

Abstract
Necrotizing enterocolitis (NEC) is a devastating disease of premature infants whose pathogenesis remains incompletely understood, although activation of the gram-negative bacterial receptor Toll-like receptor-4 (TLR4) on the intestinal epithelium plays a critical role. Patients with NEC typically display gastrointestinal dysmotility before systemic disease is manifest, suggesting dysmotility could drive NEC development. Both intestinal motility and inflammation are governed by the enteric nervous system, a network of enteric neurons and glia. We hypothesized here that enteric glia loss in the premature intestine could lead to dysmotility, exaggerated TLR4 signaling and NEC development. We found that intestinal motility is reduced early in NEC in mice, preceding the onset of intestinal inflammation, whereas pharmacologic restoration of intestinal motility reduced NEC severity. Ileal samples from mouse, piglet and human NEC presented enteric glia depletion, and three strains of glia-deficient mice (Plp1ΔDTR, Sox10ΔDTR, BdnfΔDTR) showed increased NEC severity compared with wildtype control animals. Mice lacking TLR4 on enteric glia (Sox10-Tlr4ko) did not show NEC-induced enteric glia depletion and were protected from NEC. Mechanistically, brain-derived neurotrophic factor (BDNF) from enteric glia restrained TLR4 signaling on the intestine to prevent NEC. BDNF was reduced in mouse and human NEC, and BDNF administration reduced both TLR4 signaling and NEC severity in enteric glia-deficient mice. Finally, we identified an agent (J11) that enhanced enteric glial BDNF release, inhibited intestinal TLR4, restored motility and prevented NEC in mice. Thus, enteric glia loss might contribute to NEC through intestinal dysmotility and increased TLR4 activation, suggesting enteric glia therapies for this disorder.
One Sentence Summary:
TLR4-induced enteric glia loss leads to experimental NEC through BDNF loss, which can be treated with by compound J11.
Introduction
Necrotizing enterocolitis (NEC) is a devastating disease of premature infants that is characterized by the acute development of intestinal necrosis (1), leading to early death in up to a third of patients (2, 3). The precise mechanisms that lead to NEC remain incompletely understood, although current thinking suggests that proinflammatory signaling in response to colonizing microbes within the intestine of the premature host plays a key role in its development (4). The gram-negative bacterial receptor Toll-like receptor 4 (TLR4) (5) plays a critical role in the pathogenesis of NEC (6) by inducing intestinal epithelial cell death (7–10), the recruitment of pro-inflammatory leukocytes (11–13), and the development of intestinal ischemia (14) in response to luminal bacteria. However, the upstream factors that initiate this pro-inflammatory TLR4-mediated signaling cascade within the mucosa of the premature intestine remain largely unexplored.
In seeking to understand the proinflammatory signaling responses leading to NEC, we now draw attention to the clinical symptoms that develop before full clinical manifestations of NEC; patients universally display a period of gastrointestinal dysmotility, which is manifest by the combination of abdominal distention, feeding intolerance and emesis (15, 16). Gastrointestinal motility is normally governed by the enteric nervous system (ENS), a network of neurons and enteric glia which not only coordinate peristalsis, but have also roles in the regulation of intestinal inflammation (17, 18). The enteric glia are abundant cells within the intestinal mucosa, where they both surround and support the enteric neurons (19), and express the markers S100 calcium-binding protein B (S100B), glial fibrillary acidic protein (GFAP), and Sex determining region Y (SRY) transcription factor 10 (Sox10) (20, 21). Current dogma suggests that the impaired motility seen in patients with NEC is a consequence of NEC, rather than a factor in disease development (22). However, the finding that intestinal dysmotility is often seen before any systemic manifestations of NEC are present (23) raises the intriguing possibility that abnormalities in the enteric nervous system could represent a previously unrecognized trigger for NEC development.
Here, we tested the hypothesis that impairments in the enteric nervous system of the premature gut could play critical roles in the development of NEC. In support of this hypothesis, we showed that NEC development in mice requires the TLR4-mediated disruption of the enteric glia of the newborn intestine, which results in both impaired intestinal motility, and a loss of a critical anti-TLR4 feedback loop in the premature gut through the impaired release of brain-derived neurotropic factor (BDNF). These findings provide insights into the role of the enteric nervous system in NEC development, and reveal that enteric glia-based strategies may offer approaches for the management of patients with this devastating disease.
Results
Impaired gastrointestinal motility and enteric glia loss occur early in the development of NEC.
