

Keywords: autosomal dominant polycystic kidney disease, kidney-gut axis, metabolomics, tryptophan metabolism
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited kidney disease and affects 1 in 1,000 individuals. There is accumulating evidence suggesting that there are shared cellular mechanisms responsible for cystogenesis in human and murine PKD and that reprogramming of metabolism is a key disease feature. In this study, we used a targeted metabolomics approach in an orthologous mouse model of PKD (Pkd1RC/RC) to investigate the metabolic modifications a cystic kidney undergoes during disease progression. Using the Kyoto Encyclopedia of Genes and Genomes pathway database, we identified several biologically relevant metabolic pathways that were altered early in this disease (in 3-mo-old Pkd1RC/RC mice), the most highly represented being arginine biosynthesis and metabolism and tryptophan and phenylalanine metabolism. During the next 6 mo of disease progression, multiple uremic solutes accumulated in the kidney of cystic mice, including several established markers of oxidative stress and endothelial dysfunction (allantoin, asymmetric dimethylarginine, homocysteine, malondialdehyde, methionine sulfoxide, and S-adenosylhomocysteine). Levels of kynurenines and polyamines were also augmented in kidneys of Pkd1RC/RC versus wild-type mice, as were the levels of bacteria-produced indoles, whose increase within PKD kidneys suggests microbial dysbiosis. In summary, we confirmed previously published and identified novel metabolic markers and pathways of PKD progression that may prove helpful for diagnosis and monitoring of cystic kidney disease in patients. Furthermore, they provide targets for novel therapeutic approaches that deserve further study and hint toward currently understudied pathomechanisms.
NEW & NOTEWORTHY This report delineates the evolution of metabolic changes occurring during autosomal dominant polycystic kidney disease (ADPKD) progression. Using an orthologous model, we performed kidney metabolomics and confirmed dysregulation of metabolic pathways previously found altered in nonorthologous or rapidly-progressive PKD models. Importantly, we identified novel alterations, including augmentation of kynurenines, polyamines, and indoles, suggesting increased inflammation and microbial dysbiosis that provide insights into PKD pathomechanisms and may prove helpful for diagnosing, monitoring, and treating ADPKD.
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inheritable kidney disease. The disease is characterized by an uncontrolled formation and growth of kidney cysts, resulting in a decline of kidney function (1–3). Ultimately, most patients with ADPKD reach kidney failure necessitating kidney replacement therapy (4). However, the age at which patients with ADPKD reach kidney failure shows large interindividual variability (5), even between family members that share the same mutation (6). Thus, it appears that mutations to ADPKD or modifying genes (germline or somatic), epigenetic mechanisms, and environmental factors have a considerable influence on the clinical course of the disease.
Over the past years, several in vitro and animal studies have shown that metabolic reprogramming might be a general feature of PKD (7). Glucose metabolism is defective in ADPKD, with cystic cells reprogrammed to favor aerobic glycolysis (i.e., Warburg effect) as is common in cancer cells (7–12). Higher glucose concentration increased kidney cyst growth (13), and hyperglycemia promoted cystogenesis and functional and structural kidney damage in a nonorthologous rodent model of PKD (14). Conversely, inhibition of glycolysis with 2-deoxyglucose, a glucose analog that is not metabolized by cells, reduced cell proliferation in human PKD cells and kidney cyst growth in various murine models (11, 12, 15).
In addition to glucose, amino acid metabolism is also altered in PKD. Both Pkd1 mutant cells and primary ADPKD cyst-lining cells, as opposed to normal cells, depend on glutamine for growth (16, 17). Furthermore, glutaminase-1 is upregulated in cyst-lining epithelia of human ADPKD kidneys and murine models, and its inhibition slowed cyst growth in the Pkhd1-Cre;Pkd1flx/flx model, although this therapeutic effect could not be confirmed using a collecting duct-specific Pkd1 knockout model (Aqp2-Cre;Pkd1flx/flx) (16, 17). In addition, kidney cyst growth has also been proposed to be arginine dependent since arginosuccinate synthase 1 (ASS1) expression is reduced in human and murine ADPKD and arginine depletion results in a dose-dependent increase of ASS1 and reduced cystogenesis ex vivo (18). Finally, elevated levels of methionine, S-adenosylmethionine (SAM), and methyltransferase-3 (Mettl3) were recently reported in PKD mouse models and human ADPKD kidney tissue (19). Correlatively, Mettl3 deletion in orthologous ADPKD mouse models slowed cyst growth, similar to dietary methionine restriction (19).
Furthermore, reduced fatty acid oxidation and dysregulated lipid metabolism have also been identified as a key PKD feature in different ADPKD models (20–22). Interestingly, treatment with the peroxisome proliferator-activated receptor-α (PPAR-α) agonist fenofibrate enhanced fatty acid oxidation and reduced cystic disease in an orthologous ADPKD model (22).
Finally, there are several lines of evidence to suggest that the gut microbiome is likely altered in patients with chronic kidney disease (CKD) and ADPKD (23, 24). Metabolomics can identify known gut-derived uremic toxins including tryptophan-derived indoxyl sulfate and indole acetic acid, p-cresyl sulfate (phenylalanine and tyrosine catabolism), trimethylamine-N-oxide (TMAO; metabolism of quaternary amines), and phenylacetylglutamine (phenylalanine fermentation). These uremic toxins are produced by bacteria, circulated within the body, absorbed, or further metabolized by tissues, and have been found to associate with cardiovascular diseases and progression of CKD (25, 26).
Although metabolic alterations in PKD are well described, and metabolic profiling has been carried out in nonorthologous PKD models (27, 28) and in the kidneys of a mouse model with kidney-specific Pkd1 loss (19, 20), information on the evolution of changes and their interconnections is still lacking. In this study, we aimed to verify established and discover new metabolic changes and interactions between metabolic pathways that a kidney undergoes during the progression of PKD. For this purpose, we used well-characterized orthologous Pkd1RC/RC mice carrying a homozygous hypomorphic Pkd1 gene mutation found in patients, Pkd1 p.R3277C (29–31). Pkd1RC/RC mice develop slowly progressing PKD until adulthood in the C57Bl/6J background as used for this study. Targeted mass spectrometry-based metabolic profiling was performed in kidney tissue of aging Pkd1RC/RC female and male mice and compared with age/strain-matched healthy wildtype (WT) controls.
