

Abstract
Background
Monoamine oxidase B (MAO‐B) inhibitors slow disease progression in Parkinson's disease (PD) but clinical trials have produced conflicting results.
Objectives
To assess the evidence from randomised controlled trials for the effectiveness and safety of long‐term use of MAO‐B inhibitors in early PD.
Search methods
We searched the following electronic databases: Cochrane Central Register of Controlled trials (CENTRAL) (The Cochrane Library Issue 11, 2011), MEDLINE (last searched 8th November 2011) and EMBASE (last searched 8th November 2011); and handsearched neurology and movement disorders conference proceedings, checked reference lists of relevant studies and contacted other researchers.
Selection criteria
We included all unconfounded randomised controlled trials that compared a MAO‐B inhibitor with control, in the presence or absence of levodopa or dopamine agonists, in patients with early PD where treatment and follow up lasted at least one year.
Data collection and analysis
Two reviewers independently selected trials for inclusion, assessed the methodological quality, and extracted the data. Some additional data were provided by the original authors. Random‐effects models were used to analyse results, where appropriate.
Main results
Twelve trials were included (2514 patients, average follow‐up six years), 11 using selegiline. The methodological quality was reasonable although concealment of allocation was definitely adequate in only five trials. MAO‐B inhibitors were not associated with a significant increase in deaths (odds ratio (OR) 1.12; 95% confidence interval (CI) 0.90 to 1.41). They provided small benefits over control in impairment (weighted mean difference (WMD) for change in motor UPDRS score 3.79 points less with MAO‐B inhibitors; 95% CI 2.27 to 5.30) and disability (WMD for change in UPDRS ADL score 1.49 less; 95% CI 0.49 to 2.49) at one year which may not be clinically significant. There was a levodopa‐sparing effect with MAO‐B inhibitors, which was associated with a significant reduction in motor fluctuations (OR 0.73; 95% CI 0.58 to 0.91) but not dyskinesia (OR 0.96; 95% CI 0.76 to 1.22). The reduction in motor fluctuations was, however, not robust in sensitivity analyses. There was a trend to more withdrawals due to adverse events with MAO‐B inhibitors (OR 1.72; 95% CI 0.98 to 3.01).
Authors' conclusions
MAO‐B inhibitors (more specifically selegiline which contributes most of the data) do not appear to delay disease progression in terms of improved survival but may reduce later motor fluctuations. At present, we do not feel these drugs can be recommended for routine use in the treatment of early Parkinson's disease.
Plain language summary
Monoamine oxidase B inhibitors for early Parkinson's disease
Parkinson's disease is a disabling condition of the brain characterized by slowness of movement, shaking, stiffness, and in the later stages, loss of balance. Many of these symptoms are due to the loss of certain nerves in the brain, which results in the lack of a chemical called dopamine. Current treatments for Parkinson's are designed to increase dopamine by using levodopa (Sinemet or Madopar), which is converted in the brain into dopamine, or drugs that mimic dopamine (dopamine agonists). Although useful, these treatments do not slow the progression of the disease and can be associated with side‐effects e.g. after a while levodopa use can cause involuntary movements (dyskinesia), painful leg cramps (dystonia) and a shortened response to each dose (motor fluctuations). Monoamine oxidase B (MAO‐B) inhibitors such as selegiline (Eldepryl or Selgene) boost the levels of dopamine by a different mechanism, which may reduce the risk of these complications and slow disease progression. We reviewed 11 controlled trials with a total of 2514 patients that compared giving MAO‐B inhibitors with not giving them in people with early Parkinson's to see if it was safe and effective. The results show that, although MAO‐B inhibitors do improve symptoms of Parkinson's and delay the need for levodopa by a few months, they are too weak to have a major effect and do not seem to delay the progression of the condition. They may, however, reduce motor fluctuations although more information is needed to be certain of this. Although they can cause some side‐effects, these are generally mild.
Background
Parkinson's disease is a progressive neurodegenerative disorder characterized by a combination of bradykinesia, tremor, rigidity and postural instability. In most cases the cause remains unknown but there are characteristic changes in the brain including loss of dopaminergic neurons in regions of the brainstem and neuronal inclusions called Lewy bodies. The incidence (Twelves 2003) and prevalence (Tanner 1992) of Parkinson's disease increases dramatically with age and so its impact is set to increase as the population ages.
Since its introduction in 1967, the mainstay of treatment for Parkinson's disease has been levodopa, a dopamine precursor, which replenishes the depleted dopamine and alleviates many of the symptoms and signs (Watts 1997). However, after several years of treatment with levodopa many patients develop unpleasant and potentially disabling motor fluctuations ("wearing‐off" and "on‐off" phenomena) and dyskinesias (abnormal involuntary movements). These adverse effects occur in approximately 40% of patients after five years of levodopa treatment (Ahlskog 2001), and are more common in patients with early‐onset Parkinson's disease (Lang 1998). This, and the fact that levodopa probably does not alter the underlying disease progression have led researchers to look for alternative treatments that actually delay the pathogenesis of the disease and cause fewer long‐term side‐effects.
Monoamine oxidase B (MAO‐B) inhibitors block one of the enzymes that breaks down dopamine in the brain and so enhance its effects. Interest in their use in early Parkinson's disease arose from a retrospective observational study which showed improved survival of patients with Parkinson's disease when treated with the MAO‐B inhibitor selegiline (Birkmayer 1985). Around the same time, a group of Californian heroin addicts developed severe parkinsonism, due to a heroin contaminant, 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP), that was metabolised by MAO‐B to a substance that was neurotoxic to dopaminergic neurons (Langston 1983). MAO‐B inhibitors were shown to protect against MPTP toxicity in animal models of Parkinson's disease (Heikkila 1984). It has, therefore, been proposed that a similar mechanism of neurotoxicity may exist in Parkinson's disease due to an, as yet, unidentified environmental toxin against which MAO‐B inhibitors may be effective. Furthermore, MAO‐B inhibitors may be neuroprotective in Parkinson's disease by reducing the oxidative stress that exists in dopaminergic neurons (Olanow 1996) and some may have anti‐apoptotic effects that are independent of inhibition of MAO‐B (Maruyama 2002).
The potential benefit of the early use of MAO‐B inhibitors was supported by initial clinical trials that showed a delay in the need for levodopa in patients started on selegiline (PSG 1989). This was initially interpreted as a delay in disease progression but was later thought to be simply due to a symptomatic response from the weak dopaminergic activity of selegiline (Calne 1995). The pendulum swung against the early use of MAO‐B inhibitors following the publication of another trial that showed worse survival (Lees 1995), although this may have been a chance finding (Breteler 1998). With such conflicting evidence, a systematic review of all relevant trials is required to establish the efficacy and safety of MAO‐B inhibitors in early Parkinson's disease. This review updates our previous Cochrane review (Macleod 2005), which was itself based on a published review of all trials of MAO‐B inhibitors in early disease (Ives 2004).
Objectives
The objective of this review was to review the evidence from randomised controlled trials for the efficacy and safety of long‐term use of MAO‐B inhibitors in patients with early Parkinson's disease.
Methods
Criteria for considering studies for this review
Types of studies
We sought to identify all truly randomised, properly concealed, controlled trials comparing MAO‐B inhibitors with control interventions in early Parkinson's disease. We also included studies in which the method of randomisation or concealment was unknown. Cross‐over studies were excluded as they are not designed to assess long‐term effects.
Types of participants
We included trials that recruited patients with early Parkinson's disease, that is trials in which patients were starting parkinsonian treatment for the first time or had started treatment within the last 12 months and where the majority of patients were classified as Hoehn‐Yahr stage II or less (i.e. no impairment of balance). Trials including a significant proportion of patients with motor fluctuations (greater than 10%) were excluded. No strict diagnostic criteria for Parkinson's disease were set but the definition used in each trial was recorded.
Types of interventions
We included long‐term unconfounded trials comparing any dose of MAO‐B inhibitor (currently selegiline, rasagiline, lazabemide and safinamide) with no treatment or placebo. Thus, trials comparing MAO‐B inhibitor and levodopa versus levodopa, or MAO‐B inhibitor and a dopamine agonist versus a dopamine agonist, were included. Trials in which additional levodopa or dopamine agonist could be introduced into both arms according to clinical need as the disease progressed were also included. Trials of MAO‐B inhibitors in a head‐to‐head comparison with another drug (for example, MAO‐B inhibitor alone versus levodopa alone) were excluded. Trials with treatment or follow up of less than one year's duration were also excluded because we were interested in the long‐term effects of treatment rather than short‐term symptomatic effects.
Types of outcome measures
-
To evaluate the effectiveness of MAO‐B inhibitors we collected data at the final available follow up on the following outcome measures in each treatment group.
Number of patients who were either dead or disabled (that is, needed help with activities of daily living) from any cause (for example, disease progression, motor fluctuations or dyskinesias). Patients who became disabled and subsequently died were counted only once in this analysis.
Number of deaths.
-
Disease progression in terms of:
severity of parkinsonian impairment, disability and quality of life provided that data were reported for the same duration of follow up and the same clinical condition (that is, on or off medication) in the treatment and control groups and data were available for >75% of those randomised;
levodopa requirements including mean levodopa dose and numbers of patients requiring levodopa;
time to introduction of levodopa or dopamine agonist.
Number of patients with motor fluctuations including wearing off, on/off fluctuations and early morning dystonia.
Number of patients with dyskinesias.
-
To assess the safety of MAO‐B inhibitors, we collected data at final follow up on the following outcome measures in each treatment group.
Number of patients with adverse events such as nausea, postural hypotension and neuropsychiatric effects.
Number of withdrawals due to adverse events.
Total number of withdrawals.
If sufficient trials recorded outcomes at the same point in time (for example, at one year) we sought to analyse outcomes at that time point. The primary outcome measure was the number of people who were dead or disabled since this assessed the overall effect of treatment on a clinically important outcome. It would capture any delay in disease progression or prevention of severe motor complications with treatment but also take into account any increase in death rate.
Search methods for identification of studies
The following sources and searches were used to identify relevant randomised controlled trials:
The Cochrane Central Register of Controlled Trials (CENTRAL), (The Cochrane Library Issue 11, 2011)[Appendix 1]
MEDLINE through OVID Gateway (last searched 8th November 2011)[Appendix 2]
EMBASE through OVID Gateway (last searched 8th November 2011)[Appendix 3]
Online searching of conference proceedings through the Conference proceedings citation index (CPCI), (last searched 11th March 2010)[Appendix 4]
-
Handsearching of the following conference proceedings:
First to 13th International Congress of Parkinson's Disease and Movement Disorders (1990 to 2010);
XII International Symposium on Parkinson's Disease (1997);
XIII to XVIII International Congress on Parkinson's Disease and Related Disorders (1999 to 2009);
XVIth to XIXth meeting of the World Congress of Neurology (1997 to 2009);
Thirty‐seventh to 62nd annual meetings of the American Academy of Neurology (1985 to 2010);
One hundred and tenth to 134th meetings of the American Neurological Association (1985 to 2009);
Sixth to 14th meetings of the European Federation of Neurological Sciences (2002 to 2010);
Tenth to 19th meetings of the European Neurological Society (2001 to 2009);
Twenty‐ninth to 33rd meetings of the Scandinavian Congress of Neurology (1990 to 2002);
First and 2nd World Parkinson Congress (2006, 2010).
Checking reference lists: we sought to identify any additional references to trials in the published reports of relevant trials.
Previous reviews were checked to identify any missing trials.
Personal communication − we contacted other researchers in the field.
Data collection and analysis
For this update, two reviewers (KAT, RC) independently assessed the titles and abstracts identified from the electronic and other searches and the full text of potentially relevant articles was obtained. The final eligibility of each article, on the basis of the inclusion and exclusion criteria, was checked with a third reviewer (CC). There were no disagreements over which studies should be included. Articles reporting the same study were grouped together and the most up‐to‐date results for each outcome were used for this review.
The methodological quality of each trial was assessed according to method of randomisation, the blinding of treatment and outcome assessment, the use of placebo control, the completeness of follow‐up and whether intention‐to‐treat analysis was carried out or possible from the published data. The risk of bias table was prepared by one author (KAT) and checked by another (CC). We also assessed whether the treatment groups were comparable with regard to demographics, clinical characteristics, the number of patients excluded or lost to follow up within each trial and whether the definitions of outcomes and inclusion and exclusion criteria were comparable across the different trials. Data on the number of patients with each outcome event were sought by allocated treatment group, irrespective of compliance and whether or not the patient was subsequently deemed ineligible or otherwise excluded from follow up, to allow an intention‐to‐treat analysis. All data were extracted by two reviewers and cross checked. We attempted to contact the authors of the studies for further details if details of randomisation or concealment were unclear or if any data on the outcomes were missing.
Formal meta‐analysis was performed using RevMan 5 and where we could not combine outcome data from different studies (for example, because the outcomes recorded were too variable) we gave a descriptive summary of the results. For the main analyses the denominator was the number of patients in whom the outcome was assessed. For each outcome we calculated mean duration of follow up weighted for trial size by multiplying the mean duration of follow up for each study by the total trial size then dividing by the total number of participants included for that outcome. We calculated odds ratios (and 95% confidence intervals) for the binary outcomes and, if the results were statistically significant and the duration of follow up was similar between trials, we calculated the absolute risk reductions or increases for a variety of baseline absolute risks. Some trials had significantly different periods of follow up in the two treatment arms and, therefore, we adjusted the results of binary outcomes to take account of this by increasing the number of events in the arm with the shorter follow up by a proportional amount. However, we have also reported the unadjusted results. For continuous outcome measures (for example, impairment), we calculated a mean weighted difference where possible (or a standardized mean difference where different scales for the same outcome were used). To assess the change in impairment and disability data from baseline to one year, or to the end of the washout period, we had to impute the standard deviations from studies which only reported the standard deviations for the baseline and final scores. To do this we used the following formula to estimate the variance of the change in score:
vardiff = varpre + varpost ‐ 2r√(varprevarpost)
where vardiff is the variance of the change in score; varpre is the variance of the baseline score; varpost is the variance of the final score and r is the correlation between the pre‐ and post‐treatment scores. We assumed a correlation co‐efficient of 0.5, which is a conservative estimate, to reduce the chance of false positive results. Continuous data that were obviously skewed were excluded from meta‐analysis because the statistical techniques used assume a normal distribution.
For each outcome, the primary analysis was reported from a random‐effects model. A fixed‐effect model was also used but this did not significantly alter any of the results. Heterogeneity was assessed with both the I2 statistic (Higgins 2003) (0% indicates no heterogeneity and a value greater than 50% indicates substantial heterogeneity), and the chi squared test. The latter was regarded as significant if the probability value was less than 0.1 because of the low power of the test. There were no discrepancies between these two measures of heterogeneity for any outcomes and so we have only reported the I2 value in the text. If significant heterogeneity was found, we attempted to identify possible causes for this by carrying out planned subgroup analyses based on:
trial quality ‐ high quality trials (truly randomised, well concealed randomisation and double‐blind) versus those of lower quality;
the use of levodopa or a dopamine agonist at the beginning of trial ‐ trials with patients on levodopa or a dopamine agonist at the start of a trial versus those studies where patients were started only on an MAO‐B inhibitor or placebo/no treatment;
the different MAO‐B inhibitors;
the duration of follow up ‐ trials with a follow up period greater than five years versus those with follow up less than five years;
the type of early patients recruited ‐ trials using a more strict definition of early disease (that is all patients Hoehn and Yahr II or less and previous treatment less than six months) versus those using less strict criteria.
We assessed the difference between subgroups by calculating a two‐tailed z‐score using the following formulae for binary and continuous data respectively (Fleiss 1993): z = (lnOR1 ‐ lnOR2 ) / √(var[lnOR1] + var[lnOR2 ]); z = (SMD1 ‐ SMD2) / √(var[SMD1] + var[SMD2]) where OR1/2 and SMD1/2 are the combined odds ratios or standardised mean differences from each subgroup and var is the variance of each which was calculated from the 95% confidence intervals by the formula var[lnOR] = {(ln[upper 95%CI] ‐ ln[lower95%CI])/(2*1.96)}2.
For this update we had planned to perform a meta‐analysis of hazard ratios for survival from each trial but unfortunately there were insufficient data in the published reports to do this.
