

Abstract
Across the human genome, there are nearly 500 ‘ultraconserved’ elements: regions of at least 200 contiguous nucleotides that are perfectly conserved in both the mouse and rat genomes. Remarkably, the majority of these sequences are non-coding, and many can function as enhancers that activate tissue-specific gene expression during embryonic development. From their first description more than 15 years ago, their extreme conservation has both fascinated and perplexed researchers in genomics and evolutionary biology. The intrigue around ultraconserved elements only grew with the observation that they are dispensable for viability. Here, we review recent progress towards understanding the general importance and the specific functions of ultraconserved sequences in mammalian development and human disease and discuss possible explanations for their extreme conservation.
Introduction
The early 2000s marked an inflection point in comparative genomics, with researchers racing to mine for similarities and differences between the newly sequenced genomes of various vertebrate species. The Human Genome Project published an initial draft reference in 20011, with the first ‘finished’ version in 20042. In parallel, complementary efforts sequenced the genomes of other vertebrates to facilitate research performed in common laboratory models and because it was well-recognized that sequence conservation between humans and other organisms is a powerful tool for identifying loci with important functions in the human genome3–5. During this time, the rapid succession of sequenced reference genomes included mouse (2002)3, pufferfish (2002)6, rat (2004)5, chicken (2004)7, dog (2005)8, chimpanzee (2005)9, and others. Against this backdrop, early comparisons between the human, mouse, and rat genomes led to the identification of so-called ‘ultraconserved’ elements (UCEs), originally defined in 2004 as sequences with at least 200 base pairs (bp) of perfect, uninterrupted conservation between these three species10 (Fig 1a). In total, 481 sequences fulfilled these criteria, and the sequencing of genomes for additional vertebrate species has identified strong conservation of these loci beyond the mammalian lineage11–15. The majority of ultraconserved sequences were found outside of known or predicted gene exons and were thus non-coding10. This was surprising given the expectation that coding sequences would display higher levels of constraint than non-coding loci, a presumption predicated on the well-known importance of many protein-encoding genes for organismal viability, development, and overall health10.
Figure 1. Extreme conservation of ultraconserved elements.
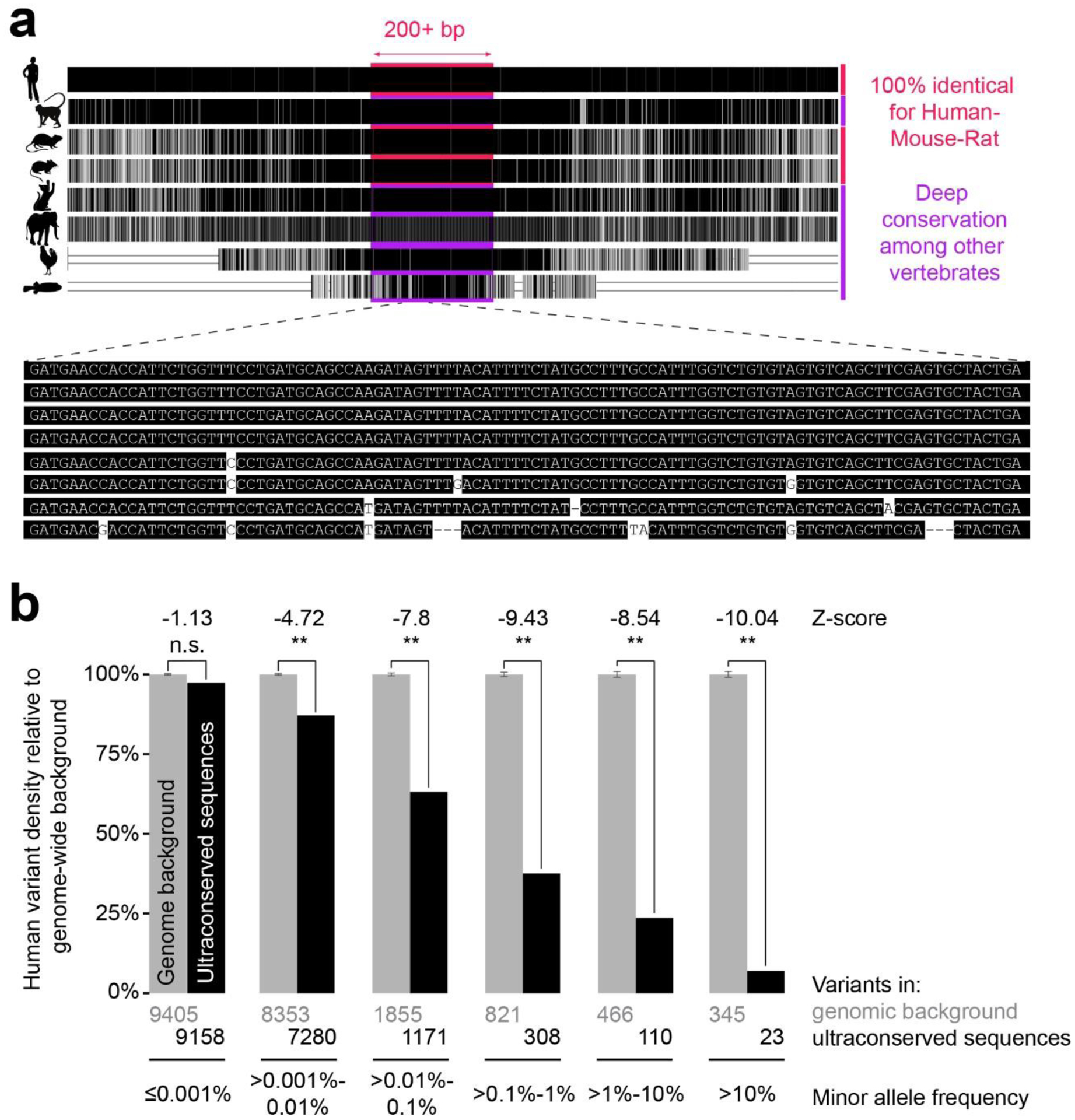
a. Schematic visualization of a multispecies sequence alignment illustrating the original definition of human–mouse–rat ultraconserved sequences (at least 200 bp of perfect sequence conservation in these three species), along with the deep conservation of these elements in other vertebrate genomes. b. Ultraconserved elements are strongly depleted for human sequence variants, with common variants showing the most severe depletion. Shown are human population variants that overlap ultraconserved elements compared to genome background. This analysis is similar to that performed in Ref.16 but was updated to use more recent population sequencing data, namely the 463 million variants observed in 62,784 individuals from the Trans-Omics for Precision Medicine (TOPMed) Whole Genome Sequencing Program97 and available through the BRAVO variant browser (https://bravo.sph.umich.edu/freeze5/hg38/). The genome background bar shows the median of 1000 iterations of random genomic sequences of the same size as the ultraconserved elements, with error bars showing the standard deviation. n.s., not significant; **, p-value < 0.001.
In addition to their strong conservation between species, ultraconserved elements show reduced sequence variation within the human population. Upon their initial discovery, it was observed that human single-nucleotide polymorphisms known at the time were heavily depleted (by ~20-fold) in these elements, further suggesting that mutations in these sites are selectively disadvantageous10. However, some speculated that highly conserved non-coding sequences could instead be mutation cold spots, presumably resulting from a theoretical molecular mechanism that repressed mutagenesis16. Subsequent larger-scale human population sequencing confirmed an overall depletion of variants in human ultraconserved sites and also showed that variants that are found are skewed towards those with rare derived allele frequencies16,17 (Fig. 1b shows similar analyses using more recent human population data). In other words, common variants are particularly depleted in ultraconserved loci, while extremely rare variants occur at levels close to those observed elsewhere in the genome, excluding the possibility that ultraconserved elements are generally protected from mutation. Furthermore, ultraconserved elements have been consistently shown to be depleted within copy number variants found in healthy individuals18–21. Collectively, these findings strongly support that these sites are under purifying selection, reinforcing their importance in human biology.
