

Summary
Cancer incidence and survival are different between men and women. Indeed, females have a lesser risk and a better prognosis than males in many tumors unrelated to reproductive functions. Although the reasons for these disparities are still unknown, they constitute an important starting point for the development of personalized cancer therapies. One of the mechanisms that fuels carcinogenesis is the accumulation of defects in DNA damage response (DDR) pathways, a complex signaling cascade that senses DNA lesions and, depending on the severity, coordinates transient cell-cycle arrest, DNA replication, repair, apoptosis, and senescence, preventing genomic instability and cancer. Recently, evidence of sexual dimorphisms is emerging in these pathways, therefore providing new opportunities for precision medicine. Here, we will discuss current knowledge about sexual disparities in the DDR, their role in tumorigenesis and cancer progression, and the importance of considering sex contribution in both research and cancer therapies.
Subject areas: Cellular physiology, Cell biology, Cancer
Graphical abstract
Cellular physiology; Cell biology; Cancer;
Introduction
Sex differences are evident in both incidence and survival in many different cancer types unrelated to reproductive functions (Sung et al., 2021). In fact, males have a higher probability to develop cancer and a worst prognosis than females for many somatic tumors like lung, bladder, colon, skin, liver, and brain cancer (Figure 1). Reasons for these differences are still not well understood, but, besides environment and lifestyle, it is known that they partially rely on circulating sex hormones. Indeed, these molecules are involved in both tumorigenesis and cancer susceptibility through different mechanisms affecting the renewal of cancer stem cell, the metabolism, the tumor microenvironment, and the immune system (Clocchiatti et al., 2016).
Figure 1.

Sexual dimorphism in the incidence of different cancer types unrelated to reproductive functions
The pie charts represent the percentages of new diagnosed cancer cases in 2020 among men and women. The % values have been calculated using data retrieved from the Global Cancer Observatory GLOBOCAN 2020 (Sung et al., 2021).
However, it is important to note that steroid hormones action represents only one of the mechanisms responsible for sexual dimorphisms in cancer, that are instead the result of combined and independent effects of multiple sex-biasing factors. Accordingly, sex disparities can be found in many different pathways important for tumor initiation, sustainment, and progression, such as regulation of senescence, angiogenesis, metabolism, immunity, epigenetic modifications (Rubin et al., 2020; Yuan et al., 2016) and, unexpectedly, in the DNA damage response (DDR).
The DDR is a complex but efficient network of pathways that cells developed to counteract DNA lesions and prevent the onset of chromosomal instability that finally can lead to cancer (Carusillo and Mussolino, 2020). Upon genotoxic stress, the DDR detects the damages and boosts DNA repair, but in case of irreparable lesions it induces premature cellular senescence or apoptosis, finally preventing the proliferation of damaged and potentially dangerous cells. The importance of DDR in cancer prevention is highlighted by the discovery in patients of mutations in genes involved in these pathways. However, it should be considered that even if defects in these signaling cascades promote tumorigenesis, members of the DDR can also be useful targets for therapy, because cancer cells, which are defective in these pathways, are more sensitive to DNA-damaging drugs than normal cells that can repair the lesions (Pearl et al., 2015).
The DDR is mainly constituted by the ATM-CHK2 and the ATR-CHK1 signaling cascades (Figure 2; Smith et al., 2010). Upon DNA lesions, ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3 related (ATR) phosphorylate a plethora of substrates collaborating with mediator proteins and the transducer kinases that are, respectively, CHK2 for ATM and CHK1 for ATR. These phosphorylations promote the appropriate cellular response to DNA damage, but, while mediator proteins stimulate ATM and ATR activation, the transducer kinases CHK2 and CHK1 propagate, modulate, or eventually redirect the DNA damage signal (Zannini et al., 2014). The ATM-CHK2 pathway is mainly involved in the response to double-strand breaks (DSBs), while the ATR-CHK1 signaling is primarily activated upon single-strand breaks (SSBs) and replication stress. However, because single-strand DNA can be produced during the repair of DSBs and, conversely, DSBs can be generated during the replication of damaged DNA, cells frequently activate both these signaling cascades (Smith et al., 2010). Moreover, beside the response to many different types of DNA lesions, the DDR is also involved in physiological cellular processes, such as telomere length maintenance, mitochondrial DNA repair, cell cycle, mitosis and meiosis regulation, circadian clock control, viral DNA processing, stem cell maintenance, and V(D)J recombination during antibodies production (Zannini et al., 2014).
Figure 2.

Graphical representation of the ATM-CHK2 and ATR-CHK1 signaling cascades
In response to DSBs or SSBs, the kinases ATM and ATR becomes activated and, collaborating with the transducer kinases CHK2 and CHK1 promote the phosphorylation of a plethora of targets. Phosphorylation of these substrates finally induces the appropriate cellular response to the lesions, that could be the induction of senescence or apoptosis, the activation of cell cycle checkpoints, and the promotion of DNA repair.
Given the complexity of these mechanisms and their importance for the maintenance of genome integrity, many efforts are continuously employed to better understand the regulation of these pathways. Until now, the DDR was considered to act equally in both male and female cells. However, recent discoveries suggest that sexual dimorphisms may exist also in these signaling cascades and may contribute to the different predisposition of men and women to cancer development and to sexual disparities in the response to treatments. Advances in these fields will be hereafter discussed.
Sex differences in the response to physiological DNA damage
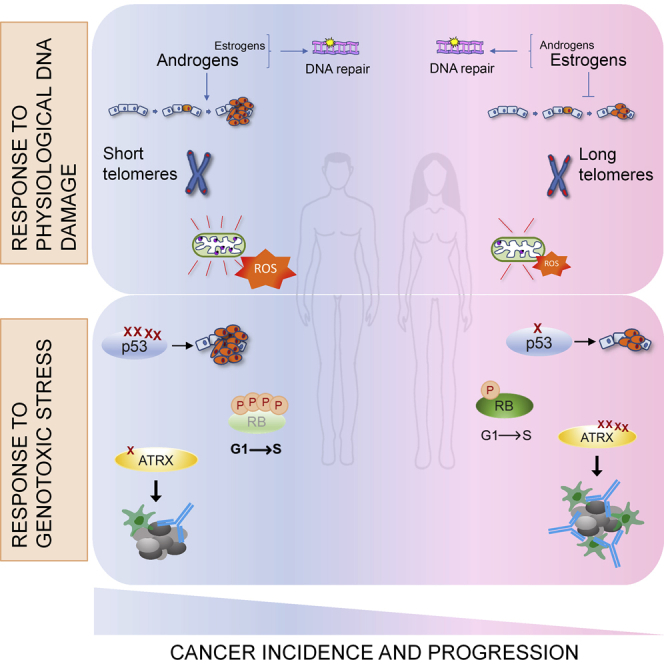
As described above, the DDR participates also to physiological cellular processes, and in some of these functions recently emerged unexpected sexual dimorphisms. Indeed, sex differences can be detected not only in sex hormones functions related to the DDR but, importantly, also in the regulation of aging and in the repair of mitochondrial DNA, two processes that were up to now considered conserved in both men and women (Figure 3).
