

Summary
Radiation-induced lung injury is a common late side effect of thoracic radiotherapy. Endothelial dysfunction following leukocytes infiltration is a prominent feature in this process. Here, we established a clinical-mimicking mouse model of radiation-induced lung injury and found the activity of phosphatase Shp2 was elevated in endothelium after injury. Endothelium-specific Shp2 deletion mice showed relieved collagen deposition along with disrupted radiation-induced Jag1 expression in the endothelium. Furthermore, endothelium-derived Jag1 activated the alternative activation of macrophages in vitro and in vivo by paracrine Notch signaling. Consistently, the Notch pathway was significantly activated by chest irradiation in the peripheral blood leukocytes of patients with cancer. Collectively, our work demonstrates that Shp2 participates in the radiation-induced endothelial dysfunction and subsequently inflammatory microenvironment producing during radiation-induced lung injury. Our findings indicate Shp2 as a potential target for radiation-induced lung injury and provide another way for endothelium to participate in the pathological process of radiation-induced lung injury.
Subject areas: Biological sciences, Immunology, Molecular biology, Transcriptomics
Graphical abstract
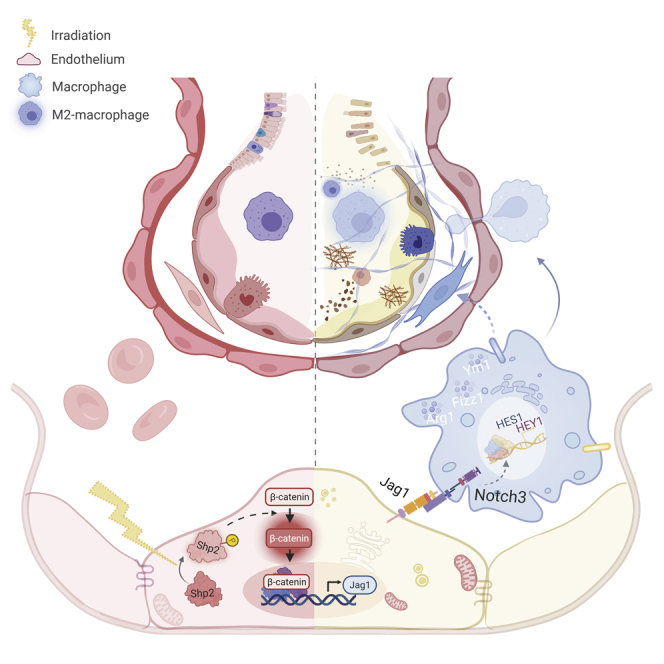
Highlights
-
•
Phosphatase activity of endothelial Shp2 is elevated by irradiation in vitro and in vivo
-
•
Radiation-induced Jag1 is blocked in Shp2-deficient endothelium
-
•
Loss of Shp2 in endothelium relieves radiation-induced pulmonary injury
-
•
Shp2-deficient endothelium restrains macrophage activation via Notch signaling
Biological sciences; Immunology; Molecular biology; Transcriptomics
Introduction
Radiotherapy is an important and effective treatment for malignant tumors of the chest, but patients usually experience varying degrees of radiation-induced lung injury after chest radiotherapy (Hanania et al., 2019). Radiation-induced lung injury is a complex dynamic response process involving the dysfunction of multiple cell types, constituting an inflammatory microenvironment in which endothelium and macrophages play indispensable functions (Xu et al., 2018). The role of macrophage in this process has been well studied because M1 macrophages contribute to acute radiation-induced lung injury while M2-macrophages lead to radiation-induced lung fibrosis (Bickelhaupt et al., 2017; Hu et al., 2020; Huang et al., 2017). Recently, increasing attention has been paid to the role of endothelium in the repair of tissue and organ injury caused by radiotherapy (Baselet et al., 2019; Wiesemann et al., 2019).
In addition to maintaining blood flow homeostasis, the integrity of the gas–blood barrier and other physiological functions, endothelium can synthesize and release a large number of adhesion molecules and cytokines that drives the immune-inflammatory response after lung injury (Wiesemann et al., 2019). It is thought that radiation-induced lung injury is preceded by vascular damage within a few weeks after radiation, such as the radiation-induced oxidative burst and mesenchymal transition (Choi et al., 2016a, 2016b). After radiation exposure, endothelial pro-inflammatory response occurs, followed by the vascular infiltration of immune cells of myeloid and lymphoid origin, leading to tissue inflammation and fibrosis (Guipaud et al., 2018). The endothelium–mesenchymal transition (EndMT) has recently been reported to contribute to radiation-induced lung injury, which is blocked by HSPB1 and HIF1α deletion in the endothelium (Choi et al., 2015b, 2016b). However, the mechanism of vascular endothelial dysfunction in the radiation-induced inflammation microenvironment remains unclear.
Reversible protein phosphorylation is the molecular basis for the regulation of cell biological functions. Shp2 is a non-receptor protein tyrosine phosphatase that participates in the regulation of various physiological and pathological processes (Xiao et al., 2018). Shp2 mutations have been detected in several diseases, such as A72G in Noonan syndrome (Yu et al., 2014). Shp2 also has two important tyrosine sites (Y542 and Y580), which are significantly associated with its phosphatase activity (Guo and Xu, 2020). Our recent studies showed that the absence of Shp2 triggers pathological characteristics of lung injury in mice (Zhang et al., 2012). The absence of Shp2 promotes macrophage type II activity, thereby accelerating the occurrence of bleomycin-induced lung injury (Tao et al., 2014). Moreover, Shp2 inhibition inhibited the LPS and cigarette extract-induced lung inflammation (Zhang et al., 2016a; Li et al., 2012). Recently, Tzouvelekis et al. found that Shp2-specific activation in fibroblasts leads to a decrease in TGF-β signaling to reduce lung injury; in contrast, Zehender’s later study showed that the absence of Shp2 leads to a decrease of the JAK/STAT signal, which caused an alleviation in the symptoms of injury in the skin and lungs of mice (Tzouvelekis et al., 2017; Zehender et al., 2018). Shp2 is widely involved in the inflammation and repair process after lung injury, but its role in radiation-induced lung injury is not yet known. Here, we investigate the potential of Shp2 to be a drug target for radiation-induced lung injury, one of the important side effects of tumor radiotherapy.
Vascular endothelium is the main component of pulmonary microvessels. It has been demonstrated that Shp2 mediates chronic endothelial inflammation by reducing the production of nitric oxide (Giri et al., 2012). Shp2 also plays an important role in mediating recovery of the vascular endothelial (VE)-cadherin-related endothelial barrier (Ukropec et al., 2000). Moreover, our previous research found that mice with endothelium-specific knockout of Shp2 show embryonic lethality, suggesting the importance of Shp2 in the maintenance of vascular function (Zhang et al., 2019). However, the relationship between Shp2 in the vascular endothelium and lung injury is still elusive. Our research provides an intrinsic mechanism for irradiated endothelium to regulate radiation-induced lung injury, as well as a potential therapeutic target in the treatment of radiation-induced lung injury.
Results
The phosphatase activity of Shp2 in endothelium is positively correlated with radiation in vitro and in vivo
First, we established a clinical-mimicking mouse model of radiation-induced lung injury with the SARRP used for small animals. Radiotherapy localization was established by a thoracic CT scan followed by a single dose of 50 Gy or 0 Gy (sham-irradiation) delivered to the whole lung (Figure S1A). After two months, leukocytes infiltration and collagen deposition in the irradiated lungs were significantly increased (Figure S1B). α-SMA, Col1α, and Col2α were also elevated (Figures S1C and S1D).
Y542 phosphorylation of Shp2 (pShp2 Y542) is positively associated with its phosphatase activity (Guo and Xu, 2020). In the irradiated lungs, we found that pShp2 Y542 was significantly increased while the total Shp2 level remained unchanged (Figures 1A and 1B), suggesting that pShp2 Y542 contributes to radiation-induced lung injury. Then Shp2 was immunoprecipitated from control and irradiated lungs and its phosphatase activity was evaluated using p-NPP as a substrate (Figures 1C and 1D). Consistently, Shp2 phosphatase activity was increased in the irradiated lungs. Radiation-induced lung injury is a complex dynamic reaction process involving multiple cell types, including fibroblasts, endothelial cells, epithelial cells, and macrophages, together constituting an inflammatory microenvironment (Xu et al., 2018). Thus, the pShp2 Y542 level and Shp2 activity in the primary cells isolated from lungs or cell lines of fibroblasts (MLF, HFL-1), endothelial cells (MLECs, HUVECs), epithelial cells (MLE-12), and macrophages (Macrophages, U937) were evaluated 40 min after irradiation (Figures 1E and 1F). Western blotting, phosphatase activity assays, and immunofluorescence results showed that the pShp2 Y542 level and Shp2 activity were specifically increased in endothelium after 10-Gy irradiation (Figures 1E–1H and S1G-L). In addition, a significant elevation of co-localization of pShp2 Y542 with the endothelial marker IB4 was evident in irradiated lung sections (Figure 1I). Moreover, in endothelium, pShp2 Y542 showed the highest level 40 min after irradiation in a dose-dependent manner (Figures 1J–1M). Interestingly, collagen deposition in this model mainly occurred around blood vessels rather than airways. Moreover, the expression levels of endothelial markers (CD31 and VE-cadherin) were reduced in injured mouse lungs (Figures S1E and S1F). Taken together, radiation increases Shp2 phosphorylation and phosphatase activity in pulmonary endothelium of radiation-injured mice.
Figure 1.

Shp2 activation is induced by irradiation in endothelium
(A) Representative western blotting images of proteins in whole lung lysates (n = 6 per condition) showing the levels of total Shp2 and pShp2 Y542. β-actin was used as a loading control. C57BL/6 mice were left untreated (control) or received 50 Gy of radiation in both lungs, and were sacrificed after two months.