In recent years, experimental studies have painstakingly explored the reasons for ultraconservation. In parallel, the widespread adoption of human whole genome sequencing (WGS) has ignited new interest in both elucidating how mutations in ultraconserved sequences contribute to human disease and in the identification of loci with extreme evolutionary signatures. Here, we review research on ultraconserved sequences found in the human genome. We first focus on our current understanding of the functions and biological importance of the original 481 human-mouse-rat ultraconserved sequences, in particular their role in regulating gene expression during embryogenesis. We highlight their importance in establishing normal development while having no obvious impact on viability, and we investigate possible explanations for their perfect sequence conservation. Next, we discuss how sequence comparisons to additional vertebrate species have identified thousands of non-coding human loci with conservation levels similar to ultraconserved sequences and explore how studies of ultraconserved elements can inform our understanding of the functional importance of this much larger collection of extremely conserved sequences. We next describe variants in extremely well-conserved gene regulatory sequences that are associated with human phenotypes, especially neurodevelopmental disorders. Finally, we articulate outstanding questions that remain about ultraconserved elements and how these sequences could aid interpretation of newly identified variants from emerging whole-genome sequencing studies of human cohorts.
Functions of ultraconserved sequences
Human–rodent ultraconserved sequences are reported to play a role in various aspects of gene function and expression regulation, from activation of transcription to exon splicing (Fig. 2a). When first defined in 2004, ultraconserved sequences were shown to overlap exons of protein-coding genes (111/481, 23%), sequences that were possibly exons of protein-coding genes (114/481, 24%), and non-coding regions (256/481, 53%) in the human genome10. Because human genome annotation has improved substantially since 2004, we used the latest UCSC RefSeq gene annotation set to update these classifications (Fig. 2b). Consistent with the original study, 23% (110) of ultraconserved elements overlap exons of protein-coding genes, while the vast majority (371, or 77%) do not. Ultraconserved sequences that overlap exons of coding genes are enriched in genes associated with RNA processing, including regulation of RNA splicing10. Ultraconserved elements in 5’ untranslated regions (5’UTRs) of coding genes likely regulate the cell-type-specific translation of the resulting protein22. Some ultraconserved exons are alternatively spliced as poison exons, which interrupt the reading frames of genes, for example by introducing an in-frame early stop codon, and target the resulting messenger RNAs for degradation23 (Fig. 2a). Functional dissection of ultraconserved poison exons revealed them to be crucial for cultured cell growth, with some exhibiting tumor-suppressor activity24,25.
Figure 2. Ultraconserved sequence functions.

a. Cartoons showing functions attributed to ultraconserved elements (UCEs; pink boxes). b. Pie chart showing the functional classification of ultraconserved elements using current human RefSeq98 genome annotations. ‘Coding’ includes all types of exons for protein-coding genes, including untranslated regions (UTRs). c. For the 371 non-coding elements shown in b, the breakdown of those with validated or predicted enhancer activity. Outer circle: Transgenic mouse enhancer assay results for each element (positive, negative, or not tested). Enhancer activity was tested at a single mouse developmental stage (typically embryonic day (E)11.5). These results were obtained from the VISTA Enhancer Browser96 (https://enhancer.lbl.gov/) and were originally reported as part of two large-scale screens of ultraconserved elements for enhancer activity26,27. Inner circle: The fraction of ultraconserved elements in each category with (+) or without (−) histone H3K27ac, a strong predictor of enhancer activity, at some point during mouse embryonic development. Ultraconserved elements were intersected with H3K27ac data generated by the Encyclopedia of DNA Elements (ENCODE) project for a panel of mouse tissues covering E10.5 to birth28 (H3K27ac data available at https://www.encodeproject.org/). ncRNA, non-coding RNA.
The largest category of ultraconserved sequences comprises those that lie in the non-coding genome (Fig. 2b). These often cluster together and are enriched in the vicinity of developmental transcription factor (TF) genes10, which suggested a role for non-coding ultraconserved sequences in regulating the expression of key patterning genes during organismal development. This role was first confirmed for ultraconserved sites in the vicinity of Irx genes using in vivo transgenic enhancer-reporter assays15 (Box 1). Subsequent systematic screens using transgenic assays showed that many non-coding ultraconserved sequences activate tissue-specific reporter gene expression during mouse embryogenesis26,27. Out of 245 non-coding ultraconserved sequences tested in these two studies, 123 (50%) were reproducibly positive for enhancer activity (Fig. 2c), with the majority active in neural tissues (67/123, or 54%). These assays tested enhancer activity only at a single developmental time point (embryonic day 11.5 (E11.5)), so those that were negative could, in principle, activate gene expression at a different stage. Indeed, intersecting the 122 sequences negative for enhancer activity in transgenic assays at E11.5 with chromatin data from ENCODE’s high-resolution series of mouse tissues and developmental stages28 shows that 71 (58%) of these ‘negative’ sites are marked by H3K27ac, a strong predictor of enhancer activity, at some point between E10.5 and birth (Fig. 2c). Furthermore, of the 126 non-coding ultraconserved sequences not tested to date using transgenic enhancer assays, 93 display H3K27ac during mouse embryogenesis. Thus, 77% (287/371) of non-coding ultraconserved sequences have validated in vivo enhancer activity and/or harbor enhancer-associated histone modifications during mouse embryogenesis, consistent with the vast majority of non-coding ultraconserved elements regulating gene expression during development.
Box 1: Transgenic assays to study enhancer function of ultraconserved elements.
Transgenic assays, first developed over 30 years ago99,100, have been used extensively to characterize the enhancer functions of non-coding ultraconserved elements (see the figure). Transgenic methods have been developed for a variety of model organisms, and mouse26,27,55, Xenopus15, and zebrafish14,15 assays have all been used to examine the activities of human-mouse-rat ultraconserved enhancers. Historically, transgenic mouse assays have involved the cloning of a candidate regulatory element of interest in front of a minimal promoter (e.g., Hsp68 or β-globin) and a reporter gene (e.g., lacZ or GFP). The resulting DNA plasmid is linearized and microinjected into the pronucleus of fertilized mouse eggs, where it will randomly integrate into the genome, typically in a tandem, multi-copy configuration101. This method yields transgenic mice at any desired developmental stage, which can then be collected and visualized for the expression of the reporter gene, a proxy for the activity of the tested enhancer.
Despite the utility of a whole-organism readout, traditional mouse transgenesis suffers from several drawbacks, including 1) position effects, due to the transgene integrating at a random and unknown genomic locus, frequently result in ectopic reporter gene expression or silencing; 2) low integration efficiency (on average only ~12% of mouse embryos successfully take up the transgene construct); 3) the requirement for highly trained staff to perform pronuclear injections; 4) cost; and 5) limited throughput.
More recently, Kvon et al.52 used CRISPR–Cas9 techniques to develop a locus-specific transgenic mouse assay to reduce these shortcomings. Targeting transgenes to a specific genomic location necessitated flanking the traditional enhancer-promoter-reporter transgene with homology arms for an endogenous mouse locus, here the transcriptionally-neutral H11 locus. The resulting DNA plasmid, along with Cas9 protein and a single-guide RNA (sgRNA) targeting H11, is then injected into fertilized mouse eggs, resulting in reproducible site-specific integration (i.e., knock-in) of the transgene into the mouse H11 locus52. While the major goal of this approach was to eliminate undesirable position effects from random transgenesis, it unexpectedly also resulted in much higher integration efficiency (~50% of resulting mice are transgenic). This advance has led to a 4-fold increase in throughput for mouse-based gene regulatory assays and far less ectopic activity or silencing of the transgene. These improvements have enabled in vivo study designs not previously possible, including the ability to systematically assess the effects of mutations on enhancer activity. Indeed, this technology directly enabled a recent large-scale mutagenesis study of ultraconserved enhancers that assessed reasons for their extreme conservation55 (described in ‘Explanations for Enhancer Ultraconservation‘ in the main text).

Figure is adapted from REF52.
In addition to enhancer activity, non-coding ultraconserved sequences can have other roles in gene expression regulation. One element upstream of the HoxD locus has been reported with dual functions, acting as an enhancer or a repressor of gene expression depending on context and assay27,29. Non-coding ultraconserved sequences have been reported to be transcribed into non-coding RNAs (ncRNAs), especially in cancer cells30. Although less is known about the role of ultraconserved ncRNAs in development, anecdotally one long non-coding RNA (lncRNA) containing an ultraconserved element has been shown to play a role in genome organization during neurogenesis31. Recent work reported that noncoding ultraconserved sites are considerably depleted at topologically associating domain (TAD) boundaries and enriched inside domains, suggesting that they do not generally function as boundary elements in establishing high-order, three-dimensional genome organization32. In addition to the validated roles described above, alternative theoretical functions have been proposed for non-coding ultraconserved sequences. The most prominent of these is a chromosome counting function, whereby ultraconserved sequences are hypothesized to ensure accurate counting of chromosomes during cell division19. However, this proposed function currently lacks experimental support. In summary, 60% (287/481) of all ultraconserved sequences show characteristics of enhancer activity during embryonic development, 23% (110/481) fall within exons of coding genes, and anecdotal examples are associated with other functions.