The effect of publication bias was analysed with a funnel plot (Egger 1997) and by calculating the size of an imaginary null trial (OR = 1) that would be required to turn any statistically significant result into a non‐significant result, assuming an event rate equal to the average event rate in the control groups. Finally, for statistically significant results from dichotomous data we assessed the effect of missing outcomes from patients excluded or lost after randomisation by performing a modified worst‐case or best‐case sensitivity analysis. For a true worst‐case analyses (that is most weighted against treatment) it is usually assumed that all excluded patients in the treatment group had an adverse outcome and all those in the control group had a positive outcome, and vice‐versa for a best‐case analysis. However, it is often unrealistic to expect this to be the case; so, for a more realistic assessment we performed a modified worst‐case or best‐case analysis. For the former we assumed the lost patients in the treatment group had the highest rate of poor outcome found in any trial whilst those in the control group had the lowest rate in any individual trial, and vice versa for the modified best‐case analysis.
Results
Description of studies
Twelve randomised trials, with a total of 2514 patients, met the inclusion criteria and were included in the review. Summary details of these trials are given in the Characteristics of included studies table. Sixteen studies were excluded (see Characteristics of excluded studies table) for the following reasons (NB some trials had more than one reason): period of treatment and follow up less than one year (13 trials); confounding by another treatment, for example comparing MAO‐B inhibitor alone versus levodopa or bromocriptine alone (two trials); too many exclusions post‐randomisation (one trial); and no useful outcome data (two trials). No relevant ongoing trials have been identified to date.
All of the included trials recruited only patients with a clinical diagnosis of idiopathic Parkinson's disease, although the precise diagnostic criteria were not usually given. Eight studies excluded patients with dementia; and all except two (UK‐PDRG (RR) 1998; UK‐PDRG 2001) excluded severe or unstable concomitant diseases. Four studies (California 1989; DATATOP 1993; Norway‐Denmark 1999; PARJUPAR 1996) excluded both very young and elderly patients, two excluded only elderly patients (SELEDO 1999; Swedish PSG 1998), and one (UK 1996) included only patients aged 65 or over. Mean age, adjusted for study size, was 62.7 years. All studies except one (Finland 1997) enrolled more males than females, five studies with a 2:1 ratio or greater. The mean stage of disease, on the Hoehn and Yahr scale was about two.
Some participants had previously been treated with levodopa although not for more than one year. In four studies the percentage of patients that had previously been treated varied between 15% and 34% (California 1989; Norway‐Denmark 1999; PSG 1996; SELEDO 1999). Four other studies (DATATOP 1993; UK‐PDRG 2001; UK‐PDRG (RR) 1998; US 1995) included only patients who were not on treatment for Parkinson's disease at the time of randomisation of whom a small unspecified proportion of participants had previously been treated with levodopa. Four studies (Finland 1997; PARJUPAR 1996; Swedish PSG 1998; UK 1996) included only patients who had never been treated with levodopa or any other dopaminergic medication. Only three studies met the criteria for a strict definition of early disease (that is restricted to patients in Hoehn and Yahr grades I or II with prior treatment of less than six months (DATATOP 1993; PARJUPAR 1996; PSG 1996).
In eleven studies the MAO‐B inhibitor used was selegiline and in one study lazabemide was used (PSG 1996). No studies of rasagiline or safinamide met the inclusion criteria. All the selegiline studies used either 10 mg daily or 5 mg twice daily. The PSG study used lazabemide at four different doses twice daily (12.5 mg, 25 mg, 50 mg, 100 mg) and for the purposes of this review the results from these dosage groups were combined because no dose‐response relationship was found.
In six trials the treated patients received selegiline (California 1989; DATATOP 1993; Finland 1997; Swedish PSG 1998; UK 1996) or lazabemide (PSG 1996) alone from the outset. In the Californian and UK trials patients received selegiline or placebo until levodopa was clinically indicated, at which point the participants were withdrawn from further evaluation. The Finnish and Swedish studies consisted of two phases: the first, until the patients required levodopa, was treatment with just selegiline or placebo; the second phase involved the addition of levodopa to both groups. The DATATOP study was a 2 by 2 factorial design with selegiline and tocopherol (vitamin E). In this review we have recorded outcomes for selegiline and placebo regardless of the use of tocopherol which was found to be ineffective. Levodopa was added to the treatment regimen, as required, following washout. After an interim analysis at two years, the protocol was modified and all participants received selegiline. Subsequently the design was modified again and the participants were re‐randomised to remain on selegiline or to have it withdrawn. Because of these changes, and the multiple publications, analysis of DATATOP by initial randomisation was problematic although we have tried to record outcomes by the original randomisation.
Four trials used levodopa and selegiline treatment from the outset of the trials (Norway‐Denmark 1999; UK‐PDRG (RR) 1998; UK‐PDRG 2001; SELEDO 1999). Patients in the Norwegian‐Danish trial and the SELEDO study received either selegiline or placebo in addition to levodopa until the endpoint or up to five years. In the Norwegian‐Danish study the endpoint was the need for additional medication whilst in SELEDO it was a 50% increase in the levodopa dose. The UK‐PDRG study was an open trial with three arms: levodopa alone, selegiline and levodopa, and bromocriptine alone. In this review we have excluded the bromocriptine patients except for a small number who were quickly re‐randomised into the first two arms because they were unable to tolerate bromocriptine (UK‐PDRG (RR) 1998). The numbers of deaths in these patients were reported separately and have been included as a separate trial. Ten years after the UK‐PDRG trial began (mean follow up of 6.8 years) the selegiline arm was discontinued due to concerns about increased mortality. Follow up continued for another mean 2.4 years. One trial (PARJUPAR 1996) compared the combination of selegiline and bromocriptine with bromocriptine alone with time to levodopa the primary endpoint at which point follow up stopped.
The final study (US 1995) used a 2 by 2 factorial design to compare selegiline with placebo in the presence of either levodopa or bromocriptine. For the main analyses we have considered the results from the levodopa arms and the bromocriptine arms separately.
Six studies included a washout period after the endpoint to try to assess the effect of MAO‐B inhibitors on actual disease progression by attempting to eliminate the confounding of any symptomatic effect. The washout period varied from two weeks (PSG 1996) to two months (Swedish PSG 1998; US 1995).
Five studies had follow up lasting between one and three years (California 1989; PARJUPAR 1996; PSG 1996; UK 1996; US 1995); two a maximum follow up duration of five years ‐ Norway‐Denmark 1999 (mean follow up 2.9 years in selegiline arm, 3.1 years in placebo arm) and SELEDO 1999 (mean follow up 3.9 years in selegiline arm, 3.6 years in placebo arm); two had follow up of six to seven years (Finland 1997; Swedish PSG 1998), whilst the DATATOP and UK‐PDRG studies both had longer duration of follow up for mortality ‐ mean 8.2 years (DATATOP 1993) and median 11.4 years (UK‐PDRG 2001).
Risk of bias in included studies
Randomization Methods of randomisation were poorly described in most of the studies. Three studies did not provide any details of generation of randomisation sequence or of concealment of allocation (PSG 1996; SELEDO 1999; UK 1996) despite requesting this information from the authors. The Californian study (California 1989) used a "biased coin" and another study used a computer to generate the randomisation sequence (US 1995), but each gave no details about concealment. Five studies gave sufficient information for us to classify the concealment as adequate (DATATOP 1993; Norway‐Denmark 1999; PARJUPAR 1996; UK‐PDRG (RR) 1998; UK‐PDRG 2001).
Baseline differences between treatment groups All the studies had similar severity scores in the intervention and control groups at baseline.
Blinding Blinding of patients, doctors and outcome assessors took place in ten of the trials. The only unblinded trial was the UK study (UK‐PDRG (RR) 1998; UK‐PDRG 2001). This leaves open the possibility of intervention bias (systematic differences in the care between groups) or measurement bias (systematic differences in the measurement of outcomes). Although the latter should have had no effect on the number of deaths, we cannot discount the possibility that bias was introduced in subjective measures such as disease impairment or disability scales, or in decisions to introduce levodopa.
Exclusions and losses to follow up Many trials did not clearly report withdrawals so it was difficult to calculate the total numbers of exclusions following randomisation and losses to follow up, which also varied depending on the time point at which the outcome was measured. As a maximum estimate of the participants not included in the analyses, the total number of withdrawals in all studies was 422 out of 2514 (16.8%), 229 of 1363 (16.8%) in the intervention group and 193 of 1151 (16.8%) in the control group. However, losses to mortality follow up were fewer because some of the withdrawn participants were included in the analysis of deaths although the precise numbers were not always reported.
Intention‐to‐treat analysis Most studies did not include withdrawn patients in the analyses. Only two trials (UK‐PDRG (RR) 1998; UK‐PDRG 2001) reported results based on a true intention‐to‐treat analysis; DATATOP 1993 reported mortality data and PSG 1996 reported results on numbers requiring levodopa on an intention‐to‐treat basis.
Effects of interventions
The 12 included trials varied substantially in measuring particular outcomes so we have more complete data for some outcome measures than for others (Table 1). All dichotomous outcomes are adverse events so an odds ratio less than one favours treatment with MAO‐B inhibitors. For the continuous outcomes a weighted mean difference less than zero favours MAO‐B inhibitors.
1. Reported outcomes in each trial.
| Study ID | Dead or disabled | Deaths | UPDRS | Need for levodopa | Time to levodopa | Levodopa dose | Motor fluctuations | Dyskinesias | Adverse events | Total withdrawals |
| California 1989 | N | Y | Y | Y | Y | N | N | N | N | Y |
| DATATOP 1993 | N | Y | Y | Y | Y | N | Y | Y | N | Y |
| Finland 1997 | N | Y | N | Y | Y | Y | Y | Y | N | Y |
| Norway‐Denmark 1999 | N | Y | Y | N/A | N/A | Y | Y | Y | Y | Y |
| PARJUPAR 1996 | N | Y | N | Y | Y | N | N | N | Y | Y |
| PSG 1996 | N | Y | Y | Y | Y | N | N | N | Y | Y |
| SELEDO 1999 | N | Y | N | N/A | N/A | Y | Y | N | Y | Y |
| Swedish PSG 1998 | N | Y | NU | Y | Y | Y | Y | Y | Y | Y |
| UK 1996 | N | Y | N | N | N | N | N | N | Y | Y |
| UK‐PDRG 2001 | N | Y | N | N/A | N/A | Y | Y | Y | N | Y |
| UK‐PDRG (RR) 2001 | N | Y | N | N/A | N/A | N | N | N | N | Y |
| US 1995 | N | Y | Y | Y | Y | Y | N | N | N | Y |
UPDRS ‐ Unified Parkinson's Disease Rating Scale; N ‐ No; Y ‐ Yes; N/A ‐ not applicable; NU ‐ not used (reported but not used in this review)
Dead or disabled at end of follow‐up No trial reported data on the primary outcome.
Deaths at end of follow up (Analysis 1.1) All the studies reported data on deaths at the end of follow up. Data were available for 2430 patients (97% of all those randomised). Duration of follow up varied considerably between the trials from one year to 11.4 years with a mean weighted follow up of 6.0 years. Two studies had data available after the end of official follow up: one study (Finland 1997) reported deaths after the trial ended in the final study publication (Myllylä 1997) and further data were available from one study (US 1995) in an individual patient data meta‐analysis (Olanow 1998a). One study had a significant difference in the duration of follow up in the two treatment arms, which was adjusted for in the main analysis (UK‐PDRG (RR) 1998).
1.1. Analysis.
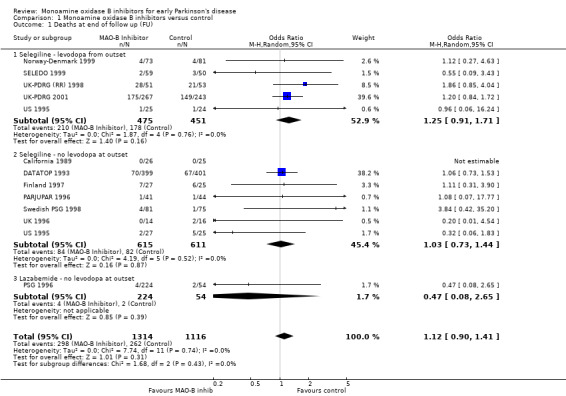
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 1 Deaths at end of follow up (FU).
Overall there was a non‐significant increase in deaths amongst patients treated with MAO‐B inhibitors compared with those given control (odds ratio (OR) 1.12; 95% confidence interval (CI) 0.90 to 1.41, P value 0.31) with no significant heterogeneity (I2 value 0%). The odds ratio remained non‐significant when the unadjusted data from the UK‐PDRG (RR) 1998 study were used (OR 1.07; 95% CI 0.86 to 1.35).
A funnel plot (Figure 1) did not show convincing evidence of publication bias and analysis of only those trials with more than 150 patients (that is removing the potentially biased smaller trials) did not alter the results (OR 1.13; 95% CI 0.88 to 1.44).
1.
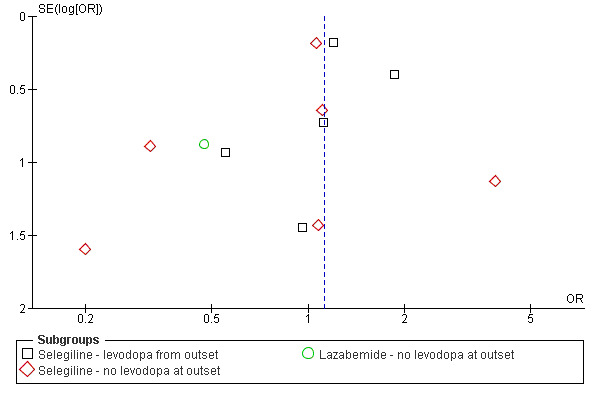
Funnel plot of deaths at end of follow up
The following pre‐specified subgroup analyses showed no significant differences (NB P value is the value from the test for heterogeneity between the subgroups):
High quality (well concealed randomisation, double‐blind trials) versus lower quality trials: OR 1.06 versus OR 1.17 respectively, P value 0.68.
Trials which used levodopa or a dopamine agonist from the beginning of the trial versus those that used MAO‐B inhibitors alone: OR 1.25 versus 1.00, P value 0.34.
Trials of selegiline versus lazabemide: OR 1.14 versus 0.47, P value 0.32.
Trials with follow up greater than five years versus those with shorter follow up: OR 1.20 versus 0.60, P value 0.08.
Trials with a strict definition of early disease versus those with a less strict definition: OR 1.02 versus 1.20, P value 0.49.
Severity of parkinsonism All the studies reported data on disease progression in some form but this lacked consistency. For example, a variety of different scales were used. Parkinsonian impairment was most commonly reported using the Unified Parkinson's Disease Rating Scale (UPDRS) motor score; other impairment scales used included the Webster Rating Scale and the Columbia University Rating Scale. Parkinsonian disability was mainly reported using the UPDRS activities of daily living (ADL) score, but the Schwab and England, and the Northwestern University Disability Scales were also used. We have used the UPDRS motor and UPDRS ADL scales in our analysis because these had the most data. Some trials reported mean final scores while others reported the mean change in scores from baseline. All trials measured patients while on treatment, and some studies also measured patients after a washout period. In addition, trials measured severity after different follow up durations. Assessment of impairment and disability data at follow up greater than one year was hampered by large numbers of withdrawals or losses to follow up. No study measured quality of life.
One study (PSG 1996) used different doses of lazabemide. There was no dose‐response relationship and so we used the arithmetic mean of the impairment and disability scores and their standard deviations across the four treatment doses.
Mean change in UPDRS motor score from baseline to one year on treatment (Analysis 1.2) For parkinsonian impairment we analysed change in UPDRS motor scores from baseline to one year. Data from five studies (1305 patients, 52% of all patients and 91% of patients randomised in those five trials) were available for this analysis. One other study also reported UPDRS motor scores (Swedish PSG 1998) but losses and withdrawals at one year (61%) were too high to allow inclusion in the meta‐analysis. Inclusion of this trial, however, did not alter the results.
