

Abstract
The small conductance calcium-activated potassium channel (KCa2.3) has long been recognized for its role in mediating vasorelaxation through the endothelium-derived hyperpolarization (EDH) response. Histone deacetylases (HDACs) have been implicated as potential modulators of blood pressure and histone deacetylase inhibitors (HDACi) are being explored as therapeutics for hypertension. Herein, we show that HDACi increase KCa2.3 expression when heterologously expressed in HEK cells and endogenously expressed in primary cultures of human umbilical vein endothelial cells (HUVECs) and human intestinal microvascular endothelial cells (HIMECs). When primary endothelial cells were exposed to HDACi, KCa2.3 transcripts, subunits, and functional current are increased. Quantitative RT-PCR (qPCR) demonstrated increased KCa2.3 mRNA following HDACi, confirming transcriptional regulation of KCa2.3 by HDACs. By using pharmacological agents selective for different classes of HDACs, we discriminated between cytoplasmic and epigenetic modulation of KCa2.3. Biochemical analysis revealed an association between the cytoplasmic HDAC6 and KCa2.3 in immunoprecipitation studies. Specifically inhibiting HDAC6 increases expression of KCa2.3. In addition to increasing the expression of KCa2.3, we show that nonspecific inhibition of HDACs causes an increase in the expression of the molecular chaperone Hsp70 in endothelial cells. When Hsp70 is inhibited in the presence of HDACi, the magnitude of the increase in KCa2.3 expression is diminished. Finally, we show a slower rate of endocytosis of KCa2.3 as a result of exposure of primary endothelial cells to HDACi. These data provide the first demonstrated approach to increase KCa2.3 channel number in endothelial cells and may partially account for the mechanism by which HDACi induce vasorelaxation.
Keywords: endothelial cells, HDAC, histone deacetylase, HUVECs, KCa2.3, SK3
INTRODUCTION
Hypertension afflicts nearly half of American adults and leads to cardiovascular disease such as atherosclerosis, peripheral artery disease (PAD), and stroke (1). The high prevalence of this condition indicates the need to identify novel pharmacological targets, which can serve as the basis for the development of new therapeutics. Of particular interest are vasoactive compounds that can decrease arterial and vascular tone. Endothelial-dependent regulation of vascular tone is altered in a host of pathologies, including sepsis, aging, heart failure, ischemia-reperfusion, hypertension, atherosclerosis, preeclampsia, and diabetes (2).
The endothelial small conductance Ca2+-activated potassium channel, KCa2.3 (syn. SK3) and the intermediate conductance Ca2+-dependent potassium channel, KCa3.1 (syn. IK1, SK4) are known to contribute to vasorelaxation through the endothelium-derived hyperpolarization (EDH) pathway (3, 4). The EDH response is a nitric oxide- and prostaglandin-independent signaling pathway in which the endothelium directs vascular smooth muscle to hyperpolarize, relax, and therefore decrease vascular tone (2, 5). When induced by application of acetylcholine, this response appears to be mediated by both KCa2.3 and KCa3.1, and the suppression of both KCa2.3 and KCa3.1 results in the elevation of arteriole blood pressure (6). Interestingly, the relative contribution of KCa2.3 and KCa3.1 to the EDH response appears to differ according to the type of vessel being investigated or mechanism of EDH activation, such as shear stress (7, 8).
Tangentially, other molecular targets showing promise for ameliorating hypertension are histone deacetylases (HDACs). Histone deacetylase inhibitors (HDACi) have shown clear therapeutic potential in cardiovascular disease (9–13), including hypertension. Indeed, HDACi attenuated hypertension in deoxy-corticosterone (DOCA)-salt hypertensive rats, angiotensin II-induced, and spontaneously hypertensive rats(14–16), as well as high-fat-induced hypertensive mice (17). The pan HDACi trichostatin-A (TCS-A) has also shown benefit for the vascular pathology of sickle cell anemia, at least in part by preventing vascular stasis (18). HDACi have already shown promise in numerous preclinical models of heart disease (9–12), including in regulating blood pressure. Importantly, HDACi, including vorinostat (Zolinza), romidepsin (Istodax), belinostat (Belodaq), and panobinostat (Farydak) have been FDA-approved for T cell lymphoma and multiple myeloma. It is therefore possible that HDACi may be of immediate clinical benefit in other diseases, such as cardiovascular disease, via off-label usage.
Previously, we identified 1-EBIO as a novel activator of KCa2.3 channels, which increases the open probability (Po) of the channel by shifting its calcium dependence (19, 20). This compound has subsequently been shown to reduce vascular tone in rodents (21) and blood pressure in a conscious canine model (22). In addition to modulating Po, KCa2.3-dependent effects on vascular tone can potentially be mediated by altering the number of KCa2.3 channels (N) in the plasma membrane (6, 23–25). For example, genetic evidence taken from mice conditionally overexpressing KCa2.3, specifically in the endothelium, has demonstrated vasorelaxation (6, 24). We have also shown that KCa2.3 is rapidly endocytosed in endothelia via Rab35-positive recycling endosomes in a Rab5, dynamin, and caveolin-1 dependent manner (26, 27). These findings led us to postulate that pharmacological regulation of KCa2.3 channel number can be obtained.
In this report, we show that the pan HDACi, TCS-A, significantly increases plasma membrane KCa2.3 expression. In a heterologous system where KCa2.3 is stably expressed, we show an approximate twofold increase in both KCa2.3 channel protein and current density. In primary cultures of human umbilical vein endothelial cells (HUVECs), overnight exposure to TCS-A increases KCa2.3 mRNA expression roughly sixfold. Unsurprisingly, KCa2.3 protein expression is increased by TCS-A exposure in both HUVECs and primary cultures of human intestinal microvascular endothelial cells (HIMECs). Whole cell patch-clamp electrophysiology in HUVECs reveals an eightfold increase in current density as a result of 24-h exposure to TCS-A. In addition, we show that the TCS-A-dependent increase in KCa2.3 expression correlates with Hsp70 expression. Finally, HDACi alter the trafficking of KCa2.3, resulting in increased plasma membrane expression. Given the critical role of KCa2.3 in controlling vascular tone, and hence blood pressure, defining how these channels may be pharmacologically manipulated is highly relevant to further our understanding of KCa2.3 as a therapeutic target. The data provided in this report suggest that HDACi represent the first pharmacological agents capable of regulating the number of plasma membrane KCa2.3 channels.
METHODS
Cell Culture
HEK293 cells were cultured as previously described (28). For patch-clamp experiments, cells were removed from flasks using 0.05% Trypsin-EDTA (Gibco) and seeded onto poly-l-lysine coated coverslips in either control media or media containing 1 µM TCS-A for 24 h before use. The electrophysiologist was blind to drug treatment during experiments.
Human umbilical cords were obtained immediately following delivery by the staff of the Obstetrical Specimen Procurement Unit (OSPU) at Magee Womens Hospital (MWH). The OSPU at MWH is designed for procurement of specimens (including umbilical cord segments). It is located within the labor and delivery suite of Magee-Womens Hospital. The unit is funded by investigators’ grants and by contributions from MWH and Magee-Womens Research Institute and Foundation. The cords were provided in a deidentified manner to the research staff in sterile DMEM media. The cords were transported to the laboratory within 2 h of delivery for isolation of cells. A total of 10 umbilical cords were used to isolate HUVECs for this work. Three of these cords were used to generate HUVECs from which experiments were carried out before any cell passaging. The project did not meet the definition of human subject research because the specimens were not associated with any identifiers.
The umbilical cord was kept in sterile culture medium and HUVECs extracted immediately upon arrival. Briefly, the umbilical vein was cannulated and thoroughly rinsed with Hanks’ balanced salt solution (HBSS) (Corning Cellgro, Manassas, VA) to remove blood. The vein was then perfused for 15 min at room temperature with a 0.2% collagenase type II solution (Worthington Biochemical Corp., Freehold, NJ) to dissociate endothelial cells. The recovered endothelial cells were pelleted and grown using Medium 200 plus low serum growth supplement (LSGS; Gibco) and penicillin/streptomycin. Approximately 1.5 × 106 cells were seeded per T25 flask and cultured at 37°C in a 5% CO2 humidified atmosphere. Cells were passaged twice per week when confluent. All studies were carried out on cells from passage 1–8, unless otherwise noted.
For patch-clamp experiments, HUVECs were exposed to 1 µM TCS-A for 24 h and lifted from flasks using 0 Ca2+ and Mg2+ PBS containing 100 µM EDTA the day of experiments. Cells were plated onto poly-l-lysine-coated coverslips and allowed to attach for 10 min after which time media was exchanged to remove cell debris, and cells were used for recordings. For TCS-A treatment, cells from passages 1–6 were maintained on coverslips in 1 µM TCS-A up until they were used for recordings.
HIMEC isolation (IRB PRO08120314) was performed using a technique adapted from dermal microvascular endothelium, as we previously described (29). Briefly, surgical specimens were rinsed with HBSS and full thickness samples of intestinal tissue were selected. Mucosal strips were dissected and washed to remove debris and contaminating bacteria, minced, and digested in a type II collagenase solution (Worthington Biochemical Corp., Freehold, NJ). Mechanical compression was used to release microvascular endothelial cells, which were then pelleted and plated onto fibronectin-coated tissue culture dishes, and grown in MCDB 131 media (GenDepot, Katy, TX) supplemented with 10% fetal bovine serum and endothelial cell growth factor (ECGF, EMD Millipore, Burlington, MA). After 7–10 days of culture, microvascular endothelial cell clusters were further purified using PECAM-1-coated magnetic beads (Dynabeads, Thermo Fisher Scientific). Endothelial cultures were confirmed by modified lipoprotein uptake (Dil-ac-LDL, Biomedical Technology, Inc., Stoughton, MA) and expression of Factor VIII-associated antigen. HIMEC isolation was carried out four separate times to generate primary cells for the experiments detailed. All studies were carried out on cells from passage 2–8.
Patch-Clamp Electrophysiology
Whole cell patch-clamp electrophysiology of HEK293 and HUVECs was performed under identical conditions. Borosilicate glass electrodes (1.65 mm outer diameter, World Precision Instruments) were pulled with a Narishige puller (model PP-830). After fire polishing with a World Precision Instruments microforge (MF-200), pipettes had a resistance of 2–3 MΩ, when filed with a pipette solution containing the following (mM): 145 K-gluconate, 9.5 CaCl2, 10 EGTA, 10 Hepes, 2 MgCl2, 0.3 NaATP (pH = 7.4), 3 µM free Ca2+. Cells were bathed in a recording solution containing (in mM): 155 Na-gluconate, 4.5 KCl, 0.002 CaCl2, 10 Hepes, and 1 MgCl2 (pH = 7.2). Recordings were performed using an Axopatch 200B integrating patch-clamp amplifier (Axon Instruments) with low pass Bessel filtering set at 2 kHz. Clampex data acquisition software (Axon Instruments, v. 9.2) was used to capture recordings, which were then analyzed using Clampfit (Axon Instruments, v. 9.2). After obtaining a whole cell configuration, a peristaltic pump (Gilson, Minipuls 3) was used to continuously perfuse bath solution throughout the recording, during which apamin (EMD Millipore, Burlington, MA) was applied to determine the extent of KCa2.3 current. According to the IUPHAR/BPS Guide to Pharmacology, the half maximal inhibitory concentration (IC50) of apamin for KCa2.3 is between 1 and 10 nM (30). As we were interested in obtaining total apamin sensitive current density, a concentration well above this value (25 nM) was used during whole cell patch-clamp experiments. Apamin sensitivity of KCa2.X channels can vary widely and involves amino acids in both the outer vestibule of the pore and in the extracellular S3-S4 loop (31). Furthermore, splice variants of KCa2.3 exist which are apamin-insensitive (32). Because the exact molecular composition of endogenous KCa2.3 in endothelial cells was not determined, we used a concentration of 100 nM apamin to ensure that total apamin sensitive current density was obtained in HUVECs.