The primary focus of the remainder of this Review will be on enhancer functions of ultraconserved sequences since they are the most extensively studied function to date and likely contribute to the conservation of the majority of ultraconserved sequences. Nonetheless, it is important to keep in mind that other functions may also be associated with some or most ultraconserved sequences.
Does ultraconserved equal ultra-important?
The studies described above were instrumental in identifying developmental enhancer activity as a predominant functional category of ultraconserved elements. However, these observations did not provide an immediate answer to the core evolutionary question surrounding these elements: does their extreme conservation signal that these sites are especially critical for viability? To begin to answer this question, Ahituv and colleagues selected four ultraconserved sequences and individually deleted each from the mouse genome33. Importantly, all four ultraconserved sites had previously been shown to act as enhancers in vivo, and all were in the vicinity of, and most had activity patterns similar to, genes that are critical for embryonic development (e.g., Dmrt1/2/3, Pax6, Arx and Sox3). Remarkably, mice that were homozygous-null for individual ultraconserved enhancers (or hemizygous-null males for enhancers on the X chromosome) showed no indication of increased prenatal lethality, were fully viable at birth, and lived well into adulthood. Assessments of pathology, growth, and gene expression did not reveal obvious detrimental phenotypes resulting from the loss of these enhancers. Given the extreme sequence conservation of these loci, the lack of apparent phenotypes was quite surprising to many in the field and led to various hypotheses as to the reason for this observation. Possible explanations included: the presence of other enhancers with functions partially redundant to ultraconserved enhancers12,27,33, the presence of phenotypes that are subtle and/or not readily apparent under laboratory conditions12,33, a susceptibility of ultraconserved sequences to gain-of-function mutations33, and the speculative possibility of unconventional drivers of conservation that would be sensitive to sequence changes but tolerant of homozygous deletion, such as the theoretical molecular copy counting function19.
Subsequent mouse studies, including the targeted deletion of additional ultraconserved enhancers and more in-depth phenotyping, have shed some light on this issue34–36. To date, a total of eight ultraconserved enhancers have been individually deleted from the mouse genome (Table 1). Some of the enhancers consist of 2–3 ultraconserved elements in close proximity, so, in total, 11 distinct ultraconserved elements from the original Bejerano et al. list have been deleted. Consistent with the Ahituv et al. results, all mice with homozygous/hemizygous-null deletions of ultraconserved enhancers to date have been viable and fertile33–35. However, detailed phenotyping revealed the presence of developmental phenotypes in several of the ultraconserved enhancer deletion lines (Table 1). Individual loss of two ultraconserved enhancers at different genomic loci and with different tissue-restricted activity patterns, one in forebrain (enhancer hs119, near Arx) and one in limb (enhancer hs280, near Tmem53), both resulted in overall body mass reduction34,35. Neither showed obvious phenotypes specific to the tissue in which they are active. Individual deletion of two additional ultraconserved enhancers (hs121 and hs122) near the Arx transcription factor gene, which is critical for the development of the brain and other tissues, resulted in brain abnormalities34. Loss of enhancer hs121 resulted in altered densities of specific cortical interneuron types, including cholinergic neurons. Loss of enhancer hs122, which is active in the dorsal forebrain, led to the reduced expression of Arx specifically in the dorsal forebrain, along with changes to the size and morphology of the hippocampus34. Importantly, these phenotypes have definitively confirmed the role of these ultraconserved enhancers in the regulation of Arx. Both the cholinergic neuron alterations and hippocampal defects observed mimic a subset of the changes observed when the Arx gene itself is deleted in mice34,37,38. Overall, non-lethal but potentially detrimental developmental phenotypes have been identified in half (4 of 8) of the mouse knockout lines where a single ultraconserved enhancer has been deleted (Table 1). There have thus far been no published experiments directly assessing if these phenotypes lead to decreased lifespan or are selected against in wild environments, but it is plausible to assume that such phenotypes would be detrimental in living conditions outside of a laboratory. For example, the hippocampus has a well-established role in memory formation and learning39, and reduction of cholinergic neurons is associated with Alzheimer disease in humans40.
Table 1.
Ultraconserved enhancers deleted from the mouse genome.
| Enhancer name (VISTA) | Ultraconserved elements (Bejerano et al.) | Tissue-specific activity | Neighboring genes | Phenotype(s) observed | Reference(s) |
|---|---|---|---|---|---|
| Single enhancer deletions | |||||
| hs112 | uc248 | Forebrain | Dmrt1/2/3 | None | 33 |
| hs113 | uc329 | Dorsal root ganglion | Pax6, Rcn1, Wt1 | None | 33 |
| hs427 | uc482 | Forebrain, hindbrain | Sox3, Atp11c | None | 33,36 |
| hs121 | uc467 | Forebrain | Arx, Pola1 | None in original study, altered densities of several neuron classes in the brain identified in subsequent study | 33,34 |
| hs119 | uc463-464-465 | Forebrain | Arx, Pola1 | Reduced body mass | 34 |
| hs122 | uc468–469 | Forebrain | Arx, Pola1 | Defects to hippocampus, decreased Arx expression in developing forebrain | 34 |
| hs123 | uc470 | Forebrain | Arx, Pola1 | None | 34 |
| hs280 | uc019 | Limb | Tmem53, Rnf220, Eri3 | Reduced body mass and overall body size, decreased expression of Tmem53 and Dmap1 in the limb | 35 |
| Compound enhancer deletions | |||||
| hs119+hs121 | uc463-464-465–467 | Forebrain | Arx, Pola1 | Altered densities of several neuron classes in the brain, reduced body mass, decreased Arx expression in developing forebrain, phenotypes more severe than loss of hs119 or hs121 alone | 34 |
| hs122+hs123 | uc468-469-470 | Forebrain | Arx, Pola1 | Defects to hippocampus, decreased Arx expression in developing forebrain, similar to loss of hs122 alone | 34 |
In all cases, homozygous-null (or hemizygous-null for X-linked loci) mice are viable, fertile, and born at Mendelian-expected frequencies. VISTA indicates the name of the enhancer in the VISTA Enhancer Browser96.
The high density of ultraconserved forebrain enhancers at the Arx locus has been critical for studying potential functional redundancy between these elements. At this locus, two distinct ultraconserved enhancers (hs119 and hs121) are active in overlapping spatial domains within the ventral forebrain, while two others (enhancers hs122 and hs123) drive expression in similar areas of the dorsal forebrain41. Like mice missing individual ultraconserved enhancers, mice with pairs of these potentially redundant enhancers (hs119+hs121 or hs122+hs123) deleted on the same haplotype were found to be viable34. In one case (hs119+hs121), hemizygous-null mice displayed a combination of the growth and neuronal phenotypes observed when either enhancer is deleted individually34. However, the phenotypes were generally more severe for the hs119+hs121 double knockout than either individual deletion. For the other double knockout (hs122+hs123), hemizygous-null mice showed a phenotype of similar severity to loss of enhancer hs122 alone (no phenotypes resulting from loss of enhancer hs123 alone have been described to date). Combined, these results suggested some limited functional redundancy between enhancers hs119 and hs121, since their combined loss resulted in a more severe phenotype. However, these results also highlighted that individual enhancers at this locus have their own distinct roles in regulating Arx expression and development. Collectively, mouse knockout studies of ultraconserved enhancers suggest that these loci are generally well conserved because they are often individually necessary for proper development, and their loss results in phenotypes that, while subtle or possibly not apparent under laboratory conditions, are likely to be selectively disadvantageous in the wild34.
Explanations for enhancer ultraconservation
Deletion experiments and associated developmental phenotypes addressed why ultraconserved enhancers are generally conserved but do not explain why they show uninterrupted sequence conservation. Mutagenesis studies of various less well-conserved elements indicate that enhancers can typically withstand some change to their nucleotide sequences without affecting their function, due to the presence of redundant binding sites for TFs and the degeneracy of TF recognition sequences42–46. For example, single nucleotide changes introduced into twenty-five different mammalian enhancers as part of massively parallel reporter assays (MPRAs) performed in cell culture and mouse liver showed only modest effects43–45. Individual sequence variants often had no measurable effect on enhancer activity, and those that did rarely changed the activity more than two-fold. Nucleotides that were more critical to enhancer activity tended to be more conserved, but that was not always the case. Changes to some conversed nucleotides did not alter enhancer activity, and not all nucleotides that altered activity were under purifying selection.
