

Abstract
Aims
The F-actin-binding protein Drebrin inhibits smooth muscle cell (SMC) migration, proliferation, and pro-inflammatory signalling. Therefore, we tested the hypothesis that Drebrin constrains atherosclerosis.
Methods and results
SM22-Cre+/Dbnflox/flox/Ldlr−/− (SMC-Dbn−/−/Ldlr−/−) and control mice (SM22-Cre+/Ldlr−/−, Dbnflox/flox/Ldlr−/−, and Ldlr−/−) were fed a western diet for 14–20 weeks. Brachiocephalic arteries of SMC-Dbn −/−/Ldlr−/− mice exhibited 1.5- or 1.8-fold greater cross-sectional lesion area than control mice at 14 or 20 weeks, respectively. Aortic atherosclerotic lesion surface area was 1.2-fold greater in SMC-Dbn−/−/Ldlr−/− mice. SMC-Dbn−/−/Ldlr−/− lesions comprised necrotic cores that were two-fold greater in size than those of control mice. Consistent with their bigger necrotic core size, lesions in SMC-Dbn−/− arteries also showed more transdifferentiation of SMCs to macrophage-like cells: 1.5- to 2.5-fold greater, assessed with BODIPY or with CD68, respectively. In vitro data were concordant: Dbn−/− SMCs had 1.7-fold higher levels of KLF4 and transdifferentiated to macrophage-like cells more readily than Dbnflox/flox SMCs upon cholesterol loading, as evidenced by greater up-regulation of CD68 and galectin-3. Adenovirally mediated Drebrin rescue produced equivalent levels of macrophage-like transdifferentiation in Dbn−/− and Dbnflox/flox SMCs. During early atherogenesis, SMC-Dbn−/−/Ldlr−/− aortas demonstrated 1.6-fold higher levels of reactive oxygen species than control mouse aortas. The 1.8-fold higher levels of Nox1 in Dbn−/− SMCs were reduced to WT levels with KLF4 silencing. Inhibition of Nox1 chemically or with siRNA produced equivalent levels of macrophage-like transdifferentiation in Dbn−/− and Dbnflox/flox SMCs.
Conclusion
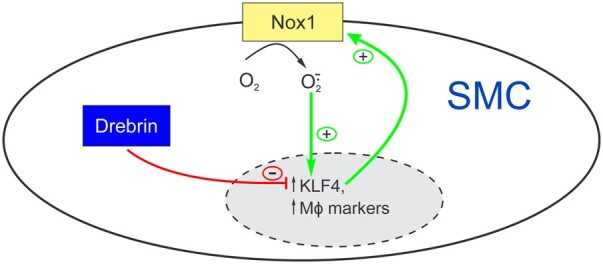
We conclude that SMC Drebrin limits atherosclerosis by constraining SMC Nox1 activity and SMC transdifferentiation to macrophage-like cells.
Keywords: Drebrin, Vascular smooth muscle cells, VSMC, Reactive oxygen species, NADPH oxidase, Nox1, Atherosclerosis, Foam cell
Graphical Abstract
1. Introduction
A hallmark of atherosclerotic lesions is the accumulation of foam cells: cholesteryl ester-laden cells that express macrophage markers and secrete pro-inflammatory cytokines as well as matrix-degrading proteases that contribute to plaque instability.1–3 It was previously believed that foam cells derived exclusively from circulating monocytes; however, lineage tracing and other approaches have identified transdifferentiated SMCs as an important subpopulation of foam cells.4–7 Indeed, SMC transdifferentiation produces ∼40–50% of foam cells in advanced human atherosclerotic lesions5,8 and up to 75% of aortic arch foam cells in advanced mouse atherosclerotic lesions.9 SMC-derived foam cells down-regulate ABCA1,9 lack expression of certain macrophage-specific genes, and exhibit poor phagocytic and efferocytic function; consequently, SMC-derived foam cells appear to augment atherosclerosis and contribute to necrotic core formation.5 Furthermore, SMC-derived foam cells are deficient with regard to collagen synthesis required to maintain a fibrous cap, and thereby may contribute to plaque instability.7,10 Therefore, molecular mechanisms that reduce SMC-to-foam-cell transdifferentiation have implications for both limiting atherosclerosis progression and promoting plaque stability.
The transcription factor Kruppel-like factor 4 (KLF4) is required for SMC de-differentiation11 and has been shown to regulate SMC transdifferentiation to macrophage-like cells during atherogenesis.7 Lineage tracing revealed that SMC-specific KLF4 deletion during atherogenesis results in decreased SMC-to-macrophage-like-cell transdifferentiation, decreased atherosclerotic lesion size, and increased fibrous cap thickness.7 To identify gene targets of KLF4 in the context of atherogenesis, Shankman et al.7 performed chromatin immunoprecipitation-sequencing on brachiocephalic artery specimens from Klf4+/+/Apoe−/− and Klf4−/−/Apoe−/− mice fed a western diet; with this approach, KLF4 was shown to bind to the promoter of the gene encoding Drebrin (Dbn1).
Drebrin was identified first as a neuronal actin-binding protein that stabilizes actin filaments and thereby contributes to synaptic plasticity and memory.12 Drebrin was later found to be abundantly expressed in SMCs, wherein it is up-regulated in response to arterial injury in mice and to atherosclerosis in humans.13 Comparing wild-type (WT) with Dbn-/+ mice, we found that Drebrin inhibits SMC migration and proliferation, both in vitro and in vivo, through stabilization of actin filaments.13 In studies with SMC-specific Dbn−/− (SMC-Dbn−/−) mice, we found that SMC Drebrin limits angiotensin II-induced remodelling of the ascending aorta, and that Drebrin-dependent effects correlate with down-regulation of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase 1 (Nox1), decreased SMC reactive oxygen species (ROS) production, and reduced vascular inflammation.14 Thus, Drebrin constrains not only the migratory/proliferative SMC phenotype evoked by vascular injury but also the pro-inflammatory SMC phenotype evoked by angiotensin II. For these reasons, we tested whether SMC Drebrin attenuates KLF4-dependent SMC-to-foam-cell transition and atherosclerosis.
2. Methods
Detailed methods are available in the Supplementary material online.
2.1 Mice
All animal studies were approved by the Duke Institutional Animal Care and Use Committee and were performed in accordance with NIH guidelines. All mice used in this study were congenic to C57BL/6J. Mice with SMC-specific Drebrin deficiency (SM22-Cre+/Dbnflox/flox or SMC-Dbn−/−) were described previously.14 These mice were crossed with Ldlr−/− mice (Jackson Laboratory) to generate SMC-Dbn−/−/Ldlr−/− mice. Control ‘Dbn+/+/Ldlr−/−’ mice comprised Dbnflox/flox/Ldlr−/−, SM22-Cre+/Ldlr−/−, and Ldlr−/− mice. Both male and female mice were used in atherosclerosis studies; they were analysed in a sex-stratified manner, as indicated.
2.2 Atherosclerosis studies
These experiments adhered to the guidelines for experimental atherosclerosis studies described in the AHA Statement,15 and performed as reported previously.16 ‘Pre-atherosclerotic’ aortas were harvested from 9-week-old mice that had been fed a western diet for 1 week. Mice fed a western diet for 14–20 weeks from age 8 weeks were used for studies of atherosclerotic aortas and brachiocephalic arteries, which were processed as described. Mice were euthanized by CO2 asphyxia followed by exsanguination; blood was used for serum cholesterol measurements, as described.16 Atherosclerotic lesion areas were measured by observers blinded to specimen identity.16
2.3 Carotid interposition grafting
This procedure was performed as we described.17 Mice were anaesthetized with pentobarbital (50 mg/kg, i.p.). Right common carotid arteries from male 8-week-old Dbnflox/flox and SMC-Dbn−/− mice were orthotopically transplanted into age-matched male Apoe−/− mice, which were fed normal chow and harvested 4 weeks post-operatively after euthanasia with pentobarbital (100 mg/kg, i.p.). Carotid grafts were embedded and frozen in Neg-50™ frozen section medium (Thermo FisherScientific, Waltham, MA, USA).
2.4 Statistical analyses
To compare two groups with regard to one parameter, we used an unpaired t test or the Mann–Whitney test, respectively, for normally or non-normally distributed data. To compare >2 groups with regard to ≥2 parameters, we used two-way ANOVA followed by the Sidak post hoc test for multiple comparisons. To compare two groups with regard to ≥2 parameters for which measurements differed greatly in magnitude, we used multiple t tests and the Holm–Sidak correction for multiple comparisons. To ascertain normality of the data, we used the D’Agostino–Pearson omnibus normality test for n ≥ 7/group, and the Kolmogorov–Smirnov test for n = 5–6/group. For datasets with n < 5/group, data were considered normally distributed if the mean and median values were within 10% of each other. Analyses were performed with GraphPad Prism 8.3.1 software (GraphPad, Inc., San Diego, CA, USA). A P < 0.05 was considered significant.