Shown in Fig. 1, A and B are representative abdominal radiographs of age-matched infants without (Fig. 1, A) and with NEC (Fig. 1, B). These images reveal the presence of intestinal dysmotility as manifest by intestinal dilation (arrows in Fig. 1, B) in infants with NEC that was not seen in infants without NEC. A similar radiographic appearance of the intestine was seen in mice at the end of a four-day NEC model (Fig. 1, C and D), in which NEC was induced in 7 day old mice using a combination of formula gavage (see Materials and Methods for details), intermittent hypoxia, and administration of stool from an infant with severe NEC, as we have previously described and validated (fig. S1). (24).
Fig. 1. Impaired gastrointestinal motility and enteric glia loss are early events in the development of NEC.
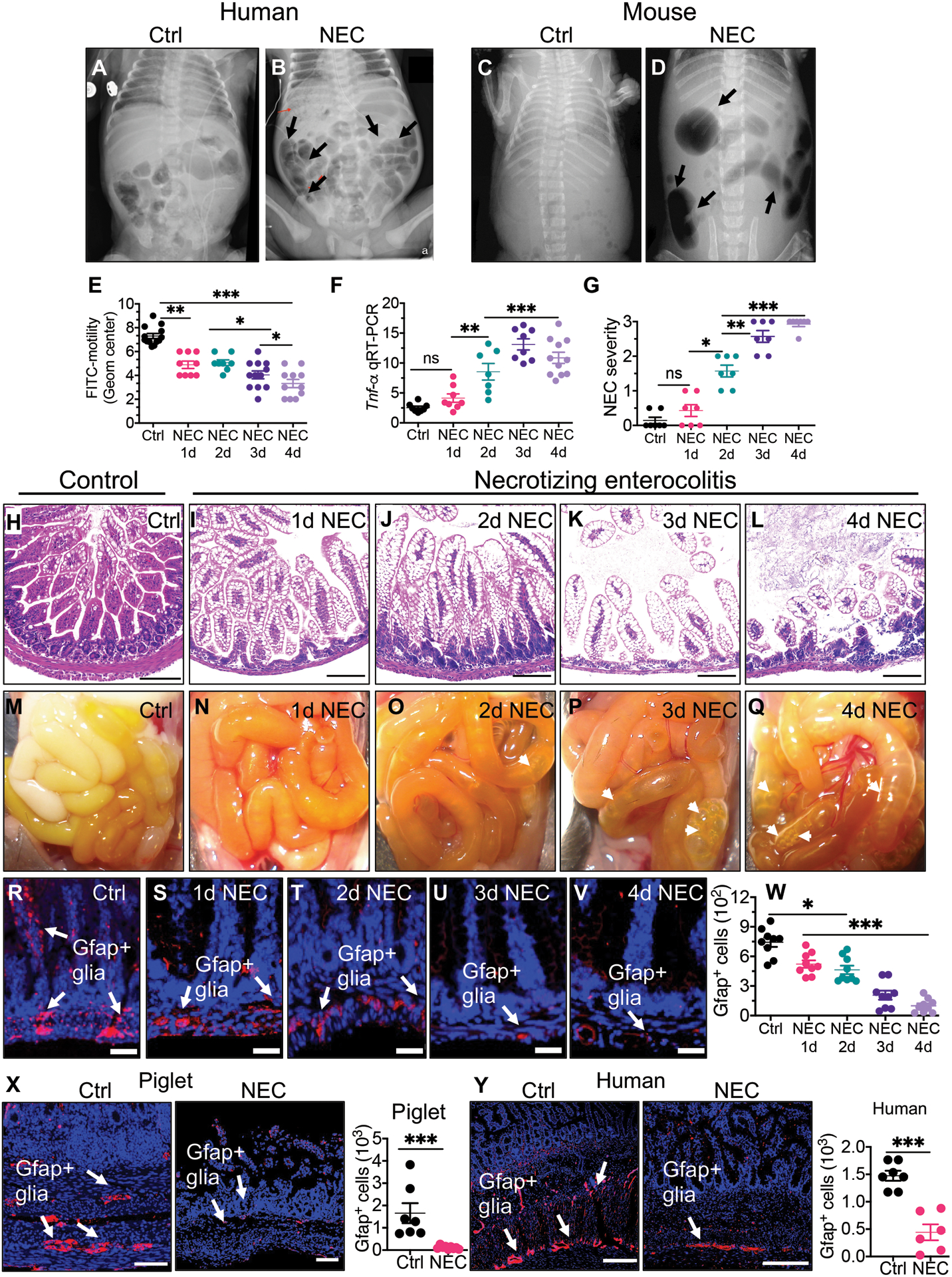
(A to B) representative abdominal radiographs of age matched infant without (A) and with NEC (B); representative abdominal radiographs of mice without (C) and with NEC (D). Arrows show areas of bowel dilation in NEC. (E) Dot plot chart of FITC-dextran transit time from stomach to the colon as an indicator of intestinal motility (n>8 per group). (F) qRT-PCR expression of Tnf-α in the intestinal mucosa in control mice and on each day of the NEC model (n>8 per group), (G) NEC severity score (n=7 per group), (H to L) representative H&E-stained histological images showing the intestinal mucosa of the terminal ileum in control mice and on each day of the NEC model. (M to Q) gross images of the small bowel of control mice and on each day of the NEC model (white arrowheads show intestinal edema and air-filled loops of bowel). (R to V) representative confocal images of the terminal ileum of control mice and mice in the NEC model, stained for DAPI (blue) and the glial marker GFAP (red, white arrows) (W) quantification of GFAP+ cells, of mice without NEC (Ctrl) and each day of the four-day NEC model (n=9 per group). (X, Y) representative confocal images of glial marker protein GFAP and quantification of GFAP+ cells in small intestine of piglets (n≥7 per group) and humans (n=7 ctrl specimens and n=6 NEC specimens) in the absence (Ctrl) and presence (NEC) of NEC as shown; quantification is provided in which each dot in dot graphs represents data from individual mice, piglets or human specimen. Statistical significance was determined by one-way ANOVA, followed by Tukey’s multiple comparisons tests or unpaired t-test using GraphPad prism 9 software. *p<0·05, **p<0·01, ***p<0·001. Scale bars: 100μm panels H to L, X, Y and 25μm panels R to V. Mouse and piglet experiments were repeated three times with at least 5–6 mice per group and 2–3 piglets/group in each experiment. Data of 6–7 de-identified control and NEC human specimens were used for quantification.