Quantitative PCR
RNA from primary HUVEC cells was isolated using the mRNeasy RNA isolation kit (Qiagen, Germantown, MD) according to the manufacturer’s protocol. Total RNA concentration and quality were evaluated for inclusion in subsequent in vitro transcription assays based on a spectrophotometric absorption ratio of 260/280 > 1.8 (NanoDrop, Wilmington, DE). Primer pairs used for PCR reactions were are follows: KCa2.3 (forward 5′- TGGCTCATCTCCATCACATTC, reverse 5′- CCTCTGGTGTTTCCTCACTTT), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (forward 5′- GATTCCACCCATGGCAAATTC, reverse 5′- GTCATGAGTCCT TCCACGATAC) and actin (forward 5′- TCCACGAAACTACCTTCAACTC, reverse 5′- AGCCATGCCAATCTCATCTT). Primers were chosen to produce single amplification products of similar lengths (∼300 base pairs), melting temperatures (∼62°C), and GC content (∼50%). The EvaGreen mastermix (MidSci, St Louis, MO) was used for the qPCR reactions. Real-time PCR was carried out using a Bio-Rad CFX96 real-time PCR detection system (Bio-Rad, Hercules, CA). Detected signals from the PCR amplifications were normalized to the relative expression GAPDH message from each sample, and expression is presented as a fold change from control untreated samples (ΔΔCT).
Molecular Biology
Wild-type Flag-tagged, catalytically inactive (DC), and ubiquitin binding domain (UBD) deleted (ΔBuz) HDAC6 were purchased from Addgene (Watertown, MA). As previously described, HEK293 were engineered to stably express a KCa2.3 construct (28), and these cells were transfected with the aforementioned HDAC6 mutants using Lipofectamine (Thermo Fisher Scientific) per the manufacturer’s instructions.
Immunoprecipitation and Immunoblots
Our immunoprecipitation (IP) and immunoblots (IB) protocols have been previously described in detail (26, 33). Succinctly, cells were lysed, and equal amounts of total protein were precleared with protein G-agarose beads (Life Technologies) and incubated with the desired antibody. As negative control, a nonspecific IgG was used. Precipitations of immune complexes were carried out with protein G-agarose beads, and proteins were resolved by SDS-PAGE followed by IB. Relative protein levels were quantified by densitometry using ImageJ software (NIH; http://rsb.info.nih.gov/ij/).
For analysis of plasma membrane expression, cell surface proteins, including KCa2.3, were biotinylated using EZ-Link Sulfo-NHS-SS-Biotin (Thermo Scientific, Rockford, IL) for 30 min at 4°C, after which the unreacted biotin was quenched (PBS plus 1% BSA). Samples were then immediately subjected to cell lysis (50 mM HEPES pH 7.4, 150 mM NaCl, 1% vol/vol Triton X-100, 1 mM EDTA containing Complete EDTA-free protease inhibitor cocktail mix, Roche Applied Science, Indianapolis, IN) and streptavidin-agarose (Sigma-Aldrich) pulldown following normalization of protein concentrations and finally SDS-PAGE analysis.
Our methods to determine endocytosis of KCa2.3 have been previously reported in detail (26, 27). Briefly, biotinylated HUVECs were allowed to warm to 37°C to promote subunit internalization for 20 min. Residual cell surface biotin was stripped using MESNA (100 mM sodium 2-mercaptoethanesulfonate, 50 mM Tris, 100 mM NaCl, 1 mM EDTA, 0.2% BSA) and then cells were lysed. Once samples were normalized for protein concentration, the remaining biotin-tagged channels were subjected to pulldown using streptavidin-agarose. These samples were compared with those which were either biotinylated and promptly subjected to lysis and streptavidin pulldown (total cell surface KCa2.3) or biotinylated and immediately stripped to verify the efficiency MESNA stripping (strip control). As mentioned previously, proteins were separated by SDS-PAGE (8% gel) and transferred to nitrocellulose for immunoblotting.
To determine total KCa2.3 protein half-life, HUVECs were incubated in cycloheximide (400 µg/mL) for the indicated times at 37°C, after which they were cooled to 4°C by washing in ice-cold PBS and prepared for IB, as previously described (26).
Antibodies
The α-KCa2.3 antibody was obtained from EMD-Millipore (Burlington, MA, AB5350). We previously verified that this antibody produces a band of appropriate molecular weight (26, 27). α-tubulin (ABT170), α-ac-tubulin (T-7451), and α-Flag (F3165) were purchased from Sigma (St. Louis, MO). α-HSP70 (4872 T) and α-HSP90 (4874S) were bought from Cell Signaling Technologies (Danvers, MA). All antibodies were used at a 1:1,000 dilution.
Chemicals
All chemicals were obtained from Sigma-Aldrich, unless otherwise stated. VER155008 was a generous gift from Dr. Joseph Brodsky (University of Pittsburgh).
Immunostaining and Image Quantification
For immunofluorescence labeling, endothelial cells were grown on poly-l-lysine (Sigma)-coated glass coverslips for 24 h before labeling. Endothelial cells were washed with ice-cold PBS and immediately fixed and permeabilized with 2% paraformaldehyde plus 0.1% Triton X-100. Cells were then blocked with 1% BSA (3 × 5 min), followed by 10% goat serum in PBS for 30 min at 4°C. Cells were then incubated with α-KCa2.3 antibody for 1 h at 4°C. Following extensive washing in 1% BSA (6 × 5 min), to remove unbound α-KCa2.3 antibody, the cells were incubated in goat α-rabbit-Alexa594 for 1 h at 4°C. Cells were again washed with PBS/1% BSA (6 × 5 min), followed by PBS after which nuclei were labeled with DAPI (Sigma-Aldrich). Cells were imaged by laser scanning confocal microscopy (Olympus FluoView 1000 system) using a ×63 oil immersion lens in which X-Y images were scanned at 1,024 × 1,024 pixels. Z-stacks were taken to cover the entire thickness of the cell in a step size of 0.5 µm.
Immunofluorescent images were analyzed using ImageJ software. Briefly, RGB immunofluorescent images were separated into individual color channels. An auto-threshold method was chosen for red and blue channels that effectively reduced background and this threshold was applied uniformly across all images in a given channel. After converting individual channels to binary images via the appropriate threshold, particle analysis of each image yielded the percent area of the image occupied by blue or red pixels. For each composite image, the area pertaining to red pixels, which corresponds to KCa2.3 staining, was divided by the area occupied by blue pixels, which corresponds to the nuclear stain. The latter step allows for normalization of each image to cell number, which may differ in each field of view. Statistical analysis was performed on the ratio of percent areas expressed as a fraction to compare experimental groups.
Statistics
All data are presented as means ± SEM, where n indicates the number of experiments. Statistical analysis was performed using a Student’s t test. To compare the normalized values of the IB band intensities, statistical analysis was performed using the nonparametric Kruskal–Wallis test. A value of P < 0.05 is considered statistically significant and is reported.
RESULTS
HDACi have been shown to modulate the expression of a number of ion channels, including the cystic fibrosis transmembrane conductance regulator (CFTR) (34) and the epithelial Na+ channel (ENaC) (35). As there are currently no pharmacological means to increase KCa2.3 expression, we determined whether the number of plasma membrane KCa2.3 channels could be similarly increased following HDAC inhibition. Initially, we determined the effect of HDACi on KCa2.3 expression in HEK293 cells stably expressing KCa2.3. For these studies, HEK cells were exposed to the pan-HDACi, TSC-A (1 µM) overnight, and the effect on total KCa2.3 expression was assessed by IB. As shown in Fig. 1A, TCS-A increased total KCa2.3 expression, averaging 2.6 ± 0.3-fold (n = 4; P < 0.05). To determine whether this increase in total KCa2.3 protein expression was reflected in an increase in functional plasma membrane expression, we carried out whole cell patch-clamp studies. Whole cell patch-clamp recordings were performed using 3 µM internal Ca2+ to activate heterologously expressed KCa2.3. Once outward currents achieved maximal activation (Fig. 1B), the SK channel inhibitor, apamin (25 nM), was applied to the bath solution (Fig. 1C) to determine the magnitude of KCa2.3 currents. Following normalization to cell capacitance, an approximately twofold increase in KCa2.3 current density was observed in cells treated for 24 h with TCS-A at all voltages analyzed (Fig. 1D). This increase in current density correlates well with the increase in total protein observed by IB, demonstrating HDAC inhibition increases functional plasma membrane expression of heterologously expressed KCa2.3.
Figure 1.