3. Results
3.1 SMC Drebrin attenuates atherosclerosis
To investigate the effects of SMC Drebrin on atherosclerosis, we bred our SM22-Cre+/Dbnflox/flox (SMC-Dbn−/−) mice14 with Ldlr−/− mice, to create congenic cohorts of SMC-Dbn−/−/Ldlr−/− and Dbnflox/flox/Ldlr−/− mice. We previously demonstrated SMC-specific knockout of Drebrin and showed that SMC-Dbn−/− and Dbnflox/flox mice have equivalent blood pressures, heart rates, and aortic dimensions.14 After 14–20 weeks on a western diet, SMC-Dbn−/−/Ldlr−/− and Dbnflox/flox/Ldlr−/− mice were indistinguishable with regard to body weight, serum total cholesterol concentration, and serum high-density lipoprotein cholesterol concentration (Table 1).
Table 1.
Mouse weights and serum cholesterol levels
| Sex | Weeks on western diet | Weight (gm) |
Serum (cholesterol), mmol/L (mg/dL) |
||
|---|---|---|---|---|---|
| Dbn flox/flox/Ldlr−/− (n = 20) | SMC-Dbn−/−/Ldlr−/− (n = 19) |
Dbn
flox/flox/Ldlr−/− (n = 14) |
SMC-Dbn−/−/Ldlr−/− (n = 14) |
||
| Male | 14 | 37 ± 1 | 37 ± 1 | 22 ± 1 (830 ± 50) | 21 ± 1 (820 ± 60) |
| 20 | 40 ± 2 | 40 ± 1 | 28 ± 1 (1090 ± 40) | 29 ± 1 (1130 ± 40) | |
| Female | 14 | 25 ± 1 | 26 ± 1 | 31 ± 1 (1190 ± 60) | 31 ± 1 (1200 ± 100) |
Mice of the indicated genotype and sex were fed a western diet for the indicated number of weeks, euthanized, and then subjected to the indicated measurements.
Prior to the infiltration of the arterial neointima by monocyte/macrophages, arterial inflammation in atherosclerosis initiates with the expression of cytokines and adhesion molecules by endothelial cells and SMCs, substantially under the control of the pro-inflammatory transcription factor NFκB.16In vivo, SMC Drebrin reduces angiotensin II-induced activation of NFκB and expression of VCAM-1 (vascular cell adhesion molecule-1).14 Therefore, we tested whether SMC Drebrin reduces NFκB activation and VCAM-1 expression in the earliest stages of atherogenesis. To do so, we fed SMC-Dbn−/−/Ldlr−/− and Dbnflox/flox/Ldlr−/− mice a western diet for just one week starting at the age of 8 weeks; aortas harvested at this time point were devoid of intimal monocyte/macrophages.16,18 Compared with aortas from control Dbnflox/flox/Ldlr−/− mice, aortas from SMC-Dbn−/−/Ldlr−/− mice demonstrated equivalent levels of the NFκB subunit p65, but 19 ± 3% higher levels of Ser536-phosphorylated NFκB p65, which is transcriptionally activated14,16 (Supplementary material online, Figure S2). Congruent with this increased activation of NFκB in pre-atherosclerotic aortas, SMC-Dbn−/−/Ldlr−/− aortas also demonstrated 35 ± 5% and 30 ± 5% higher levels of the NFκB-dependent19 gene product VCAM-1 in the tunica media and tunica intima, respectively (Supplementary material online, Figure S2). Thus, Drebrin deficiency in SMCs augments arterial inflammation in the earliest stages of atherogenesis.
To determine whether the increased inflammatory signalling in SMC-Dbn−/−/Ldlr−/− aortas augmented atherosclerosis, we fed our mice a western diet for 14 weeks prior to examining brachiocephalic arteries and aortas. Control ‘Dbn+/+/Ldlr−/−’ mice for these experiments comprised Dbnflox/flox/Ldlr−/−, SM22-Cre+/Ldlr−/−, and Ldlr−/− mice. Each of these three groups yielded equivalent results; consequently, we pooled results from all of these groups (Supplementary material online, Figures S3 and S4). In brachiocephalic arteries, SMC-Dbn−/−/Ldlr−/− mice showed ∼1.5-fold greater lesion cross-sectional area than control mice, in both males (Supplementary material online, Figure S3) and females (Supplementary material online, Figure S4). By contrast, en face analyses of Sudan IV-stained aortas showed that SMC-Dbn−/−/Ldlr−/− and control mice had equivalent surface areas of cholesteryl ester-rich lesions after 14 weeks on a western diet (Supplementary material online, Figures S3 and S4). These data thus presented an apparent paradox between cross-sectional analyses showing anti-atherogenic effects of Drebrin, on the one hand, and en face analyses showing no effect of Drebrin, on the other. To resolve this apparent paradox, we tested whether, in the setting of more advanced atherosclerosis, SMC-Dbn−/−/Ldlr−/− mouse atherosclerotic lesions would be larger not only in cross-sectional area but also in surface area, assessed en face.
Atherosclerotic lesion size in brachiocephalic arteries was greater in male mice fed a western diet for 20 weeks, as compared with 14 weeks: by 1.4 ± 0.3-fold in Dbn+/+/Ldlr−/− mice and by 1.6 ± 0.5-fold in SMC-Dbn−/−/Ldlr−/− mice (P < 0.05; Figure 1 and Supplementary material online, Figure S3). Congruently, aortic lesion surface area was also greater after 20 weeks than after 14 weeks on western diet: 1.7 ± 0.4-fold greater in Dbn+/+/Ldlr−/− mice, and 2.0 ± 0.5-fold greater in SMC-Dbn−/−/Ldlr−/− mice (P < 0.001; Figure 1 and Supplementary material online, Figure S3). At this 20-week time point, furthermore, SMC-Dbn−/−/Ldlr−/− mice demonstrated larger atherosclerotic lesions than Dbn+/+/Ldlr−/− mice in both cross-sectional and surface area analyses: 1.8 ± 0.6-fold greater in brachiocephalic cross sections and 1.2 ± 0.1-fold greater in aortas analysed en face (Figure 1). Thus, as it did in mice fed a western diet for 14 weeks, SMC Drebrin activity attenuated atherosclerosis in mice fed a western diet for 20 weeks. However, even after 20 weeks on a western diet, SMC Drebrin deficiency augmented atherosclerotic lesion cross-sectional area more extensively than lesion surface area. To gain insight into this difference, we examined lesion composition.
Figure 1.
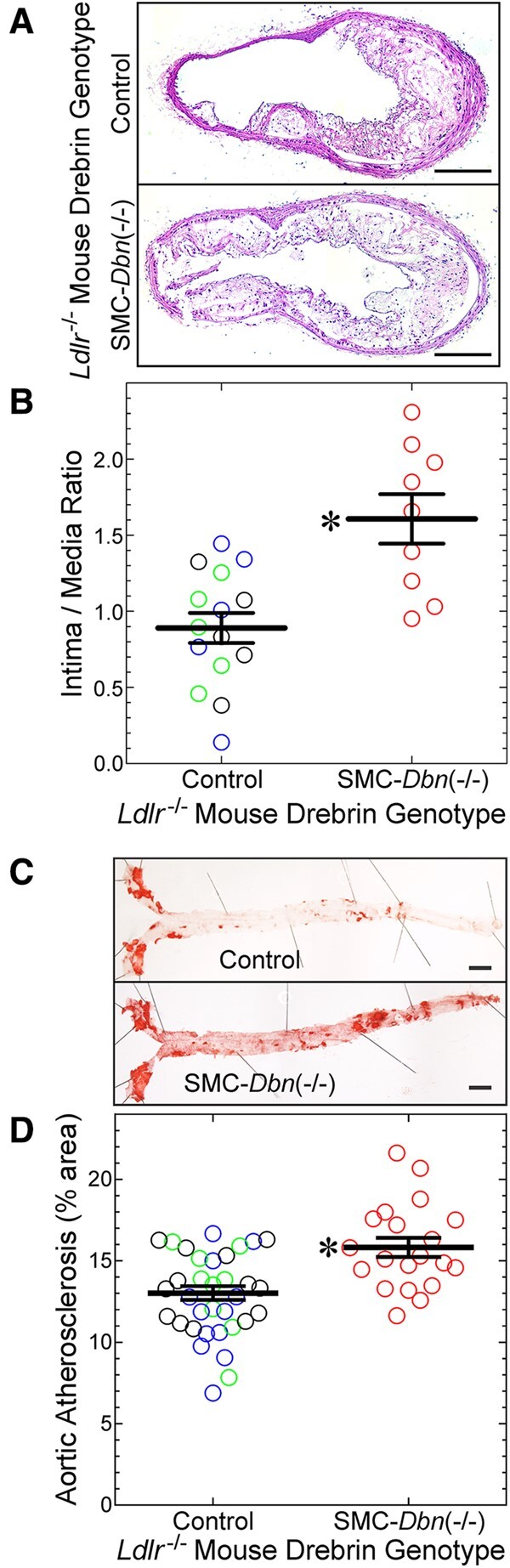
SMC Drebrin attenuates atherosclerosis. The indicated male mice were fed a western diet for 20 weeks, from age 8 weeks: SMC-Dbn−/−/Ldlr−/− mice, and ‘control’ mice [SM22-Cre+/Ldlr−/− (green), Dbnflox/flox/Ldlr−/− (blue), and Dbn+/+/Ldlr−/− (black)]. All control groups yielded congruent data; therefore, data from these groups were pooled. (A) Brachiocephalic artery sections from the indicated mice were stained with H&E; one specimen of each group is shown, representing 9–15 examined per group. Scale bars = 200 μm. (B) Brachiocephalic cross-sectional neointimal and medial areas were measured by planimetry for control (n = 15) and SMC-Dbn−/−/Ldlr−/− mice (n = 9). For each artery, neointimal area was divided by medial area; these ratios for each mouse were plotted along with group means ± S.E. Compared with control mice: *P < 0.001 by t test. (C) Aortas from these mice were excised from the root to the iliac bifurcation, stained with Sudan IV, pinned and photographed en face. Scale bars = 2 mm. (D) The percentage of aortic surface area comprising Sudanophilic lesions was quantitated and plotted for each mouse [control (n = 33) and SMC-Dbn−/−/Ldlr−/− (n = 20)], along with group means ± SE. Compared with control: *P < 0.001 (t test).
