

Abstract
Suppressing mineralocorticoid receptor (MR) activity with MR antagonists is therapeutic for chronic skeletal muscle pathology in Duchenne muscular dystrophy (DMD) mouse models. Although mechanisms underlying clinical MR antagonist efficacy for DMD cardiomyopathy and other cardiac diseases are defined, mechanisms in skeletal muscles are not fully elucidated. Myofiber MR knockout improves skeletal muscle force and a subset of dystrophic pathology. However, MR signaling in myeloid cells is known to be a major contributor to cardiac efficacy. To define contributions of myeloid MR in skeletal muscle function and disease, we performed parallel assessments of muscle pathology, cytokine levels, and myeloid cell populations resulting from myeloid MR genetic knockout in muscular dystrophy and acute muscle injury. Myeloid MR knockout led to lower levels of C-C motif chemokine receptor 2 (CCR2)-expressing macrophages, resulting in sustained myofiber damage after acute injury of normal muscle. In acute injury, myeloid MR knockout also led to increased local muscle levels of the enzyme that produces the endogenous MR agonist aldosterone, further supporting important contributions of MR signaling in normal muscle repair. In muscular dystrophy, myeloid MR knockout altered cytokine levels differentially between quadriceps and diaphragm muscles, which contain different myeloid populations. Myeloid MR knockout led to higher levels of fibrosis in dystrophic diaphragm. These results support important contributions of myeloid MR signaling to skeletal muscle repair in acute and chronic injuries and highlight the useful information gained from cell-specific genetic knockouts to delineate mechanisms of pharmacological efficacy.
Keywords: dystrophy, inflammation, muscle, myeloid, transgenic
INTRODUCTION
Chronic myofiber damage and inflammation diminish the regenerative potential of skeletal muscle in patients with Duchenne muscular dystrophy (DMD), resulting in fibrosis and complete loss of ambulation in the teens (1–3). Advances in the development of micro-dystrophin gene therapies that effectively transduce skeletal and cardiac muscle to functionally replace the missing dystrophin protein have yielded promising preclinical and early clinical results (4–6). Despite the success, novel immunomodulatory and antifibrotic drugs will likely be necessary as adjunct medications to optimize patient outcomes because of low-grade persistent muscle inflammation (7).
The current standard of care for DMD patients is long-term treatment with prednisone, a commonly prescribed corticosteroid with severe side effects, which moderately increases skeletal muscle strength and delays ventilator usage (8–13). Intermittent treatment with prednisone is found to be as effective as daily dosing, which helps dampen adverse side effects (14–16). However, many of the side effects persist, necessitating the development of different therapies.
Mineralocorticoid receptor (MR) antagonists are safe, regularly prescribed medications that exhibit therapeutic properties in multiple diseases, including for delaying cardiomyopathy and heart failure in dystrophic mice and DMD patients (17–19). Additionally, MR antagonists prevent myofiber damage and fibrosis in the limb and respiratory (diaphragm) muscles of dystrophic mice, but the complete mechanism of action for improvements in skeletal muscle is unknown (18). Prednisolone treatment has been found to dampen MR antagonist benefits in dystrophic mice, also supporting the necessity for alternative treatment options to corticosteroids (20). Since MR is expressed in many distinct cell types present in dystrophic skeletal muscle, including myofibers and immune cells, our laboratory has taken a systematic, genetics-based approach for determining the cell types on which MR antagonists are exerting the most beneficial effects (21–23).
Our laboratory previously demonstrated that myofiber-specific knockout of MR in dystrophin-deficient (mdx) mice recapitulates some benefits of MR antagonist treatment, including improvements in respiratory muscle force and fatigue, reductions in ongoing myofiber degeneration, and alterations of collagen packaging; however, it does not account for all the positive effects (24). Conversely, MR antagonist treatment and myofiber MR knockout appear to delay regeneration in barium chloride (BaCl2)-induced acute muscle injury of wild-type (WT) tibialis anterior muscles (TAs), indicating dynamic regulation of MR activity dependent upon the magnitude of damage (25). In addition to delayed regeneration, protein levels of aldosterone synthase [cytochrome P-450 group 11, subgroup B, gene 2 (CYP11B2)], the enzyme that produces the endogenous MR ligand aldosterone, are elevated in TAs of injured myofiber myeloid mineralocorticoid receptor knockout (MRcko) mice (25). Both in vivo and in vitro treatment with MR antagonists, independent of MR, stabilize dystrophic myofiber sarcolemmas assessed by laser injury assays, also contributing to the reduction of dystrophic myofiber damage (24, 26).
Innate, or myeloid, immune cells such as monocytes, macrophages, and neutrophils are the predominant leukocytes found in dystrophic skeletal muscles, and substantial evidence indicates that genetically or pharmacologically inhibiting myeloid MR signaling reduces pathology in rodent disease models (3). Myeloid MR knockout or treatment with steroidal MR antagonists (spironolactone and eplerenone) prevents poor in vivo outcomes in diseases and injuries of the cardiovascular system, heart, kidney, blood, and brain (27–36). Overall benefits conferred by both MR knockout and MR antagonists include a reduction in inflammatory cytokine signaling, upregulation of immunosuppressive M2-like macrophage activity, and prevention of fibrosis. Usage of the nonsteroidal MR antagonist finerenone exhibits effects similar to steroidal counterparts in kidney disease, suggesting that the effects are specific to MR blockade (29, 37). Additionally, in vitro treatment with MR antagonists of isolated immune cells including mouse peritoneal macrophages and human peripheral blood mononuclear cells causes similar anti-inflammatory effects (27, 38). Myeloid MR knockout recapitulates many improvements of MR antagonist therapies, indicating that MR signaling within innate immune cells contributes most to the drugs’ successes in various pathologies (27–36). We have previously shown that specific and nonspecific MR antagonists are equivalent in effectiveness in dystrophic mice and that MR antagonists do not affect aging and exercise phenotypes (39, 40). Given our previous studies and the extensive literature supporting the inhibition of myeloid MR signaling to lessen tissue injury and disease, we hypothesized that MR antagonists may be exerting benefits in dystrophic skeletal muscle by inhibiting MR in innate immune cells responding to contractile-induced myofiber damage. Our recently improved immune cell isolation techniques enable analysis of skeletal muscle myeloid cell population distributions that may be altered because of diminished MR signaling (41). In the present study we targeted MR expression in myeloid cells, utilizing models of chronic (mdx) and acute (BaCl2-injured WT) muscle injury.
MATERIALS AND METHODS
Myeloid Mineralocorticoid Receptor Knockout in mdx and WT Mice
All mouse protocols were approved by the Institutional Animal Care and Use Committee of the Ohio State University, comply with all laws of the United States of America, and conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All mice used in experiments were euthanized by cervical dislocation to avoid chemical contamination of muscle tissues, as approved by the above guidelines. To generate myeloid mineralocorticoid receptor (MR) knockout mouse models, mdx (Dmdmdx C57BL/10) and mixed C57BL/6-C57BL/10 (WT) mice containing a floxed MR allele were crossed to hemizygous, lysozyme M-driven Cre recombinase transgenic (LysM-Cre) mice (Jackson Laboratories, Bar Harbor, ME) (24, 25, 42, 43). Homozygous MR floxed (MRFl/Fl) mice containing the LysM-Cre transgene were used as experimental mice, and MR floxed mice lacking the LysM-Cre transgene (Cre−) were used as control mice for both the WT and mdx backgrounds.
Primers used for Cre genotyping were Cre-For 5′- ATGTCCAATTTACTGACCG-3′, Cre-Rev 5′- CGCCGCATAACCAGTGAAAC-3′. Primers used for excision PCR of the MR null allele were MRflox-7 5′- CTGGAGATCTGAACTCCAGGCT-3′, MRflox-8 5′- CCTAGAGTTCCTGAGCTGCTGA-3′. Primers used for WT and floxed allele genotyping were MR flox-7 5′- CTGGAGATCTGAACTCCAGGCT-3′, MR flox-10 5′- TAGAAACACTTCGTAAAGTAGAGCT-3′. To validate the myeloid mineralocorticoid receptor knockout (MRcko) mdx model, the null allele of MR resulting from Cre excision was detected by PCR of genomic DNA extracted from 4-wk-old MRcko mdx quadriceps (n: MRcko = 2 M, 1 F; Cre− = 3M), diaphragms (n: MRcko = 2 M, 1 F; Cre− = 1 M, 2 F), spleens (n: MRcko = 1 F; Cre− = 1 M), and protein tyrosine phosphatase receptor type C (CD45)+ cluster of differentiation 11b (CD11b)+ myeloid cells sorted from MRcko mdx quadriceps [n: MRcko = 1 (pooled 2 M, 1 F); Cre− = 1 (pooled 3 M)] and mdx Cre− controls. PCR validation from MRcko WT mice was performed on DNA extracted from tibialis anterior muscles (TAs) 4 days after barium chloride (BaCl2)-induced injury in 8- to 10-wk-old MRcko WT (n: MRcko = 2 M, 2 F; Cre− = 2 M, 2 F) and WT Cre− controls. The DNA was extracted from tissues and sorted cells with the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany; catalog no. 69504) according to the manufacturer’s instructions. Genotyping primers were previously used to validate myofiber-specific MR-knockout mice on the mdx and WT backgrounds (24, 25). All PCRs were conducted with a ProFlex PCR System (ThermoFisher Scientific, Waltham, MA).
Experimental Design
Validation of myeloid MR knockout was achieved by performing PCR with primers to specifically identify the excised MR null allele using DNA extracted from 4-wk-old MRcko mdx quadriceps, diaphragms, spleens, and cell-sorted CD45+ CD11b+ myeloid cells, as well as from MRcko WT TAs 4 days after acute injury with BaCl2. Myofiber MR-knockout mdx and WT mice were generated previously under the control of the muscle creatine kinase promoter and used as positive (+) controls for the respective myeloid MRcko mouse models (24, 25). Since 4 wk of age is the peak of myeloid cell infiltration into mdx muscles, this time point was chosen to validate knockout of MR in the MRcko mdx model (41, 44). Excision of the MR null allele in the MRcko WT model was examined at 4 days after acute injury because of the influx of macrophages present in the injured TA at that time point (45).
After model validation, we performed an unbiased assessment of muscle cytokine and chemokine levels in MRcko mdx and acutely injured MRcko WT muscles at the peak of myeloid infiltration in both models. For the dystrophic model, we additionally compared diaphragm, which is known to become more fibrotic, with quadriceps muscles, which continue to regenerate throughout the mouse life span and which we have shown to contain different compositions of myeloid cells (41). TAs were used for acute injury to be consistent with the large body of literature using this muscle (46, 47), whereas quadriceps were used as the limb muscle for mdx to be consistent with the muscular dystrophy literature and all of our previous studies on MR antagonism (18, 39). No direct comparisons were made between acute injury and mdx, only comparisons between Cre+ and Cre− controls for each mouse line and muscle.