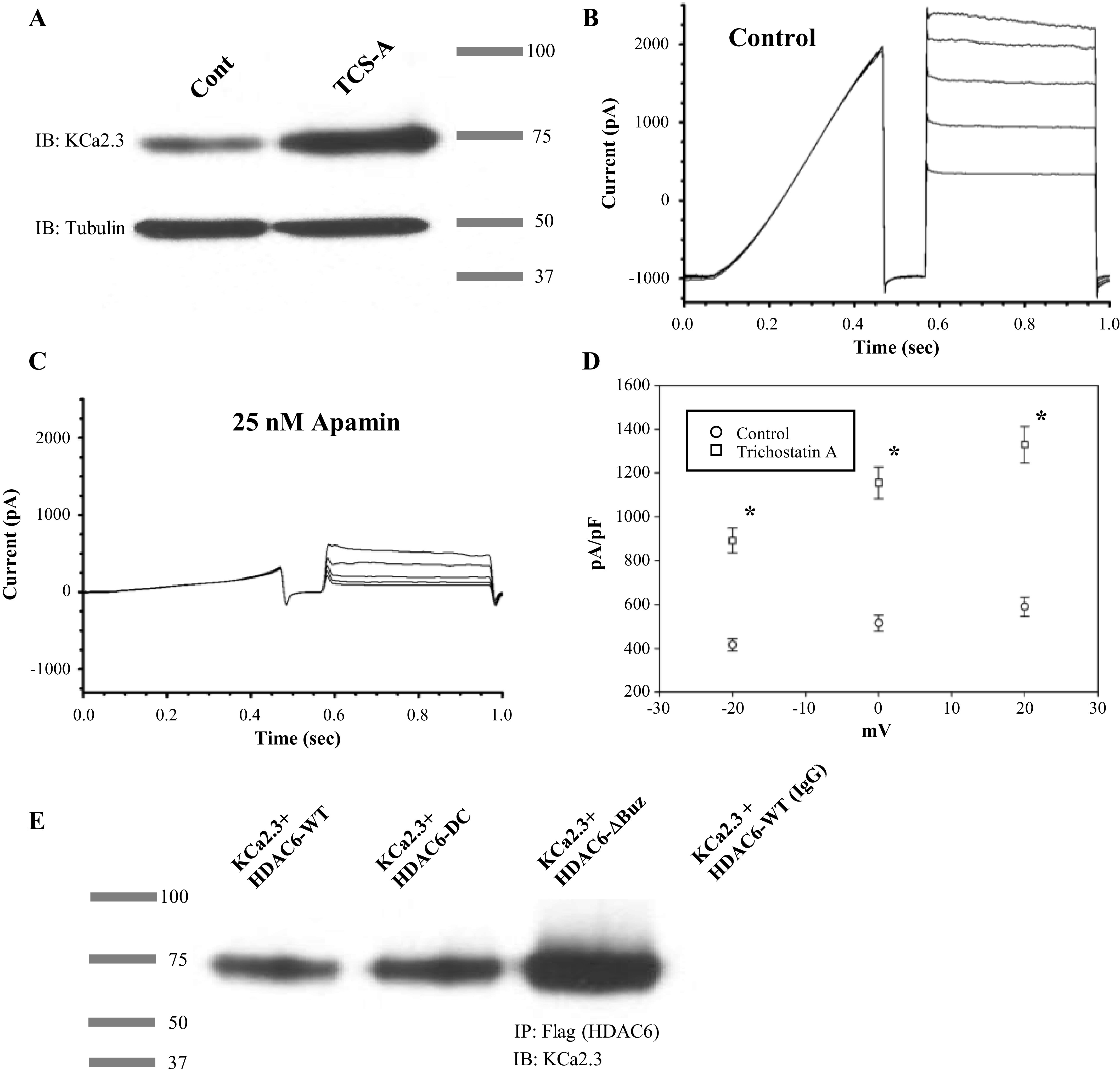
A: TCS-A increases heterologous KCa2.3 expression in HEK293T cells, as assessed by immunoblot. B: representative whole cell currents are presented showing maximal activation of KCa2.3 in HEK293T cells. Voltage sweeps consisted of 400 ms ramp from −120 mV to 0 mV, followed by a return to −120 mv for 100 ms, and then a pulse from −60 to 20 mV in 20 mV increments. C: representative whole cell currents are presented in the same cell treated with 25 nM apamin. D: average current density and SEM are displayed for control (open circles, n = 10) and cells treated with 1 µM TCS-A for 24 h (open squares, n = 10). An asterisk indicates statistical significance (P < 0.05) with a one tailed Student’s t test assuming independent means. E: a representative blot is presented exemplifying one of three coimmunoprecipitation experiments of KCa2.3 with HDAC6 in HEK cells. KCa2.3 was coexpressed with Flag-tagged WT (lane 1), catalytically inactive (DC; lane 2) or an ubiquitin binding-domain deleted (ΔBuz; lane 3) HDAC6. HDAC6 was IPed with α-FLAG Ab (nonspecific IgG was used as control; lane 4). Immunoblot was carried out using α-KCa2.3 Ab. KCa2.3, calcium-activated potassium channel; TCS-A, trichostatin-A.
Since heterologous expression of KCa2.3 in HEK293 cells is driven by a CMV promoter and is therefore not subjected to nuclear transcriptional regulation, we hypothesized the increase in KCa2.3 expression observed by exposure to HDACi was mediated by a cytosolic protein. As HDAC6 is a purely cytosolic protein that has been shown to alter the trafficking of various proteins to the plasma membrane by acetylating tubulin (36–38), we sought to determine whether HDAC6 was closely associated with KCa2.3 by co-IP. To carry out these studies, we transiently expressed either Flag-tagged WT, catalytically deficient (DC), or ubiquitin binding domain (UBD)-deleted (ΔBuz) HDAC6 into our stable KCa2.3 HEK cell line. Immunoprecipitation using an anti-Flag antibody, followed by IB for KCa2.3 showed an association between HDAC6 and KCa2.3 (Fig. 1E). This association was independent of HDAC6 catalytic activity (DC) or expression of the HDAC6 UBD (ΔBuz). Similar results were observed in three separate co-IP experiments.
As stated previously, HDACi and modulation of KCa2.3 N and Po have been observed to induce vasorelaxation. Having confirmed that heterologous KCa2.3 subunit and functional expression can be enhanced by inhibition of HDACs, we proceeded to investigate whether endogenous KCa.2.3 expression could similarly be affected by HDACi in the endothelium. For these studies we used two distinct primary endothelial cell culture systems. First, we employed primary cultures of HUVECs, as these have been extensively studied. In addition, we used primary cultures of human intestinal microvascular endothelial cells (HIMECs). These cells are ideal because the contribution of KCa2.3 to the EDH pathway appears to be more pronounced in the microvasculature (6). Both HUVECs and HIMECs were treated with TCS-A (1 µM), valproic acid (VPA; 3 mM), or tubastatin-A (TUB-A; 3 µM) overnight. TCS-A was used as a pan-HDACi. VPA was used a class I HDAC inhibitor, which does not increase cytosolic protein acetylation, whereas TUB-A was used as a specific cytosolic HDAC6 inhibitor. TCS-A induced an ∼15–20-fold increase in total KCa2.3 expression in both HUVECs and HIMECs (Fig. 2, A and B). This is a significantly larger increase than observed in HEK293 cells heterologously expressing KCa2.3. Valproic acid, which alters histone acetylation, but not acetylation of cytosolic proteins, induced a slightly smaller increase in total KCa2.3 expression. Finally, TUB-A induced an approximately twofold increase in total KCa2.3 expression, similar to what we observed in HEK cells exposed to TCS-A. These data are summarized in Fig. 2, I and J. We confirmed the relative specificity of these HDACi by demonstrating that total α-tubulin expression remained unaffected (Fig. 2, E and F), whereas TCS-A and TUB-A significantly increased acetylated α-tubulin and VPA had no effect (Fig. 2, G and H), as expected. Finally, immunostaining for total KCa2.3 expression in HUVECs (Fig. 2K) and HIMECs (Fig. 2L) similarly demonstrated an increase in the immunolabeled KCa2.3 following TCS-A treatment (compare left and right panels). Quantification of immunofluorescent images reveals a significant increase in KCa2.3 labeling for both HUVECs (Fig. 2M) and HIMECs (Fig. 2N) following TCS-A treatment. These results demonstrate that HDACi increase total expression of KCa2.3 in two distinct primary endothelial cell cultures.
Figure 2.
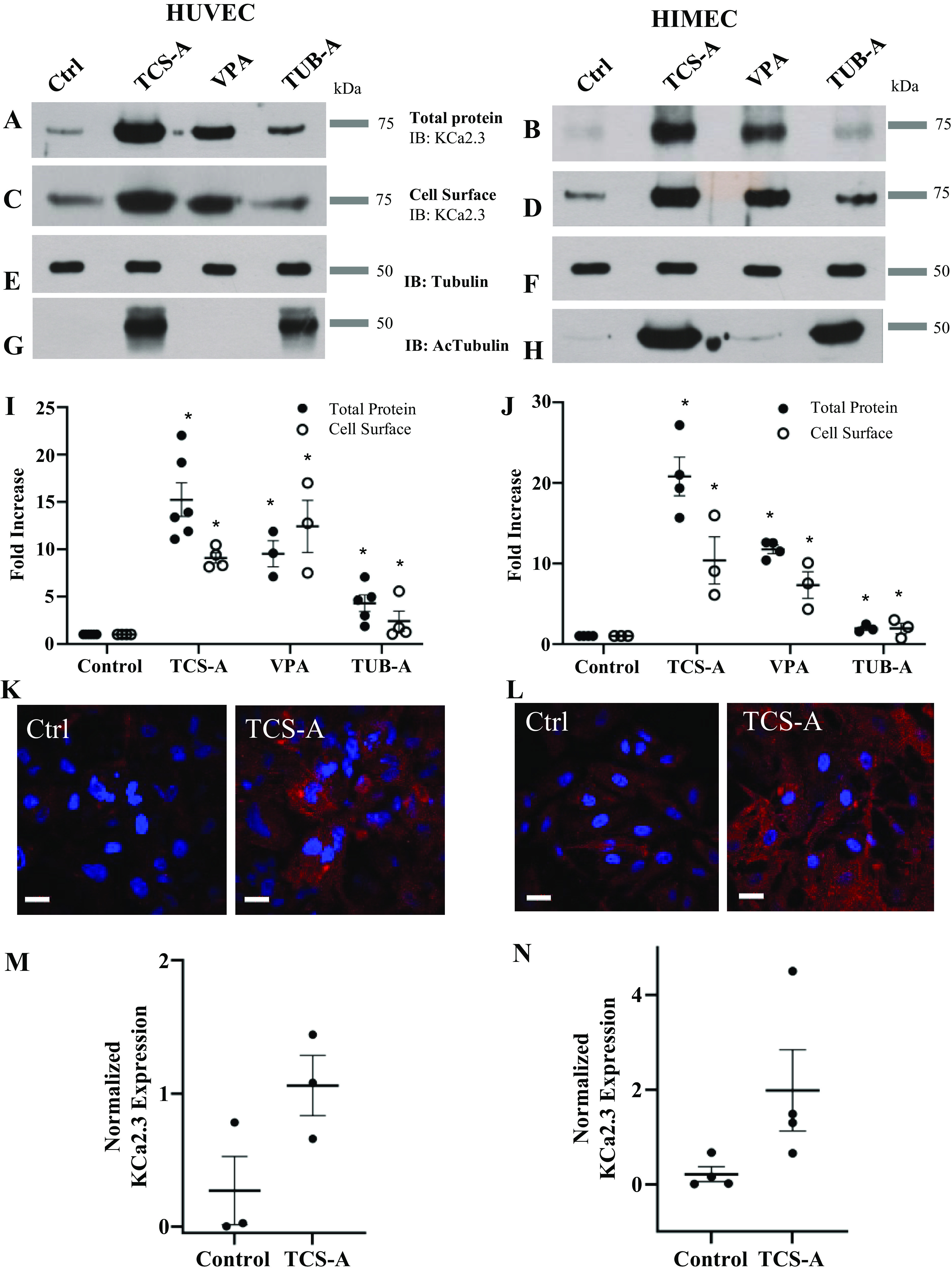
Effect of HDACi on KCa2.3 expression is shown in HUVECs (left) and HIMECs (right). TCS-A (1 µM,) VPA (3 mM), and TUB-A (3 µM) increased both total KCa2.3 (A and B) and plasma membrane (C and D) expression. Quantitation of immunoblots are provided showing average increases with SEM for total (closed circles) and plasma membrane (PM; open circles) KCa2.3 expression (n = 4, *P < 0.05) (I and J). TCS-A and TUB-A increased acetylated tubulin (G and H), whereas total tubulin expression was unchanged by HDACi (E and F). TCS-A also caused an increase in KCa2.3 expression as assessed by immunofluorescence labeling (K and L). KCa2.3 is labeled in red and nuclei are in blue. Quantification of immunoblots was performed in ImageJ to obtain the percent area of red and blue channels in each image. The percent area of red pixels, which corresponds to KCa2.3 staining, was divided by the percent area of blue staining, which corresponds to a nuclear stain and, ipso facto cell number, to obtain a readout of normalized KCa2.3 expression per image. When HUVECs were treated with 1 µM TCS-A, normalized KCa2.3 expression (P < 0.05, n = 3) was increased significantly vs. controls (M). Likewise, when HIMECs were treated with 1 µM TCS-A, normalized KCa2.3 (P < 0.05, n = 4) expression was also increased significantly compared with controls (N). HDACi, histone deacetylase inhibitors; HIMECs, human intestinal microvascular endothelial cells; HUVECs, human umbilical vein endothelial cells; KCa2.3, calcium-activated potassium channel; TCS-A, trichostatin-A; TUB-A, tubastatin-A; VPA, valproic acid.