Comparative functional genomics studies have used complementary approaches to explore the effect of nucleotide variation on enhancer properties. For example, multiple studies have used ChIP-seq to enhancer-associated TFs to profile tissue or cells from different mouse strains and/or rodent species to determine whether and how sequence changes in TF recognition sites alter TF binding47,48. As expected, these studies show there is typically an enrichment of sequence variants in the TF binding sites (TFBSs) in loci where TF binding is not conserved between species. However, many loci that are functionally divergent in TF binding frequently have no sequence differences between the strains/species being compared, and many sites of conserved TF binding do have sequence differences in the TFBS. These studies have highlighted the importance of combinatorial binding of multiple different TF proteins in preserving the functions of putative enhancer regions. For example, binding of a TF is more likely to be functionally conserved between species if it occurs in close proximity to binding by other TFs47, and TF binding can be disrupted by sequence variants in neighboring binding sites48. Together, MPRA and comparative genomic studies reveal a complex relationship between genetic variation and functional changes in enhancer activity, with many individual variants having little impact on the functions of enhancers that are not ultraconserved.
In contrast, extended blocks of perfect sequence identity suggest that ultraconserved enhancers may be uniquely susceptible to the effects of mutagenesis and that even single base pair changes could alter their function. Mapping of in vitro protein–DNA interactions has revealed a high degree of TF occupancy throughout ultraconserved enhancers, which is attributed to overlapping conserved TFBSs49. Therefore, one possible explanation of ultraconservation at enhancers is that, unlike less well-conserved enhancers, every base pair in the sequence may be essential for enhancer activity (Fig. 3, Model 1). In this case, any mutation would result in the partial or complete loss of enhancer function and reduced expression of the enhancer’s target gene. Alternatively, ultraconserved enhancers may be uniquely susceptible to gain-of-function mutations that result in new spatiotemporal gene expression33 (Fig. 3, Model 2), a phenomenon observed in at least one less well-conserved enhancer50–52. Ultraconserved sequences may also not constitute single enhancer units but be composed of multiple overlapping enhancers that are active in different spatial domains or at different developmental stages (Fig. 3, Model 3). Finally, the function of transcriptional enhancers alone might be insufficient to explain the phenomenon of non-coding ultraconservation, and it has been proposed that multiple superimposed functional constraints could result in uninterrupted blocks of conserved nucleotide sequences53. For example, one ultraconserved element has been shown to have coding and enhancer activity, and both functions are likely under selective pressure54. Purely non-coding ultraconserved sites may harbor additional, unspecified, non-coding functions overlapping nucleotides responsible for enhancer activity (Fig. 3, Model 4). These proposed models are not mutually exclusive, and it is conceivable, or even likely, that a combination of two or more of these possibilities underlies the phenomenon of ultraconservation.
Figure 3. Possible drivers of enhancer ultraconservation.
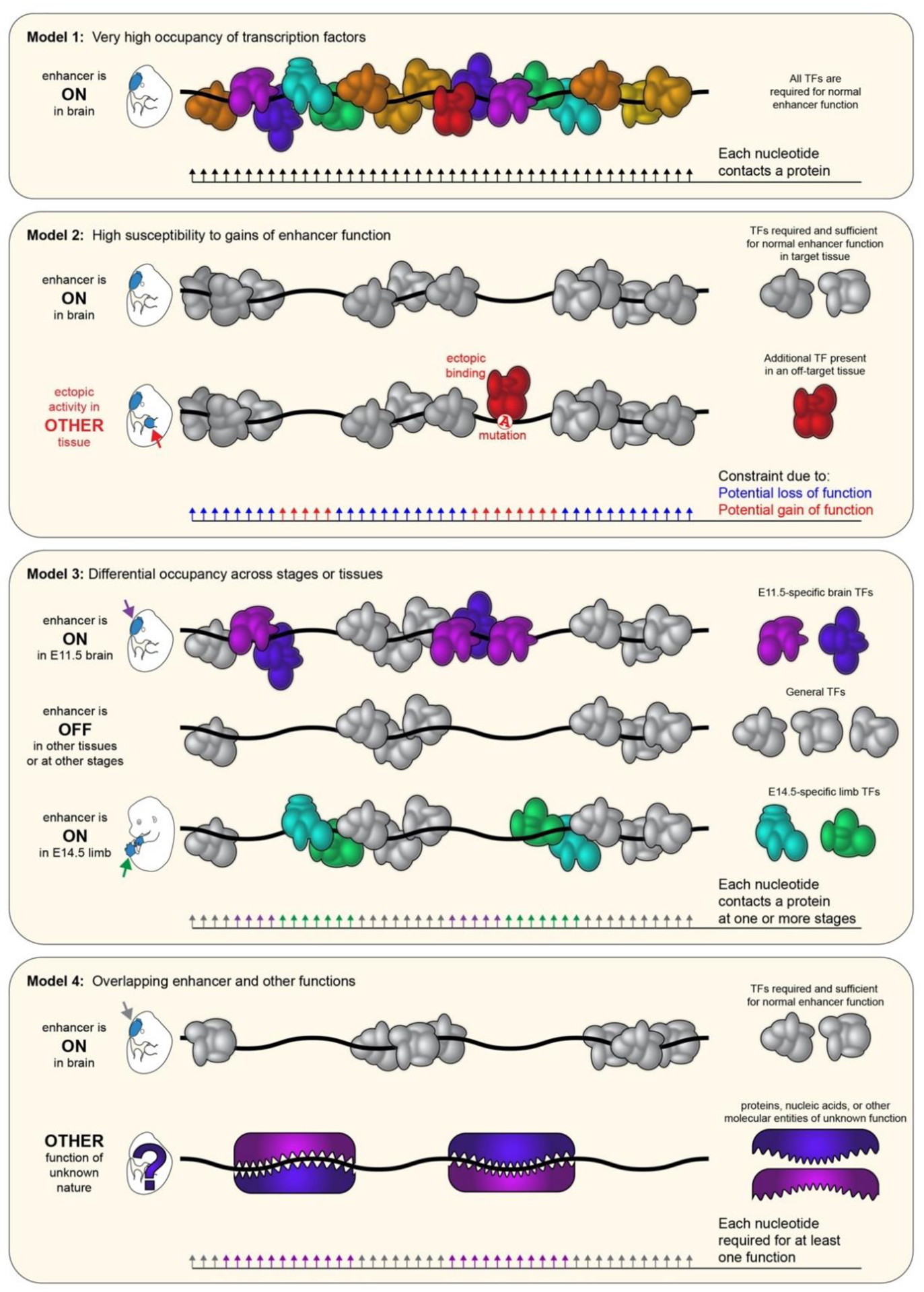
Schematic illustrations of molecular interactions that may result in the extreme evolutionary constraint observed at ultraconserved enhancers, including: extremely dense occupancy by transcription factors (TFs) and/or other proteins in the active state (Model 1); high susceptibility to gain of enhancer function through creation of TF binding sites (TFBSs) that act in concert with pre-existing TFBSs (Model 2); differential occupancy of enhancer subregions by TFs in different tissues and/or at different time points (Model 3); and a combination of known enhancer function and other molecular interactions and functions embedded in the same sequence (Model 4). See main text for details.
If ultraconserved enhancers indeed constitute a binding hub for interdependent, non-redundant transcription factors, their mutagenesis would be expected to result in significant changes to gene regulatory function. To address how mutations affect enhancer function of ultraconserved sequences, a recent study used transgenic mouse assays combined with large-scale mutagenesis of nearly two dozen ultraconserved enhancers in vivo, mutating from 2 to 20% of ultraconserved base pairs within each enhancer55. Initial experiments, aimed at testing the first hypothesis (Fig. 3, Model 1) were performed at a single developmental stage midway through mouse gestation (typically E11.5). Nearly all (83%) of the ultraconserved enhancers showed no decrease in enhancer activity upon mutation of at least 2% of their ultraconserved base pairs. Surprisingly, even at a substantial mutation rate of 5%, nearly half of them (44%) remained active. In one extreme case, an ultraconserved enhancer showed residual tissue-specific activity upon mutation of 20% of ultraconserved base pairs. These results indicated that ultraconserved enhancers do not commonly lose their enhancer function upon even significant levels of mutation. These data also enabled the testing of the second hypothesis (Fig. 3, Model 2), i.e. the gain of additional, ectopic enhancer activity upon mutation. In only rare cases (5% of all tested alleles) did mutations lead to a gain of enhancer activity, which included cases of stronger activity in the same tissue as well as activity in a new tissue. Together, these results suggest that ultraconserved enhancers are surprisingly robust to sequence changes and that susceptibility to either loss or gain of the known enhancer function at E11.5 is unlikely to be the sole explanation for ultraconservation.