MATERIALS AND METHODS
Animals
Pkd1RC/RC mice have a hypomorphic Pkd1 gene mutation orthologous to that of the human ADPKD disease variant PKD1 p.R3277C (Pkd1RC/RC) (29). Pkd1RC/RC mice in the C57Bl/6J background have mild but established cystic disease at 3 mo of age (30–32). Cyst expansion and size correlate with increased tubular cell proliferation (29). WT C57Bl/6J mice (Stock No. 000664, Jackson Laboratory) were purchased directly from the vendor at 3–4 wk of age. To minimize the impact of environmental factors on metabolism/microbiota, WT and Pkd1RC/RC mice were cohoused and aged together as well as received the same diet and care. Kidney samples undergoing metabolomics of all animals within the same age group (WT and Pkd1RC/RC mice) were obtained at the same time.
All experiments were conducted with adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The animal protocol was approved by the Animal Care and Use Committee of the University of Colorado, Anschutz Medical Campus. Mice were maintained on a standard diet (No. 2920, Envigo) under standard pathogen-free housing conditions, with food and water freely available.
Kidney Function Analyses
Blood urea nitrogen (BUN) was measured following the manufacturer’s protocol (QuantiChrom Urea Assay Kit, No. 501078333, BioAssay Systems, Hayward, CA). Each sample was measured in duplicate. Plasma creatinine concentration was analyzed with HPLC-MS/MS (Applied Biosystems 3200 Qtrap) as previously published (30).
Histomorphometric Analyses
The kidney cystic index, cyst size, and cyst number were analyzed using a custom-built NIS-Elements AR v4.6 macro (Nikon, Minato, Tokyo, Japan). Fibrotic area was analyzed from picrosirius red-stained kidney sections and visualized using an Olympus BX41 microscope (Olympus, Center Valley, PA) with a linear polarizer. Images were obtained and quantified as previously described (30).
Metabolomics
Sample analysis was performed based on a validated approach (33). Briefly, animals were euthanized by lethal isoflurane exposure and cervical dislocation. Kidneys were perfused with ice-cold PBS/heparin, dissected, and snap frozen in liquid nitrogen. Afterward, kidney tissue samples (∼50–100 mg) were homogenized in adequate volume of 80% (vol/vol) ice-cold methanol in water, incubated for protein precipitation, dried in a SpeedVac concentrator centrifuge (Savant, Thermo Fisher, Waltham, MA), reconstituted in water/methanol, and subjected to a modified semiquantitative targeted metabolomics assay and analysis.
LC-MS/MS analysis.
Eight microliters of sample were injected onto an Amide XBridge HPLC column (3.5 μm, 4.6-mm inner diameter × 100-mm length, Waters). The mobile phase consisted of HPLC buffer A [pH 9.0, 95% (vol/vol) water, 5% (vol/vol) acetonitrile, 20 mM ammonium hydroxide, and 20 mM ammonium acetate] and HPLC buffer B (100% acetonitrile). The HPLC settings were as follows: from 0 to 3 min, the mobile phase was kept at 85% solvent B, and from 3 to 22 min, the percentage of solvent B was decreased from 85% to 2% and was kept at 2% for an additional 3 min. At minute 26, solvent B was increased again back to 85%, and the column was flushed for an additional 7 min at 85% solvent B.
Data acquisition.
The Q1 (precursor ion) and Q3 (fragment ion) transitions, metabolite names, dwell times, and appropriate collision energies for both positive and negative ion modes were adapted from a study by Yuan et al. (33) with several specific transitions added by our group. Q1 and Q3 transitions were set to unit resolution for optimal metabolite ion isolation and selectivity. In addition, the polarity switching (settling) time was set to 50 ms; in 1.42 s using a 3-ms dwell time, we were able to obtain 6–14 scans per metabolite peak. The source temperature was set at 500°C, with curtain gas (nitrogen) at 20, collision gas (nitrogen) at high, ion source gases 1 and 2 at 33, declustering potential at +93/−93, entrance potential at +10/−10, and collision cell exit potential at +10/−10 for positive and negative ion modes, respectively. To minimize the batch-to-batch variability, all samples were analyzed within one batch. Once the data were acquired, MultiQuant software (v. 2.1) was used for data processing and relative quantitation (integration) of metabolites in kidney tissues.
Confirmation experiments.
Positive identification of the metabolites of interest was performed through injection of corresponding pure compound standards using the earlier-described LC-MS/MS platform [confirmation of the fragmentation pattern (MS/MS) and retention time].
Statistical Analyses
PKD phenotyping.
Data were analyzed using PRISM9 (GraphPad Software, La Jolla, CA) and are presented as means ± SD. Analyses were performed as one-way ANOVA with Tukey’s multiple comparison test. P values are denoted by *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 in the figures. The data presented in Supplemental Table S1 (all Supplemental Material is available at https://doi.org/10.6084/m9.figshare.14959659) were reanalyzed differentiating by sex from a previously published study (30).
Targeted metabolomics.
MetaboAnalyst 5.0 (University of Alberta) was used for statistical analysis of metabolomics data (34). Peak areas (analyte areas) were initially normalized to the deuterated internal standards followed by the summing of all integrals and correction for tissue weight. After that, the data were log transformed and then Paretto scaled (mean centered and divided by the square root of the SD of each variable). ANOVA with a post hoc Tukey honest significant difference test was used to compare group differences. Analysis of changes in metabolites between different animal groups was performed using partial least squares-discriminant analysis (PLS-DA). False discovery rate (FDR) correction was applied to correct for multiple comparisons (FDR < 0.05 for statistical significance). Pathway analysis was performed using the pathway analysis tool in MetaboAnalyst 5.0 (and the small molecule pathways database). This tool uses both pathway enrichment analysis through the R package GlobalTest based on compound concentration values as well as pathway topological analysis accounting for the impact of individual measured metabolites within the pathway. The goal of assessing pathway impact is to account for pathway structure and the intuitive concept that central or nodal positions in a pathway will have a greater impact than marginal or isolated positions. Total or maximal importance for each pathway is designated as 1, whereas the importance of measured metabolites to that pathway is designated as the cumulative percentage from matched metabolite nodes.
