, 2009. doi: 10.1210/jcem.94.2.9988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rhee Y, Bivi N, Farrow E, Lezcano V, Plotkin LI, White KE, Bellido T. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 49: 636–643, 2011. doi: 10.1016/j.bone.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Simic P, Kim W, Zhou W, Pierce KA, Chang W, Sykes DB, Aziz NB, Elmariah S, Ngo D, Pajevic PD, Govea N, Kestenbaum BR, de Boer IH, Cheng Z, Christov M, Chun J, Leaf DE, Waikar SS, Tager AM, Gerszten RE, Thadhani RI, Clish CB, Jüppner H, Wein MN, Rhee EP. Glycerol-3-phosphate is an FGF23 regulator derived from the injured kidney. J Clin Invest 130: 1513–1526, 2020. doi: 10.1172/JCI131190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Huang J, Romero-Suarez S, Lara N, Mo C, Kaja S, Brotto L, Dallas SL, Johnson ML, Jähn K, Bonewald LF, Brotto M. Crosstalk between MLO-Y4 osteocytes and C2C12 muscle cells is mediated by the Wnt/beta-catenin pathway. JBMR Plus 1: 86–100, 2017. doi: 10.1002/jbm4.10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gorski JP, Huffman NT, Vallejo J, Brotto L, Chittur SV, Breggia A, Stern A, Huang J, Mo C, Seidah NG, Bonewald L, Brotto M. Deletion of Mbtps1 (PCSK8, S1P, SKI-1) in osteocytes stimulates soleus muscle regeneration and increased size and contractile force with age. J Biol Chem 291: 4308–4322, 2016. doi: 10.1074/jbc.M115.686626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mo C, Zhao R, Vallejo J, Igwe O, Bonewald L, Wetmore L, Brotto M. Prostaglandin E2 promotes proliferation of skeletal muscle myoblasts via EP4 receptor activation. Cell Cycle 14: 1507–1516, 2015. doi: 10.1080/15384101.2015.1026520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wood CL, Pajevic PD, Gooi JH. Osteocyte secreted factors inhibit skeletal muscle differentiation. Bone Rep 6: 74–80, 2017. doi: 10.1016/j.bonr.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kitase Y, Vallejo JA, Gutheil W, Vemula H, Jähn K, Yi J, Zhou J, Brotto M, Bonewald LF. β-aminoisobutyric acid, l-BAIBA, is a muscle-derived osteocyte survival factor. Cell Rep 22: 1531–1544, 2018. doi: 10.1016/j.celrep.2018.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sato AY, Peacock M, Bellido T. Glucocorticoid excess in bone and muscle. Clin Rev Bone Miner Metab 16: 33–47, 2018. doi: 10.1007/s12018-018-9242-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Weinstein RS. Clinical practice. Glucocorticoid-induced bone disease. N Engl J Med 365: 62–70, 2011. doi: 10.1056/NEJMcp1012926. [DOI] [PubMed] [Google Scholar]
- 203.Weinstein RS. Glucocorticoid-induced osteoporosis. Rev Endocr Metab Disord 2: 65–73, 2001. doi: 10.1023/A:1010007108155. [DOI] [PubMed] [Google Scholar]
- 204.Aarden EM, Wassenaar AM, Alblas MJ, Nijweide PJ. Immunocytochemical demonstration of extracellular matrix proteins in isolated osteocytes. Histochem Cell Biol 106: 495–501, 1996. doi: 10.1007/BF02473312. [DOI] [PubMed] [Google Scholar]
- 205.O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology 145: 1835–1841, 2004. doi: 10.1210/en.2003-0990. [DOI] [PubMed] [Google Scholar]
- 206.Gohel A, McCarthy MB, Gronowicz G. Estrogen prevents glucocorticoid-induced apoptosis in osteoblasts in vivo and in vitro. Endocrinology 140: 5339–5347, 1999. doi: 10.1210/endo.140.11.7135. [DOI] [PubMed] [Google Scholar]
- 207.Plotkin LI, Manolagas SC, Bellido T. Glucocorticoids induce osteocyte apoptosis by blocking focal adhesion kinase-mediated survival: evidence for inside-out signaling leading to anoikis. J Biol Chem 282: 24120–24130, 2007. doi: 10.1074/jbc.M611435200. [DOI] [PubMed] [Google Scholar]
- 208.Weinstein RS, Chen JR, Powers CC, Stewart SA, Landes RD, Bellido T, Jilka RL, Parfitt AM, Manolagas SC. Promotion of osteoclast survival and antagonism of bisphosphonate-induced osteoclast apoptosis by glucocorticoids. J Clin Invest 109: 1041–1048, 2002. doi: 10.1172/JCI0214538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Necela BM, Cidlowski JA. Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proc Am Thorac Soc 1: 239–246, 2004. doi: 10.1513/pats.200402-005MS. [DOI] [PubMed] [Google Scholar]
- 210.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 353: 1711–1723, 2005. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 211.Druilhe A, Létuvé S, Pretolani M. Glucocorticoid-induced apoptosis in human eosinophils: mechanisms of action. Apoptosis 8: 481–495, 2003. doi: 10.1023/A:1025590308147. [DOI] [PubMed] [Google Scholar]
- 212.Limbourg FP, Liao JK. Nontranscriptional actions of the glucocorticoid receptor. J Mol Med (Berl) 81: 168–174, 2003. doi: 10.1007/s00109-003-0418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Chauhan D, Pandey P, Ogata A, Teoh G, Treon S, Urashima M, Kharbanda S, Anderson KC. Dexamethasone induces apoptosis of multiple myeloma cells in a JNK/SAP kinase independent mechanism. Oncogene 15: 837–843, 1997. doi: 10.1038/sj.onc.1201253. [DOI] [PubMed] [Google Scholar]