General support for the notion that a combination of activities across tissues and time points (Figure 3, Model 3) may explain ultraconservation comes from the observation that many noncoding ultraconserved elements display the H3K27ac chromatin mark at multiple stages of embryonic development and in multiple tissues55. While this mark is associated with enhancer activity56, it is not a perfect predictor of regulatory function28,57. Therefore, this hypothesis was further tested by using the transgenic mouse assay approach to assess the activity of mutated ultraconserved enhancers at more than one developmental stage55. Out of 9 enhancers that had been impervious to loss-of-function for 5% mutagenesis at E11.5, five showed the same robustness at E14.5, while four showed reduced activity. These results offered partial support for Model 3. To explore this further, three of the tested 5% mutation alleles were used to replace the wild-type enhancers at the endogenous locations in the mouse genome. This included mutated alleles of enhancers hs121 and hs122 that lost regulatory activity in the transgenic mouse assay, along with an allele of hs121 that was robust to mutagenesis at both E11.5 and E14.5. As expected, alleles that had no enhancer function in transgenic assays resulted in brain phenotypes in postnatal mice, similar to those resulting from the complete loss of the enhancers. By contrast, one mutated allele that had normal function in transgenic assays resulted in normal brain development, suggesting that the introduced mutations did not substantially impact the enhancer’s activity at any preceding developmental stage. Therefore, while differential enhancer activities across tissues and stages exist, they are unlikely to be the sole explanation for ultraconservation.
While offering general support for three of the proposed models contributing to the selective constraint acting on ultraconserved sequences, it remains unclear if these are collectively sufficient to explain the perfect conservation. Notably, enhancer activity was only assessed at one or two developmental stages, and while it is likely that examining additional stages will reveal further detrimental effects of mutagenesis, the full quantitative extent of these stage-specific effects remains unclear. Furthermore, in vivo mouse transgenic assays provide data on enhancer activity throughout an organism but can miss more subtle quantitative changes to reporter gene expression. This study also did not assess the effect of mutations on possible functions aside from enhancer activity (Fig. 3, Model 4), which is conceptually challenging since these may include hitherto unidentified molecular mechanisms. These limitations notwithstanding, the collective findings from this study provide direct evidence for three of the proposed models, supporting that non-coding ultraconservation is likely to be driven by the cumulative effects of multiple forces, including loss- and gain-of-function effects, as well as negative selection to maintain stage- and tissue-specific enhancer activities.
This conclusion is consistent with a study of sequences that are ultraconserved across Drosophila species, which are distinct from those found in vertebrates but have similar overall characteristics, such as enrichment in the vicinity of developmental genes. This study reported multiple forces acting to maintain perfect conservation of ultraconserved exons58 by identifying that >70% of coding ultraconserved elements in Drosophila species are associated with at least two distinct functions: protein coding, alternative splicing, RNA editing, and/or TF binding. To confirm these multiple roles, the authors mutated one alternatively spliced ultraconserved exon in the Hox gene Ultrabithorax in a way that would not alter its protein coding potential. This resulted in a decrease in the expression of the corresponding mRNA isoform, most likely by altering the binding of splicing factors. Collectively, these studies illustrate that ultraconservation is likely to be maintained by multiple forces.
Extreme but not unparalleled constraint
The original definition of ultraconserved elements was striking in its simplicity10. It imposed the most extreme possible conservation threshold (perfect, uninterrupted conservation) across a considerable length (≥200 bp) between three mammalian reference genomes that were available at the time (human, mouse and rat). As such, it served as a valuable starting point for exploring the most extremely conserved subset of human non-coding sequences. However, the original description of ultraconserved sequences already noted that this definition is likely to capture only a small subset of strongly conserved sites in the human genome. For example, lowering the length threshold for human–mouse–rat comparisons to 100 bp increased the number of identified elements by an order of magnitude, to more than 5,00010. The use of comparison species other than mouse and rat, or even the use of different individuals to generate the human, mouse, and rat reference genomes, likely would have resulted in somewhat different lists of ultraconserved elements.
In parallel to the identification of ultraconserved elements, the availability of whole reference genomes for a growing number of mammalian and vertebrate species sparked the development of a variety of other strategies to identify extremely conserved sequences in the human genome. These approaches can be broadly grouped into three categories (Fig. 4). First, building on the concept of ultraconservation, searches for perfectly conserved stretches of sequence across varying sets of species and with different length thresholds resulted in partially overlapping but distinct sets of other types of ‘ultraconserved’ sequences12,59,60. Second, approaches imposing a sub-perfect, but considerable, sequence identity threshold across a small genomic window61,62 identified extremely conserved sequences, especially when applied to comparisons across phylogenetically distant species, such as humans and fish26,63–67. Third, methods building on more statistically advanced models that carefully measure nucleotide substitution events within the context of known phylogenetic distances and relationships of the species being compared provided more rigorous measures of actual evolutionary constraint53,68–70. These methods identified more than a million sequences in the human genome that are under negative selection. However, using stringent score and size thresholds they can be used to identify the most constrained subset of elements genome-wide from comparisons across a wide range of evolutionary distances.
Figure 4. Conservation-based approaches for identification of extremely conserved non-coding sequences.
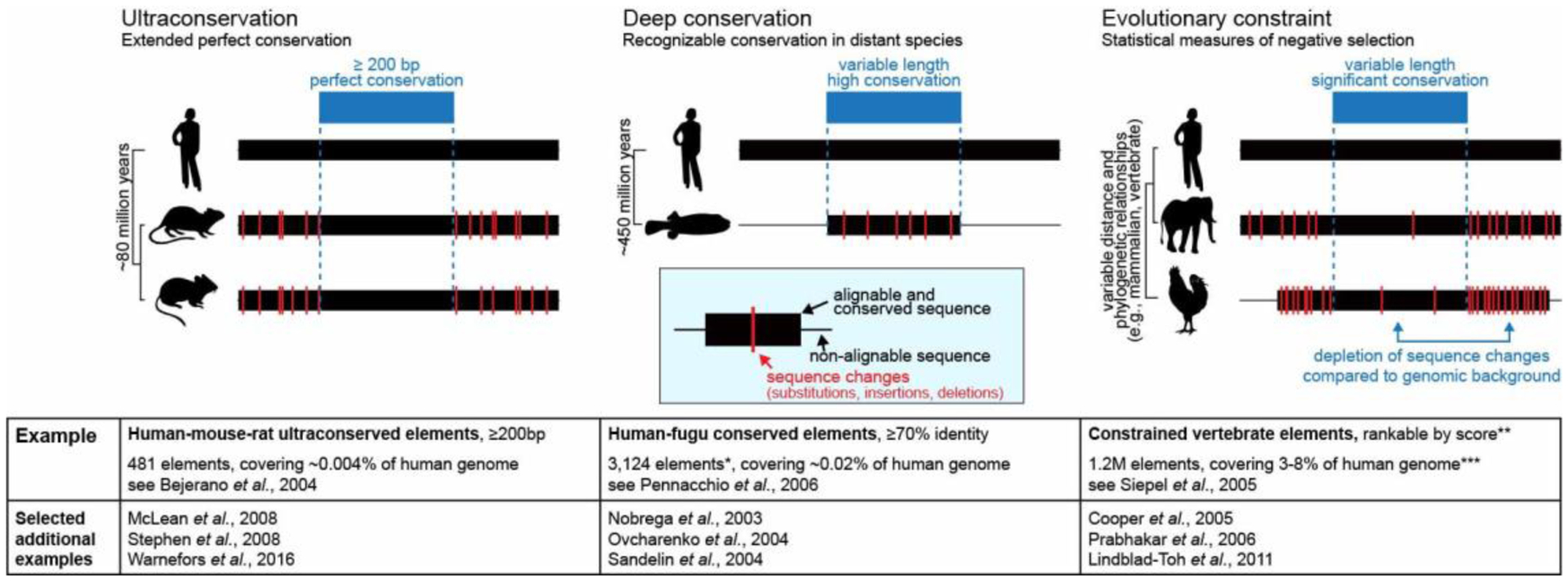
Left: Ultraconservation is based on defining a minimum length of contiguous nucleotides with perfect conservation across a selected set of species. Center: Deep conservation relies on the conservation of non-coding sequences across extremely long phylogenetic distances. Right: More-advanced statistical models quantify the strength of selection acting on conserved sequences and can be flexibly applied across a wide range of phylogenetic distances. *, non-coding elements only; **, scores range from extremely to moderately constrained elements; ***, depending on calibration and settings. Cited studies are REFS10,12,26,53,58,59,64,66–70.