(B) Protein levels of pShp2 Y542 normalized to Shp2 in (A) and repeat images were quantified by ImageJ.
(C) Phosphatase activity of Shp2 in whole lung lysates (n = 6 per condition) measured by determining the hydrolysis of p-NPP.
(D) Equal Shp2 was immunoprecipitated from lung lysates (n = 6 per condition) using a complex of protein A and Shp2 antibody to measure Shp2 activity.
(E) Representative western blotting images of total Shp2 and pShp2 Y542 in lysates of fibroblasts (HFL-1), endothelial cells (HUVEC), epithelial cells (MLE-12), and macrophages (U937). β-actin was used as a loading control. Cells were irradiated with 10 Gy X-ray and then incubated for 40 min.
(F) Protein level of pShp2 Y542 normalized to Shp2 in (E) and repeat images were quantified by ImageJ, pshp2/Shp2 represented for the value of pShp2 level compared to Shp2 level.
(G) Phosphatase activity of Shp2 in lysates of primary macrophages, endothelial cells (MLECs), epithelial cells (MLE-12), and fibroblasts (MLF) measured by determining the hydrolysis of p-NPP.
(H) Immunofluorescence staining of pShp2 Y542 (red) and Shp2 (green) in HUVECs with or without irradiation. Nuclei were counterstained with DAPI (blue). Scale bar, 20 μm.
(I) Immunofluorescence staining of pShp2 Y542 (green) and IB4 (red) in lung sections from control and irradiated mice, co-localization is shown in yellow. Nuclei were counterstained with DAPI (blue). Scale bar, 20 μm.
(J) Western blotting showing total Shp2 and pShp2 Y542 levels in lysates of HUVECs irradiated with 10 Gy X-ray and incubated for 0, 20, 40, 60, and 120 min. β-actin was used as a loading control.
(K) Protein levels of pShp2 Y542 normalized to Shp2 in (J) from three independent experiments were quantified by ImageJ.
(L) Western blotting showing total Shp2 and pShp2 Y542 levels in lysates from HUVECs irradiated with 0, 2.5, 5, 10, or 20 Gy X-ray and incubated for 40 min. β-actin was used as a loading control.
(M) Protein levels of pShp2 Y542 normalized to Shp2 in (L) from three independent experiments were quantified by ImageJ.
Data were shown as the mean ± SEM of three independent experiments. p-value was calculated using one-way ANOVA or two-tailed Student's t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S1.
Radiation-induced endothelial dysfunction is independent of EndMT, ROS, and apoptosis in Shp2-deficient endothelium
To further explore the physiological relevance of radiation-induced Shp2 activation in endothelium, HUVECs were exposed to 10-Gy irradiation and then incubated for 48 h. Western blotting assays showed that radiation elevated the level of α-SMA, a fibroblastic marker, while the level of the endothelial marker CD31 reduced. However, Shp2 deficiency did not involve in this process (Figure S2A). Similarly, Shp2 was not involved in the radiation-induced endothelial oxidative burst or apoptosis (Figures S2B–S2E). Therefore, RNA-seq analysis was performed in irradiated HUVECs transduced with shScr or shShp2 lentivirus. Venn diagrams showed three independent biological replicates in two conditions (Figure S2F). Through the DisGeNET and DO database, we found that the genes differentially expressed between irradiated control and Shp2-deletion HUVECs were significantly related to lung injury and vascular disease (Figures S2H and S2J). GO enrichment analysis revealed a significant change in signal transduction (Figure S2I). In addition, volcano plots and heatmaps revealed that the Jag1 level was notably reversed by Shp2 deletion in HUVECs (Figures 2A and S2G). These data imply that Jag1 is involved in radiation-induced endothelial dysfunction.
Figure 2.

Jag1 is elevated by radiation-induced activation of Shp2
(A) Heatmap showing clusters of top 20 genes induced (red) or repressed (blue) by Shp2 deletion.
(B) Relative Jag1 mRNA levels were analyzed by qRT-PCR in HUVECs 24 h after irradiation. shShp2 lentivirus were administered to HUVECs 24 h prior to irradiation.
(C) Jag1 levels were assessed by western blotting in HUVECs lysates 48 h after irradiation. β-actin was used as a loading control. shShp2 lentivirus were administered to HUVECs 24 h prior to irradiation.
(D) Protein levels of Jag1 normalized to β-actin in (C) from four independent experiments were quantified by ImageJ.
(E) Immunofluorescence staining of Jag1 (red) and Shp2 (green) in MLECs 48 h after irradiation. Nuclei were counterstained by DAPI (blue). Scale bar, 50 μm.
(F) Relative Jag1 and Jag2 mRNA levels were analyzed by qRT-PCR in HUVECs overexpressing Shp2.
(G) Jag1 levels in Shp2 overexpressed HUVECs were assessed by western blotting. 3xFlag labeled Shp2 WT, Shp2 A72G, or Shp2 C459S lentivirus were administrated for 48h. β-actin was used as a loading control.
(H) Protein levels of Jag1 normalized to β-actin in (G) from three independent experiments were quantified by ImageJ.
(I) Immunofluorescence staining of Jag1 (red) and Shp2 (green) in HUVECs 48 h after treatment with Shp2 WT, Shp2 A72G, or Shp2 C459S lentivirus. Nuclei were counterstained with DAPI (blue). Scale bar, 50 μm
(J) Relative Jag1 and Jag2 mRNA levels were analyzed by qRT-PCR in HUVECs 24 h after irradiation. Shp099 (10 μM) was added to HUVECs 2 h prior to irradiation.
(K) Jag1 levels were assessed by western blotting using proteins from HUVECs lysates 48 h after irradiation. β-actin was used as a loading control. Shp099 (10 μM) was added to HUVECs 2 h prior to irradiation.
(L) Protein levels of Jag1 normalized to β-actin in (K) from three independent experiments were quantified by ImageJ. Data were shown as the mean ± SEM of three or more independent experiments. p-value was calculated using one-way or two-way ANOVA. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figures S2 and S3.
Radiation-induced Jag1 expression is blocked in Shp2-deficient endothelium
To confirm the RNA-seq results, qRT-PCR was performed in irradiated HUVECs. Shp2 deficiency reduced radiation-induced Jag1, however, other significantly changed gene levels were independent of irradiation or Shp2 deletion (Figure S3A). Moreover, the mRNA level of Jag1, rather than other Notch ligands, was markedly elevated in a time-dependent way after irradiation (Figures S3B and S3C), confirming that Shp2 deletion specifically blocked the radiation-induced Jag1 level in endothelium. Furthermore, western blotting analysis showed radiation-induced Jag1 expression particularly in endothelium (Figure S3D), and it was disrupted by shShp2 lentivirus treatment (Figures 2B–2D). Next, we generated transgenic mice with endothelial conditional Shp2 knockout (Shp2 iECKO) in which the Cdh5-ERT2 construct reduced Shp2 expression after injection of tamoxifen. Knockout efficiency was detected in the isolated primary mouse lung endothelial cells (MLECs) (Figures S3E–S3I). Immunofluorescence results revealed that the shShp2 administrated or Shp2 knockout in endothelium suppressed the radiation-induced Jag1 level (Figures 2E, S3J, and S3K).
Shp2 phosphatase activity is tuned by an auto-inhibitory mechanism that requires a conformational change to expose its conserved protein domain. The disease-associated coding variants Shp2 A72G and Shp2 C459S are considered to be gain-of-function and loss-of-function mutants, respectively (Kontaridis et al., 2006; Yu et al., 2014). To further evaluate the effect of Shp2 activation on Jag1, Shp2 WT, Shp2 A72G, or Shp2 C459S was overexpressed in HUVECs by lentivirus. The results showed that only Shp2 A72G overexpression significantly upregulated Jag1 (Figures 2F–2I), suggesting that the phosphatase activity of Shp2 is associated with the level of Jag1. Pretreatment with Shp099, an allosteric inhibitor of Shp2 (Dardaei et al., 2018), reduced radiation-induced Jag1 level in irradiated HUVECs (Figures 2J–2L). In addition, similar findings were obtained in MLECs (Figure S3L–S3N), confirming that inactivated Shp2 reduced the radiation-induced Jag1. Overall, Jag1 in endothelium is induced by radiation and is positively correlated with the phosphatase activity of Shp2.
Shp2 deletion inhibits radiation-induced Jag1 expression via the β-catenin pathway
We next dissected the molecular mechanism by which Shp2 regulated radiation-induced Jag1. First, we analyzed the sequence of the Jag1 promoter in three transcription factor prediction websites (Figure 3A); β-catenin/LEF was identified as a potential transcription factor of Jag1. Upon Wnt signal activation, β-catenin accumulates in the cytoplasm and is transferred to the nucleus by transactivation of TCF/LEF, thereby resulting in the expression of target genes (Clevers, 2006). After irradiation, increased nuclear accumulation of β-catenin in endothelium was detected by western blotting, while Shp2 knockdown clearly reduced the nuclear translocation of β-catenin (Figures 3B and 3C). Moreover, the active β-catenin exhibited the same trend (Figure S4A). Therefore, mutant β-catenin (active β-catenin) was overexpressed in β-catenin knockdown HUVECs, Jag1 level was found to be markedly raised by mutant β-catenin (Figures S4C and S4D).
Figure 3.

Shp2 deletion blocks the radiation-induced Jag1 through β-catenin
(A) Results of three transcription factor prediction websites showing five common transcription factors of Jag1.
(B) Western blotting shows β-catenin level. Nuclear and cytoplasmic separation assay was performed in HUVECs 6 h after irradiation. Lamin-B1 was used as a loading control for nuclei, and GAPDH for cytoplasm. shShp2 was administered to HUVECs 48 h prior to irradiation.