Figure 3.

Sex disparities in physiological cellular processes regulated by DDR
Sexual dimorphisms are evident in at least three aspects of normal cell physiology regulated also by DDR proteins. i) There is a reciprocal regulation between steroid hormones and DDR proteins: both androgens and estrogens promote DNA repair, but, while male hormones seem to be pro-tumorigenic, estrogens appear to halt cancer formation. ii) Telomeres shortening activate the DDR and it has been found that men are characterized by shorter telomeres than women. iii) Sexual disparities can also be detected in mitochondria; indeed, in males these organelles display high levels of oxidative stress which is instead reduced in females' mitochondria that therefore demonstrate a better oxidative balance.
Hormones regulation
Steroid hormones are small molecules that, circulating in the human body and regulating nuclear receptors function, promote autocrine, paracrine, and endocrine signaling in both physiological and pathological conditions. However, steroid hormones (estrogens, androgens, and glucocorticoids) may also produce genotoxic stress and it is not yet clear if they can be considered carcinogenic or tumor protective (Joosten et al., 2004). Given the widespread use of hormonal steroids also in medicine (for contraceptive and therapy purpose), their genotoxic activity has been widely investigated and it has been found that their signaling may cause mutations, DNA damages, chromosome breakage, and polyploidy. Nonetheless, it has been reported that sex hormones contribute differently, and possibly in an antagonistic manner, to tumor formation and progression. Indeed, studies in hepatocellular carcinoma, but also in other types of cancer, demonstrated that estrogens counteract tumorigenesis while, on the contrary, androgens promote cellular proliferation and metabolism activation (Clocchiatti et al., 2016). In general, males and females share the same hormones, but differ in their production site, blood concentrations, and interplay with organs, systems, and apparatuses. Indeed, estrogens and androgens have important roles in both male and female biology and their physiologic response depends on the ratio of estrogen and androgen receptors signaling and on the amount of steroid metabolism (Hammes and Levin 2019). This suggests that sexual dimorphism in cancer is not simply the result of the presence of estrogens in females and androgens in males, but their levels and combinations contribute to the differences between sexes.
It should be, however, noted that sex steroid receptors are also involved in DDR modulation by regulating the activity and expression of genes involved in these pathways (Joosten et al., 2004). For example, the activity and expression of p53, one of the most important players of these signaling cascades (see below), was reported to be positively regulated by estrogens and progesterons (Dunphy et al., 2008), while, on the contrary, male sex hormones decrease p53 function (Ma et al., 2008). Therefore, these functional associations should be kept in consideration when combining hormone and DNA damaging therapy in tumors with wild-type p53.
In addition, steroid hormones have also been reported to regulate DNA repair (Wengner et al., 2020), especially non-homologous end-joining (NHEJ) and homologous recombination (HR), the two main pathways involved in DSBs repair (Carusillo and Mussolino, 2020). Indeed, in prostate cancer models, androgen receptors (ARs) stimulate the activity and expression of DNA-dependent protein kinase (DNA-PK), a serine/threonine kinase that constitutes one of the main components of the NHEJ machinery (Mohiuddin and Kang, 2019). Similarly, in breast cancer, estrogens positively regulate the expression of nibrin (NBS1), the protein mutated in the radiation-sensitive Nijmegen breakage syndrome that, early after damage, accumulates at DSBs and induces the recruitment of DDR proteins favoring the repair of DNA (Komatsu, 2016). In addition, steroid hormones also regulate HR both positively and negatively (Bowen et al., 2015; Wilk et al., 2012), demonstrating complexity in the modulation of these pathways and suggesting that therapeutic inhibition of nuclear receptors may result in non-reparable DNA damages in different types of tumor.
Conversely, there are also members of DDR pathways involved in the regulation of nuclear receptors functions. For example, poly(ADP-ribose)-polymerase-1 (PARP1), a sensor of DNA damage important for the recruitment at DSBs of DNA repair proteins (Pascal, 2018), has been reported to regulate estrogen receptors (ERs), ARs, and progesterone receptors (PRs) functions and to foster their binding to target genes promoters (Weaver and Yang, 2013). Therefore, the sex-specific effect of PARP1 functions should be considered in clinical trials using PARP inhibitors to treat patients with breast cancer or metastatic castration-resistant prostate cancer.
Mediator of DNA damage checkpoint 1 (MDC1), a scaffold protein that amplifies the DNA damage signal in the initial steps of the DDR (Ruff et al., 2020), has also been found to bind ERs and increase its transcriptional activity, finally suppressing breast cancer cells growth (Zou et al., 2015). This finding, together with the observation that MDC1 levels decrease during tumor progression, indicates that regulation of ERs by MDC1 could have an important role in preventing breast cancer advancement. Similarly, MDC1 has been shown to support ARs function in prostate cancer (Wang et al., 2015) and, considering the important role of ERs and ARs in breast and prostate tumors, it should be interesting to investigate the mechanisms underlying the regulation of the antitumorigenic functions of nuclear receptors by MDC1.
Additionally, other DDR proteins associated with post-translational modifications have been found to regulate ARs function. For example, PIAS1, a sumo E3-ligase involved in DSBs repair (Galanty et al., 2009) increases ARs stability and transcriptional function (Puhr et al., 2016), while DNA-PK establishes a positive feedback loop of clinical relevance with ARs (Goodwin et al., 2015).
Because ERs and ARs are also involved in the development and progression of somatic tumors, such as hepatocellular carcinoma, non-small cell lung cancer, and thyroid cancer, differences in sex steroid hormone levels could contribute to disparities in cancer susceptibility between males and females and the feedforward loop between DDR and hormone signaling should be considered when analyzing the results of clinical trials or in the development of novel combinations for therapy. Importantly, hormones effect should be considered also for cancer therapy in transgender people who are exposed to high levels of estrogens or testosterone to, respectively, promote feminization or masculinization. However, the findings that women are more protected than men from different types of cancer even in the post-menopausal period (Chlebowski et al., 2004) and that sexual disparities in cancer incidence and survival can be as well found in pre-pubescent children (Williams et al., 2019) indicate that, as better detailed in the next sections, also hormone-independent mechanisms are responsible for sex differences in both DDR and cancer.