These methods retrieved different sets of extremely conserved sequences that ranged in element size (from tens to thousands of nucleotides) and total number across the genome (from hundreds of ultraconserved elements to tens of thousands of sequences with statistically rigorous constraint scores in vertebrates). Despite these differences, some common patterns began to emerge. In most comparisons across different sets of categories, a strong correlation between perfect or near-perfect conservation in closely-related species and weaker conservation across extremely long phylogenetic distances was noted. For example, many human non-coding elements are conserved within the tetrapod and vertebrate linages, while non-coding sites that are conserved between humans and more distantly related non-chordate species (e.g., sea urchin, insects, sea anemone) are rare71,72. Like the human–mouse–rat ultraconserved sequences10, classes of extremely highly conserved sequences defined by other criteria are predominantly non-coding, and their genome-wide distribution is not random. They, too, show marked enrichment in the vicinity of certain categories of genes, such as transcriptional regulators and developmentally active genes53. Indeed, developmental regulatory genes are commonly embedded in genomic regulatory blocks, regions of extended interspecies synteny that contain the gene and a collection of highly conserved distal elements that regulate its expression73,74. Topologically associating domains that contain conserved regulatory elements have high conservation levels throughout the whole domain, suggesting that sequence conservation is preserved in blocks of these self-interacting domains75. Like human–mouse–rat ultraconserved sequences, systematic testing through large-scale in vivo reporter studies in transgenic mouse and fish models confirmed that many other types of extremely conserved non-coding sequences also act as reproducible, tissue-specific regulators of gene expression during vertebrate embryonic development14,26,65,76,77. However, these studies did not directly answer the question whether there is any difference with respect to this functional property between ultraconserved elements and the larger number of extremely, but imperfectly, conserved sequences present in the human genome.
A targeted investigation27 of this question tested the in vivo enhancer activity of more than 200 elements that showed extreme but imperfect evolutionary constraint in human–mouse–rat genome comparisons69 alongside more than 200 human–mouse–rat ultraconserved sequences. These ‘ultra-like’ sequences were identified using statistical measures of evolutionary constraint and showed similar characteristics to ultraconserved sequences in terms of branch length and rejected substitution counts in other mammalian species. Mouse transgenic assays revealed that half of the tested regions in both categories (115 of 231 regions, containing a total of 245 ultraconserved elements; and 102 of 206 ‘ultra-like’ regions tested) activated highly reproducible reporter expression in specific tissues or organs of developing mouse embryos27. Additionally, the distribution of embryonic tissues in which ultraconserved and the ‘ultra-like’-conserved sequences activated expression were virtually identical, with subregions of the central nervous system being the most frequent sites of reporter expression for both categories of elements. Likewise, both groups of elements showed strong enrichment near genes associated with the same set of biological functions, such as regulation of transcription (6.9-fold vs 5.3-fold enrichment in related gene ontology [GO] terms in ultraconserved vs ‘ultra-like’ sequences), development (4.8-fold vs 4.3-fold enrichment), and nervous system development (6.5-fold vs 6.3-fold enrichment). Cumulatively, these studies indicate that the initial definition of ultraconserved elements identified only a small subset of a much larger collection of similarly well-conserved sequences, with both similar distributions throughout the genome and functional properties, at least with respect to their likelihood of being developmentally active enhancers. Ultraconserved enhancers have been a useful model class for studying the functions, necessity, and other characteristics of this much larger category of highly conserved enhancers.
Phenotypic impacts of human variation
In addition to their functions and genomic properties, there has been considerable interest in understanding the role of ultraconserved and extremely conserved sequences in human phenotypes, especially in cancer and developmental phenotypes. Ultraconserved elements are enriched within somatic copy number alterations found in cancerous cells21, and common germline sequence variants in ultraconserved elements are associated with prostate cancer survival and recurrence78. These examples suggest that DNA sequence variants, both inherited and somatic, within ultraconserved elements can influence cancer pathogenesis. Additionally, transcription of ultraconserved elements into ncRNAs has been studied in cancer. Changes in the expression levels of certain ultraconserved ncRNAs was found to characterize some cancer types30, and this topic has been thoroughly reviewed previously79,80.
In addition to cancer, there is growing evidence that sequence variants in ultraconserved and extremely conserved enhancers may contribute to developmental phenotypes. One example comes from aniridia81, a severe eye condition that typically results from haploinsufficient mutations in the gene PAX6. Resequencing of conserved enhancers in the vicinity of PAX6 in patients with aniridia that could not be explained by PAX6 coding mutations identified a de novo variant in the nearby SIMO enhancer in a single individual. This enhancer is active in the eye and other tissues during embryogenesis, and it harbors an ultraconserved core sequence (uc325). Although the de novo variant falls outside of the core ultraconserved sequence, it changes an otherwise highly conserved base within the enhancer and abolishes SIMO activity specifically in the eye, suggesting that it could contribute to the development of aniridia by leading to a reduction in PAX6 expression.
More recently, copy number analyses and higher-throughput sequencing of increasingly large human cohorts are signaling that variants in or encompassing highly conserved non-coding regulatory sequences could underlie neurodevelopmental phenotypes. For example, ultraconserved sequences are enriched both within de novo copy number variants and in the vicinity of balanced structural rearrangements found in subjects with neurodevelopmental disorders82,83. Additionally, the Deciphering Developmental Disorders (DDD) study resequenced the most highly conserved ~4,400 non-coding sequences in the human genome in a cohort of nearly 8,000 individuals with genetically unexplained developmental disorders84. This study identified an enrichment of de novo variants from individuals with neurodevelopmental disorders specifically in highly conserved non-coding regulatory sequences predicted to be active in the brain. Collectively, these studies suggest that variants that disrupt the gene regulatory activity of ultraconserved and extremely conserved non-coding sequences may underlie some cases of developmental disorders, particularly those with neural phenotypes.
Conclusions and perspectives
In the more than 15 years since their initial description, ultraconserved elements have remained a fascinating, perplexing, and important class of DNA sequences. These elements, originally defined as 481 sites with perfect sequence conservation between three available mammalian reference genomes, have grown to a larger group of thousands of extremely conserved sites in the human genome that, while generally imperfectly conserved in human–mouse–rat alignments, show overall similar levels of evolutionary constraint in other vertebrates and mammals. The majority of these elements, both ultraconserved and extremely conserved, have clear and often critical roles in regulating the expression of genes during embryogenesis. In the intervening years, interest in ultraconserved elements has also extended beyond humans and into ecology. Ultraconserved and other extremely conserved sites have been defined for numerous other species. For example, in addition to vertebrates, Siepel et al. identified highly conserved noncoding sequences for insects, yeasts, and worms53. Additionally, targeted capture probes to ultraconserved elements have been used to sequence selected genomic regions from non-model species85, a method that is commonly used to map phylogenetic relationships of wild species and museum specimens, including birds86, insects87, and fishes88.
Currently, unanswered questions remain about why ultraconserved sequences are so highly conserved and exactly what function evolutionary selection is acting on to maintain this conservation. To date, half of all ultraconserved enhancers deleted in mice have not been shown to result in a potentially detrimental phenotype, and there has been no direct demonstration that loss of any ultraconserved enhancer results in reduced viability, fertility, or fecundity. Nevertheless, evolution acts over generations, and selection against even small fitness defects will readily remove deleterious variants from the population. Therefore, effects on these traits may be very subtle or not observable under typical laboratory conditions. To explore if the loss of ultraconserved enhancers affects viability and reproductive success over generations, population-based field experiments, like those that have been used to study selection in wild mouse populations89, would be insightful.
Additionally, ultraconserved enhancers appear to be surprisingly robust to mutagenesis given their extreme level of sequence constraint, and no single hypothesis experimentally examined to date seems to fully explain their conservation. Since ultraconserved constraint is likely to be due to a combination of factors, future work should explore evidence for all potential drivers more fully, including the following lines of research. First, highly quantitative massively parallel reporter assays could be used in an appropriate cell type to determine whether ultraconserved enhancer mutagenesis results in modest changes to gene expression below the detection level of transgenic assays. Second, the effects of mutations on in vivo enhancer activity could be assessed across a larger number of developmental stages. Finally, epigenomic data and experimentation could be used to elucidate whether non-coding ultraconserved elements broadly have additional functions (e.g., repressor activity).