RESULTS
Metabolomic Profiling Shows Multiple Changes in Homozygous Pkd1 p.R3277C Mice
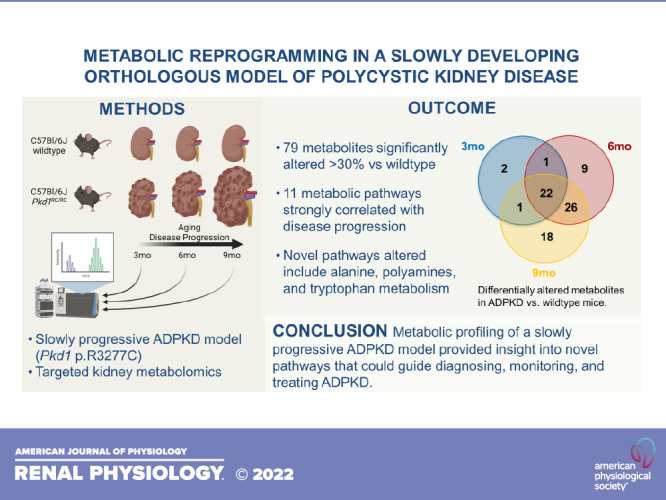
As previously shown, Pkd1RC/RC animals develop mild but progressive PKD. The increase in the percentage of kidney weight per body weight (%KW/BW) was shown to directly correlate with the progressive increase in cyst burden seen histologically at 3, 6, and 9 mo (29–31).
At 3 mo of age, the Pkd1RC/RC animals used in this study had significantly increased %KW/BW but unchanged BUN or fibrotic burden compared with strain-, age-, and sex-matched WT mice (Supplemental Table S1) (30). PKD progressed slowly from 3 to 9 mo of age, with the cystic index and cyst size or count increasing significantly from one study end point to the next (3 mo vs. 6 mo and 6 mo vs. 9 mo). At 9 mo age, Pkd1RC/RC females presented with significantly worse PKD compared with Pkd1RC/RC males, which in addition to increased %KW/BW and cystic index was marked by significantly increased fibrotic burden and kidney function decline (BUN and plasma creatinine; Supplemental Table S1) (30). Given these parameters, the chosen study end points (3, 6, and 9 mo) seemed well suited to study the progression of PKD metabolically.
Metabolic changes early in the disease.
Metabolically, at 3 mo, PLS-DA revealed a separation between WT and Pkd1RC/RC mouse kidneys (males and females combined; Fig. 1A), with 26 metabolites identified as significantly different between the two groups (FDR < 0.05; Supplemental Table S2). Well-known uremic and cardiovascular toxins, including trimethylamine oxide (TMAO), indole-3-acetic acid (IAA), asymmetric dimethylarginine (ADMA), 5-hydroxyindoleacetic acid (5-HIAA), hippurate, and allantoin, were significantly increased in Pkd1RC/RC versus WT mouse kidneys (Fig. 1B and Supplemental Table S2). The predominant metabolic pathways responsible for the separation between cystic and WT mice at 3 mo were spermine and spermidine biosynthesis, arginine, phenylalanine and tryptophan metabolism, and arginine and nitric oxide (NO) biosynthesis (Fig. 1B).
Figure 1.

Mildly cystic kidneys of 3-mo-old autosomal dominant polycystic kidney disease type 1 animals show dysregulated metabolism compared with wildtype (WT) controls. A: the plot depicts the separation of Pkd1RC/RC vs. WT male (M) and female (F) mouse metabolic profiles (n = 5/sex/group) at 3 mo of age using the first two components of the partial least square discriminant analysis. The peak areas (normalized to the internal standards and tissue weight) were log transformed and Paretto scaled (mean centered and divided by the square root of the SD of each variable). Colored areas (red and green) represent 95% confidence intervals of the respective groups. B: heat map of all metabolites that were significantly different between Pkd1RC/RC and WT mice at 3 mo. The scaled expression value of each feature (as group average) is plotted in red-blue color scale. The red color of the tile indicates higher abundance; the blue color of the tile indicates lower abundance. The corresponding pathways are presented in parentheses next to the metabolites. Data were analyzed using MetaboAnalyst 5.0 software. ADMA, asymmetric dimethylarginine; Arg, arginine; GSH, glutathione; Lys, lysine; Met, methionine; NO, nitric oxide; Orn, ornthine; Phe, phenylalanine; PKD, polycystic kidney disease; Pro, proline; PtdCho, phosphatidylcholine; PtdInsP3, phosphatidylinositol phosphate; TCA cycle, tricarboxylic acid cycle; TMAO, trimethylamine oxide; Trp, tryptophan; Tyr, tyrosine.
Metabolic changes at 6 and 9 mo.
At 6 mo, we identified 58 metabolites as significantly different between WT and Pkd1RC/RC mouse kidneys (Supplemental Table S2). Approximately half of the altered metabolites were the same as previously identified in 3-mo-old Pkd1RC/RC versus WT control mice, suggesting their relevance as potential markers for disease progression (Fig. 2A and Supplemental Table S2). Additional metabolites that became significant in Pkd1RC/RC mice at 6 mo and remained changed at 9 mo included homoserine, homocysteine, dimethylglycine and methionine sulfoxide (methionine and betaine metabolism), inosine, guanosine, xanthine, hypoxanthine and xanthosine (purine metabolism), and uridine, uracil, cytidine, and N-carbamoyl aspartate (pyrimidine metabolism).
Figure 2.

Metabolic dysregulation becomes more pronounced as polycystic kidney disease (PKD) severity progresses. A: Venn diagram depicting the number of kidney metabolites common/different between 3-, 6-, and 9-mo-old Pkd1RC/RC mice significantly altered by more than 30% compared with wildtype (WT) controls. B: partial least square discriminant analysis (PLS-DA) visualization of Pkd1RC/RC versus WT male (M) and female (F) mouse metabolic profiles (n = 5 in each group) based on the progression of the disease (3, 6, and 9 mo). C: PLS-DA visualization of Pkd1RC/RC male and female mouse metabolic profiles (n = 5 in each group) normalized to the group average of the corresponding WT controls (at 3, 6, and 9 mo, respectively). D: pathway analysis of metabolites that significantly changed with time in Pkd1RC/RC mice. For A, details on the individual metabolites are provided in Supplemental Table S2. For B and C, peak areas (analyte areas normalized to the internal standards and tissue weight) were log transformed and Paretto scaled (mean centered and divided by the square root of the SD of each variable). Colored areas represent 95% confidence intervals of the respective groups. For D, analysis was based on 77 unique metabolites from Supplemental Table S3 that were identified as significantly changed in Pkd1RC/RC male and female mice with time = 3, 6, and 9 mo (after two-way ANOVA, type I, between 3-, 6-, and 9-month-old males and female and normalized to the corresponding WT control value, respectively). The y-axis shows −log P values from pathway enrichment analysis. The x-axis shows pathway impact values from pathway topology analysis. The node color and radius are based on P values and pathway impact values, respectively. Data were analyzed using MetaboAnalyst 5.0 software; pathway analysis used the Mus musculus small molecule pathways database. Ala, alanine; Glc, glucose; Met, methionine; PtdCho, phosphatidylcholine.