The larger atherosclerotic lesions of SMC-Dbn−/−/Ldlr−/− brachiocephalic arteries were similar to those of Dbn+/+/Ldlr−/− brachiocephalic lesions in terms of (i) percent cross-sectional area comprising macrophages and SMCs, as judged by CD68 and smooth muscle (SM) α-actin immunostaining, and (ii) fibrous cap thickness (Figure 2A and B; Supplementary material online, Figure S5). In accord with their larger size, SMC-Dbn−/−/Ldlr−/− brachiocephalic lesions contained ∼50% more SM α-actin-positive cells (SMCs, Figure 2C). However, when we identified monocyte/macrophages and SMCs by immunostaining for CD11b and smooth muscle myosin heavy chain (SMMHC), respectively, SMC-Dbn−/−/Ldlr−/− brachiocephalic atheromata comprised ∼10% more monocyte/macrophage area and ∼50% less SMC area than Dbn+/+/Ldlr−/− brachiocephalic lesions (Supplementary material online, Figure S6). Nonetheless, proliferation of SM α-actin-positive cells was 1.7 ± 0.2-fold greater in SMC-Dbn−/−/Ldlr−/− than in Dbn+/+/Ldlr−/− brachiocephalic lesions after 14 weeks on western diet (Supplementary material online, Figure S5)—a finding consistent with data obtained with Dbn-/+ and WT SMCs in vitro and in injured arteries.13 The prevalence of apoptotic cells in brachiocephalic atheromata was 30 ± 3% greater in SMC-Dbn−/−/Ldlr−/− than in Dbn+/+/Ldlr−/− mice after 14 weeks of western diet (Supplementary material online, Figure S5); furthermore, the necrotic core cross-sectional areas were 2.0 ± 0.1- to 2.4 ± 0.4-fold larger in SMC-Dbn−/−/Ldlr−/− than in Dbn+/+/Ldlr−/− mice after 20 weeks of western diet (Supplementary material online, Figure S6 and Figure 2, respectively). Thus, aside from promoting larger atherosclerotic lesion size, the most notable effect of SMC Drebrin deficiency was promoting atherosclerotic lesion cell apoptosis and enlarging the necrotic core area of atherosclerotic lesions.
Figure 2.

SMC Drebrin deficiency augments atherosclerotic lesion necrotic core size and the prevalence of SMC-derived foam cells. (A) Brachiocephalic arteries from Dbnflox/flox/Ldlr−/− (control) and SMC-Dbn−/−/Ldlr−/− mice used in Figure 1 were immunostained with IgG specific for macrophages (CD68, green) and smooth muscle (SM) α-actin (red); all specimens were counterstained for DNA (blue). Serial sections stained with isotype control IgG yielded no green or red colour (not shown). Scale bars = 200 µm. (B) The percentages of atheroma area comprising macrophages, SMCs, and necrotic core were measured with ImageJ for five discrete mice of each group. The thickness of the atheroma SM α-actin+ fibrous cap was measured at six locations, which were averaged and plotted for each artery (n = 8–9), along with means ± SE for each genotype. Compared with control: *P < 0.01 (multiple t tests, Holm–Sidak correction for multiple comparisons). (C) SM α-actin-positive cells in each brachiocephalic atheroma were counted manually, and plotted with mean ± SE of 6–7 mice/genotype. Compared with control: *P < 0.03 (t test). (D, E) Serial sections of brachiocephalic arteries from A and B were incubated simultaneously with BODIPY® 493/503 (for cholesteryl ester), Hoechst 33342 (DNA), and either Cy3-conjugated IgG specific for SM α-actin or for no known protein. Confocal microscopy used an optical slice thickness of 1 µm. Serial sections stained with Cy3-control IgG yielded no red colour (not shown). The dotted white lines indicate the internal elastic lamina (IEL). The dashed boxes indicate areas enlarged further in the adjacent panels. L, lumen. Scale bars = 50 µm. Co-localization of green (BODIPY) with either blue (Hoechst) or red (SM α-actin) was performed using Imaris 9.2 software, to yield white or yellow, respectively. BODIPY-stained material in the neointima or media was judged to be cellular (as opposed to extracellular), and therefore foam cells, if there was co-localization of green BODIPY with blue DNA fluorescence (designated white). (D) Within several neointimal microscopic fields, the number of BODIPY+ (foam) cells (≥100 per artery) was divided by the total number of cells to obtain neointimal foam cell prevalence; the number of foam cells showing yellow SM α-actin co-localization was divided by the total number of neointimal foam cells to obtain ‘% of total foam cells’. Data were plotted for distinct brachiocephalic arteries from control (n = 8) and SMC-Dbn−/−/Ldlr−/− mice (n = 9), along with means ± SE. Compared with control: *P < 0.02 (t test).
3.2 Drebrin constrains SMC-to-foam-cell transdifferentiation
To begin to understand how SMC Drebrin deficiency could engender greater atherosclerotic lesion necrotic core size, we considered the possibility that in SMC-Dbn−/−/Ldlr−/− mice a greater proportion of atherosclerotic lesion macrophages derive from SMCs than in Dbn+/+/Ldlr−/− mice. Approximately 40–50% of foam cells in advanced human atherosclerotic lesions derive from SMCs,5,8 and up to 75% of aortic arch foam cells in advanced mouse atherosclerotic lesions derive from SMCs.9 SMC-derived foam cells down-regulate ABCA1,9 lack expression of certain macrophage-specific genes, and exhibit poor phagocytic/efferocytic function; consequently, SMC-derived foam cells appear to augment atherosclerosis5,7 and atherosclerotic lesion necrotic core size.5
In brachiocephalic arteries from mice fed a western diet for 20 weeks, we identified foam cells with the fluorescent dye BODIPY 493/503, which stains cholesteryl ester. Foam cells were designated as cholesteryl ester fluorescence that with confocal microscopy co-localized with DNA fluorescence, to ensure that we focused upon intracellular lipid, as opposed to extracellular lipid9 (Figure 2D and E). We quantitated SMC-derived foam cells by co-localizing smooth muscle α-actin with BODIPY20 (Figure 2D and E). The prevalence of foam cells among all neointimal cells was 1.6-fold greater in SMC-Dbn−/−/Ldlr−/− than in Dbn+/+/Ldlr−/− brachiocephalic arteries (Figure 2D and E). Furthermore, the prevalence of cells staining for both smooth muscle α-actin and BODIPY was 1.5-fold greater in SMC-Dbn−/−/Ldlr−/− than in Dbn+/+/Ldlr−/− brachiocephalic arteries (Figure 2D and E). By these measures, SMC Drebrin deficiency appears to engender a higher prevalence of SMC-derived foam cells.