1.2. Analysis.
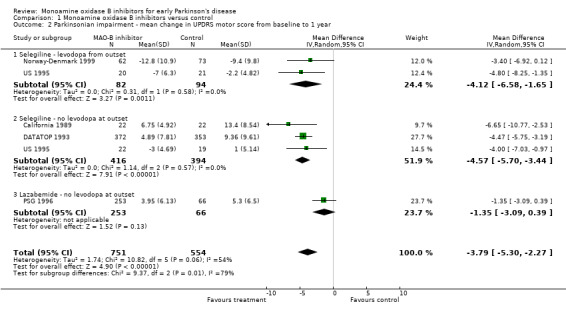
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 2 Parkinsonian impairment ‐ mean change in UPDRS motor score from baseline to 1 year.
All the studies reporting this outcome favoured treatment with MAO‐B inhibitors. The weighted mean difference (WMD) was ‐3.79 (95% CI ‐5.30 to ‐2.27) i.e. the mean decline in the motor impairment score at one year was nearly four points (out of a total scale of 108 points) less in participants treated with MAO‐B inhibitors than in those treated with control. Although this result is highly statistically significant (P value < 0.00001) its clinical significance is unclear and the results should be treated with caution because we cannot be certain these data were normally distributed.
There was significant heterogeneity (I2 54%) amongst the studies in this analysis. However, this heterogeneity was entirely attributable to the PSG 1996 study, which used lazabemide. When this study was excluded from the analysis, the I2 value was 0%. The results suggest that lazabemide (WMD ‐1.35; 95% CI ‐3.09 to 0.39) has a significantly weaker effect than selegiline (WMD ‐4.49; 95% CI ‐5.52 to ‐3.46, P value 0.002).
UPDRS motor scores at one year follow up (Analysis 1.3) In addition to analysing the change in impairment scores over one year, we also looked at the actual scores at one year follow up. Only two studies reported data that we could use in this analysis (217 patients, 9% of all patients). Both studies favoured treatment with MAO‐B inhibitors, the difference varied between ‐1.30 and ‐3.00. We did not carry out meta‐analysis, however, because the data were skewed. Three other trials recorded motor impairment with scales other than the UPDRS (Finland 1997; UK‐PDRG 2001; SELEDO 1999) and all showed small benefits in favour of MAO‐B inhibitors.
1.3. Analysis.
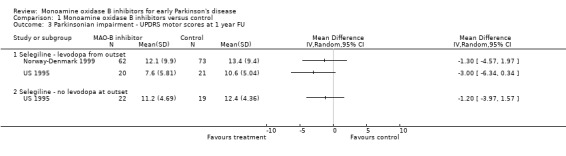
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 3 Parkinsonian impairment ‐ UPDRS motor scores at 1 year FU.
Mean change in UPDRS ADL score form baseline to one year (Analysis 1.4) Six studies reported UPDRS ADL scores but once again we omitted data from one study because of large losses to follow up (Swedish PSG 1998). Data from 1306 participants were available (52% of all patients, 91% of those randomised in the five studies). All the studies favoured treatment with MAO‐B inhibitors. The WMD was ‐1.49 (95% CI ‐2.49 to ‐0.49, P value 0.004), that is the scores were about one and a half points better (out of a total score of 52 points) after one year in patients treated with MAO‐B inhibitors. Again, we cannot discount the possibility that some of the disability data were skewed.
1.4. Analysis.
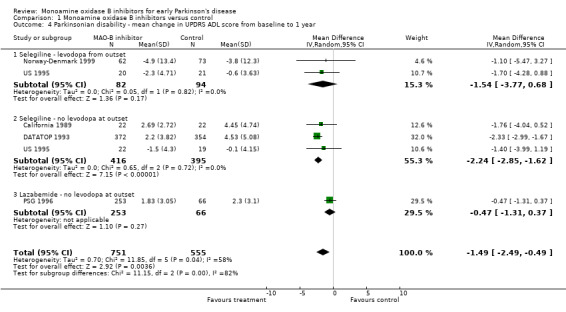
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 4 Parkinsonian disability ‐ mean change in UPDRS ADL score from baseline to 1 year.
As with the impairment scores, there was an apparent difference in effect between the studies using selegiline (WMD ‐2.19; 95% CI ‐2.78 to ‐1.60) and the study that used lazabemide (WMD ‐0.47; 95% CI ‐1.31 to 0.37). This accounted for the substantial heterogeneity (I2 58%) in this analysis. Subgroup analysis showed a significant difference between the two types of MAO‐B inhibitor used (P value 0.0006).
UPDRS ADL scores at one year follow up (Analysis 1.5) Only two studies reported this outcome (217 patients, 9% of all patients). Meta‐analysis could not be performed because some data were skewed but there was no good evidence of significant improvements with MAO‐B inhibitors.
1.5. Analysis.
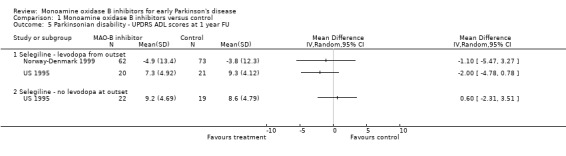
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 5 Parkinsonian disability ‐ UPDRS ADL scores at 1 year FU.
Mean change in UPDRS total score from baseline to end of washout (Analysis 1.6) Six studies incorporated a washout period into the study design to minimise the effects of drug therapy on symptoms compared with any effects on disease progression. Two studies (DATATOP 1993; Norway‐Denmark 1999) reported data on insufficient numbers of patients to allow inclusion in this analysis and one did not report the total UPDRS score (California 1989). Data from the other three studies (PSG 1996; Swedish PSG 1998; US 1995) were combined in meta‐analysis with a total of 429 patients (17% of all patients, 74% of those included in the three studies). The mean duration of follow up for this analysis was 1.1 years. The length of the washout was between two weeks and two months. Meta‐analysis yielded a weighted mean difference of ‐3.15 (95% CI ‐5.48 to ‐0.82, P value 0.008), that is the increase in severity score from baseline to the end of washout was about three points less in the treatment group. Heterogeneity was low (I2 20%) and there was no obvious trend between the duration of the washout and result.
1.6. Analysis.
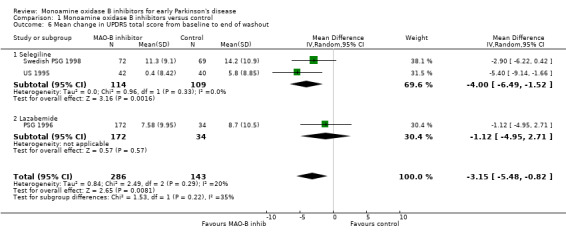
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 6 Mean change in UPDRS total score from baseline to end of washout.
Levodopa requirements Participants requiring levodopa (Analysis 1.7) All seven studies in which MAO‐B inhibitors were initially compared with control in the absence of levodopa reported the number of participants who subsequently required levodopa. Four studies (DATATOP 1993; PARJUPAR 1996; PSG 1996; UK 1996) assessed this outcome at a comparable follow up period of about one year (1138 patients, 75% of patients in studies without levodopa from the beginning, 92% of those randomised in the four studies). The combined OR was 0.48 (95% CI 0.37 to 0.62), significantly in favour of MAO‐B inhibitors (P value < 0.00001) and with no significant heterogeneity (I2 0%). The absolute rate of requiring levodopa at one year in the control groups of the four trials varied from about 15% (UK 1996) to 60% (DATATOP 1993), whereas baseline severity in terms of the UPDRS were similar. This implies either different rates of progression in the four trials or, perhaps more likely, different thresholds for starting levodopa. The number needed to treat with MAO‐B inhibitors to avoid one person requiring levodopa at one year, therefore, varied between 14 (95% CI 11 to 21) at a control event rate of 15% to 6 (95% CI 4 to 9) at a control rate of 60%.
1.7. Analysis.
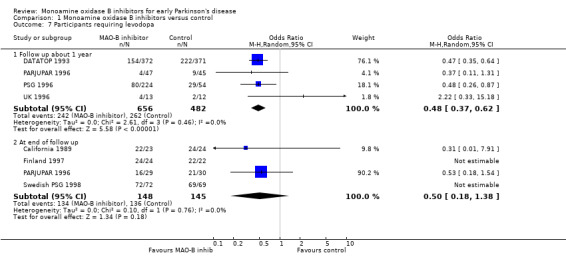
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 7 Participants requiring levodopa.
At the end of three years follow up, the reduction in the odds of needing levodopa was maintained in one study, which gave bromocriptine to both groups (PARJUPAR 1996), but another study that did not use a dopamine agonist (California 1989) reported that only one patient (in the selegiline arm) still did not require levodopa. In the other two studies without dopamine agonist use all the patients were receiving levodopa at the end of about four years of follow up (Finland 1997; Swedish PSG 1998).
Time until levodopa was required (Table 2) Six studies reported this outcome (1480 patients, 98% of patients in trials without levodopa from the outset). Because the data from these studies were skewed it was not possible to use formal meta‐analysis. However, the data from all these studies showed a delay in the median time to introduce levodopa with MAO‐B inhibitors of between 4.1 and 8.7 months.
2. Time to levodopa.
| Study ID | Measure | MAOB inhibitor | Control |
| California 1989 | Mean ± SD | 548.9 ± 286.2 days | 312.1 ± 208.6 days |
| DATATOP 1993 | Median | 719 days | 454 days |
| Finland 1997 | Median ± SE Mean ± SE | 545 ± 90 days 686.7 ± 73.7 days | 372 ± 28 days 487.0 ± 74.4 days |
| PARJUPAR 1996 | Median | 965 days | 804 days |
| PSG 1996 | Mean ± SE | 310.2 ± 13.1 days | 276 ± 15.7 days |
| Swedish PSG 1998 | Median ± quartile | 386.3 ± 276.8 days | 261.6 ± 243.3 days |
SD ‐ standard deviation; SE ‐ standard error
Mean levodopa dose (Table 3) Data from six trials were available for this outcome. We could not calculate the total number of patients included in this analysis because one study (UK‐PDRG 2001) did not report this figure. A meta‐analysis was not done because the data were skewed and there was substantial heterogeneity. The latter was partly attributable to varying durations of follow up, ranging from one to four years. All these studies showed higher levodopa doses in the control groups than in patients treated with MAO‐B inhibitors. The difference varied between 30 and 185 mg/day of levodopa and generally increased as the duration of follow up increased up to five years (Finland 1997; Norway‐Denmark 1999; SELEDO 1999; UK‐PDRG 2001).
3. Daily levodopa dose (mean or median).
| Study ID | Length of follow up | N (MAO‐B inhibitor) | Dose (MAO‐B inhib) | N (control) | Dose (control) | Difference |
| US 1995 (bromocriptine arms) | 1 year | 22 | Mean 85 mg (SD 198) | 19 | Mean 117 mg (SD 173) | 32 mg/day |
| US 1995 (levodopa arms) | 1 year | 20 | Mean 382 mg (SD 155) | 21 | Mean 426 mg (SD 110) | 44 mg/day |
| Finland 1997 | About 3 years | 23 | Mean 358 mg (SD 117) | 21 | Mean 543 mg (SD 150) | 185 mg/day |
| SELEDO 1999 | About 3.5 years | 26 | Mean 338 mg | 20 | Mean 521 mg | 183 mg/day |
| UK‐PDRG 2001 | 4 years | Unknown | Median 375 mg | Unknown | Median 625 mg | 250 mg/day |
| Norway‐Denmark 1999 | 5 years | 38 | Mean 424 mg (SD 113) | 43 | Mean 506 mg (SD 184) | 82 mg/day |
| Swedish PSG 1998 | 7 years | 19 | Mean 529 mg (SD 145) | 28 | Mean 631 (SD 186) | 102 mg/day |
SD ‐ standard deviation
Development of motor complications Motor fluctuations (Analysis 1.8) Data from six trials (1461 patients, 58% of all patients, 81% of those randomised in the six trials) were available with a mean weighted duration of follow up of 3.4 years. There were three issues that created difficulty in analysing these data. Firstly, raw data from the UK‐PDRG 2001 study did not show any difference in the number of patients developing motor fluctuations between the selegiline and non‐selegiline arms at the end of follow up (161 of 268 (60%) versus 146 of 240 (61%) respectively). However, the rate of developing motor fluctuations was lower in the selegiline arm (157.5 per 1,000 patient‐years versus 179.7 per 1,000 patient‐years), implying that the selegiline patients had longer follow up (mean 3.8 years versus 3.4 years). In the main analysis we adjusted for this difference although we also performed an analysis with the raw data. Secondly, owing to the changes in the study design over time, data on fluctuations from the DATATOP study did not include a substantial number of patients (36%); we felt justified in including DATATOP in the main analysis because the losses were similar in both arms of the study. However, we also performed an analysis excluding DATATOP. Thirdly, the definitions of motor fluctuations varied: two trials included "end of dose deterioration" (Finland 1997; Norway‐Denmark 1999), one included patients taking more than four doses of levodopa per day (Swedish PSG 1998), one "severe end of dose deterioration and random on/off fluctuations" (SELEDO 1999), one "on/off fluctuations" (UK‐PDRG 2001), and one "wearing off" (DATATOP 1993).
1.8. Analysis.
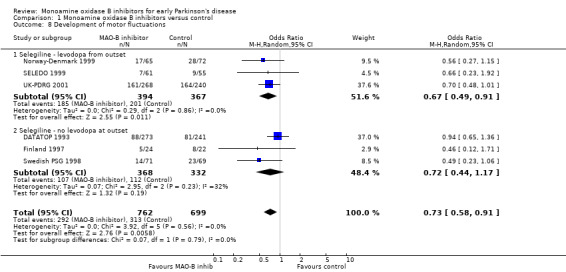
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 8 Development of motor fluctuations.
The overall effect was significantly in favour of MAO‐B inhibitors (OR 0.73; 95% CI 0.58 to 0.91, P value 0.006) with no evidence of heterogeneity (I2 0%). However, this result was dependent on the adjusted results of the UK‐PDRG study and if the unadjusted figures were used the overall result became non‐significant (OR 0.83; 95% CI 0.0.66 to 1.03). Excluding the DATATOP study, increased the observed benefit with selegiline (OR 0.63; 95% CI 0.47 to 0.83).
The rate of developing fluctuations varied in the control groups from 16% to 68%. This variation may have been due to differences in definitions since the mean duration of follow up was very similar. The number needed to treat with selegiline to prevent one person developing motor fluctuations over about 3.4 years varied from 13 (95% CI 8 to 43) to 25 (95% CI 17 to 73) across these baseline risks.
Results were not reported for 351 patients in these six studies. A modified worst‐case analysis, in which excluded subjects in the treatment group were given the highest event rate in any individual study (164/240, 68%) whilst excluded patients in the control group were given the lowest (7/61, 11%), made the results non‐significant (OR 0.97; 95% CI 0.55 to 1.72). A null study of over 1500 patients with an event rate of 45% would be required to make the motor fluctuations data non‐significant. As there was no heterogeneity, no subgroup analyses were performed.
Dyskinesias (Analysis 1.9) Data on the incidence of dyskinesias were reported in only five trials (1362 participants, 54% of all randomised patients, 80% of those randomised in the five trials). The mean weighted duration of follow up of 3.5 years. Adjustments were again made to the data from the UK‐PDRG study to account for the slightly shorter follow up in the control group. The result showed no difference between intervention and control (OR 0.96; 95% CI 0.76 to 1.22). As with the motor fluctuations data, re‐analysis using the raw data from the UK‐PDRG study did not alter the results (OR 0.98), nor did exclusion of the DATATOP trial, which again had missing data for this outcome (OR 0.94).
1.9. Analysis.
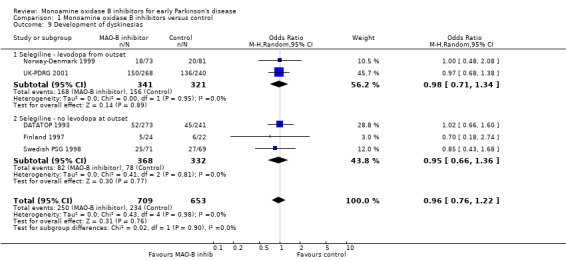
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 9 Development of dyskinesias.