Interestingly, the increase in age from 6 to 9 mo did not lead to extensive changes in the kidney metabolic profiles of Pkd1RC/RC mice (Fig. 2, A and B). We identified 67 metabolites as significantly changed between WT and cystic animals at 9 mo (Fig. 2A and Supplemental Table S2), with only 18 of them being uniquely different in 9-mo-old Pkd1RC/RC mice versus 3 or 6 mo. Of note, when normalized to their respective WT controls (average of n = 5 at 3, 6, or 9 mo in males and female, respectively), male 9-mo-old Pkd1RC/RC mice diverged the most from the rest of the groups (Fig. 2C). The top 10 metabolites responsible for this separation (based on their variable of importance projection score) were quinolinic acid, shikimate, cysteine, hippurate, methyl nicotinate, methionine sulfoxide, methyl-5-thioadenosine, xanthurenic acid, hydroxyphenylacetic acid, and allenoate, all of which were higher in 9-mo-old Pkd1RC/RC males compared with the rest of the groups, including 9-mo-old Pkd1RC/RC females.
Disease progression.
Two-way ANOVA of WT-normalized data revealed that 77 metabolites were significantly changed as a function of time (in Pkd1RC/RC mice at 3, 6, and 9 mo, FDR < 0.05; Supplemental Table S3). The pathways most affected by the progression of PKD were (from highest to lowest ranking pathway impact) alanine metabolism, spermine and spermidine biosynthesis, phosphatidylcholine biosynthesis, urea cycle, methionine metabolism, and glycolysis as well as gluconeogenesis (Fig. 2D and Supplemental Table S3). To investigate the relationship between altered metabolites and disease progression even further, we performed stepwise linear regression of normalized metabolite areas (as detected in kidneys of 3-, 6-, and 9-mo-old male and female mice and normalized to the group average of the corresponding WT metabolite areas, respectively) to %KW/BW. Thirty-two metabolites were identified to correlate with %KW/BW, a marker commonly used for disease severity/progression (Supplemental Table S4). Of these, 20 metabolites were common to both our “disease progression” analyses; the most highly associated being metabolites of arginine biosynthesis as well as arginine, tryptophan, and phenylalanine metabolism.
As could be expected based on what is known about PKD pathomechanisms, known markers of oxidative stress including malondialdehyde and methionine sulfoxide (Fig. 3A and Supplemental Table S3) as well as uremic toxins allantoin, hippurate, and homocysteine increased with the progression of the disease (data normalized to WT controls, from 3 to 9 mo in Pkd1RC/RC mice; Fig. 3B and Supplemental Table S3). Interestingly, uremic toxins TMAO and betaine aldehyde only increased in male but not in female Pkd1RC/RC mouse kidneys over time (Fig. 3B).
Figure 3.

Uremic toxins and oxidative stress markers change significantly with time in the kidneys of Pkd1RC/RC male and female mice. A: oxidative stress markers. B′–B′′′: uremic toxins. C: arginine metabolites. Two-way repeated-measures ANOVA with a false discovery rate of <0.05 was used for statistical analysis (variables of time: 3, 6, and 9 mo and group: male and female). Data are presented as box and whisker plots. Peak normalized areas (analyte areas normalized to the internal standards and tissue weight) were log transformed and Paretto scaled (mean centered and divided by the square root of the SD of each variable). The lines in the boxes represent the median (50th percentile), the boxes represent the 25th and 75th percentile (lower and upper quartiles), and the whiskers represent the minimum and maximum values. Data were analyzed using MetaboAnalyst 5.0 software. TMAO, trimethylamine oxide.
Pathway spotlight.
From the measured glycolysis intermediates, we did not observe a steady change (over at least two subsequent time points) in any of them (Supplemental Table S2). Aconitate was the only tricarboxylic acid (TCA) cycle metabolite that was higher in Pkd1RC/RC mouse kidneys at all time points compared with WT controls, whereas citrate and oxaloacetate were higher at only one time point (at 9 and 6 mo, respectively; Supplemental Table S2), similar to glutamate, which was lower in the kidneys of 9-mo-old Pkd1RC/RC mice compared with WT mice (Supplemental Table S2).
Within arginine metabolism, citrulline levels remained elevated during all evaluated stages of PKD progression, and so were the levels of ADMA, which serves as a negative regulator of the arginine to citrulline converting enzyme NO synthase (Supplemental Table S2). At 9 mo of age, the kidneys of Pkd1RC/RC mice presented with significantly lower arginosuccinate compared with WT kidneys, suggesting a reduction of ASS1 activity (Supplemental Table S2). Ornithine was elevated at 3 and 6 mo in Pkd1RC/RC kidneys. At 9 mo, its level declined due to increased decarboxylation processes (via ornithine decarboxylase) leading to an accumulation of putrescine and spermidine and a reduction of its conversion to glutamate (Fig. 3C and Supplemental Table S2).
Kynurenine, kynurenic acid, quinolinic acid, and xanthurenic acid, all part of the tryptophan to kynurenine metabolism pathway, were not only higher in male and female Pkd1RC/RC mouse kidneys compared with their corresponding WT controls (Fig. 4, A and B, and Supplemental Table S2) but also increased over time in Pkd1RC/RC mice and correlated with %KW/BW (from 3 to 9 mo; Fig. 4C and Supplemental Tables S3 and S4). Nicotinamide, on the other hand, declined over time in both female and male Pkd1RC/RC mice, but the decline only reached significance in females (Fig. 4, A and C). Within the tryptophan/indole metabolism pathway, IAA and 5-HIAA were both increased (in Pkd1RC/RC mice compared with WT controls as well during the progression of PKD from 3 to 9 mo; Fig. 4, B and C, and Supplemental Tables S2–S4).
Figure 4.