Telomere length regulation
Telomeres are nucleoprotein structures that protect the end of chromosomes from being recognized as DSBs and ensure proper chromosome replication (Kong et al., 2013). Among eukaryotes, the enzyme responsible for telomere length maintenance is telomerase reverse transcriptase (hTERT) and upregulation of hTERT activity has been found in many tumors, suggesting a cancer promoter role. However, during aging or in response to genotoxic stress (e.g. oxidative damage), telomeres shorten and induce DDR activation and NHEJ-mediated DNA repair. These events may lead to end-end chromosome fusions that, during the next mitotic cycles, could break and generate chromosomal aberrations, finally fueling malignant transformation. Paradoxically, critically short telomeres also trigger cell-cycle arrest, senescence, or apoptosis, preventing cancer cells proliferation (Kong et al., 2013). Therefore, telomere attrition has a dual role in tumorigenesis: it can promote genome instability and carcinogenesis while, on the contrary, it can also suppress tumor progression.
Nonetheless, the relationship between age, sex, and telomere length is complex and even if there are few exceptions, different meta-analysis studies demonstrated that, in physiological conditions, adult women have longer telomeres than men (Gardner et al., 2014). This association becomes stronger with the increasing of age and suggests that females may be more protected from telomere-shortening effects. However, it is still unclear if these differences appear soon after conception or later in life (Factor-Litvak et al., 2016; Zhu et al., 2011) and also the reasons for sex disparities in telomeres are still undefined. It has been suggested that larger body size is paired with more cell divisions and therefore with shorter telomeres (Barrett and Richardson, 2011). Because generally males have larger bodies than females, the reduced length of telomeres in men could be a natural consequence of this disparity. However, the most supported hypotheses foresee the involvement of sex hormones. Indeed, estrogen-responsive elements have been found in hTERT promoter and treatments with 17-β-estradiol result in upregulation of telomerase activity (Mayer et al., 2006). Moreover, telomeres that are particularly sensitive to oxidative stress and estrogen, which has antioxidant properties (Carrero et al., 2008), could help the metabolism of reactive oxygen species (ROS). On the contrary, women may also have longer telomeres because of the low levels of testosterone (Alonso-Alvarez et al., 2007), which is known to increase the deleterious effects of oxidative stress. However, because steroid hormones also regulate the function of DDR proteins (see above), it should be investigated whether these circulating molecules could be involved in telomere length maintenance by modulating DDR pathways.
Anyhow, it is important to note that short telomeres are generally associated with increased mortality and morbidity in humans and that cancer incidence increases more rapidly in males than in females with increasing age. Therefore, the presence of longer telomeres in female cells could contribute to explain why, worldwide, women live longer than men and are less affected by cancer.
Repair of mitochondrial DNA
Oxidative stress activates the DDR and one of the main sources of ROS is mitochondria. These organelles provide most of the cell energy through oxidative phosphorylation and contain their own DNA (mtDNA) that lacks histones and encodes for proteins important for mitochondrial functions (Zong et al., 2016). Therefore, mtDNA is more susceptible than nuclear DNA to genotoxic stress because besides being assaulted by exogenous stresses, such as UV radiations, it lies in the proximity of the oxidative phosphorylation sites and moreover it is not protected by histones. Additionally, excessive mtDNA damages, if not properly repaired, increase the production of ROS, finally promoting mitochondrial dysfunction and the pathogenesis of many human diseases including cancer (Rong et al., 2021). It was previously thought that mtDNA might not be repaired and that these organelles, when damaged, are simply degraded (Druzhyna et al., 2008). Now, it is clear that repair pathways exist also in mitochondria and the most important is base excision repair (BER) that removes non-bulky DNA lesions mostly produced by oxidation. However, ROS may also cause DSBs in mtDNA that, similarly to nuclear DNA, are repaired by HR and NHEJ (Rong et al., 2021).
Mitochondria present a strong sex-specific behavior and differ, between males and females, for morphology, function, and oxidative stress regulation (Ventura-Clapier et al., 2017). Studies performed in rat brain demonstrated that females have higher mitochondrial activity (Guevara et al., 2009) and increased respiration rate (Khalifa et al., 2017) than age-matched males. Similarly, also in human brain, the activity of mitochondrial enzymes is higher in females than in males (Harish et al., 2013). However, even if their respiratory rate is increased, both mouse and rat female brains accumulate less ROS than males, suggesting a better oxidative balance. Moreover, also oxidative damage is reduced in female mouse brains, if compared with equal in age males, and the same happens also in humans, where biomarkers of oxidative stress are lower in women than in men (Harish et al., 2013; Khalifa et al., 2017). These differences in mitochondrial activity suggest that ROS accumulation, regulation, and sensitivity are sexually dimorphic. Therefore, it seems that female mitochondria are better optimized than male mitochondria and this difference could explain the sexual dimorphism detectable in different types of cancer. Indeed, besides brain, sexual dimorphism in mitochondrial function has been found also in other tissues, such as white and brown adipose tissues, liver, and skeletal muscles, and, therefore, it is possible that men, accumulating more ROS than women, may have a higher risk to develop cancer. Because ROS accumulation increases with age, this well correlates also with the increased cancer incidence rate found in males. Importantly, one of the main players in the response to ROS is p53 (Shi and Dansen, 2020) that, as described below, is more frequently mutated in men than in women, possibly contributing to the sex differences in ROS accumulation.
Sexual dimorphisms in the response to genotoxic stress
During their lifetime, men accumulate more somatic mutations than women. These genetic alterations could derive from errors in DNA replication, but also from defects in repairing DNA lesions. Therefore, they could reflect important differences between males and females in the response to chemical and physical genotoxic agents and in DDR pathways activation. However, nowadays only few evidence of sexual dimorphism in these signaling cascades does exist and these disparities are generally restricted to specific inactivation or increased mutation rate of DDR genes (Figure 4).
Figure 4.

Sex disparities in the response to genotoxic stress
Sexual dimorphisms have been found in the behavior of some DDR-related proteins: i) p53, which has been found to mutate more frequently in men than in women, ii) RB, that in male cancer cells is kept in a hyper-phosphorylated and inactive state also upon DNA damage, therefore favoring G1-S transition and the propagation of potentially dangerous cells, and iii) ATRX, that is more frequently mutated in women than in men and these mutations seem to increase the immune response toward cancer. Evidence of sexual dimorphism has been found also in DNA repair pathways, but the results of these disparities are still unclear.
p53
The main player of cancer defense mechanisms is the tumor suppressor protein p53 (Hafner et al., 2019). Upon DNA damage, replicative, and oxidative stress, p53 acts as a sensor for the cell, promoting either transient or permanent cell-cycle arrest, allowing the repair or inducing apoptosis or senescence. Recently, evidence is emerging that some sexual disparities in cancer could be ascribed to different p53 activities and regulations in men and women (Haupt et al., 2019).