- 214.Blaukat A, Ivankovic-Dikic I, Grönroos E, Dolfi F, Tokiwa G, Vuori K, Dikic I. Adaptor proteins Grb2 and Crk couple Pyk2 with activation of specific mitogen-activated protein kinase cascades. J Biol Chem 274: 14893–14901, 1999. doi: 10.1074/jbc.274.21.14893. [DOI] [PubMed] [Google Scholar]
- 215.Tokiwa G, Dikic I, Lev S, Schlessinger J. Activation of Pyk2 by stress signals and coupling with JNK signaling pathway. Science 273: 792–794, 1996. doi: 10.1126/science.273.5276.792. [DOI] [PubMed] [Google Scholar]
- 216.Chauhan D, Hideshima T, Pandey P, Treon S, Teoh G, Raje N, Rosen S, Krett N, Husson H, Avraham S, Kharbanda S, Anderson KC. RAFTK/PYK2-dependent and -independent apoptosis in multiple myeloma cells. Oncogene 18: 6733–6740, 1999. doi: 10.1038/sj.onc.1203082. [DOI] [PubMed] [Google Scholar]
- 217.Xiong W, Parsons JT. Induction of apoptosis after expression of PYK2, a tyrosine kinase structurally related to focal adhesion kinase. J Cell Biol 139: 529–539, 1997. doi: 10.1083/jcb.139.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Sasaki H, Nagura K, Ishino M, Tobioka H, Kotani K, Sasaki T. Cloning and characterization of cell adhesion kinase beta, a novel protein-tyrosine kinase of the focal adhesion kinase subfamily. J Biol Chem 270: 21206–21219, 1995. doi: 10.1074/jbc.270.36.21206. [DOI] [PubMed] [Google Scholar]
- 219.Avraham H, Park SY, Schinkmann K, Avraham S. RAFTK/Pyk2-mediated cellular signalling. Cell Signal 12: 123–133, 2000. doi: 10.1016/S0898-6568(99)00076-5. [DOI] [PubMed] [Google Scholar]
- 220.Giancotti FG, Ruoslahti E. Integrin signaling. Science 285: 1028–1032, 1999. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 221.Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science 268: 233–239, 1995. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 222.Frisch SM, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol 9: 701–706, 1997. doi: 10.1016/S0955-0674(97)80124-X. [DOI] [PubMed] [Google Scholar]
- 223.Ginsberg MH, Partridge A, Shattil SJ. Integrin regulation. Curr Opin Cell Biol 17: 509–516, 2005. doi: 10.1016/j.ceb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 224.Hynes R. Integrins: bidirectional, allosteric signaling machines. Cell 110: 673–687, 2002. doi: 10.1016/S0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 225.Vanden Berghe W, Francesconi E, De Bosscher K, Resche-Rigon M, Haegeman G. Dissociated glucocorticoids with anti-inflammatory potential repress interleukin-6 gene expression by a nuclear factor-kappaB-dependent mechanism. Mol Pharmacol 56: 797–806, 1999. [PubMed] [Google Scholar]
- 226.Cheng SL, Zhang SF, Mohan S, Lecanda F, Fausto A, Hunt AH, Canalis E, Avioli LV. Regulation of insulin-like growth factors I and II and their binding proteins in human bone marrow stromal cells by dexamethasone. J Cell Biochem 71: 449–458, 1998. doi: 10.1002/(SICI)1097-4644(19981201)71:3<449::AID-JCB13>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 227.Chang DJ, Ji C, Kim KK, Casinghino S, McCarthy TL, Centrella M. Reduction in transforming growth factor beta receptor I expression and transcription factor CBFa1 on bone cells by glucocorticoid. J Biol Chem 273: 4892–4896, 1998. doi: 10.1074/jbc.273.9.4892. [DOI] [PubMed] [Google Scholar]
- 228.Doherty WJ, DeRome ME, McCarthy MB, Gronowicz GA. The effect of glucocorticoids on osteoblast function. The effect of corticosterone on osteoblast expression of beta 1 integrins. J Bone Joint Surg Am 77: 396–404, 1995. doi: 10.2106/00004623-199503000-00009. [DOI] [PubMed] [Google Scholar]
- 229.Sato AY, Cregor M, McAndrews K, Li T, Condon KW, Plotkin LI, Bellido T. Glucocorticoid-induced bone fragility is prevented in female mice by blocking Pyk2/anoikis signaling. Endocrinology 160: 1659–1673, 2019. doi: 10.1210/en.2019-00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Almeida M, Han L, Ambrogini E, Weinstein RS, Manolagas SC. Glucocorticoids and tumor necrosis factor (TNF) alpha increase oxidative stress and suppress WNT signaling in osteoblasts. J Biol Chem 286: 44326–44335, 2011. doi: 10.1074/jbc.M111.283481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633, 2003. doi: 10.1016/S1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 232.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086, 2011. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 233.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307: 935–939, 2005. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 234.Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332: 91–94, 2011. doi: 10.1126/science.1201396. [DOI] [PubMed] [Google Scholar]