Tryptophan metabolism is dysregulated in Pkd1RC/RC mice. A: diagram of the tryptophan-nicotinamide pathway. Arrows represent a significant increase or decrease of a specific metabolite in both sexes of Pkd1RC/RC mice (dark arrows) or in females only (light arrows). Significance levels were determined using ANOVA with a post hoc Tukey honest significant difference analysis (P < 0.05). For the sake of clarity, not all reactants and products are shown. B: metabolites from the tryptophan-nicotinamide pathways identified as significantly changed in the kidneys of 3-mo-old Pkd1RC/RC versus wildtype (WT) mice (ANOVA with Tukey’s honest significant difference post hoc analysis, false discovery rate < 0.05, n = 5). M and F, male and female, respectively. C: changes in tryptophan metabolism intermediates within the kynurenine and serotonin pathway with polycystic kidney disease progression in Pkd1RC/RC mice. Two-way repeated-measures ANOVA with a false discovery rate of <0.05 was used for statistical analysis (variables of time: 3, 6, and 9 mo and group: male and female). Data are presented as box and whisker plots. Peak normalized areas (analyte areas normalized to the internal standards and tissue weight) were log transformed and Paretto scaled (mean centered and divided by the square root of the SD of each variable). The lines in the boxes represent the median (50th percentile), the boxes represent the 25th and 75th percentile (lower and upper quartiles), and the whiskers represent the minimum and maximum values. For B, the y-axis shows log-transformed and Paretto-scaled normalized peak areas; for C, the normalized peak areas quantified in the kidneys of Pkd1RC/RC mice were further normalized to the corresponding WT group average normalized peak areas, followed by log transformation and Paretto scaling. 5-HT, 5-hydroxytryptophan; 5-HIAA, 5-hydroxyindoleacetic acid; DDC, dopa decarboxylase; IAA, indoleacetic acid; IDO, indoleamine 2,3-dioxygenase; KATs, kynurenine amino transferases; KMO, kynurenine 3-monooxygenase; KYNU, kynureninase; MAOA/B, monoamine oxidase A/B; QPRT, quinolinate phosphoribosyltransferase; TDO, tryptophan 2,3-dioxygenase; TPH, tryptophan hydroxylase. Data were analyzed using MetaboAnalyst 5.0 software.
DISCUSSION
Metabolic reprogramming describes the rewiring of intracellular metabolic pathways occurring in response to specific needs of the cell in physiological or pathological conditions. In ADPKD, a broad reprogramming of metabolic pathways has been described with most of these pathways being involved in central carbon metabolism.
Previous studies have shown that murine and human Pkd1/PKD1 mutant kidneys exhibit a metabolic shift toward glycolysis with cells producing more lactate from glucose accompanied by an increased expression of glycolytic enzymes (8, 11, 12). Overall, cystic kidney cells divert glucose into glycolysis and the pentose phosphate pathway and compensate with glutamine usage (16, 17). This substrate redistribution is used to generate citrate for fatty acid synthesis and lipid production (8, 35). The increase in de novo synthesis of fatty acids is accompanied by a decline in fatty acid oxidation and increased acetylcarnitine urinary excretion (8, 20, 21, 36).
Our study supported only some of the observed redistributions in glucose consumption via TCA cycle and glutamine/glutamate metabolism. Several of the pathway intermediates were only significantly changed at one or two of the investigated time points (e.g., higher citrate and lower glutamate levels in 9-mo-old Pkd1RC/RC vs. WT animals) but failed to remain significantly different throughout all three time points of PKD progression. Aconitate was the only metabolite increased in the kidneys of all Pkd1RC/RC animals versus their corresponding WT controls.
The arginine biosynthesis pathway was differentially regulated in kidney tissues of 3-mo-old Pkd1RC/RC mice, where increased arginase-mediated arginine metabolism resulted in elevated citrulline and ornithine concentrations. Between 3 and 6 mo of age, higher arginase activity and increased kidney levels of citrulline and ornithine remained present in Pkd1RC/RC mice; at 9 mo, however, decarboxylation of ornithine strongly increased (via ornithine decarboxylase) leading to declining ornithine levels and a more than threefold rise in the production of the polyamines putrescine and spermidine. Polyamines are involved in many cellular processes, including cell proliferation and the immune response (37, 38). Polyamine levels are often elevated in proliferative diseases including cancer (39) and acute kidney injury and CKD (40, 41). At the same time, arginosuccinate strongly declined, suggesting the previously described reduction in ASS1 activity (18).
The dysregulation of the tryptophan/kynurenine metabolism pathway (Fig. 4) is a novel finding in an animal model of PKD but confirms our results obtained in children and young adults with ADPKD (42). Although kynurenine, kynurenic acid, and xanthurenic and quinolinic acids were all higher in the kidneys of 3-mo-old Pkd1RC/RC versus WT mice and continued to rise with disease progression, the production of the pathway’s end product, nicotinamide, was impaired, providing an indication of why supplementation with niacin has been shown to alleviate the disease (43). Levels of kynurenine are elevated in patients with kidney diseases (44–46). Because kynurenines play an important role in the regulation of immunity and have been implicated in comorbid atherosclerosis and neuropsychiatric symptoms, patients with CKD are at particularly high risk of kynurenine-associated pathophysiologies (47, 48). Furthermore, kynurenine, kynurenic acid, and quinolinic acid may promote atherosclerosis in patients with kidney failure by activating oxidative stress in endothelial and vascular smooth muscle cells (49).
Regarding indoles, on the serotonin-producing side of tryptophan metabolism (Fig. 4), kidney tissue concentrations of the indole derivatives 5-HIAA and IAA were increased in cystic versus control mice and continued to increase with time. Both are uremic solutes (50, 51) and have been shown to contribute to activation of the aryl hydrocarbon receptor (AhR) (52). The aryl hydrocarbon receptor, which can also be induced by polyamines, has been shown to aggravate kidney damage (53–55) by increasing inflammation, oxidative stress, and atherosclerosis (56, 57).
In addition to the immunomodulating tryptophan metabolites (kynurenines, indoles, and polyamines), kidney levels of the microbiota-derived branched-chain ketoacid ketoisovalerate declined with the progression of the disease (from ages of 3 to 9 mo).
Consistent with a recent publication (19), we found dysregulation of the methionine/homocysteine pathway at later stages of PKD, with significantly elevated levels of methionine or its oxidized form methionine sulfoxide, SAH, SAM, and homocysteine in 6- and 9-mo-old Pkd1RC/RC versus age-matched WT mice. The rise of these metabolites at ages when kidney function declines in our PKD animal model is in line with impaired kidney function resulting in reduced elimination of these metabolites from the kidney. Furthermore, the uremic milieu in kidneys with CKD has been proposed to inhibit crucial metabolizing enzymes, further accentuating the accumulation of these metabolites (58). Whether accumulation of these metabolites directly fuels cyst growth needs further exploration, but recent data suggest that methionine/SAM levels drive cyst proliferation in an ex vivo model of PKD (19). Furthermore, levels of betaine and choline were significantly reduced in 9-mo-old Pkd1RC/RC versus age-matched WT mice. Both of these metabolites feed into the methionine/homocysteine pathway, which can be regulated by dietary intake. Interestingly, a low-methionine diet has been shown to slow PKD progression in an orthologous model of ADPKD (19).