The gene encoding for p53, TP53, is the most mutated gene in patients with cancer and these mutations are often followed by loss of heterozygosity and complete p53 deficiency, which promotes cancer initiation and progression (Levine, 2020). It has been established that mutations in TP53 affect more males than females in non-reproductive cancers. Indeed, statistical analysis conducted among 12 most common sporadic tumors in the US population proved that men are more frequently characterized by cancers with mutated TP53 than women (Haupt et al., 2019). Reasons for this difference could be attributed to a network of p53 regulatory genes on the X chromosome that mutate more frequently in males than in females. Importantly, this disparity can be observed only in genes linked to p53 pathway, therefore suggesting that females can specifically inactivate mutant genes on the X chromosome finally protecting p53 signaling. In addition, p53 demonstrates sex-specific functions also in patients with Li Fraumeni who bear p53 mutations and are characterized by increased predisposition to cancer. However, the outcome is still not clear since some groups demonstrated that p53 mutations increase the risk of cancer formation mostly in female carriers (Gonzalez et al., 2009; Wu et al., 2006), while other laboratories contradicted these findings (Olivier et al., 2003). However, studies with mouse models demonstrated that p53 mutant males develop more aggressive tumors and have reduced lifespan than females (Kfoury et al., 2018).
In unstressed condition, p53 is kept at low levels by its major E3 ubiquitin ligase MDM2 through an autoregulatory negative feedback loop. Upon stress signals, MDM2-p53 interaction is disrupted inducing p53 accumulation and stabilization (Karni-Schmidt et al., 2016). A single nucleotide polymorphism (SNP, SNP309 G/G) in MDM2 produces high mRNA and protein levels and the consequent reduction of p53 tumor suppressor function (Bond et al., 2004). This SNP, that is mostly disadvantageous for women, is associated with increased predisposition for multiple cancers and the higher risk has been attributed to the female Asian population, in the context of non-small cell lung cancer (Luan et al., 2019).
Another level of p53 regulation is represented by the ATM-, ATR-, and CHK2-dependent phosphorylation cascade. Sexual dimorphism among these regulators has been observed in mouse models in contrast with the sex differences characterizing human cancers. In C57BL/6 mice, p53 functions after γ-irradiation decrease with age, due to a reduction of ATM kinase activity, and the decline occurs later in males than in females (Feng et al., 2007). In addition, the great majority of mice that carry the CHEK2∗1100delC allele, lacking the kinase activity, and that develop tumors were females (Bahassi El et al., 2009). Similarly, germline CHEK2 mutations increase the risk of developing breast cancer for human females (Bahassi El et al., 2009), while in contrast, lung tissue developed mechanisms to counteract CHK2 inactivity and prevent tumor formation (van Jaarsveld et al., 2020). These results indicate the existence of sex and tissue-specific risks for carcinogenesis and that evidence obtained with murine models cannot be always translated to humans.
RB
Another protein with important roles in cancer development is RB which negatively regulates cellular proliferation (Dick and Rubin, 2013); in fact, hyperphosphorylation or loss of RB results in higher levels of E2F1-dependent transcription and a more rapid G1-S progression. Recently, RB has been linked to the sexual dimorphism that can be detected in glioblastoma (GBM), which is known to affect more males than females. This sex disparity has been observed in mesenchymal GBM, which frequently shows loss of neurofibromin (Nf1) and mutation in TP53 (Verhaak et al., 2010). Studies with male and female Nf1−/−DNp53 astrocytes demonstrated that male cells have a higher proliferation rate than females. At the molecular basis, higher levels of phosphorylated and inactive RB were found upon serum addition in male cells, indicating a more rapid G1 to S transit (Sun et al., 2014), and similar results were obtained also in response to the DNA-damaging drug etoposide (Kfoury et al., 2018). Moreover, complete inactivation of p53 and RB by SV40-TAg expression prevented sexual differences in the transformation process, indicating for RB a sexually dimorphic role (Sun et al., 2014). Interestingly, retinoblastoma, in which RB is completely inactivated, is not a sexually dimorphic tumor (Broaddus et al., 2009). Further analyses also demonstrated that, upon serum withdrawal or etoposide treatment, there was a significant increase of the RB regulators p16 and p21 in female GBM astrocytes, compared to male cells (Kfoury et al., 2018). These results suggest that female astrocytes are protected by the cooperative induction of the CDK inhibitors p16 and p21, arresting cell cycle. On the contrary, male cells do not arrest their growth and continue to acquire DNA damage-induced chromosomal aberrations, possibly explaining the increase incidence and the poorer outcome of GBM in males. Notably, p21 expression regulation is achieved also through sexually dimorphic epigenetic modifications. Indeed, the lysine demethylase KDM6A, which escapes X-inactivation and is more frequently mutated in male tumors, was found to regulate the expression of p53 and RB target genes, including p21, finally protecting females from bladder cancer (Kaneko and Li, 2018). These findings demonstrate that sex-specific epigenetic modifications differently regulate gene expression in males and females and could contribute to sex disparities in DDR and cancer.
ATRX
ATRX is an X-linked gene encoding a tumor suppressor protein involved in chromatin remodeling, maintenance of genetic stability, and X chromosome inactivation (XCI) through its binding to XIST (Ren et al., 2020). ATRX has been found mutated or absent in many cancer types, including gastric cancer (GC), in which ATRX mutations occur more frequently in female compared to male patients (Ge et al., 2021). Among patients with GC, ATRX mutations have been associated with a higher overall survival in both sexes. However, it has been observed that especially female ATRX mutants are characterized by stronger anticancer immunity and favorable clinical response when compared to male ATRX mutants. This effect is mainly due to a higher activation of DNA repair pathways and increased sensitivity to DNA damage (Ge et al., 2021), as previously seen in glioma (Han et al., 2018) and murine pancreatic cancer (Young et al., 2018). Therefore, ATRX mutations could be a predictive biomarker of favorable clinical outcome for female patients and together with other markers could contribute to identify those women that possibly could benefit of specific therapy.
DNA repair
In accordance with the increased cancer incidence found in males, chromosomal abnormalities and somatic mutations have been found earlier and to a greater extent in men than in women (Podolskiy et al., 2016). The mechanisms involved in this difference are still unknown, but it was suggested that DNA repair efficacy could be different between males and females. However, the studies investigating the role of sex on DNA repair have been up to now inconclusive. In fact, no sex disparities have been found in the repair of DSBs in human peripheral blood mononuclear cells or in a meta-analysis of DNA damage studies (Garm et al., 2013; Soares et al., 2014) therefore indicating that DNA repair effectiveness is not different between men and women. On the contrary, another study (Wei et al., 2000) reported that females have reduced capacity to repair tobacco-induced DNA damages by nucleotide excision repair (NER) and lower NER activity has been associated with increased risk of lung and non-melanoma skin cancer (Spitz et al., 2003; Wei et al., 1993). In addition, Trzeciak et al., using a modified form of comet assay, were able to demonstrate that the fast component of SSBs repair, involving DNA ligation and polymerization steps of BER, is lower in females than in males (Trzeciak et al., 2008). These findings suggest that at least the initial phases of NER and BER pathways could be reduced in women compared to men, even if in mice it was demonstrated that DNA repair works better in females than in males (Winkelbeiner et al., 2020).
