

Abstract
Membrane proteins are essential components in cell membranes and enable cells to communicate with their outside environment and to carry out intracellular signaling. Functional reconstitution of complex membrane proteins using cell-free expression (CFE) systems has been proved to be challenging mainly due to the lack of necessary machinery for proper folding and translocation of nascent membrane proteins and their delivery to the supplied synthetic bilayers. Here, we provide protocols for detergent-free cell-free reconstitution of functional membrane proteins using HeLa-based CFE system and outline assays for studying their membrane insertion, topology, and their orientation upon incorporation into the supported lipid bilayers or bilayers of giant unilamellar vesicles as well as methods to isolate functional translocated cell-free produced membrane proteins.
Keywords: Cell-free expression, membrane proteins, in vitro reconstitution, HeLa-based cell-free expression, SUN, linker of nucleoskeleton and cytoskeleton complex, encapsulation, giant unilamellar vesicle
1. Introduction
Bottom-up in vitro reconstitution into biological membranes is a promising approach to demystify complex interactions between different proteins in isolated cellular functions (1). Despite recent advances, using purified membrane proteins or detergent-mediated reconstitution approaches still have certain drawbacks (2–5). For instance, the expression of recombinant proteins in conventional cell culture systems might cause growth retardation or lysis of the host cells. Also, toxic effects might be attributed to the overproduction of heterologous proteins. Additionally, residual detergents in membranes can affect protein function and membrane integrity of liposomes. Moreover, arbitrary protein insertion into the lipid membrane hinders the probing of intracellular and extracellular structure and functions of membrane proteins. To overcome these challenges, cell-free methods that focus on production of proteins outside the cellular environment offer promising solutions. Cell-free expression (CFE) systems that rely on de novo synthesis of proteins using the translation-transcription (TXTL) machinery of cell lysates (6) are becoming popular. Using such platforms, TXTL machineries are fully devoted to expressing the protein(s) of interest. This yields an efficient expression system and as an in vitro system, there is a high degree of control over its components. Therefore, CFE systems have significant advantages over detergent-free reconstitution and can circumvent the limitations mentioned above (7–10).
Membrane proteins play essential roles in different cellular contexts by providing communication pathways between the intra- and extra-cellular environments through receptors, transporters, and channels. Despite their importance, CFE platforms for studying membrane proteins have not been straightforward. The main barrier is the inability for common CFE systems, including E. coli, to produce functional eukaryotic membrane proteins due to the lack of necessary components such as molecular chaperons and post-translational modifications. Even though prokaryotic CFE systems have gained significant interest and are well established in literature, there is increasing attention to eukaryotic CFE systems (11, 12). A few eukaryotic CFE systems have been utilized for detergent-free production and incorporation of membrane proteins. For instance, microsomal bodies in the cell-free lysates of Sf-21 insect cells have been isolated for the incorporation of pH-sensitive bacterial membrane protein KcsA by Dondopati et al. (13). Among mammalian CFE systems, HeLa-based cell-free protein synthesis system (available commercially) is a great candidate for de novo production of complex proteins since it contains endogenous microsomal structures that mediate the direct translocation and post-translational modifications of nascent proteins.
While the cell-free production of membrane proteins is a milestone towards a robust platform for in vitro reconstitution of functional membrane proteins, establishing a proper environment in which the cell-free synthesized membrane proteins reside remains challenging. Depending on the application, several platforms have been developed to study membrane proteins topology, function, and biophysical properties (14). Supported lipid bilayers, for instance, are popular platforms for analyzing the functionality of reconstituted membrane proteins, specifically ion channels since they feature lipid tunability and easy channel recording process (15). Also, recently, giant unilamellar vesicles (GUVs) have gained attention as they can confine purified components or CFE reactions in cell-sized compartments that allows reconstitution of cellular mechanisms in a micron-sized space (16–18) as well as creation of synthetic cells that can sense and respond to external stimuli through their membrane proteins (19, 20).
Here, we outline several platforms for reconstitution of the linker of nucleoskeleton and cytoskeleton complex (LINC) assembly membrane proteins SUN1 and SUN2 as our model membrane proteins (21). The approaches are translatable to other membrane proteins. We describe a HeLa-based CFE system and illustrate that endogenous microsomes that contain translocated membrane protein can be isolated. Additionally, we offer a strategy to investigate the localization of membrane proteins on the supplied lipid membranes by supplementing the CFE reaction with supported lipid bilayers with excess reservoirs (SUPER templates) (22). The orientation of membrane proteins inserted into the lipid bilayer is further analyzed using protease digestion. Moreover, we introduce a method to encapsulate the CFE reactions in GUVs to visualize the localization of membrane proteins in the membrane of GUVs (Figure 1).