(C) Protein levels of nuclear β-catenin normalized to Lamin-B1 in (B) from four independent experiments were quantified by ImageJ.
(D) Immunofluorescence staining of β-catenin (red) and Shp2 (green) in MLECs 6 h after irradiation. Nuclei were counterstained with DAPI (blue). Scale bar, 50 μm.
(E and F) Luciferase activity of TCF/LEF reporter was measured in 293T cells 24 h after irradiation. Luciferase activity is normalized to Renilla activity.
(G) Western blotting showing Jag1 levels in HUVECs lysates. β-actin was used as a loading control. shβ-catenin was administered 24 h before overexpressing Shp2 A72G.
(H) Protein levels of Jag1 normalized to β-actin in (G) from four independent experiments were quantified by ImageJ.
(I) Western blotting showing Jag1 levels in HUVECs lysates. β-actin was used as a loading control. HUVECs were treated with shShp2 lentivirus for 36 h. Wnt agonist 1 (2 μM) was added for 24 h.
(J) Protein levels of Jag1 normalized to β-actin in (I) from four independent experiments were quantified by ImageJ.
(K) Co-immunoprecipitation of Shp2 in 293T cells exposed to 10 Gy for 24 h assessed for the presence of β-catenin. The expression of β-catenin and Shp2 were detected in the corresponding lysates.
Data were shown as the mean ± SEM of three or more independent experiments. p-value was calculated using one-way ANOVA. ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S4.
Immunofluorescence staining of irradiated control or Shp2 iECKO MLECs and HUVECs also revealed that Shp2 deletion disrupted the nuclear accumulation of β-catenin after irradiation (Figures 3D and S4B). Transcriptional activation assays using a luciferase reporter driven by a consensus TCF/LEF response element showed that radiation-induced reporter activity was markedly suppressed upon Shp2 knockdown or inhibition, indicating that Shp2 mediates endothelial irradiation responses by activating β-catenin signaling (Figures 3E and 3F).
Next, to confirm that Shp2 deletion blocked the radiation-induced Jag1 through β-catenin, Shp2 A72G was overexpressed in β-catenin knockdown (KD) HUVECs. As shown in Figures 3G and 3H, β-catenin-KD disrupted the elevated Jag1 induced by Shp2 A72G. Wnt agonist 1 is a cell-permeable Wnt signaling activator that induces β-catenin-dependent transcriptional activity (Kim et al., 2017). The Jag1 level was significantly increased in Wnt agonist 1-treated HUVECs (Figure S4E). In addition, the reduced Jag1 level was rescued by Wnt agonist 1 in Shp2-deficient HUVECs (Figures 3I, 3J, and S4F), indicating that activated Shp2 elevates the Jag1 level through the β-catenin pathway. Moreover, co-immunoprecipitation assays showed that radiation promoted the interaction between Shp2 and β-catenin (Figure 3K). Taken together, Shp2 regulates the radiation-induced Jag1 level via the β-catenin pathway in endothelium.
Radiation-induced lung injury is relieved in Shp2 iECKO mice
Radiation-induced lung injury includes acute inflammation and subsequent chronic fibrotic phenotypes. To further explore the role of endothelial Shp2 in radiation-induced lung injury in vivo, Shp2 iECKO and control mice received a single dose of 20-Gy whole lung irradiation a week after tamoxifen injection for 5 days (Figure 4A). As evidenced by H&E and Masson’s trichrome staining (Figures 4B and 4C), endothelial-specific Shp2 deletion ameliorated the radiation-induced leukocytes infiltration and collagen deposition. Col1α protein (Figures 4D and 4F), as well as Col1α and Col2α mRNA levels (Figures 4G and 4H) were reduced in irradiated Shp2 iECKO lungs compared to those in control lungs. In addition, the hydroxyproline content (Figure 4I), leukocytes infiltration in BALF, and blood were also decreased (Figures 4J and 4K). In the early stage, inflammation and mild fibrosis are revealed in lungs, which contribute to later chronic fibrosis. To evaluate the effect of endothelial Shp2 in the early stage, 50-Gy whole lung irradiation was delivered to Shp2 iECKO and control mice, lung sections and tissue were obtained two months or two weeks later (Figures S5A and S6A). The inflammation level, collagen deposition (Figures S5B–S5G, and S6B–S6E), and hydroxyproline content (Figure S5H) induced by radiation in Shp2 iECKO mice were significantly lower than those in irradiated control mice. Inflammatory factors such as IL-1β, IL-6, and IL-10 production were notably reduced in irradiated Shp2 iECKO mice (Figures S5I–S5L, S6G, and S6H). In line with in vitro results, the Jag1 mRNA and protein levels were upregulated in irradiated control lungs but not in Shp2 iECKO lungs (Figures 4L–4N, S5M, and S5N). Moreover, Jag1 and IB4 displayed co-localization in lung tissues, indicating that Jag1 was specifically elevated in endothelium (Figure S5O). These results indicate that Shp2 exacerbates radiation-induced lung injury by supporting Jag1 expression.
Figure 4.

Shp2 iECKO mice exhibit reduced lung injury in response to radiation
(A) Schematic illustrating the radiation-induced lung injury mouse model. Shp2 iECKO and control mice were injected with 20 mg/kg tamoxifen for 5 days, then subjected to thoracic radiation with 20 Gy a week later. Lungs (n = 6 per condition) were obtained from mice 6 months after irradiation.
(B) Lung sections from Shp2 iECKO and control mice with or without 20-Gy irradiation stained with H&E (left) or Masson’s trichrome (right) to determine lung injury. Scale bar, 50 μm.
(C) Fibrotic areas in (B) were measured by randomly selecting microscopic fields in ImageJ.
(D) Col1α and α-SMA levels were assessed by western blotting using proteins of whole lung lysates. β-actin was used as a loading control. All samples were biologically independent and three independent experiments were performed.
(E and F) Protein levels of Col1α and α-SMA normalized to β-actin in (D) were quantified by ImageJ.
(G and H) Relative Col1α and Col2α mRNA levels in irradiated lung tissues were measured by qRT-PCR.
(I) Total hydroxyproline content in irradiated lungs from Shp2 iECKO and control mice.
(J and K) Total leukocyte counts in BALF and blood from control and Shp2 iECKO mice with or without 20-Gy irradiation.
(L) Relative Jag1 mRNA levels in irradiated lungs by qRT-PCR.
(M) Jag1 levels were assessed by western blotting using proteins from irradiated lung lysates. β-actin was used as a loading control. All samples were biologically independent and three independent experiments were performed.
(N) Protein levels of Jag1 normalized to β-actin in (M) were quantified by ImageJ.
Data were shown as the mean ± SEM of three independent experiments. p-value was calculated using one-way ANOVA. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figures S5 and S6.
Alternative activation of macrophages is inhibited in irradiated Shp2 iECKO mice
It has been increasingly recognized that endothelium is a critical determinant of radiotoxicity in healthy tissues, and the microvascular niche is one of the most sensitive after radiation (Choi et al., 2018). In irradiated mice, we found that macrophages accumulated around the endothelium, implying that endothelium may affect macrophages in the process of radiation-induced lung injury (Figure 5A).
Figure 5.

Specific Shp2 knockout in endothelium suppresses M2 activation of macrophages in injured lungs
(A) Immunofluorescence staining of irradiated lung sections with IB4 (endothelial marker, red) and F4/80 (macrophage marker, green), F4/80 positive cells that observed nearly to (≤5 μm) IB4 positive cells were considered to be accumulated. Nuclei were counterstained with DAPI (blue). Scale bar, 20 μm.
(B) qRT-PCR analysis of relative Arg1, Fizz1, and Ym1 mRNA levels in lung tissues from irradiated control and Shp2 iECKO mice.
(C) Immunofluorescence staining of Arg1 (red) and CD68 (green) in lung sections from control and Shp2 iECKO mice 6 months after irradiation. Nuclei were counterstained with DAPI (blue). Scale bar, 20 μm.
(D) qRT-PCR analysis of relative Arg1, Fizz1, and Ym1 mRNA levels in AMs from the BALF of irradiated control and Shp2 iECKO mice.
(E and F) qRT-PCR analysis of relative HES1, HEY1, Notch1, Notch2, and Notch3 mRNA levels in AMs from the BALF of irradiated control and Shp2 iECKO mice.
Data were shown as the mean ± SEM of three independent experiments. p-value was calculated using one-way or two-way ANOVA. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S6.
During lung regeneration after radiation, macrophages in a chronic inflammation microenvironment undergo alternative (M2) activation to acquire pro-fibrosis functions (Vannella and Wynn, 2017). Thus, we next examined whether Shp2-deficient endothelium affects macrophage activation in injured mice. qRT-PCR results showed that the levels of several M2 macrophage markers, such as Arg1, Fizz1, and Ym1, were significantly higher in irradiated control mice than in Shp2 iECKO mice (Figures 5B and S6F). The co-localization of CD68 (a macrophage marker) and Arg1 was markedly elevated in irradiated control mice but not in Shp2 iECKO mice (Figure 5C). To further confirm the role of endothelial Shp2 in macrophage activation in vivo, alveolar macrophages (AMs) were separated from the BALF of mice. Activation of AMs was apparently greater in irradiated control mice than in Shp2 iECKO mice (Figures 5D, S6I, and S6J).
Jag1, a ligand of Notch signaling, has been reported to regulate macrophage polarization contributing to diseases such as tissue inflammation and regeneration (Huang et al., 2018; Yin et al., 2018). Ligand-receptor binding activates Notch signaling and subsequently promotes the transcription of HES1 and HEY1 (Artavanis-Tsakonas et al., 1999). Thus, we next detected the Notch receptors Notch1, Notch2, and Notch3, and the HES1 and HEY1 levels in AMs, and found that HES1 and HEY1 exhibited the same trend as the M2 activation marker genes (Figure 5E). In addition, the Notch3 mRNA level, but not Notch1 or Notch2, was elevated in the AMs of irradiated control mice, and this was impaired by Shp2 knockout in endothelium (Figure 5F). Altogether, our data indicate that Shp2 knockout in endothelium inhibits the alternative activation of macrophages induced by radiation, thus relieving the radiation-induced inflammation microenvironment in mice lungs.