The increased use of human WGS, including for both healthy controls and patients, offers additional exciting avenues for exploring the functions and importance of non-coding ultraconserved and other extremely conserved sequences, particularly for developmental phenotypes. As ultraconserved elements were identified by sequence comparisons between species, non-coding elements with extreme conservation within the human population could be identified by comparing genomes from millions of healthy human controls. Do such elements show similar or different functional profiles as human–mouse–rat ultraconserved elements? Are there non-coding regions of the genome that are highly conserved within humans but not between humans and other species? Initial analyses in this vein indicate that while some non-coding ultraconserved sites are under extreme purifying selection against sequence variants within the human population, the majority are subject to weaker selection in humans90. Finally, many human genetics studies are performing WGS on thousands of subjects with developmental phenotypes or birth defects to identify the genetic drivers of these traits (e.g., Refs.91,92). However, identifying pathogenic non-coding sequence variants from among the vast majority that are benign remains extraordinarily challenging92–94 and often requires extensive experimental testing52,95. As suggested by the Deciphering Developmental Disorders study described above, one strategy to ameliorate this challenge is to focus first on variants that change base pairs of very highly conserved non-coding sequences with known regulatory functions. Given the established role of ultraconserved and other extremely conserved enhancers in regulating developmental gene expression, their high degree of conservation, and that their mutation or loss is already implicated in developmental phenotypes in both mice and humans, experimental interrogation of variants in these enhancers could be an effective initial strategy for interpreting WGS studies of human developmental phenotypes.
Acknowledgements
D.E.D. and L.A.P. are Weill Neurohub Investigators, and this work was supported by U.S. National Institutes of Health grants R01HG003988 (to L.A.P.), UM01HG009421 (to L.A.P. and A.V.), R01MH117106 (to D.E.D.), and R01DE028599 (to A.V.). Research was conducted at the E.O. Lawrence Berkeley National Laboratory and performed under U.S. Department of Energy Contract DE-AC02-05CH11231, University of California.
Glossary
- Coding
The portion of the genome that encodes proteins.
- Derived allele frequency
A derived allele is a variant that occurs to change a sequence from its previous (ancestral) state. The frequency of an allele is the percentage of the allele in a given population, relative to all observed alleles present at a specific site.
- Comparative genomics
Comparing DNA sequences of different organisms to identify similarities and differences. Unexpectedly high sequence similarities of loci between species indicate conservation due to negative evolutionary selection and, therefore, often pinpoint regions of the genome that have important functions.
- Ectopic
In the context of this review, ectopic refers to expression of a gene in an incorrect location. For example, ectopic reporter gene expression can occur as a result of mutating an enhancer linked to the reporter gene or from a reporter transgene integrating into the genome near an active regulatory element.
- Functional redundancy
When two or more genomic elements (e.g., genes or enhancers) perform the same function. When one element is removed, the remaining element is sufficient to carry out this function alone.
- Haploinsufficient
Here, this refers to a dominant mutation that alters the expression of a gene such that the remaining wild-type copy of the gene does not produce sufficient quantities of the encoded protein to prevent a phenotypic change.
- Non-coding
The portion of the genome that does not encode proteins. Approximately 98% of the human genome is non-coding2.
- Purifying selection
An evolutionary pressure to remove deleterious sequence variants from a population. Also known as negative selection.
- Synteny
Here, this refers to the conservation in ordering of several blocks of sequence between species.
- Ultraconserved elements
Originally defined as regions of the human reference genome of at least 200 base pairs that are perfectly conserved to both the mouse and rat reference genomes, which is the definition used throughout this Review. In the literature, this term has been more broadly used to refer to highly conserved sequences identified using various definitions of conservation and different combinations of species, which we refer to here instead as “extremely conserved” sequences.
Footnotes
Competing Interests
The authors declare no competing interests.
Bibliography
- 1.Lander ES et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 2.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Mouse Genome Sequencing Consortium et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Jacob HJ & Kwitek AE Rat genetics: attaching physiology and pharmacology to the genome. Nat. Rev. Genet 3, 33–42 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Gibbs RA et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428, 493–521 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Aparicio S et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297, 1301–1310 (2002). [DOI] [PubMed] [Google Scholar]
- 7.International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Lindblad-Toh K et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438, 803–819 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Bejerano G et al. Ultraconserved elements in the human genome. Science 304, 1321–1325 (2004). [DOI] [PubMed] [Google Scholar]; This is the first work to describe ultraconserved elements in the human genome.
- 11.Hecker N & Hiller M A genome alignment of 120 mammals highlights ultraconserved element variability and placenta-associated enhancers. Gigascience 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLean C & Bejerano G Dispensability of mammalian DNA. Genome Res. 18, 1743–1751 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ovcharenko I Widespread ultraconservation divergence in primates. Mol. Biol. Evol 25, 1668–1676 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navratilova P et al. Systematic human/zebrafish comparative identification of cis-regulatory activity around vertebrate developmental transcription factor genes. Dev. Biol 327, 526–540 (2009). [DOI] [PubMed] [Google Scholar]
- 15.de la Calle-Mustienes E et al. A functional survey of the enhancer activity of conserved non-coding sequences from vertebrate Iroquois cluster gene deserts. Genome Res. 15,1061–1072 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the first detailed experimental analysis of an ultraconserved enhancer.
- 16.Drake JA et al. Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat. Genet 38, 223–227 (2006). [DOI] [PubMed] [Google Scholar]; This study used nascent human population sequencing data from the International HapMap Project to show that extremely conserved non-coding elements display higher rates of depletion for common human variants than rare variants, consistent with negative selection acting to maintain sequence conservation at these sites.
- 17.Habic A et al. Genetic Variations of Ultraconserved Elements in the Human Genome. OMICS 23, 549–559 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Derti A, Roth FP, Church GM & Wu C-T Mammalian ultraconserved elements are strongly depleted among segmental duplications and copy number variants. Nat. Genet 38, 1216–1220 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Chiang CWK et al. Ultraconserved elements: analyses of dosage sensitivity, motifs and boundaries. Genetics 180, 2277–2293 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conrad DF et al. Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCole RB, Fonseka CY, Koren A & Wu C-T Abnormal dosage of ultraconserved elements is highly disfavored in healthy cells but not cancer cells. PLoS Genet. 10, e1004646 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Byeon GW et al. Functional and structural basis of extreme conservation in vertebrate 5’ untranslated regions. Nat. Genet 53, 729–741 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leclair NK et al. Poison Exon Splicing Regulates a Coordinated Network of SR Protein Expression during Differentiation and Tumorigenesis. Mol. Cell 80, 648–665.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas JD et al. RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat. Genet 52, 84–94 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lareau LF, Inada M, Green RE, Wengrod JC & Brenner SE Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446, 926–929 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Pennacchio LA et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502 (2006). [DOI] [PubMed] [Google Scholar]; This paper describes the first systematic characterization of ultraconserved elements for enhancer function and definitively established that many regulate gene expression during embryonic development.
- 27.Visel A et al. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat. Genet 40, 158–160 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorkin DU et al. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583, 744–751 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kushawah G & Mishra RK Ultraconserved Sequences Associated with HoxD Cluster Have Strong Repression Activity. Genome Biol. Evol 9, 2049–2054 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calin GA et al. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 12, 215–229 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Cajigas I et al. The Evf2 Ultraconserved Enhancer lncRNA Functionally and Spatially Organizes Megabase Distant Genes in the Developing Forebrain. Mol. Cell 71, 956–972.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCole RB, Erceg J, Saylor W & Wu C-T Ultraconserved Elements Occupy Specific Arenas of Three-Dimensional Mammalian Genome Organization. Cell Rep. 24, 479–488 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahituv N et al. Deletion of ultraconserved elements yields viable mice. PLoS Biol. 5, e234 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work describes the first mouse knockout studies of ultraconserved enhancers, which stunningly found that mice missing individual ultraconserved elements were viable and had no obvious phenotypes.
- 34.Dickel DE et al. Ultraconserved Enhancers Are Required for Normal Development. Cell 172, 491–499.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; With Nolte et al (2014) this study established that loss of ultraconserved enhancers in mice commonly results in developmental phenotypes that are likely to be selectively disadvantageous, partially explaining their extreme conservation.
- 35.Nolte MJ et al. Functional analysis of limb transcriptional enhancers in the mouse. Evol. Dev 16, 207–223 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work was the first to identify a phenotype resulting from the deletion of an ultraconserved enhancer in mice.