Figure 1:
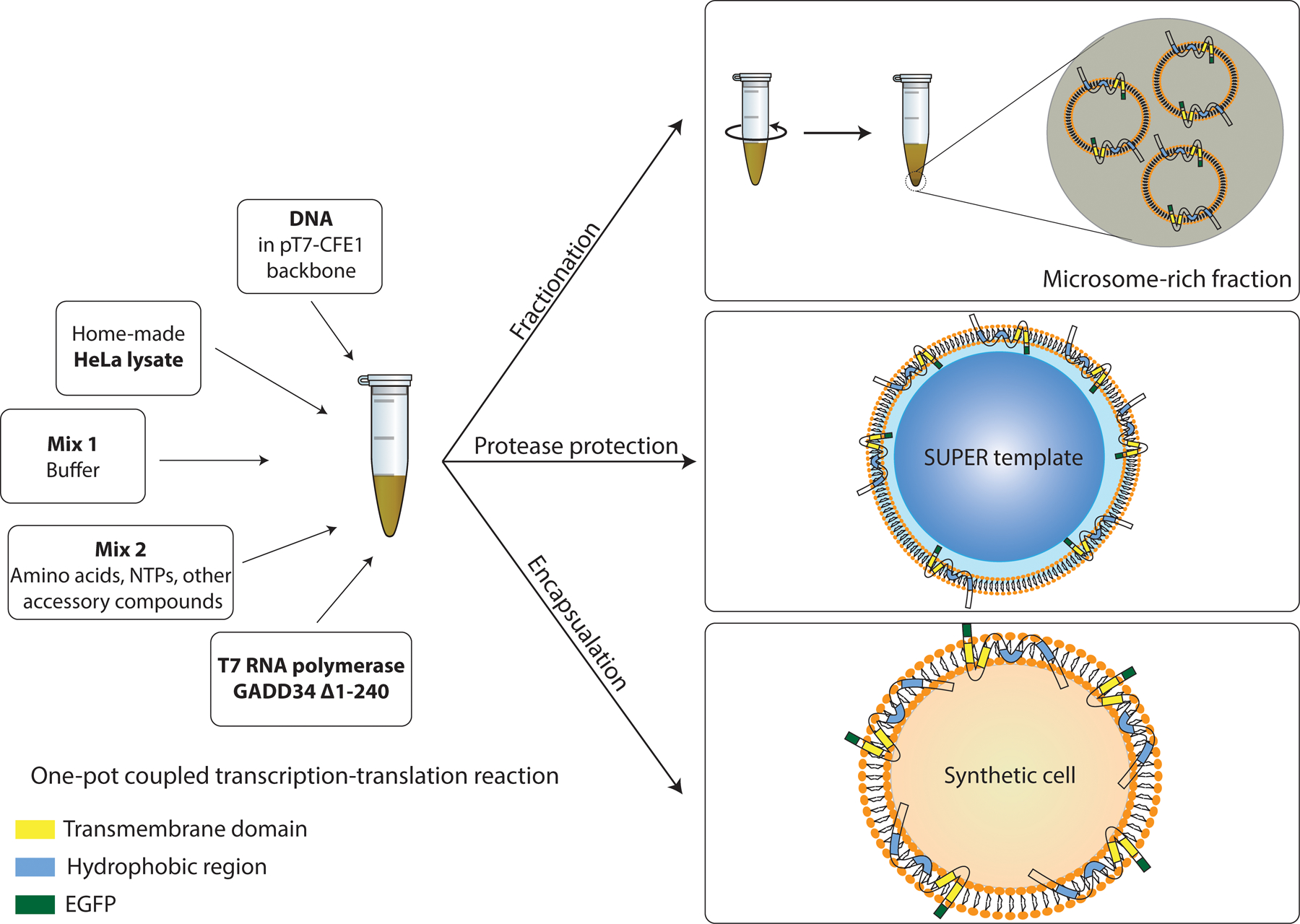
Schematics of HeLa CFE system depicting in vitro platforms for isolating translocated membrane proteins and reconstituting the cell-free synthesized membrane proteins into membranes.
The protocols described here are not unique to the HeLa-based CFE system and can provide rapid assays to investigate the ability of other mammalian CFE systems in the reconstitution of membrane proteins. Given the rising attention towards utilizing CFE technologies to recreate certain cellular processes in cell-sized compartments, the need for functional reconstitution of membrane proteins is inevitable. The assays we present here provide a powerful toolbox for characterizing cell-free membrane protein synthesis and their functional reconstitution.
2. Materials
2.1. Cell-free expression
2.1.1. HeLa cell culture
Cell culture medium: DMEM, 10% fetal bovine serum, 1X GlutaMAX, 100 IU/ml penicillin, 100 µg/ml streptomycin. Store at 4 °C.
Mg- and Cl-free PBS (DPBS).
0.05% Trypsin/EDTA. Store at 4 °C.
Tissue culture petri dishes (150 mm diameter).
2.1.2. HeLa lysate preparation
Washing buffer: 35 mM HEPES-KOH, pH 7.5, 140 mM NaCl, and 11 mM glucose. Store at 4 °C.
Extraction buffer: 20 mM HEPES-KOH, pH 7.5, 45 mM potassium acetate, 45 mM KCl, 1.8 mM magnesium acetate, 1 mM dithiothreitol (DTT). Store at 4 °C without DTT. Add DTT just before use.
High K buffer: 20 mM HEPES-KOH, pH 7.5, 945 mM potassium acetate, 945 mM KCl, 1.8mM magnesium acetate, 1 mM DTT. Store at 4 °C without DTT. Add DTT just before use.
BeadBug bead-beater (Benchmark Scientific) or equivalent.
Bead-beating tubes containing 0.1 mm zirconium beads (Benchmark Scientific, item number: D1032–01, conical bottom if using BeadBug).
Bench-top microcentrifuge.
2.1.3. Assembling TX-TL coupled CFE reaction
HeLa lysate.
Plasmid DNA containing the gene of interest after an internal ribosome entry site (IRES) sequence. The recommended commercial product is the expression vector pT7-CFE1 from Thermo Fisher Scientific (Note 1).
Amino acid mixture: 5–100 mM amino acid mixture (Sigma Aldrich). Store in aliquots at −80 °C.
Mixture 1: 50 mM magnesium acetate (Mg(OAc)2), 170 mM K-HEPES (pH 7.5). Store in aliquots at −80 °C.
Mixture 2: 7.8 mM K-HEPES (pH 7.5), 12.5 mM ATP, 8.36 mM GTP, 8.36 mM CTP, 8.36 mM UTP, 200 mM creatine phosphate, 0.6 mg/ml creatine kinase, 1X amino acid mixture, 5 mM spermidine. Store in aliquots at −80 °C.
T7 RNA polymerase and GADD34 are expressed in E. coli and purified as previously reported (23). Store in aliquots at −80 °C.
2.2. Airfuge fractionation assay
Plasmid DNA containing the gene of interest under the T7 promoter after an IRES sequence.
Extraction buffer: 20 mM HEPES-KOH, pH 7.5, 45 mM potassium acetate, 45 mM KCl, 1.8 mM magnesium acetate. Store at 4°C.
Low volume clear flat bottom 384-well plate (Corning, product number: 3542)
Polyacrylamide gels, use precast polyacrylamide gels with a gradient concentration of 4–20% if desired.
Beckman Coulter Airfuge ultracentrifuge A-100.
2.3. Membrane protein incorporation assay
2.3.1. SUPER template preparation
Lipids: 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt) (DOPS), L-α-phosphatidic acid (egg, chicken) (sodium salt) (Egg-PA), and cholesterol.
100 nm liposome extruder (Avanti or T&T Scientific)
5M NaCl solution (filtered)
Silica beads (preferably 5 µm, Bangs Laboratories)
2.3.2. Incorporation and protease protection assay
DNA plasmids (stock concentration of 500 ng/ml): pT7-CFE1-EGFP-SUN1 and pT7-CFE1-EGFP-SUN2
1X phosphate buffer saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4)
Glass slides and coverslips
Double-sided tape
Lyophilized S. griseus pronase (Roche)
2.4. Vesicle encapsulation system
DNA plasmids (stock concentration of 500 ng/ml): pT7-CFE1-EGFP-SUN2 and pT7-CFE1-EGFP-YAP
Opti-Prep (Sigma-Aldrich)
Lipids: stock concentration of 0.4 mM with 69.9% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 30% cholesterol, and 0.1% 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rhod-PE) in a 4:1 mixture of silicone oil (Sigma Aldrich, catalog number: 317667) and mineral oil (Thermo Fisher Scientific, catalog number: AC415080010).