The absence of Shp2 in irradiated endothelium restrains macrophage alternative activation through Notch signaling
To confirm the effect of Shp2-deficient endothelium on macrophages in vitro, irradiated Shp2-deficient HUVECs were co-cultured with macrophages (Figure 6A). Immunofluorescence staining showed that macrophages co-cultured with irradiated HUVECs exhibited an M2-like phenotype, which was disrupted by Shp2 deficiency (Figures 6B and S7A). Then, macrophages were isolated to assess the levels of marker genes of M2 activation. The levels of Arg1, Fizz1, and Ym1 were markedly increased in macrophages co-cultured with irradiated control HUVECs but not those co-cultured with Shp2-deficient HUVECs (Figures 6C, S7B, and S7C). HES1, HEY1, and Notch3 showed the same trend (Figures 6D, 6E, and S7D). The overexpression of Shp2 A72G in endothelium also activated macrophages while the control or Shp2 C459S had no change (Figures 6F and 6G). DAPT, a γ-secretase inhibitor, suppresses Notch signaling by preventing the cleavage of Notch receptors (Qian et al., 2019). IL-4 is known to promote the M2 activation of macrophages. To further establish that Notch signaling positively regulates macrophage M2 activation, DAPT was added to IL-4 treated macrophages, and the FCM and qRT-PCR results showed that DAPT inhibited the M2 activation of macrophages (Figures S7E–S7H). Moreover, DAPT inhibited the M2 activation of macrophages that were directly treated with rhJag1 or co-cultured with irradiated endothelium (Figures 6H, 6L, and S8A). Similarly, HES1 and HEY1 showed the same trend in these macrophages (Figures 6I, S8B, and S8C). Nevertheless, when Jag1 was immunodepleted by an anti-Jag1 antibody, the activation of macrophages was disrupted (Figures 6J and 6K).
Figure 6.

The absence of Shp2 in irradiated endothelium restrains M2 macrophage activation via Notch signaling
(A) Scheme illustrating the experimental setup.
(B) Immunofluorescence staining of Arg1 (red) in PMs co-cultured with irradiated Shp2-deficient HUVECs. Nuclei counterstained with DAPI (blue). Scale bar, 50 μm.
(C) qRT-PCR analysis of relative Arg1, Fizz1, and Ym1 mRNA levels in PMs co-cultured with irradiated Shp2-deficient HUVECs for 24 h.
(D and E) qRT-PCR analysis of relative HES1, HEY1, Notch1, Notch2, and Notch3 mRNA levels in PMs co-cultured with irradiated Shp2-deficient HUVECs for 24 h.
(F and G) qRT-PCR analysis of relative qRT-PCR analysis of relative Arg1, Fizz1, and Ym1 (F), HES1, HEY1, and Notch3 (G) mRNA levels in PMs co-cultured with overexpressed Shp2-mutant MLECs for 24 h.
(H and I) qRT-PCR analysis of relative Arg1, Ym1, HES1, and HEY1 mRNA levels in BMDMs treated with rhJag1 (250 ng/mL) for 24 h. DAPT (20 μM) and rhJag1 were added when endothelium co-cultured with macrophages.
(J and K) qRT-PCR analysis of relative Arg1, Fizz1, and Ym1 (J), HES1, HEY1, and Notch3 (K) mRNA levels in PMs co-cultured with irradiated HUVECs for 24 h. Anti-Jag1 antibody (2 μg/mL) was added before co-culture.
(L) qRT-PCR analysis of relative Arg1, Fizz1, and Ym1 mRNA levels in PMs co-cultured with irradiated HUVECs for 24 h. DAPT (20 μM) was added when co-culture started.
(M) Jag1 level was measured 48 h after 10-Gy irradiation by western blotting. H460 CM was added 6 h before irradiation.
(N) qRT-PCR analysis of relative Fizz1, HES1, and Notch3 mRNA levels in peripheral blood leukocytes from patients before (n = 17) or after (n = 23) chest irradiation.
Data were shown as the mean ± SEM of three independent experiments. p-value was calculated using Student's t test and one-way or two-way ANOVA. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figures S7 and S8.
Next, we found that endothelium under tumor microenvironment was more sensitive to irradiation, as H460 conditional medium (CM) promoted radiation-induced Jag1 level in HUVECs (Figure 6M). Given this, we then verified our findings during clinical tumor radiotherapy. Peripheral blood leukocytes were extracted from patients with tumor before or after chest radiotherapy, in line with our findings, M2 and Notch signaling activation were significantly elevated in peripheral blood leukocytes from patients after chest radiotherapy (Figure 6N). In summary, endothelial Shp2 contributes to radiation-induced inflammation microenvironment by promoting the Notch signaling-mediated M2 activation of macrophages (Figure 7).
Figure 7.

Proposed mechanism by which endothelial Shp2 mediates radiation-induced lung injury
Schematic illustration showing the mechanism and the role of Shp2 in regulating radiation-induced lung injury.
Discussion
Radiation-induced lung injury is increasingly becoming a dose-limiting factor for radiation therapy with the progress of tumor therapy. Endothelium is a critical determinant of radiotoxicity in healthy tissues, as radiation induces vascular damage and subsequent leukocyte recruitment, resulting in alteration of the microenvironment (Choi et al., 2018; Klein et al., 2016; Korpela and Liu, 2014). However, the mechanisms by which endothelium is involved in radiation-induced lung injury are not fully understood. Previous studies have reported that Shp2 is highly involved in the process of lung injury caused by LPS, cigarette extract, or bleomycin (Zhang et al., 2016a; Li et al., 2012; Tao et al., 2014). However, the role of Shp2 in radiation-induced lung injury remains unclear. Here, we firstly found that Shp2 is a key regulator of radiation-induced lung injury.
The phosphatase activity of Shp2 is closely related to its phosphorylation, Y542 and Y580 are the main phosphorylation sites at the tail end of Shp2 (Guo and Xu, 2020). In the analysis of irradiated mouse lungs, pShp2 Y542 and Shp2 phosphatase activity were dramatically increased, further stressing that this process particularly occurred in endothelium, and suggesting that activated Shp2 in endothelium is associated with radiation-induced lung injury.
To further explore how endothelial Shp2 is involved in radiation-induced lung injury, RNA-seq analysis was performed in irradiated control and Shp2-deficient HUVECs. The results showed that Jag1, a ligand of Notch signaling, is likely to be associated with radiation-induced endothelial dysfunction. In line with the RNA-seq results, we confirmed that the radiation-induced Jag1 level was notably reduced by Shp2 deficiency in endothelium. Moreover, the Jag1 level in endothelium was closely related to the phosphatase activity of Shp2.
To elucidate the mechanism of how Shp2 regulates the Jag1 level in endothelium, we found β-catenin as a transcription factor using bioinformatics methods. Further results indicated that radiation-induced nuclear accumulation of β-catenin was disrupted by Shp2 deficiency. In addition, Shp2 activation failed to elevate the Jag1 level when β-catenin was lost. Moreover, Wnt agonist 1, an activator of β-catenin-dependent transcription, restored the Jag1 level in Shp2-deficient HUVECs, indicating the critical role of β-catenin in this process.
Radiation causes endothelial activation, inflammation with subsequent mild fibrosis in the early stage, and endothelial dysfunction with chronic inflammation and moderate fibrosis in the long stage (Wiesemann et al., 2019). To verify the pathological effects of endothelial Shp2 in the mouse model of radiation-induced lung injury, we created two short-term (2 weeks and 2 months) models and a long-term (6 months) model of radiation-induced lung injury in Shp2 iECKO mice. The Shp2 iECKO mice showed less inflammation and collagen deposition after irradiation than control mice. In line with our findings in vitro, the radiation-induced Jag1 level was lower in the lung endothelium of irradiated Shp2 iECKO mice.
Radiation induces vascular damage and subsequent leukocyte recruitment, thus leading to an inflammation microenvironment (Korpela and Liu, 2014). As a key regulator of lung regeneration, macrophages exhibit an M1 phenotype contributing to inflammation, rapidly thereafter, this turns to a more pro-regenerative M2 phenotype leading to chronic fibrosis. It has been reported that endothelium-derived lactate promotes macrophage polarization contributing to muscle regeneration (Zhang et al., 2020). Another report indicats that radio-resistant tumor endothelium recruits SDF-1+ tumor-associated macrophages and stimulates their M2 activation, thus promoting anti-tumor immune responses (Choi et al., 2018). In support, we showed here that macrophages were closely co-localized with endothelial cells in irradiated lung sections, furthermore, macrophages showed an M2 phenotype in irradiated lungs. We further demonstrated that M2 activation of AMs was impaired in Shp2 iECKO mice. Besides, macrophages co-cultured with irradiated Shp2-deficient endothelial cells showed a markedly decreased M2 phenotype. Notch signaling is strongly associated with regeneration as it has been reported to be markedly elevated in renal regeneration, systemic sclerosis, cardiac fibrosis, and pulmonary regeneration (Huang et al., 2018; Nemir et al., 2014; Dees et al., 2011; Yin et al., 2018). In our study, endothelium-derived Jag1 stimulation by activating Notch signaling in macrophages, and DAPT, an inhibitor of Notch signaling, effectively inhibited endothelium-derived Jag1-activated macrophages, demonstrated that the process of endothelium-activated macrophages is dependent on paracrine Notch signaling contributing to the radiation-induced inflammation microenvironment and lung injury.