The induction of NEC in mice resulted in the progressive impairment in gut motility over the four-day model, as revealed by the impaired passaged of orally administered FITC-dextran throughout the gastrointestinal tract (Fig. 1, E). The initial reduction in motility was accompanied by an increase in NEC severity, as measured by an increase in the expression of Tnf-α in the intestinal epithelium (Fig. 1, F), and a parallel increase in mucosal injury as reflected in the daily increase in the NEC severity score (Fig. 1, G to L). The progressive development of intestinal injury was also observed on gross inspection of the small intestine, as revealed by edema and air-filled loops of bowel (Fig. 1, M to Q), consistent with the x-ray findings (Fig. 1, C and D).
To interrogate the link between impaired motility and the progression of NEC, we first focused on a potential role for the enteric glia, which have critical roles in both the regulation of intestinal motility and inflammation (25). As shown in Fig. 1, R, S, enteric glia in the submucosal layer of the intestine were markedly reduced on the first day of the mouse NEC model (Fig. 1, W), and then decreased further as measured on the final day of the model (Fig. 1, T to W, fig. S2). Neither enteric neurons nor interstitial cells of Cajal were depleted in NEC, indicating that the cell loss was limited to enteric glia (fig. S3). The reduction in enteric glia corresponded with the loss in intestinal motility (Fig. 1, E) and preceded the development of both intestinal inflammation (Fig. 1, F to G) and epithelial injury (Fig. 1, H to J), raising the possibility that the reduction in enteric glia could predispose to the development of NEC. Myenteric glia were also reduced in mice with NEC, potentially contributing to the impaired motility (fig. S4).
Finally, we sought to assess the number of enteric glia in the small intestinal mucosa of both piglets and humans with and without NEC. We induced NEC in premature piglets at 92% gestation as described in Methods, resulting in intestinal inflammation that resembles that seen in mouse and human NEC (fig. S1). As shown in Fig. 1, X, Y, the quantity of enteric glia was significantly reduced in NEC as compared with piglets (p<0.001) and human infants (p<0.001) without NEC, consistent with the reduction in enteric glia that we observed in mice with NEC (P<0.05 to 0.001; Fig. 1, R to W). Myenteric glia were also reduced in piglets with NEC, in a pattern that resembled the reduction in human infants with NEC, which confirms the findings seen in mice (fig. S4).
Taken together, these data indicate that a loss of enteric glia is generalizable across species and precedes intestinal injury in NEC. We therefore next sought to investigate whether reduced intestinal motility and the loss of enteric glia could drive NEC development.