To determine whether HDAC inhibition also increases plasma membrane KCa2.3 expression in endothelial cells, we carried out cell surface biotinylations on HUVECs and HIMECs treated overnight with TCS-A, VPA, or TUB-A, as above. In each case, there was a tight correlation between the observed increase in total KCa2.3 protein expression and plasma membrane expression (Fig. 2, C and D). These data are summarized in Fig. 2, I and J.
Because the expression of some proteins appears to be lost through repeated passaging of endothelial cells(39), we confirmed that the effect of TCS-A on KCa2.3 expression is reproducible in freshly isolated HUVECs at passage 0. TCS-A induced a similar increase in total KCa2.3 expression in freshly isolated HUVECs to that observed in passages 1–8 (Fig. 3). Based on the similar effects of HDACi on HUVECs and HIMECs and the greater ease of obtaining HUVECs, the remaining studies have been carried out on HUVECs.
Figure 3.

A: immunoblot showing the effects of the HDACi TCS-A, VPA, and TUB-A on freshly isolated (P0) HUVECs. Tubulin was used as a loading control. B: quantification of immunoblot data comparing expression of KCa2.3 among control, TCS-A, TUB-A, and VPA treated P0 HUVECs. Application of the Kruskal–Wallis test indicates a statistical difference between the mean ranks of experimental groups (P < 0.05, n = 3). HDACi, histone deacetylase inhibitors; HUVECs, human umbilical vein endothelial cells; TCS-A, trichostatin-A; TUB-A, tubastatin-A; VPA, valproic acid.
Our biochemical results above demonstrate HDAC inhibition significantly increases both total and plasma membrane KCa2.3 expression. To determine whether the increase in plasma membrane protein observed is reflected in an increase in functional KCa2.3 channels at the plasma membrane, we carried out whole cell patch-clamp recordings, as above. Whole cell current profiles in HUVECs reveal a range of conductances, including fast inward transient currents when stepping to positive potentials and slowly inactivating outward currents, presumably mediated by Kv channels (Fig. 4A). Application of 100 nM apamin to the bath solution results in a reduction of outward current and reveals the SK component of whole cell currents in HUVECs (Fig. 4B). When normalized for current density, TCS-A-treated cells show a significant difference in the magnitude of apamin-sensitive current, when compared with controls, in which apamin-sensitive currents are not readily observable (Fig. 4C). The roughly 8–10-fold increase in apamin-sensitive current density correlates well with the observed increase in KCa2.3 subunit expression, as previously shown via cell surface biotinylation (Fig. 2).
Figure 4.

A: representative whole cell currents from HUVECs treated with 1 µM TCS-A for 24 h are shown. Voltage sweeps consisted of 400 ms ramp from −120 mV to 0 mV, followed by a return to −120 mv for 100 ms, and then a pulse from −60 to 20 mV in 20 mV increments. B: whole cell currents from the same cell as in A following application of 100 nM apamin are displayed. C: average current density and SEM for control (open circles, n = 17) and HUVECs (open squares, n = 17). TCS-A-treated HUVECs exhibited significantly more apamin-sensitive currents across a range of voltages as indicated by a single (P < 0.05) or double (P < 0.005) asterisk. HUVECs, human umbilical vein endothelial cells; TCS-A, trichostatin-A.
As shown above, the magnitude of the effects of TCS-A on endothelial cell KCa2.3 subunit expression is significantly larger than that observed in HEK cells, whereas the effect of the cytosolic HDACi, TUB-A, was very similar. As TCS-A and VPA are well-known to alter protein expression via effects on transcription (40), whereas TUB-A only affects the cytosolic HDAC6, we hypothesized the difference in the magnitude of the effects observed for these HDACi is due to effects on transcription. To assess this possibility, we carried out quantitative PCR (qPCR) on HUVECs treated overnight with TCS-A (1 µM), VPA (3 mM), or TUB-A (3 µM). As shown in Fig. 5, both TCS-A and VPA induced a 5–6-fold increase in KCa2.3 expression, when normalized to GAPDH. In contrast, the HDAC6 inhibitor TUB-A had no effect on KCa2.3 mRNA, as expected. The expression of actin was unaffected by HDACi, also as expected. These data are consistent with our biochemical and electrophysiological results and suggest at least two components to the total increase in KCa2.3 expression observed: a transcriptional component and a cytosolic component.
Figure 5.

Quantitative PCR was performed on total RNA isolated from HUVECs which were either untreated or treated for 24 h with 1 µM TCS-A (filled squares), 3 mM VPA (filled circles), or 3 µM TUB-A (filled triangles). Detected signals from the PCR amplifications were normalized to the relative expression GAPDH message from each sample, and expression is presented as a fold change from control untreated samples (ΔΔCT). Actin mRNA was amplified and detected as a control for each treatment. Three experiments were carried out on subsequent passages of HUVECs and each result indicated by a separate symbol. A statistically significant difference was observed between VPA and TUB-A and their respective actin controls (n = 3, P < 0.005), which is denoted by an asterisk. HUVECs, human umbilical vein endothelial cells; TCS-A, trichostatin-A; TUB-A, tubastatin-A; VPA, valproic acid.
Given that enhancement of KCa2.3 expression was reliably reproduced in endothelial cells, we sought to determine the concentration- and time-dependency of this effect. We exposed HUVECs to a range of concentrations of TCS-A for 24 h and blotted for KCa2.3 protein. A significant increase in KCa2.3 protein expression was first observed with 100 nM TCS-A and exhibited its largest increase at 1 µM (Fig. 6, A and B). After establishing TCS-A dependency on KCa2.3 expression, we sought to determine the onset of its effects. HUVECs were exposed to 1 µM TCS-A over a 24-h period and total protein was obtained at 2, 4, 8, 16, and 24 h. The onset of TCS-A mediated KCa2.3 expression begins at 8 h and increases steadily at 16 and 24 h (Fig. 6, C and D).
Figure 6.

A: TCS-A increases KCa2.3 expression in a concentration-dependent manner in HUVECs. B: average response for five experiments normalized to KCa2.3 in the absence of TCS-A are shown. C: a time course for TCS-A-dependent increase in KCa2.3 expression in HUVECs is presented. TCS-A was used at 1 µM tubulin was used as a loading control. D: average response for three experiments normalized to KCa2.3 at T = 0 h represented as a scatter plot. An asterisk indicates significant increase in KCa2.3 expression compared with control (P < 0.05). HUVECs, human umbilical vein endothelial cells; KCa2.3, calcium-activated potassium channel; TCS-A, trichostatin-A.
Given the large increase in total KCa2.3 protein induced by HDAC inhibition, we postulated there would be a similar increase in a chaperone protein required for the proper folding and assembly of KCa2.3. However, to date, no molecular chaperones have been described for KCa2.3 or any other member of the KCNN gene family. Importantly, HDACi are known to affect the expression of the molecular chaperone, heat shock protein 70 (Hsp70) but not Hsp90, via transcriptional regulation (35, 41–46). Thus, we postulated that Hsp70 may play a role in the HDACi-mediated effects on KCa2.3. For these studies, HUVECs were exposed to 1 µM TCS-A for 24 h and total KCa2.3, Hsp70, and Hsp90 protein were assessed by IB. As before, there was a large increase in KCa2.3 expression. Importantly, an ∼12-fold increase in Hsp70 expression was observed, whereas Hsp90 was unaffected (Fig. 7, A and B). Next, we asked whether inhibition of Hsp70 activity would affect the TCS-A-induced expression of KCa2.3. HUVECs were exposed to 1 µM TCS-A for 24 h in the presence or absence of the selective Hsp70 inhibitor, VER155008 (VER). In control cells, VER decreased endogenous KCa2.3 expression, implicating Hsp70 activity in the proper folding/assembly of KCa2.3 (Fig. 7C). Importantly, VER significantly reduced the TCS-A-induced increase in KCa2.3 expression to fourfold (Fig. 7D), indicating the HDACi-mediated increase in Hsp70 expression is required to maintain folding of the increased KCa2.3 protein induced by HDAC inhibition.
Figure 7.

A: TCS-A (1 µM) increases KCa2.3 and HSP70 expression while having no effect on HSP90 expression in HUVECs. B: average responses for five HSP70 experiments and three HSP90 experiments are represented by a dot plot. Results were normalized to the relevant HSP in the absence of TCS-A and the mean ranks are statistically significant vial the Kruskal–Wallis test (P < 0.01). C: the HSP70 inhibitor, VER155008 (VER, 40 µM) decreases KCa2.3 expression under control conditions and inhibits the TCS-A-induced increase in KCa2.3 expression. D: average response for four experiments normalized to control is represented by a dot plot. Tubulin was used as a loading control. *Significant change in KCa2.3 expression compared with control (P < 0.05). #A decrease in expression relative to TCS-A (P < 0.05). HUVECs, human umbilical vein endothelial cells; HSP70, heat shock protein 70; KCa2.3, calcium-activated potassium channel; TCS-A, trichostatin-A.