We next performed flow cytometry to assess myeloid MRcko effects on muscle myeloid cell distribution in the contexts of muscular dystrophy and acute injury. Changes in myeloid cells were quantified in single-cell suspensions derived from digested 4-wk-old MRcko mdx quadriceps and diaphragms, as well as in MRcko WT TAs 4 days after acute injury with the corresponding Cre− controls.
To determine whether changes in muscle cytokines and chemokines or alterations of myeloid populations in dystrophic and injured myeloid MRcko skeletal muscle translated into changes in muscle pathology, we then quantified all the steps of muscle injury and repair including myofiber damage, regeneration, and fibrosis in both models. We quantified degenerating myofibers at the peak of myeloid infiltration in each injury model, 4 wk of age for mdx and 4 days after acute injury (41, 44). The presence of serum immunoglobulin G (IgG) within myofibers serves as a marker of myofiber damage. Myofibers were outlined by staining for laminin-2, an extracellular matrix protein found in the basement membrane (48). We also assessed the role of myeloid MRcko on muscle regeneration in muscular dystrophy and acute injury. Central nucleated fibers are a hallmark of regeneration and start out small and grow larger. Embryonic myosin heavy chain (eMHC) is a marker of early regenerating myofibers, with expression for a short time after myoblast fusion (49, 50). To evaluate whether MRcko mdx and injured MRcko WT skeletal muscle contain different percentages of regenerating fibers and/or exhibit a delay in regenerating fiber growth, laminin-2 and nuclei were stained, imaged, and analyzed with semiautomatic muscle analysis using segmentation of histology (SMASH) software (24, 25, 51). The percentage and area of early regenerating eMHC+ myofibers were quantified, as was the percentage of centrally nucleated fibers (CNFs). Early (4 days) and late (14 days) regeneration time points were chosen for injured MRcko WT TAs, whereas 8 wk of age was chosen for MRcko mdx quadriceps to investigate peak regeneration after the early necrotic period in mdx pathology. Fibrosis was then analyzed by fibronectin staining at 8 wk of age in MRcko mdx and at 4 days post-acute injury in MRcko WT because damaged myofibers from peak necrosis should be regenerated or replaced with fibrosis by these time points.
Aldosterone synthase (CYP11B2) is an intracellular enzyme that synthesizes the last three steps in production of the endogenous ligand for MR, aldosterone, that we have previously detected in dystrophic skeletal muscles and particularly in infiltrated myeloid cells (22, 25). We have also shown that CYP11B2 levels correlate with intramuscular aldosterone levels. To determine whether myeloid MRcko in mdx and injured WT alters production of CYP11B2, we analyzed localization of CYP11B2 relative to CD11b+ myeloid cells in MRcko WT TAs 4 days after acute injury and in MRcko mdx muscles.
Barium Chloride-Induced Acute Muscle Injury
Male and female myeloid MRcko WT and WT Cre− control mice between 8 and 10 wk of age were anesthetized with isoflurane during the procedure as previously described (25). Hair was removed from the anterior portion of both lower legs with Baby Oil Nair Lotion (Church and Dwight Co., Ewing, NJ). After hair removal, the legs were rinsed with sterile water and dried completely. Next, the mouse TAs were injected intramuscularly (Becton Dickinson, Franklin Lakes, NJ; 3/10 mL U-100 Insulin syringe, 30 gauge × 3/8-in. needle) with either 50 µL of 1.2% barium chloride (Sigma-Aldrich, St. Louis, MO; catalog no. B0750) diluted in sterile water or 50 µL of sterile saline. Mice were euthanized at 4 or 14 days after acute injury for all the experiments performed, and entire litters were injected at once. Muscles injected with sterile saline were confirmed to lack an acute injury phenotype, except for along the needle track (not shown). Injections for mice used for immunohistochemistry and cytokine analyses were not all injected simultaneously because of high sample numbers but were performed by the same person at a similar time of day. Injections for flow cytometric analysis were performed on the same day, using only littermates.
Histological Staining and Analyses
Myeloid MRcko mdx (quadriceps and diaphragm) and WT (TA) skeletal muscles and the respective Cre− controls were dissected and cut in half as previously described (24, 25). Half of the muscle tissue was flash-frozen in liquid nitrogen for protein isolation, and the other half was embedded in optimal cutting compound and frozen with liquid nitrogen-cooled isopentane. Sectioning (8 µm) was performed with a cryostat (Bright, Huntingdon, UK).
Actively degenerating myofibers were quantified in 4-wk-old MRcko mdx quadriceps (n: MRcko = 6 M, 4 F; Cre− = 7 M, 7 F) and diaphragms (n: MRcko = 6 M, 8 F; Cre− = 3 M, 10 F) as well as in MRcko WT TAs 4 days after acute injury (n: MRcko = 9 M, 7 F; Cre− = 8 M, 9 F) as previously described (24, 25). Sections from both mouse models were incubated with 1:200 Alexa Fluor 488-conjugated goat anti-mouse immunoglobulin G (IgG) antibody (Invitrogen, Waltham, MA; catalog no. A11029) to stain serum IgG leaking into damaged myofibers. Fibrosis was quantified in 8-wk-old MRcko mdx diaphragms (n: MRcko = 7 M, 8 F; Cre− = 7 M, 8 F) and 4-days post-acute injury MRcko WT TAs (n: MRcko = 7 M, 7 F; Cre− = 6 M, 7 F) as previously described (24, 25). All immunofluorescence, unless otherwise specified, was performed with primary antibody incubations of 2 h and secondary antibody incubations of 1 h at room temperature with antibodies diluted in 1% normal goat serum and potassium phosphate-buffered saline. Sections from both mouse models were incubated with 1:40 rabbit anti-mouse fibronectin (Abcam, Cambridge, MA; catalog no. 23750) primary antibody and 1:200 goat anti-rabbit Alexa Fluor 555-conjugated (Invitrogen, Waltham, MA; catalog no. A21429) secondary antibody. Quadriceps from MRcko mdx were not analyzed because mdx quadriceps accumulate little fibrosis during pathology. Embryonic myosin heavy chain (eMHC)+, early regenerating myofibers, and eMHC+ myofiber area were quantified in MRcko WT TAs 4 days after acute injury (n: MRcko = 6 M, 3 F; Cre− = 7 M, 4 F) by immunofluorescence staining with 1:2.5 monoclonal anti-mouse eMHC (Departmental Studies Hybridoma Bank, The University of Iowa, Iowa City, IA) primary antibody and 1:200 goat anti-mouse Alexa Fluor 488-conjugated (Invitrogen, Waltham, MA; catalog no. A11029) secondary antibody. Centrally nucleated fibers (CNFs) and CNF area were quantified in 8-wk-old MRcko mdx quadriceps and MRcko WT TAs 14 days after acute injury as previously described (24, 25). Sections from both mouse models were incubated with 1:500 rat anti-mouse laminin-2 (α-2 chain) (Sigma-Aldrich, St. Louis, MO; catalog no. L0663) primary antibody and 1:200 Cy3-conjugated goat anti-rat (Jackson Immuno Research Laboratories, West Grove, PA; catalog no. 112-165-167) secondary antibody to outline the myofibers for analysis (24, 25). To quantify myofiber CYP11B2 in MRcko WT TAs 4 days after acute injury, sections were costained with 1:250 rabbit anti-mouse CYP11B2 (AS 2084) with 1:50 rat anti-mouse CD11b (BD PharMingen, San Jose, CA; catalog no. 550282) primary antibodies overnight at 4°C and 1:200 goat anti-rabbit Alexa Fluor 555-conjugated (Invitrogen, Waltham, MA; catalog no. A21429) with 1:200 goat anti-rat Alexa Fluor 488-conjugated (Jackson Immuno Research Laboratories, West Grove, PA; catalog no. 112-165-167) secondary antibodies as previously described (22, 24, 25). All immunofluorescence sections were mounted with Vectashield (Vector, Burlingame, CA; catalog no. H1000) containing 2 ng/µL of 4′,6-diamidino-2-phenylindole (DAPI) to stain nuclei.
Composite images for all immunostaining experiments were taken on a Nikon Eclipse 800 microscope under a ×10 objective with a Nikon DS-Ri2 camera driven by Nikon Br Elements software (Melville, NY). The percent areas of IgG staining and numbers of IgG+ fibers, percent areas of fibronectin staining, and percent areas of CYP11B2 staining were quantified with Adobe Photoshop CS6 as previously described (24, 25, 40). Percentages of centrally nucleated myofibers (CNFs) and their area were determined with the semiautomatic muscle analysis using segmentation of histology (SMASH) MATLAB (MathWorks, Natick, MA) plug-in as previously described (24, 25). Percent eMHC+ and eMHC+ area were also determined with the SMASH MATLAB plug-in (24, 25). The myofiber maximum area for quadriceps data was set to 5,700 µm2 and the maximum area for TA data was set to 3,600 µm2 for SMASH analysis. All histological analyses were conducted by an individual blinded to sample genotypes.
Skeletal Muscle Protein Isolation and Cytokine Proteome Profiler Array
Skeletal muscle protein isolation and cytokine and chemokine analysis were performed as previously described (41). Briefly, 4-wk-old MRcko mdx quadriceps and diaphragms (n: MRcko = 3 M, 3 F; Cre− = 3 M, 3 F) and MRcko WT TAs 4 days after acute injury (n: MRcko = 3 M, 3 F; Cre− = 3 M, 3 F) were flash-frozen in liquid nitrogen and homogenized in Dulbecco’s phosphate-buffered saline (DPBS)-solubilized 1% Triton X-100 (Thermo Fisher, Waltham, MA; catalog no. 14190-144) containing protease inhibitors. Sample protein concentrations were determined with the DC protein assay (Bio-Rad Laboratories, Hercules, CA; catalog no. 5000166). Lysate supernatants (protein loaded: quadriceps = 5 mg, diaphragms = 2.5 mg, TAs = 1.5 mg) were incubated with membranes from Proteome Profiler Mouse Cytokine Array Kit A (R&D Systems, Minneapolis, MN; catalog no. ARY006), and the assay was completed according to the manufacturer’s instructions. Pixel densitometry was performed with HL Image++ Quick Spots Tool version 25.0.0r (Western Vision Software, Salt Lake City, UT). Pixel densitometry analytics are presented in two tables (Table 1 and Table 2), separating chemokines and cytokines quantified from the assay.
Table 1.