Importantly, it was also found that polymorphism in DNA repair genes may influence their repair ability, finally increasing the risk of cancer development. For example, XPC Gln939Gln was found to increase the risk of developing lung cancer in women but not in men (Letkova et al., 2013).
In addition, analysis of molecular differences in 13 cancers of The Cancer Genome Atlas (TCGA) database revealed that DNA repair genes are expressed at higher levels in female patients than in males, possibly because of the increased methylation that can be detected in male genomes (Yuan et al., 2016). Collectively, these reports indicate that there is still a lot to investigate to understand the role of sex in DNA repair and that the effect could be different depending on the tissue and on the type of lesions.
Different sex effects of cancer therapies
The main goal of cancer therapy is the complete eradication of the tumor either via selective killing of cancer cells or surgical procedures. A common feature of cancer cells is the presence of defects in DDR pathways that make them more susceptible to DNA damaging agents and therefore radio- and chemotherapy are frequently used to treat these diseases (Pearl et al., 2015). Although sex is an important factor in determining the sensitivity to cancer therapy, sex-based studies are still insufficient mostly because of the under-representation of females in clinical trials (Murthy et al., 2004). However, it seems clear that generally cancer therapies work better in females than in males. Indeed, it was shown that women respond differently from men to many drugs and develop more frequently adverse reactions (Yu et al., 2016). For different types of chemotherapy agents, it was reported a greater cancer cell death and worst side effects for females than males and this means that women require lower doses of drug than men (Wagner et al., 2019). This disparity finally leads females to better survive to therapy even if the reasons for these differences are still unknown. Possible explanations could be that adult women have stronger innate and immune response than men (Klein and Flanagan, 2016), and that males and females differ in their body composition (Wagner et al., 2019). In fact, males have a higher percentage of fat-free body mass than females and this diversity could lead to differences in drugs adsorption, metabolism, and excretion. In addition, body composition also influences drugs distribution, with females being more responsive to lipophilic compounds, because of the higher proportion of fat, and males to water-soluble drugs. Therefore, the greater cancer cell death detectable in females could be attributed to slow drugs clearance and the subsequent prolonged survival for women has been detected in response to alkylating platinum for esophageal cancers (Davidson et al., 2019), upon paclitaxel for NSCLC (Wheatley-Price et al., 2010), and upon temozolomide treatment of glioblastoma (Yang et al., 2019). Radiotherapy was also found to extend the survival of females affected by esophageal squamous cell carcinoma (Luo et al., 2019), compared to men, and this increase may be due to the augmented radiosensitivity that can be detected in women and that can lead to improved therapeutic effect. Accordingly, the median toxic dose of ionizing radiation is lower in females than in males and the number of DNA damages is greater in women than in men. Nonetheless, understanding the reasons for these disparities is of extreme importance, in the aim of cancer treatment optimization, and, therefore, in the future, there will be an urgent need to expand these studies to better define personalized cancer therapies.
Conclusions and perspectives
In the last years, many efforts have been employed with the purpose of developing personalized cancer therapies. These studies mainly exploited the presence of specific mutations and only recently sex has begun to be considered. In fact, male and female patients with different cancer types are currently treated in a similar manner without dealing with the factor of sex. However, for therapeutic targets with strong sex bias signature, sex-specific trials should be employed to succeed.
Therefore, understanding the basis for cancer sex disparities is fundamental to achieve comparable outcomes for both male and female patients with cancer. Indeed, it is now essential that we consider that sex differences may significantly affect cell biology of cancer and how males and females respond to therapy. Therefore, it is extremely important that in the future both basic research and clinical studies consider the sex component during data analysis and trials design, a procedure that was up to now complicated also because of the low representation of women in clinical testing.
Moreover, the finding that males develop cancer earlier than females (Podolskiy et al., 2016), if confirmed, could constitute an important information also for the national health systems, suggesting that men should be included in cancer screening at younger age. However, reasons for sex differences in cancer incidence are still not clear and even if initially the lifestyle of males has been blamed for greater exposure to carcinogens, it is now commonly accepted that both environmental and genetic factors, among which sex, may impact tumor formation and progression.
DDR pathways are involved in cancer development and therapy (Carusillo and Mussolino, 2020), but until now they were considered common signaling cascades for both males and females. Only recently, evidence of sexual dimorphisms in the DDR are emerging and given the importance of these pathways for many aspects of the cell’s life, we expect that in the future many studies will be developed with the purpose of clarifying the sex-specific aspects of these signaling cascades. Nowadays, these analyses and considerations are sometimes complicated by the lack of information on the sex of the cell lines and of the animals used during experimentations. In addition, another obstacle is posed by the absence of valuable databases with the levels of protein expression in human biopsies. Indeed, only comprehensive data of gene expression are available, but they are not always sufficient for the comprehension of sex differences in DDR pathways, because the stability of proteins implicated in these signaling cascades are generally regulated through post-translational modifications. Therefore, while the mRNA of a gene of interest could be equally expressed in male and female cells, the levels of the corresponding protein in the same cellular systems could be significantly different.
However, we expect that in the future the importance of sex impact in DDR studies will be recognized and these complications will be overcome, finally allowing a better awareness of the molecular mechanisms underlying sex disparities in cancer. These results will hopefully put the basis for the full understanding of the molecular mechanisms underlying cancer formation and for the development and refinement of novel personalized cancer therapies for both male and female patients.