Several reports indicate that endothelium contributes to radiation-induced lung injury by the mesenchymal transition, the oxidative burst, and apoptosis (Choi et al., 2015b, 2016a, 2016b). We conclude that the activation of macrophages in irradiated endothelium by paracrine Notch signaling which provides another way for endothelium to participate in the pathological process of radiation-induced lung injury, suggesting that targeting endothelium is a potential strategy for lung injury.
Our findings firstly demonstrate that endothelial Shp2 is a key regulator of radiation-induced lung injury. Recently, the successful development of Shp2 inhibitors with oral bioavailability indicates that targeting Shp2 become a promising strategy for clinical cancer treatment (Chen et al., 2016). These inhibitors inhibited the proliferation of cancer cells driven by receptor tyrosine kinase in vitro and in vivo (Fedele et al., 2018; Ruess et al., 2018; Dardaei et al., 2018; Mainardi et al., 2018). What’s more, they have entered phase Ⅱ clinical trials, and are expected to have broad prospects in translational therapy (Song et al., 2021). Shp2 also plays an important role in regulating blood vessels, as Shp2 inhibitors relieve pulmonary arterial hypertension and atherosclerosis (Cheng et al., 2018; Chen et al., 2018). Our results provide unique insight into the mechanisms by which endothelial Shp2 regulates radiation-induced lung injury, demonstrate that endothelial Shp2 deficiency relieves radiation-induced injury in mice lungs, and strongly argue for Shp2 inhibition as a potential strategy for lung injury and cancers.
Limitations of the study
Our study demonstrates that endothelial Shp2 regulates the pathological process of radiation-induced lung injury by activation of β-catenin and Notch signaling. Nevertheless, the interaction of Shp2 and β-catenin may be indirect, some other substrates of Shp2 such as CDC73 may involve in the process, and the underlying mechanism of this interaction needs further study. In addition, our results suggest that the administration of Shp099 or Shp099 and the Notch inhibitor combination is potential therapy for clinical radiation-induced lung injury. As its aim to solve the side effects of chemotherapy, whether this therapy will benefit malignant tumors control, requires further research.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Shp2 | Cell Signaling Technology |
Cat. # 3397 |
| Anti-Shp2 | Sigma | Cat. # PA5-17956 |
| Anti-Shp2 | Santa cruz | Cat. # sc-7384 |
| Anti-Shp2 (phospho Y542) | Abcam | Cat. # ab62322 |
| Anti-Jag1 | diagbio | Cat. # db8690 |
| Anti-Jag1 | Cell Signaling Technology |
Cat. # 70109 |
| Anti-Jag1 | R&D | Cat. # AF-1277 |
| Anti-β-catenin | Abcam | Cat. # ab32572 |
| Anti-β-catenin | Abcam | Cat. # ab223075 |
| Anti-Active-β-catenin | Cell Signaling Technology |
Cat. # 8814S |
| Anti-Col1α | Proteintech | Cat. # 14695-1-AP |
| Anti-α-SMA | Cell Signaling Technology |
Cat. # 19245 |
| Anti-CD31 | Abcam | Cat. # ab28364 |
| Anti-CD31 | R&D | Cat. # AF3628 |
| Anti-VE-cadhenrin | eBioscience | Cat. # AB2864994 |
| Anti-Lamin-B1 | Proteintech | Cat. # 12987-1-AP |
| Anti-GAPDH | diagbio | Cat. # bd106 |
| Anti-β-tubulin | Huabio | Cat. # M1305-2 |
| Anti-β-actin | Huabio | Cat. # AC026 |
| Anti-Arg1 | Proteintech | Cat. # 16001-1-AP |
| Anti-CD68 | BioLegend | Cat. # 137006 |
| Anti-CD206 | BioLegend | Cat. # 141720 |
| Anti-CD11b | BioLegend | Cat. # 101206 |
| Anti-IB4-Alexa Fluor 594 | Invitrogen | Cat. # I21413 |
| Anti-F4/80 | Cell Signaling Technology |
Cat. # 70076 |
| Rabbit Control IgG | Abclonal | Cat. # AC005 |
| Goat anti Mouse IgG (H+L) Alexa Fluor Plus 488 | Invitrogen | Cat. # A32723 |
| Goat anti Rabbit IgG (H+L) Alexa Fluor Plus 488 | Invitrogen | Cat. # A32731 |
| Goat anti Mouse IgG (H+L) Alexa Fluor Plus 555 | Invitrogen | Cat. # A32727 |
| Goat anti Rabbit IgG (H+L) Alexa Fluor Plus 555 | Invitrogen | Cat. # A32732 |
| Donkey anti Goat IgG (H+L) Alexa Fluor Plus 647 | Invitrogen | Cat. # A32849 |
| Goat anti mouse IgG HRP | MultiSciences | Cat. # 70-GAM007 |
| Goat anti rabbit IgG HRP | MultiSciences | Cat. # 70-GAR007 |
| 800CW Goat anti-Rabbit | LI-COR Biosciences | Cat. # 925-32211 |
| 680RD Goat anti-Mouse | LI-COR Biosciences | Cat. # 925-68070 |
| Bacterial and virus strains | ||
| shShp2 #1 | this manuscript | N/A |
| shShp2 #2 | this manuscript | N/A |
| shShp2 #3 | this manuscript | N/A |
| shβ-catenin #1 | this manuscript | N/A |
| shβ-catenin #2 | this manuscript | N/A |
| Shp2 WT | this manuscript | N/A |
| Shp2 A72G | this manuscript | N/A |
| Shp2 C459S | this manuscript | N/A |
| Mutant-β-catenin | Addgene | Cat. # 82176 |
| pGL4.49(luc2P/TCF-LEF RE/Hygro) | Promega | Cat. # E4611 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human Active Shp2 protein | R&D | Cat. # 1894-SH-01M |
| Recombinant Human Jag1 protein | Novoprotein | Cat. # CB95 |
| IL-4 | Novoprotein | Cat. # CK15 |
| MCS-F | Novoprotein | Cat. # CJ46 |
| Human TGF-β1 | Perprotech | Cat. # 96-100-21-2 |
| Critical commercial assays | ||
| Shp099 | Selleck | Cat. # S6388 |
| DAPI | Thermo Fisher | Cat. # 62248 |
| DAPT | Selleck | Cat. # 208255-80-5 |
| Wnt agonist 1 | Selleck | Cat. # S8178 |
| Deposited data | ||
| RNA-Seq | this study | GEO:GSE191056 |
| Software and algorithms | ||
| GraphPad Prism8 | Graphpad | https://www.graphpad.com/ |
| ImageJ | Schneider et al., 2012 (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Biorender | Biorender | http://biorender.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Xue Zhang (zhangxue@zju.edu.cn).
Material availability
This study did not generate new unique reagents.
Experimental models and subjects details
Mice
Shp2 flox/flox mice were generated as previously described (Xiao et al., 2018). Shp2 flox/+; Cdh5-CreERT2/+ mice were obtained by mating Shp2 flox/flox mice with Cdh5-CreERT2 mice. Then, Cdh5-CreERT2/+ Shp2 flox/flox mice were generated by intercrossing Cdh5-CreERT2/+ Shp2 flox/+ mice. Female mice were used within 6-8 weeks. CreERT2 recombinase activity and gene deletion in these transgenic mice were induced by daily injection of tamoxifen (Sigma-Aldrich) for 5 days. All mice were housed under specific pathogen-free conditions, and the experimental protocols were approved by the Zhejiang University Institutional Animal Care and Use Committee.
Radiation-induced lung injury mouse model
For whole lung irradiation, mice received irradiation with a small-animal radiation research platform (SARRP, XStrahl Medical and Life Sciences) in the Zhejiang Key Laboratory of Radiation Oncology. The procedures were as described previously (Zeng et al., 2020). Briefly, mice (anesthetized with 1% sodium pentobarbital, i.p.) were laid on the apparatus, then, radiotherapy localization was established by thoracic CT scan (with 50 KV X-ray at 0.8 mA and filtered with 1.0 mm aluminum). Whole-lung irradiation was implemented with isocentric ARC-field irradiation (220 KV X-ray, 13 mA, SSD 345 mm and a 0.15 mm Cu filter). A single dose of 50 Gy, 20 Gy, or 0 Gy (sham-irradiation) was administered to the whole lung. The field size was 10 mm × 10 mm. Before irradiation, the accurate position of the lungs was ensured by a digital radiograph. The dose for normal tissue (heart and spinal cord) was constrained to the lowest level.
Method details
Lung histopathology
Mice were euthanized, the lungs were harvested, fixed in 4% neutral buffered formalin, and paraffin sections were prepared. Paraffin-embedded sections were deparaffinized as previously described (Gong et al., 2019), then stained with hematoxylin and eosin (H&E, Beyotime) to evaluate changes in lung morphology or with a Masson's trichrome staining kit (Nanjing Jiancheng) to visualize collagen in lung tissue.
Broncho-alveolar lavage fluid (BALF) and alveolar macrophage (AMs) isolation
Irradiated and control mice were sacrificed by pentobarbital injection. BALF from mice was collected as described previously and centrifuged at 500x g for 5 min at 4°C, supernatants were stored at –80°C for ELISA (Zhang et al., 2016b).
AMs were isolated as described previously (Guo et al., 2017). Briefly, cell pellets from BALF were re-suspended in RIPM 1640 (Gibco) with 10% heat-inactivated fetal bovine serum (FBS, Biowest) and 50 units/ml penicillin and streptomycin (Gibco), then plated for 2 h; non-adherent cells were washed out with PBS, and macrophages were enriched ∼90%.