Chemokine quantification in MRcko mdx and WT skeletal muscle
| CCL2 | CCL3 | CCL4 | CCL5 | CCL12 | CXCL1 | CXCL2 | CXCL9 | CXCL10 | CXCL13 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Coordinate | C5 | C8 | C9 | C11 | C6 | C3 | C10 | C14 | C1 | A1 |
| MRcko | ||||||||||
| mdx 4WK QUAD | ↓29% | ↓80% | ↑4.9× | ↓5% | ↓47% | ↑1.9× | N.D. | ↑2.0× | ↑3.6× | ↓9% |
| mdx 4WK DIA | ↓11% | ↓65% | ↓52% | ↓72% | ↓20% | ↓15% | ↓66% | ↓53% | ↓30% | ↓18% |
| WT AID4 TA | === | ↓54% | N.D. | ↓18% | ↓9% | ↓87% | N.D. | ↓14% | N.D. | ↓14% |
Quantification of relative chemokine levels detected in myeloid mineralocorticoid receptor knockout (MRcko) mdx and injured MRcko wild-type (WT) skeletal muscle by pixel densitometry. Results are displayed as fold increases and % decreases in MRcko skeletal muscle relative to the appropriate Cre− controls. Coordinates listed in the table correspond to the individual chemokine’s location (in duplicate) on the immunoblot. Some quantified chemokines were equivalent (===) or not detected (N.D.) compared with Cre− controls. AID4, 4 days after acute injury; DIA, diaphragm; QUAD, quadriceps; TA, tibialis anterior; 4WK, 4 wk of age.
Table 2.
Cytokine quantification in MRcko mdx and WT skeletal muscle
| TNF-α | IL-1β | M-CSF | C5/C5a | IL-4 | IL-10 | sICAM-1 | TIMP-1 | IL-1α | IL-1ra | |
|---|---|---|---|---|---|---|---|---|---|---|
| Coordinate | D3 | A10 | C4 | A2 | B2 | B6 | A7 | D2 | A9 | A11 |
| MRcko | ||||||||||
| mdx 4WK QUAD | +++ | +++ | ↑2.8× | ↑1.2× | +++ | +++ | ↓1% | ↑1.2× | +++ | ↓6% |
| mdx 4WK DIA | ↓20% | ↓64% | ↓10% | ↓1% | ↓45% | ↓50% | === | === | ↓50% | ↓11% |
| WT AID4 TA | ↑1.6× | N.D. | ↑1.3× | ↑1.1× | N.D. | N.D. | === | ↑1.1× | N.D. | === |
Quantification of relative cytokine levels detected in myeloid mineralocorticoid receptor knockout (MRcko) mdx and injured MRcko wild-type (WT) skeletal muscle by pixel densitometry. Results are displayed as fold increases and % decreases in MRcko skeletal muscle relative to the appropriate Cre− controls. Coordinates listed in the table correspond to the individual chemokine’s location (in duplicate) on the immunoblot. Some quantified cytokines were equivalent (===) or not detected (N.D.) compared with Cre− controls. Other cytokines were upregulated (+++) but, however, undetected in Cre− controls and therefore excluded from quantification. AID4, 4 days after acute injury; DIA, diaphragm; M-CSF, macrophage colony-stimulating factor; QUAD, quadriceps; sICAM-1, soluble ICAM-1; TA, tibialis anterior; TIMP-1, tissue inhibitor of metalloproteinase-1; 4WK, 4 wk of age.
Flow Cytometric Analysis and Fluorescence-Activated Cell Sorting of Skeletal Muscle-Derived Myeloid Cells
Single-cell suspensions for flow cytometric analysis were generated as previously described from MRcko mdx quadriceps and diaphragms (n: MRcko = 3 M, 3 F; Cre− = 5 M, 1 F) and 4 days post-acute injury MRcko WT TAs (n: MRcko = 3 M, 3 F; Cre− = 5 M, 1 F) (41). Diaphragms from two mice were pooled to obtain sufficient immune cells for analysis. After muscle digestion and generation of single-cell suspensions, samples were fixed in DPBS-solubilized 1% paraformaldehyde (Sigma-Aldrich, St. Louis, MO; catalog no. P6148) and kept at 4°C for up to 3 days before flow cytometry. The staining methodology for flow cytometry was performed as previously described (41). Antibodies used for staining are as follows: CD45 (phycoerythrin-Cy7; Thermo Fisher, Waltham, MA; catalog no. 25045182), CD11b (allophycocyanin; BioLegend, San Diego, CA; catalog no. 101212), Fc fragment of IgG receptor la (CD64) (BV605; BioLegend, San Diego, CA; catalog no. 139323), mannose receptor C-type 1 (CD206) (peridinin-chlorophyll-protein (PerCP)-eFluor710; Thermo Fisher, Waltham, MA; catalog no. 46206182), lymphocyte antigen 6 complex family member G6C (LY6C) (eFluor450; Thermo Fisher, Waltham, MA; catalog no. 48593282), lymphocyte antigen 6 complex family member G6D (LY6G) (allophycocyanin/FIRE750; BioLegend, San Diego, CA; catalog no. 127652), and C-C motif chemokine receptor 2 (CCR2) (phycoerythrin; R&D Systems, Minneapolis, MN; catalog no. FAB5538P). These markers were used to quantify the following cell populations: leukocytes (CD45+), myeloid (CD45+ CD11b+), neutrophils (CD45+ CD11b+ LY6G+), infiltrating monocytes (CD45+ CD11b+ LY6G− CD64− LY6CHi), macrophages (CD45+ CD11b+ LY6G− CD64+), CD206+ macrophages (CD45+ CD11b+ LY6G− CD64+ CD206+), and CCR2+ macrophages (CD45+ CD11b+ LY6G− CD64+ CCR2+). Intracellular CD206 staining was performed by permeabilizing the cells with DPBS-solubilized 0.5% Tween 20, quantifying macrophages with both membrane CD206 and vesicular CD206 being actively trafficked to the membrane. Cellular analysis experiments were conducted in the Analytical Cytometry Shared Resource at The Ohio State University with a Becton Dickinson LSRFortessa Flow Cytometer (Becton Dickinson, Franklin Lakes, NJ), and all data were analyzed with FlowJo software version 10.7.1 (Becton Dickinson). Samples used for FACS were prepared in the same way but without paraformaldehyde fixation. CD45+ CD11b+ myeloid cells were stained and sorted with a Becton Dickinson FACSAria III the same day they were isolated for MRcko mdx model validation.
Statistical Analysis
Data are shown as dot plots displaying all biological replicates for the given experiment and a horizontal line depicting the mean or bar graphs displaying all biological replicates and means ± SE. The two-tailed, unpaired Student’s t test was utilized for all statistical analysis comparing MRcko mdx and WT data to their respective Cre− controls. Significance testing was performed in GraphPad Prism software version 9.3.1 (GraphPad Prism Software, San Diego, CA). Means were considered significantly different if P ≤ 0.05.
RESULTS
Validation of Myeloid MR Conditional Knockout Models (MRcko mdx and WT)
To understand the role of myeloid MR signaling in skeletal muscle injury, we developed two myeloid MR conditional knockout (MRcko) mice, using Cre-Lox recombination under control of the lysozyme M promoter. Quadriceps, diaphragm, spleen, and sorted myeloid cell DNA from MRcko mdx tissues displayed the PCR product for the excised MR null allele, whereas DNA isolated from mdx Cre− (MRFl/Fl) controls did not (Fig. 1A). The MR null allele was also robustly detected in MRcko wild-type TAs 4 days after acute injury in mice expressing Cre and not in the muscles lacking Cre expression (Fig. 1B).
Figure 1.

Validation of myeloid mineralocorticoid receptor knockout (MRcko) mdx and wild-type (WT) mouse models. A: excision PCR enabled detection of the mineralocorticoid receptor (MR) null allele in 4-wk-old (4WK) MRcko mdx quadriceps (QUAD, Q; n: MRcko = 2 M, 1 F; Cre− = 3 M), diaphragms (DIA, D; n: MRcko = 2 M, 1 F; Cre− = 1 M, 2 F), spleens (SPN, S; n: MRcko = 1 F; Cre− = 1 M), and fluorescence-activated cell sorting (FACS)-derived CD45+ CD11b+ myeloid cells [n: MRcko = 1 (pooled 2 M, 1 F); Cre− = 1 (pooled 3 M)] but not in the respective mdx Cre− controls. The positive control (+) utilized was a 4-wk-old myofiber MRcko mdx quadriceps DNA sample previously validated. B: the MR null allele was also detected in MRcko WT tibialis anterior muscles (TAs) 4 days after acute injury (AI) but not in the WT Cre− controls (n: MRcko = 2 M, 2 F; Cre− = 2 M, 2 F). The positive control (+) utilized was a myofiber MRcko WT TA 4 days after acute injury DNA sample previously validated. Negative controls (−) were run with Tris-EDTA buffer instead of DNA template.
Cytokine and Chemokine Signaling Is Differentially Altered by Myeloid MRcko in Different Injuries and Muscle Types
Quantification of unbiased assessments of muscle chemokine (Table 1) and cytokine (Table 2) levels in MRcko mdx and acutely injured MRcko WT muscles compared with their respective Cre− controls at the peak of myeloid infiltration for both models is shown in Tables 1 and 2. Interestingly, 4-wk-old MRcko mdx quadriceps muscles exhibited elevation of some cytokines and chemokines while demonstrating a reduction in others compared with mdx Cre− controls (Fig. 2A). Cytokines and chemokines that were upregulated in MRcko mdx quadriceps but undetectable in the mdx Cre− quadriceps are listed but not quantified. In MRcko mdx quadriceps, cytokines and chemokines that increased were CCL4 (4.9-fold); CXCL1 (1.9-fold); CXCL9 (2.0-fold); CXCL10 (3.6-fold); TNF-α, IL-1β, macrophage colony-stimulating factor (M-CSF; 2.8-fold); C5/C5a (1.2-fold); IL-4, IL-10, tissue inhibitor of metalloproteinase-1 (TIMP-1; 1.2-fold); and IL-1α. Cytokines or chemokines that decreased 20% or more in MRcko mdx quadriceps were CCL2 (29%), CCL3 (80%), and CCL12 (47%). Conversely, most cytokine and chemokine signaling was reduced in 4-wk-old MRcko mdx diaphragms relative to mdx Cre− controls. In MRcko mdx diaphragms the cytokines and chemokines that decreased 20% or more were CCL3 (65%), CCL4 (52%), CCL5 (72%), CCL12 (20%), CXCL2 (66%), CXCL9 (53%), CXCL10 (30%), TNF-α (20%), IL-1β (64%), IL-4 (45%), IL-10 (50%), and IL-1α (50%). The only cytokine or chemokine that remained at the same level was soluble ICAM-1 (sICAM-1).
Figure 2.