To determine whether Drebrin affects SMC-to-foam-cell transdifferentiation in an independent model of atherosclerosis, we employed a surgically chimeric Apoe−/− mouse model: common carotid arteries from Dbnflox/flox or SMC-Dbn−/− mice were orthotopically transplanted (as interposition grafts) into the right common carotid of Apoe−/− mice.17 The foam cell-rich atherosclerosis that develops in these carotid grafts over 4–8 weeks mirrors aortic atherosclerosis with regard to the effects of various gene deletions on atherosclerosis severity.16,17,21,22 However, because these congenic carotid grafts are Apoe+/+, the apoE protein expressed by carotid graft-derived cells can be used as a sort of lineage marker in the Apoe−/− carotid graft recipients—for example, to show that fibrous cap SMCs derive from tunica media SMCs in these carotid grafts,21 as they do in brachiocephalic arteries of Apoe−/− mice.23
SMC-Dbn−/− carotid grafts demonstrated 1.7 ± 0.3-fold greater neointimal atheroma cross-sectional area than Dbnflox/flox carotid grafts (Figure 3A and B)—thereby recapitulating results obtained with brachiocephalic atherosclerotic lesions in Dbn+/+/Ldlr−/− and SMC-Dbn−/−/Ldlr−/− mice (Figure 1). Furthermore, as a percentage of neointimal lesion area, the CD68+ (macrophage) cross-sectional area was 2 ± 0.4-fold greater in SMC-Dbn−/− than in Dbnflox/flox carotid grafts (Figure 3C and D); the prevalence of macrophages among all neointimal cells was 1.7 ± 0.3-fold greater in SMC-Dbn−/− than in Dbnflox/flox carotid grafts (32 ± 9% vs. 19 ± 1%). Consistent with the greater mass and prevalence of macrophages (and consequently greater outward arterial remodelling14), SMC-Dbn−/− carotid grafts also demonstrated 1.5 ± 0.4-fold greater cross-sectional area than Dbnflox/flox carotid grafts (Figure 3B). We identified foam cells in the carotid grafts as we did for Figure 2, and found that the prevalence of foam cells was greater in SMC-Dbn−/− than in Dbnflox/flox carotid grafts: by 1.4 ± 0.2-fold and 1.3 ± 0.2-fold in the neointima and media, respectively (Figure 3E and F).
Figure 3.

SMC Drebrin inhibits atherosclerosis and SMC-to-foam cell transdifferentiation. Common carotid arteries from Dbnflox/flox and SMC-Dbn−/− mice were transplanted into the right common carotid of congenic Apoe−/− mice as interposition grafts, and harvested 4 weeks later. (A) Sections were stained with a modified connective tissue stain to facilitate planimetry of neointima and media. Scale bars = 100 μm. (B) Neointimal, medial, and total arterial cross-sectional areas were measured by planimetry (ImageJ), and plotted for nine distinct carotid grafts/genotype along with means ± SE. Compared with Dbnflox/flox: *P < 0.01 (Mann–Whitney). (C) Serial sections of carotid grafts from A were incubated with anti-CD68 or isotype control IgG, along with Hoechst 33342 (DNA). Negative control IgG yielded no colour (not shown). Scale bars = 50 μm. L, lumen; IEL, internal elastic lamina. (D) In cross sections from C, the CD68-positive neointimal area was divided by the cognate total neointimal area, and plotted for nine distinct carotid grafts/genotype, along with means ± SE. Compared with Dbnflox/flox: *P < 10−3 (t test). (E) Serial sections of carotid grafts were incubated simultaneously with BODIPY® 493/503 and goat anti-apoE, followed by Hoechst 33342 (DNA) and anti-goat/Alexa 546 IgG. Confocal microscopy used an optical slice thickness of 1 μm. Serial sections stained with non-immune primary IgG yielded no colour (not shown). The dotted white lines indicate the IEL. The dashed boxes indicate areas enlarged further in the adjacent panels. L, lumen. Scale bars = 20 μm. Co-localization of red (apoE) with either blue (Hoechst) or green (BODIPY) was performed as in Figure 2. (F) BODIPY-stained material was judged to be cellular as in Figure 2. Foam cell prevalence was determined as in Figure 2 and plotted for 9 distinct carotids/genotype, along with means ± SE. Compared with Dbnflox/flox: *P < 10−3 (Mann–Whitney). (G) BODIPY+ neointimal or medial cells (≥100 per layer per carotid graft) were scored as containing yellow (apoE+/BODIPY+) or not; the percentage of apoE+/BODIPY+ in each layer was plotted for nine distinct carotids per genotype, along with means ± SE. Compared with Dbnflox/flox: *P < 10−3 (t test).
In order to use apoE expression as a marker for foam cells that derive from carotid graft medial SMCs, we first tested whether apoE was expressed at equivalent levels in Dbnflox/flox and SMC-Dbn−/− carotid arteries. Immunofluorescence of carotid arteries demonstrated equivalent apoE expression in Dbnflox/flox and SMC-Dbn−/− arteries, predominantly (if not exclusively) in SMCs (Supplementary material online, Figure S7). To identify foam cells derived from the carotid artery graft, we co-localized these foam cells (as defined above) with apoE immunofluorescence (Figure 3E and G). Using this approach, we found that the prevalence of apoE+/BODIPY+, carotid graft-derived foam cells in the atherosclerotic neointima was 1.8 ± 0.2-fold greater in SMC-Dbn−/− than in Dbnflox/flox carotid grafts (Figure 3G). Carotid graft-derived foam cells constituted 33 ± 4% and 59 ± 9% of all foam cells in the neointima of Dbnflox/flox and SMC-Dbn−/− carotid grafts, respectively. In the tunica media, however, carotid graft-derived foam cells constituted 100% of all foam cells in both Dbnflox/flox and SMC-Dbn−/− carotid grafts (Figure 3G). Thus, SMC Drebrin deficiency increased not only the size of atherosclerotic lesions but also the proportion of foam cells derived from the carotid graft.
To verify these findings, we took a parallel approach that identified carotid graft-derived intimal macrophages by co-localizing apoE and CD68 immunofluorescence (Supplementary material online, Figure S8). With this approach, the prevalence of carotid graft-derived macrophage-like cells in the atherosclerotic neointima was 2.5 ± 0.6-fold greater in SMC-Dbn−/− than in Dbnflox/flox carotid grafts (Supplementary material online, Figure S8B). Thus, our CD68 analysis comported with our BODIPY analysis regarding Drebrin-mediated reduction in carotid graft-derived foam cells. Furthermore, as judged by CD68 expression in vitro (Figures 4A and B and 5A and B) or in vivo (Supplementary material online, Figure S8), SMC transdifferentiation to macrophage-like cells is constrained substantially by Drebrin activity.
Figure 4.

Drebrin inhibits SMC-to-foam cell transdifferentiation in a KLF4-dependent manner. (A) WT SMCs were treated with vehicle (control) or cholesterol-methyl-β-cyclodextrin (10 µg/mL) for 72 h and stained with anti-CD68 IgG. (Isotype control IgG yielded no colour.) Scale bar = 10 µm. Results represent three independent experiments with >100 SMCs evaluated per experiment. (B) Dbnflox/flox and SMC-Dbn−/− SMCs were treated ± cholesterol (as in A) for 24 h before mRNA isolation and qRT-PCR for CD68 and GAPDH. Threshold CD68 counts were normalized to cognate GAPDH values and plotted from four experiments (means ± SE) using three sets of independently isolated Dbnflox/flox and SMC-Dbn−/− SMC lines. Compared with cognate Dbnflox/flox: *P < 0.05 (two-way ANOVA with Sidak post hoc test). (C) SMCs from B were treated (Tx) as above with vehicle or cholesterol (‘Chol’) for 48 h; soluble SMC extracts were immunoblotted sequentially for Galectin-3 (Gal-3) and β-actin. (D) Band densities for Gal-3 were normalized to cognate β-actin band densities; ratios were plotted from three independent experiments (means ± SE) with distinct pairs of Dbnflox/flox and SMC-Dbn−/− SMC lines. Compared with vehicle control: *P < 0.01 (two-way ANOVA with Sidak post hoc test). (E) The mRNA of SMCs from B was subjected to qRT-PCR for SMMHC and GAPDH; data were obtained and processed as in B. Compared with vehicle-treated SMCs: *P < 0.001; compared with Dbnflox/flox: #, P < 0.05 (two-way ANOVA with Sidak post hoc test, n = 4/group). (F) Dbnflox/flox and SMC-Dbn−/− SMCs were grown to confluence, serum starved for 24 h, and then solubilized. SMC lysates were immunoblotted sequentially for KLF4 and β-actin. KLF migrates as a doublet due to its known post-translational modifications.24 (G) Band densities for KLF4 were normalized to cognate β-actin band densities; the ratios were plotted as arbitrary units (a. u.) from 3 independent experiments with three sets of independently isolated Dbnflox/flox and SMC-Dbn−/− SMC lines. Compared with Dbnflox/flox: *P < 0.05 (t test). (H) SMC-Dbn−/− SMCs were transfected with siRNAs targeting KLF4 or no known mRNA (‘control’). Forty-eight hours later, SMCs were treated for a further 48 h with cholesterol-methyl-β-cyclodextrin (10 µg/mL, ‘cholesterol’) or vehicle, and then solubilized. SMC lysates were immunoblotted sequentially for Galectin-3, KLF4 and β-actin. (I) Band densities for Gal-3 or KLF4 were normalized to cognate β-actin band densities; the ratios were plotted as means ± SE from three independent experiments with two independently isolated primary SMC-Dbn−/− SMC lines. Compared with cognate control siRNA-transfected SMCs: *P < 0.01 (Gal-3) or P < 0.05 (KLF4), two-way ANOVA with Sidak post hoc test.