Cell culture and treatment
Human umbilical vein endothelium cells (HUVECs) were isolated and identified as described previously (Baudin et al., 2007; Zhang et al., 2019). HUVECs (passages 2-6) were cultured in 53% M199 (Corning)/ 37% human endothelial serum-free mixed medium (Gibco) with 10% FBS (Biowest) and endothelial cell growth supplement (Millipore). Cells were exposed to different doses of radiation using an X-ray linear accelerator (Rad Source Technologies) at a dose rate of 1.201 Gy/min. Lentiviral particles were directly delivered into cells in OPTI-MEM (Gibco). Shp099 (10 μM) was added to HUVECs 2 h prior to irradiation. H460 conditional medium was added to HUVECs 6 h prior to irradiation.
Bone marrow-derived macrophages (BMDMs) were isolated from wild type C57B/L6 mice as described previously (Guo et al., 2016). Peritoneal macrophages (PMs) were isolated as described previously (Guo et al., 2018). Briefly, wild-type C57B/L6 mice were injected with 4% thioglycolate (i.p., Merck), 4 days later, peritoneal cells were rinsed and inoculated on the culture plate, the adherent cells were ∼95% CD11b+F4/80+ macrophages, confirmed by flow cytometry. In the following experiments, BMDMs or PMs were co-cultured with HUVECs, or treated with interleukin (IL)-4 (20 ng/ml; Peprotech) and DAPT (20 μM, Selleck). In addition, recombinant human Jag1 (rhJag1, 250 ng/mL, Novoprotein) and anti-Jag1 antibody (2 μg/ml, R&D) were directly added to BMDMs or PMs before co-culture.
MLECs were isolated as described previously (Huang et al., 2020). Briefly, lung tissues from control and Shp2 iECKO mice (4-6 weeks) were cut into 1 mm3 pieces and digested in solution which containing 2 mg/mL collagenase type I (Sigma-Aldrich), 1 mg/mL dispase (Roche), and 10 μg/mL DNase (Roche) in DPBS (Dulbecco’s PBS, Corning) for 45 min with 80 rpm shaking at 37°C. Enzymes were neutralized with Dulbecco’s modified Eagle’s medium (DMEM) containing 20% FBS and a 40 μm cell strainer (Thermo Fisher Scientific) was used to collect cells, which were further purified using magnetic beads (Thermo Fisher Scientific) coated with anti-mouse CD31 (BD Bioscience). Cells were cultured in DMEM (Gibco) containing 20% FBS (Biowest), 15 mg/L endothelial cell growth supplement (Millipore), 1% nonessential amino-acids (Thermo Fisher Scientific), and 0.1 mg/mL heparin (Sangon Biotech). Cells were ∼90% CD31+ or IB4+ endothelial cells, confirmed by immunofluorescence staining as CD31 and IB4 are specific markers of endothelium (Wang et al., 2016; van Berlo et al., 2014; Chen et al., 2012; Choi et al., 2015a).
In vitro co-culture system
MLECs from Control and Shp2 iECKO mice lungs were exposed to 10 Gy irradiation, HUVECs were administrated with shScr or shShp2 lentivirus for 24 h, then exposed to 10 Gy irradiation. Next, BMDMs or PMs were isolated and added to co-cultured with MLECs or HUVECs, the number of co-cultured macrophages and endothelium were almost the same. After co-culture for 24 h, BMDMs or PMs were separated to detect the level of M2 or Notch signaling activation. 250 ng/mL rhJag1 or 2 μg/mL anti-Jag1 antibody was added before co-culture.
Human blood samples
The blood samples were provided by the Affiliated Hangzhou Cancer Hospital, Zhejiang University School of Medicine (Hangzhou, China) and the research has been approved by Medical Ethics Committee of Hangzhou Cancer Hospital. Leukocytes were isolated from the peripheral blood of tumor patients undergoing thoracic radiotherapy (n=23) or the control group (n=17), and TRIzol was used to extract RNA. Detailed information of patients were shown in Supplementary Table S1. Written informed consent was obtained from all patients.
Phosphatase activity assay
Proteins from cells and lung tissue were isolated in RIPA buffer, and Shp2 was immunoprecipitated as described above. The catalytic activity of Shp2 was monitored using the surrogate substrate paranitrophenyl phosphate (p-NPP, Sigma-Aldrich). The immune complexes of Shp2 were washed five times in PBST buffer and once in phosphatase buffer (30 mM HEPES pH 7.4, 120 mM NaCl). Then, the proteins were re-suspended in 200 μl phosphatase assay buffer (30 mM HEPES pH 7.4, 120 mM NaCl, 5 mM p-NPP, 1 mM DTT and 65 ng/μl bovine serum albumin) and incubated at 30°C for 30–120 min. Hydrolysis of p-NPP was measured by reading the absorbance at 405 nm on a microplate reader. A standard curve (0, 0.5, 1, 2, 4, and 8 ng) was generated using recombinant human Shp2 (Abcam).
RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNA from cells or lungs was extracted using TRIzol according to the manufacturer’s instructions. RNA was then reverse-transcribed to cDNA using the ReverTraAce qPCR RT kit (Toyobo). Gene expression was quantified by qRT-PCR using an SYBR Green kit on the CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad). The primer sequences are listed in Supplementary Table S2. Quantification was performed using the 2−ΔΔCt method relative to the expression of ACTB or 18s rRNA.
RNA-seq analysis
RNA from irradiated HUVECs treated with shShp2 lentivirus or shScr lentivirus was extracted using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies). Then RNA-seq analysis was performed on the Hiseq 4000 platform by Novogene (China). One microgram of RNA per sample was used for the RNA sample preparations. The differential expression under two conditions (three biological replicates per condition) was analyzed using the DESeq2 R package. Genes with an adjusted p-value < 0.05 and a fold change < –1.5 or > 1.5 were assigned as differentially expressed. Gene Ontology (GO) enrichment analysis was implemented using the clusterProfiler in R package. GO terms with corrected p-value < 0.05 were considered significantly enriched. Disease Ontology (DO) terms with corrected p-value < 0.05 were considered significantly enriched. DisGeNET pathways with corrected p-value < 0.05 were considered significantly enriched in differentially-expressed genes.
Western blotting
Cells or lung tissue were lysed in RIPA buffer (Beyotime) containing Na3VO4 (1 mM), protease inhibitor cocktail, and phosphatase inhibitor (phosSTOP, Roche), and the protein concentration was quantified by bicinchoninic acid assay (Beyotime). Equal amounts of protein were separated by SDS-PAGE, then transferred onto a nitrocellulose membrane (Pall Corp). The membranes were incubated at 4°C overnight with primary antibodies against Jag1 (Cell Signaling Technology), Shp2 (Cell Signaling Technology), pShp2 Y542 (Abcam), β-catenin (Abcam), active-β-catenin (Cell Signaling Technology), Col1α (Proteintech Group), α-SMA (Cell Signaling Technology), CD31 (Abcam), VE-cadherin (Ebioscience), Lamin-B1 (Proteintech Group), GAPDH (Daige), β-tubulin (Huabio), and β-actin (Huabio). The matched fluorescein-linked secondary antibodies (LI-COR Biosciences) were used to visualize proteins by incubation at room temperature for 1 h. The membranes were scanned with Odyssey (LI-COR Biosciences) and quantified by ImageJ.
Co-immunoprecipitation
For co-immunoprecipitation, cells were lysed in NP-40 lysis buffer containing Na3VO4 (1 mM), protease inhibitor cocktail, and phosSTOP for 1 h at 4°C. The lysates were then incubated with protein A magnetic bead (Bio-Rad)-conjugated Shp2 antibody (Cell Signaling Technology) overnight at 4°C. Protein levels were determined by western blotting as described above.
Immunofluorescence staining
Lung sections were incubated at 65°C for 1 h, then heated in citrate antigen-retrieval solution for 30 min (Solarbio). 0.5% TritonX-100 was used for permeabilization and 5% FBS was used for blocking. Sections were incubated with primary antibodies against Jag1 (Cell Signaling Technology), Shp2 (Sigma-Aldrich), pShp2 Y542 (Abcam), IB4 (Invitrogen), α-SMA (Cell Signaling Technology), Arg1 (Proteintech Group), and CD68 (Ebioscience) overnight at 4°C. The matched fluorescein-linked secondary antibodies (Invitrogen) were used for visualization by incubation at room temperature for 1 h. Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI, Beyotime). Images were acquired with an Olympus IX81-FV1000 microscope.
Cells on slides were fixed with 4% paraformaldehyde for 20 min, then permeabilized with 0.1% TritonX-100 for 20 min.
Flow cytometry (FCM)
Cells were washed with cold PBS and stained with fluorochrome-conjugated monoclonal antibodies against mouse CD206 (BioLegend) and CD11b (BioLegend) in the dark for 30 min at 4°C. Then cells were subjected to FCM analysis using an AECA NovoCyte TM system (ACEA Biosciences). Data were analyzed by FlowJo.
ELISA
IL-1β, IL-6 and IL-10 cytokine concentrations in BALF from irradiated and control mice were quantified using ELISA kits (EBioscience).
Quantification and statistical analysis
Statistics
Data were shown as the mean ± SEM of three or more independent experiments. Statistical analysis was performed using Student’s t test (two-tailed unpaired) for two groups, ANOVA for multi-group comparison (one-way or two-way ANOVA followed by Tukey’s post hoc tests or multi-comparisons) by GraphPad Prism, p<0.05 was considered to be statistically significant.
Power analysis
Power analysis, which was performed by “pwr” package in the G Power statistical analysis program, was applied with an analysis of variance (ANOVA) to determine the sample size for animal model. The results indicated that at least 5 samples for each group were needed. The curve between the required minimum sample size and the effect size was made under different statistical power. Statistical power=0.9 was selected to create a plot, which indicated the minimum sample size required for effect size=1.0 is n=5. Thus 5 samples per group is required to reach 90% power in this study.