- 235.Sato AY, Tu X, McAndrews KA, Plotkin LI, Bellido T. Prevention of glucocorticoid induced-apoptosis of osteoblasts and osteocytes by protecting against endoplasmic reticulum (ER) stress in vitro and in vivo in female mice. Bone 73: 60–68, 2015. doi: 10.1016/j.bone.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sato AY, Cregor M, Delgado-Calle J, Condon KW, Allen MR, Peacock M, Plotkin LI, Bellido T. Protection from glucocorticoid-induced osteoporosis by anti-catabolic signaling in the absence of Sost/sclerostin. J Bone Miner Res 31: 1791–1802, 2016. doi: 10.1002/jbmr.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Fowler TW, Acevedo C, Mazur CM, Hall-Glenn F, Fields AJ, Bale HA, Ritchie RO, Lotz JC, Vail TP, Alliston T. Glucocorticoid suppression of osteocyte perilacunar remodeling is associated with subchondral bone degeneration in osteonecrosis. Sci Rep 7: 44618, 2017. doi: 10.1038/srep44618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 40: 1434–1446, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Hodsman AB, Bauer DC, Dempster DW, Dian L, Hanley DA, Harris ST, Kendler DL, McClung MR, Miller PD, Olszynski WP, Orwoll E, Yuen CK. Parathyroid hormone and teriparatide for the treatment of osteoporosis: a review of the evidence and suggested guidelines for its use. Endocr Rev 26: 688–703, 2005. doi: 10.1210/er.2004-0006. [DOI] [PubMed] [Google Scholar]
- 240.Rhee Y, Lee EY, Lezcano V, Ronda AC, Condon KW, Allen MR, Plotkin LI, Bellido T. Resorption controls bone anabolism driven by PTH receptor signaling in osteocytes. J Biol Chem 288: 29809–29820, 2013. doi: 10.1074/jbc.M113.485938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kim SW, Pajevic PD, Selig M, Barry KJ, Yang JY, Shin CS, Baek WY, Kim JE, Kronenberg HM. Intermittent PTH administration converts quiescent lining cells to active osteoblasts. J Bone Miner Res 27: 2075–2084, 2012. doi: 10.1002/jbmr.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Bellido T, Saini V, Divieti Pajevic P. Effects of PTH on osteocyte function. Bone 54: 250–257, 2013. doi: 10.1016/j.bone.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.O'Brien CA, Plotkin LI, Galli C, Goellner J, Gortazar AR, Allen MR, Robling AG, Bouxsein M, Schipani E, Turner CH, Jilka RL, Weinstein RS, Manolagas SC, Bellido T. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 3: e2942, 2008. doi: 10.1371/journal.pone.0002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ben-Awadh AN, Delgado-Calle J, Tu X, Kuhlenschmidt K, Allen MR, Plotkin LI, Bellido T. Parathyroid hormone receptor signaling induces bone resorption in the adult skeleton by directly regulating the RANKL gene in osteocytes. Endocrinology 155: 2797–2809, 2014. doi: 10.1210/en.2014-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Delgado-Calle J, Hancock B, Likine EF, Sato AY, McAndrews K, Sanudo C, Bruzzaniti A, Riancho JA, Tonra JR, Bellido T. MMP14 is a novel target of PTH signaling in osteocytes that controls resorption by regulating soluble RANKL production. FASEB J 32: 2878–2890, 2018. doi: 10.1096/fj.201700919RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Delgado-Calle J, Tu X, Pacheco-Costa R, McAndrews K, Edwards R, Pellegrini G, Kuhlenschmidt K, Olivos N, Robling A, Peacock M, Plotkin LI, Bellido T. Control of bone anabolism in response to mechanical loading and PTH by distinct mechanisms downstream of the PTH receptor. J Bone Miner Res 32: 522–535, 2017. doi: 10.1002/jbmr.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Saini V, Marengi DA, Barry KJ, Fulzele KS, Heiden E, Liu X, Dedic C, Maeda A, Lotinun S, Baron R, Pajevic PD. Parathyroid hormone (PTH)/PTH-related peptide type 1 receptor (PPR) signaling in osteocytes regulates anabolic and catabolic skeletal responses to PTH. J Biol Chem 288: 20122–20134, 2013. doi: 10.1074/jbc.M112.441360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Wein MN. Parathyroid hormone signaling in osteocytes. JBMR Plus 2: 22–30, 2018. doi: 10.1002/jbm4.10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Rhee Y, Allen MR, Condon K, Lezcano V, Ronda AC, Galli C, Olivos N, Passeri G, O'Brien CA, Bivi N, Plotkin LI, Bellido T. PTH receptor signaling in osteocytes governs periosteal bone formation and intra-cortical remodeling. J Bone Miner Res 26: 1035–1046, 2011. doi: 10.1002/jbmr.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Parfitt AM. The actions of parathyroid hormone on bone: relation to bone remodeling and turnover, calcium homeostasis, and metabolic bone diseases. II. PTH and bone cells: bone turnover and plasma calcium regulation. Metabolism 25: 909–955, 1976. doi: 10.1016/0026-0495(76)90124-4. [DOI] [PubMed] [Google Scholar]
- 251.Bélanger LF. Osteocytic osteolysis. Calcif Tissue Res 4: 1–12, 1969. doi: 10.1007/BF02279101. [DOI] [PubMed] [Google Scholar]
- 252.Qing H, Ardeshirpour L, Pajevic PD, Dusevich V, Jähn K, Kato S, Wysolmerski J, Bonewald LF. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J Bone Miner Res 27: 1018–1029, 2012. doi: 10.1002/jbmr.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Tsourdi E, Jähn K, Rauner M, Busse B, Bonewald LF. Physiological and pathological osteocytic osteolysis. J Musculoskelet Neuronal Interact 18: 292–303, 2018. [PMC free article] [PubMed] [Google Scholar]