Acknowledgments
This work is supported by AIRC under IG 2018 - ID. 21535 project – P.I. Zannini Laura
Declaration of interests
The authors declare no competing interest
References
- Alonso-Alvarez C., Bertrand S., Faivre B., Chastel O., Sorci G. Testosterone and oxidative stress: the oxidation handicap hypothesis. Proc. Biol. Sci. 2007;274:819–825. doi: 10.1098/rspb.2006.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahassi El M., Robbins S.B., Yin M., Boivin G.P., Kuiper R., van Steeg H., Stambrook P.J. Mice with the CHEK2∗1100delC SNP are predisposed to cancer with a strong gender bias. Proc. Natl. Acad. Sci. U S A. 2009;106:17111–17116. doi: 10.1073/pnas.0909237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett E.L., Richardson D.S. Sex differences in telomeres and lifespan. Aging Cell. 2011;10:913–921. doi: 10.1111/j.1474-9726.2011.00741.x. [DOI] [PubMed] [Google Scholar]
- Bond G.L., Hu W., Bond E.E., Robins H., Lutzker S.G., Arva N.C., Bargonetti J., Bartel F., Taubert H., Wuerl P., et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Bowen C., Zheng T., Gelmann E.P. NKX3.1 suppresses TMPRSS2-ERG gene rearrangement and mediates repair of androgen receptor-induced DNA damage. Cancer Res. 2015;75:2686–2698. doi: 10.1158/0008-5472.CAN-14-3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broaddus E., Topham A., Singh A.D. Survival with retinoblastoma in the USA: 1975-2004. Br. J. Ophthalmol. 2009;93:24–27. doi: 10.1136/bjo.2008.143842. [DOI] [PubMed] [Google Scholar]
- Carrero J.J., Stenvinkel P., Fellstrom B., Qureshi A.R., Lamb K., Heimburger O., Barany P., Radhakrishnan K., Lindholm B., Soveri I., et al. Telomere attrition is associated with inflammation, low fetuin-A levels and high mortality in prevalent haemodialysis patients. J. Intern. Med. 2008;263:302–312. doi: 10.1111/j.1365-2796.2007.01890.x. [DOI] [PubMed] [Google Scholar]
- Carusillo A., Mussolino C. DNA damage: from threat to treatment. Cells. 2020;9 doi: 10.3390/cells9071665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlebowski R.T., Wactawski-Wende J., Ritenbaugh C., Hubbell F.A., Ascensao J., Rodabough R.J., Rosenberg C.A., Taylor V.M., Harris R., Chen C., et al. Estrogen plus progestin and colorectal cancer in postmenopausal women. N. Engl. J. Med. 2004;350:991–1004. doi: 10.1056/NEJMoa032071. [DOI] [PubMed] [Google Scholar]
- Clocchiatti A., Cora E., Zhang Y., Dotto G.P. Sexual dimorphism in cancer. Nat. Rev. Cancer. 2016;16:330–339. doi: 10.1038/nrc.2016.30. [DOI] [PubMed] [Google Scholar]
- Davidson M., Wagner A.D., Kouvelakis K., Nanji H., Starling N., Chau I., Watkins D., Rao S., Peckitt C., Cunningham D. Influence of sex on chemotherapy efficacy and toxicity in oesophagogastric cancer: a pooled analysis of four randomised trials. Eur. J. Cancer. 2019;121:40–47. doi: 10.1016/j.ejca.2019.08.010. [DOI] [PubMed] [Google Scholar]
- Dick F.A., Rubin S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 2013;14:297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druzhyna N.M., Wilson G.L., LeDoux S.P. Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 2008;129:383–390. doi: 10.1016/j.mad.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy K.A., Blackburn A.C., Yan H., O'Connell L.R., Jerry D.J. Estrogen and progesterone induce persistent increases in p53-dependent apoptosis and suppress mammary tumors in BALB/c-Trp53+/- mice. Breast Cancer Res. 2008;10:R43. doi: 10.1186/bcr2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Factor-Litvak P., Susser E., Kezios K., McKeague I., Kark J.D., Hoffman M., Kimura M., Wapner R., Aviv A. Leukocyte telomere length in newborns: implications for the role of telomeres in human disease. Pediatrics. 2016;137 doi: 10.1542/peds.2015-3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z., Hu W., Teresky A.K., Hernando E., Cordon-Cardo C., Levine A.J. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc. Natl. Acad. Sci. U S A. 2007;104:16633–16638. doi: 10.1073/pnas.0708043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanty Y., Belotserkovskaya R., Coates J., Polo S., Miller K.M., Jackson S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009;462:935–939. doi: 10.1038/nature08657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner M., Bann D., Wiley L., Cooper R., Hardy R., Nitsch D., Martin-Ruiz C., Shiels P., Sayer A.A., Barbieri M., et al. Gender and telomere length: systematic review and meta-analysis. Exp. Gerontol. 2014;51:15–27. doi: 10.1016/j.exger.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garm C., Moreno-Villanueva M., Burkle A., Petersen I., Bohr V.A., Christensen K., Stevnsner T. Age and gender effects on DNA strand break repair in peripheral blood mononuclear cells. Aging Cell. 2013;12:58–66. doi: 10.1111/acel.12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y., Wei F., Du G., Fei G., Li W., Li X., Chu J., Wei P. The association of sex-biased ATRX mutation in female gastric cancer patients with enhanced immunotherapy-related anticancer immunity. BMC Cancer. 2021;21:240. doi: 10.1186/s12885-021-07978-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez K.D., Noltner K.A., Buzin C.H., Gu D., Wen-Fong C.Y., Nguyen V.Q., Han J.H., Lowstuter K., Longmate J., Sommer S.S., et al. Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J. Clin. Oncol. 2009;27:1250–1256. doi: 10.1200/JCO.2008.16.6959. [DOI] [PubMed] [Google Scholar]
- Goodwin J.F., Kothari V., Drake J.M., Zhao S., Dylgjeri E., Dean J.L., Schiewer M.J., McNair C., Jones J.K., Aytes A., et al. DNA-PKcs-Mediated transcriptional regulation drives prostate cancer progression and metastasis. Cancer Cell. 2015;28:97–113. doi: 10.1016/j.ccell.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guevara R., Santandreu F.M., Valle A., Gianotti M., Oliver J., Roca P. Sex-dependent differences in aged rat brain mitochondrial function and oxidative stress. Free Radic. Biol. Med. 2009;46:169–175. doi: 10.1016/j.freeradbiomed.2008.09.035. [DOI] [PubMed] [Google Scholar]
- Hafner A., Bulyk M.L., Jambhekar A., Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019;20:199–210. doi: 10.1038/s41580-019-0110-x. [DOI] [PubMed] [Google Scholar]
- Hammes S.R., Levin E.R. Impact of estrogens in males and androgens in females. J. Clin. Invest. 2019;129:1818–1826. doi: 10.1172/JCI125755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B., Cai J., Gao W., Meng X., Gao F., Wu P., Duan C., Wang R., Dinislam M., Lin L., et al. Loss of ATRX suppresses ATM dependent DNA damage repair by modulating H3K9me3 to enhance temozolomide sensitivity in glioma. Cancer Lett. 2018;419:280–290. doi: 10.1016/j.canlet.2018.01.056. [DOI] [PubMed] [Google Scholar]
- Harish G., Venkateshappa C., Mahadevan A., Pruthi N., Bharath M.M., Shankar S.K. Mitochondrial function in human brains is affected by pre- and post mortem factors. Neuropathol. Appl. Neurobiol. 2013;39:298–315. doi: 10.1111/j.1365-2990.2012.01285.x. [DOI] [PubMed] [Google Scholar]