Acknowledgments
We thank Dr. Gensheng Feng (University of California San Diego, La Jolla, CA, USA) for providing the Shp2 flox/flox mice, Dr. Luyang Yu (Zhejiang University) for providing the Cdh5-ERT2 cre mice, Xiaojian Wang for providing the mutant β-catenin plasmid, Peng Xiao (Sir Run Run Shaw Hospital School of Medicine, Zhejiang University) for assistance with flow cytometry analysis, Dacheng Gong, Shihao Zhao (Zhejiang University School of medicine), Xueping Zhou (laboratory animal center of Zhejiang University), Ming Chen and Peng Zhang (Zhejiang Key Laboratory of Radiation Oncology) for assistance with the mouse model, Wei Yin (Zhejiang University School of Medicine) for assistance with confocal microscopy, Xuliang Zhang (laboratory animal center of Zhejiang University) for assistance with cell irradiation. This work was supported by the National Natural Science Foundation of China (81873418 to YK, 31870901 to XZ, 81530001 to YK and 32000799 to JZ).
Author contributions
XZ and YK designed the work, PL, ML, YL, HZ, HZ, YZ, and YX performed experiments and analyzed data, JX, JZ, BX, HC, YK, and XZ provided funding, technical, or material support, XZ and PL wrote the paper.
Declaration of interests
The authors declare no conflict of interest.
Published: March 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103867.
Contributor Information
Yuehai Ke, Email: yke@zju.edu.cn.
Xue Zhang, Email: zhangxue@zju.edu.cn.
Supplemental information
Data and code availability
-
•
All data are included in the published article and the supplemental information files are available from the lead contact upon request. The RNA sequence datasets during this study are available at the Gene Expression Omnibus (GEO) site with accession number GEO: GSE191056.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Artavanis-Tsakonas S., Rand M.D., Lake R.J. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Baselet B., Sonveaux P., Baatout S., Aerts A. Pathological effects of ionizing radiation: endothelial activation and dysfunction. Cell Mol. Life Sci. 2019;76:699–728. doi: 10.1007/s00018-018-2956-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin B., Bruneel A., Bosselut N., Vaubourdolle M. A protocol for isolation and culture of human umbilical vein endothelial cells. Nat. Protoc. 2007;2:481–485. doi: 10.1038/nprot.2007.54. [DOI] [PubMed] [Google Scholar]
- Bickelhaupt S., Erbel C., Timke C., Wirkner U., Dadrich M., Flechsig P., Tietz A., Pfohler J., Gross W., Peschke P., et al. Effects of CTGF blockade on attenuation and reversal of radiation-induced pulmonary fibrosis. J. Natl. Cancer Inst. 2017;109 doi: 10.1093/jnci/djw339. [DOI] [PubMed] [Google Scholar]
- Chen J., Cao Z., Guan J. SHP2 inhibitor PHPS1 protects against atherosclerosis by inhibiting smooth muscle cell proliferation. BMC Cardiovasc. Disord. 2018;18:72. doi: 10.1186/s12872-018-0816-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.X., Zeng H., Reese J., Aschner J.L., Meyrick B. Overexpression of angiopoietin-2 impairs myocardial angiogenesis and exacerbates cardiac fibrosis in the diabetic db/db mouse model. Am. J. Physiol. Heart Circ. Physiol. 2012;302:H1003–H1012. doi: 10.1152/ajpheart.00866.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.N., Lamarche M.J., Chan H.M., Fekkes P., Garcia-Fortanet J., Acker M.G., Antonakos B., Chen C.H., Chen Z., Cooke V.G., et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535:148–152. doi: 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- Cheng Y., Yu M., Xu J., He M., Wang H., Kong H., Xie W. Inhibition of Shp2 ameliorates monocrotaline-induced pulmonary arterial hypertension in rats. BMC Pulm. Med. 2018;18:130. doi: 10.1186/s12890-018-0700-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H.J., Zhang H., Park H., Choi K.S., Lee H.W., Agrawal V., Kim Y.M., Kwon Y.G. Yes-associated protein regulates endothelial cell contact-mediated expression of angiopoietin-2. Nat. Commun. 2015;6:6943. doi: 10.1038/ncomms7943. [DOI] [PubMed] [Google Scholar]
- Choi S.H., Hong Z.Y., Nam J.K., Lee H.J., Jang J., Yoo R.J., Lee Y.J., Lee C.Y., Kim K.H., Park S., et al. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin. Cancer Res. 2015;21:3716–3726. doi: 10.1158/1078-0432.CCR-14-3193. [DOI] [PubMed] [Google Scholar]
- Choi S.H., Kim A.R., Nam J.K., Kim J.M., Kim J.Y., Seo H.R., Lee H.J., Cho J., Lee Y.J. Tumour-vasculature development via endothelial-to-mesenchymal transition after radiotherapy controls CD44v6(+) cancer cell and macrophage polarization. Nat. Commun. 2018;9:5108. doi: 10.1038/s41467-018-07470-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.H., Kim M., Lee H.J., Kim E.H., Kim C.H., Lee Y.J. Effects of NOX1 on fibroblastic changes of endothelial cells in radiationinduced pulmonary fibrosis. Mol. Med. Rep. 2016;13:4135–4142. doi: 10.3892/mmr.2016.5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.H., Nam J.K., Kim B.Y., Jang J., Jin Y.B., Lee H.J., Park S., Ji Y.H., Cho J., Lee Y.J. HSPB1 inhibits the endothelial-to-mesenchymal transition to suppress pulmonary fibrosis and lung tumorigenesis. Cancer Res. 2016;76:1019–1030. doi: 10.1158/0008-5472.CAN-15-0952. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Dardaei L., Wang H.Q., Singh M., Fordjour P., Shaw K.X., Yoda S., Kerr G., Yu K., Liang J., Cao Y., et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat. Med. 2018;24:512–517. doi: 10.1038/nm.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dees C., Tomcik M., Zerr P., Akhmetshina A., Horn A., Palumbo K., Beyer C., Zwerina J., Distler O., Schett G., Distler J.H. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Ann. Rheum. Dis. 2011;70:1304–1310. doi: 10.1136/ard.2010.134742. [DOI] [PubMed] [Google Scholar]
- Fedele C., Ran H., Diskin B., Wei W., Jen J., Geer M.J., Araki K., Ozerdem U., Simeone D.M., Miller G., et al. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 2018;8:1237–1249. doi: 10.1158/2159-8290.CD-18-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri H., Muthuramu I., Dhar M., Rathnakumar K., Ram U., Dixit M. Protein tyrosine phosphatase SHP2 mediates chronic insulin-induced endothelial inflammation. Arteriosclerosis Thromb. Vasc. Biol. 2012;32:1943–U508. doi: 10.1161/ATVBAHA.111.239251. [DOI] [PubMed] [Google Scholar]
- Gong H., Ni J., Xu Z., Huang J., Zhang J., Huang Y., Zeng C., Zhang X., Cheng H., Ke Y. Shp2 in myocytes is essential for cardiovascular and neointima development. J. Mol. Cell Cardiol. 2019;137:71–81. doi: 10.1016/j.yjmcc.2019.09.014. [DOI] [PubMed] [Google Scholar]
- Guipaud O., Jaillet C., Clement-Colmou K., Francois A., Supiot S., Milliat F. The importance of the vascular endothelial barrier in the immune-inflammatory response induced by radiotherapy. Br. J. Radiol. 2018;91 doi: 10.1259/bjr.20170762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C., Chi Z., Jiang D., Xu T., Yu W., Wang Z., Chen S., Zhang L., Liu Q., Guo X., et al. Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity. 2018;49:842–856.e7. doi: 10.1016/j.immuni.2018.08.021. [DOI] [PubMed] [Google Scholar]
- Guo C., Xie S., Chi Z., Zhang J., Liu Y., Zhang L., Zheng M., Zhang X., Xia D., Ke Y., et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. 2016;45:944. doi: 10.1016/j.immuni.2016.10.009. [DOI] [PubMed] [Google Scholar]
- Guo W., Xu Q. Phosphatase-independent functions of SHP2 and its regulation by small molecule compounds. J. Pharmacol. Sci. 2020;144:139–146. doi: 10.1016/j.jphs.2020.06.002. [DOI] [PubMed] [Google Scholar]
- Guo X., Li T., Xu Y., Xu X., Zhu Z., Zhang Y., Xu J., Xu K., Cheng H., Zhang X., Ke Y. Increased levels of Gab1 and Gab2 adaptor proteins skew interleukin-4 (IL-4) signaling toward M2 macrophage-driven pulmonary fibrosis in mice. J. Biol. Chem. 2017;292:14003–14015. doi: 10.1074/jbc.M117.802066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanania A.N., Mainwaring W., Ghebre Y.T., Hanania N.A., Ludwig M. Radiation-induced lung injury: assessment and management. Chest. 2019;156:150–162. doi: 10.1016/j.chest.2019.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D., Zhang Y., Cao R., Hao Y., Yang X., Tian T., Zhang J. The protective effects of granulocyte-macrophage colony-stimulating factor against radiation-induced lung injury. Transl. Lung Cancer Res. 2020;9:2440–2459. doi: 10.21037/tlcr-20-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Cai C., Zheng T., Wu X., Wang D., Zhang K., Xu B., Yan R., Gong H., Zhang J., et al. Endothelial scaffolding protein ENH (enigma homolog protein) promotes PHLPP2 (pleckstrin homology domain and leucine-rich repeat protein phosphatase 2)-mediated dephosphorylation of AKT1 and eNOS (endothelial NO synthase) promoting vascular remodeling. Arterioscler Thromb. Vasc. Biol. 2020;40:1705–1721. doi: 10.1161/ATVBAHA.120.314172. [DOI] [PubMed] [Google Scholar]