Patients with adverse events (Analysis 1.10) Six trials (847 patients, 34% of all patients, 96% of those randomised in the six trials) reported the numbers of patients with any significant adverse events. The precise definitions probably varied between trials. Overall, there were more adverse events with MAO‐B inhibitors (65% vs 58%, OR 1.45; 95% CI 1.04 to 2.02), which just reached statistical significance (P value 0.03). The reporting of individual side‐effects (for example nausea, confusion, hallucinations, postural hypotension) varied significantly between trials, with many trials not reporting any information on specific side‐effects and others only reporting those that were significantly different between treatment groups. This introduced a reporting bias. Eight studies (1436 patients, 57% of all patients, 98% of those randomised in the eight trials) reported data on nausea (Analysis 1.11). Significantly more patients in the MAO‐B inhibitor group reported nausea (OR 1.76; 95% CI 1.10 to 2.82) and another study reported significantly higher withdrawals due to upper gastrointestinal side‐effects (UK‐PDRG 2001).
1.10. Analysis.
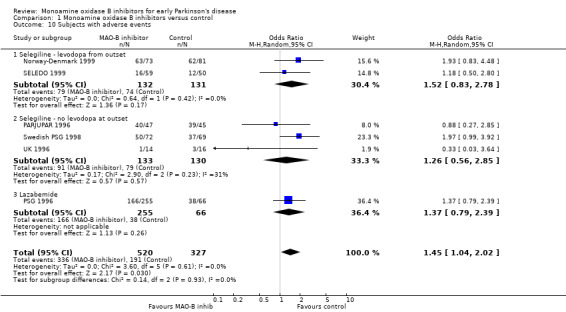
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 10 Subjects with adverse events.
1.11. Analysis.
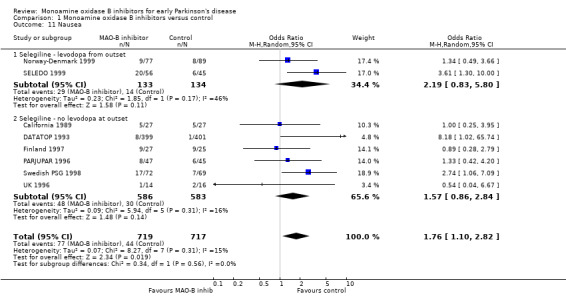
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 11 Nausea.
Concerns have been raised about potentially harmful cardiovascular side‐effects of MAO‐B inhibitors including postural hypotension and cardiac arrhythmias. However, the studies in this review that reported data on blood pressure (DATATOP 1993; Finland 1997; Swedish PSG 1998; UK 1996) did not find lower mean blood pressures in patients in the MAO‐B arms. DATATOP 1993 reported significantly more arrhythmias with selegiline than with control (eight versus one); one other study reported more ECG abnormalities with selegiline (Norway‐Denmark 1999); and one other study reported no difference in development of ECG abnormalities between lazabemide and control (PSG 1996). The arrhythmias that were reported were not considered life threatening.
Two studies reported significantly higher numbers of patients in the MAO‐B inhibitor groups with elevated serum aminotransferases (DATATOP 1993; PSG 1996). However, these enzyme abnormalities were not thought to pose serious health risks. There was insufficient information to evaluate neuropsychiatric side‐effects.
Withdrawals Eight trials (1479 patients, 59% of all randomised patients, 100% of those randomised in the eight trials) reported the numbers of withdrawals due to adverse events at the end of follow up (Analysis 1.12). There was a non‐significant trend for more withdrawals with MAO‐B inhibitors (13% vs 9%, OR 1.72; 95% CI 0.98 to 3.01, P value 0.06) with significant heterogeneity (I2 51%).
1.12. Analysis.
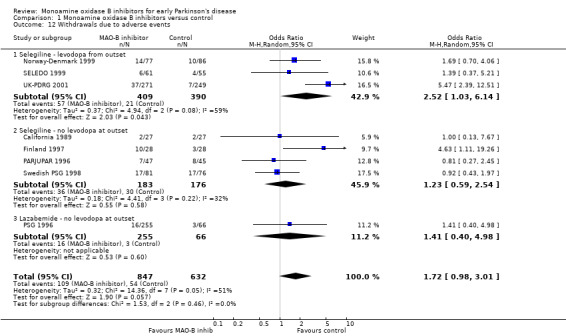
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 12 Withdrawals due to adverse events.
All 11 trials reported data on total numbers of withdrawals by end of follow up (Analysis 1.13). Data were reported from 2410 participants, all patients except the re‐randomised patients from the UK‐PDRG study. We have included losses to follow up in the total number of withdrawals (losses to follow up were not consistently reported) but have not included deaths. There was no significant difference in the numbers of withdrawals between the MAO‐B inhibitor and control arms (OR 0.95; 95% CI 0.78 to 1.17).
1.13. Analysis.
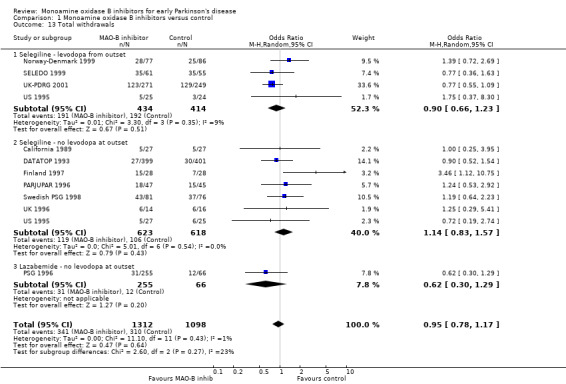
Comparison 1 Monoamine oxidase B inhibitors versus control, Outcome 13 Total withdrawals.
Discussion
Since most data in this review were from studies using selegiline our conclusions predominately relate to selegiline. Given that one of the aims of treatment of Parkinson's disease should be to reduce disability and mortality in the long term, it was disappointing that none of the trials assessed our primary outcome, which was chosen to capture both of these factors. We feel that future studies should incorporate this clinically important outcome into the study design, particularly for trials of possible neuroprotective agents that would be expected to delay disability.
Death There has been much debate about whether selegiline increases the risk of death after the finding that it was associated with excess mortality in the UK‐PDRG trial. This review and a similar review (Ives 2004) of all the available randomised evidence showed no significant increase in death with MAO‐B inhibitors in general or selegiline specifically. Only the UK‐PDRG trial showed an excess of deaths with selegiline, which we feel was probably a chance finding exacerbated by early stopping of the trial. However, publication of this trial resulted in a dramatic reduction in the prescription of selegiline (in the UK at least) (Clarke 2001), which highlights the dangers of changing practice on the basis of a single trial's results. Whilst the confidence interval does not exclude a small increase in death rates with MAO‐B inhibitors it does exclude any significant reduction in mortality as was suggested by the observational data from the 1980s (Birkmayer 1985).
Disease progression, impairment and disability Many claims and counter claims have been made with regard to the effect of MAO‐B inhibitors on the progression of Parkinson's disease. Some researchers have argued that MAO‐B inhibitors confer a neuroprotective effect and delay disease progression, whilst others have considered any beneficial effects of MAO‐B inhibitors to be merely symptomatic improvements due to their dopaminergic effects. It is difficult clinically to separate out a symptomatic effect from an effect on disease progression. It might be expected that slowed disease progression would be reflected in improved survival, which we have not shown to be the case. An alternative way is to assess outcome following the withdrawal of medication to try to remove any symptomatic effect. However, this requires precise information about the duration of a symptomatic effect, which is often lacking. Moreover, it becomes difficult to withdraw treatment for long periods of time in patients with Parkinson's disease who require treatment to control their symptoms. Another strategy to differentiate a symptomatic from a disease‐modifying effect would be to establish whether differences in impairment and disability on treatment or control medication diverge over time, as would be expected with a disease modifying effect, or whether they remain static, suggesting a symptomatic effect. Divergence was demonstrated in the seven year data from the Swedish trial but unfortunately this analysis is impossible to interpret due to large losses to follow‐up over time (Pålhagen 2006). Recent studies of rasagiline using a delayed‐start design, which aims to separate symptomatic and neuroprotective effects have shown conflicting evidence (TEMPO 2002; ADAGIO 2009). The validity of this study design in detecting neuroprotection is also debatable because it assumes that the symptomatic effect is constant over time, which may not be true.
This review showed that MAO‐B inhibitors did reduce impairment and disability in Parkinson's patients in the short term (one year) but the validity and clinical significance of these benefits is less clear because the changes were small, the data were limited both in terms of quantity (analyses only included about 50% of all randomised patients) and quality, and the washout periods were short. Analysis was hindered by the use of several different measures of impairment and disability and by trials reporting the data in various ways (for example absolute scores or change in scores from baseline), in various states (that is on or off medication) and at various time points. Few trials were analysed on an intention‐to‐treat basis so patients who dropped out were not included in these analyses. Finally, it was often unclear whether the data were normally distributed, which can invalidate the statistical methods used for meta‐analysis.
Whilst the analysis of those studies that included a washout phase to minimise any symptomatic effects did show a significant benefit in favour of MAO‐B inhibitors, we would caution against interpreting this as demonstrating a significant slowing of disease progression for several reasons. Firstly, the absolute difference was small (about three points out of a total UPDRS score of 176, which combines motor, ADL and mentation components). Secondly, it was based on limited data (only 582 patients). And thirdly, it remains unclear whether the washout periods (a mean of 1.3 months) were long enough to ensure complete elimination of the study drug. Some researchers have suggested that a washout of two months may be insufficient, citing a half‐life of 40 days for selegiline (Fowler 1996). If this is correct some if not all of the residual effect of MAO‐B inhibitors observed after washout could have been an ongoing symptomatic effect. Taken overall, the results from the mortality, impairment and disability outcomes do not provide any strong evidence for a delay in disease progression with MAO‐B inhibitors.
Levodopa requirements The reduction in the need for additional levodopa at one year is consistent with a symptomatic effect of MAO‐B inhibitors. However, as expected given the weak nature of this symptomatic effect, as the disease progressed over three to four years almost all patients eventually required levodopa. The exact duration of this levodopa sparing effect in any given patient may vary depending on several factors including how early in the disease MAO‐B inhibitors are started. If treatment is started early when the symptoms are very mild levodopa may not be required for many months whilst if started later, when symptoms are more severe, levodopa may be required much sooner. What is more interesting is that once patients started levodopa the dose required in those taking MAO‐B inhibitors remained significantly lower than for those who were not taking them and that this difference increased over time in some trials. Whether this implies a slowing in disease progression remains unclear as does the importance of minimising the levodopa dose in delaying long term complications (see below). Motor complications One of the complications of long‐term levodopa use is the development of motor fluctuations and dyskinesias. Some data have shown that lower doses of levodopa are associated with a lower risk of these complications (Poewe 1986) and previous guidelines have recommended minimising the dose of levodopa because of this (Olanow 1998; PDCWG 2001). However, it may be that using lower levodopa doses reflects less severe striatonigral neurodegeneration and it may be that it is the latter that is the most important determinant of motor complications.
We found that, as well as being associated with lower levodopa doses, selegiline was associated with a 25% reduction in the odds of motor fluctuations but no reduction in dyskinesias. There may be several reasons for this apparent discrepancy between these two types of motor complication. Firstly, the reduction in motor fluctuations may be spurious since it was based on a relatively small number of patients; was largely dependent on the result from the UK‐PDRG trial which had to be adjusted for differences in the duration of follow up; and was not robust to sensitivity analysis based on losses to follow up. However, a similar reduction in motor fluctuations was found in a study comparing selegiline with levodopa that was not eligible for this review (Italian PDSG 2001). Secondly, it may be that MAO‐B inhibitors do also reduce dyskinesias and that the present result for dyskinesias is a false negative one. For example, the lower confidence limit for dyskinesias is compatible with a 20% to 25% reduction in the odds of dyskinesias. Thirdly, it may be that the effect of MAO‐B inhibitors on motor fluctuations is purely a symptomatic one rather than due to any fundamental effect on the mechanisms that cause motor complications. The earliest and most common motor fluctuation is end‐of‐dose wearing off. MAO‐B inhibitors may reduce this by prolonging the dopamine half‐life in the synapse but may not have any effect on more random on/off oscillations and dyskinesias. There is increasing clinical and experimental evidence to suggest that the latter are triggered by the intermittent stimulation of the striatal dopamine receptors (Katzenschlager 2002; Olanow 1998) so that it may be that pulsatile exogenous dopamine, largely independent of the actual levodopa dose, is the main cause of these motor complications.
If the reduction in motor fluctuations with selegiline is real the next issue would be to determine whether it is clinically relevant. This is unclear from the data in this review as we were unable to determine the severity of the fluctuations.
Safety In general the side‐effects reported by patients receiving MAO‐B inhibitors were mild. However, analyses were limited by incomplete and non‐standardised reporting of specific side‐effects. There were more side‐effects in general with MAO‐B inhibitors and, in particular, more nausea. Although there may have been a greater incidence of cardiac arrhythmias and elevated hepatic enzymes these were rare and not thought to be life threatening. We were unable to assess whether there were more psychiatric complications with MAO‐B inhibitors and could not confirm the higher risk of postural hypotension that some have found (Churchyard 1999). Given that we have not shown clear evidence of an increase in deaths with selegiline we believe that MAO‐B inhibitors are safe in early Parkinson's disease.
Authors' conclusions
Implications for practice.
We did not find any convincing evidence that MAO‐B inhibitors significantly delay disease progression in early Parkinson's disease, although currently most of the data in this review relate to selegiline. Our data on parkinsonian impairment and disability scores were consistent with a small symptomatic effect but the clinical relevance of this was uncertain. Similarly, whilst there is good evidence that MAO‐B inhibitors have a levodopa sparing effect, whether this results in fewer long‐term, clinically relevant motor complications is unclear although there are promising data on motor fluctuations. The existing data do not exclude the possibility that MAO‐B inhibitors cause an increase in mortality but, given that only one trial has suggested this, we consider it very unlikely. Other side‐effects were generally mild and infrequent. Overall we do not feel the present evidence supports the routine use of selegiline or any other MAO‐B inhibitor in early Parkinson's disease although clinicians may wish to consider it in situations where they feel it is important to delay or limit levodopa exposure, for example in young patients.
Implications for research.
More research is required, particularly to determine:
whether selegiline or other MAO‐B inhibitors do increase death rates. Further analysis of the existing data may help clarify this (for example, survival analysis using hazard ratios) but this will require individual patient data from each trial;
whether the reduction in motor fluctuations is real, clinically relevant, and is also associated with a reduction in dyskinesias;
the relative benefits of the newer MAO‐B inhibitors (especially rasagiline) over selegiline.
A large ongoing trial of selegiline in early Parkinson's disease (PD MED, http://www.pdmed.bham.ac.uk/) will provide additional important data although it will not be eligible for this review since it compares initiating treatment with selegiline alone versus levodopa alone or dopamine agonists alone (that is it is not comparing selegiline with no selegiline). We believe that further trials in early Parkinson's disease comparing the combination of MAO‐B inhibitors with either levodopa or dopamine agonists versus monotherapy with levodopa or agonists alone are merited.
What's new
| Date | Event | Description |
|---|---|---|
| 8 November 2011 | New search has been performed | Searches updated; safinamide added as a new MAO‐B inhibitor (no relevant trial found); one new trial included (PARJUPAR, n=92); additional data added from latest publications for two trials (Swedish PSG 1998, UKPDRG 2001) and minor corrections to previous data; risk of bias table added. Conclusions unchanged. |
History
Protocol first published: Issue 3, 2004 Review first published: Issue 3, 2005
| Date | Event | Description |
|---|---|---|
| 8 February 2007 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
We thank Professor Cristina Sampaio for providing the unpublished Parjupar data for this updated review. If there are any relevant completed or ongoing trials that we have omitted from the review please contact Carl Counsell.