The pan HDACi, Trichostatin-A, increased KCa2.3 total protein expression approximately twofold in HEK cells (Fig. 1), and this cannot be attributed to a transcriptional effect given that KCa2.3 is heterologously expressed via a CMV promoter. Similarly, TUB-A, which specifically inhibits cytosolic HDAC6, increased total KCa2.3 protein expression approximately twofold in HUVECs and HIMECs (Fig. 2). These results suggest an additional component is involved in the total change in plasma membrane KCa2.3 expression observed. We demonstrate that both TCS-A and TUB-A significantly increase acetylated α-tubulin expression in HUVECs and HIMECs (Fig. 2, G and H). Importantly, it is well-known that acetylation of α-tubulin affects trafficking of a number of proteins both into and out of the plasma membrane (36–38, 47). Thus, we hypothesized that KCa2.3 trafficking would be affected by exposure to HDACi. Previously, we demonstrated that KCa2.3 is rapidly endocytosed and recycled at the plasma membrane, with a time constant for endocytosis of ∼5 min, in both HEK and endothelial cell lines [HMEC-1 (26)]. Using membrane protein biotinylation/stripping methods published previously, we investigated whether HDACi would affect KCa2.3 endocytosis. For these studies, we used TCS-A, as it induces a more robust increase in plasma membrane KCa2.3 expression, thereby facilitating our studies. In control HUVECs, 21 ± 1% (n = 3) of KCa2.3 was endocytosed in 20 min (Fig. 8A), similar to what we previously reported in HEK and HMEC-1 cells (26). In contrast, when treated with TCS-A, only 6 ± 1% (n = 3) of KCa2.3 protein was endocytosed from the plasma membrane of HUVECs in 20 min (Fig. 8, A and C). As shown in Fig. 8B, TCS-A increased total KCa2.3 expression, as expected. These data demonstrate HDAC inhibition alters the trafficking of KCa2.3 via an effect on plasma membrane endocytosis. This decreased endocytic rate would result in an increase in plasma membrane KCa2.3 in both HEK and HUVEC cells in the absence of transcriptional effects.
Figure 8.

A: blot showing endocytosis of PM KCa2.3 in HUVECs. Plasma membrane KCa2.3 was biotinylated at 4°C and either immediately pulled down (T = 0) with streptavidin beads, immediately stripped in MESNA (Strip) or incubated at 37°C for 20 min to allow endocytosis, followed by MESNA stripping of the remaining plasma membrane biotin and pulled down. The blot was then probed with α-KCa2.3 Ab. B: IB of total KCa2.3 showing equivalent expression in each condition. KCa2.3 expression was increased in TCS-A-treated cells (1 µM). Different amounts of total protein were used for the control and TCS-A pulldown to facilitate observing endocytosed KCa2.3 in control conditions. C: dot plot comparing percent endocytosis of control and TCS-A-treated HUVECs after 20 min. D: the results of a CHX (400 µg/mL) chase assay are shown for control and TCS-A-treated HUVECS at 0, 3, 6, 9, and 12 h. E: comparison of relative expression levels between TCS-A and control HUVECs (n = 4). Immunoblots were converted to line plots in ImageJ and subsequently quantified. Data from each time point were normalized to expression level at T = 0. The Kruskal–Wallis test was applied at 3, 6, 9, and 12 h and no statistical difference was revealed (P = 1, 0.2482, 0.2482, and 0.5127, respectively). CHX, cycloheximide; IB, immunoblot; HUVECs, human umbilical vein endothelial cells; KCa2.3, calcium-activated potassium channel; TCS-A, trichostatin-A.
Given that the rate of KCa2.3 endocytosis is decreased and its overall expression is increased in HUVECs exposed to HDACi, we postulated that KCa2.3 degradation might also be affected. To determine whether degradation of total KCa2.3 protein was impacted by HDACi, we performed a cycloheximide (CHX) chase experiment. HUVECs were treated with 400 µg/mL CHX to prevent new protein synthesis, and levels of KCa2.3 protein, in both control and TCS-A treated cells, were assessed every 3 h for 12 h (Fig. 8D). As shown in Fig. 8E, total KCa2.3 protein remained higher at 9 and 12 h in TCS-A-treated HUVECs when compared with control (n = 4); however, the data are not statistically significant at any time point. This slightly diminished rate of degradation is consistent with a decreased endocytic rate and trafficking to the lysosome for KCa2.3.
Previously, we have shown that insertion of KCa2.3 into the plasma membrane is dependent on the SNARE proteins syntaxin-4 and SNAP23 (48). Others have demonstrated a functional coupling between TRPV4 and KCa2.3 within caveolin-rich microdomains in endothelial cells (24, 49, 50). To determine whether HDACi enhances the expression of these proteins, as it does KCa2.3, HUVECs were exposed to TCS-A and total protein was resolved and blotted for syntaxin-4, SNAP23, and TRPV4. No difference in the expression levels of these proteins could be observed as a result of TCS-A treatment (Fig. 9). These data clearly show that HDACi leads to an increase in plasma membrane KCa2.3 subunit expression in primary endothelial cells, which is specific and does not extend to other proteins involved in KCa2.3 trafficking and function.
Figure 9.

Shown is an analysis of relative expression levels for proteins relevant to the EDH response and KCa2.3 trafficking in endothelial cells. A: immunoblot showing expression of TRPV4, SNAP-23, and Syntaxin-4 in control and TCS-A treated HUVECs. B: immunoblot showing expression of TRPV4, SNAP-23, and Syntaxin-4 in control and TCS-A treated HIMECs. None of these proteins exhibit the enhancement of expression seen in KCa2.3 in either HUVECs or HIMECs. EDH, endothelium-derived hyperpolarization; HIMECs, human intestinal microvascular endothelial cells; HUVECs, human umbilical vein endothelial cells; KCa2.3, calcium-activated potassium channel; TCS-A, trichostatin-A.
DISCUSSION
KCa2.3 has been shown to play an important role in myoendothelial coupling by contributing to the endothelial-derived hyperpolarization response (3–5). It is therefore unsurprising that agonists of KCa channels have shown promise for pharmacologically inducing vasorelaxation and attenuating hypertension in vivo (22). KCa channel openers, such as the benzimidazolinone, 1-EBIO, are known to shift calcium dependence to lower concentrations, thereby increasing open probability (PO). Although increasing KCa2.3 PO is an attractive means by which KCa2.3-mediated vasorelaxation can be achieved in vivo, pharmacologically targeting KCa2.3 channel N is a comparable strategy with unexplored potential. Previously, our group has examined the KCa2.3 recycling loop in detail (26, 27, 48, 51) but as of yet, no pharmacological approach to increasing N of KCa2.3 has been proposed.
Histone deacetylase inhibitors (HDACi) are being explored for their utility in cardiovascular pathologies and there is evidence to suggest that they may have use in alleviating hypertension. Given that HDACi have been shown to increase plasma membrane expression of ion channels such as CFTR (34) and ENaC (35), we asked whether KCa2.3 expression could be altered in a similar manner, as a potential explanatory mechanism by which HDACi lowers blood pressure. In this report, we demonstrate HDACi drastically increase the expression levels of KCa2.3 in both a heterologous expression system (Fig. 1) and two types of primary endothelial cells (Fig. 2). We also reveal complex, multilevel regulation of KCa2.3 by HDACi. A cell model detailing this regulation is shown in Fig. 10.
Figure 10.

Cell model illustrating effects of HDACi on KCa2.3 transcription, folding, trafficking, and plasma membrane expression. Starting in the nucleus, an increase in histone acetylation by HDACi results in increased transcriptional expression of KCa2.3. HDACi also increases HSP70 expression which facilitates the folding of KCa2.3. Inhibition of HSP70 by VER155008 results in degradation of KCa2.3, presumably in the proteasome. Inhibition of dynamin- and Rab5-dependent KCa2.3 endocytosis by HDACi would result in an increase in KC2.3 plasma membrane (PM) expression. Whether this is a direct result of increased tubulin acetylation or acetylation of KCa2.3 remains to be elucidated. The effects of HDAC inhibition on the Rab35-dependent recycling and Golgi-to-PM trafficking of KCa2.3 remain to be determined. HDACi, histone deacetylase inhibitors; KCa2.3, calcium-activated potassium channel.
For example, the fact that KCa2.3 expression is increased in a heterologously expressing HEK cell line (Fig. 1) suggests a nontranscriptional component to the effects of HDACi on KCa2.3. In this nonclonal, stable cell line, WT KCa2.3 subunits are transcribed from a cytomegalovirus promoter containing plasmid, which should not be subjected to epigenetic regulation by HDACs. Indeed, coimmunoprecipitation experiments revealed an association of heterologously expressed KCa2.3 subunit protein with the exclusively cytosolic HDAC6 in HEK cells (Fig. 1E), supporting the notion of nontranscriptional control. Importantly, HDAC6 has been shown to interact with a plethora of other proteins (52). For example, HDAC6 has been shown to bind to tyrosine-protein phosphatase nonreceptor type 1, PTPN1, which promotes the proliferation, colony formation, and migration of melanoma cells (53). In thyroid cancer cells, HDAC6 has been suggested to stabilize the transcriptional complex of RUNX2 – an embryonic transcription factor believed to be reactivated in certain forms of cancer (54). With respect to ion channels, HDAC6 has been suggested to deacetylate a key lysine residue on Pannexin1, which activates the channel downstream of GPCR activation (55). Furthermore, inhibition of HDAC6 has been shown to stabilize a mutant form of the hERG channel responsible for long QT syndrome type 2, presumably through blocking its ubiquitination (56). Our studies further indicate the association of HDAC6 with KCa2.3 does not require either catalytic activity (DC) or the presence of an ubiquitin-binding domain (ΔBuz) on HDAC6 (Fig. 1E). Although it would be interesting to determine whether these HDAC6 domains regulate channel function of endogenously expressed KCa2.3 in endothelial cells, overexpression of HDAC6 in HUVECs or HIMECs would be expected to decrease plasma membrane expression, based on our HDACi studies. As shown in Fig. 3, baseline expression of endogenous KCa2.3 is extremely low, such that any decrease in expression would be beyond our limits of detection.
Our studies further suggest the HDACi-mediated increase in KCa2.3 expression also involves a cytoplasmic component in HUVECs and HIMECs (Fig. 2). That is, the HDAC6 inhibitor TUB-A induced a similar increase in KCa2.3 expression to that observed in HEK cells. Both TCS-A and TUB-A exposure resulted in an increase in acetylated tubulin suggesting that increasing microtubule acetylation may enhance accumulation of KCa2.3 at the plasma membrane beyond what would be expected from the typical anterograde trafficking we previously described (28). In this regard, in the endothelium, KCa2.3 is known to be localized in caveolin-1-rich microdomains (27, 57).
Previously, we have shown that KCa2.3 is rapidly endocytosed from the plasma membrane (time constant ∼ 5 min) and have elucidated the domains of the channel and binding partners critical to its recycling (26, 27, 48). Using similar methods employed in our previous work, we demonstrate KCa2.3 endocytosis follows a similar time frame in primary HUVECs as we previously determined in heterologous expression systems [HEK, HMEC-1; (26)]. Furthermore, we demonstrate that KCa2.3 endocytosis is at least three times slower in HUVECs exposed to HDACi, relative to control cells (Fig. 8). Given that α-tubulin deacetylation results in dysregulation of the microtubule network, this may explain the cytoplasmic effects of HDACi on KCa2.3. Indeed, an increase in α-tubulin acetylation results in the increased binding of kinesin-1 and dynein to microtubules leading to an increased rate of vesicle trafficking for several different proteins (36–38, 47). In cystic fibrosis human airway epithelial cells, which have higher levels of acetylated tubulin, inhibition of HDAC6 broadly restores cholesterol homeostasis by increasing organelle transport along microtubules (58). Although we demonstrate that global HDAC inhibition results in a decreased rate of KCa2.3 endocytosis (Fig. 8), which would result in an increase in plasma membrane expression (Fig. 10), we cannot exclude the possibility that, following increased expression and HSP-70-dependent folding, anterograde trafficking of KCa2.3 is also enhanced, partially explaining the increased plasma membrane expression observed.