- 254.Lotinun S, Ishihara Y, Nagano K, Kiviranta R, Carpentier VT, Neff L, Parkman V, Ide N, Hu D, Dann P, Brooks D, Bouxsein ML, Wysolmerski J, Gori F, Baron R. Cathepsin K-deficient osteocytes prevent lactation-induced bone loss and parathyroid hormone suppression. J Clin Invest 129: 3058–3071, 2019. doi: 10.1172/JCI122936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Tawfeek H, Bedi B, Li JY, Adams J, Kobayashi T, Weitzmann MN, Kronenberg HM, Pacifici R. Disruption of PTH Receptor 1 in T cells protects against PTH-induced bone loss. PLoS ONE 5: e12290, 2010. doi: 10.1371/journal.pone.0012290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Gao Y, Wu X, Terauchi M, Li JY, Grassi F, Galley S, Yang X, Weitzmann MN, Pacifici R. T cells potentiate PTH-induced cortical bone loss through CD40L signaling. Cell Metab 8: 132–145, 2008. doi: 10.1016/j.cmet.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Li JY, Yu M, Tyagi AM, Vaccaro C, Hsu E, Adams J, Bellido T, Weitzmann MN, Pacifici R. IL-17 receptor signaling in osteoblasts/osteocytes mediates PTH-induced bone loss and enhances osteocytic RANKL production. J Bone Miner Res 34: 349–360, 2019. doi: 10.1002/jbmr.3600. [DOI] [PubMed] [Google Scholar]
- 258.Wein MN, Foretz M, Fisher DE, Xavier RJ, Kronenberg HM. Salt-inducible kinases: physiology, regulation by cAMP, and therapeutic potential. Trends Endocrinol Metab 29: 723–735, 2018. doi: 10.1016/j.tem.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res 25: 178–189, 2010. doi: 10.1359/jbmr.090730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ben-Awadh A, Delgado-Calle J, Tu X, Kuhlenschmidt K, Allen MR, Plotkin LI, Bellido T. Parathyroid hormone receptor signaling induces bone resorption in the adult skeleton by directly regulating the RANKL gene in osteocytes. Endocrinology 155: 2797–2809, 2014. doi: 10.1210/en.2014-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Parfitt AM. Targeted and nontargeted bone remodeling: relationship to basic multicellular unit origination and progression. Bone 30: 5–7, 2002. doi: 10.1016/s8756-3282(01)00642-1. [DOI] [PubMed] [Google Scholar]
- 262.Manolagas SC, Parfitt AM. What old means to bone. Trends Endocrinol Metab 21: 369–374, 2010. doi: 10.1016/j.tem.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Tiede-Lewis LM, Xie Y, Hulbert MA, Campos R, Dallas MR, Dusevich V, Bonewald LF, Dallas SL. Degeneration of the osteocyte network in the C57BL/6 mouse model of aging. Aging (Albany NY) 9: 2190–2208, 2017. doi: 10.18632/aging.101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev 31: 266–300, 2010. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O'Brien CA, Thostenson J, Roberson PK, Boskey AL, Clemens TL, Manolagas SC. Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in 21-month-old mice. Aging Cell 9: 147–161, 2010. doi: 10.1111/j.1474-9726.2009.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Davis HM, Aref MW, Aguilar-Perez A, Pacheco-Costa R, Allen K, Valdez S, Herrera C, Atkinson EG, Mohammad A, Lopez D, Harris MA, Harris SE, Allen M, Bellido T, Plotkin LI. Cx43 overexpression in osteocytes prevents osteocyte apoptosis and preserves cortical bone quality in aging mice. JBMR Plus 2: 206–216, 2018. doi: 10.1002/jbm4.10035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Plotkin LI, Bellido T. Beyond gap junctions: connexin43 and bone cell signaling. Bone 52: 157–166, 2013. doi: 10.1016/j.bone.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Zhang Y, Paul EM, Sathyendra V, Davison A, Sharkey N, Bronson S, Srinivasan S, Gross TS, Donahue HJ. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS ONE 6: e23516, 2011. doi: 10.1371/journal.pone.0023516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Grimston SK, Brodt MD, Silva MJ, Civitelli R. Attenuated response to in vivo mechanical loading in mice with conditional osteoblast ablation of the Connexin43 gene (Gja1). J Bone Miner Res 23: 879–886, 2008. doi: 10.1359/jbmr.080222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Qiu S, Rao DS, Palnitkar S, Parfitt AM. Age and distance from the surface but not menopause reduce osteocyte density in human cancellous bone. Bone 31: 313–318, 2002. doi: 10.1016/S8756-3282(02)00819-0. [DOI] [PubMed] [Google Scholar]
- 271.Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—a meta-analysis. Osteoporos Int 18: 427–444, 2007. doi: 10.1007/s00198-006-0253-4. [DOI] [PubMed] [Google Scholar]
- 272.Hofbauer LC, Brueck CC, Singh SK, Dobnig H. Osteoporosis in patients with diabetes mellitus. J Bone Miner Res 22: 1317–1328, 2007. doi: 10.1359/jbmr.070510. [DOI] [PubMed] [Google Scholar]
- 273.Jiao H, Xiao E, Graves DT. Diabetes and its effect on bone and fracture healing. Curr Osteoporos Rep 13: 327–335, 2015. doi: 10.1007/s11914-015-0286-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Maycas M, McAndrews KA, Sato AY, Pellegrini GG, Brown DM, Allen MR, Plotkin LI, Gortazar AR, Esbrit P, Bellido T. PTHrP-derived peptides restore bone mass and strength in diabetic mice: additive effect of mechanical loading. J Bone Miner Res 32: 486–497, 2017. doi: 10.1002/jbmr.3007. [DOI] [PubMed] [Google Scholar]