Appendices
Appendix 1. The Cochrane Central Register of Controlled Trials (CENTRAL) search strategy
#1. MeSH descriptor Parkinsonian Disorders explode all trees #2. parkinson* in Clinical Trials #3. (#1 OR #2) #4. MeSH descriptor Monoamine Oxidase Inhibitors, this term only #5. monoamine oxidase inhibitor* in Clinical Trials #6. MAO B inhibitor* in Clinical Trials #7. selegiline in Clinical Trials #8. deprenyl in Clinical Trials #9. deprenil in Clinical Trials #10. eldepryl in Clinical Trials #11. jumex in Clinical Trials #12. movergan in Clinical Trials #13. anipryl in Clinical Trials #14. antiparkin in Clinical Trials #15. atapryl in Clinical Trials #16. deprenaline in Clinical Trials #17. egibren in Clinical Trials #18. eldeprine in Clinical Trials #19. emsam in Clinical Trials #20. jumexal in Clinical Trials #21. parkryl in Clinical Trials #22. plurimen in Clinical Trials #23. seledat in Clinical Trials #24. zelapar in Clinical Trials #25. rasagiline in Clinical Trials #26. agilect in Clinical Trials #27. azilect in Clinical Trials #28. lazabemide in Clinical Trials #29. pakio in Clinical Trials #30. tempium in Clinical Trials #31. isocarboxazid in Clinical Trials #32. ladostigil in Clinical Trials #33. mofegiline in Clinical Trials #34. pargyline in Clinical Trials #35. phenelzine in Clinical Trials #36. safinamide in Clinical Trials #37. tranylcypromine in Clinical Trials #38.(#4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR #37)
Appendix 2. MEDLINE search strategy
#1. Parkinson$.tw. #2. exp Parkinsonian Disorders/ #3. 1 or 2 #4. randomised controlled trial.pt. #5. controlled clinical trial.pt. #6. randomised.ab. #7. placebo.ab. #8. drug therapy.fs. #9. randomly.ab. #10. trial.ab. #11. groups.ab. #12. 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 #13. Monoamine Oxidase Inhibitors/ #14. selegiline/ #15. selegiline.tw. #16. deprenyl.tw. #17. deprenil.tw. #18. eldepryl.tw. #19. jumex.tw. #20. movergan.tw. #21. anipryl.tw. #22. antiparkin.tw. #23. atapryl.tw. #24. deprenaline.tw. #25. egibren.tw. #26. eldeprine.tw. #27. emsam.tw. #28. jumexal.tw. #29. parkryl.tw. #30. plurimen.tw. #31. seledat.tw. #32. zelapar.tw. #33. rasagiline.tw. #34. agilect.tw. #35. azilect.tw. #36. lazabemide.tw. #37. pakio.tw. #38. tempium.tw. #39. isocarboxacid.tw. #40. ladostigil.tw. #41. mofegiline.tw. #42. pargyline.tw. #43. phenelzine.tw. #44. safinamide.tw. #45. tranylcypromine.tw. #46. or/13‐45 #47. 3 and 12 and 46 #48. exp animals/ not humans.sh. #49. 47 not 48 #50. 49
Appendix 3. EMBASE search strategy
#1. parkinson disease/ #2. parkinsonism/ #3. parkinson$.tw. #4. or/1‐3 #5. selegiline/ #6. selegiline.tw. #7. deprenyl.tw. #8. deprenil.tw. #9. eldepryl.tw. #10. jumex.tw. #11. movergan.tw. #12. anipryl.tw. #13. antiparkin.tw. #14. atapryl.tw. #15. deprenaline.tw. #16. egibren.tw. #17. eldeprine.tw. #18. emsam.tw. #19. jumexal.tw. #20. parkryl.tw. #21. plurimen.tw. #22. seledat.tw. #23. zelapar.tw. #24. rasagiline/ #25. rasagiline.tw. #26. agilect.tw. #27. azilect.tw. #28. lazabemide/ #29. lazabemide.tw. #30. pakio.tw. #31. tempium.tw. #32. monoamine oxidase B inhibitor/ #33. monoamine oxidase inhibitor/ or isocarboxazid/ or ladostigil/ or lazabemide/ or mofegiline/ or pargyline/ or phenelzine/ or rasagiline/ or safinamide/ or selegiline/ or tranylcypromine/ #34. or/5‐33 #35. clinical tral/ #36. multicenter study/ #37. phase 2 clinical trial/ #38. phase 3 clinical trial/ #39. phase 4 clinical trial/ #40. randomised controlled trial/ #41. controlled study/ #42. meta analysis/ #43. double blind procedure/ #44. single blind procedure/ #45. randomisation/ #46. major clinical study/ #47. placebo/ #48. drug comparison/ #49. clinical study/ #50. (clin$ adj25 trial$).tw. #51. ((singl$ or doubl$ or tripl$ or trebl$) adj25 (blind$ or mask$)).tw. #52. placebo$.tw. #53. random$.tw. #54. control$.tw. #55. or/35‐54 #56. human/ #57. nonhuman/ #58. 56 and 57 #59. 57 not 58 #60. 55 not 59 #61. 4 and 34 and 60 #62. 61
Appendix 4. Conference Proceedings Citation Index (CPCI)
#1 TS=(selegiline OR deprenyl OR deprenil OR eldepryl OR lazabemide OR rasagiline OR azilect) #2 TS=(TRIAL OR RANDOM* OR PLACEBO* OR CONTROL*) #3 #1 AND #2
Data and analyses
Comparison 1. Monoamine oxidase B inhibitors versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Deaths at end of follow up (FU) | 12 | 2430 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.90, 1.41] |
| 1.1 Selegiline ‐ levodopa from outset | 5 | 926 | Odds Ratio (M‐H, Random, 95% CI) | 1.25 [0.91, 1.71] |
| 1.2 Selegiline ‐ no levodopa at outset | 7 | 1226 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.73, 1.44] |
| 1.3 Lazabemide ‐ no levodopa at outset | 1 | 278 | Odds Ratio (M‐H, Random, 95% CI) | 0.47 [0.08, 2.65] |
| 2 Parkinsonian impairment ‐ mean change in UPDRS motor score from baseline to 1 year | 5 | 1305 | Mean Difference (IV, Random, 95% CI) | ‐3.79 [‐5.30, ‐2.27] |
| 2.1 Selegiline ‐ levodopa from outset | 2 | 176 | Mean Difference (IV, Random, 95% CI) | ‐4.12 [‐6.58, ‐1.65] |
| 2.2 Selegiline ‐ no levodopa at outset | 3 | 810 | Mean Difference (IV, Random, 95% CI) | ‐4.57 [‐5.70, ‐3.44] |
| 2.3 Lazabemide ‐ no levodopa at outset | 1 | 319 | Mean Difference (IV, Random, 95% CI) | ‐1.35 [‐3.09, 0.39] |
| 3 Parkinsonian impairment ‐ UPDRS motor scores at 1 year FU | 2 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3.1 Selegiline ‐ levodopa from outset | 2 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Selegiline ‐ no levodopa at outset | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Parkinsonian disability ‐ mean change in UPDRS ADL score from baseline to 1 year | 5 | 1306 | Mean Difference (IV, Random, 95% CI) | ‐1.49 [‐2.49, ‐0.49] |
| 4.1 Selegiline ‐ levodopa from outset | 2 | 176 | Mean Difference (IV, Random, 95% CI) | ‐1.54 [‐3.77, 0.68] |
| 4.2 Selegiline ‐ no levodopa at outset | 3 | 811 | Mean Difference (IV, Random, 95% CI) | ‐2.24 [‐2.85, ‐1.62] |
| 4.3 Lazabemide ‐ no levodopa at outset | 1 | 319 | Mean Difference (IV, Random, 95% CI) | ‐0.47 [‐1.31, 0.37] |
| 5 Parkinsonian disability ‐ UPDRS ADL scores at 1 year FU | 2 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 5.1 Selegiline ‐ levodopa from outset | 2 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Selegiline ‐ no levodopa at outset | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Mean change in UPDRS total score from baseline to end of washout | 3 | 429 | Mean Difference (IV, Random, 95% CI) | ‐3.15 [‐5.48, ‐0.82] |
| 6.1 Selegiline | 2 | 223 | Mean Difference (IV, Random, 95% CI) | ‐4.00 [‐6.49, ‐1.52] |
| 6.2 Lazabemide | 1 | 206 | Mean Difference (IV, Random, 95% CI) | ‐1.12 [‐4.95, 2.71] |
| 7 Participants requiring levodopa | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Follow up about 1 year | 4 | 1138 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.37, 0.62] |
| 7.2 At end of follow up | 4 | 293 | Odds Ratio (M‐H, Random, 95% CI) | 0.50 [0.18, 1.38] |
| 8 Development of motor fluctuations | 6 | 1461 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.58, 0.91] |
| 8.1 Selegiline ‐ levodopa from outset | 3 | 761 | Odds Ratio (M‐H, Random, 95% CI) | 0.67 [0.49, 0.91] |
| 8.2 Selegiline ‐ no levodopa at outset | 3 | 700 | Odds Ratio (M‐H, Random, 95% CI) | 0.72 [0.44, 1.17] |
| 9 Development of dyskinesias | 5 | 1362 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.76, 1.22] |
| 9.1 Selegiline ‐ levodopa from outset | 2 | 662 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.71, 1.34] |
| 9.2 Selegiline ‐ no levodopa at outset | 3 | 700 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.66, 1.36] |
| 10 Subjects with adverse events | 6 | 847 | Odds Ratio (M‐H, Random, 95% CI) | 1.45 [1.04, 2.02] |
| 10.1 Selegiline ‐ levodopa from outset | 2 | 263 | Odds Ratio (M‐H, Random, 95% CI) | 1.52 [0.83, 2.78] |
| 10.2 Selegiline ‐ no levodopa at outset | 3 | 263 | Odds Ratio (M‐H, Random, 95% CI) | 1.26 [0.56, 2.85] |
| 10.3 Lazabemide | 1 | 321 | Odds Ratio (M‐H, Random, 95% CI) | 1.37 [0.79, 2.39] |
| 11 Nausea | 8 | 1436 | Odds Ratio (M‐H, Random, 95% CI) | 1.76 [1.10, 2.82] |
| 11.1 Selegiline ‐ levodopa from outset | 2 | 267 | Odds Ratio (M‐H, Random, 95% CI) | 2.19 [0.83, 5.80] |
| 11.2 Selegiline ‐ no levodopa at outset | 6 | 1169 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [0.86, 2.84] |
| 12 Withdrawals due to adverse events | 8 | 1479 | Odds Ratio (M‐H, Random, 95% CI) | 1.72 [0.98, 3.01] |
| 12.1 Selegiline ‐ levodopa from outset | 3 | 799 | Odds Ratio (M‐H, Random, 95% CI) | 2.52 [1.03, 6.14] |
| 12.2 Selegiline ‐ no levodopa at outset | 4 | 359 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.59, 2.54] |
| 12.3 Lazabemide ‐ no levodopa at outset | 1 | 321 | Odds Ratio (M‐H, Random, 95% CI) | 1.41 [0.40, 4.98] |
| 13 Total withdrawals | 11 | 2410 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.78, 1.17] |
| 13.1 Selegiline ‐ levodopa from outset | 4 | 848 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.66, 1.23] |
| 13.2 Selegiline ‐ no levodopa at outset | 7 | 1241 | Odds Ratio (M‐H, Random, 95% CI) | 1.14 [0.83, 1.57] |
| 13.3 Lazabemide ‐ no levodopa at outset | 1 | 321 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.30, 1.29] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
California 1989.
| Methods | G: "biased coin" C: no details given Patients/doctors/assessors blind Not intention‐to‐treat | |
| Participants | California, USA 54 randomised 27 to selegiline; 27 to placebo Incl: untreated idiopathic PD or treatment < 1 yr, H&Y I/II, duration < 5 yrs Excl: age <30 or >80; major medical or psychiatric disorders, dementia Baseline mean UPDRS (motor): selegiline 22; placebo 21 | |
| Interventions | Treatment: selegiline 5 mg twice a day Control: Placebo Duration of treatment: up to 3 years (mean 312 days in selegiline group; 549 days in placebo group) | |
| Outcomes | Deaths UPDRS Time to levodopa & levodopa requirement Withdrawals due to side‐effects Total withdrawals | |
| Notes | Mean FU: selegiline 0.9 yrs placebo 1.5 yrs 1 month washout | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “patients were randomised... using a biased coin randomisation method” |
| Allocation concealment (selection bias) | Unclear risk | Comment: No information provided. |
| Blinding (performance bias and detection bias) Doctors, All outcomes | Low risk | Quote: “The patients, and all clinic personnel who came in contact with them, were blinded as to treatment status.” Quote: “Deprenyl and identical (except for the deprenyl) placebo tablets were provided” |
| Blinding (performance bias and detection bias) Patients, All outcomes | Low risk | |
| Blinding (performance bias and detection bias) Outcome assessors, All outcomes | Low risk | |
| Incomplete outcome data (attrition bias) Mortality | Low risk | Rx: 27 randomised, 26 analysed (1 required early levodopa) C: 27 randomised, 25 analysed (2 required early levodopa) |
| Incomplete outcome data (attrition bias) Parkinsonian impairment & disability | Low risk | Rx: 27 randomised, 22 analysed (1 early treatment, 2 lost to FU, 2 side‐effect withdrawals) C: 27 randomised, 22 analysed (2 early treatment, 2 side‐effect withdrawals, 1 incidental surgery) |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Low risk | Rx: 27 randomised, 23 analysed (2 lost to FU, 2 side‐effects withdrawals) C: 27 randomised, 24 analysed (2 side‐effects withdrawals, 1 incidental surgery) |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | Low risk | Outcome not assessed |
| Selective reporting (reporting bias) | Unclear risk | Comment: No protocol available |
| Other bias | High risk | Different mean durations of FU (longer for selegiline arm). |
DATATOP 1993.
| Methods | G: computer C: central allocation 2x2 factorial design Patients/doctors/assessors blind Intention‐to‐treat for deaths only | |
| Participants | USA 800 randomised 399 to selegiline; 401 to placebo Incl: untreated idiopathic PD not requiring symptomatic treatment at present, H&Y I / II, duration < 5 yrs Excl: age <30 or > 79, dementia, severe tremor, certain drugs, unstable medical or psychiatric problems, previous neurosurgery Baseline mean UPDRS: selegiline 25; placebo 25 | |
| Interventions | Treatment: selegiline 10 mg daily Control: placebo Also tocopherol | |
| Outcomes | Deaths UPDRS Time to levodopa & levodopa requirement Motor fluctuations Dyskinesias Total withdrawals | |
| Notes | Mean FU (for mortality): 8.2 yrs 2 month washout | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “computer generated randomisation.” |
| Allocation concealment (selection bias) | Low risk | Quote “central allocation.” |
| Blinding (performance bias and detection bias) Doctors, All outcomes | Low risk | Quote: “double‐blind” Quote: “identical‐appearing placebo capsules” |
| Blinding (performance bias and detection bias) Patients, All outcomes | Low risk | |
| Blinding (performance bias and detection bias) Outcome assessors, All outcomes | Low risk | |
| Incomplete outcome data (attrition bias) Mortality | Low risk | Rx: 399 randomised, 399 analysed C: 401 randomised, 401 analysed |
| Incomplete outcome data (attrition bias) Parkinsonian impairment & disability | Unclear risk | Rx: 399 randomised, 372 analysed (27 withdrawals [reasons unclear]) C: 401 randomised, 353 analysed (30 withdrawals [reasons unclear], 18 not followed for sufficiently long to be included) |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Low risk | Rx: 399 randomised, 372 analysed (27 withdrawals [reasons unclear]) C: 401 randomised, 371 analysed (30 withdrawals [reasons unclear]) |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | High risk | Rx: 399 randomised, 273 analysed (126 withdrawals [reasons unclear]) C: 401 randomised, 241 analysed (160 withdrawals [reasons unclear]) |
| Selective reporting (reporting bias) | High risk | Study protocol is available. Primary outcome of time to levodopa is reported. Several secondary outcomes not reported (eg some functional and cognitive assessments). |
| Other bias | High risk | Initial phase of trial stopped early after interim analysis at about 12 months showed benefit in delaying time to levodopa with selegiline: benefits for time to levodopa and numbers requiring levodopa may therefore be overestimated. All remaining patients then put on open‐label selegiline after mean of 23 months FU. After another two years, there was a further randomisation of those on levodopa & open‐label selegiline to either continue or stop the selegiline. Hence, significant cross contamination of treatment and control groups affecting results for deaths and motor complications. |
Finland 1997.