- Huang S., Park J., Qiu C., Chung K.W., Li S.Y., Sirin Y., Han S.H., Taylor V., Zimber-Strobl U., Susztak K. Jagged1/Notch2 controls kidney fibrosis via Tfam-mediated metabolic reprogramming. PLoS Biol. 2018;16 doi: 10.1371/journal.pbio.2005233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Zhang W., Yu F., Gao F. The cellular and molecular mechanism of radiation-induced lung injury. Med. Sci. Monit. 2017;23:3446–3450. doi: 10.12659/MSM.902353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W., Khan S.K., Gvozdenovic-Jeremic J., Kim Y., Dahlman J., Kim H., Park O., Ishitani T., Jho E.H., Gao B., Yang Y. Hippo signaling interactions with Wnt/beta-catenin and Notch signaling repress liver tumorigenesis. J. Clin. Invest. 2017;127:137–152. doi: 10.1172/JCI88486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D., Schmetter A., Imsak R., Wirsdoerfer F., Unger K., Jastrow H., Stuschke M., Jendrossek V. Therapy with multipotent mesenchymal stromal cells protects lungs from radiation-induced injury and reduces the risk of lung metastasis. Antioxid. Redox Signaling. 2016;24:53–69. doi: 10.1089/ars.2014.6183. [DOI] [PubMed] [Google Scholar]
- Kontaridis M.I., Swanson K.D., David F.S., Barford D., Neel B.G. PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J. Biol. Chem. 2006;281:6785–6792. doi: 10.1074/jbc.M513068200. [DOI] [PubMed] [Google Scholar]
- Korpela E., Liu S.K. Endothelial perturbations and therapeutic strategies in normal tissue radiation damage. Radiat. Oncol. 2014;9:266. doi: 10.1186/s13014-014-0266-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.F., Shen J., Shen H.J., Zhang X., Cao R., Zhang Y., Qui Q., Lin X.X., Xie Y.C., Zhang L.H., et al. Shp2 plays an important role in acute cigarette smoke-mediated lung inflammation. J. Immunol. 2012;189:3159–3167. doi: 10.4049/jimmunol.1200197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi S., Mulero-Sanchez A., Prahallad A., Germano G., Bosma A., Krimpenfort P., Lieftink C., Steinberg J.D., De Wit N., Goncalves-Ribeiro S., et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018;24:961–967. doi: 10.1038/s41591-018-0023-9. [DOI] [PubMed] [Google Scholar]
- Nemir M., Metrich M., Plaisance I., Lepore M., Cruchet S., Berthonneche C., Sarre A., Radtke F., Pedrazzini T. The Notch pathway controls fibrotic and regenerative repair in the adult heart. Eur. Heart J. 2014;35:2174–2185. doi: 10.1093/eurheartj/ehs269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian D., Li L., Rong Y., Liu W., Wang Q., Zhou Z., Gu C., Huang Y., Zhao X., Chen J., et al. Blocking Notch signal pathway suppresses the activation of neurotoxic A1 astrocytes after spinal cord injury. Cell Cycle. 2019;18:3010–3029. doi: 10.1080/15384101.2019.1667189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruess D.A., Heynen G.J., Ciecielski K.J., Ai J., Berninger A., Kabacaoglu D., Gorgulu K., Dantes Z., Wormann S.M., Diakopoulos K.N., et al. MUTANT KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018;24:954–960. doi: 10.1038/s41591-018-0024-8. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z., Wang M., Ge Y., Chen X.-P., Xu Z., Sun Y., Xiong X.-F. Tyrosine phosphatase SHP2 inhibitors in tumor-targeted therapies. Acta Pharmaceutica Sinica B. 2021;11:13–29. doi: 10.1016/j.apsb.2020.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao B., Jin W., Xu J., Liang Z., Yao J., Zhang Y., Wang K., Cheng H., Zhang X., Ke Y. Myeloid-specific disruption of tyrosine phosphatase Shp2 promotes alternative activation of macrophages and predisposes mice to pulmonary fibrosis. J. Immunol. 2014;193:2801–2811. doi: 10.4049/jimmunol.1303463. [DOI] [PubMed] [Google Scholar]
- Tzouvelekis A., Yu G., Lino Cardenas C.L., Herazo-Maya J.D., Wang R., Woolard T., Zhang Y., Sakamoto K., Lee H., Yi J.S., et al. SH2 domain-containing phosphatase-2 is a new antifibrotic regulator in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2017;195:500–514. doi: 10.1164/rccm.201602-0329OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukropec J.A., Hollinger M.K., Salva S.M., Woolkalis M.J. SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J. Biol. Chem. 2000;275:5983–5986. doi: 10.1074/jbc.275.8.5983. [DOI] [PubMed] [Google Scholar]
- van Berlo J.H., Kanisicak O., Maillet M., Vagnozzi R.J., Karch J., Lin S.C., Middleton R.C., Marban E., Molkentin J.D. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature. 2014;509:337–341. doi: 10.1038/nature13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannella K.M., Wynn T.A. Mechanisms of organ injury and repair by macrophages. Annu. Rev. Physiol. 2017;79:593–617. doi: 10.1146/annurev-physiol-022516-034356. [DOI] [PubMed] [Google Scholar]
- Wang J.M., Chen A.F., Zhang K. Isolation and primary culture of mouse aortic endothelial cells. J. Vis. Exp. 2016:52965. doi: 10.3791/52965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesemann A., Ketteler J., Slama A., Wirsdorfer F., Hager T., Rock K., Engel D.R., Fischer J.W., Aigner C., Jendrossek V., Klein D. Inhibition of radiation-induced Ccl2 signaling protects lungs from vascular dysfunction and endothelial cell loss. Antioxid. Redox Signal. 2019;30:213–231. doi: 10.1089/ars.2017.7458. [DOI] [PubMed] [Google Scholar]
- Xiao P., Guo Y., Zhang H., Zhang X., Cheng H., Cao Q., Ke Y. Myeloid-restricted ablation of Shp2 restrains melanoma growth by amplifying the reciprocal promotion of CXCL9 and IFN-gamma production in tumor microenvironment. Oncogene. 2018;37:5088–5100. doi: 10.1038/s41388-018-0337-6. [DOI] [PubMed] [Google Scholar]
- Xu T., Zhang Y., Chang P., Gong S., Shao L., Dong L. Mesenchymal stem cell-based therapy for radiation-induced lung injury. Stem Cell Res. Ther. 2018;9:18. doi: 10.1186/s13287-018-0776-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q., Wang W., Cui G., Yan L., Zhang S. Potential role of the Jagged1/Notch1 signaling pathway in the endothelial-myofibroblast transition during BLM-induced pulmonary fibrosis. J. Cell Physiol. 2018;233:2451–2463. doi: 10.1002/jcp.26122. [DOI] [PubMed] [Google Scholar]
- Yu Z.-H., Zhang R.-Y., Walls C.D., Chen L., Zhang S., Wu L., Liu S., Zhang Z.-Y. Molecular basis of gain-of-function LEOPARD syndrome-associated SHP2 mutations. Biochemistry. 2014;53:4136–4151. doi: 10.1021/bi5002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehender A., Huang J., Gyorfi A.H., Matei A.E., Trinh-Minh T., Xu X., Li Y.N., Chen C.W., Lin J., Dees C., et al. The tyrosine phosphatase SHP2 controls TGFbeta-induced STAT3 signaling to regulate fibroblast activation and fibrosis. Nat. Commun. 2018;9:3259. doi: 10.1038/s41467-018-05768-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z.-M., Du H.-Y., Xiong L., Zeng X.-L., Zhang P., Cai J., Huang L., Liu A.-W. BRCA1 protects cardiac microvascular endothelial cells against irradiation by regulating p21-mediated cell cycle arrest. Life Sci. 2020;244:117342. doi: 10.1016/j.lfs.2020.117342. [DOI] [PubMed] [Google Scholar]
- Zhang J., Huang J., Qi T., Huang Y., Lu Y., Zhan T., Gong H., Zhu Z., Shi Y., Zhou J., et al. SHP2 protects endothelial cell barrier through suppressing VE-cadherin internalization regulated by MET-ARF1. FASEB J. 2019;33:1124–1137. doi: 10.1096/fj.201800284R. [DOI] [PubMed] [Google Scholar]
- Zhang J., Muri J., Fitzgerald G., Gorski T., Gianni-Barrera R., Masschelein E., D'hulst G., Gilardoni P., Turiel G., Fan Z., et al. Endothelial lactate controls muscle regeneration from ischemia by inducing M2-like macrophage polarization. Cell Metab. 2020;31:1136–1153.e7. doi: 10.1016/j.cmet.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zhang Y., Tao B., Teng L., Li Y., Cao R., Gui Q., Ye M., Mou X., Cheng H., et al. Loss of Shp2 in alveoli epithelia induces deregulated surfactant homeostasis, resulting in spontaneous pulmonary fibrosis. FASEB J. 2012;26:2338–2350. doi: 10.1096/fj.11-200139. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu H., Yao J., Huang Y., Qin S., Sun Z., Xu Y., Wan S., Cheng H., Li C., et al. Manipulating the air-filled zebrafish swim bladder as a neutrophilic inflammation model for acute lung injury. Cell Death Dis. 2016;7 doi: 10.1038/cddis.2016.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Xu Y., Liu S., Guo X., Cen D., Xu J., Li H., Li K., Zeng C., Lu L., et al. Scaffolding protein Gab1 regulates myeloid dendritic cell migration in allergic asthma. Cell Res. 2016;26:1226–1241. doi: 10.1038/cr.2016.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data are included in the published article and the supplemental information files are available from the lead contact upon request. The RNA sequence datasets during this study are available at the Gene Expression Omnibus (GEO) site with accession number GEO: GSE191056.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.