- Haupt S., Caramia F., Herschtal A., Soussi T., Lozano G., Chen H., Liang H., Speed T.P., Haupt Y. Identification of cancer sex-disparity in the functional integrity of p53 and its X chromosome network. Nat. Commun. 2019;10:5385. doi: 10.1038/s41467-019-13266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten H.F., van Acker F.A., van den Dobbelsteen D.J., Horbach G.J., Krajnc E.I. Genotoxicity of hormonal steroids. Toxicol. Lett. 2004;151:113–134. doi: 10.1016/j.toxlet.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Kaneko S., Li X. X chromosome protects against bladder cancer in females via a KDM6A-dependent epigenetic mechanism. Sci. Adv. 2018;4:eaaw7317. doi: 10.1126/sciadv.aar5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karni-Schmidt O., Lokshin M., Prives C. The roles of MDM2 and MDMX in cancer. Annu. Rev. Pathol. 2016;11:617–644. doi: 10.1146/annurev-pathol-012414-040349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kfoury N., Sun T., Yu K., Rockwell N., Tinkum K.L., Qi Z., Warrington N.M., McDonald P., Roy A., Weir S.J., et al. Cooperative p16 and p21 action protects female astrocytes from transformation. Acta Neuropathol. Commun. 2018;6:12. doi: 10.1186/s40478-018-0513-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalifa A.R., Abdel-Rahman E.A., Mahmoud A.M., Ali M.H., Noureldin M., Saber S.H., Mohsen M., Ali S.S. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol. Rep. 2017;5 doi: 10.14814/phy2.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S.L., Flanagan K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016;16:626–638. doi: 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- Komatsu K. NBS1 and multiple regulations of DNA damage response. J. Radiat. Res. 2016;57:i11–i17. doi: 10.1093/jrr/rrw031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong C.M., Lee X.W., Wang X. Telomere shortening in human diseases. Febs J. 2013;280:3180–3193. doi: 10.1111/febs.12326. [DOI] [PubMed] [Google Scholar]
- Letkova L., Matakova T., Musak L., Sarlinova M., Krutakova M., Slovakova P., Kavcova E., Jakusova V., Janickova M., Drgova A., et al. DNA repair genes polymorphism and lung cancer risk with the emphasis to sex differences. Mol. Biol. Rep. 2013;40:5261–5273. doi: 10.1007/s11033-013-2626-z. [DOI] [PubMed] [Google Scholar]
- Levine A.J. P53: 800 million years of evolution and 40 Years of discovery. Nat. Rev. Cancer. 2020;20:471–480. doi: 10.1038/s41568-020-0262-1. [DOI] [PubMed] [Google Scholar]
- Luan L., Wang H., Zhao B., Wang F., Shi J., Xu X. Association of MDM2 gene SNP 309 polymorphism and human non-small cell lung cancer susceptibility: a meta-analysis. Pathol. Res. Pract. 2019;215:152538. doi: 10.1016/j.prp.2019.152538. [DOI] [PubMed] [Google Scholar]
- Luo H.S., Xu H.Y., Du Z.S., Li X.Y., Wu S.X., Huang H.C., Lin L.X. Impact of sex on the prognosis of patients with esophageal squamous cell cancer underwent definitive radiotherapy: a propensity score-matched analysis. Radiat. Oncol. 2019;14:74. doi: 10.1186/s13014-019-1278-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W.L., Hsu C.L., Wu M.H., Wu C.T., Wu C.C., Lai J.J., Jou Y.S., Chen C.W., Yeh S., Chang C. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology. 2008;135:947–955. doi: 10.1053/j.gastro.2008.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer S., Bruderlein S., Perner S., Waibel I., Holdenried A., Ciloglu N., Hasel C., Mattfeldt T., Nielsen K.V., Moller P. Sex-specific telomere length profiles and age-dependent erosion dynamics of individual chromosome arms in humans. Cytogenet. Genome Res. 2006;112:194–201. doi: 10.1159/000089870. [DOI] [PubMed] [Google Scholar]
- Mohiuddin I.S., Kang M.H. DNA-PK as an emerging therapeutic target in cancer. Front. Oncol. 2019;9:635. doi: 10.3389/fonc.2019.00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy V.H., Krumholz H.M., Gross C.P. Participation in cancer clinical trials: race-, sex-, and age-based disparities. Jama. 2004;291:2720–2726. doi: 10.1001/jama.291.22.2720. [DOI] [PubMed] [Google Scholar]
- Olivier M., Goldgar D.E., Sodha N., Ohgaki H., Kleihues P., Hainaut P., Eeles R.A. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003;63:6643–6650. [PubMed] [Google Scholar]
- Pascal J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair (Amst) 2018;71:177–182. doi: 10.1016/j.dnarep.2018.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl L.H., Schierz A.C., Ward S.E., Al-Lazikani B., Pearl F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer. 2015;15:166–180. doi: 10.1038/nrc3891. [DOI] [PubMed] [Google Scholar]
- Podolskiy D.I., Lobanov A.V., Kryukov G.V., Gladyshev V.N. Analysis of cancer genomes reveals basic features of human aging and its role in cancer development. Nat. Commun. 2016;7:12157. doi: 10.1038/ncomms12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhr M., Hoefer J., Eigentler A., Dietrich D., van Leenders G., Uhl B., Hoogland M., Handle F., Schlick B., Neuwirt H., et al. PIAS1 is a determinant of poor survival and acts as a positive feedback regulator of AR signaling through enhanced AR stabilization in prostate cancer. Oncogene. 2016;35:2322–2332. doi: 10.1038/onc.2015.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W., Medeiros N., Warneford-Thomson R., Wulfridge P., Yan Q., Bian J., Sidoli S., Garcia B.A., Skordalakes E., Joyce E., et al. Disruption of ATRX-RNA interactions uncovers roles in ATRX localization and PRC2 function. Nat. Commun. 2020;11:2219. doi: 10.1038/s41467-020-15902-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Z., Tu P., Xu P., Sun Y., Yu F., Tu N., Guo L., Yang Y. The mitochondrial response to DNA damage. Front. Cell. Dev. Biol. 2021;9:669379. doi: 10.3389/fcell.2021.669379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin J.B., Lagas J.S., Broestl L., Sponagel J., Rockwell N., Rhee G., Rosen S.F., Chen S., Klein R.S., Imoukhuede P., et al. Sex differences in cancer mechanisms. Biol. Sex. Differ. 2020;11:17. doi: 10.1186/s13293-020-00291-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff S.E., Logan S.K., Garabedian M.J., Huang T.T. Roles for MDC1 in cancer development and treatment. DNA Repair (Amst) 2020;95:102948. doi: 10.1016/j.dnarep.2020.102948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T., Dansen T.B. Reactive oxygen species induced p53 activation: DNA damage, redox signaling, or both? Antioxid. Redox Signal. 2020;33:839–859. doi: 10.1089/ars.2020.8074. [DOI] [PubMed] [Google Scholar]
- Smith J., Tho L.M., Xu N., Gillespie D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- Soares J.P., Cortinhas A., Bento T., Leitao J.C., Collins A.R., Gaivao I., Mota M.P. Aging and DNA damage in humans: a meta-analysis study. Aging (Albany NY) 2014;6:432–439. doi: 10.18632/aging.100667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz M.R., Wei Q., Dong Q., Amos C.I., Wu X. Genetic susceptibility to lung cancer: the role of DNA damage and repair. Cancer Epidemiol. Biomarkers Prev. 2003;12:689–698. [PubMed] [Google Scholar]