| Methods | G: computer C: sealed envelopes Patients/doctors/assessors blind Not intention‐to‐treat | |
| Participants | Finland 56 randomised 28 to selegiline; 28 to placebo Incl: untreated idiopathic PD, H&Y I ‐ III Excl: other neurological diseases, active psychosis, severe medical diseases Baseline mean WRS: selegiline 8; placebo 8 | |
| Interventions | Treatment: selegiline 10 mg daily Control: placebo Levodopa combination therapy in phase II of study | |
| Outcomes | Deaths
Time to levodopa & levodopa requirement
Motor fluctuations Dyskinesia Withdrawals due to side‐effects Total withdrawals |
|
| Notes | Mean FU: selegiline 6.0 yrs ; placebo 5.7 yrs No washout No significant differences in blood pressure found | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Patients were grouped randomly (by a computer)” |
| Allocation concealment (selection bias) | Unclear risk | Comment: No information provided. |
| Blinding (performance bias and detection bias) Doctors, All outcomes | Low risk | Quote: “double‐blind” Quote: “Individual patient data and randomisation codes have not been shown to the clinical investigators or staff” |
| Blinding (performance bias and detection bias) Patients, All outcomes | Low risk | Quote: “matching placebo controlled” |
| Blinding (performance bias and detection bias) Outcome assessors, All outcomes | Low risk | |
| Incomplete outcome data (attrition bias) Mortality | Low risk | Rx: 28 randomised, 27 analysed (one excluded because drug induced park) C: 28 randomised, 25 analysed (three excluded, one with post traumatic park, two reasons unclear) |
| Incomplete outcome data (attrition bias) Parkinsonian impairment & disability | Low risk | Outcome not assessed |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Low risk | Rx: 28 randomised, 24 analysed (one excluded because drug induced park, three withdrew because of co‐morbidities) C: 28 randomised, 22 analysed (three excluded [one with post traumatic park, two reasons unclear], two withdrew with co‐morbidities, one withdrew for personal reasons) |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | Low risk | Rx: 28 randomised, 24 analysed (one excluded because drug induced park, three withdrew because of co‐morbidities) C: 28 randomised, 22 analysed (three excluded [one with post traumatic park, two reasons unclear], two withdrew with co‐morbidities, one withdrew for personal reasons) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Different mean durations of FU: slightly longer in selegiline arm (6.0 yrs vs 5.7 yrs). Three of the five authors affiliated with Orion Pharma. |
Norway‐Denmark 1999.
| Methods | G: unknown C: central allocation Patients/doctors/assessors blind Not intention‐to‐treat | |
| Participants | Norway and Denmark 163 randomised 77 to selegiline; 86 to placebo Incl: untreated idiopathic PD or levodopa < 6 months, H&Y I ‐ III Excl: age <35 or >75, dementia, psychosis, unstable diseases Baseline mean UPDRS: selegiline 37; placebo 35 | |
| Interventions | Treatment: selegiline 10 mg daily Control: placebo Both groups also received levodopa Duration of treatment: up to 5 years | |
| Outcomes | Deaths
UPDRS
Mean levodopa dose
Motor fluctuations Dyskinesia Withdrawals due to side‐effects Total withdrawals |
|
| Notes | Mean FU: selegiline 2.9 yrs; placebo 3.1 yrs 1 month washout | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomisation was done centrally by the study monitor in each country" Comment: Probably random |
| Allocation concealment (selection bias) | Low risk | Quote: "randomisation was done centrally by the study monitor in each country" Comment: Central allocation. |
| Blinding (performance bias and detection bias) Doctors, All outcomes | Unclear risk | Quote: “Double‐blind” Quote: "Sealed envelopes containing the treatment code were added to the study material. The code was broken by the data analyst after the analyses were done.” Unclear if envelopes were opaque and placebo identical. |
| Incomplete outcome data (attrition bias) Mortality | Low risk | Rx: 77 randomised, 73 analysed C: 86 randomised, 81 analysed 9 excluded (three protocol violations, six not deemed to have Parkinson’s disease after one year FU). Data not given by randomised group. |
| Incomplete outcome data (attrition bias) Parkinsonian impairment & disability | Unclear risk | Rx: 77 randomised, 62 analysed (reason for withdrawals unclear) C: 86 randomised, 73 analysed (reasons for withdrawlas unclear) |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Low risk | Outcome not assessed |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | High risk | Rx: 77 randomised, 65 analysed (reasons for withdrawal not given) C; 86 randomised, 72 analysed (reasons for withdrawal not given) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Different mean durations of FU: slightly longer in control arm (2.9 vs 3.1 yrs). Quote: “supported by Orion Pharma” |
PARJUPAR 1996.
| Methods | G: Random number table C: central allocation of numbered medication packages in each centre Patients/doctors/assessors blind Not intention‐to‐treat | |
| Participants | Portugal 92 randomised patients included (exact number randomised unclear) 47 to selegiline and bromocriptine; 45 to bromocriptine Incl: untreated idiopathic PD, H&Y I ‐ II, age 30‐75 yrs Excl: severe tremor, depression Baseline mean UPDRS: seleg & bromo 31, bromo 31 | |
| Interventions | Treatment: selegiline 10 mg daily plus bromocriptine up to 30 mg daily Control: Bromocriptine up to 30 mg daily Duration of treatment: mean 1.7 yrs | |
| Outcomes | Deaths Time to levodopa & levodopa requirement Withdrawals due to side‐effects Total withdrawals | |
| Notes | Mean FU: 1.7 years (max 3 yrs) No washout | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "a table of random numbers, blocked in units of six" |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomisation was centralised. The centres received numbered medication packages". |
| Blinding (performance bias and detection bias) Doctors, All outcomes | Unclear risk | Quote: “Placebo were used in a double‐dummy way”. Unclear if identical placebos. |
| Incomplete outcome data (attrition bias) Mortality | Unclear risk | Unclear how many patients randomised in each group. 165 patients randomised in total to three groups (one group not eligible for this review) but 20 not analysed because had not reached time for first assessment and six excluded post‐randomisation because failed inclusion/exclusion criteria. No details given for which groups these patients were in. Rx: at least 47 randomised, 41 analysed: six lost to FU C: at least 45 randomised, 44 analysed: 1 lost to FU |
| Incomplete outcome data (attrition bias) Parkinsonian impairment & disability | Low risk | Not assessed |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Unclear risk | Rx: at least 47 randomised, 29 analysed: six lost to FU, 12 withdrawals (10 adverse events, 2 protocol violations) C: at least 45 randomised, 30 analysed: one lost to FU, 14 withdrawals (10 adverse events, 4 protocol violations) One year requirements estimated from Kaplan Meier curves |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | Low risk | Outcome not assessed |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Low risk | |
PSG 1996.
| Methods | G: unknown C: unknown Patients/doctors/assessors blind Not intention‐to‐treat | |
| Participants | USA 321 randomised 60 to 25 mg group, 67 to 50 mg group, 64 to 100 mg group, 64 to 200 mg group, 66 to placebo Incl: untreated idiopathic PD, H&Y I ‐ II, <7 yrs duration Excl: severe tremor, unstable medical or psychiatric problems, dementia Baseline mean UPDRS: 25 mg group 23; 50 mg group 23; 100 mg group 21; 200 mg group 21; placebo 20 | |
| Interventions | Treatment: lazabemide 12.5 / 25 / 50 / 100mg twice a day Control: placebo Duration of treatment: 52‐54 weeks | |
| Outcomes | Deaths UPDRS Levodopa requirement Total withdrawals | |
| Notes | FU: 56 weeks 2‐4 week washout | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding (performance bias and detection bias) Doctors, All outcomes | Low risk | Quote: “double‐blind” Quote: “All patients, investigators and co‐ordinating staff were kept unaware of treatment assignments.” |
| Blinding (performance bias and detection bias) Patients, All outcomes | Low risk | Quote: “matching tablets of placebo” |
| Blinding (performance bias and detection bias) Outcome assessors, All outcomes | Low risk | |
| Incomplete outcome data (attrition bias) Mortality | Unclear risk | Rx: 255 randomised, 224 (88%) analysed (16 withdrew because of side‐effects, 15 reasons for withdrawal not stated) C: 66 randomised, 54 (82%) analysed (three withdrew because of side‐effects, nine reasons for withdrawal not stated) |
| Incomplete outcome data (attrition bias) Parkinsonian impairment & disability | Unclear risk | Rx: 255 randomised, 253 analysed using last outcome carried forward analysis C: 66 randomised, 66 analysed using last outcome carried forward analysis |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Unclear risk | Rx: 255 randomised, 224 (88%) analysed (16 withdrew because of side‐effects, 15 reasons for withdrawal not stated) C: 66 randomised, 54 (82%) analysed (three withdrew because of side‐effects, nine reasons for withdrawal not stated) |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | Unclear risk | Outcome not assessed |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Comment: Hoffmann‐ La Roche contributed to the authorship. |
SELEDO 1999.
| Methods | G: unknown C: unknown Patients/doctors/assessors blind Not intention‐to‐treat | |
| Participants | Germany 116 randomised 61 to selegiline; 55 to placebo Incl: untreated idiopathic PD or levodopa < 1 yr, H&Y I ‐ III Excl: age >75, dementia, serious concomitant diseases, certain drugs Baseline mean CURS: selegiline 23; placebo 26 | |
| Interventions | Treatment: selegiline 5 mg twice a day Control: placebo Both groups also received levodopa Duration of treatment: up to 5 years | |
| Outcomes | Deaths Withdrawals due to side‐effects Total withdrawals | |
| Notes | Mean FU: selegiline 3.9 yrs ; placebo 3.6 yrs No washout | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details given. Quote: "randomized (stratified by trial centre, block size four)". |
| Allocation concealment (selection bias) | Unclear risk | No details given. |
| Blinding (performance bias and detection bias) Doctors, All outcomes | Low risk | Quote: “double‐blind” Quote: “placebo tablets of identical appearance.” |
| Blinding (performance bias and detection bias) Patients, All outcomes | Low risk | |
| Blinding (performance bias and detection bias) Outcome assessors, All outcomes | Low risk | |
| Incomplete outcome data (attrition bias) Mortality | Low risk | 116 randomised Rx: 61 randomised, 59 analysed C: 55 randomised, 50 analysed 7 excluded (2 consent withdrawals, 2 misdiagnoses, 1 inclusion violation, 2 loss to FU) but data not given by group. |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Low risk | Outcome not assessed |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | Low risk | Outcome not assessed: time to motor fluctuations was reported but not numbers of patients. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Low risk | Quote: “Costs of the study were covered by ASTA Germany” |
Swedish PSG 1998.
| Methods | G: unknown C: sequentially numbered identical packs Patients/doctors/assessors blind Not intention‐to‐treat | |
| Participants | Sweden 157 randomised 81 to selegiline; 76 to placebo Incl: untreated idiopathic PD, H&Y I‐III Excl: unstable medical or psychiatric diseases; age > 75 Baseline mean UPDRS selegiline 24; placebo 21 | |
| Interventions | Phase I: Treatment: selegiline 10 mg daily Control: placebo Phase II: Levodopa added to all maintaining original blinded allocation after median of 12.7 months in selegiline arm and 8.6 months placebo arm) | |
| Outcomes | Deaths UPDRS Time to levodopa requirement Motor fluctuations Dyskinesias Withdrawals due to side‐effects Total withdrawals | |
| Notes | FU: max 7 yrs, mean unknown 2 month washout period before starting levodopa | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote from paper: "randomized" Quote from correspondence with principal investigator: "Each study site had their own randomisation lists" |
| Allocation concealment (selection bias) | Unclear risk | Quote from correspondence with principal investigator: "Investigators were instructed to give a subject number to subjects in the order they entered the study. The subject numbers were the same as the medication number. Both packages and tablets for selegiline and placebo were identical." |
| Blinding (performance bias and detection bias) Doctors, All outcomes | Low risk | Quote from paper: “placebo‐controlled, double‐blind”, “investigators and patients unaware of treatment assignments” Quote from correspondence with principal investigator: "Both packages and tablets for selegiline and placebo were identical." |
| Blinding (performance bias and detection bias) Patients, All outcomes | Low risk | |
| Blinding (performance bias and detection bias) Outcome assessors, All outcomes | Low risk | |
| Incomplete outcome data (attrition bias) Mortality | Unclear risk | Rx: 81 randomised, 81 analysed C: 76 randomised, 75 analysed (one lost to FU) However, only deaths occurring on study treatment or within 2 weeks of withdrawing from treatment included. |
| Incomplete outcome data (attrition bias) Parkinsonian impairment & disability | High risk | Rx: 81 randomised, 37 (46%) analysed at one year, 19 at six years C: 76 randomised, 24 (32%) analysed at one year, 28 at six years Data not included in meta‐analysis because of amount of missing data. |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Unclear risk | Rx: 81 randomised, 72 analysed (4 withdrew because of side‐effects, 5 other reasons) C: 76 randomised, 69 analysed (1 withdrew because of side‐effects, 1 lost to FU, 5 other reasons) |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | Unclear risk | Rx: 81 randomised, 71 analysed (5 withdrew because of side‐effects, 5 other reasons) C: 76 randomised, 69 analysed (1 withdrew because of side‐effects, 1 lost to FU, 5 other reasons) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Different mean durations of FU: about 4 months longer in selegiline arm. Baseline severity of PD slightly worse in selegiline arm. Quote: “sponsored by Orion Pharma” |
UK 1996.
| Methods | G: unknown C: unknown Patients/doctors/assessors blind Not intention‐to‐treat | |
| Participants | UK 30 randomised 14 to selegiline; 16 to placebo Incl: untreated idiopathic PD, H&Y I ‐ III Excl: age < 65, mental or physical disorders affecting performance or evaluation, medication with effect on CNS Baseline median H&Y: selegiline 2; placebo 2 | |
| Interventions | Treatment: selegiline 5 mg twice a day Control: placebo Duration of treatment and FU: 54 weeks | |
| Outcomes | Deaths Levodopa requirements Total withdrawals | |
| Notes | FU: 54 weeks No washout | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details given. |
| Allocation concealment (selection bias) | Unclear risk | No details given. |
| Blinding (performance bias and detection bias) Doctors, All outcomes | Unclear risk | Quote: "double‐blind" Quote: "matched placebo tablets" |
| Blinding (performance bias and detection bias) Patients, All outcomes | Low risk | |
| Blinding (performance bias and detection bias) Outcome assessors, All outcomes | Low risk | |
| Incomplete outcome data (attrition bias) Mortality | Low risk | Rx: 14 randomised, 14 analysed C: 16 randomised, 16 analysed |
| Incomplete outcome data (attrition bias) Parkinsonian impairment & disability | Unclear risk | Outcome not assessed |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Unclear risk | Rx: 14 randomised, 13 analysed (1 withdrawal due to side‐effects or lack of benefit) C: 16 randomised, 12 analysed (4 withdrawals due to side‐effects or lack of benefit). |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | Unclear risk | Outcome not assessed |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Quote: "we wish to thank Dr Esa Heinonen, Orion‐Farmos Pharmaceuticals, and Mr Derek Woodcock, Britannia Pharmaceuticals, for their help and advice." |
UK‐PDRG (RR) 1998.
| Methods | G: random number tables C: central allocation Patients/doctors/assessors all unblinded Intention‐to‐treat | |
| Participants | UK 104 patients from arm 3 (bromocriptine) re‐randomised 51 to selegiline; 53 to control Incl and excl criteria as for UK‐PDRG 2001 | |
| Interventions | Treatment: selegiline 5 mg twice a day Control: no selegiline Both groups received levodopa | |
| Outcomes | Deaths | |
| Notes | Patients who initially could not tolerate bromocriptine were subsequently re‐randomised to selegiline or no selegiline Mean duration of FU: 6.8 years No washout | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "random number tables" |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization was carried out by an independent coordinator" Quote: "the investigators telephoned the coordinator for the treatment code, which was subsequently confirmed in writing." |
| Blinding (performance bias and detection bias) Doctors, All outcomes | High risk | Quote: “An open, randomised trial” Comment: Not blinded. Unlikely to bias mortality data but may influence other more subjective outcomes. |
| Blinding (performance bias and detection bias) Patients, All outcomes | High risk | |
| Blinding (performance bias and detection bias) Outcome assessors, All outcomes | High risk | |
| Incomplete outcome data (attrition bias) Mortality | Low risk | Rx: 51 re‐randomised, 51 analysed C: 53 re‐randomised, 53 analysed No reasons for loss documented. |
| Incomplete outcome data (attrition bias) Parkinsonian impairment & disability | Unclear risk | Outcome not assessed |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Unclear risk | Outcome not assessed |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | Unclear risk | Outcome not assessed |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Quote: "sponsorship from Britannia Pharmaceuticals and Sandoz Products" |
UK‐PDRG 2001.