Outer solution stock solution: 115 mM HEPES, 23 mM MgCl2, 1.15M KCl, 770 mM glucose.
3. Methods
3.1. Preparation of HeLa-based CFE system
We describe how to produce and utilize a HeLa-derived cell-free protein synthesis system. The procedure described herein is a modification of the protocol described by Mikami et al. (23) The method we present does not require specialized equipment (e.g., spinner flasks) for cell culture, and can be carried out in conventional culture dishes. The use of a bead-beater during the cell lysis step can be substituted by any other non-chemical lysis procedure (e.g., French press, nitrogen cavitation).
3.1.1. HeLa cell culture
-
Pre-warm all media and solutions to 37 °C.
If cells are frozen in a liquid nitrogen stock:- Thaw the frozen cells in a 37 °C water bath by swirling the tube until thawed. (Note 2)
- Transfer the content of the cryotube into a 15 ml tube.
- Add 1 ml of warm cell culture medium drop by drop to the 15 ml tube.
- Add warm medium to 10 ml.
- Centrifuge at 250 g for 5 minutes and discard supernatant.
- Resuspend the cell pellet into 20 ml of warm cell medium.
- Transfer the cell suspension to a 150 mm petri dish. (Note 3)
- Cross-shake the dish and place in the incubator at 37 °C with 5% CO2.
When the cells reach ~80% confluency, aspirate the medium and wash with DPBS once. Aspirate the washing solution.
Add 2 ml 0.05% trypsin/EDTA and incubate at 37 °C for 10 minutes.
Add 10 ml of warm cell culture medium and pipette up and down to break down clumps of cells. Gently swirl the dish to detach all the cells.
Add 0.5 ml of the cell suspension in each of 4 x 150 mm petri dishes.
Add 5 ml of culture medium, cross-shake and incubate at 37 °C with 5% CO2.
After cells reach ~80% confluency, repeat the steps 3–6 and split cells in a total of 8 x 150 mm dishes. Grow cells at 37 °C with 5% CO2 until each dish reaches ~90% confluency. (Note 4)
3.1.2. HeLa lysate preparation
Pre-chill washing buffer, extraction buffer and high K buffer on ice for 20 minutes.
Aspirate the medium from 150 mm dishes and wash the cells with DPBS once.
Add 2 ml 0.05% trypsin/EDTA to each dish and incubate at 37 °C for 10 minutes.
Add 10 ml per dish of warm culture medium and break cell clumps by pipetting up and down several times.
Pool the content of the 8 petri dishes into two 50 ml conical tubes. Centrifuge tubes at 250 g for 5 minutes and use 20 ml of cold washing buffer to resuspend and pool cells into a single tube.
Centrifuge at 1200 g for 5 minutes and discard the supernatant. Wash for a total of three times using the cold washing buffer. (Note 5)
Resuspend the cell pellet in 10 ml of cold extraction buffer, and check cell viability with trypan blue (expect ~95% viability). Centrifuge at 1200 g for 5 minutes and discard the supernatant. Weigh the cell pellet.
Add extraction buffer to the pellet in a 1:1 ratio of buffer (in ml):pellet weight (measured in grams).
Fill a bead-beating tube with the cell suspension. Place some suspension in the cap of the bead-beating tube and tightly screw the cap, make sure that no bubbles are present. (Note 6)
Place the bead-beating tubes in the bead-beater and start the apparatus at a speed of 3500 rpm for 20 seconds, then keep the tubes on ice for 2 minutes.
Repeat step 10 for 7 times.
Add high-K buffer of a volume of 1/29 of the homogenate volume and carefully mix by pipetting.
Transfer the homogenate to 1.5 ml microcentrifuge tubes and centrifuge at 1200 g for 10 minutes at 4 °C. Recover the supernatant and repeat the centrifugation once more.
Divide the supernatant (cell lysate) into aliquots and snap-freeze them in liquid nitrogen. Store at −80 °C. (Note 7)
3.1.3. Assembling coupled cell-free expression reaction
Pre-chill tubes on ice. Thaw aliquots of HeLa lysate, mixture 1 (Mix 1), mixture 2 (Mix 2), T7 RNA polymerase and GADD34 and keep them on ice.
Assemble a reaction by mixing 5 µl of HeLa lysate, 1 µl of Mix 1, 1 µl GADD34 (final concentration of 310 nM), 1 µl Mix 2, 0.5 µl T7 RNA polymerase (final concentration of 450 nM), 5 nM plasmid DNA, and RNase-free water to a total volume of 10 µl in a 1.5 ml microcentrifuge tube. Keep the tubes on ice while assembling the reaction, and carefully mix by pipetting after each addition. (Note 8)
Incubate the reaction at 30 °C for 4–5 h. Synthesized protein can be detected either in a plate reader in real time if using a fluorescence protein or using Western blot analysis. (Note 9)
3.2. Airfuge fractionation assay
Assemble a coupled CFE reaction as described in section 3.1.3 (Note 10). Run two control reactions one with EGFP and one without DNA. Transfer the reactions to the low-volume 384 well-plate and use a fluorescence plate reader to monitor protein synthesis.
Once the CFE reaction is complete (as assessed by a plateau in fluorescence reading), remove the well-plate and add 30 µl of extraction buffer to the CFE reaction. This ensures that the concentrations of soluble proteins in both pellet and supernatant fractions subsequently are the same.
Use the Airfuge to centrifuge the mixture at ~100,000 g for 15 minutes at room temperature (refer to the manual for the exact pressure settings). Recover 20 µl of the supernatant and transfer to a 1.5 ml microcentrifuge tube. Pipette up and down the remaining 20 µl in the CFE reaction tube to make sure the pellet (Note 11) is resuspended in the pellet fraction. Transfer the pellet fraction to another 1.5 ml microcentrifuge tube (Note 12).
Following the recovery of fractions, cell-free expressed proteins in each fraction can be visualized using Western blot using appropriate antibodies after SDS-PAGE or directly on an SDS-PAGE gel for a fluorescent protein. The difference in the intensity of lanes containing supernatant and pellet indicates the amount of membrane proteins that are translocated into the membrane of microsomes. (Note 13). Figure 2 shows the presence of microsomes in the pellet fraction by comparing the localization of ER-tracker stain in the puncta in pellet fraction and no localization of the stain in the supernatant fraction.