Did SMC-Dbn−/− carotid grafts demonstrate more carotid graft-derived foam cells than Dbnflox/flox carotid grafts because of greater SMC-to-foam cell transdifferentiation? To address this question, we replicated our brachiocephalic artery strategy from Figure 2, and identified SMC-derived foam cells by co-localizing smooth muscle α-actin (instead of apoE) with BODIPY. This approach yielded data congruent with those obtained using apoE as a marker of carotid graft-derived cells: the prevalence of SMC-derived foam cells in the atherosclerotic neointima was 1.5 ± 0.1-fold greater in SMC-Dbn−/− than in Dbnflox/flox carotid grafts (Supplementary material online, Figure S9). SMC-derived foam cells constituted 19 ± 3% and 28 ± 4% of all foam cells in the neointima of Dbnflox/flox and SMC-Dbn−/− carotid grafts, respectively, and SMC-derived foam cells constituted 100% of all foam cells in the tunica media of both Dbnflox/flox and SMC-Dbn−/− carotid grafts (Supplementary material online, Figure S9).
3.3 Drebrin inhibits SMC-to-foam-cell transdifferentiation by reducing Nox1 expression
To discern mechanisms by which Drebrin attenuates SMC-to-foam-cell transdifferentiation, we loaded Dbnflox/flox and Dbn−/− SMCs with soluble cholesterol in vitro.4 Although Dbnflox/flox and Dbn−/− SMCs accumulated equivalent levels of cholesteryl ester (Supplementary material online, Figure S10), Dbn−/− SMCs up-regulated the macrophage marker CD68 1.7 ± 0.2-fold more than Dbnflox/flox SMCs did (Figure 4A and B). Congruently, with cholesterol loading Dbn−/− SMCs up-regulated the macrophage marker galectin-3 protein ∼10-fold, while Dbnflox/flox SMCs did not up-regulate galectin-3 at all (Figure 4C and D). In contrast, cholesterol loading down-regulated the SMC contractile phenotype marker SMMHC (Myh11) more in Dbn−/− than in Dbnflox/flox SMCs (by 1.9 ± 0.2-fold, Figure 4E). Thus, cholesterol loading up-regulated macrophage markers more in Dbn−/− than in Dbnflox/flox SMCs, and down-regulated SMMHC more in Dbn−/− than in Dbnflox/flox SMCs.
The transcription factor KLF4 appears to be required for SMC-to-foam-cell transdifferentiation.7 Accordingly, we asked whether Dbn−/− SMCs express more KLF4 than Dbnflox/flox SMCs. Consistent with their greater SMC-to-foam-cell transdifferentiation, Dbn−/− SMCs demonstrated 1.7 ± 0.2-fold greater KLF4 protein expression than Dbnflox/flox SMCs (Figure 4F–G). To determine if up-regulation of KLF4 in Dbn−/− SMCs is responsible for greater SMC-to-foam-cell transdifferentiation in Dbn−/− SMCs, we used an RNAi approach. Dbn−/− SMCs transfected with KLF4-targeted siRNA demonstrated ∼50–60% less KLF4 than control-transfected Dbn−/− SMCs (approximately the same level of KLF4 seen in Dbnflox/flox SMCs). This degree of KLF4 silencing abrogated galectin-3 up-regulation induced by cholesterol loading in Dbn−/− SMCs (Figure 4H and I), and thus restored the Dbnflox/flox (‘WT’) SMC phenotype. Thus, Drebrin activity in SMCs appears to inhibit SMC-to-foam-cell transdifferentiation triggered by cholesterol loading in a KLF4-dependent manner. Consistent with KLF4’s ability to bind to the Drebrin promoter and act as a transcriptional repressor,7 cholesterol loading also reduced the expression of Drebrin in WT SMCs, by >10-fold (Supplementary material online, Figure S11A and B).
Loss of Drebrin results in actin depolymerization, which in turn promotes SMCs to transdifferentiate from a contractile to a proliferative phenotype.13 For this reason, we asked whether the enhanced SMC-to-foam-cell transdifferentiation observed in Dbn−/− SMCs could be restored to WT SMC levels by stabilizing filamentous actin. To address this question, we cholesterol-loaded Dbn−/− SMCs and simultaneously treated them with the F-actin stabilizer jasplakinolide.25 As judged by up-regulation of the macrophage marker galectin-3, SMC-to-foam-cell transdifferentiation in Dbn−/− SMCs was virtually abolished by jasplakinolide (Supplementary material online, Figure S11C and D). Thus, in Dbn−/− SMCs, the F-actin destabilization engendered by the absence of Drebrin may, by itself, substantially augment cholesterol-induced SMC-to-foam-cell transdifferentiation.
To confirm the role of Drebrin in constraining SMC-to-foam-cell transdifferentiation, we restored physiological levels of Drebrin in Dbn−/− SMCs and then tested whether doing so would engender a WT phenotype with regard to SMC-to-foam-cell transdifferentiation. In response to cholesterol loading, the prevalence of CD68-positive cells doubled in both WT and Dbn−/− SMCs (Figure 5A and B). However, with control adenovirus transduction, the prevalence of CD68-positive cells was 1.6- to 1.9-fold higher in Dbn−/− than in WT SMCs (Figure 5A and B). This difference diminished significantly with Drebrin-encoding adenovirus transduction: indeed, Dbn−/− SMCs transduced with Drebrin-encoding adenovirus were indistinguishable from WT SMCs transduced with control adenovirus (Figure 5A and B). Concordant data obtained when we examined the macrophage marker galectin-3 instead of CD68 (Figure 5C and D): cholesterol up-regulated galectin-3 levels by ∼2.5-fold in Dbn−/− SMCs, and galectin-3 levels were significantly higher in Dbn−/− than in WT SMCs when both SMC types were infected with control adenovirus. However, when SMCs were transduced with the Drebrin-encoding adenovirus, galectin-3 levels in Dbn−/− SMCs no longer differed significantly from those in WT SMCs. Thus, rescuing physiological levels of Drebrin expression in Dbn−/− SMCs does engender WT levels of SMC-to-foam-cell transdifferentiation as assessed by macrophage markers in vitro.
Figure 5.

Drebrin inhibits SMC up-regulation of CD68 and Galectin-3. (A) Aortic SMCs from Dbnflox/flox and SMC-Dbn−/− mice were transduced with empty vector or Drebrin-encoding adenoviruses and then exposed to medium containing vehicle or solubilized cholesterol for 48 h (37°C). SMCs were then immunostained with IgG specific for CD68 or Drebrin (or isotype control IgG), as indicated, counter-stained for DNA, and imaged at 200× (original magnification). Scale bars = 50 μm. (B) For each SMC group, the number of CD68+ SMCs was divided by the total number of SMCs and multiplied by 100; the resulting percentages are plotted (along with means ± SE) for four experiments with four distinct pairs of Dbnflox/flox and Dbn−/− SMCs. P < 0.05 for comparisons of (i) Dbn−/− vs. cognate Dbnflox/flox SMCs (*); (ii) Drebrin adenovirus-transduced vs. cognate control adenovirus-transduced SMCs (§); (iii) cholesterol-treated vs. vehicle-treated SMCs (#) (two-way ANOVA with Sidak post hoc test). (C) Dbnflox/flox (f/f) and Dbn−/− SMCs were transduced and treated ± cholesterol just as in A. After the 48-h incubation ± cholesterol; however, SMCs were solubilized; protein extracts were immunoblotted serially for the indicated proteins. (D) The band intensities for Galectin-3 were normalized to cognate β-actin band densities, and these ratios were plotted (along with means ± SE) for three experiments with three distinct pairs of Dbnflox/flox and Dbn−/− SMCs. P < 0.05 for comparisons of (i) Dbn−/− vs. cognate Dbnflox/flox SMCs (*); (ii) cholesterol-treated vs. vehicle-treated SMCs (#); (iii) Drebrin adenovirus-transduced vs. cognate control adenovirus-transduced SMCs (§) (two-way ANOVA with Sidak post hoc test).
To elucidate molecular mechanisms promoting greater SMC-to-foam-cell transdifferentiation in Dbn−/− SMCs, we began by examining SMC redox reactions—because in the context of angiotensin II-induced hypertension, Dbn−/− aortic SMCs express higher levels of the NADPH oxidase subunit Nox1 than Dbnflox/flox SMCs,14 and produce higher levels of reactive oxygen species (ROS) than Dbnflox/flox aortic SMCs.14 We first asked whether Dbn−/− arteries have higher levels of ROS in the context of atherogenesis. To address this question, we again examined ascending aortas from SMC-Dbn−/−/Ldlr−/− and Dbnflox/flox/Ldlr−/− mice fed a western diet for just 1 week; as described for Supplementary material online, Figure S2, these aortas lacked intimal monocyte/macrophages,16 and allowed us to focus on the ROS signal from SMCs of the tunica media. CellROX® Orange staining demonstrated 1.6 ± 0.3-fold greater ROS levels in SMC-Dbn−/−/Ldlr−/− than in Dbnflox/flox/Ldlr−/− aortas (Figure 6A). Concordantly, tunica media ROS levels were 1.4 ± 0.2-fold greater in SMC-Dbn−/− than in Dbnflox/flox carotid grafts (Figure 6B). We ascertained that all tunica media cells originated from the carotid graft (and were thus either Dbnflox/flox or Dbn−/− SMCs) by immunostaining with apoE (Figure 6B). Consistent with these ex vivo ROS data from arteries’ tunica media, Dbn−/− SMCs in vitro demonstrated 1.8 ± 0.5-fold higher levels of Nox1 than Dbnflox/flox SMCs (Figure 6C).