Furthermore, although our co-IP experiments reveal an association between KCa2.3 and HDAC6, we cannot rule out the possibility that other cytosolic HDACs associate with the channel to modulate endocytosis, anterograde trafficking, or recycling. Both HDAC8 and HDAC5 have been observed to shuttle between the nucleus and cytoplasm (59) with the latter translocating to the cytoplasm in response to shear stress (60). Importantly, shear stress is known to result in both an increased expression and activation of KCa2.3 in endothelial cells (49, 61–63). In addition, the intracellular trafficking of HDAC4 has been shown to regulate neuronal cell death (64). Finally, overexpression of HDAC8 in cervical cancer cell leads to functional redundancy in tubulin deacetylation with HDAC6 (65). Our current work establishes a role for HDACi in modulating PM KCa2.3 levels and introduces the concept of pharmacologically regulating KCa2.3 N as a strategy for promoting EDH-mediated vasorelaxation. However, future gene silencing studies in HUVECs will be required to identify the specific HDACs responsible for the transcriptional control of KCa2.3 expression, as well as its proper folding and anterograde/retrograde trafficking.
Beyond effects on acetylated tubulin, we cannot rule out the possibility that KCa2.3 is, itself, directly acetylated in such a way that its trafficking is affected. However, we have noted that SNAP23 and Syntaxin-4, which our group has demonstrated as essential for KCa2.3 recycling, are not affected by HDAC inhibition (Fig. 9). The association of HDAC6 with KCa2.3 in the same microdomain could mean that the channel is itself deacetylated, thereby altering its function and recycling. Furthermore, does HDAC6 act specifically on KCa2.3 or on a larger complex? KCa2.3 is known to associate with TRPV4 in microdomains (61) within the endothelia and impairment of this interaction has been proposed as a possible strategy for attenuating hypertension (66). Given that HDACi had no effect on TRPV4 expression, HDAC inhibition could allow for differential modulation of gene expression between the two coupled channels as well as their ability to associate into microdomains within endothelial cells.
Despite the clear contribution of a cytoplasmic HDACi to enhancing KCa2.3 N at the plasma membrane, our data suggest this only accounts for a small proportion of the total increase observed following more general HDAC inhibition. Indeed, endothelial KCa2.3 subunit expression was significantly elevated by VPA, a class I HDACi that acts only in the nucleus, epigenetically, as well as by the pan-HDACi, TCS-A. This suggested transcriptional regulation at the KCNN3 locus by HDACi, and qPCR verified both VPA and the pan HDACi, TSC-A, induced elevated KCa2.3 transcript levels in HUVECs (Fig. 5). Unsurprisingly, TUB-A, which again acts on the cytosolic HDAC6, failed to elevate KCa2.3 transcripts above actin controls. We therefore conclude that HDAC inhibition results in increased KCa2.3 expression via both cytoplasmic and nuclear mechanisms (Fig. 10).
As previously mentioned, the extent to which HDACi enhances KCa2.3 expression correlates well across the various methods of analysis employed here. The existence of enhanced apamin-sensitive current density in endothelial cells exposed to HDACi indicates that a greater number of correctly folded KCa2.3 subunits are being inserted into the plasma membrane following HDACi treatment. In addition to upregulating KCa2.3, endothelial cells treated with HDACi demonstrate elevated levels of the molecular chaperone Hsp70, but not Hsp90. Both HSP70 and HSP90 are directly acetylated following HDAC inhibition (41, 44, 46), and acetylation of HSP70 has been shown to increase its affinity for hypoxia-inducible factor 1, HIF-1, in coimmunoprecipitation studies (67). Inhibition of class I HDACs has been proposed to result in the increased acetylation of Hsp70 in cortical neurons (42) and HDAC5, which shuttles between the nucleus and cytoplasm, has been demonstrated to deacetylate Hsp70 in various cell lines (67). Like HIF-1, the affinity of Hsp70 for KCa2.3 may be increased if either protein remains acetylated as a result of HDACi treatment, and this may affect their localization. Importantly, inhibition of Hsp70 reduces KCa2.3 expression in HUVECs and diminishes the increased expression of KCa2.3 induced by HDACi (Fig. 7). These data suggest that Hsp70 plays a role in the correct folding of KCa2.3 and represents, to our knowledge, the first molecular chaperone identified as being associated with the biogenesis of any member of the KCNN gene family (Fig. 10). More work is needed to determine if either the channel or chaperone are directly acetylated in a way that alters their affinity for each other and possibly their position within the cell.
HDACi have found some success in the treatment of noncardiovascular-related disease (68–70). The class I HDACi, valproic acid (VPA) is a well-known anticonvulsant and has been approved to treat seizures (71), mania (72), and prevent migraines (73). The precise mechanism by which valproic acid exerts its effects is not known, and in addition to HDAC inhibition, blocking sodium channels and altering GABA levels have both been proposed (74). In the brain, SK channels underly the medium afterhyperpolarization (mAHP) (75–77) and ipso facto contribute to modulating firing frequency and neuronal excitability (78–80). The data presented here suggest that valproic acid may, through its inhibition of HDACs, upregulate SK channel subunits in the brain, thereby dampening neural excitability.
In this work, we have demonstrated that the inhibition of HDACs reliably increases the expression of KCa2.3 in a heterologous expression system (HEK) as well as primary endothelial cells (HUVECs, HIMECs). Taking advantage of the differential selectivity of various HDACi, we show that this enhancement of KCa2.3 expression is the result of genetic and cytoplasmic components. KCa2.3 transcripts are more abundant in HUVECs treated with HDACi, whereas in the cytoplasm HCACi enhances the expression of the molecular chaperone Hsp70, which, when inhibited, diminishes the increase in KCa2.3 expression observed by HDACi exposure. Furthermore, we have observed an association with KCa2.3 and HDAC6 in the cytoplasm and slower endocytosis of KCa2.3 when HDACs are inhibited. Together, these three components – transcription, folding and trafficking – combine to increase functional plasma membrane KCa2.3 expression following HDAC inhibition (Fig. 10).
Taken together, our data suggest the HDAC-mediated effects on vasorelaxation and blood pressure may be the result of increased KCa2.3 expression – particularly at the myoendothelial junction where KCa2.3 is known to contribute to the EDH response (3, 5, 81). To date, pharmacological manipulation of KCa2.3 channel number has not yet been proposed as a therapeutic strategy, although the effects of genetic manipulation of this channel on vasorelaxation and blood pressure are clear (6, 23, 25). Given that some HDACi have been approved to treat blood cancer and that the SK opener, Riluzole (82), has been approved for the treatment of amyotrophic lateral sclerosis (83), there may be immediate therapeutic off label use for these drugs, either alone or in combination, for the treatment of cardiovascular disease such as hypertension or PAD. Clinically, approved HDACi possess a host of negative side effects (84), and further elucidation of the mechanism by which KCa2.3 is upregulated by these compounds could lead to a more targeted approach to promote vasorelaxation in vivo. In addition, such work would establish the viability of the strategy for increasing KCa2.3 channel number to treat cardiovascular disease or other indications.
GRANTS
Dr. Gandley received funding from the National Institutes of Health (R21 HD083659) and the American Heart Association (16SFRN27810001). Dr. Straub received funding from the National Institutes of Health (R01 HL 128304, R01 HL153532) and the American Heart Association (19EIA34770095). Drs. Binion and Wilson were supported by the Department of Defense (W81XWH-14-1-0376 and W81XWH-17-1-0502). Dr. Butterworth was supported by the National Institutes of Health (DK102843). Dr. Devor was supported by the Cystic Fibrosis Foundation (DEVOR20GO).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.K-A., C.M.B., C.A.B., A.C.S., M.B.B., D.B., and D.C.D. conceived and designed research; A.K-A., C.M.B., C.A.B., A.S.W., W.M.R., X.L., R.E.G., and D.C.D. performed experiments; A.K-A., C.M.B., C.A.B., M.B.B., and D.C.D. analyzed data; A.K-A., C.M.B., C.A.B., and D.C.D. interpreted results of experiments; A.K-A., C.M.B., and D.C.D. prepared figures; A.K-A. and D.C.D. drafted manuscript; A.K-A. edited and revised manuscript; A.K-A., C.M.B., C.A.B., A.S.W., W.M.R., X.L., R.E.G., A.C.S., M.B.B., D.B., and D.C.D. approved final version of manuscript.
REFERENCES
- 1.American Heart Association. The Facts About High Blood Pressure (Online). https://www.heart.org/en/health-topics/high-blood-pressure/the-facts-about-high-blood-pressure [2022 Jan 23].