Figure 2:
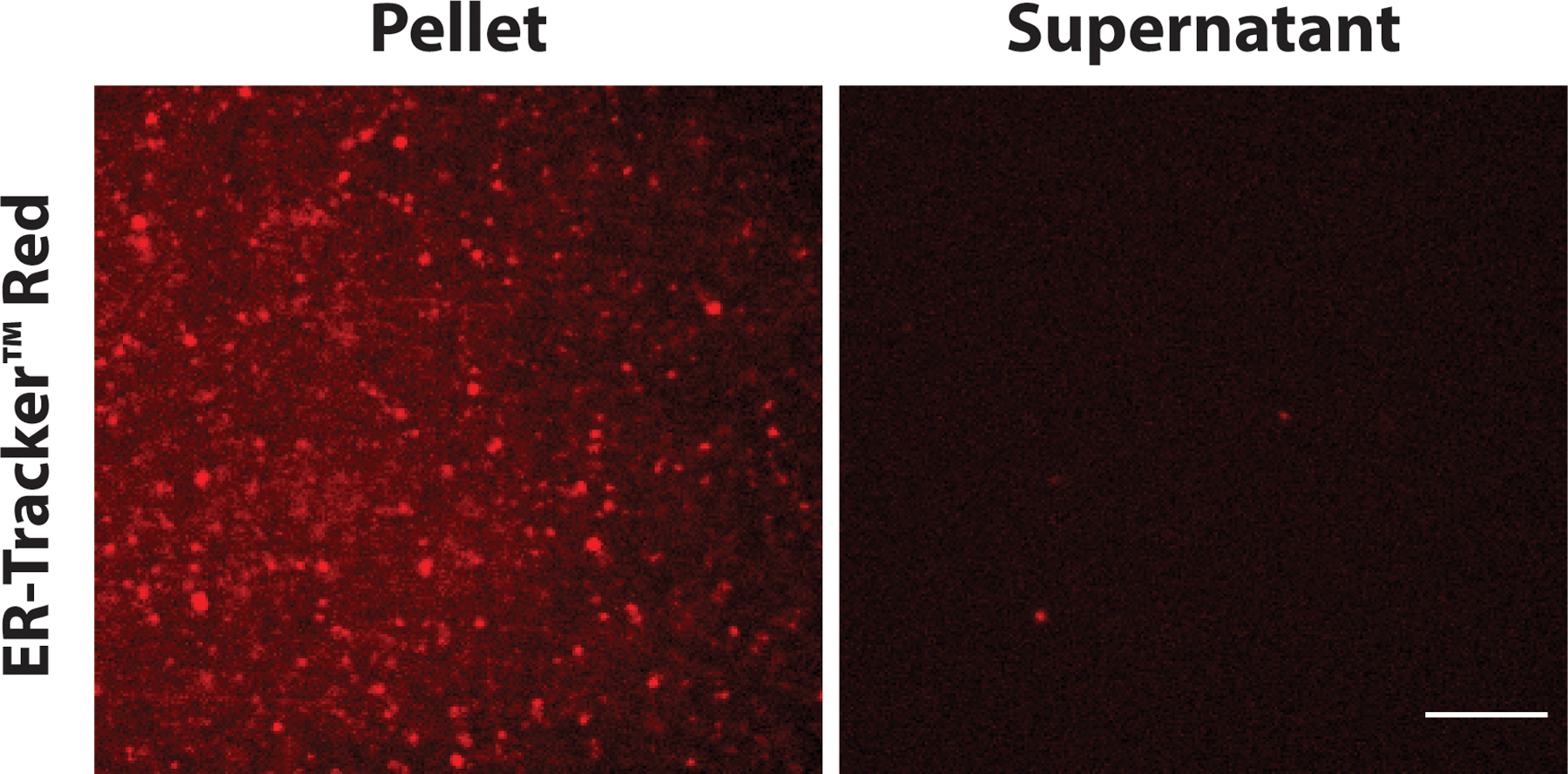
The enrichment of microsomes in the pellet fraction after ultracentrifugation can be detected by staining with a fluorescent ER-tracker. The localization of ER-tracker signals shows the presence of microsomes in the pellet fraction in HeLa lysate. Scale bar: 10 µm.
3.3. Membrane protein incorporation assay
3.3.1. SUPER template preparation
Mix appropriate aliquots of lipids with the mole percentage of 45% DOPC, 27% DOPE, 9% DOPS, 2.2% Egg-PA, and 16.8% cholesterol for a final concentration of 1 mM in a glass test tube. (Note 14)
Dry the lipid film under a gentle stream of nitrogen gas.
To make sure that any residuals of organic solvent are removed from the mixture, desiccate the dried lipid film for at least one hour under vacuum.
Add Milli-Q water to a final lipid concentration of 1 mM and mix thoroughly by vortexing.
-
Pass the mixture through the liposome-extruder (Note 15) 11 times to generate SUV solution. Store the 1 mM SUV stock solution at 4 °C. The stock solution is stable at 4 °C for about two weeks.
The following steps can be done while the CFE reaction is being incubated.
Using a Neubauer counting chamber, calculate the concentration of bead stock solution.
In a 1.5 ml microcentrifuge tube, mix 20 µl of SUV stock solution, 20 µl of 5 M NaCl solution, and 5 × 106 beads. Bring the mixture to a final volume of 100 µl with Milli-Q water.
Incubate the mixture at room temperature for 30 minutes. Flick the tube gently and occasionally to make sure that the beads are suspended and mixed.
Add 900 µl of Milli-Q water to the mixture, and centrifuge at 100 g for 2 minutes. Remove 970 µl of supernatant carefully (Note 16) and mix by gently flicking until the pellet is completely resuspended.
Repeat step 4 three times. The final mixture is the stock solution of SUPER templates (30 µl) that is stable at room temperature for 2–3 hours. (Note 17)
3.3.2. Membrane protein incorporation
Assemble a coupled CFE reaction as described in section 3.1.3. Run two control reactions one with EGFP and one without DNA. Use a fluorescence plate reader to monitor protein synthesis.
Once the CFE reaction has reached completion (typically in about 4 hours), add 1 µl of the SUPER template stock solution to the reaction in the well-plate (if assembling a CFE reaction with higher volumes, the SUPER template to the reaction volume ratio is kept at 1:10). Alternatively, the solution can be transferred to a 1.5 ml microcentrifuge tube and the beads are added.
Incubate the plate at 30 °C for 30 minutes or on a heat block if using microcentrifuge tubes.
Transfer the CFE reaction to a 1.5 ml microcentrifuge tube and centrifuge at 200 g for 5 minutes. Carefully remove the supernatant (the pellet is hardly visible, and care should be taken when recovering the supernatant, as it can be used for other purposes such as Airfuge fractionation assay or an SDS-PAGE).