Cytokine and chemokine signaling in 4-wk-old myeloid mineralocorticoid receptor knockout (MRcko) mdx skeletal muscles and MRcko wild-type (WT) tibialis anterior muscles (TAs) 4 days after acute injury (AI). Proteome profiler cytokine array immunoblots incubated with lysates from 4-wk-old (4WK) MRcko mdx quadriceps (QUAD; 5 mg of protein; n: MRcko = 3 M, 3 F; Cre− = 3 M, 3 F) and diaphragms (DIA; 2.5 mg of protein; n: MRcko = 3 M, 3 F; Cre− = 3M, 3 F) (A) as well as with MRcko WT TAs (1.5 mg of protein; n: MRcko = 3 M, 3 F; Cre− = 3 M, 3 F) 4 days after acute injury (B) compared with the genotypic Cre− controls. Blots are labeled with coordinates that specifically correspond to changes in chemokine and cytokine expression quantified by densitometry in Table 1 and Table 2, respectively.
Cytokine and chemokine signaling compared with Cre− controls during peak inflammation was not changed to the same extent in MRcko WT TAs 4 days after acute injury (Fig. 2B) as in 4-wk-old MRcko mdx skeletal muscle. The cytokines or chemokines that increased in MRcko WT TAs 4 days after acute injury were TNF-α (1.6-fold), M-CSF (1.3-fold), C5/C5a (1.1-fold), and TIMP-1 (1.1-fold). Cytokines or chemokines that decreased 20% or more were CCL3 (54%) and CXCL1 (87%).
MRcko Reduces Proportions of Muscle CCR2+ Macrophages in Acute Injury
We next quantified skeletal muscle myeloid leukocytes to assess MRcko effects in the contexts of muscular dystrophy and acute injury. Gating strategies for the seven-color flow cytometry used to quantify myeloid cells (Mye), neutrophils (Nφ), infiltrating monocytes (MO), macrophages (MΦ), CD206+ macrophages, and CCR2+ macrophages from MRcko mdx and MRcko WT skeletal muscle are displayed in Fig. 3, A and B, respectively. Flow cytometry analysis demonstrated that myeloid immune cell populations were remarkably similar between dystrophic and acutely injured normal skeletal muscle, except for a shift toward macrophage differentiation (CD45+ CD11b+ LY6G− CD64+) in 4 days post-acute injury MRcko WT TAs (Fig. 3).
Figure 3.

Flow cytometry gating of myeloid immune cells in 4-wk-old (4WK) myeloid mineralocorticoid receptor knockout (MRcko) mdx skeletal muscle and MRcko wild-type (WT) tibialis anterior muscles (TAs) 4 days after acute injury (AI). Representative flow cytometry dot plots displaying the gating strategy used to identify all immune cells (CD45+), myeloid cells (CD45+ CD11b+), neutrophils (CD45+ CD11b+ LY6G+), infiltrating monocytes (CD45+ CD11b+ LY6G− CD64− LY6CHi), macrophages (CD45+ CD11b+ LY6G− CD64+), CD206+ macrophages (CD45+ CD11b+ LY6G− CD64+ CD206+), and CCR2+ macrophages (CD45+ CD11b+ LY6G− CD64+ CCR2+) in 4-wk-old MRcko mdx skeletal muscle [A; quadriceps (QUAD) shown] and MRcko WT TAs 4 days after acute injury (B).
Representative gating dot plots for MRcko mdx quadriceps (Fig. 4A) and diaphragms (Fig. 5A) did not indicate discernible shifts in immune cells at 4 wk of age. Cells per milligram of muscle tissue were not changed in 4-wk-old MRcko mdx quadriceps compared with mdx Cre− littermate controls (Fig. 4C): myeloid cells (2,397 ± 709 vs. 1,717 ± 601 cells/mg; P = 0.481), neutrophils (234 ± 97 vs. 173 ± 57 cells/mg; P = 0.600), infiltrating monocytes (212 ± 77 vs. 171 ± 57 cells/mg; P = 0.673), macrophages (1,619 ± 440 vs. 1,150 ± 421 cells/mg; P = 0.459), CD206+ macrophages (349 ± 102 vs. 237 ± 98 cells/mg; P = 0.450), or CCR2+ macrophages (202 ± 57 vs. 129 ± 50 cells/mg; P = 0.359). No differences were observed in immune cell population density in 4-wk-old MRcko mdx diaphragms relative to mdx Cre− controls (Fig. 5B): myeloid cells (1,358 ± 530 vs. 1,018 ± 221 cells/mg; P = 0.585), neutrophils (141 ± 13 vs. 209 ± 88 cells/mg; P = 0.494), infiltrating monocytes (106 ± 22 vs. 169 ± 69 cells/mg; P = 0.431), macrophages (873 ± 442 vs. 441 ± 72 cells/mg; P = 0.390), CD206+ macrophages (88 ± 39 vs. 46 ± 3 cells/mg; P = 0.349), or CCR2+ macrophages (96 ± 43 vs. 55 ± 9 cells/mg; P = 0.411).
Figure 4.

Flow cytometric analysis of myeloid populations in 4-wk-old (4WK) myeloid mineralocorticoid receptor knockout (MRcko) mdx quadriceps (QUAD) and MRcko wild-type (WT) tibialis anterior muscles (TAs) 4 days after acute injury (AI). A and B: representative flow cytometry dot plots of 4-wk-old MRcko mdx quadriceps and mdx Cre− controls (A; n: MRcko = 3 M, 3 F; Cre− = 5 M, 1 F) and MRcko WT TAs 4 days after acute injury (B; n: MRcko = 2 M, 5 F; Cre− = 2 M, 5 F) for myeloid cell (Mye), neutrophil (Nϕ), infiltrating monocyte (MO), macrophage (MΦ), CD206+ macrophage, and CCR2+ macrophage gating displayed with sample population percentages. C and D: quantification of cells per milligram of muscle tissue for the described populations in 4-wk-old MRcko mdx quadriceps and mdx Cre− controls (C) and MRcko WT TAs 4 days after acute injury and WT Cre− controls (D). Data displayed in dot plots with mean horizontal line and x-axes labeled with “CRE NEG” for control and “CRE POS” for MRcko. E and F: quantification of the percentage of total cells (%CD45+ or %CD45+ CD11b+ LY6G− CD64+) for the described populations in 4-wk-old MRcko mdx quadriceps and mdx Cre− controls (E) and MRcko WT TAs 4 days after acute injury and WT Cre− controls (F). Data displayed in bar graphs depicting mean ± SE with individual data points shown; x-axes labeled “CRE NEG” for control and “CRE POS” for MRcko. Student’s t test: *P ≤ 0.05.
Figure 5.
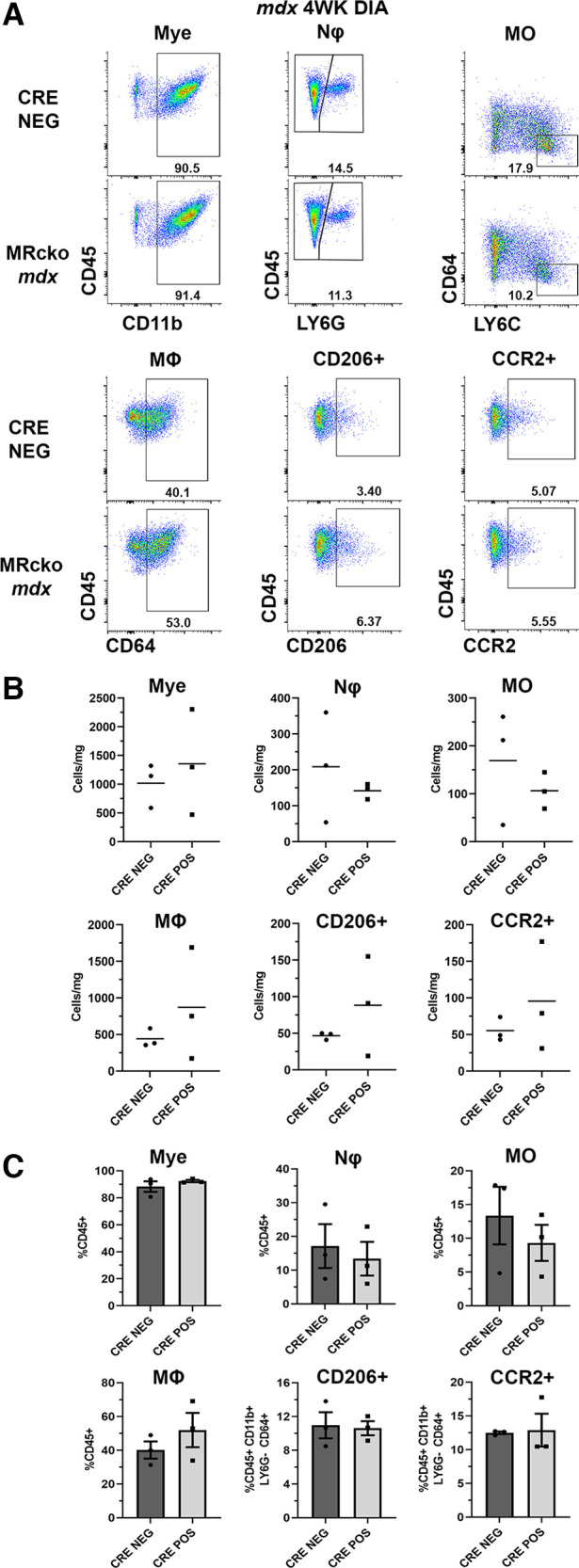
Flow cytometric analysis of myeloid populations in 4-wk-old (4WK) myeloid mineralocorticoid receptor knockout (MRcko) mdx diaphragm (DIA). A: representative flow cytometry dot plots of 4-wk-old MRcko mdx diaphragms and mdx Cre− controls (n: MRcko = pooled 3 M, 3 F; Cre− = pooled 5 M, 1 F) for myeloid cell (Mye), neutrophil (Nϕ), infiltrating monocyte (MO), macrophage (MΦ), CD206+ macrophage, and CCR2+ macrophage gating displayed with sample population percentages. B: quantification of cells per milligram of muscle tissue for the described populations in 4-wk-old MRcko mdx diaphragms and mdx Cre− controls. Data displayed in dot plots with x-axes labeled with mean horizontal line and “CRE NEG” for control and “CRE POS” for MRcko. C: quantification of the percentage of total cells (%CD45+ or %CD45+ CD11b+ LY6G− CD64+) for the described populations in 4-wk-old MRcko mdx diaphragms and mdx Cre− controls. Data displayed in bar graphs depicting mean ± SE with individual data points shown; x-axes labeled “CRE NEG” for control and “CRE POS” for MRcko. Student’s t test.