- 2.Vanhoutte PM. Endothelium-dependent hyperpolarizations: the history. Pharmacol Res 49: 503–508, 2004. doi: 10.1016/j.phrs.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 3.Köhler R, Eichler I, Schönfelder H, Grgic I, Heinau P, Si H, Hoyer J. Impaired EDHF-mediated vasodilation and function of endothelial Ca-activated K channels in uremic rats. Kidney Int 67: 2280–2287, 2005. doi: 10.1111/j.1523-1755.2005.00331.x. [DOI] [PubMed] [Google Scholar]
- 4.Félétou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler Thromb Vasc Biol 26: 1215–1225, 2006. doi: 10.1161/01.ATV.0000217611.81085.c5. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt K, de Wit C. Endothelium-derived hyperpolarizing factor and myoendothelial coupling: the in vivo perspective. Front Physiol 11: 602930, 2020. doi: 10.3389/fphys.2020.602930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brähler S, Kaistha A, Schmidt VJ, Wölfle SE, Busch C, Kaistha BP, Kacik M, Hasenau AL, Grgic I, Si H, Bond CT, Adelman JP, Wulff H, de Wit C, Hoyer J, Köhler R. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 119: 2323–2332, 2009. doi: 10.1161/CIRCULATIONAHA.108.846634. [DOI] [PubMed] [Google Scholar]
- 7.Dora KA, Gallagher NT, McNeish A, Garland CJ. Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res 102: 1247–1255, 2008. doi: 10.1161/CIRCRESAHA.108.172379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McNeish AJ, Sandow SL, Neylon CB, Chen MX, Dora KA, Garland CJ. Evidence for involvement of both IKCa and SKCa channels in hyperpolarizing responses of the rat middle cerebral artery. Stroke 37: 1277–1282, 2006. doi: 10.1161/01.STR.0000217307.71231.43. [DOI] [PubMed] [Google Scholar]
- 9.Bagchi RA, Weeks KL. Histone deacetylases in cardiovascular and metabolic diseases. J Mol Cell Cardiol 130: 151–159, 2019. doi: 10.1016/j.yjmcc.2019.04.003. [DOI] [PubMed] [Google Scholar]
- 10.McKinsey TA. Targeting inflammation in heart failure with histone deacetylase inhibitors. Mol Med 17: 434–441, 2011. doi: 10.2119/molmed.2011.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol 52: 303–319, 2012. doi: 10.1146/annurev-pharmtox-010611-134712. [DOI] [PubMed] [Google Scholar]
- 12.Romanick SS, Ferguson BS. The nonepigenetic role for small molecule histone deacetylase inhibitors in the regulation of cardiac function. Future Med Chem 11: 1345–1356, 2019. doi: 10.4155/fmc-2018-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chun P. Therapeutic effects of histone deacetylase inhibitors on heart disease. Arch Pharm Res 43: 1276–1296, 2020. doi: 10.1007/s12272-020-01297-0. [DOI] [PubMed] [Google Scholar]
- 14.Cardinale JP, Sriramula S, Pariaut R, Guggilam A, Mariappan N, Elks CM, Francis J. HDAC inhibition attenuates inflammatory, hypertrophic, and hypertensive responses in spontaneously hypertensive rats. Hypertension 56: 437–444, 2010. doi: 10.1161/HYPERTENSIONAHA.110.154567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iyer A, Fenning A, Lim J, Le GT, Reid RC, Halili MA, Fairlie DP, Brown L. Antifibrotic activity of an inhibitor of histone deacetylases in DOCA-salt hypertensive rats. Br J Pharmacol 159: 1408–1417, 2010. doi: 10.1111/j.1476-5381.2010.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang G, Lee YR, Joo HK, Park MS, Kim CS, Choi S, Jeon BH. Trichostatin A modulates angiotensin II-induced vasoconstriction and blood pressure via inhibition of p66shc activation. Korean J Physiol Pharmacol 19: 467–472, 2015. doi: 10.4196/kjpp.2015.19.5.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoon GE, Jung JK, Lee YH, Jang BC, In Kim J. Histone deacetylase inhibitor CG200745 ameliorates high-fat diet-induced hypertension via inhibition of angiotensin II production. Naunyn Schmiedebergs Arch Pharmacol 393: 491–500, 2020. doi: 10.1007/s00210-019-01749-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hebbel RP, Vercellotti GM, Pace BS, Solovey AN, Kollander R, Abanonu CF, Nguyen J, Vineyard JV, Belcher JD, Abdulla F, Osifuye S, Eaton JW, Kelm RJ Jr, Slungaard A. The HDAC inhibitors trichostatin A and suberoylanilide hydroxamic acid exhibit multiple modalities of benefit for the vascular pathobiology of sickle transgenic mice. Blood 115: 2483–2490, 2010. doi: 10.1182/blood-2009-02-204990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devor DC, Singh AK, Frizzell RA, Bridges RJ. Modulation of Cl- secretion by benzimidazolones. I. Direct activation of a Ca(2+)-dependent K+ channel. Am J Physiol 271: L775–L784, 1996. doi: 10.1152/ajplung.1996.271.5.L775. [DOI] [PubMed] [Google Scholar]
- 20.Singh S, Syme CA, Singh AK, Devor DC, Bridges RJ. Benzimidazolone activators of chloride secretion: potential therapeutics for cystic fibrosis and chronic obstructive pulmonary disease. J Pharmacol Exp Ther 296: 600–611, 2001. [PubMed] [Google Scholar]
- 21.Adeagbo AS. 1-Ethyl-2-benzimidazolinone stimulates endothelial K(Ca) channels and nitric oxide formation in rat mesenteric vessels. Eur J Pharmacol 379: 151–159, 1999. doi: 10.1016/s0014-2999(99)00489-6. [DOI] [PubMed] [Google Scholar]
- 22.Damkjaer M, Nielsen G, Bodendiek S, Staehr M, Gramsbergen JB, de Wit C, Jensen BL, Simonsen U, Bie P, Wulff H, Köhler R. Pharmacological activation of KCa3.1/KCa2.3 channels produces endothelial hyperpolarization and lowers blood pressure in conscious dogs. Br J Pharmacol 165: 223–234, 2012. doi: 10.1111/j.1476-5381.2011.01546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, Adelman JP, Nelson MT. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res 93: 124–131, 2003. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- 24.Yap FC, Weber DS, Taylor MS, Townsley MI, Comer BS, Maylie J, Adelman JP, Lin MT. Endothelial SK3 channel-associated Ca2+ microdomains modulate blood pressure. Am J Physiol Heart Circ Physiol 310: H1151–H1163, 2016. doi: 10.1152/ajpheart.00787.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yap FC, Taylor MS, Lin MT. Ovariectomy-induced reductions in endothelial SK3 channel activity and endothelium-dependent vasorelaxation in murine mesenteric arteries. PLoS One 9: e104686, 2014. doi: 10.1371/journal.pone.0104686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao Y, Balut CM, Bailey MA, Patino-Lopez G, Shaw S, Devor DC. Recycling of the Ca2+-activated K+ channel, KCa2.3, is dependent upon RME-1, Rab35/EPI64C, and an N-terminal domain. J Biol Chem 285: 17938–17953, 2010. doi: 10.1074/jbc.M109.086553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao Y, Bertuccio CA, Balut CM, Watkins SC, Devor DC. Dynamin- and Rab5-dependent endocytosis of a Ca2+ -activated K+ channel, KCa2.3. PLoS One 7: e44150, 2012. e44150doi: 10.1371/journal.pone.0044150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones HM, Hamilton KL, Devor DC. Role of an S4-S5 linker lysine in the trafficking of the Ca(2+)-activated K(+) channels IK1 and SK3. J Biol Chem 280: 37257–37265, 2005. doi: 10.1074/jbc.M508601200. [DOI] [PubMed] [Google Scholar]
- 29.Binion DG, West GA, Ina K, Ziats NP, Emancipator SN, Fiocchi C. Enhanced leukocyte binding by intestinal microvascular endothelial cells in inflammatory bowel disease. Gastroenterology 112: 1895–1907, 1997. doi: 10.1053/gast.1997.v112.pm9178682. [DOI] [PubMed] [Google Scholar]
- 30.Harding SD, Armstrong JF, Faccenda E, Southan C, Alexander SPH, Davenport AP, Pawson AJ, Spedding M, Davies JA; NC-IUPHAR. The IUPHAR/BPS guide to PHARMACOLOGY in 2022: curating pharmacology for COVID-19, malaria and antibacterials. Nucleic Acids Res 50: D1282–D1294, 2021. doi: 10.1093/nar/gkab1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nolting A, Ferraro T, D'Hoedt D, Stocker M. An amino acid outside the pore region influences apamin sensitivity in small conductance Ca2+-activated K+ channels. J Biol Chem 282: 3478–3486, 2007. doi: 10.1074/jbc.M607213200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wittekindt OH, Visan V, Tomita H, Imtiaz F, Gargus JJ, Lehmann-Horn F, Grissmer S, Morris-Rosendahl DJ. An apamin- and scyllatoxin-insensitive isoform of the human SK3 channel. Mol Pharmacol 65: 788–801, 2004. doi: 10.1124/mol.65.3.788. [DOI] [PubMed] [Google Scholar]
- 33.Balut CM, Gao Y, Murray SA, Thibodeau PH, Devor DC. ESCRT-dependent targeting of plasma membrane localized KCa3.1 to the lysosomes. Am J Physiol Cell Physiol 299: C1015–C1027, 2010. doi: 10.1152/ajpcell.00120.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hutt DM, Herman D, Rodrigues AP, Noel S, Pilewski JM, Matteson J, Hoch B, Kellner W, Kelly JW, Schmidt A, Thomas PJ, Matsumura Y, Skach WR, Gentzsch M, Riordan JR, Sorscher EJ, Okiyoneda T, Yates JR 3rd, Lukacs GL, Frizzell RA, Manning G, Gottesfeld JM, Balch WE. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol 6: 25–33, 2010. doi: 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butler PL, Staruschenko A, Snyder PM. Acetylation stimulates the epithelial sodium channel by reducing its ubiquitination and degradation. J Biol Chem 290: 12497–12503, 2015. doi: 10.1074/jbc.M114.635540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci 27: 3571–3583, 2007. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu W, Fan LX, Zhou X, Sweeney WE Jr, Avner ED, Li X. HDAC6 regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation in renal epithelial cells. PLoS One 7: e49418, 2012. doi: 10.1371/journal.pone.0049418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol 16: 2166–2172, 2006. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 39.Sato GH, Sato JD, Okamoto T, McKeehan WL, Barnes DW. Tissue culture: the unlimited potential. In Vitro Cell Dev Biol Anim 46: 590–594, 2010. doi: 10.1007/s11626-010-9315-1. [DOI] [PubMed] [Google Scholar]
- 40.Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20: 6969–6978, 2001. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiménez-Canino R, Lorenzo-Díaz F, Jaisser F, Farman N, Giraldez T, Alvarez de la Rosa D. Histone deacetylase 6-controlled Hsp90 acetylation significantly alters mineralocorticoid receptor subcellular dynamics but not its transcriptional activity. Endocrinology 157: 2515–2532, 2016. doi: 10.1210/en.2015-2055. [DOI] [PubMed] [Google Scholar]
- 42.Marinova Z, Leng Y, Leeds P, Chuang DM. Histone deacetylase inhibition alters histone methylation associated with heat shock protein 70 promoter modifications in astrocytes and neurons. Neuropharmacology 60: 1109–1115, 2011. doi: 10.1016/j.neuropharm.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marinova Z, Ren M, Wendland JR, Leng Y, Liang MH, Yasuda S, Leeds P, Dm C. Valproic acid induces functional heat-shock protein 70 via class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem 111: 976–987, 2009. doi: 10.1111/j.1471-4159.2009.06385.x.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mawatari T, Ninomiya I, Inokuchi M, Harada S, Hayashi H, Oyama K, Makino I, Nakagawara H, Miyashita T, Tajima H, Takamura H, Fushida S, Ohta T. Valproic acid inhibits proliferation of HER2-expressing breast cancer cells by inducing cell cycle arrest and apoptosis through Hsp70 acetylation. Int J Oncol 47: 2073–2081, 2015. doi: 10.3892/ijo.2015.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 62: 670–684, 2005. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Panella S, Marcocci ME, Celestino I, Valente S, Zwergel C, Li Puma DD, Nencioni L, Mai A, Palamara AT, Simonetti G. MC1568 inhibits HDAC6/8 activity and influenza A virus replication in lung epithelial cells: role of Hsp90 acetylation. Future Med Chem 8: 2017–2031, 2016. doi: 10.4155/fmc-2016-0073. [DOI] [PubMed] [Google Scholar]