- 275.Motyl KJ, McCauley LK, McCabe LR. Amelioration of type I diabetes-induced osteoporosis by parathyroid hormone is associated with improved osteoblast survival. J Cell Physiol 227: 1326–1334, 2012. doi: 10.1002/jcp.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Ozgurel Sato US, McAndrews AY, Halladay K, Cregor D, Villasenor M, Marmani-Huanca A, Barbas M, Gortazar C, Bellido T. Abaloparatide is more effective than PTH in restoring bone formation, but both agents correct the impaired bone structural and material properties and citrate content in mice with Type 1 diabetes (Abstract). J Bone Miner Res 34, Suppl 1: Abstract P-707, 2020. [Google Scholar]
- 277.Ucer Ozgurel S, McAndrews K, Halladay D, Sato AY, Cregor M, Bellido T. Abaloparatide is more effective than PTH in promoting bone gain under physiological conditions and in established Type 1 Diabetes in male mice (Abstract). J Bone Miner Res 33, Suppl 1: Abstract LB-1159, 2019. [Google Scholar]
- 278.Brun J, Berthou F, Trajkovski M, Maechler P, Foti M, Bonnet N. bone regulates browning and energy metabolism through mature osteoblast/osteocyte PPARgamma expression. Diabetes 66: 2541–2554, 2017. doi: 10.2337/db17-0116. [DOI] [PubMed] [Google Scholar]
- 279.Baroi S, Czernik PJ, Chougule A, Griffin PR, Lecka-Czernik B. PPARG in osteocytes controls sclerostin expression, bone mass, marrow adiposity and mediates TZD-induced bone loss. Bone 147: 115913, 2021. doi: 10.1016/j.bone.2021.115913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Croucher PI, McDonald MM, Martin TJ. Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer 16: 373–386, 2016. doi: 10.1038/nrc.2016.44. [DOI] [PubMed] [Google Scholar]
- 281.Delgado-Calle J. Osteocytes and their messengers as targets for the treatment of multiple myeloma. Clin Rev Bone Miner Metab 15: 49–56, 2017. doi: 10.1007/s12018-017-9227-7. [DOI] [Google Scholar]
- 282.Delgado-Calle J, Bellido T, Roodman GD. Role of osteocytes in multiple myeloma bone disease. Curr Opin Support Palliat Care 8: 407–413, 2014. doi: 10.1097/SPC.0000000000000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Giuliani N, Ferretti M, Bolzoni M, Storti P, Lazzaretti M, Dalla Palma B, Bonomini S, Martella E, Agnelli L, Neri A, Ceccarelli F, Palumbo C. Increased osteocyte death in multiple myeloma patients: role in myeloma-induced osteoclast formation. Leukemia 26: 1391–1401, 2012. doi: 10.1038/leu.2011.381. [DOI] [PubMed] [Google Scholar]
- 284.Delgado-Calle J, Anderson J, Cregor MD, Hiasa M, Chirgwin JM, Carlesso N, Yoneda T, Mohammad KS, Plotkin LI, Roodman GD, Bellido T. Bidirectional Notch signaling and osteocyte-derived factors in the bone marrow microenvironment promote tumor cell proliferation and bone destruction in multiple myeloma. Cancer Res 76: 1089–1100, 2016. doi: 10.1158/0008-5472.CAN-15-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Toscani D, Palumbo C, Dalla Palma B, Ferretti M, Bolzoni M, Marchica V, Sena P, Martella E, Mancini C, Ferri V, Costa F, Accardi F, Craviotto L, Aversa F, Giuliani N. The proteasome inhibitor bortezomib maintains osteocyte viability in multiple myeloma patients by reducing both apoptosis and autophagy: a new function for proteasome inhibitors. J Bone Miner Res 31: 815–827, 2016. doi: 10.1002/jbmr.2741. [DOI] [PubMed] [Google Scholar]
- 286.McDonald MM, Delgado-Calle J. Sclerostin: an emerging target for the treatment of cancer-induced bone disease. Curr Osteoporos Rep 15: 532–541, 2017. doi: 10.1007/s11914-017-0403-y. [DOI] [PubMed] [Google Scholar]
- 287.Zhu M, Liu C, Li S, Zhang S, Yao Q, Song Q. Sclerostin induced tumor growth, bone metastasis and osteolysis in breast cancer. Sci Rep 7: 11399, 2017. doi: 10.1038/s41598-017-11913-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Delgado-Calle J, Anderson J, Cregor MD, Condon KW, Kuhstoss SA, Plotkin LI, Bellido T, Roodman GD. Genetic deletion of Sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia 31: 2686–2694, 2017. doi: 10.1038/leu.2017.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.McDonald MM, Reagan MR, Youlten SE, Mohanty ST, Seckinger A, Terry RL, Pettitt JA, Simic MK, Cheng TL, Morse A, Le LM, Abi-Hanna D, Kramer I, Falank C, Fairfield H, Ghobrial IM, Baldock PA, Little DG, Kneissel M, Vanderkerken K, Bassett JH, Williams GR, Oyajobi BO, Hose D, Phan TG, Croucher PI. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood 129: 3452–3464, 2017. doi: 10.1182/blood-2017-03-773341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Eda H, Santo L, Wein MN, Hu DZ, Cirstea DD, Nemani N, Tai YT, Raines SE, Kuhstoss SA, Munshi NC, Kronenberg HM, Raje NS. Regulation of sclerostin expression in multiple myeloma by Dkk-1; a potential therapeutic strategy for myeloma bone disease. J Bone Miner Res 31: 1225–1234, 2016. doi: 10.1002/jbmr.