Next, we quantified each population as a percentage of total CD45+ immune cells or as a percentage of total CD45+ CD11b+ LY6G− CD64+ macrophages for analyzing CCR2+ and CD206+ macrophage populations. No significant differences were observed in MRcko mdx quadriceps (myeloid cells: 94.9 ± 0.4% vs. 93.4 ± 1.0%; P = 0.199, neutrophils: 8.4 ± 1.0% vs. 9.2 ± 0.9%; P = 0.524, infiltrating monocytes: 8.3 ± 0.9% vs. 9.1 ± 1.0%; P = 0.578, macrophages: 65.1 ± 2.5% vs. 62.2 ± 1.7%; P = 0.367, CD206+ macrophages: 21.3 ± 1.2% vs. 19.7 ± 1.0%; P = 0.323, CCR2+ macrophages: 12.6 ± 0.9% vs. 10.9 ± 0.3%; P = 0.097) (Fig. 4E) or MRcko mdx diaphragms (myeloid cells: 92.4 ± 0.9% vs. 88.3 ± 3.9%; P = 0.364, neutrophils: 13.4 ± 5.0% vs. 17.1 ± 6.5%; P = 0.671, infiltrating monocytes: 9.3 ± 2.7% vs. 13.4 ± 4.3%; P = 0.467, macrophages: 52.0 ± 10.2% vs. 40.1 ± 5.1%; P = 0.355, CD206+ macrophages: 10.6 ± 0.8% vs. 11.0 ± 1.5%; P = 0.552, CCR2+ macrophages: 12.9 ± 2.4% vs. 10.9 ± 0.3%; P = 0.166) (Fig. 5C) compared with Cre− mdx controls.
Myeloid cells from MRcko WT TAs 4 days after acute injury were analyzed identically to cells derived from MRcko mdx skeletal muscle. Representative gating dot plots are depicted in Fig. 4B. No significant differences were found in cells per milligram of muscle tissue in MRcko WT TAs 4 days after acute injury compared with WT Cre− controls for myeloid cells (4,206 ± 792 vs. 3,866 ± 1,321 cells/mg; P = 0.828), neutrophils (86 ± 19 vs. 66 ± 20 cells/mg; P = 0.464), infiltrating monocytes (55 ± 22 vs. 68 ± 27 cells/mg; P = 0.718), macrophages (3,838 ± 724 vs. 3,535 ± 1,232 cells/mg; P = 0.835), CD206+ macrophages (889 ± 132 vs. 962 ± 317 cells/mg; P = 0.836), or CCR2+ macrophages (181 ± 35 vs. 234 ± 65 cells/mg; P = 0.482) (Fig. 4D).
However, CCR2+ macrophages represented a significantly smaller percentage of total CD45+ CD11b+ LY6G− CD64+. macrophages (4.8 ± 0.3% vs. 9.2 ± 1.9%; P = 0.036) in MRcko WT TAs 4 days after acute injury compared with WT Cre− controls (Fig. 4F). Other quantified population percentages were not different relative to WT Cre− controls, including myeloid cells (93.3 ± 0.4% vs. 93.5 ± 0.8%; P = 0.806), neutrophils (1.9 ± 0.2% vs. 1.7 ± 0.2%; P = 0.476), infiltrating monocytes (1.1 ± 0.2% vs. 1.4 ± 0.4%; P = 0.495), macrophages (85.0 ± 0.6% vs. 84.5 ± 1.2%; P = 0.694), or CD206+ macrophages (24.5 ± 2.0% vs. 29.9 ± 2.4%; P = 0.114).
At the Peak of Myeloid Inflammation, MRcko Temporally Increases Degeneration in Acute Injury but Not Muscular Dystrophy
Representative immunofluorescence images of IgG staining for myofiber damage in 4-wk-old MRcko mdx and mdx Cre− quadriceps and diaphragms show no overt differences between genotypes (Fig. 6A). No differences in IgG staining per area (%IgG) were present in 4-wk-old MRcko mdx quadriceps (2.7 ± 0.8%IgG+ vs. 3.6 ± 0.8%IgG+; P = 0.479) or diaphragms (12.1 ± 2.7%IgG+ vs. 9.1 ± 1.9%IgG+; P = 0.394) compared with mdx Cre− controls (Fig. 6C).
Figure 6.

Myofiber degeneration in 4-wk-old (4WK) myeloid mineralocorticoid receptor knockout (MRcko) mdx skeletal muscles and MRcko wild-type (WT) tibialis anterior muscles (TAs) 4 days after acute injury (AI). A: representative merged images of 4-wk-old MRcko mdx quadriceps (QUAD) (n: MRcko = 6 M, 4 F; Cre− = 7 M, 7 F) and diaphragms (DIA) (n: MRcko = 6 M, 8 F; Cre− = 3 M, 10 F) and mdx Cre− controls stained for immunoglobulin G (IgG, green) and laminin-2 (red) to visualize and quantify degenerating myofibers. Scale bars, 100 µm. B: representative composite IgG images of MRcko WT TAs 4 days after acute injury (n: MRcko = 9 M, 7 F; Cre− = 8 M, 9 F) and zoomed representative IgG and laminin-2 merged images. Composite scale bars, 500 µm; zoomed scale bars, 100 µm. C: quantification of tissue area from 4-wk-old MRcko mdx quadriceps and diaphragm muscles containing IgG staining (%IgG) compared with mdx Cre− controls. D: the number of IgG+ fibers in MRcko WT TAs 4 days after acute injury compared with WT Cre− controls. Data displayed in dot plots with mean horizontal line and x-axes labeled with “CRE NEG” for control and “CRE POS” for MRcko. Student’s t test: *P ≤ 0.05.
More myofiber damage was observed in MRcko WT TAs 4 days after acute injury compared with WT Cre− controls (Fig. 6B). Quantification of IgG staining is represented as total number of IgG+ fibers in entire TA cross sections (25). MRcko WT TAs contained more degenerating myofibers compared with controls (244 ± 61 vs. 71 ± 13 IgG+ myofibers; P = 0.007) (Fig. 6D), supporting a temporal role of myeloid MRcko in increasing the prevalence of necrotic fibers.
Muscle Regeneration Is Not Directly Affected by Myeloid MRcko in Muscular Dystrophy or Acute Injury
No obvious differences were observed in myofiber size or the amount of regenerated fibers in 8-wk-old MRcko mdx quadriceps versus mdx Cre− controls (Fig. 7A). Quantification verified the absence of differences in percentages of centrally nucleated fibers (75.4 ± 2.3%CNF vs. 76.8 ± 3.6%CNF; P = 0.786) or the average regenerating fiber area (1,403 ± 100 vs. 1,504 ± 62 µm2; P = 0.381) (Fig. 7D), supporting the lack of effect of MRcko on the regeneration process in muscular dystrophy.
Figure 7.

Analysis of regeneration and fibrosis in muscles from myeloid mineralocorticoid receptor knockout (MRcko) mdx mice and MRcko wild-type (WT) mice after acute injury (AI). A and B: representative merged images of 8-week-old (8WK) MRcko mdx quadriceps (QUAD) and mdx Cre− controls (A; n: MRcko = 5 M, 1 F; Cre− = 6 M, 4 F), and MRcko WT tibialis anterior muscles (TAs) 14 days after acute injury (B; n: MRcko = 7 M, 7 F; Cre− = 8 M, 7 F) stained for nuclei (blue) and laminin-2 (red) to quantify and measure centrally nucleated fibers (CNFs). Scale bars, 100 µm. C: representative merged images of MRcko WT TAs 4 days after acute injury (n: MRcko = 6 M, 3 F; Cre− = 7 M, 4 F) stained for embryonic myosin heavy chain (eMHC; green) and laminin-2 (red) used to quantify and measure early regenerating myofibers. D and E: quantification of the percentage of CNF (%CNF) and average CNF size (µm2) in 8-wk-old MRcko mdx quadriceps compared with mdx Cre− controls (D) and MRcko WT TAs 14 days after -acute injury compared with WT Cre− controls (E). F: quantification of the total number (#) of eMHC+ fibers in an entire TA and eMHC+ fiber size (µm2) in MRcko WT TAs 4 days after acute injury compared with WT Cre− controls. G and H: representative images of 8-wk-old MRcko and Cre− mdx diaphragms (DIA) (G; n: MRcko = 7 M, 8 F; Cre− = 7 M, 8 F) and MRcko and WT Cre− TAs 4 days after acute injury (H; n: MRcko = 7 M, 7 F; Cre− = 6 M, 7 F) stained for fibronectin (red). Scale bars, 100 µm. I and J: quantification of fibronectin staining of section area (%Fibronectin) in 8-wk-old MRcko mdx diaphragms and mdx Cre− controls (I) and MRcko WT TAs 4 days after acute injury and WT Cre− controls (J). Data displayed in dot plots with mean horizontal line and x-axes labeled with “CRE NEG” for control and “CRE POS” for MRcko. Student’s t test: *P ≤ 0.05.
To evaluate changes in early regenerating myofibers in MRcko WT TAs 4 days after acute injury, sections were stained for eMHC. Representative images from MRcko WT TAs displayed fewer eMHC+ myofibers than WT Cre− controls (Fig. 7C). Quantification supported these results, although the difference was not statistically significant (1,664 ± 195 vs. 2,157 ± 147; P = 0.054) (Fig. 7F). The size of the eMHC+ myofibers was not affected in MRcko WT TAs 4 days after acute injury (263 ± 13 vs. 282 ± 8 µm2; P = 0.436), supporting that the myoblast fusion process to form new fibers does not appear to be affected.
To determine whether mild effects of myeloid MRcko on early stages of regeneration amplified into larger effects at later stages, MRcko TAs from 14 days after acute injury at the end of the muscle regeneration process were compared to Cre− WT controls (Fig. 7B). Blinded quantification of MRcko WT TAs 14 days after acute injury showed no differences in percentages of centrally nucleated fibers (73.9 ± 2.3%CNF vs. 69.1 ± 1.2%CNF; P = 0.074) or regenerated myofiber area (1,744 ± 33 vs. 1,645 ± 63 µm2; P = 0.187) compared with Cre− WT controls (Fig. 7E).
MRcko Leads to Increased Fibrosis in mdx Diaphragms but Not in Acute Injury
Since myeloid cells and cytokine levels are known to affect fibrosis, we assessed fibrosis by immunostaining for fibronectin, which accumulates in the extracellular matrix between myofibers after muscle injuries. Eight-week-old MRcko mdx diaphragms appeared to display more fibronectin staining than Cre− controls (Fig. 7G). Quantification showed that MRcko mdx diaphragms contain a significantly higher area of fibronectin staining (%fibronectin) than mdx Cre− controls (17.4 ± 1.4% fibronectin vs. 12.3 ± 1.4%fibronectin; P = 0.013) (Fig. 7I). Quadriceps from 8-wk-old mdx mice do not accumulate quantifiable fibrosis; therefore this tissue was excluded from this analysis. MRcko WT TAs 4 days after acute injury (Fig. 7H) did not accumulate more fibrosis than Cre− littermates (68.6 ± 2.5%fibronectin vs. 65.5 ± 3.0%fibronectin; P = 0.432) (Fig. 7J).