Resuspend the pellet in 1 ml of 1X PBS and wash the beads by centrifuging at 200 g for 5 minutes. Repeat once.
Resuspend the pellet in 40 µl of 1X PBS.
Prepare an imaging flow chamber by adhering a glass coverslip and a glass slide using double-sided tape and transfer the mixture to the chamber for imaging. The solution will creep into the chamber by capillary action. Fluorescence imaging can be carried out by confocal or epifluorescence imaging. Figure 3A shows membrane incorporation of membrane proteins EGFP-SUN1 and EGFP-SUN2.
Figure 3:
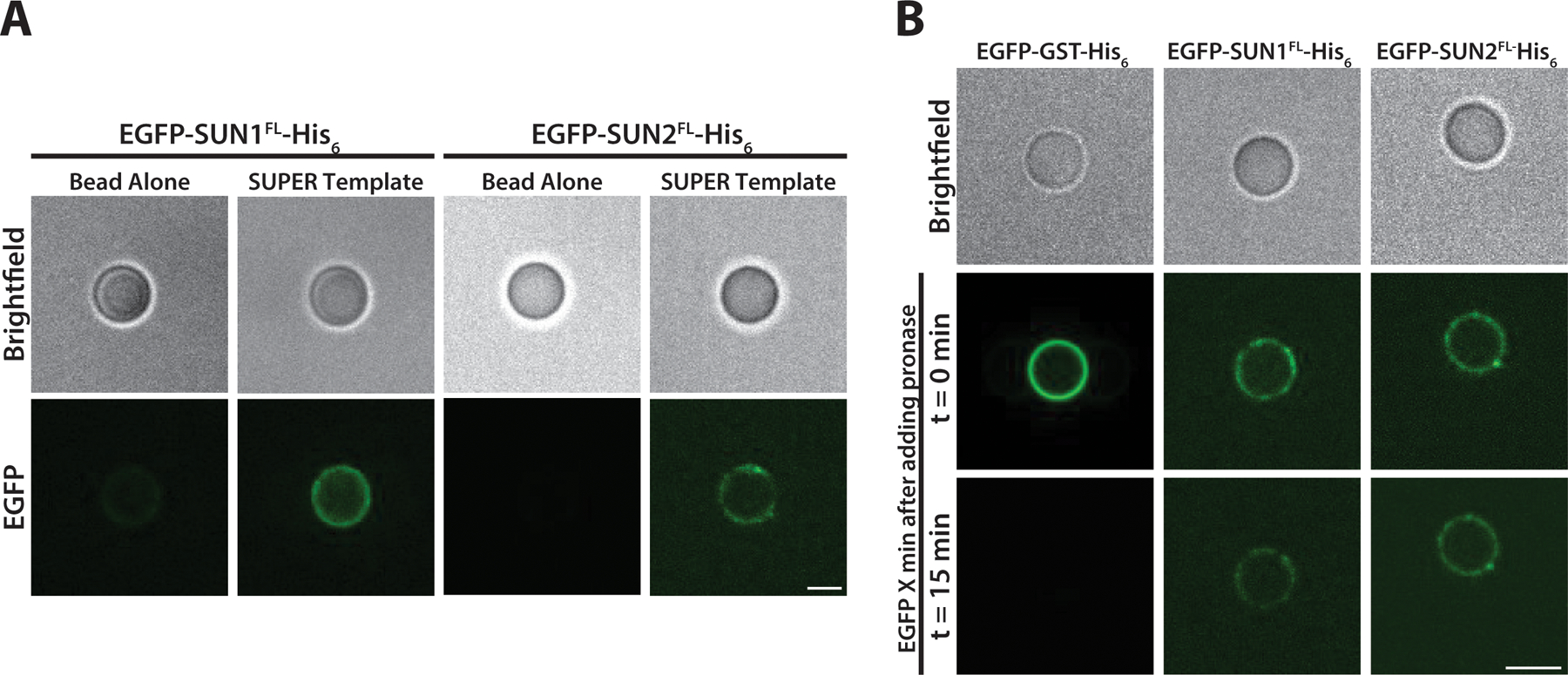
A) Localization of cell-free expressed EGFP-SUN1 and EGFP-SUN2 on the SUPER templates provides visual evidence of membrane protein incorporation into the supported lipid bilayer B) The protease protection assay can help reveal membrane protein topology. The EGFP signal for SUN1 and SUN2 after addition of protease remains on SUPER templates, indicating that the N-terminal EGFP is in the space between the bead and the lipid bilayer where the protease cannot reach. Adapted with permission from Majumder et al. (21) from Journal of Cell Science. Scale bar: 5 µm.
3.3.3. Protease protection assay
Protease protection assay can be utilized to study the topology of membrane protein insertion into the lipid bilayer. Additionally, protease protection assay can confirm the incorporation of the integral membrane protein into the bilayer and determine if the protein is peripherally associated with the membrane. To investigate the topology of membrane protein insertion, the protein of interest is labeled fluorescently. In the case of fluorescent protein fusion, the reporter (e.g., EGFP) can be fused to either terminus of the protein to investigate the topology of insertion. Using this strategy, we have previously identified an additional transmembrane domain in SUN1 and a membrane-associated hydrophobic region in both SUN1 and SUN2 (21).
Prepare a 6 mg/ml stock solution of lyophilized S. griseus pronase in Milli-Q water. Store at 4 °C for a maximum of 3 days.
After resuspending beads in 20 µl of 1X PBS and loading the sample into an imaging flow chamber as described in the previous section, add the appropriate amount of pronase to the final concentration of 2 mg/ml.
Incubate for at least 15 minutes at room temperature before imaging. Figure 3B shows strong association of EGFP-SUN1 and EGFP-SUN2 with SUPER template which implies that N-terminal EGFP is located between the bead and the lipid membrane and thus protected from protease digestion. However, the loss of fluorescence after addition of protease to the His6-GST-EGFP (bound to Ni-NTA containing SUPER template) shows its peripheral association with the membrane of SUPER templates.
3.4. Vesicle encapsulation system
Vesicles were generated by a modification of the continuous droplet interface crossing encapsulation (cDICE) method (18, 24) (Figure 4). A rotor chamber was 3D-printed with clear resin by using a 3D printer (Formlabs) and mounted on the servo motor of a benchtop stir plate and rotated at 700 rpm. The custom chamber has a capped hole near the outer edge where samples can be retrieved.
Figure 4: The schematic for generating vesicles encapsulating CFE reactions by cDICE method.