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Hesse E, Schröder S, Brandt D, Pamperin J, Saito H, Taipaleenmäki H. Sclerostin inhibition alleviates breast cancer-induced bone metastases and muscle weakness. JCI Insight 5: e125543, 2019. doi: 10.1172/jci.insight.125543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Mulcrone PL, Edwards SK, Petrusca DN, Haneline LS, Delgado-Calle J, Roodman GD. Osteocyte Vegf-a contributes to myeloma-associated angiogenesis and is regulated by Fgf23. Sci Rep 10: 17319, 2020. doi: 10.1038/s41598-020-74352-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Suvannasankha A, Tompkins DR, Edwards DF, Petyaykina KV, Crean CD, Fournier PG, Parker JM, Sandusky GE, Ichikawa S, Imel EA, Chirgwin JM. FGF23 is elevated in multiple myeloma and increases heparanase expression by tumor cells. Oncotarget 6: 19647–19660, 2015. doi: 10.18632/oncotarget.3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Sabol HM, Amorim T, Kurihara N, Anderson J, Cregor M, Roodman GD, Bellido T, Delgado-Calle J. Autocrine and paracrine Notch receptor 3 signaling in the myeloma niche stimulates tumor growth and bone destruction (Abstract). J Bone Miner Res 34, Suppl 1: Abstract P-158, 2020. [Google Scholar]
- 295.Sottnik JL, Dai J, Zhang H, Campbell B, Keller ET. Tumor-induced pressure in the bone microenvironment causes osteocytes to promote the growth of prostate cancer bone metastases. Cancer Res 75: 2151–2158, 2015. doi: 10.1158/0008-5472.CAN-14-2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Sottnik JL, Campbell B, Mehra R, Behbahani-Nejad O, Hall CL, Keller ET. Osteocytes serve as a progenitor cell of osteosarcoma. J Cell Biochem 115: 1420–1429, 2014. doi: 10.1002/jcb.24793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Cui YX, Evans BA, Jiang WG. New roles of osteocytes in proliferation, migration and invasion of breast and prostate cancer cells. Anticancer Res 36: 1193–1201, 2016. [PubMed] [Google Scholar]
- 298.Hiasa M, Okui T, Allette YM, Ripsch MS, Sun-Wada GH, Wakabayashi H, Roodman GD, White FA, Yoneda T. Bone pain induced by multiple myeloma is reduced by targeting V-ATPase and ASIC3. Cancer Res 77: 1283–1295, 2017. doi: 10.1158/0008-5472.CAN-15-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Pathak JL, Bravenboer N, Klein-Nulend J. The osteocyte as the new discovery of therapeutic options in rare bone diseases. Front Endocrinol (Lausanne) 11: 405, 2020. doi: 10.3389/fendo.2020.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, Gardner JC, Galas D, Schatzman RC, Beighton P, Papapoulos S, Hamersma H, Brunkow ME. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet 110: 144–152, 2002. doi: 10.1002/ajmg.10401. [DOI] [PubMed] [Google Scholar]
- 301.Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 39: 91–97, 2002. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet 10: 537–543, 2001. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 303.Balemans W, Van Den Ende J, Freire Paes-Alves A, Dikkers FG, Willems PJ, Vanhoenacker F, de Almeida-Melo N, Alves CF, Stratakis CA, Hill SC, Van Hul W. Localization of the gene for sclerosteosis to the van Buchem disease-gene region on chromosome 17q12-q21. Am J Hum Genet 64: 1661–1669, 1999. doi: 10.1086/302416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Van Hul W, Balemans W, Van Hul E, Dikkers FG, Obee H, Stokroos RJ, Hildering P, Vanhoenacker F, Van Camp G, Willems PJ. Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12-q21. Am J Hum Genet 62: 391–399, 1998. doi: 10.1086/301721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Dixon JM, Cull RE, Gamble P. Two cases of Van Buchem's disease. J Neurol Neurosurg Psychiatry 45: 913–918, 1982. doi: 10.1136/jnnp.45.10.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Carpenter TO. The expanding family of hypophosphatemic syndromes. J Bone Miner Metab 30: 1–9, 2012. doi: 10.1007/s00774-011-0340-2. [DOI] [PubMed] [Google Scholar]
- 307.Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, Kawakami T, Ito T, Zhang X, Humphrey J, Insogna KL, Peacock M. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest 124: 1587–1597, 2014. doi: 10.1172/JCI72829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Martin E, Shapiro JR. Osteogenesis imperfecta: epidemiology and pathophysiology. Curr Osteoporos Rep 5: 91–97, 2007. doi: 10.1007/s11914-007-0023-z. [DOI] [PubMed] [Google Scholar]
- 309.Zimmerman SM, Dimori M, Heard-Lipsmeyer ME, Morello R. The osteocyte transcriptome is extensively dysregulated in mouse models of osteogenesis imperfecta. JBMR Plus 3: e10171, 2019. doi: 10.1002/jbm4.10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Zimmerman SM, Heard-Lipsmeyer ME, Dimori M, Thostenson JD, Mannen EM, O’Brien CA, Morello R. Loss of RANKL in osteocytes dramatically increases cancellous bone mass in the osteogenesis imperfecta mouse (oim). Bone Rep 9: 61–73, 2018. doi: 10.1016/j.bonr.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Plotkin LI, Manolagas SC, Bellido T. Dissociation of the pro-apoptotic effects of bisphosphonates on osteoclasts from their anti-apoptotic effects on osteoblasts/osteocytes with novel analogs. Bone 39: 443–452, 2006. doi: 10.1016/j.bone.2006.02.060. [DOI] [PubMed] [Google Scholar]