Myeloid MRcko Leads to Elevated Myofiber Aldosterone Synthase (CYP11B2) after Acute Injury
The localization of CYP11B2 appeared unchanged in 4-wk-old MRcko mdx quadriceps and diaphragms relative to mdx Cre− controls, and expression was largely present in myeloid cells (not shown), as previously reported for dystrophic muscles (22, 25). In contrast, CYP11B2 localization was more prevalent in injured MRcko WT TA muscles relative to WT Cre− controls, and the increased localization appeared within myofibers (Fig. 8A). By costaining with CYP11B2 and CD11b to visualize aldosterone synthase expression relative to myeloid cells, CYP11B2 was present in myofibers, particularly in regions infiltrated with CD11b+ myeloid cells (Fig. 8B). Degenerating myofibers (IgG+) did not show any CYP11B2 localization (not shown). In contrast, WT Cre− control CYP11B2 staining colocalized with CD11b+ myeloid cells but not myofibers. Quantification of total area of CYP11B2 staining (%CYP11B2) showed that MRcko WT TA muscles had more CYP11B2 staining than WT Cre− controls (9.1 ± 2.4%CYP11B2 vs. 1.9 ± 0.5%CYP11B2; P = 0.011) (Fig. 8C).
Figure 8.
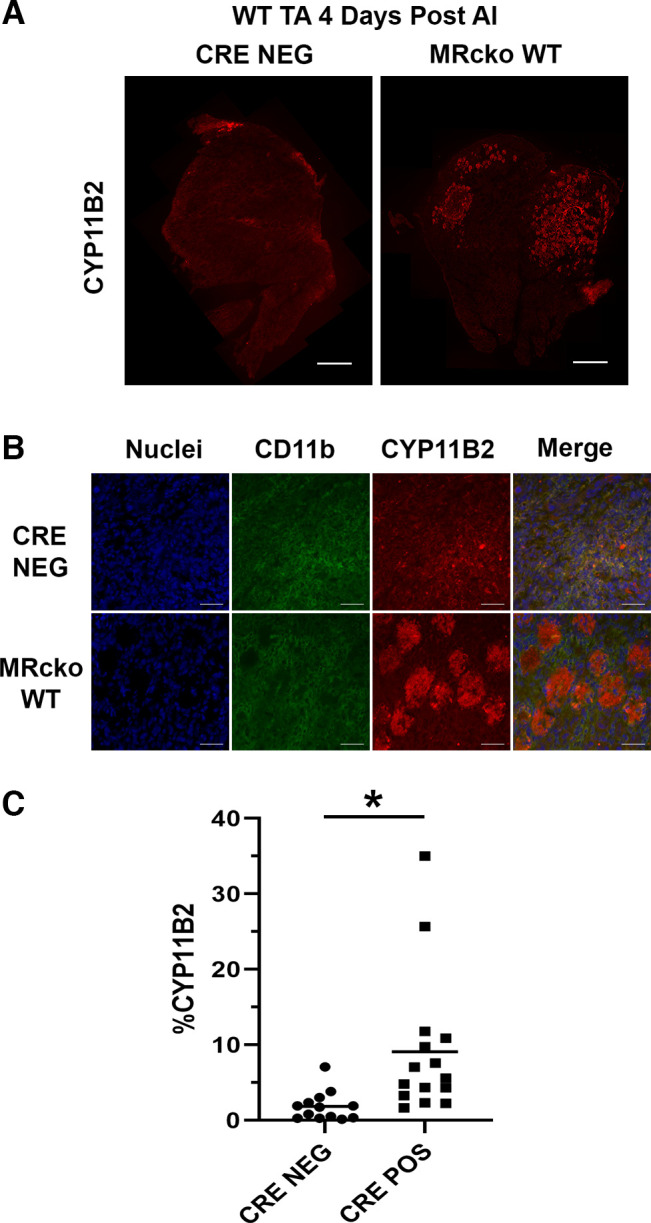
Analysis of CYP11B2 levels in myeloid mineralocorticoid receptor knockout (MRcko) wild-type (WT) tibialis anterior muscles (TAs) 4 days after acute injury (AI). A: representative composite CYP11B2 (red) images of MRcko WT TAs 4 days after acute injury and WT Cre− controls (n: MRcko = 8 M, 7 F; Cre− = 7 M, 6 F). Scale bars, 500 µm. B: representative zoomed images of MRcko WT TAs 4 days after acute injury and WT Cre− controls, including nuclei (blue), CD11b (green), CYP11B2 (red), and merged images. Scale bars, 100 µm. C: quantification of total area of CYP11B2 staining (%CYP11B2) from MRcko and WT Cre− control TAs 4 days after acute injury. Data displayed in dot plots with mean horizontal line and x-axes labeled with “CRE NEG” for control and “CRE POS” for MRcko. Student’s t test: *P ≤ 0.05.
DISCUSSION
To understand the role of myeloid MR signaling in skeletal muscle injury, we developed two myeloid MR conditional knockout (MRcko) mice, using Cre-Lox recombination under control of the lysozyme M promoter to broadly knock out MR in numerous cell types of the innate immune system in muscular dystrophy and acute muscle injury. Differences in cytokine and chemokine levels as indicators of inflammatory signaling were assessed with unbiased proteome profiler arrays. We then used our recently optimized muscle immune cell isolation techniques to quantify differences in myeloid leukocytes isolated from skeletal muscle of both models (41). Moreover, we employed immunohistochemistry to quantify parameters of myofiber damage, regeneration, and fibrosis for both MRcko models. Identifying effects of myeloid MR ablation on skeletal muscles will contribute to optimizing use of MR antagonists, such as spironolactone, for treating DMD and other muscle diseases containing considerable tissue inflammation.
Although MR-independent membrane stabilizing effects were previously observed with MR antagonist treatment of mdx muscles, these effects would not be predicted in the myeloid MRcko mdx mice (24). Instead, reductions in cellular and molecular markers of inflammation due to the purported role of MR in proinflammatory macrophage polarization were expected. In contrast to the reported improvements in pathology across numerous rodent models, including diseases of the renal, cardiac, liver, vascular, and nervous systems, we observed mildly enhanced pathology in several indicators of skeletal muscle dysfunction in both mouse models (27–36).
Although past studies indicate incomplete, variable excision of myeloid cell lox-flanked genes in Cre+ animals recombined under control of the lysozyme M promoter, we detected robust excision of the null MR allele in DNA extracted from 4-wk-old MRcko mdx tissues and cells, as well as in MRcko WT 4 days after acute injury (52). Since CD45+ CD11b+ myeloid cells represent >90% of total immune cells during peak inflammation in both injury models according to the flow cytometry data, the strong excision is reasonable.
In the absence of myeloid MR, cytokine and chemokine signaling changed dramatically in the quadriceps and diaphragm muscles of MRcko mdx mice. Quantified chemokines in MRcko mdx quadriceps displayed both activation and suppression of multiple chemotactic signaling pathways. Cytokines present in MRcko mdx quadriceps were mostly upregulated, including proinflammatory cytokines that are known to contribute to dystrophic skeletal muscle pathology, including TNF-α, IL-1β, IL-1α, and C5/C5a (53). Interestingly, proregenerative (and profibrotic) cytokines were also upregulated, such as M-CSF, IL-4, IL-10, and TIMP-1. Upregulation of both categories of cytokines may be evidence of dynamic cross talk between proinflammatory and immunosuppressive factors to coordinate wound healing and fibrotic responses. Loss of MR may inhibit important myeloid cell functions in regenerating mdx quadriceps, which would be reflected by the changes in inflammatory signaling. The observed signaling changes did not improve or exacerbate degeneration at 4 wk or myofiber regeneration at 8 wk in MRcko mdx quadriceps.
In contrast, all cytokines and chemokines were reduced in MRcko mdx diaphragms, which may be important evidence for differences in immune-mediated regenerative potential between limb and respiratory muscles. We have shown that myeloid cells expressing F4/80 are different between dystrophic quadriceps and diaphragms, which may also have implications for the present study (41). Reductions in diaphragm inflammation may lead to worse outcomes, since more fibrosis appears to accumulate by 8 wk of age in MRcko mdx diaphragms compared with mdx Cre− controls. Although excessive inflammation may be detrimental to skeletal muscle regeneration in specific contexts, decreasing inflammation in muscles with less regenerative potential may obstruct healing, in which myeloid MR appears to be integrally involved. Although no obvious differences in fiber size or centrally nucleated fibers were present in MRcko mdx diaphragms, one limitation is that we did not perform blinded quantification for these parameters. This decision was made because of the large amount of fibrosis that would have likely made numbers of measurable myofibers in the diaphragm small enough to not be statistically valid. These measurements in diaphragms are more useful in older mdx animals after the necrotic/highly regenerative and inflammatory phases have passed, but in this article we focus on earlier time points to assess effects from lack of myeloid MR signaling during the peak of inflammation. Since we did not observe quantifiable differences in diaphragm fiber sizes at 12 wk in myofiber MR-knockout mice, we would not predict diaphragm regeneration changes independent from fibrosis in myeloid MR-knockout mice.
The density and distribution of myeloid populations quantified in MRcko mdx skeletal muscles were surprisingly unchanged compared with mdx Cre− controls. The percentage of total macrophages expressing CCR2 increased slightly in MRcko mdx quadriceps, which we previously demonstrated to represent a more transcriptionally active macrophage population expressing pathological genes, although the difference was not significant (P = 0.097) (41). Quantifying skeletal muscle myeloid populations in MRcko mdx mice at 4 wk of age during peak inflammation is most relevant for understanding the role of myeloid MR in DMD pathology, despite the limitation of high variability. We and others have shown that the numbers of immune cells drop dramatically after this time point, and although inflammation is less variable, analysis at later time points requires much larger numbers of pooled mice (41).
Knockout of myeloid MR in injured WT mice resulted in more degenerating myofibers 4 days after acute injury in the TA, whereas most necrotic myofibers were already cleared in the WT Cre− TA controls. Cytokine and chemokine signaling was slightly changed in MRcko WT TAs compared with the larger changes in MRcko mdx skeletal muscle. Delayed clearance of damaged myofibers, coupled with the small changes in cytokine and chemokine expression, suggests that the loss of myeloid MR affects the functions of monocytes and neutrophils that infiltrate the tissue within the first 4 days after acute injury. Loss of function in monocytes and neutrophils may delay the regenerative process in acute muscle injury, which is reinforced by nonsignificant increases in the number and size of centrally nucleated fibers, lower percentages of macrophages expressing CD206 (P = 0.114) or CCR2, and elevated CYP11B2 localization within MRcko WT TA myofibers to compensate for the loss of myeloid MR signaling.