- 47.Li J, Song J, Cassidy MG, Rychahou P, Starr ME, Liu J, Li X, Epperly G, Weiss HL, Townsend CM Jr, Gao T, Evers BM. PI3K p110α/Akt signaling negatively regulates secretion of the intestinal peptide neurotensin through interference of granule transport. Mol Endocrinol 26: 1380–1393, 2012. doi: 10.1210/me.2012-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertuccio CA, Wang TT, Hamilton KL, Rodriguez-Gil DJ, Condliffe SB, Devor DC. Plasma membrane insertion of KCa2.3 (SK3) is dependent upon the SNARE proteins, syntaxin-4 and SNAP23. PLoS One 13: e0196717, 2018. doi: 10.1371/journal.pone.0196717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu T, Wang XL, Chai Q, Sun X, Sieck GC, Katusic ZS, Lee HC. Role of the endothelial caveolae microdomain in shear stress-mediated coronary vasorelaxation. J Biol Chem 292: 19013–19023, 2017. doi: 10.1074/jbc.M117.786152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma X, Du J, Zhang P, Deng J, Liu J, Lam FF, Li RA, Huang Y, Jin J, Yao X. Functional role of TRPV4-KCa2.3 signaling in vascular endothelial cells in normal and streptozotocin-induced diabetic rats. Hypertension 62: 134–139, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01500. [DOI] [PubMed] [Google Scholar]
- 51.Gao Y, Chotoo CK, Balut CM, Sun F, Bailey MA, Devor DC. Role of S3 and S4 transmembrane domain charged amino acids in channel biogenesis and gating of KCa2.3 and KCa3.1. J Biol Chem 283: 9049–9059, 2008. doi: 10.1074/jbc.M708022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng K, Jiang Y, He Z, Kitazato K, Wang Y. Cellular defence or viral assist: the dilemma of HDAC6. J Gen Virol 98: 322–337, 2017. doi: 10.1099/jgv.0.000679. [DOI] [PubMed] [Google Scholar]
- 53.Liu J, Luan W, Zhang Y, Gu J, Shi Y, Yang Y, Feng Z, Qi F. HDAC6 interacts with PTPN1 to enhance melanoma cells progression. Biochem Biophys Res Commun 495: 2630–2636, 2018. doi: 10.1016/j.bbrc.2017.12.145. [DOI] [PubMed] [Google Scholar]
- 54.Manzotti G, Torricelli F, Donati B, Sancisi V, Gugnoni M, Ciarrocchi A. HDACs control RUNX2 expression in cancer cells through redundant and cell context-dependent mechanisms. J Exp Clin Cancer Res 38: 346, 2019. doi: 10.1186/s13046-019-1350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chiu YH, Medina CB, Doyle CA, Zhou M, Narahari AK, Sandilos JK, Gonye EC, Gao HY, Guo SY, Parlak M, Lorenz UM, Conrads TP, Desai BN, Ravichandran KS, Bayliss DA. Deacetylation as a receptor-regulated direct activation switch for pannexin channels. Nat Commun 12: 4482, 2021. doi: 10.1038/s41467-021-24825-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li P, Kurata Y, Endang M, Ninomiya H, Higaki K, Taufiq F, Morikawa K, Shirayoshi Y, Horie M, Hisatome I. Restoration of mutant hERG stability by inhibition of HDAC6. J Mol Cell Cardiol 115: 158–169, 2018. doi: 10.1016/j.yjmcc.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 57.Absi M, Burnham MP, Weston AH, Harno E, Rogers M, Edwards G. Effects of methyl β-cyclodextrin on EDHF responses in pig and rat arteries; association between SK(Ca) channels and caveolin-rich domains. Br J Pharmacol 151: 332–340, 2007. doi: 10.1038/sj.bjp.0707222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rymut SM, Harker A, Corey DA, Burgess JD, Sun H, Clancy JP, Kelley TJ. Reduced microtubule acetylation in cystic fibrosis epithelial cells. Am J Physiol Lung Cell Mol Physiol 305: L419–L431, 2013. doi: 10.1152/ajplung.00411.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ververis K, Hiong A, Karagiannis TC, Licciardi PV. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics 7: 47–60, 2013. doi: 10.2147/BTT.S29965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang W, Ha CH, Jhun BS, Wong C, Jain MK, Jin ZG. Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood 115: 2971–2979, 2010. doi: 10.1182/blood-2009-05-224824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goedicke-Fritz S, Kaistha A, Kacik M, Markert S, Hofmeister A, Busch C, Bänfer S, Jacob R, Grgic I, Hoyer J. Evidence for functional and dynamic microcompartmentation of Cav-1/TRPV4/K(Ca) in caveolae of endothelial cells. Eur J Cell Biol 94: 391–400, 2015. doi: 10.1016/j.ejcb.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 62.Li G, Yang Q, Yang Y, Yang G, Wan J, Ma Z, Du L, Sun Y, Ζhang G. Laminar shear stress alters endothelial KCa2.3 expression in H9c2 cells partially via regulating the PI3K/Akt/p300 axis. Int J Mol Med 43: 1289–1298, 2019. doi: 10.3892/ijmm.2019.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takai J, Santu A, Zheng H, Koh SD, Ohta M, Filimban LM, Lemaitre V, Teraoka R, Jo H, Miura H. Laminar shear stress upregulates endothelial Ca2+ -activated K+ channels KCa2.3 and KCa3.1 via a Ca2+/calmodulin-dependent protein kinase kinase/Akt/p300 cascade. Am J Physiol Heart Circ Physiol 305: H484–H493, 2013. doi: 10.1152/ajpheart.00642.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bolger TA, Yao TP. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J Neurosci 25: 9544–9553, 2005. doi: 10.1523/JNEUROSCI.1826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vanaja GR, Ramulu HG, Kalle AM. Overexpressed HDAC8 in cervical cancer cells shows functional redundancy of tubulin deacetylation with HDAC6. Cell Commun Signal 16: 20, 2018. doi: 10.1186/s12964-018-0231-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He D, Pan Q, Chen Z, Sun C, Zhang P, Mao A, Zhu Y, Li H, Lu C, Xie M, Zhou Y, Shen D, Tang C, Yang Z, Jin J, Yao X, Nilius B, Ma X. Treatment of hypertension by increasing impaired endothelial TRPV4-KCa2.3 interaction. EMBO Mol Med 9: 1491–1503, 2017. doi: 10.15252/emmm.201707725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen S, Yin C, Lao T, Liang D, He D, Wang C, Sang N. AMPK-HDAC5 pathway facilitates nuclear accumulation of HIF-1α and functional activation of HIF-1 by deacetylating Hsp70 in the cytosol. Cell Cycle 14: 2520–2536, 2015. doi: 10.1080/15384101.2015.1055426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karagiannis D, Rampias T. HDAC inhibitors: dissecting mechanisms of action to counter tumor heterogeneity. Cancers (Basel) 13: 3575, 2021. doi: 10.3390/cancers13143575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shetty MG, Pai P, Deaver RE, Satyamoorthy K, Babitha KS. Histone deacetylase 2 selective inhibitors: a versatile therapeutic strategy as next generation drug target in cancer therapy. Pharmacol Res 170: 105695, 2021. doi: 10.1016/j.phrs.2021.105695. [DOI] [PubMed] [Google Scholar]
- 70.Wawruszak A, Halasa M, Okon E, Kukula-Koch W, Stepulak A. Valproic acid and breast cancer: state of the art in 2021. Cancers (Basel) 13: 3409, 2021. doi: 10.3390/cancers13143409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pinder RM, Brogden RN, Speight TM, Avery GS. Sodium valproate: a review of its pharmacological properties and therapeutic efficacy in epilepsy. Drugs 13: 81–123, 1977. doi: 10.2165/00003495-197713020-00001. [DOI] [PubMed] [Google Scholar]
- 72.Emrich HM, von Zerssen D, Kissling W, Möller HJ, Windorfer A. Effect of sodium valproate on mania. The GABA-hypothesis of affective disorders. Arch Psychiatr Nervenkr (1970) 229: 1–16, 1980. doi: 10.1007/BF00343800. [DOI] [PubMed] [Google Scholar]
- 73.Sorensen KV. Valproate: a new drug in migraine prophylaxis. Acta Neurol Scand 78: 346–348, 1988. doi: 10.1111/j.1600-0404.1988.tb03667.x.. [DOI] [PubMed] [Google Scholar]
- 74.Ghodke-Puranik Y, Thorn CF, Lamba JK, Leeder JS, Song W, Birnbaum AK, Altman RB, Klein TE. Valproic acid pathway: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics 23: 236–241, 2013. doi: 10.1097/FPC.0b013e32835ea0b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bond CT, Herson PS, Strassmaier T, Hammond R, Stackman R, Maylie J, Adelman JP. Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J Neurosci 24: 5301–5306, 2004. doi: 10.1523/JNEUROSCI.0182-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dwivedi D, Upinder SB. Physiology and therapeutic potential of SK, H, and M medium afterhyperpolarization ion channels. Front Mol Neurosci 14: 658435, 2021. doi: 10.3389/fnmol.2021.658435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Villalobos C, Shakkottai VG, Chandy KG, Michelhaugh SK, Andrade R. SKCa channels mediate the medium but not the slow calcium-activated afterhyperpolarization in cortical neurons. J Neurosci 24: 3537–3542, 2004. doi: 10.1523/JNEUROSCI.0380-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen QH, Toney GM. Excitability of paraventricular nucleus neurones that project to the rostral ventrolateral medulla is regulated by small-conductance Ca2+-activated K+ channels. J Physiol 587: 4235–4247, 2009. doi: 10.1113/jphysiol.2009.175364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pedarzani P, McCutcheon JE, Rogge G, Jensen BS, Christophersen P, Hougaard C, Strøbaek D, Stocker M. Specific enhancement of SK channel activity selectively potentiates the afterhyperpolarizing current I(AHP) and modulates the firing properties of hippocampal pyramidal neurons. J Biol Chem 280: 41404–41411, 2005. doi: 10.1074/jbc.M509610200. [DOI] [PubMed] [Google Scholar]
- 80.Wolfart J, Roeper J. Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci 22: 3404–3413, 2002. doi: 10.1523/JNEUROSCI.22-09-03404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marrelli SP, Eckmann MS, Hunte MS. Role of endothelial intermediate conductance KCa channels in cerebral EDHF-mediated dilations. Am J Physiol Heart Circ Physiol 285: H1590–H1599, 2003. doi: 10.1152/ajpheart.00376.2003. [DOI] [PubMed] [Google Scholar]
- 82.Cao YJ, Dreixler JC, Couey JJ, Houamed KM. Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur J Pharmacol 449: 47–54, 2002. doi: 10.1016/s0014-2999(02)01987-8. [DOI] [PubMed] [Google Scholar]
- 83.Xu X, Shen D, Gao Y, Zhou Q, Ni Y, Meng H, Shi H, Le W, Chen S, Chen S. A perspective on therapies for amyotrophic lateral sclerosis: can disease progression be curbed? Transl Neurodegener 10: 29, 2021. doi: 10.1186/s40035-021-00250-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Owens MJ, Nemeroff CB. Pharmacology of valproate. Psychopharmacol Bull 37: 17–24, 2003. [PubMed] [Google Scholar]