- 312.Plotkin LI, Bivi N, Bellido T. A bisphosphonate that does not affect osteoclasts prevents osteoblast and osteocyte apoptosis and the loss of bone strength induced by glucocorticoids in mice. Bone 49: 122–127, 2011. doi: 10.1016/j.bone.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Appelman-Dijkstra NM, Papapoulos SE. Sclerostin Inhibition in the Management of Osteoporosis. Calcif Tissue Int 98: 370–380, 2016. doi: 10.1007/s00223-016-0126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Padhi D, Allison M, Kivitz AJ, Gutierrez MJ, Stouch B, Wang C, Jang G. Multiple doses of sclerostin antibody romosozumab in healthy men and postmenopausal women with low bone mass: a randomized, double-blind, placebo-controlled study. J Clin Pharmacol 54: 168–178, 2014. doi: 10.1002/jcph.239. [DOI] [PubMed] [Google Scholar]
- 315.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res 26: 19–26, 2011. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 316.Recknor CP, Recker RR, Benson CT, Robins DA, Chiang AY, Alam J, Hu L, Matsumoto T, Sowa H, Sloan JH, Konrad RJ, Mitlak BH, Sipos AA. The effect of discontinuing treatment with blosozumab: follow-up results of a phase 2 randomized clinical trial in postmenopausal women with low bone mineral density. J Bone Miner Res 30: 1717–1725, 2015. doi: 10.1002/jbmr.2489. [DOI] [PubMed] [Google Scholar]
- 317.Saag KG, Petersen J, Brandi ML, Karaplis AC, Lorentzon M, Thomas T, Maddox J, Fan M, Meisner PD, Grauer A. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med 377: 1417–1427, 2017. doi: 10.1056/NEJMoa1708322. [DOI] [PubMed] [Google Scholar]
- 318.Lewiecki EM, Blicharski T, Goemaere S, Lippuner K, Meisner PD, Miller PD, Miyauchi A, Maddox J, Chen L, Horlait S. A Phase III randomized placebo-controlled trial to evaluate efficacy and safety of romosozumab in men with osteoporosis. J Clin Endocrinol Metab 103: 3183–3193, 2018. doi: 10.1210/jc.2017-02163. [DOI] [PubMed] [Google Scholar]
- 319.McClung MR. Emerging therapies for osteoporosis. Endocrinol Metab (Seoul) 30: 429–435, 2015. doi: 10.3803/EnM.2015.30.4.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.McClung MR, Lewiecki EM, Cohen SB, Bolognese MA, Woodson GC, Moffett AH, Peacock M, Miller PD, Lederman SN, Chesnut CH, Lain D, Kivitz AJ, Holloway DL, Zhang C, Peterson MC, Bekker PJ; AMG 162 Bone Loss Study Group. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 354: 821–831, 2006. doi: 10.1056/NEJMoa044459. [DOI] [PubMed] [Google Scholar]
- 321.Papapoulos S, Lippuner K, Roux C, Lin CJ, Kendler DL, Lewiecki EM, Brandi ML, Czerwinski E, Franek E, Lakatos P, Mautalen C, Minisola S, Reginster JY, Jensen S, Daizadeh NS, Wang A, Gavin M, Libanati C, Wagman RB, Bone HG. The effect of 8 or 5 years of denosumab treatment in postmenopausal women with osteoporosis: results from the FREEDOM Extension study. Osteoporos Int 26: 2773–2783, 2015. doi: 10.1007/s00198-015-3234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.McClung MR, Wagman RB, Miller PD, Wang A, Lewiecki EM. Observations following discontinuation of long-term denosumab therapy. Osteoporos Int 28: 1723–1732, 2017. doi: 10.1007/s00198-017-3919-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.McDonald MM, Khoo WH, Ng PY, Xiao Y, Zamerli J, Thatcher P, Kyaw W, et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell 184: 1330–1347.e13, 2021. doi: 10.1016/j.cell.2021.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Raje N, Terpos E, Willenbacher W, Shimizu K, García-Sanz R, Durie B, Legiec W, Krejčí M, Laribi K, Zhu L, Cheng P, Warner D, Roodman GD. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: an international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol 19: 370–381, 2018. doi: 10.1016/S1470-2045(18)30072-X. [DOI] [PubMed] [Google Scholar]
- 325.Mansinho A, Ferreira AR, Casimiro S, Alho I, Vendrell I, Costa AL, Sousa R, Abreu C, Pulido C, Macedo D, Pacheco TR, Correia L, Costa L. Levels of circulating fibroblast growth factor 23 (FGF23) and prognosis in cancer patients with bone metastases. Int J Mol Sci 20: 695, 2019. doi: 10.3390/ijms20030695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Feng S, Wang J, Zhang Y, Creighton CJ, Ittmann M. FGF23 promotes prostate cancer progression. Oncotarget 6: 17291–17301, 2015. doi: 10.18632/oncotarget.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Kim HJ, Kim KH, Lee J, Oh JJ, Cheong HS, Wong EL, Yang BS, Byun SS, Myung SC. Single nucleotide polymorphisms in fibroblast growth factor 23 gene, FGF23, are associated with prostate cancer risk. BJU Int 114: 303–310, 2014. doi: 10.1111/bju.12396. [DOI] [PubMed] [Google Scholar]