Figure 6.

Drebrin regulates SMC ROS levels and foam cell transdifferentiation via Nox1. (A) Serial frozen sections of Dbnflox/flox/Ldlr−/− and SMC-Dbn−/−/Ldlr−/− pre-atherosclerotic aortas used in Supplementary material online, Figure S2 were incubated without (negative control) or with CellROX® Orange in the absence (total signal) or presence (nonspecific, Nox1-independent signal) of the Nox1 inhibitor ML171 (see Section 2). Fluorescence photomicrographs from single aortas of each genotype are shown; negative control samples yielded no red fluorescence (not shown). Scale bars = 50 μm. (Right), Specific CellROX® fluorescence was calculated as the fluorescence (red pixels/mm2) in aortic slices incubated with CellROX® minus that obtained from slices incubated with CellROX® plus ML171; values from five distinct aortas of each genotype were plotted, along with means ± SE. Compared with Dbnflox/flox/Ldlr−/−: *P < 0.01 (t test). (B) Frozen sections from atherosclerotic carotid interposition grafts used in Figure 3 were processed like the aortas of A; serial sections were immunostained with goat IgG specific for apoE or for no known protein (control). ‘L’, lumen. Arrows indicate the internal and external elastic laminae (and thus the tunica media). Scale bars = 50 μm. (Right) Specific CellROX® fluorescence in the tunica media was quantitated as in B. Values for four discrete carotid grafts per genotype are plotted, with means ± SE. Compared with Dbnflox/flox carotid grafts: *P < 0.03 (Mann–Whitney test). (C) SMCs from age- and sex-matched Dbn−/− and Dbnflox/flox (fl/fl) mice were solubilized and immunoblotted serially for Nox1 and β-actin. Nox1 band densities were normalized to cognate β-actin band densities; ratios were plotted as arbitrary units for four independent pairs of Dbnflox/flox and Dbn−/− SMC lines, with means ± SE. Compared with Dbnflox/flox: *P < 0.03 (Mann–Whitney test). (D) Dbn−/− and Dbnflox/flox (‘f/f’) SMCs were treated without or with the Nox1-selective inhibitor ML171 (1 μmol/L) in the presence or absence of cholesterol-methyl-β-cyclodextrin (10 µg/mL) or vehicle for 24 h and then solubilized. SMC lysates were immunoblotted sequentially for Galectin-3 and β-actin. Band densities for galectin-3 were normalized to cognate β-actin band densities; the ratios were plotted as means ± SE from three independent experiments with two independently isolated SMC lines of each genotype. Compared with cognate Dbnflox/flox SMCs: *P < 0.05 (two-way ANOVA with Sidak post hoc test).
Because Dbn−/− SMCs express higher levels of KLF4 than Dbnflox/flox SMCs (Figure 4), and because the expression of KLF4 and cellular ROS levels promote SMC-to-foam cell transdifferentiation,20 we asked whether KLF4 regulates Nox1 expression in Dbn−/− SMCs. For this purpose, we used Dbn−/− SMCs subjected to KLF4 silencing, from Figure 4. When KLF4 protein levels in Dbn−/− SMCs were reduced to WT levels by siRNA transfection, Nox1 protein levels were reduced by 55 ± 6%—to levels prevailing in WT SMCs (Supplementary material online, Figure S12A and B; Figure 6C). Thus, Drebrin deficiency appears to up-regulate Nox1 in a manner that depends substantially upon KLF4.
To determine whether higher Nox1-dependent ROS levels engendered greater SMC-to-foam-cell transdifferentiation in Dbn−/− SMCs, we examined the effects of Nox1 inhibition on cholesterol-induced up-regulation of the macrophage marker galectin-3.26 Cholesterol loading induced ∼9-fold more galectin-3 expression in Dbn−/− than in Dbnflox/flox SMCs (Figure 6D). However, the Nox1-selective inhibitor ML17114 abrogated galectin-3 up-regulation by cholesterol loading, and rendered equivalent galectin-3 expression in Dbn−/− and Dbnflox/flox SMCs (Figure 6D). In parallel experiments, we used siRNA transfection to reduce Nox1 levels by ∼60% in Dbn−/− SMCs. Whereas cholesterol loading up-regulated galectin-3 by 4 ± 1-fold in control-transfected Dbn−/− SMCs, cholesterol loading failed to up-regulate galectin-3 in Nox1-silenced Dbn−/− SMCs (Supplementary material online, Figure S12C–E). Taken together, these data demonstrate that Drebrin limits SMC-to-foam-cell transdifferentiation at least in part by constraining the expression of Nox1 and steady-state ROS levels in SMCs.
4. Discussion
This study demonstrates that SMC Drebrin attenuates atherosclerosis, assessed in the aorta, brachiocephalic artery, and in either Ldlr−/− or Apoe−/− mouse models. Mechanistically, Drebrin substantially reduces SMC-to-foam-cell transdifferentiation both in vitro and in vivo, and does so by down-regulating KLF4, Nox1 and SMC ROS levels. SMC-derived macrophage-like cells are deficient in efferocytosis,5 and impairment of macrophage efferocytosis aggravates atherosclerosis.27 Consequently, SMC Drebrin could reduce atherosclerosis both by reducing SMC ROS levels28 and by reducing the prevalence of SMC-derived macrophage-like cells in atherosclerotic lesions.20,27
4.1 Drebrin effects on ROS and SMC phenotype
Previously, Drebrin was shown to reduce SMC phenotypic plasticity: in response to platelet-derived growth factor BB, Dbn-/+ and Dbn−/− SMCs convert from the contractile to the synthetic phenotype more readily than WT SMCs, and this plasticity is restored to WT levels with the rescue of physiologic levels of Drebrin expression.13,14 Our current work demonstrates that this Drebrin-mediated constraint of SMC plasticity also obtains in the presence of atherogenic stimuli, which provoke SMC-to-foam-cell transdifferentiation as an extreme on the continuum of inflammatory SMC phenotypes.4,5,7–9 Control of SMC-to-foam-cell transdifferentiation fundamentally involves the transcriptional regulator KLF4,5,7,29 which we found up-regulates in Dbn−/− SMCs (Figure 4). However, as our work with Nox1 inhibition and silencing demonstrates, KLF4-dependent SMC transdifferentiation is itself dependent upon SMC ROS levels. In this way, our findings accord with previous work demonstrating that SMC ROS levels determine KLF4 expression levels.20,30 KLF4 may down-regulate Drebrin7 and, in so doing, augment levels of KLF4. In turn, elevated levels of KLF4 apparently up-regulate Nox1 (Supplementary material online, Figure S12)—a mechanism revealed for the first time, to our knowledge, in this work. Nox1 expression has been shown to be transcriptionally regulated by alternative promoters. When SMCs transition away from the contractile phenotype, Nox1 expression transitions from a ‘type a’ transcript to a ‘type c’ transcript, which possesses a 5’ untranslated region distinct from that of the type a transcript.31 PROMO in silico analysis of the type c promoter reveals a putative KLF4 binding motif absent from the type a promoter.32 By up-regulating Nox1, thereby augmenting SMC ROS levels, KLF4 activity may be facilitating its own expression in a positive-feedback manner (Supplementary material online, Figure S13).