- 36.Gaynor KU et al. Studies of mice deleted for Sox3 and uc482: relevance to X-linked hypoparathyroidism. Endocr Connect 9, 173–186 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colasante G et al. ARX regulates cortical intermediate progenitor cell expansion and upper layer neuron formation through repression of Cdkn1c. Cereb. Cortex 25, 322–335 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitamura K et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat. Genet 32, 359–369 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Squire LR Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol. Rev 99, 195–231 (1992). [DOI] [PubMed] [Google Scholar]
- 40.Schliebs R & Arendt T The cholinergic system in aging and neuronal degeneration. Behav. Brain Res 221, 555–563 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Visel A et al. A high-resolution enhancer atlas of the developing telencephalon. Cell 152, 895–908 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jindal GA & Farley EK Enhancer grammar in development, evolution, and disease: dependencies and interplay. Dev. Cell 56, 575–587 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patwardhan RP et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol 30, 265–270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melnikov A et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol 30, 271–277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kircher M et al. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun 10, 3583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Canver MC et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stefflova K et al. Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell 154, 530–540 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heinz S et al. Effect of natural genetic variation on enhancer selection and function. Nature 503, 487–492 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Viturawong T, Meissner F, Butter F & Mann M A DNA-centric protein interaction map of ultraconserved elements reveals contribution of transcription factor binding hubs to conservation. Cell Rep. 5, 531–545 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Lettice LA, Hill AE, Devenney PS & Hill RE Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum. Mol. Genet 17, 978–985 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Lettice LA et al. Opposing functions of the ETS factor family define Shh spatial expression in limb buds and underlie polydactyly. Dev. Cell 22, 459–467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kvon EZ et al. Comprehensive In Vivo Interrogation Reveals Phenotypic Impact of Human Enhancer Variants. Cell 180, 1262–1271.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siepel A et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper introduced phastCons, one of the most widely used methods to identify highly conserved sequences with multiple species alignments.
- 54.Lampe X et al. An ultraconserved Hox-Pbx responsive element resides in the coding sequence of Hoxa2 and is active in rhombomere 4. Nucleic Acids Res. 36, 3214–3225 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Snetkova V et al. Ultraconserved enhancer function does not require perfect sequence conservation. Nat. Genet 53, 521–528 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the most comprehensive examination of how sequence changes alter the activity of ultraconserved enhancers, finding that they are surprisingly robust to mutation. Collectively, the results suggest that there are likely multiple molecular drivers behind ultraconservation.
- 56.Rada-Iglesias A et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.ENCODE Project Consortium et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warnefors M, Hartmann B, Thomsen S & Alonso CR Combinatorial Gene Regulatory Functions Underlie Ultraconserved Elements in Drosophila. Mol. Biol. Evol 33, 2294–2306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stephen S, Pheasant M, Makunin IV & Mattick JS Large-scale appearance of ultraconserved elements in tetrapod genomes and slowdown of the molecular clock. Mol. Biol. Evol 25, 402–408 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Christley S, Lobo NF & Madey G Multiple organism algorithm for finding ultraconserved elements. BMC Bioinformatics 9, 15 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mayor C et al. VISTA : visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16, 1046–1047 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Schwartz S et al. PipMaker--a web server for aligning two genomic DNA sequences. Genome Res. 10, 577–586 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nobrega MA & Pennacchio LA Comparative genomic analysis as a tool for biological discovery. J. Physiol 554, 31–39 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sandelin A et al. Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics 5, 99 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Woolfe A et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 3, e7 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ovcharenko I, Stubbs L & Loots GG Interpreting mammalian evolution using Fugu genome comparisons. Genomics 84, 890–895 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Nobrega MA, Ovcharenko I, Afzal V & Rubin EM Scanning human gene deserts for long-range enhancers. Science 302, 413 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Cooper GM et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15, 901–913 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prabhakar S et al. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res. 16, 855–863 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lindblad-Toh K et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Royo JL et al. Transphyletic conservation of developmental regulatory state in animal evolution. Proc. Natl. Acad. Sci. U. S. A 108, 14186–14191 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clarke SL et al. Human Developmental Enhancers Conserved between Deuterostomes and Protostomes. PLoS Genetics vol. 8 e1002852 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kikuta H et al. Genomic regulatory blocks encompass multiple neighboring genes and maintain conserved synteny in vertebrates. Genome Res. 17, 545–555 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Touceda-Suárez M et al. Ancient Genomic Regulatory Blocks Are a Source for Regulatory Gene Deserts in Vertebrates after Whole-Genome Duplications. Mol. Biol. Evol 37, 2857–2864 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harmston N et al. Topologically associating domains are ancient features that coincide with Metazoan clusters of extreme noncoding conservation. Nat. Commun 8, 441 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Poulin F et al. In vivo characterization of a vertebrate ultraconserved enhancer. Genomics 85, 774–781 (2005). [DOI] [PubMed] [Google Scholar]
- 77.Ragvin A et al. Long-range gene regulation links genomic type 2 diabetes and obesity risk regions to HHEX, SOX4, and IRX3. Proc. Natl. Acad. Sci. U. S. A 107, 775–780 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bao B-Y et al. Genetic variants in ultraconserved regions associate with prostate cancer recurrence and survival. Sci. Rep 6, 22124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Terracciano D et al. The role of a new class of long noncoding RNAs transcribed from ultraconserved regions in cancer. Biochim. Biophys. Acta Rev. Cancer 1868, 449–455 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Fabris L & Calin GA Understanding the Genomic Ultraconservations: T-UCRs and Cancer. Int. Rev. Cell Mol. Biol 333, 159–172 (2017). [DOI] [PubMed] [Google Scholar]
- 81.Bhatia S et al. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am. J. Hum. Genet 93, 1126–1134 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martínez F et al. Enrichment of ultraconserved elements among genomic imbalances causing mental delay and congenital anomalies. BMC Med. Genomics 3, 54 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McCole RB et al. Structural disruption of genomic regions containing ultraconserved elements is associated with neurodevelopmental phenotypes. Cold Spring Harbor Laboratory 233197 (2017) doi: 10.1101/233197. [DOI] [Google Scholar]
- 84.Short PJ et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature 555, 611–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Faircloth BC et al. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst. Biol 61, 717–726 (2012). [DOI] [PubMed] [Google Scholar]
- 86.Winker K, Glenn TC & Faircloth BC Ultraconserved elements (UCEs) illuminate the population genomics of a recent, high-latitude avian speciation event. PeerJ 6, e5735 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blaimer BB, Lloyd MW, Guillory WX & Brady SG Sequence Capture and Phylogenetic Utility of Genomic Ultraconserved Elements Obtained from Pinned Insect Specimens. PLoS One 11, e0161531 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gilbert PS et al. Genome-wide ultraconserved elements exhibit higher phylogenetic informativeness than traditional gene markers in percomorph fishes. Mol. Phylogenet. Evol 92, 140–146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barrett RDH et al. Linking a mutation to survival in wild mice. Science 363, 499–504 (2019). [DOI] [PubMed] [Google Scholar]
- 90.Dukler N, Mughal MR, Ramani R, Huang Y-F & Siepel A Extreme purifying selection against point mutations in the human genome. bioRxiv 2021.08.23.457339 (2021) doi: 10.1101/2021.08.23.457339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Richter F et al. Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat. Genet 52, 769–777 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Werling DM et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet 50, 727–736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boycott KM et al. A Diagnosis for All Rare Genetic Diseases: The Horizon and the Next Frontiers. Cell 177, 32–37 (2019). [DOI] [PubMed] [Google Scholar]
- 94.Lappalainen T, Scott AJ, Brandt M & Hall IM Genomic Analysis in the Age of Human Genome Sequencing. Cell 177, 70–84 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tewhey R et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 165, 1519–1529 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Visel A, Minovitsky S, Dubchak I & Pennacchio LA VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–92 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Taliun D et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.O’Leary NA et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kothary R et al. Inducible expression of an hsp68-lacZ hybrid gene in transgenic mice. Development 105, 707–714 (1989). [DOI] [PubMed] [Google Scholar]
- 100.Zakany J, Tuggle CK, Patel MD & Nguyen-Huu MC Spatial regulation of homeobox gene fusions in the embryonic central nervous system of transgenic mice. Neuron 1, 679–691 (1988). [DOI] [PubMed] [Google Scholar]
- 101.Rijkers T, Peetz A & Rüther U Insertional mutagenesis in transgenic mice. Transgenic Res. 3, 203–215 (1994). [DOI] [PubMed] [Google Scholar]