The study presents with some limitation. First, the WT animals whose kidneys were analyzed were not littermates of the ADPKD mice, nor were they born in the same facility as the ADPKD mice. However, the strain-matched WT mice were obtained at a very young age (3–4 wk) and cohoused with the Pkd1RC/RC mice for the duration of the study, receiving an identical diet and care to minimize any effect environment may have on metabolism/microbiota. Second, acknowledged challenges for metabolomic approaches include analyses and interpretation of large and complex data sets and an inability of the technology to separate structural isomers. Analytic issues include the fact that metabolomic data violate the assumption of independence assumed by the general linear model. Finally, the present data are limited to whole kidney tissue extracts and therefore do not provide information on metabolites within the cellular and subcellular fractions.
Perspectives and Significance
We identified several abnormal metabolic pathways in a slowly progressive orthologous mouse model of ADPKD. We confirmed changes in TCA cycle, but not glycolysis, and arginine synthesis and metabolites that have been shown to contribute to the development of PKD. Furthermore, we identified significant changes in in-part microbiota-derived/regulated tryptophan metabolism via kynurenine and indole pathways. The coordinated alterations within the interconnected metabolic pathways offer a unique opportunity for therapeutic targeting at multiple entry points to reduce the substrate supply in cystic epithelial cells and thus reduce their proliferation and survival.
SUPPLEMENTAL DATA
Supplemental Tables S1–S4: https://doi.org/10.6084/m9.figshare.14959659.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants K01DK114164 (to K. Hopp), R01DK114424 (to J. Klawitter), R01DK114424-03S1 (to J. Klawitter), and T325T32DK007135 (to E. K. Kleczko) and by the Zell Family Foundation (to K. Hopp).
DISCLOSURES
K.H. receives royalties for industry use of the Pkd1RC/RC mouse model in concordance with Mayo Clinic Ventures regulations (Mayo Technology Case No. 2012-144). None of the other authors have any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
K.H. and J.K. conceived and designed research; K.H. and E.K.K. performed experiments; K.H. and J.K. analyzed data; K.H. and J.K. interpreted results of experiments; K.H. and J.K. prepared figures; K.H. and J.K. drafted manuscript; K.H., E.K.K., B.Y.G., M.C., J.K., U.C., and J.K. edited and revised manuscript; K.H., E.K.K., B.Y.G., M.C., J.K., U.C., and J.K. approved final version of manuscript.
REFERENCES
- 1.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 369: 1287–1301, 2007. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 2.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med 60: 321–337, 2009. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest 124: 2315–2324, 2014. doi: 10.1172/JCI72272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gabow PA, Johnson AM, Kaehny WD, Kimberling WJ, Lezotte DC, Duley IT, Jones RH. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 41: 1311–1319, 1992. doi: 10.1038/ki.1992.195. [DOI] [PubMed] [Google Scholar]
- 5.Grantham JJ, Torres VE, Chapman AB, Guay-Woodford LM, Bae KT, King BF Jr, Wetzel LH, Baumgarten DA, Kenney PJ, Harris PC, Klahr S, Bennett WM, Hirschman GN, Meyers CM, Zhang X, Zhu F, Miller JP, Investigators C. Volume progression in polycystic kidney disease. N Engl J Med 354: 2122–2130, 2006. doi: 10.1056/NEJMoa054341. [DOI] [PubMed] [Google Scholar]
- 6.Cornec-Le Gall E, Audrezet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau E, Jousset P, Guillodo MP, Grall-Jezequel A, Saliou P, Ferec C, Le Meur Y. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol 24: 1006–1013, 2013. doi: 10.1681/ASN.2012070650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nowak KL, Hopp K. Metabolic reprogramming in autosomal dominant polycystic kidney disease: evidence and therapeutic potential. Clin J Am Soc Nephrol 15: 577–584, 2020. doi: 10.2215/CJN.13291019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Podrini C, Rowe I, Pagliarini R, Costa ASH, Chiaravalli M, Di Meo I, Kim H, Distefano G, Tiranti V, Qian F, di Bernardo D, Frezza C, Boletta A. Dissection of metabolic reprogramming in polycystic kidney disease reveals coordinated rewiring of bioenergetic pathways. Commun Biol 1: 194, 2018. doi: 10.1038/s42003-018-0200-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Priolo C, Henske EP. Metabolic reprogramming in polycystic kidney disease. Nat Med 19: 407–409, 2013. doi: 10.1038/nm.3140. [DOI] [PubMed] [Google Scholar]
- 10.Seeger-Nukpezah T, Geynisman DM, Nikonova AS, Benzing T, Golemis EA. The hallmarks of cancer: relevance to the pathogenesis of polycystic kidney disease. Nat Rev Nephrol 11: 515–534, 2015. doi: 10.1038/nrneph.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, Song XW, Xu H, Mari S, Qian F, Pei Y, Musco G, Boletta A. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med 19: 488–493, 2013. doi: 10.1038/nm.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riwanto M, Kapoor S, Rodriguez D, Edenhofer I, Segerer S, Wuthrich RP. Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PLoS One 11: e0146654, 2016. doi: 10.1371/journal.pone.0146654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kraus A, Schley G, Kunzelmann K, Schreiber R, Peters DJ, Stadler R, Eckardt KU, Buchholz B. Glucose promotes secretion-dependent renal cyst growth. J Mol Med (Berl) 94: 107–117, 2016. doi: 10.1007/s00109-015-1337-4. [DOI] [PubMed] [Google Scholar]