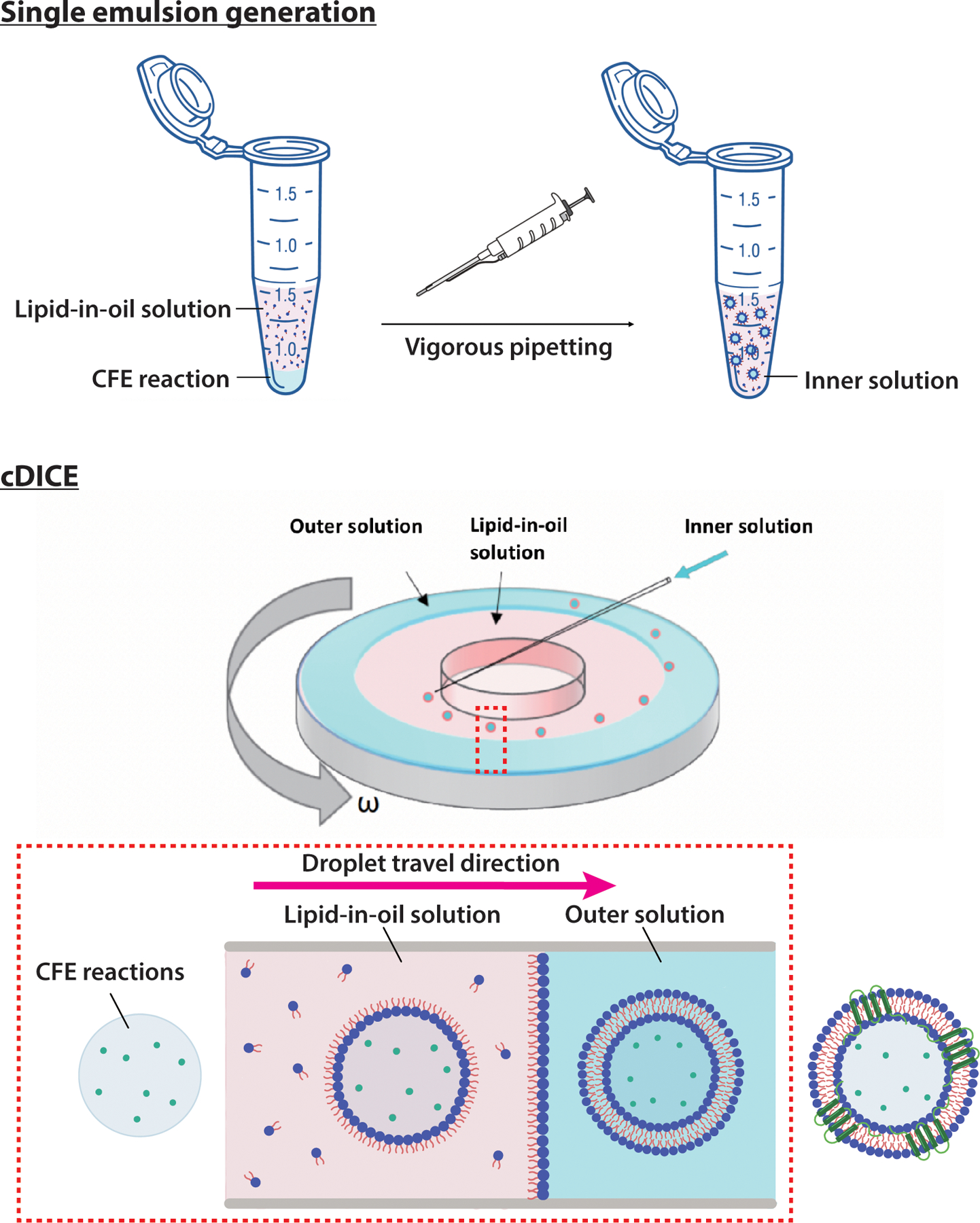
(Top) The inner solution is made by vigorous pipetting of the CFE reaction with lipid-in-oil solution (LOS) solution. (Bottom) The cDICE contains a cylindrical chamber with a lid. Through the hole in the lid, the outer solution (OS) and LOS are sequentially introduced. The IS droplets are ejected from the pipette tip into the chamber and are then centrifuged radially towards the LOS/OS interface to form the final lipid bilayer vesicles containing CFE reactions. The top schematics were sourced from https://scidraw.io/, and the cDICE device schematic is adapted with permission from Bashirzadeh et al. (18).
Assemble a coupled CFE reaction as described in section 3.1.3. Run two control reactions one with EGFP and one without DNA.
Dilute 1 µl of CFE reaction with 9 µl Milli-Q water and measure the osmolarity. Use triplicates to determine the final osmolarity of the saturated CFE reaction before encapsulation. Add 5% OptiPrep to the CFE reaction and keep in ice as the inner solution (IS).
Make 1 ml of aqueous outer solution (OS) by diluting OS stock with DI water until the osmolarity matches that of the IS. (Note 18)
Make lipid-in-oil solution (LOS) by mixing lipids together in a glass test tube with the desired mole percentage and chloroform (e.g., DOPC:DOPE:cholesterol = 40:30:30) and a total of 0.4 mM lipids (Note 19). Mix the lipids with the desired volume of 1:4 silicon oil:mineral oil by vortexing for at least 10 seconds. (Note 20)
Gently add 500 µl LOS on top of encapsulated CFE reaction in the 1.5 ml microcentrifuge tube. The interface between the LOS and the encapsulated CFE reaction should be clearly visible because the difference in density between the two solutions results in the formation of two distinct layers with a vertical water/oil interface at their boundary.
Make water-in-oil emulsion by vigorously pipetting ~8 µl of CFE reaction in 500 µl of LOS. (Note 21)
Sequentially add 700 µl aqueous OS, 5 ml LOS and the water-in-oil emulsion generated in step 6 into the cDICE chamber rotating at 700 rpm (Figure 4). (Note 22)
After 3 minutes of rotation, there are two layers of solutions inside the chamber. Vesicles will accumulate in the OS near the chamber wall. Carefully collect the vesicles by gently retrieving the OS (Note 23). Figure 5 shows the localization of membrane proteins EGFP-SUN1 in the lipid bilayer membrane of vesicles generated by cDICE method. EGFP-YAP (Yes-Associated Protein), a transcription factor involved in mechanotransduction, is shown as a negative control as it does not localize to the membrane.
Figure 5: Localization of cell-free expressed EGFP-SUN2 and EGFP-YAP in vesicles created with cDICE.