In the setting of SMC Drebrin deficiency, increased Nox1 expression and SMC ROS levels engender increased arterial inflammation not only in the context of atherosclerosis (Supplementary material online, Figure S2) but also in the context of angiotensin II-induced hypertension.14 Increased SMC ROS levels exert pro-atherogenic effects by oxidizing LDL in the subendothelial space,33 increasing NFκB-dependent inflammatory gene expression in SMCs,14,20 and augmenting SMC-to-foam-cell transdifferentiation,20 among other mechanisms. In addition to entailing KLF4, the mechanism of Nox1 up-regulation in Dbn−/− SMCs may involve a positive-feedback loop that is constrained not only by Drebrin but also by other actin-stabilizing proteins.34 Excess NADPH oxidase activity augments pro-inflammatory signalling pathways, such as NFκB,34 which, in a positive-feedback manner, further enhance expression of NADPH oxidases.35 In Dbn−/− SMCs, for example, TNF evokes more NFκB p65 activation than in WT SMCs; this excess NFκB activation is ROS-dependent.14
Drebrin may reduce ROS-dependent inflammatory signalling not only by reducing Nox1 expression but also by reducing Nox1 endocytosis.36 Nox1 endocytosis is required for ROS generation in response to TNF,37 and endosomal ROS is required for activation of NFκB.38 Drebrin inhibits dynamin-mediated endocytosis via its interaction with cortactin, which links dynamin 2-tethered vesicles with actin-related protein 3 (ARP3).36 Furthermore, by associating with cortactin, Drebrin may also inhibit NADPH oxidase activity by inhibiting the assembly of the NADPH oxidase complex—because cortactin mediates the interaction of the p47phox subunit of the NADPH oxidase complex with the actin cytoskeleton.39
4.2 Drebrin in human atherosclerosis
The GWAS Catalog40 demonstrates no genetic association between atherosclerosis and SNPs in the human Drebrin gene (DBN1) proper. However, there is an association between atherosclerosis and a SNP within a ∼200-kb window around DBN1: rs337366. This SNP is located in DOK3,41 a gene that resides close to DBN1 on chromosome 5. The atherosclerosis risk allele of this DOK3 SNP is associated with the expression of DBN1 in the aorta (P < 9 × 10−8) and posterior tibial artery (P = 2 × 10−13), such that DBN1 expression increases by 0.20 SD units with each copy of the risk allele (the GTEX Portal).42 Thus, publicly available human databases suggest that Drebrin expression increases with atherosclerosis—just as we found in our immunofluorescence microscopic examination of human arteries.13
4.3 Study limitations
Identifying SMC-derived foam cells ex vivo by using smooth muscle α-actin expression surely underestimates the prevalence of SMC-derived foam cells: smooth muscle α-actin expression is downregulated in activated SMCs,4 and the prevalence of SMC-derived foam cells is substantially higher when quantitated with genetic lineage-tracing methods than with smooth muscle α-actin.7 The same problem of underestimating SMC-derived foam cells likely prevails when using apoE expression to identify foam cells originating from the carotid grafts in our studies, in part because SMC apoE expression declines with SMC proliferation.43 Nonetheless, to determine relative differences between Dbnflox/flox and SMC-Dbn−/− with regard to SMC-derived foam cells, both smooth muscle α-actin and apoE provided comparable data in our analyses of two independent models. Indeed, when SMC lineage for foam cells was inferred from smooth muscle α-actin expression, SMC-derived foam cells were 1.5-fold more prevalent in SMC-Dbn−/−/Ldlr−/− brachiocephalic arteries and in SMC-Dbn−/− carotid grafts than in cognate control arteries. Furthermore, carotid graft-derived foam cells were 1.8-fold more prevalent in SMC-Dbn−/− carotids when we used apoE to distinguish carotid graft-derived cells. Because Drebrin deficiency promotes SMC de-differentiation,13 and because SMC de-differentiation downregulates the expression of smooth muscle α-actin4 and apoE,43 using these markers to identify SMC-derived foam cells may underestimate the actual differences between Dbnflox/flox and SMC-Dbn−/− atherosclerotic lesions. In addition, because apoE+ foam cells in our Apoe+/+ carotid grafts may derive from carotid resident macrophages in addition to SMCs, counting apoE+ macrophage-derived foam cells would diminish the apparent effect of SMC Drebrin on the prevalence of graft-derived foam cells: SMC-Dbn−/− carotid grafts lack Drebrin predominantly, if not exclusively, in SMCs.
The increase in plaque necrotic core size observed in SMC-Dbn−/−/Ldlr−/− mice (Figure 2 and Supplementary material online, Figure S6) may be due to (i) an increase in SMC transdifferentiation to macrophage-like cells, which are relatively poor at efferocytosis,5 or (ii) an increase in the prevalence of apoptotic plaque cells (Supplementary material online, Figure S5). Indeed, these mechanisms are likely to be interdependent. The diminished phagocytosis/efferocytosis associated with increased SMC transdifferentiation5 may increase the prevalence of apoptotic plaque cells because these cells are cleared more slowly (or not at all, with subsequent necrosis).44,45 Conversely, decreased SMC transdifferentiation engendered by KLF4 deletion diminishes the prevalence of apoptotic plaque cells, at least in part by reducing the prevalence of defective efferocytes.7 Thus, increased necrotic core size is likely attributable to a combination of reduced efferocytosis and increased apoptosis. Although Drebrin inhibits endocytosis,36 it is unlikely that Drebrin would exert any direct effect on phagocytic or efferocytic function of SMC-derived foam cells–because macrophages express very little or no Drebrin,14 and Drebrin down-regulates in cholesterol-loaded SMCs (Supplementary material online, Figure S11). Therefore, the effect of Drebrin on phagocytosis/efferocytosis in SMC-derived macrophages would be expected to be small, if not negligible.
SM22-Cre expression is not entirely specific to SMCs.46 However, of the cells in which SM22-Cre has been shown to be expressed, Drebrin protein is undetectable in myocardium,14 skeletal muscle,14 macrophages,13 and whole bone marrow (Supplementary material online, Figure S1). Therefore, possible SM22-Cre activity in these cells or tissues would seem physiologically insignificant in our studies of Drebrin. SM22-Cre has some activity in platelets,46 and Drebrin protein is expressed in human platelets.47 Furthermore, adventitial fibroblast Drebrin protein expression is ablated by SM22-Cre in our Dbnflox/flox mice.14 SM22-Cre shows activity in perivascular adipocytes and their precursors,46 but to our knowledge Drebrin is not known to be expressed in these cells. Therefore, it is conceivable that anti-atherogenic effects observed in SM22-Cre+/Dbnflox/flox mice may be mediated, in part, through effects on several cells other than SMCs.
5. Conclusions
Drebrin is expressed in SMCs at levels far higher than in other cells of the atherosclerotic plaque.13 Although Drebrin expression up-regulates in SMCs of injured or atherosclerotic arteries,13 Drebrin down-regulates profoundly in SMCs with the cholesterol loading that engenders SMC-to-foam-cell transdifferentiation (Supplementary material online, Figure S11). For this reason, strategies aimed at augmenting SMC Drebrin expression in atherosclerotic plaques may limit atherosclerosis progression and enhance plaque stability by bridling SMC-to-foam-cell transdifferentiation.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
Experimental design: B.M.S., E.R.H., F.J.M., N.J.F., and J.A.S. Experiments and data collection: J-H.W., L.Z., I.N., L.B., T.H., and K.P.S. Data analysis: J-H.W., L.Z., N.J.F., and J.A.S. Writing of manuscript: B.M.S, E.R.H, N.J.F., and J.A.S.
Conflict of interest: none declared.
Funding
This work was supported by the American Heart Association [AHA TPA34860015 to J.A.S.], National Institutes of Health [HL121531 and HL147157 to N.J.F. and J.A.S., T32HL007101 to B.M.S.], and the Edna and Fred L. Mandel Jr Foundation [N.J.F. and J.A.S.].
Data availability
The data underlying this article will be shared on reasonable request to the corresponding authors.
Supplementary Material
Translational perspective
Drebrin is abundantly expressed in vascular smooth muscle cells (SMCs) and is up-regulated in human atherosclerosis. A hallmark of atherosclerosis is the accumulation of foam cells that secrete pro-inflammatory cytokines and contribute to plaque instability. A large proportion of these foam cells in humans derive from SMCs. We found that SMC Drebrin limits atherosclerosis by reducing SMC transdifferentiation to macrophage-like foam cells in a manner dependent on Nox1 and KLF4. For this reason, strategies aimed at augmenting SMC Drebrin expression in atherosclerotic plaques may limit atherosclerosis progression and enhance plaque stability by bridling SMC-to-foam-cell transdifferentiation.