- 14.Sas KM, Yin H, Fitzgibbon WR, Baicu CF, Zile MR, Steele SL, Amria M, Saigusa T, Funk J, Bunni MA, Siegal GP, Siroky BJ, Bissler JJ, Bell PD. Hyperglycemia in the absence of cilia accelerates cystogenesis and induces renal damage. Am J Physiol Renal Physiol 309: F79–F87, 2015. doi: 10.1152/ajprenal.00652.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiaravalli M, Rowe I, Mannella V, Quilici G, Canu T, Bianchi V, Gurgone A, Antunes S, D'Adamo P, Esposito A, Musco G, Boletta A. 2-Deoxy-d-glucose ameliorates PKD progression. J Am Soc Nephrol 27: 1958–1969, 2016. doi: 10.1681/ASN.2015030231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soomro I, Sun Y, Li Z, Diggs L, Hatzivassiliou G, Thomas AG, Rais R, Parker SJ, Slusher BS, Kimmelman AC, Somlo S, Skolnik EY. Glutamine metabolism via glutaminase 1 in autosomal-dominant polycystic kidney disease. Nephrol Dial Transplant 35: 1824, 2020. doi: 10.1093/ndt/gfz109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flowers EM, Sudderth J, Zacharias L, Mernaugh G, Zent R, DeBerardinis RJ, Carroll TJ. Lkb1 deficiency confers glutamine dependency in polycystic kidney disease. Nat Commun 9: 814, 2018. doi: 10.1038/s41467-018-03036-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trott JF, Hwang VJ, Ishimaru T, Chmiel KJ, Zhou JX, Shim K, Stewart BJ, Mahjoub MR, Jen KY, Barupal DK, Li X, Weiss RH. Arginine reprogramming in ADPKD results in arginine-dependent cystogenesis. Am J Physiol Renal Physiol 315: F1855–F1868, 2018. doi: 10.1152/ajprenal.00025.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramalingam H, Kashyap S, Cobo-Stark P, Flaten A, Chang CM, Hajarnis S, Hein Kz Lika J, Warner GM, Espindola-Netto JM, Kumar A, Kanchwala M, Xing C, Chini EN, Patel V. A methionine-Mettl3-N(6)-methyladenosine axis promotes polycystic kidney disease. Cell Metab 33: 1234–1247.e7, 2021. doi: 10.1016/j.cmet.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menezes LF, Lin CC, Zhou F, Germino GG. Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine 5: 183–192, 2016. doi: 10.1016/j.ebiom.2016.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menezes LF, Zhou F, Patterson AD, Piontek KB, Krausz KW, Gonzalez FJ, Germino GG. Network analysis of a Pkd1-mouse model of autosomal dominant polycystic kidney disease identifies HNF4alpha as a disease modifier. PLoS Genet 8: e1003053, 2012. doi: 10.1371/journal.pgen.1003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lakhia R, Yheskel M, Flaten A, Quittner-Strom EB, Holland WL, Patel V. PPARalpha agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am J Physiol Renal Physiol 314: F122–F131, 2018. doi: 10.1152/ajprenal.00352.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaziri ND, Wong J, Pahl M, Piceno YM, Yuan J, DeSantis TZ, Ni Z, Nguyen TH, Andersen GL. Chronic kidney disease alters intestinal microbial flora. Kidney Int 83: 308–315, 2013. doi: 10.1038/ki.2012.345. [DOI] [PubMed] [Google Scholar]
- 24.Yacoub R, Nadkarni GN, McSkimming DI, Chaves LD, Abyad S, Bryniarski MA, Honan AM, Thomas SA, Gowda M, He JC, Uribarri J. Fecal microbiota analysis of polycystic kidney disease patients according to renal function: a pilot study. Exp Biol Med (Maywood) 244: 505–513, 2019. doi: 10.1177/1535370218818175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanholder R, Glorieux G. Gut-derived metabolites and chronic kidney disease: the forest (f)or the trees? Clin J Am Soc Nephrol 13: 1311–1313, 2018. doi: 10.2215/CJN.08200718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wikoff WR, Nagle MA, Kouznetsova VL, Tsigelny IF, Nigam SK. Untargeted metabolomics identifies enterobiome metabolites and putative uremic toxins as substrates of organic anion transporter 1 (Oat1). J Proteome Res 10: 2842–2851, 2011. doi: 10.1021/pr200093w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor SL, Ganti S, Bukanov NO, Chapman A, Fiehn O, Osier M, Kim K, Weiss RH. A metabolomics approach using juvenile cystic mice to identify urinary biomarkers and altered pathways in polycystic kidney disease. Am J Physiol Renal Physiol 298: F909–F922, 2010. doi: 10.1152/ajprenal.00722.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Natoli TA, Modur V, Ibraghimov-Beskrovnaya O. Glycosphingolipid metabolism and polycystic kidney disease. Cell Signal 69: 109526, 2020. 109526 doi: 10.1016/j.cellsig.2020.109526. [DOI] [PubMed] [Google Scholar]
- 29.Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, Rossetti S, Torres VE, Harris PC. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest 122: 4257–4273, 2012. doi: 10.1172/JCI64313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleczko EK, Marsh KH, Tyler LC, Furgeson SB, Bullock BL, Altmann CJ, Miyazaki M, Gitomer BY, Harris PC, Weiser-Evans MCM, Chonchol MB, Clambey ET, Nemenoff RA, Hopp K. CD8(+) T cells modulate autosomal dominant polycystic kidney disease progression. Kidney Int 94: 1127–1140, 2018. doi: 10.1016/j.kint.2018.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arroyo J, Escobar-Zarate D, Wells HH, Constans MM, Thao K, Smith JM, Sieben Cj, Martell MR, Kline TL, Irazabal MV, Torres VE, Hopp K, Harris PC. The genetic background significantly impacts the severity of kidney cystic disease in the Pkd1(RC/RC) mouse model of autosomal dominant polycystic kidney disease. Kidney Int 99: 1392–1407, 2021. doi: 10.1016/j.kint.2021.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hopp K, Hommerding CJ, Wang X, Ye H, Harris PC, Torres VE. Tolvaptan plus pasireotide shows enhanced efficacy in a PKD1 model. J Am Soc Nephrol 26: 39–47, 2015. doi: 10.1681/ASN.2013121312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 7: 872–881, 2012. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, Xia J. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46: W486–W494, 2018. doi: 10.1093/nar/gky310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci 39: 347–354, 2014. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hajarnis S, Lakhia R, Yheskel M, Williams D, Sorourian M, Liu X, Aboudehen K, Zhang S, Kersjes K, Galasso R, Li J, Kaimal V, Lockton S, Davis S, Flaten A, Johnson JA, Holland WL, Kusminski CM, Scherer PE, Harris PC, Trudel M, Wallace DP, Igarashi P, Lee EC, Androsavich JR, Patel V. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun 8: 14395, 2017. doi: 10.1038/ncomms14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Latour YL, Gobert AP, Wilson KT. The role of polyamines in the regulation of macrophage polarization and function. Amino Acids 52: 151–160, 2020. doi: 10.1007/s00726-019-02719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Proietti E, Rossini S, Grohmann U, Mondanelli G. Polyamines and kynurenines at the intersection of immune modulation. Trends Immunol 41: 1037–1050, 2020. doi: 10.1016/j.it.2020.09.007. [DOI] [PubMed] [Google Scholar]