Reinforcing our findings, other studies have linked MR activation to the CCL2-CCR2 chemotaxis axis. In deoxycorticosterone and salt-treated mice, a rodent model of MR activation, increases in acute cardiac inflammation, fibrosis, and blood pressure were mitigated by CCL2 knockout (54). Systemic monocyte and macrophage proinflammatory signaling also appear to be influenced by MR signaling in atherosclerosis (55). More pronounced myofiber damage or signaling differences may occur at earlier regeneration time points in acutely injured skeletal muscle, which could explain the lack of dramatic changes in the presented cytokine arrays and flow cytometry data. Protein levels of CYP11B2 were also upregulated in acutely injured muscle myofibers near CD11b+ cells, suggesting that MR is implicated in myeloid cell responses to acute muscle injury if the myofibers express more CYP11B2 to compensate for diminished MR signaling in the MRcko myeloid cells.
Contrary to much of the literature supporting knockout of myeloid MR to reduce pathology in numerous disease models, we demonstrated that myeloid MR signaling is important for skeletal muscle regeneration in dystrophic and acutely injured mice. Although myeloid MR is not essential for regeneration, the immunohistochemical, signaling, and cellular analyses conducted for this study indicated that myeloid MR signaling may influence the functions of initial responder myeloid cells, such as monocytes and neutrophils. The development of more fibrosis, with less inflammatory signaling, in myeloid MRcko mdx diaphragm indicates a restorative role of inflammation in the process of healing in this muscle type.
Although studying time points earlier than 4 days after acute injury in the MRcko WT mouse may provide some small additional insight into the role of MR in acute injury repair, the restoration of normal regeneration and myofiber size by 14 days after injury supports a limited effect from MR ablation in myeloid cells that are present transiently in this setting. However, future longitudinal studies of MRcko mdx mice will be required to assess any longer-term effects of myeloid MR knockout in chronic muscle injury where myeloid cells are continuously present. Investigating later time points in MRcko mdx mice will clarify whether there is a latent effect on skeletal muscle pathology, since most of the data indicated no significant differences with myeloid MR knockout early in the disease and the small effects observed may accumulate over time. Future studies comparing global gene expression between myeloid MRcko and myofiber MRcko skeletal muscles and individual myeloid populations will provide additional insight into the cell type-specific effects underlying the efficacy of MR antagonists in chronic muscle injuries.
GRANTS
This work was supported by National Institutes of Health Grants R01AR072574 and T32HL134616 and P30CA016058, which partially supports the Flow Cytometry Shared Resource.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Z.M.H., N.R., J.S.H., S.S.B., and J.A.R.-F. conceived and designed research; Z.M.H., N.R., J.L., J.S.H., P.I., C.G., C.E.G.-S., and E.P.G.-S. performed experiments; Z.M.H., N.R., S.S.B., and J.A.R.-F. analyzed data; Z.M.H., N.R., S.S.B., and J.A.R.-F. interpreted results of experiments; Z.M.H. and N.R. prepared figures; Z.M.H. and N.R. drafted manuscript; Z.M.H., S.S.B., and J.A.R.-F. edited and revised manuscript; Z.M.H., N.R., J.L., J.S.H., P.I., C.G., C.E.G.-S., E.P.G.-S., S.S.B., and J.A.R.-F. approved final version of manuscript.
ACKNOWLEDGMENTS
Mineralocorticoid receptor floxed mice (originally made by Dr. Stephan Berger and Dr. Günther Schütz, Heidelberg University in Germany) were obtained as a generous gift from Dr. Richard Mortensen at the University of Michigan. We thank Bryan McElwain from the Flow Cytometry Shared Resource for assistance.
REFERENCES
- 1.Rosenberg AS, Puig M, Nagaraju K, Hoffman EP, Villalta SA, Rao VA, Wakefield LM, Woodcock J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci Transl Med 7: 299rv4, 2015. doi: 10.1126/scitranslmed.aaa7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol 298: R1173–R1187, 2010. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tidball JG, Welc SS, Wehling-Henricks M. Immunobiology of inherited muscular dystrophies. Compr Physiol 8: 1313–1356, 2018. doi: 10.1002/cphy.c170052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duan D. Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol Ther 26: 2337–2356, 2018. doi: 10.1016/j.ymthe.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willcocks RJ, Forbes SC, Walter GA, Sweeney L, Rodino-Klapac LR, Mendell JR, Vandenborne K. Assessment of rAAVrh.74.MHCK7.micro-dystrophin gene therapy using magnetic resonance imaging in children with Duchenne muscular dystrophy. JAMA Netw Open 4: e2031851, 2021. doi: 10.1001/jamanetworkopen.2020.31851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendell JR, Sahenk Z, Lehman K, Nease C, Lowes LP, Miller NF, Iammarino MA, Alfano LN, Nicholl A, Al-Zaidy S, Lewis S, Church K, Shell R, Cripe LH, Potter RA, Griffin DA, Pozsgai E, Dugar A, Hogan M, Rodino-Klapac LR. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol 77: 1122–1131, 2020. doi: 10.1001/jamaneurol.2020.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kupatt C, Windisch A, Moretti A, Wolf E, Wurst W, Walter MC. Genome editing for Duchenne muscular dystrophy: a glimpse of the future? Gene Ther 28: 542–548, 2021. doi: 10.1038/s41434-021-00222-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bach JR, Martinez D, Saulat B. Duchenne muscular dystrophy: the effect of glucocorticoids on ventilator use and ambulation. Am J Phys Med Rehabil 89: 620–624, 2010. doi: 10.1097/PHM.0b013e3181e72207. [DOI] [PubMed] [Google Scholar]
- 9.Connolly AM, Schierbecker J, Renna R, Florence J. High dose weekly oral prednisone improves strength in boys with Duchenne muscular dystrophy. Neuromuscul Disord 12: 917–925, 2002. doi: 10.1016/S0960-8966(02)00180-3. [DOI] [PubMed] [Google Scholar]
- 10.Daftary AS, Crisanti M, Kalra M, Wong B, Amin R. Effect of long-term steroids on cough efficiency and respiratory muscle strength in patients with Duchenne muscular dystrophy. Pediatrics 119: e320–e324, 2007. doi: 10.1542/peds.2006-1400. [DOI] [PubMed] [Google Scholar]
- 11.Markham LW, Kinnett K, Wong BL, Woodrow Benson D, Cripe LH. Corticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophy. Neuromuscul Disord 18: 365–370, 2008. doi: 10.1016/j.nmd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 12.Merlini L, Cicognani A, Malaspina E, Gennari M, Gnudi S, Talim B, Franzoni E. Early prednisone treatment in Duchenne muscular dystrophy. Muscle Nerve 27: 222–227, 2003. doi: 10.1002/mus.10319. [DOI] [PubMed] [Google Scholar]
- 13.Hussein MR, Abu-Dief EE, Kamel NF, Mostafa MG. Steroid therapy is associated with decreased numbers of dendritic cells and fibroblasts, and increased numbers of satellite cells, in the dystrophic skeletal muscle. J Clin Pathol 63: 805–813, 2010. doi: 10.1136/jcp.2010.078204. [DOI] [PubMed] [Google Scholar]
- 14.Beenakker EA, Fock JM, Van Tol MJ, Maurits NM, Koopman HM, Brouwer OF, Van der Hoeven JH. Intermittent prednisone therapy in Duchenne muscular dystrophy: a randomized controlled trial. Arch Neurol 62: 128–132, 2005. doi: 10.1001/archneur.62.1.128. [DOI] [PubMed] [Google Scholar]
- 15.Escolar DM, Hache LP, Clemens PR, Cnaan A, McDonald CM, Viswanathan V, Kornberg AJ, Bertorini TE, Nevo Y, Lotze T, Pestronk A, Ryan MM, Monasterio E, Day JW, Zimmerman A, Arrieta A, Henricson E, Mayhew J, Florence J, Hu F, Connolly AM. Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy. Neurology 77: 444–452, 2011. doi: 10.1212/WNL.0b013e318227b164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quattrocelli M, Barefield DY, Warner JL, Vo AH, Hadhazy M, Earley JU, Demonbreun AR, McNally EM. Intermittent glucocorticoid steroid dosing enhances muscle repair without eliciting muscle atrophy. J Clin Invest 127: 2418–2432, 2017. doi: 10.1172/JCI91445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raman SV, Hor KN, Mazur W, Cardona A, He X, Halnon N, Markham L, Soslow JH, Puchalski MD, Auerbach SR, Truong U, Smart S, McCarthy B, Saeed IM, Statland JM, Kissel JT, Cripe LH. Stabilization of early Duchenne cardiomyopathy with aldosterone inhibition: results of the multicenter AIDMD trial. J Am Heart Assoc 8: e013501, 2019. doi: 10.1161/JAHA.119.013501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rafael-Fortney JA, Chimanji NS, Schill KE, Martin CD, Murray JD, Ganguly R, Stangland JE, Tran T, Xu Y, Canan BD, Mays TA, Delfín DA, Janssen PM, Raman SV. Early treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in Duchenne muscular dystrophy mice. Circulation 124: 582–588, 2011. doi: 10.1161/CIRCULATIONAHA.111.031716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raman SV, Hor KN, Mazur W, Halnon NJ, Kissel JT, He X, Tran T, Smart S, McCarthy B, Taylor MD, Jefferies JL, Rafael-Fortney JA, Lowe J, Roble SL, Cripe LH. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 14: 153–161, 2015. doi: 10.1016/S1474-4422(14)70318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janssen PM, Murray JD, Schill KE, Rastogi N, Schultz EJ, Tran T, Raman SV, Rafael-Fortney JA. Prednisolone attenuates improvement of cardiac and skeletal contractile function and histopathology by lisinopril and spironolactone in the mdx mouse model of Duchenne muscular dystrophy. PLoS One 9: e88360, 2014. doi: 10.1371/journal.pone.0088360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chadwick JA, Hauck JS, Lowe J, Shaw JJ, Guttridge DC, Gomez-Sanchez CE, Gomez-Sanchez EP, Rafael-Fortney JA. Mineralocorticoid receptors are present in skeletal muscle and represent a potential therapeutic target. FASEB J 29: 4544–4554, 2015. doi: 10.1096/fj.15-276782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chadwick JA, Swager SA, Lowe J, Welc SS, Tidball JG, Gomez-Sanchez CE, Gomez-Sanchez EP, Rafael-Fortney JA. Myeloid cells are capable of synthesizing aldosterone to exacerbate damage in muscular dystrophy. Hum Mol Genet 25: 5167–5177, 2016. doi: 10.1093/hmg/ddw331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chadwick JA, Hauck JS, Gomez-Sanchez CE, Gomez-Sanchez EP, Rafael-Fortney JA. Gene expression effects of glucocorticoid and mineralocorticoid receptor agonists and antagonists on normal human skeletal muscle. Physiol Genomics 49: 277–286, 2017. doi: 10.1152/physiolgenomics.00128.