| Methods | G: random number tables C: central allocation Patients/doctors/assessors all unblinded Intention‐to‐treat | |
| Participants | UK
520 randomised
271 to selegiline; 249 to control Incl: untreated idiopathic PD with incapacity requiring dopaminergic treatment Excl: dementia, previously failed to respond to dopaminergic drugs Baseline mean WRS: selegiline 11; control 11 |
|
| Interventions | Treatment: selegiline 5 mg twice a day Control: no selegiline Both groups received levodopa Mean mortality FU: 9.2 years |
|
| Outcomes | Deaths Mean levodopa dose Motor fluctuations Dyskinesias Withdrawals due to side‐effects Total withdrawals | |
| Notes | Mean FU: 9.2 yrs (11.4 yrs for mortality) No washout | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "random number tables" |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization was carried out by an independent coordinator" Quote: "the investigators telephoned the coordinator for the treatment code, which was subsequently confirmed in writing." |
| Blinding (performance bias and detection bias) Doctors, All outcomes | High risk | Quote: “An open, randomised trial” Comment: Not blinded. Unlikely to bias mortality data but may influence other more subjective outcomes. |
| Blinding (performance bias and detection bias) Patients, All outcomes | High risk | |
| Blinding (performance bias and detection bias) Outcome assessors, All outcomes | High risk | |
| Incomplete outcome data (attrition bias) Mortality | Low risk | Rx: 271 randomised, 267 analysed (four lost to mortality FU) C: 249 randomised, 243 analysed (six lost to mortality FU) |
| Incomplete outcome data (attrition bias) Parkinsonian impairment & disability | Low risk | Outcome not included in this review (assessed using Webster rating scale not UPDRS) |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Low risk | Outcome not assessed |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | Low risk | Rx: 271 randomised, 268 analysed (as rate per 1,000 person‐yrs FU). C: 249 randomised, 240 analysed (as rate per 1,000 person‐yrs FU) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Different mean durations of FU: slightly longer in selegiline arm (3.8 vs 3.4 yrs). Quote: "sponsorship from Britannia Pharmaceuticals and Sandoz Products" |
US 1995.
| Methods | G: computer C: unknown 2x2 factorial design Patients/doctors/assessors blind Intention‐to‐treat for mortality only | |
| Participants | US 101 randomised 52 to selegiline; 49 to placebo Incl: untreated idiopathic PD, H&Y stage I ‐ III, Excl: previous neurosurgery, clinically significant medical or laboratory abnormalities Baseline mean UPDRS: selegiline 25; placebo 21 | |
| Interventions | Treatment: selegiline 10mg daily Control: placebo Half also received levodopa (Sinemet) and half received bromocriptine (doses adjusted as clinically appropriate) Duration of treatment: 12 months | |
| Outcomes | Deaths UPDRS Mean levodopa dose Total withdrawals | |
| Notes | FU: 14 months 2 month washout | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "computer‐generated randomisation schedule" |
| Allocation concealment (selection bias) | Unclear risk | No details given. |
| Blinding (performance bias and detection bias) Doctors, All outcomes | Low risk | Quote: "all evaluations were performed by the same observer who was blinded" Quote: "Adjustment of treatment drug was carried out by a neurologist‐therapist who was unaware of the identity of the study drug" |
| Blinding (performance bias and detection bias) Patients, All outcomes | Low risk | Quote: "Identical appearing placebo" |
| Blinding (performance bias and detection bias) Outcome assessors, All outcomes | Low risk | |
| Incomplete outcome data (attrition bias) Mortality | Low risk | Rx: 52 randomised, 52 analysed C: 49 randomised, 49 analysed |
| Incomplete outcome data (attrition bias) Parkinsonian impairment & disability | Unclear risk | Rx: 52 randomised, 42 analysed (10 withdrew or lost to FU) C: 49 randomised, 40 analysed (9 withdrew or lost to FU) Reasons for withdrawal not given by treatment group: 2 side‐effects, 3 lack of efficacy, 1 death, 3 unrelated medical problems, 1 refused washout, 5 loss to FU, 4 unknown) . Quote: "there were no significant differences in the reasons for dropout among the treatment groups" |
| Incomplete outcome data (attrition bias) Participants requiring levodopa | Unclear risk | Outcome not assessed |
| Incomplete outcome data (attrition bias) Motor fluctuations & dyskinesias | Unclear risk | Outcome not assessed |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Baseline severity of PD slightly worse in selegiline arm. Comment: Supported by grants from Sandoz and Somerset Pharmaceuticals. |
G = generation of random sequence; C = concealment of randomisation; Incl = inclusion criteria; Excl = exclusion criteria; FU = follow up; PD = Parkinson's disease; yr = year; H&Y = Hoehn and Yahr stage; CURS = Columbia University Rating Scale; NUDS = Northwestern University Disability Scale; S&E = Schwab and England scale; UPDRS = Unified Parkinson's Disease Rating Scale; WRS = Webster Rating Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| ADAGIO 2009 | Too short ‐ 9 months duration for rasagiline vs placebo |
| Ahangar 2005 | High percentage (>25%) excluded from analysis post‐randomisation No useful outcomes measured |
| Biglan 2006 | Too short ‐ 6 months duration |
| Dalrymple‐Alford 95 | Too short ‐ 8 weeks duration |
| FSMT 1993 | Too short ‐ 3 months duration |
| Haapaniemi 2000 | Confounded ‐ selegiline versus levodopa versus bromocripitine Too short ‐ 6 months treatment |
| Italian PDSG 2001 | Confounded ‐ selegiline versus levodopa versus dopamine agonist |
| Mally 1995 | Too short ‐ 3 weeks duration |
| Nappi 1991 | Too short ‐ 6 months duration |
| PSG Lazabemide 1994 | Too short ‐ 8 weeks duration |
| RSG 2000 | Too short ‐ 12 weeks duration; some patients late disease |
| Stern 2004 | Too short ‐ 10 weeks duration |
| Stocchi 2004 | Too short ‐ 3 months duration |
| Su 2004 | Too short ‐ 3 months duration |
| TEMPO 2002 | Too short ‐ 26 weeks duration |
| Zhao 2005 | Sub‐therapeutic dose of selegiline used No useful outcomes measured |
Contributions of authors
Protocol: AM, CC, NI, RS. Literature search: KAT, RC, AM, RS. Literature selection: KAT, RC, AM, CC. Papers quality assessment: KAT, RC, AM, CC. Data collection from papers: KAT, RC, AM, CC. Interpretation of data: CC, NI. Review writing: KAT, RC, CC, AM, NI, RS.
Sources of support
Internal sources
University of Aberdeen, UK.
University of Birmingham, UK.
External sources
The Health Foundation, UK.
Declarations of interest
CC, NI, and RS are all involved in the PD MED trial.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
California 1989 {published data only}
- Kurth MC, Tetrud JW, Langston JW, Tanner CM. Age at diagnosis and the effect of selegiline in patients with Parkinson's disease [Abstract]. Neurology 1991;41(Suppl 1):400. [Google Scholar]
- Tetrud JW, Langston JW. The effect of deprenyl (selegiline) on the natural history of Parkinson's disease. Science 1989;245(4917):519‐22. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
DATATOP 1993 {published data only}
- Ben‐Shlomo Y, Head J, Lees AJ, Shoulson I, Oakes D, Olanow CW. Mortality in DATATOP [multiple letters]. Annals of Neurology 1999;45(1):138‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. DATATOP: A multicenter controlled clinical trial in early Parkinson's disease. Archives of Neurology 1989;46(10):1052‐60. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. Effect of deprenyl on the progression of disability in early Parkinson's disease. New England Journal of Medicine 1989;321(20):1364‐71. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. Impact of deprenyl and tocopherol treatment on Parkinson's disease in DATATOP patients requiring levodopa. Annals of Neurology 1996;39(1):37‐45. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. Impact of deprenyl and tocopherol treatment on Parkinson's disease in DATATOP subjects not requiring levodopa. Annals of Neurology 1996;39(1):29‐36. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. Mortality in DATATOP: A multicenter trial in early Parkinson's disease. Annals of Neurology 1998;43(3):318‐25. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Shoulson I for the Parkinson Study Group. The impact of extended deprenyl treatment in Parkinson's disease patients not requiring levodopa therapy [Abstract]. Annals of Neurology 1993;34:263. [Google Scholar]
- Shoulson I, Oakes D, Fahn S, Lang A, Langston JW, LeWitt P, et al. Impact of sustained deprenyl (selegiline) in levodopa‐treated Parkinson's disease: a randomised placebo‐controlled extension of the deprenyl and tocopherol antioxidative therapy of parkinsonism trial. Annals of Neurology 2002;51(5):604‐12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Shoulson I, The Parkinson Study Group. An interim report of the effect of selegiline (L‐deprenyl) on the progression of disability in early Parkinson's disease. European Neurology 1992;32(Suppl 1):46‐53. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Shoulson I, and the Parkinson Study Group. DATATOP: A decade of neuroprotective inquiry. Annals of Neurology 1998;44 Suppl 1:S160‐6. [MEDLINE: ] [PubMed] [Google Scholar]
- Shoulson I, and the Parkinson Study Group. Deprenyl and tocopherol antioxidative therapy of parkinsonism (DATATOP). Acta Neurologica Scandinavica Supplementum 1989;126:171‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Shults CW, and The Parkinson Study Group. Effect of selegiline (deprenyl) on the progression of disability in early Parkinson's disease. Acta Neurologica Scandinavica 1993;87 (Suppl 146):36‐42. [MEDLINE: ] [PubMed] [Google Scholar]
- The Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. New England Journal of Medicine 1993;328(3):176‐83. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Finland 1997 {published and unpublished data}
- Myllylä VV, Heinonen EH, Vuorinen JA, Kilkku OI, Sotaniemi KA. Early selegiline therapy reduces levodopa dose requirement in Parkinson's disease. Acta Neurologica Scandinavica 1995;91(3):177‐82. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Myllylä VV, Sontaniemi K, Hakulinen P, Mäki‐Ikola O, Heinonen EH. Early selegiline treatment has a beneficial long‐term effect on the outcome of Parkinson's disease [Abstract]. Movement Disorders 1996;11(Suppl 1):150. [Google Scholar]
- Myllylä VV, Sontaniemi KA, Tuominen J, Heinonen EH. Selegiline monotherapy in early Parkinson's disease [Abstract]. Acta Neurologica Scandinavica 1990;82(Suppl 128):31. [Google Scholar]
- Myllylä VV, Sotaniemi KA, Hakulinen P, Mäki‐Ikola O, Heinonen EH. Selegiline as the primary treatment of Parkinson's disease‐‐a long‐term double‐blind study. Acta Neurologica Scandinavica 1997;95(4):211‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Myllylä VV, Sotaniemi KA, Tuominen J, Heinonen EH. Selegiline as primary treatment in early phase Parkinson's disease ‐ an interim report. Acta Neurologica Scandinavica 1989;126:177‐82. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Myllylä VV, Sotaniemi KA, Vuorinen JA, Heinonen EH. Selegiline as initial treatment in de novo parkinsonian patients. Neurology 1992;42(2):339‐43. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Myllylä VV, Sotaniemi KA, Vuorinen JA, Heinonen EH. Selegiline in de novo parkinsonian patients: the Finnish study. Movement Disorders 1993;8(Suppl 1):S41‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Myllylä VV, Sotaniemi KA, Vuorinen JA, Heinonen EH. Selegiline as a primary treatment of Parkinson's disease. Acta Neurologica Scandinavica 1991;84(Suppl 136):70‐2. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Turkka J, Suominen K, Tolonen U, Sotaniemi K, Myllylä VV. Selegiline diminishes cardiovascular autonomic responses in Parkinson's disease. Neurology 1997;48(3):662‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Norway‐Denmark 1999 {published data only}
- Boas J. Effects of early selegiline therapy on long‐term levodopa treatment and Parkinsonian disability [Abstract]. Acta Neurological Scandinavica 1996;94(Suppl 167):5. [Google Scholar]
- Larsen JP, Boas J, Aasly J, Boas J, Boesen F, Boisen E, et al. The effects of early selegiline therapy on long‐term levodopa treatment and parkinsonian disability: An interim analysis of a Norwegian‐Danish 5‐year study. Movement Disorders 1997;12(2):175‐82. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Larsen JP, Boas J, Erdal JE and the Norwegian‐Danish Study Group. Does selegiline modify the progression of early Parkinson's disease? Results from a five‐year study. European Journal of Neurology 1999;6(5):539‐47. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
PARJUPAR 1996 {published and unpublished data}
- Castro‐Caldas A, Sampaio C, Botelho A, Gouveia A and The PARJUPAR study group. Bromocriptine or selegiline, or both, to delay the need of levodopa therapy in “de novo” Parkinson’s disease patients [Abstract]. Movement Disorders 1996;11(Suppl 1):744. [Google Scholar]
- Castro‐Caldas A, Sampaio C, Botelho MA, Gouveia A and The PARJUPAR Study Group. PARJUPAR Study: Bromocriptine, Selegeline or Both Delay the Need of Levodopa Therapy in "De Novo" Parkinson's Disease Patients. Personal communication.
- Castro‐Caldas A, Sampaio C, Botelho MA, Gouveia A for The Parjupar Study Group. Parjupar study: bromocriptine or selegiline or both, to delay the need of levodopa therapy in de novo Parkinson's disease patients [Abstract]. Neurology 1996;46:A375. [Google Scholar]
PSG 1996 {published data only}
- Kieburtz K and the Parkinson Study Group. Effect of Lazabemide on the progression of disability in early Parkinson's disease [Abstract]. Neurology 1995;45:A291. [DOI] [PubMed] [Google Scholar]
- The Parkinson Study Group. Effect of lazabemide on the progression of disability in early Parkinson's disease. Annals of Neurology 1996;40(1):99‐107. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
SELEDO 1999 {published data only}
- Kraus PH. Seledo‐study. Aktuelle Neurologie 1998;25(suppl 4):S324‐6. [MEDLINE: ] [Google Scholar]
- Przuntek H. Five years' analysis of the SELEDO study [Abstract]. Neurology 1996;46:A376. [Google Scholar]
- Przuntek H, Conrad B, Dichgans J, Kraus PH, Krauseneck P, Pergande G. SELEDO: a 5‐year long‐term trial on the effect of selegiline in early Parkinsonian patients treated with levodopa. European Journal of Neurology 1999;6(2):141‐50. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Swedish PSG 1998 {published and unpublished data}
- Pålhagen S and the Swedish Parkinson Study Group. Final results of the monotherapy part from the multicentre trial on selegiline [Abstract]. Acta Neurologica Scandinavica 1996;94(Suppl 167):5. [Google Scholar]
- Pålhagen S and the Swedish Parkinson Study Group. Selegiline delays the onset of disability in de novo patients with Parkinson's disease [Abstract]. Movement Disorders 1996;11(Suppl 1):150. [Google Scholar]
- Pålhagen S, Heinonen EH, Hägglund J, Kaugesaar T, Kontants H, Mäki‐Ikola O, et al and the Swedish Parkinson Study Group. Selegiline delays the onset of disability in de novo parkinsonian patients. Neurology 1998;51(2):520‐5. [DOI] [PubMed] [Google Scholar]
- Pålhagen S, Heinonen EH, Hägglund J, Kaugesaar T, Mäki‐Ikola O, Palm R and the Swedish Parkinson Study Group. Selegiline slows the progression of the symptoms of Parkinson disease. Neurology 2006;66(8):1200‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Pålhagen SE, Heinonen E. Use of selegiline as monotherapy and in combination with levodopa in the management of Parkinson's disease: perspectives from the MONOCOMB study. Progress in Neurotherapeutics and Neuropsychopharmacology 2008;3(1):49‐71. [Google Scholar]
UK 1996 {published data only}
- Kirollos C, Bowes SG, Charlett A, Purkiss A, Weller C, O'Neill CJ. An evaluation, with objective assessment, of selegiline monotherapy in newly diagnosed elderly sufferers from idiopathic parkinsonism [Abstract]. British Journal of Clinical Pharmacology 1994;37(1):114P. [MEDLINE: ] [Google Scholar]
- Kirollos C, Charlett A, Bowes SG, Purkiss AG, O'Neill CJ, Weller C, et al. Time course of physical and psychological responses to selegiline monotherapy in newly diagnosed, idiopathic parkinsonism. European Journal of Clinical Pharmacology 1996;50(1‐2):7‐18. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
UK‐PDRG (RR) 1998 {published data only}
- Ben‐Shlomo Y, Churchyard A, Head J, Hurwitz B, Overstall P, Ockelford J, et al. Investigation by Parkinson's Disease Research Group of United Kingdom into excess mortality seen with combined levodopa and selegiline treatment in patients with early, mild Parkinson's disease: further results of randomised trial and confidential inquiry. BMJ 1998;316(7139):1191‐6. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
UK‐PDRG 2001 {published data only}
- Ben‐Shlomo Y, Churchyard A, Head J, Hurwitz B, Overstall P, Ockelford J, et al. Investigation by Parkinson's Disease Research Group of United Kingdom into excess mortality seen with combined levodopa and selegiline treatment in patients with early, mild Parkinson's disease: further results of randomised trial and confidential inquiry. BMJ 1998;316(7139):1191‐6. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenschlager R, Head J, Ben‐Shlomo Y, Lees AJ. Update on 10‐year follow‐up of the data of the Parkinson's Disease Research Group of United Kingdom randomised trial of levodopa, levodopa in combination with selegiline or bromocriptine in patients with early, mild Parkinson's disease; 11th Meeting of the European Neurological Society. http://congress.akm.ch/abstract/abstract/abt.ausgabe?xnkon_nr=48&xssprache=ENG. (accessed 19th September 2003).