- Sun T., Warrington N.M., Luo J., Brooks M.D., Dahiya S., Snyder S.C., Sengupta R., Rubin J.B. Sexually dimorphic RB inactivation underlies mesenchymal glioblastoma prevalence in males. J. Clin. Invest. 2014;124:4123–4133. doi: 10.1172/JCI71048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA cancer. J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Trzeciak A.R., Barnes J., Ejiogu N., Foster K., Brant L.J., Zonderman A.B., Evans M.K. Age, sex, and race influence single-strand break repair capacity in a human population. Free Radic. Biol. Med. 2008;45:1631–1641. doi: 10.1016/j.freeradbiomed.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Jaarsveld M.T.M., Deng D., Ordonez-Rueda D., Paulsen M., Wiemer E.A.C., Zi Z. Cell-type-specific role of CHK2 in mediating DNA damage-induced G2 cell cycle arrest. Oncogenesis. 2020;9:35. doi: 10.1038/s41389-020-0219-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura-Clapier R., Moulin M., Piquereau J., Lemaire C., Mericskay M., Veksler V., Garnier A. Mitochondria: a central target for sex differences in pathologies. Clin. Sci. (Lond) 2017;131:803–822. doi: 10.1042/CS20160485. [DOI] [PubMed] [Google Scholar]
- Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer. Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A.D., Oertelt-Prigione S., Adjei A., Buclin T., Cristina V., Csajka C., Coukos G., Dafni U., Dotto G.P., Ducreux M., et al. Gender medicine and oncology: report and consensus of an ESMO workshop. Ann. Oncol. 2019;30:1914–1924. doi: 10.1093/annonc/mdz414. [DOI] [PubMed] [Google Scholar]
- Wang C., Sun H., Zou R., Zhou T., Wang S., Sun S., Tong C., Luo H., Li Y., Li Z., et al. MDC1 functionally identified as an androgen receptor co-activator participates in suppression of prostate cancer. Nucleic Acids Res. 2015;43:4893–4908. doi: 10.1093/nar/gkv394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver A.N., Yang E.S. Beyond DNA repair: additional functions of PARP-1 in cancer. Front. Oncol. 2013;3:290. doi: 10.3389/fonc.2013.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q., Cheng L., Amos C.I., Wang L.E., Guo Z., Hong W.K., Spitz M.R. Repair of tobacco carcinogen-induced DNA adducts and lung cancer risk: a molecular epidemiologic study. J. Natl. Cancer Inst. 2000;92:1764–1772. doi: 10.1093/jnci/92.21.1764. [DOI] [PubMed] [Google Scholar]
- Wei Q., Matanoski G.M., Farmer E.R., Hedayati M.A., Grossman L. DNA repair and aging in basal cell carcinoma: a molecular epidemiology study. Proc. Natl. Acad. Sci. U S A. 1993;90:1614–1618. doi: 10.1073/pnas.90.4.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengner A.M., Scholz A., Haendler B. Targeting DNA damage response in prostate and breast cancer. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21218273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley-Price P., Le Maitre A., Ding K., Leighl N., Hirsh V., Seymour L., Bezjak A., Shepherd F.A., NCIC Clinical Trials Group The influence of sex on efficacy, adverse events, quality of life, and delivery of treatment in National Cancer Institute of Canada Clinical Trials Group non-small cell lung cancer chemotherapy trials. J. Thorac. Oncol. 2010;5:640–648. doi: 10.1097/JTO.0b013e3181d40a1b. [DOI] [PubMed] [Google Scholar]
- Wilk A., Waligorska A., Waligorski P., Ochoa A., Reiss K. Inhibition of ERbeta induces resistance to cisplatin by enhancing Rad51-mediated DNA repair in human medulloblastoma cell lines. PLoS ONE. 2012;7:e33867. doi: 10.1371/journal.pone.0033867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams L.A., Richardson M., Marcotte E.L., Poynter J.N., Spector L.G. Sex ratio among childhood cancers by single year of age. Pediatr. Blood Cancer. 2019;66:e27620. doi: 10.1002/pbc.27620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelbeiner N., Wandt V.K., Ebert F., Lossow K., Bankoglu E.E., Martin M., Mangerich A., Stopper H., Bornhorst J., Kipp A.P., et al. A multi-endpoint Approach to base excision repair incision activity augmented by PARylation and DNA damage levels in mice: impact of sex and age. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21186600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.C., Shete S., Amos C.I., Strong L.C. Joint effects of germ-line p53 mutation and sex on cancer risk in Li-Fraumeni syndrome. Cancer Res. 2006;66:8287–8292. doi: 10.1158/0008-5472.CAN-05-4247. [DOI] [PubMed] [Google Scholar]
- Yang W., Warrington N.M., Taylor S.J., Whitmire P., Carrasco E., Singleton K.W., Wu N., Lathia J.D., Berens M.E., Kim A.H., et al. Sex differences in GBM revealed by analysis of patient imaging, transcriptome, and survival data. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aao5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young C.C., Baker R.M., Howlett C.J., Hryciw T., Herman J.E., Higgs D., Gibbons R., Crawford H., Brown A., Pin C.L. The loss of ATRX increases susceptibility to pancreatic injury and oncogenic KRAS in female but not male mice. Cell. Mol. Gastroenterol. Hepatol. 2018;7:93–113. doi: 10.1016/j.jcmgh.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Chen J., Li D., Wang L., Wang W., Liu H. Systematic analysis of adverse event reports for sex differences in adverse drug events. Sci. Rep. 2016;6:24955. doi: 10.1038/srep24955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Liu L., Chen H., Wang Y., Xu Y., Mao H., Li J., Mills G.B., Shu Y., Li L., et al. Comprehensive characterization of molecular differences in cancer between male and female patients. Cancer Cell. 2016;29:711–722. doi: 10.1016/j.ccell.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannini L., Delia D., Buscemi G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell. Biol. 2014;6:442–457. doi: 10.1093/jmcb/mju045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Wang X., Gutin B., Davis C.L., Keeton D., Thomas J., Stallmann-Jorgensen I., Mooken G., Bundy V., Snieder H., et al. Leukocyte telomere length in healthy Caucasian and African-American adolescents: relationships with race, sex, adiposity, adipokines, and physical activity. J. Pediatr. 2011;158:215–220. doi: 10.1016/j.jpeds.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong W.X., Rabinowitz J.D., White E. Mitochondria and cancer. Mol. Cell. 2016;61:667–676. doi: 10.1016/j.molcel.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou R., Zhong X., Wang C., Sun H., Wang S., Lin L., Sun S., Tong C., Luo H., Gao P., et al. MDC1 enhances estrogen receptor-mediated transactivation and contributes to breast cancer suppression. Int. J. Biol. Sci. 2015;11:992–1005. doi: 10.7150/ijbs.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]