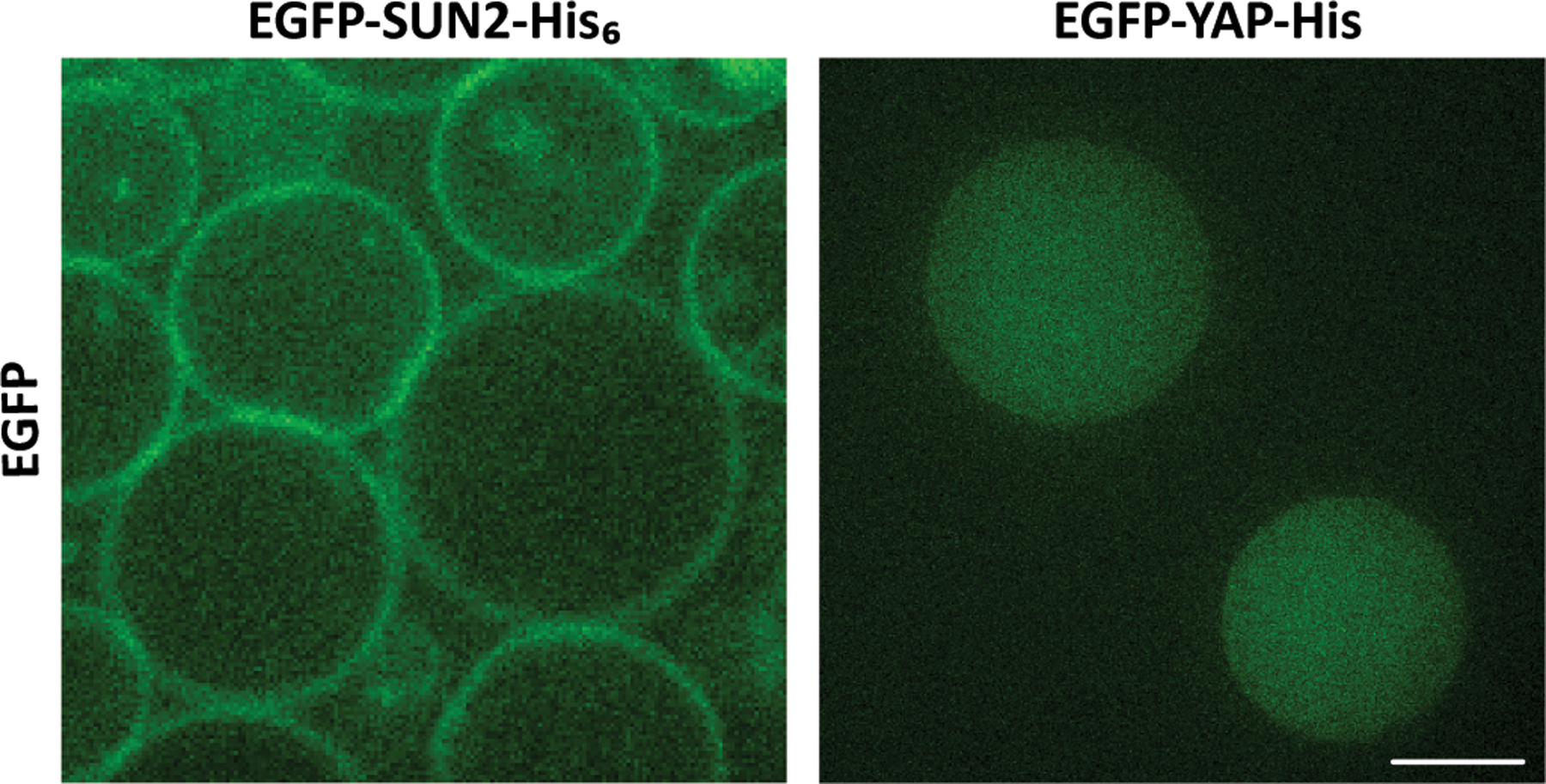
Membrane protein SUN2 localizes to the lipid bilayer vesicle membrane whereas YAP is uniformly distributed inside vesicles. Scale bar: 10 µm.
Footnotes
The use of high-quality fresh plasmid preparations is strongly encouraged.
Transfer the cryotube to the tissue culture hood when a small piece of ice is still present in the tube. It will thaw before starting the procedure.
If the initial stock concentration of cells is low, cells can be grown in a 100 mm petri dish and split when they reach ~100% confluency to two 150 mm petri dishes.
Do not let the cells overgrow, as their metabolic state may change and lower the transcription/translation efficiency of the lysate.
It is important to keep the “cold chain” in this and the following steps. Centrifuge at 4 °C if possible, keep the solutions and the tubes in ice.
It is crucial that no air bubbles are present during the bead-beating step. When the bead-beating tube is full, tap the tube on a hard surface to make the bubbles emerge to the surface, and pop the bubbles with a needle if needed. Then place a small amount of solution in the cap of the tube, tightly close the assembly, and tap on a hard surface. If bubbles appear, unscrew the cap and repeat the process.
Repeated freeze-thaw cycles severely impair the transcription/translation efficiency of the lysate. Ideally, a single aliquot should be used for no more than 2 – 3 experiments. Plan the aliquot volume accordingly.
To ensure consistency in the results in the case of multiple reactions, consider preparing a master mix. Be aware that the lysate efficiency varies from batch to batch and may be dependent on other factors such as the quality of the plasmid, the purity of T7 and GADD34, and the absence of RNase in the solutions.
Depending on the fluorescence reporter, the incubation time can vary. For instance, due to the slower folding of mCherry, CFE reactions synthesizing the protein of interest fused with mCherry take a longer time to reach completion.
We used a final DNA concentration of 5 nM in the CFE reactions. Performing a DNA titration assay to obtain the optimal concentration of the DNA to have the highest reaction efficiency is recommended.
After fractionation, the pellet fraction is not visible. Therefore, it is crucial to carefully recover the supernatant without disturbing the pellet.
Even though we have recommended the addition of the beads directly to the CFE reaction in this protocol, one can add beads to each fraction to assess the membrane incorporation efficiency of the soluble fraction of cell-free synthesized proteins.
For quantification purposes, one can use a fluorescent reporter such as EGFP with a certain concentration in the SDS-PAGE to quantify the amount of translocated cell-free produced membrane proteins.
This lipid composition mimics the composition of nuclear membrane that has been used for SUN protein reconstitution. One can use other lipid compositions as desired.
Alternatively, one can use an Avanti mini-extruder with a 100 nm filter.
The pellet is visible and if it is hard to see, make sure to flick the tube enough to resuspend the beads homogeneously, and repeat the centrifugation.
SUPER templates should be made fresh right before adding to the CFE reaction.