References
- 1. Wang N, Tabas I, Winchester R, Ravalli S, Rabbani LE, Tall A.. Interleukin 8 is induced by cholesterol loading of macrophages and expressed by macrophage foam cells in human atheroma. J Biol Chem 1996;271:8837–8842. [DOI] [PubMed] [Google Scholar]
- 2. Galis ZS, Sukhova GK, Kranzhofer R, Clark S, Libby P.. Macrophage foam cells from experimental atheroma constitutively produce matrix-degrading proteinases. Proc Natl Acad Sci U S A 1995;92:402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM.. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016;354:472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rong JX, Shapiro M, Trogan E, Fisher EA.. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A 2003;100:13531–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM, Fisher EA.. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol 2015;35:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, Feil R.. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res 2014;115:662–667. [DOI] [PubMed] [Google Scholar]
- 7. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK.. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 2015;21:628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA.. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014;129:1551–1559. [DOI] [PubMed] [Google Scholar]
- 9. Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Erh Jaw J, Sin DD, Seidman MA, Leeper NJ, Francis GA.. Smooth muscle cells contribute the majority of foam cells in ApoE (Apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol 2019;39:876–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frontini MJ, O'Neil C, Sawyez C, Chan BM, Huff MW, Pickering JG.. Lipid incorporation inhibits Src-dependent assembly of fibronectin and type I collagen by vascular smooth muscle cells. Circ Res 2009;104:832–841. [DOI] [PubMed] [Google Scholar]
- 11. Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK.. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J Biol Chem 2005;280:9719–9727. [DOI] [PubMed] [Google Scholar]
- 12. Jung G, Kim EJ, Cicvaric A, Sase S, Groger M, Hoger H, Sialana FJ, Berger J, Monje FJ, Lubec G.. Drebrin depletion alters neurotransmitter receptor levels in protein complexes, dendritic spine morphogenesis and memory-related synaptic plasticity in the mouse hippocampus. J Neurochem 2015;134:327–339. [DOI] [PubMed] [Google Scholar]
- 13. Stiber JA, Wu JH, Zhang L, Nepliouev I, Zhang ZS, Bryson VG, Brian L, Bentley RC, Gordon-Weeks PR, Rosenberg PB, Freedman NJ.. The actin-binding protein drebrin inhibits neointimal hyperplasia. Arterioscler Thromb Vasc Biol 2016;36:984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang L, Wu JH, Huang TQ, Nepliouev I, Brian L, Zhang Z, Wertman V, Rudemiller NP, McMahon TJ, Shenoy SK, Miller FJ, Crowley SD, Freedman NJ, Stiber JA.. Drebrin regulates angiotensin II-induced aortic remodeling. Cardiovasc Res 2018;114:1806–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP 3rd, Rosenfeld ME, Virmani R; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; and Council on Basic Cardiovascular Sciences. Recommendation on design, execution, and reporting of animal atherosclerosis studies: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol 2017;37:e131–e157. [DOI] [PubMed] [Google Scholar]
- 16. Jean-Charles PY, Wu JH, Zhang L, Kaur S, Nepliouev I, Stiber JA, Brian L, Qi R, Wertman V, Shenoy SK, Freedman NJ.. Ubiquitin-specific Protease 20 inhibits tumor necrosis factor-triggered smooth muscle cell inflammation and attenuates atherosclerosis. Arterioscler Thromb Vasc Biol 2018;38:2295–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang L, Peppel K, Sivashanmugam P, Orman ES, Brian L, Exum ST, Freedman NJ.. Expression of tumor necrosis factor receptor-1 in arterial wall cells promotes atherosclerosis. Arterioscler Thromb Vasc Biol 2007;27:1087–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis. Circ Res 2014;114:1757–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neish AS, Williams AJ, Palmer HJ, Whitley MZ, Collins T.. Functional analysis of the human vascular cell adhesion molecule 1 promoter. J Exp Med 1992;176:1583–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vendrov AE, Sumida A, Canugovi C, Lozhkin A, Hayami T, Madamanchi NR, Runge MS.. NOXA1-dependent NADPH oxidase regulates redox signaling and phenotype of vascular smooth muscle cell during atherogenesis. Redox Biol 2019;21:101063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu JH, Zhang L, Fanaroff AC, Cai X, Sharma KC, Brian L, Exum ST, Shenoy SK, Peppel K, Freedman NJ.. G protein-coupled receptor kinase-5 attenuates atherosclerosis by regulating receptor tyrosine kinases and 7-transmembrane receptors. Arterioscler Thromb Vasc Biol 2012;32:308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu Y, Zhang L, Liao X, Sangwung P, Prosdocimo DA, Zhou G, Votruba AR, Brian L, Han YJ, Gao H, Wang Y, Shimizu K, Weinert-Stein K, Khrestian M, Simon DI, Freedman NJ, Jain MK.. Kruppel-like factor 15 is critical for vascular inflammation. J Clin Invest 2013;123:4232–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E.. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol 2006;26:2696–2702. [DOI] [PubMed] [Google Scholar]
- 24. Zhou H, Liu Y, Zhu R, Ding F, Wan Y, Li Y, Liu Z.. FBXO32 suppresses breast cancer tumorigenesis through targeting KLF4 to proteasomal degradation. Oncogene 2017;36:3312–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bubb MR, Spector I, Beyer BB, Fosen KM.. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J Biol Chem 2000;275:5163–5170. [DOI] [PubMed] [Google Scholar]
- 26. Bentzon JF, Majesky MW.. Lineage tracking of origin and fate of smooth muscle cells in atherosclerosis. Cardiovasc Res 2018;114:492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yurdagul A Jr, Doran AC, Cai B, Fredman G, Tabas IA.. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med 2017;4:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vendrov AE, Hakim ZS, Madamanchi NR, Rojas M, Madamanchi C, Runge MS.. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arterioscler Thromb Vasc Biol 2007;27:2714–2721. [DOI] [PubMed] [Google Scholar]
- 29. Ackers-Johnson M, Talasila A, Sage AP, Long X, Bot I, Morrell NW, Bennett MR, Miano JM, Sinha S.. Myocardin regulates vascular smooth muscle cell inflammatory activation and disease. Arterioscler Thromb Vasc Biol 2015;35:817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sunaga H, Matsui H, Anjo S, Syamsunarno MR, Koitabashi N, Iso T, Matsuzaka T, Shimano H, Yokoyama T, Kurabayashi M.. Elongation of long-chain fatty acid family member 6 (Elovl6)-driven fatty acid metabolism regulates vascular smooth muscle cell phenotype through AMP-activated protein kinase/Kruppel-like factor 4 (AMPK/KLF4) signaling. J Am Heart Assoc 2016;5:e004014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arakawa N, Katsuyama M, Matsuno K, Urao N, Tabuchi Y, Okigaki M, Matsubara H, Yabe-Nishimura C.. Novel transcripts of Nox1 are regulated by alternative promoters and expressed under phenotypic modulation of vascular smooth muscle cells. Biochem J 2006;398:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Messeguer X, Escudero R, Farré D, Núñez O, Martínez J, Albà MM.. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002;18:333–334. [DOI] [PubMed] [Google Scholar]
- 33. Morel DW, DiCorleto PE, Chisolm GM.. Endothelial and smooth muscle cells alter low density lipoprotein in vitro by free radical oxidation. Arteriosclerosis 1984;4:357–364. [DOI] [PubMed] [Google Scholar]
- 34. Shen J, Yang M, Ju D, Jiang H, Zheng JP, Xu Z, Li L.. Disruption of SM22 promotes inflammation after artery injury via nuclear factor kappaB activation. Circ Res 2010;106:1351–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Salazar G, Huang J, Feresin RG, Zhao Y, Griendling KK.. Zinc regulates Nox1 expression through a NF-kappaB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic Biol Med 2017;108:225–235. [DOI] [PubMed] [Google Scholar]
- 36. Li B, Ding S, Feng N, Mooney N, Ooi YS, Ren L, Diep J, Kelly MR, Yasukawa LL, Patton JT, Yamazaki H, Shirao T, Jackson PK, Greenberg HB.. Drebrin restricts rotavirus entry by inhibiting dynamin-mediated endocytosis. Proc Natl Acad Sci U S A 2017;114:E3642–E3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller FJ Jr, Chu X, Stanic B, Tian X, Sharma RV, Davisson RL, Lamb FS.. A differential role for endocytosis in receptor-mediated activation of Nox1. Antioxid Redox Signal 2010;12:583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miller FJ Jr, Filali M, Huss GJ, Stanic B, Chamseddine A, Barna TJ, Lamb FS.. Cytokine activation of nuclear factor kappa B in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ Res 2007;101:663–671. [DOI] [PubMed] [Google Scholar]
- 39. Touyz RM, Yao G, Quinn MT, Pagano PJ, Schiffrin EL.. p47phox associates with the cytoskeleton through cortactin in human vascular smooth muscle cells: role in NAD(P)H oxidase regulation by angiotensin II. Arterioscler Thromb Vasc Biol 2005;25:512–518. [DOI] [PubMed] [Google Scholar]
- 40. Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, Suveges D, Vrousgou O, Whetzel PL, Amode R, Guillen JA, Riat HS, Trevanion SJ, Hall P, Junkins H, Flicek P, Burdett T, Hindorff LA, Cunningham F, Parkinson H.. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 2019;47:d1005–d1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van der Harst P, Verweij N.. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res 2018;122:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.GETEX Portal t. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 11/14/2020.
- 43. Majack RA, Castle CK, Goodman LV, Weisgraber KH, Mahley RW, Shooter EM, Gebicke-Haerter PJ.. Expression of apolipoprotein E by cultured vascular smooth muscle cells is controlled by growth state. J Cell Biol 1988;107:1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W.. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol 2005;25:1256–1261. [DOI] [PubMed] [Google Scholar]
- 45. Bennett MR, Sinha S, Owens GK.. Vascular smooth muscle cells in atherosclerosis. Circ Res 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chakraborty R, Saddouk FZ, Carrao AC, Krause DS, Greif DM, Martin KA.. Promoters to study vascular smooth muscle. Arterioscler Thromb Vasc Biol 2019;39:603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goodall AH, Burns P, Salles I, Macaulay IC, Jones CI, Ardissino D, de Bono B, Bray SL, Deckmyn H, Dudbridge F, Fitzgerald DJ, Garner SF, Gusnanto A, Koch K, Langford C, O'Connor MN, Rice CM, Stemple D, Stephens J, Trip MD, Zwaginga JJ, Samani NJ, Watkins NA, Maguire PB, Ouwehand WH; Bloodomics Consortium. Transcription profiling in human platelets reveals LRRFIP1 as a novel protein regulating platelet function. Blood 2010;116:4646–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding authors.