- 39.Gerner EW, Meyskens FL Jr.. Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer 4: 781–792, 2004. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 40.Zahedi K, Barone S, Soleimani M. Polyamine catabolism in acute kidney injury. Int J Mol Sci 20: 4790, 2019. doi: 10.3390/ijms20194790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng YL, Cao G, Chen DQ, Vaziri ND, Chen L, Zhang J, Wang M, Guo Y, Zhao YY. Microbiome-metabolomics reveals gut microbiota associated with glycine-conjugated metabolites and polyamine metabolism in chronic kidney disease. Cell Mol Life Sci 76: 4961–4978, 2019. doi: 10.1007/s00018-019-03155-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baliga MM, Klawitter J, Christians U, Hopp K, Chonchol M, Gitomer BY, Cadnapaphornchai MA, Klawitter J. Metabolic profiling in children and young adults with autosomal dominant polycystic kidney disease. Sci Rep 11: 6629, 2021. doi: 10.1038/s41598-021-84609-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou X, Fan LX, Sweeney WE Jr, Denu JM, Avner ED, Li X. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J Clin Invest 123: 3084–3098, 2013. doi: 10.1172/JCI64401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sekula P, Goek ON, Quaye L, Barrios C, Levey AS, Romisch-Margl W, Menni C, Yet I, Gieger C, Inker LA, Adamski J, Gronwald W, Illig T, Dettmer K, Krumsiek J, Oefner PJ, Valdes AM, Meisinger C, Coresh J, Spector TD, Mohney RP, Suhre K, Kastenmuller G, Kottgen A. A metabolome-wide association study of kidney function and disease in the general population. JASN 27: 1175–1188, 2016. doi: 10.1681/ASN.2014111099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schefold JC, Zeden JP, Fotopoulou CV, Haehling S, Pschowski R, Hasper D, Volk HD, Schuett C, Reinke P. Increased indoleamine 2,3-dioxygenase (IDO) activity and elevated serum levels of tryptophan catabolites in patients with chronic kidney disease: a possible link between chronic inflammation and uraemic symptoms. Nephrol Dial Transplant 24: 1901–1908, 2009. doi: 10.1093/ndt/gfn739. [DOI] [PubMed] [Google Scholar]
- 46.Debnath S, Velagapudi C, Redus L, Thameem F, Kasinath B, Hura CE, Lorenzo C, Abboud HE, O'Connor JC. Tryptophan metabolism in patients with chronic kidney disease secondary to type 2 diabetes: relationship to inflammatory markers. Int J Tryptophan Res 10: 117864691769460, 2017. doi: 10.1177/1178646917694600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Etienne-Mesmin L, Chassaing B, Gewirtz AT. Tryptophan: A gut microbiota-derived metabolites regulating inflammation. World J Gastrointest Pharmacol Ther 8: 7–9, 2017. doi: 10.4292/wjgpt.v8.i1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moffett JR, Namboodiri MA. Tryptophan and the immune response. Immunol Cell Biol 81: 247–265, 2003. doi: 10.1046/j.1440-1711.2003.t01-1-01177.x. [DOI] [PubMed] [Google Scholar]
- 49.Pawlak K, Brzosko S, Mysliwiec M, Pawlak D. Kynurenine, quinolinic acid—the new factors linked to carotid atherosclerosis in patients with end-stage renal disease. Atherosclerosis 204: 561–566, 2009. doi: 10.1016/j.atherosclerosis.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 50.Duranton F, Cohen G, De Smet R, Rodriguez M, Jankowski J, Vanholder R, Argiles A, European Uremic Toxin Work G, European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J Am Soc Nephrol 23: 1258–1270, 2012. [Erratum in J Am Soc Nephrol 24: 2127–2129, 2013]. doi: 10.1681/ASN.2011121175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jourde-Chiche N, Dou L, Cerini C, Dignat-George F, Brunet P. Vascular incompetence in dialysis patients–protein-bound uremic toxins and endothelial dysfunction. Semin Dial 24: 327–337, 2011. doi: 10.1111/j.1525-139X.2011.00925.x. [DOI] [PubMed] [Google Scholar]
- 52.Gondouin B, Cerini C, Dou L, Sallee M, Duval-Sabatier A, Pletinck A, Calaf R, Lacroix R, Jourde-Chiche N, Poitevin S, Arnaud L, Vanholder R, Brunet P, Dignat-George F, Burtey S. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int 84: 733–744, 2013. doi: 10.1038/ki.2013.133. [DOI] [PubMed] [Google Scholar]
- 53.Lu H, Lei X, Klaassen C. Gender differences in renal nuclear receptors and aryl hydrocarbon receptor in 5/6 nephrectomized rats. Kidney Int 70: 1920–1928, 2006. doi: 10.1038/sj.ki.5001880. [DOI] [PubMed] [Google Scholar]
- 54.Dou L, Poitevin S, Sallee M, Addi T, Gondouin B, McKay N, Denison MS, Jourde-Chiche N, Duval-Sabatier A, Cerini C, Brunet P, Dignat-George F, Burtey S. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int 93: 986–999, 2018. doi: 10.1016/j.kint.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 55.Kim JT, Kim SH, Min HK, Jeon SJ, Sung SA, Park WH, Lee HK, Choi HS, Pak YK, Lee SY. Effect of dialysis on aryl hydrocarbon receptor transactivating activity in patients with chronic kidney disease. Yonsei Med J 61: 56–63, 2020. doi: 10.3349/ymj.2020.61.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kopf PG, Walker MK. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases reactive oxygen species production in human endothelial cells via induction of cytochrome P4501A1. Toxicol Appl Pharmacol 245: 91–99, 2010. doi: 10.1016/j.taap.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dou L, Sallee M, Cerini C, Poitevin S, Gondouin B, Jourde-Chiche N, Fallague K, Brunet P, Calaf R, Dussol B, Mallet B, Dignat-George F, Burtey S. The cardiovascular effect of the uremic solute indole-3 acetic acid. J Am Soc Nephrol 26: 876–887, 2015. doi: 10.1681/ASN.2013121283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Friedman AN, Bostom AG, Selhub J, Levey AS, Rosenberg IH. The kidney and homocysteine metabolism. J Am Soc Nephrol 12: 2181–2189, 2001. doi: 10.1681/ASN.V12102181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables S1–S4: https://doi.org/10.6084/m9.figshare.14959659.