- Katzenschlager R, Head J, Schrag A, Ben‐Shlomo Y, Evans A, Lees AJ. Fourteen‐year final report of the randomised PDRG‐UK trial comparing three initial treatments in PD. Neurology 2008;71(7):474‐80. [PUBMED: 18579806] [DOI] [PubMed] [Google Scholar]
- Lees AJ, Abbott R, Banerji N, Barrie M, Boddie GH, Bradbury P, et al. Comparison of therapeutic effects and mortality data of levodopa and levodopa combined with selegiline in patients with early, mild Parkinson's disease. BMJ 1995;311(7020):1602‐7. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees AJ, Abbott R, Banerji N, Barrie M, Boddie GH, Bradbury P, et al. Comparisons of therapeutic effects of levodopa, levodopa and selegiline, and bromocriptine in patients with early, mild Parkinson's disease: Three year interim report. BMJ 1993;307(6902):469‐72. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees AJ, Katzenschlager R, Head J, Ben‐Schlomo Y on behalf of the Parkinson's Disease Research Group of the United Kingdom. Ten‐year follow‐up of three different initial treatments in de‐novo PD: A randomised trial. Neurology 2001;57(9):1687‐94. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Lees AJ, Venna N. Levodopa, selegiline, and bromocriptine for Parkinson disease. Annals of Internal Medicine 1994;120(6 Suppl 2):36. [MEDLINE: ] [Google Scholar]
- Parkinson's Disease Research Group in the United Kingdom. Comparisons of therapeutic effects of levodopa, levodopa and selegiline, and bromocriptine in patients with early, mild Parkinson's disease: three year interim report. Parkinson's Disease Research Group in the United Kingdom. BMJ 1993;307(6902):469‐72. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
US 1995 {published data only}
- Fowler JS, Fazzini E, Volkow ND. Deprenyl and levodopa and Parkinson's disease progression [Multiple letters]. Annals of Neurology 1996;40(2):267‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Olanow CW, Hauser RA, Gauger L, Malapira T, Koller W, Hubble J, et al. The effect of deprenyl and levodopa on the progression of Parkinson's disease. Annals of Neurology 1995;38(5):771‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Olanow CW, Koller W, Hauser R, Malapira T, Busenbark K. A prospective, longitudinal controlled study of deprenyl in Parkinson's disease [Abstract]. Neurology 1994;44:A258. [Google Scholar]
References to studies excluded from this review
ADAGIO 2009 {published data only}
- Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A, et al for the ADAGIO Study Investigators. A double‐blind, delayed‐start trial of rasagiline in Parkinson's disease. New England Journal of Medicine 2009;361:1268‐78. [DOI] [PubMed] [Google Scholar]
Ahangar 2005 {published data only}
- Ahangar AA, Sadraie AR, Vaghefi SBA, Ramesani MS. Comparison between bromocriptine and selegiline in treatment of Parkinson. DARU (Journal of Faculty of Pharmacy, Tehran University of Medical Sciences) 2005;13(1):23‐7. [Google Scholar]
- Sadraie A, Ashrafvaghefi SB, Ramezani MS. Comparison between bromocriptine and selegiline in treatment of Parkinson. Movement Disorders 2006;21(Suppl 15):S635. [Google Scholar]
Biglan 2006 {published data only}
- Biglan KM, Schwid S, Eberly S, Blindauer K, Fahn S, Goren T, et al. Rasagiline improves quality of life in patients with early Parkinson's disease. Movement disorders 2006;21(5):616‐23. [PUBMED: 16450340] [DOI] [PubMed] [Google Scholar]
Dalrymple‐Alford 95 {published data only}
- Dalrymple‐Alford JC, Jamieson CF, Donaldson IM. Effects of selegiline (deprenyl) on cognition in early Parkinson's disease. Clinical Neuropharmacology 1995;18(4):348‐59. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
FSMT 1993 {published data only}
- Allain H, Cougnard J, Neukirch HC. Selegiline in de novo parkinsonian patients: the French selegiline multicenter trial (FSMT). Acta Neurologica Scandinavica 1991;84(Suppl 136):73‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Allain H, Pollak P, Neukirch HC and members of the French Selegiline Multicenter Trial. Symptomatic effect of selegiline in de novo Parkinsonian patients. Movement Disorders 1993;8(Suppl 1):S36‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Haapaniemi 2000 {published data only}
- Haapaniemi TH, Kallio MA, Korpelainen JT, Suominen K, Tolonen U, Sotaniemi KA, et al. Levodopa, bromocriptine and selegiline modify cardiovascular responses in Parkinson's disease. Journal of Neurology 2000;247(11):868‐74. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Italian PDSG 2001 {published data only}
- Caraceni T, Musicco M. Levodopa or dopamine agonists, or deprenyl as initial treatment for Parkinson's disease. A randomised multicenter study. Parkinsonism & Related Disorders 2001;7(2):107‐14. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Caraceni TA, on behalf of the Italian Parkinson Disease Study Group. Dopamine agonists and deprenyl in comparison to levodopa for initial treatment of Parkinson disease. Movement Disorders 1997;12(Suppl 1):S81. [Google Scholar]
- The Italian Parkinson Study Group. A multicenter Italian randomised study on early treatment of Parkinson disease: comparison of l‐doipa, l‐deprenyl and dopaminoagonists. Study design and short term results.. Italian Journal of Neurological Sciences 1992;13:735‐9. [DOI] [PubMed] [Google Scholar]
Mally 1995 {published data only}
- Mally J, Kovacs AB, Stone TW. Delayed development of symptomatic improvement by (‐‐)‐deprenyl in Parkinson's disease. Journal of the Neurological Sciences 1995;134(1‐2):143‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Nappi 1991 {published data only}
- Nappi G, Martignoni E, Horowski R, Pacchetti C, Rainer E, Bruggi P, et al. Lisuride plus selegiline in the treatment of early Parkinson's disease. Acta Neurologica Scandinavica 1991;83(6):407‐10. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
PSG Lazabemide 1994 {published data only}
- Parkinson Study Group. A controlled trial of lazabemide (Ro 19‐6327) in levodopa‐treated Parkinson's disease. Archives of Neurology 1994;51(4):342‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. A controlled trial of lazabemide (Ro 19‐6327) in untreated Parkinson's disease. Annals of Neurology 1993;33(4):350‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
RSG 2000 {published data only}
- Rabey JM, Sagi I, Huberman M, Melamed E, Korczyn A, Giladi N, et al. Rasagiline mesylate, a new MAO‐B inhibitor for the treatment of Parkinson's disease: a double‐blind study as adjunctive therapy to levodopa. Clinical Neuropharmacology 2000;23(6):324‐30. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Stern 2004 {published data only}
- Stern MB, Marek KL, Friedman J, Hauser RA, LeWitt PA, Tarsy D, et al. Double‐blind, randomised, controlled trial of rasagiline as monotherapy in early Parkinson's disease patients. Movement Disorders 2004;19(8):916‐23. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Stocchi 2004 {published data only}
- Stocchi F, Arnold G, Onofrj M, Kwiecinski H, Szczudlik A, Thomas A, et al. Improvement of motor function in early Parkinson disease by safinamide. Neurology 2004;63(4):746‐8. [PUBMED: 15326260] [DOI] [PubMed] [Google Scholar]
Su 2004 {published data only}
- Su W, Chen H.‐B, Zhang Z.‐X, Chen B, Wang L.‐N, Sun X.‐R, Shen Y, Li Y, Geng T.‐C, Zhao W.‐Q, Zhang X.‐Y. A multi‐center, randomised, vitamin E controlled and opening clinical trial of selegiline in patients with Parkinsons disease. Chinese Journal of Neurology 2004;37(5):413‐416. [EMBASE: 2005008352] [Google Scholar]
TEMPO 2002 {published data only}
- Parkinson Study Group. A controlled trial of rasagiline in early Parkinson disease: the TEMPO Study. Archives of Neurology 2002;59(12):1937‐43. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. A controlled trial of rasagiline in early Parkinson's disease [Abstract]. Annals of Neurology 2000;48:489. [Google Scholar]
- Parkinson Study Group. A controlled, randomised, delayed‐start study of rasagiline in early Parkinson disease. Archives of Neurology 2004;61(4):561‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Zhao 2005 {published data only}
- Zhao W‐W, He X‐J, Zhang Z‐F, Kong L‐S, Su J‐J, Xie H‐J. Influence of selegiline on dopaminergic neurons in patients with early Parkinson disease. Zhongguo Linchuang Kangfu 2005; Vol. 9, issue 9:190‐2. [CN‐00569431]
Additional references
Ahlskog 2001
- Ahlskog JE, Muenter MD. Frequency of levodopa‐related dyskinesias and motor fluctuations as estimated from the cumulative literature. Movement Disorders 2001;16(3):448‐58. [DOI] [PubMed] [Google Scholar]
Birkmayer 1985
- Birkmayer W, Knoll J, Riederer P, Youdim MBH, Hars V, Marton J. Increased life expectancy resulting from addition of (‐) deprenyl to Madopar treatment in Parkinson's disease: A long term study. Journal of Neural Transmission 1985;64(2):113‐27. [DOI] [PubMed] [Google Scholar]
Breteler 1998
- Breteler MM. Selegiline, or the problem of early termination of clinical trials. The clinical questions are not well answered, and probably never will be. BMJ 1998;316(7139):1182‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Calne 1995
- Calne DB. Selegiline in Parkinson's disease. BMJ 1995;311(7020):1583‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Churchyard 1999
- Churchyard A, Mathias CJ, Lees AJ. Selegiline‐induced postural hypotension in Parkinson's disease: a longitudinal study on the effects of drug withdrawal. Movement Disorders 1999;14:246‐51. [DOI] [PubMed] [Google Scholar]
Clarke 2001
- Clarke CE. Medical management ‐ neuroprotection. In: Clarke CE editor(s). Parkinson's disease in practice. 1st Edition. London: The Royal Society of Medicine Press Limited, 2001:35‐41. [ISSN 1473‐6845] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fleiss 1993
- Fleiss JL. The statistical basis of meta‐analysis. Statistical Methods in Medical Research 1993;2(2):121‐45. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fowler 1996
- Fowler JS, Fazzini E, Volkow ND. Deprenyl and Parkinson's disease progression. Annals of Neurology 1996;40(2):267‐8. [DOI] [PubMed] [Google Scholar]
Heikkila 1984
- Heikkila RE, Manzino L, Cabbat FS, Duvoisin RC. Protection against the dopaminergic neurotoxicity of 1‐methyl‐4‐phenyl‐1,2,5,6‐tetrahydropyridine by monoamine oxidase inhibitors. Nature 1984;311(5985):467‐9. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Katzenschlager 2002
- Katzenschlager R, Lees AJ. Treatment of Parkinson's disease: levodopa as the first choice. Journal of Neurology 2002;249(Suppl 2):19‐24. [DOI] [PubMed] [Google Scholar]
Lang 1998
- Lang AE, Lozano AM. Parkinson's disease. New England Journal of Medicine 1998;339(16):1130‐43. [DOI] [PubMed] [Google Scholar]
Langston 1983
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine‐analog synthesis. Science 1983;219(4587):979‐80. [DOI] [PubMed] [Google Scholar]
Lees 1995
- Lees AJ, Abbott R, Banerji N, Barrie M, Boddie GH, Bradbury P, et al. Comparison of therapeutic effects and mortality data of levodopa and levodopa combined with selegiline in patients with early, mild Parkinson's disease. BMJ 1995;311(7020):1602‐7. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Macleod 2005
- Macleod AD, Counsell CE, Ives N, Stowe R. Monoamine oxidase B inhibitors for early Parkinson's disease. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD004898.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Maruyama 2002
- Maruyama W, Akao Y, Carrillo MC, Kitani K, Youdium MB, Naoi M. Neuroprotection by propargylamines in Parkinson's disease: suppression of apoptosis and induction of prosurvival genes. Neurotoxicology & Teratology 2002;24(5):675‐82. [DOI] [PubMed] [Google Scholar]
Myllylä 1997
- Myllylä VV, Sotaniemi KA, Hakulinen P, Mäki‐Ikola O, Heinonen EH. Selegiline as the primary treatment of Parkinson's disease‐‐a long‐term double‐blind study. Acta Neurologica Scandinavica 1997;95(4):211‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Olanow 1996
- Olanow CW. Selegiline: current perspectives on issues related to neuroprotection and mortality. Neurology 1996;47(6 Suppl 3):S210‐8. [DOI] [PubMed] [Google Scholar]
Olanow 1998
- Olanow CW, Koller WC. An algorithm (decision tree) for the management of Parkinson's disease: treatment guidelines. Neurology 1998;50(Suppl 2):S1‐S57. [DOI] [PubMed] [Google Scholar]
PDCWG 2001
- Parkinson's Disease Consensus Working Group. Updated guidelines for the management of Parkinson's disease. Hospital Medicine 2001;62(8):456‐70. [DOI] [PubMed] [Google Scholar]
Poewe 1986
- Poewe WH, Lees AJ, Stern GM. Low‐dose L‐dopa therapy in Parkinson's disease: a 6 year follow‐up study. Neurology 1986;36(11):1528‐30. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
PSG 1989
- Parkinson Study Group. Effect of deprenyl on the progression of disability in early Parkinson's disease. New England Journal of Medicine 1989;321(20):1364‐71. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pålhagen 2006
- Pålhagen S, Heinonen EH, Hägglund J, Kaugesaar T, Mäki‐Ikola O, Palm R and the Swedish Parkinson Study Group. Selegiline slows the progression of the symptoms of Parkinson disease. Neurology 2006;66(8):1200‐6. [DOI] [PubMed] [Google Scholar]
Tanner 1992
- Tanner CM. Epidemiology of Parkinson's disease. Neurologic Clinics 1992;10(2):317‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Twelves 2003
- Twelves D, Perkins KSM, Counsell C. Systematic review of incidence studies of Parkinson's disease. Movement Disorders 2003;18(1):19‐31. [DOI] [PubMed] [Google Scholar]
Watts 1997
- Watts RL. The role of dopamine agonists in early Parkinson's disease. Neurology 1997;49(1 Suppl 1):S34‐48. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Ives 2004
- Ives NJ, Stowe RL, Marro J, Counsell C, Macleod A, Clarke CE, et al. Monoamine oxidase type B inhibitors in early Parkinson's disease: meta‐analysis of 17 randomised trials involving 3523 patients. BMJ 2004;329:593‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]