The difference of the osmolarity between the OS and encapsulated CFE reaction should be less than 50 mOsm. If the osmolarity of the OS is larger than the encapsulated CFE reaction, the vesicles shrink. On the other hand, when the osmolarity of the OS is smaller than the encapsulated CFE reaction, the vesicles might burst. As compared to encapsulated CFE systems, a bit larger osmolarity of the OS is better than less osmolarity.
The LOS can be used immediately or stored at 4 °C for a maximum of 2 days.
The composition and concentration of lipids are critical for the reconstitution of membrane proteins. The insertion (solubilization) efficiency and quality of membrane protein can depend on the lipid composition of the provided membrane with a preference for negatively charged lipids.
The density of the encapsulated solution inside vesicles should be a bit larger than the OS so that the vesicles can settle down to facilitate imaging. The addition of 5% Optiprep, a density gradient medium, is helpful.
For the use of cDICE, there are some crucial factors that could determine the success of the formation of lipid bilayer vesicles. First, the thickness of the LOS layer and its corresponding flight time should be large enough so that the lipids from LOS have enough time to saturate the surface of droplets. Second, the inertia of the droplets should not significantly deform the interface and the slight density difference between the encapsulated solution and the outer fluid is needed to help the vesicles to detach from the lipid monolayer.
To collect vesicles, tilt the 3D-printed chamber at 45 degrees and the interface between LOS and OS with vesicles should be clear. When collecting vesicles, try to avoid taking the oil layer on top of the OS; otherwise, oil droplets will obstruct imaging.
Contributor Information
Hossein Moghimianavval, University of Michigan, Ann Arbor, MI..
Yen-Yu Hsu, University of Michigan, Ann Arbor, MI..
Alessandro Groaz, Baylor College of Medicine, Houston, TX..
Allen P. Liu, University of Michigan, Ann Arbor, MI..
References
- 1.Laohakunakorn N, Grasemann L, Lavickova B, et al. (2020), Bottom-Up Construction of Complex Biomolecular Systems With Cell-Free Synthetic Biology, https://pubmed.ncbi.nlm.nih.gov/32266240/ [DOI] [PMC free article] [PubMed]
- 2.Wingfield PT (2015) Overview of the purification of recombinant proteins. Curr Protoc Protein Sci 2015:6.1.1–6.1.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mancia F and Love J (2010) High-throughput expression and purification of membrane proteins. J Struct Biol 172:85–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knol J, Sjollema K, and Poolman B (1998) Detergent-mediated reconstitution of membrane proteins. Biochemistry 37:16410–16415 [DOI] [PubMed] [Google Scholar]
- 5.Rigaud J-LL and Lévy D (2003) Reconstitution of Membrane Proteins into Liposomes, Academic Press Inc. [DOI] [PubMed] [Google Scholar]
- 6.Noireaux V and Liu AP (2020) The New Age of Cell-Free Biology. Annu Rev Biomed Eng 22:51–77 [DOI] [PubMed] [Google Scholar]
- 7.Chong S (2014) Overview of cell-free protein synthesis: Historic landmarks, commercial systems, and expanding applications. Curr Protoc Mol Biol 2014:16.30.1–16.30.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu Y (2017) Cell-free synthetic biology: Engineering in an open world, KeAi Communications Co. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez JG, Stark JC, and Jewett MC (2016) Cell-free synthetic biology: Engineering beyond the cell. Cold Spring Harb Perspect Biol 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khambhati K, Bhattacharjee G, Gohil N, et al. (2019), Exploring the Potential of Cell-Free Protein Synthesis for Extending the Abilities of Biological Systems, https://pubmed.ncbi.nlm.nih.gov/31681738/ [DOI] [PMC free article] [PubMed]
- 11.Zemella A, Thoring L, Hoffmeister C, et al. (2015) Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems. 16:2420–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gregorio NE, Levine MZ, and Oza JP (2019), A user’s guide to cell-free protein synthesis, https://pubmed.ncbi.nlm.nih.gov/31164605/ [DOI] [PMC free article] [PubMed]
- 13.Dondapati SK, Kreir M, Quast RB, et al. (2014) Membrane assembly of the functional KcsA potassium channel in a vesicle-based eukaryotic cell-free translation system. Biosens Bioelectron 59:174–183 [DOI] [PubMed] [Google Scholar]
- 14.Komiya M, Kato M, Tadaki D, et al. (2020), Advances in Artificial Cell Membrane Systems as a Platform for Reconstituting Ion Channels, https://pubmed.ncbi.nlm.nih.gov/31944562/ [DOI] [PubMed]
- 15.Demarche S, Sugihara K, Zambelli T, et al. (2011) Techniques for recording reconstituted ion channels. Analyst 136:1077–1089 [DOI] [PubMed] [Google Scholar]
- 16.Bashirzadeh Y and Liu AP (2019) Encapsulation of the cytoskeleton: Towards mimicking the mechanics of a cell. Soft Matter 15:8425–8436 [DOI] [PubMed] [Google Scholar]
- 17.Bashirzadeh Y, Wubshet NH, and Liu AP (2020) Confinement Geometry Tunes Fascin-Actin Bundle Structures and Consequently the Shape of a Lipid Bilayer Vesicle. Front Mol Biosci 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bashirzadeh Y, Redford SA, Lorpaiboon C, et al. (2020) Actin crosslinker competition and sorting drive emergent GUV size-dependent actin network architecture. 2020.10.03.322354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groaz A, Moghimianavval H, Tavella F, et al. (2020), Engineering spatiotemporal organization and dynamics in synthetic cells, https://pubmed.ncbi.nlm.nih.gov/33219745/ [DOI] [PMC free article] [PubMed]
- 20.Majumder S, Garamella J, Wang YL, et al. (2017) Cell-sized mechanosensitive and biosensing compartment programmed with DNA. Chem Commun 53:7349–7352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majumder S, Willey PT, DeNies MS, et al. (2019) A synthetic biology platform for the reconstitution and mechanistic dissection of LINC complex assembly. J Cell Sci 132:234153. [DOI] [PubMed] [Google Scholar]
- 22.Neumann S, Pucadyil TJ, and Schmid SL (2013) Analyzing membrane remodeling and fission using supported bilayers with excess membrane reservoir. Nat Protoc 8:213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mikami S, Kobayashi T, and Imataka H (2010) Cell-free protein synthesis systems with extracts from cultured human cells. Methods Mol Biol 607:43–52 [DOI] [PubMed] [Google Scholar]
- 24.Abkarian M, Loiseau E, and Massiera G (2011) Continuous droplet interface crossing encapsulation (cDICE) for high throughput monodisperse vesicle design. Soft Matter 7:4610–4614 [Google Scholar]