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hauck JS, Lowe J, Rastogi N, McElhanon KE, Petrosino JM, Peczkowski KK, Chadwick AN, Zins JG, Accornero F, Janssen PM, Weisleder NL, Rafael-Fortney JA. Mineralocorticoid receptor antagonists improve membrane integrity independent of muscle force in muscular dystrophy. Hum Mol Genet 28: 2030–2045, 2019. doi: 10.1093/hmg/ddz039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hauck JS, Howard ZM, Lowe J, Rastogi N, Pico MG, Swager SA, Petrosino JM, Gomez-Sanchez CE, Gomez-Sanchez EP, Accornero F, Rafael-Fortney JA. Mineralocorticoid receptor signaling contributes to normal muscle repair after acute injury. Front Physiol 10: 1324, 2019. doi: 10.3389/fphys.2019.01324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chadwick JA, Bhattacharya S, Lowe J, Weisleder N, Rafael-Fortney JA. Renin-angiotensin-aldosterone system inhibitors improve membrane stability and change gene-expression profiles in dystrophic skeletal muscles. Am J Physiol Cell Physiol 312: C155–C168, 2017. doi: 10.1152/ajpcell.00269.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schütz G, Lumeng CN, Mortensen RM. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest 120: 3350–3364, 2010. doi: 10.1172/JCI41080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han SY, Kim CH, Kim HS, Jee YH, Song HK, Lee MH, Han KH, Kim HK, Kang YS, Han JY, Kim YS, Cha DR. Spironolactone prevents diabetic nephropathy through an anti-inflammatory mechanism in type 2 diabetic rats. J Am Soc Nephrol 17: 1362–1372, 2006. doi: 10.1681/ASN.2005111196. [DOI] [PubMed] [Google Scholar]
- 29.Belden Z, Deiuliis JA, Dobre M, Rajagopalan S. The role of the mineralocorticoid receptor in inflammation: focus on kidney and vasculature. Am J Nephrol 46: 298–314, 2017. doi: 10.1159/000480652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraccarollo D, Thomas S, Scholz CJ, Hilfiker-Kleiner D, Galuppo P, Bauersachs J. Macrophage mineralocorticoid receptor is a pleiotropic modulator of myocardial infarct healing. Hypertension 73: 102–111, 2019. doi: 10.1161/HYPERTENSIONAHA.118.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frieler RA, Meng H, Duan SZ, Berger S, Schütz G, He Y, Xi G, Wang MM, Mortensen RM. Myeloid-specific deletion of the mineralocorticoid receptor reduces infarct volume and alters inflammation during cerebral ischemia. Stroke 42: 179–185, 2011. doi: 10.1161/STROKEAHA.110.598441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li C, Zhang YY, Frieler RA, Zheng XJ, Zhang WC, Sun XN, Yang QZ, Ma SM, Huang B, Berger S, Wang W, Wu Y, Yu Y, Duan SZ, Mortensen RM. Myeloid mineralocorticoid receptor deficiency inhibits aortic constriction-induced cardiac hypertrophy in mice. PLoS One 9: e110950, 2014. doi: 10.1371/journal.pone.0110950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Montes-Cobos E, Schweingruber N, Li X, Fischer HJ, Reichardt HM, Lühder F. Deletion of the mineralocorticoid receptor in myeloid cells attenuates central nervous system autoimmunity. Front Immunol 8: 1319, 2017. doi: 10.3389/fimmu.2017.01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen ZX, Chen XQ, Sun XN, Sun JY, Zhang WC, Zheng XJ, Zhang YY, Shi HJ, Zhang JW, Li C, Wang J, Liu X, Duan SZ. Mineralocorticoid receptor deficiency in macrophages inhibits atherosclerosis by affecting foam cell formation and efferocytosis. J Biol Chem 292: 925–935, 2017. doi: 10.1074/jbc.M116.739243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun JY, Li C, Shen ZX, Zhang WC, Ai TJ, Du LJ, Zhang YY, Yao GF, Liu Y, Sun S, Naray-Fejes-Toth A, Fejes-Toth G, Peng Y, Chen M, Liu X, Tao J, Zhou B, Yu Y, Guo F, Du J, Duan SZ. Mineralocorticoid receptor deficiency in macrophages inhibits neointimal hyperplasia and suppresses macrophage inflammation through SGK1-AP1/NF-κB pathways. Arterioscler Thromb Vasc Biol 36: 874–885, 2016. doi: 10.1161/ATVBAHA.115.307031. [DOI] [PubMed] [Google Scholar]
- 36.Zhang YY, Li C, Yao GF, Du LJ, Liu Y, Zheng XJ, Yan S, Sun JY, Liu Y, Liu MZ, Zhang X, Wei G, Tong W, Chen X, Wu Y, Sun S, Liu S, Ding Q, Yu Y, Yin H, Duan SZ. Deletion of macrophage mineralocorticoid receptor protects hepatic steatosis and insulin resistance through ERα/HGF/Met pathway. Diabetes 66: 1535–1547, 2017. doi: 10.2337/db16-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrera-Chimal J, Estrela GR, Lechner SM, Giraud S, El Moghrabi S, Kaaki S, Kolkhof P, Hauet T, Jaisser F. The myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int 93: 1344–1355, 2018. doi: 10.1016/j.kint.2017.12.016. [DOI] [PubMed] [Google Scholar]
- 38.Miura R, Nakamura K, Miura D, Miura A, Hisamatsu K, Kajiya M, Nagase S, Morita H, Fukushima Kusano K, Ohe T, Ishihara K. Anti-inflammatory effect of spironolactone on human peripheral blood mononuclear cells. J Pharmacol Sci 101: 256–259, 2006. doi: 10.1254/jphs.sc0060049. [DOI] [PubMed] [Google Scholar]
- 39.Lowe J, Floyd KT, Rastogi N, Schultz EJ, Chadwick JA, Swager SA, Zins JG, Kadakia FK, Smart S, Gomez-Sanchez EP, Gomez-Sanchez CE, Raman SV, Janssen PM, Rafael-Fortney JA. Similar efficacy from specific and non-specific mineralocorticoid receptor antagonist treatment of muscular dystrophy mice. J Neuromuscul Dis 3: 395–404, 2016. doi: 10.3233/JND-160173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowe J, Kadakia FK, Zins JG, Haupt M, Peczkowski KK, Rastogi N, Floyd KT, Gomez-Sanchez EP, Gomez-Sanchez CE, Elnakish MT, Rafael-Fortney JA, Janssen PM. Mineralocorticoid receptor antagonists in muscular dystrophy mice during aging and exercise. J Neuromuscul Dis 5: 295–306, 2018. doi: 10.3233/JND-180323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Howard ZM, Lowe J, Blatnik AJ, Roberts D, Burghes AH, Bansal SS, Rafael-Fortney JA. Early inflammation in muscular dystrophy differs between limb and respiratory muscles and increases with dystrophic severity. Am J Pathol 191: 730–747, 2021. doi: 10.1016/j.ajpath.2021.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berger S, Wolfer DP, Selbach O, Alter H, Erdmann G, Reichardt HM, Chepkova AN, Welzl H, Haas HL, Lipp HP, Schütz G. Loss of the limbic mineralocorticoid receptor impairs behavioral plasticity. Proc Natl Acad Sci USA 103: 195–200, 2006. doi: 10.1073/pnas.0503878102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8: 265–277, 1999. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 44.Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet 18: 482–496, 2009. doi: 10.1093/hmg/ddn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang W, Hu P. Skeletal muscle regeneration is modulated by inflammation. J Orthop Translat 13: 25–32, 2018. doi: 10.1016/j.jot.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bentzinger CF, Wang YX, Dumont NA, Rudnicki MA. Cellular dynamics in the muscle satellite cell niche. EMBO Rep 14: 1062–1072, 2013. doi: 10.1038/embor.2013.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hardy D, Besnard A, Latil M, Jouvion G, Briand D, Thépenier C, Pascal Q, Guguin A, Gayraud-Morel B, Cavaillon JM, Tajbakhsh S, Rocheteau P, Chrétien F. Comparative study of injury models for studying muscle regeneration in mice. PLoS One 11: e0147198, 2016. doi: 10.1371/journal.pone.0147198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holmberg J, Durbeej M. Laminin-211 in skeletal muscle function. Cell Adh Migr 7: 111–121, 2013. doi: 10.4161/cam.22618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bigard AX, Janmot C, Sanchez H, Serrurier B, Pollet S, d’Albis A. Changes in myosin heavy chain profile of mature regenerated muscle with endurance training in rat. Acta Physiol Scand 165: 185–192, 1999. doi: 10.1046/j.1365-201x.1999.00487.x. [DOI] [PubMed] [Google Scholar]
- 50.Schiaffino S, Rossi AC, Smerdu V, Leinwand LA, Reggiani C. Developmental myosins: expression patterns and functional significance. Skelet Muscle 5: 22, 2015. doi: 10.1186/s13395-015-0046-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith LR, Barton ER. SMASH—semi-automatic muscle analysis using segmentation of histology: a MATLAB application. Skelet Muscle 4: 21, 2014. doi: 10.1186/2044-5040-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods 408: 89–100, 2014. doi: 10.1016/j.jim.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Paepe B, De Bleecker JL. Cytokines and chemokines as regulators of skeletal muscle inflammation: presenting the case of Duchenne muscular dystrophy. Mediators Inflamm 2013: 540370, 2013., doi: 10.1155/2013/540370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shen JZ, Morgan J, Tesch GH, Fuller PJ, Young MJ. CCL2-dependent macrophage recruitment is critical for mineralocorticoid receptor-mediated cardiac fibrosis, inflammation, and blood pressure responses in male mice. Endocrinology 155: 1057–1066, 2014. doi: 10.1210/en.2013-1772. [DOI] [PubMed] [Google Scholar]
- 55.van der Heijden CD, Deinum J, Joosten LA, Netea MG, Riksen NP. The mineralocorticoid receptor as a modulator of innate immunity and atherosclerosis. Cardiovasc Res 114: 944–953, 2018. doi: 10.1093/cvr/cvy092. [DOI] [PubMed] [Google Scholar]