- 328.Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 107: 4907–4916, 2006. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Konstantinova IM, Tsimokha AS, Mittenberg AG. Role of proteasomes in cellular regulation. Int Rev Cell Mol Biol 267: 59–124, 2008. doi: 10.1016/S1937-6448(08)00602-3. [DOI] [PubMed] [Google Scholar]
- 330.Mukherjee S, Raje N, Schoonmaker JA, Liu JC, Hideshima T, Wein MN, Jones DC, Vallet S, Bouxsein ML, Pozzi S, Chhetri S, Seo YD, Aronson JP, Patel C, Fulciniti M, Purton LE, Glimcher LH, Lian JB, Stein G, Anderson KC, Scadden DT. Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. J Clin Invest 118: 491–504, 2008. doi: 10.1172/JCI33102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Accardi F, Toscani D, Costa F, Aversa F, Giuliani N. The proteasome and myeloma-associated bone disease. Calcif Tissue Int 102: 210–226, 2018. doi: 10.1007/s00223-017-0349-1. [DOI] [PubMed] [Google Scholar]
- 332.Terpos E, Christoulas D, Katodritou E, Bratengeier C, Gkotzamanidou M, Michalis E, Delimpasi S, Pouli A, Meletis J, Kastritis E, Zervas K, Dimopoulos MA. Elevated circulating sclerostin correlates with advanced disease features and abnormal bone remodeling in symptomatic myeloma: reduction post-bortezomib monotherapy. Int J Cancer 131: 1466–1471, 2012. doi: 10.1002/ijc.27342. [DOI] [PubMed] [Google Scholar]
- 333.Sato AY, Richardson D, Cregor M, Davis HM, Au ED, McAndrews K, Zimmers TA, Organ JM, Peacock M, Plotkin LI, Bellido T. Glucocorticoids induce bone and muscle atrophy by tissue-specific mechanisms upstream of E3 ubiquitin ligases. Endocrinology 158: 664–677, 2017. doi: 10.1210/en.2016-1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Canalis E, Bridgewater D, Schilling L, Zanotti S. Canonical Notch activation in osteocytes causes osteopetrosis. Am J Physiol Endocrinol Metab 310: E171–E182, 2016. doi: 10.1152/ajpendo.00395.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Liu P, Ping Y, Ma M, Zhang D, Liu C, Zaidi S, Gao S, Ji Y, Lou F, Yu F, Lu P, Stachnik A, Bai M, Wei C, Zhang L, Wang K, Chen R, New MI, Rowe DW, Yuen T, Sun L, Zaidi M. Anabolic actions of Notch on mature bone. Proc Natl Acad Sci USA 113: E2152–E2161, 2016. doi: 10.1073/pnas.1603399113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Ferrari A, McAndrews K, Nelson JH, Bell J, Srinivasan V, Ebetino FH, Boeckman RK, Roodman GD, Bellido T, Delgado-Calle J. Bone-Targeted Inhibition of Notch Signaling Blocks Tumor Growth and Prevents Bone Loss Without Inducing Gut Toxicity In Immunodeficient and Immunocompetent Murine Models of Established Multiple Myeloma (Abstract). J Bone Miner Res 32, Suppl 1: Abstract 1091, 2019. [Google Scholar]
- 337.Schwarzer R, Nickel N, Godau J, Willie BM, Duda GN, Schwarzer R, Cirovic B, Leutz A, Manz R, Bogen B, Dörken B, Jundt F. Notch pathway inhibition controls myeloma bone disease in the murine MOPC315.BM model. Blood Cancer J 4: e217, 2014. doi: 10.1038/bcj.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM, Berridge BR, Gao H, Higgins MA, May PC, Ryan TP. Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional gamma-secretase inhibitor. J Biol Chem 278: 46107–46116, 2003. doi: 10.1074/jbc.M307757200. [DOI] [PubMed] [Google Scholar]
- 339.Delgado-Calle J, Wu G, Olson ME, McAndrews K, Nelson JH, Daniel AL, Kurihara N, Atkinson EG, Xiao L, Ebetino FH, Roodman GD, Boeckman RK, Bellido T. Bone-Targeted pharmacological inhibition of Notch signaling potentiates PTH-induced bone gain (Abstract). J Bone Miner Res 33: 80, 2018. [Google Scholar]
- 340.Colombo M, Galletti S, Garavelli S, Platonova N, Paoli A, Basile A, Taiana E, Neri A, Chiaramonte R. Notch signaling deregulation in multiple myeloma: A rational molecular target. Oncotarget 6: 26826–26840, 2015. doi: 10.18632/oncotarget.5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Colombo M, Mirandola L, Platonova N, Apicella L, Basile A, Figueroa AJ, Cobos E, Chiriva-Internati M, Chiaramonte R. Notch-directed microenvironment reprogramming in myeloma: a single path to multiple outcomes. Leukemia 27: 1009–1018, 2013. doi: 10.1038/leu.2013.6. [DOI] [PubMed] [Google Scholar]
- 342.Wu Y, Yuan X, Perez KC, Hyman S, Wang L, Pellegrini G, Salmon B, Bellido T, Helms JA. Aberrantly elevated Wnt signaling is responsible for cementum overgrowth and dental ankylosis. Bone 122: 176–183, 2019. doi: 10.1016/j.bone.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Chen J, Yuan X, Pilawski I, Liu X, Delgado-Calle J, Bellido T, Turkkahraman H, Helms JA. Molecular basis for craniofacial phenotypes caused by Sclerostin deletion. J Dent Res 100: 310–317, 2021. doi: 10.1177/0022034520963584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Aquino-Martinez R, Rowsey JL, Fraser DG, Eckhardt BA, Khosla S, Farr JN, Monroe DG. LPS-induced premature osteocyte senescence: implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone 132: 115220, 2020. doi: 10.1016/j.bone.2019.115220. [DOI] [PMC free article] [PubMed] [Google Scholar]