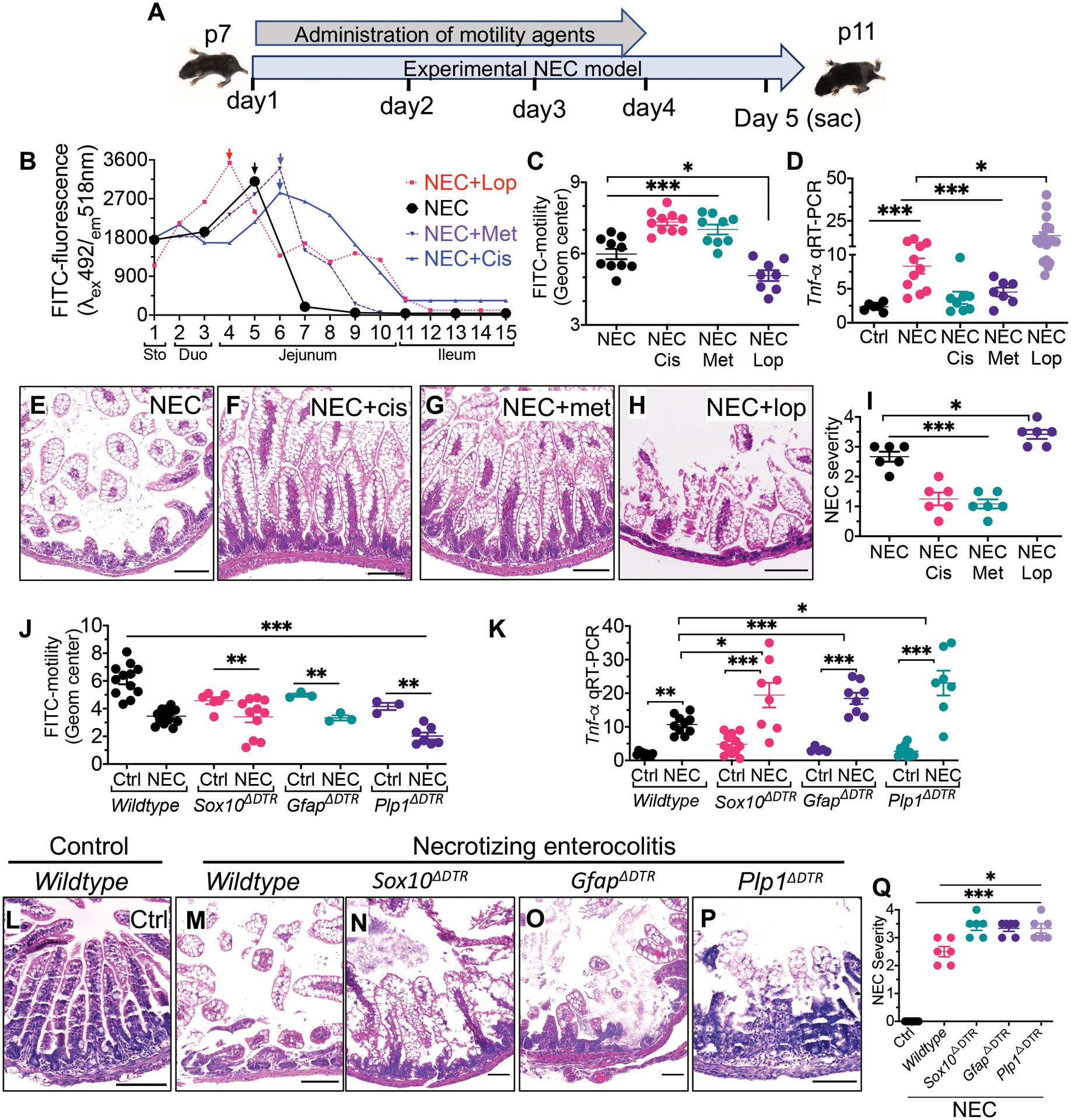
Administration of prokinetic agents reduce the severity of NEC in mice, whereas mice lacking enteric glia show increased NEC severity.
We next sought to investigate whether impaired gastrointestinal motility could be a predisposing factor in the development of NEC in mice. To do so, we treated mice with the pro-kinetic agents Cisapride (which acts as a serotonin 5-HT4 receptor agonist) and Metoclopramide (which acts as a dopamine D2 receptor antagonist), or the anti-kinetic agent Loperamide (which is a μ-opioid receptor agonist) (26) prior to and during NEC development in the mouse model (see scheme in Fig. 2, A). The effects of these motility agents on the passage of FITC-dextran in mice with NEC is shown in Fig. 2, B, C, in which loperamide delayed the passage of FITC-dextran, whereas Cisapride and Metoclopramide each accelerated FITC-dextran passage. Administration of the prokinetic agents Cisapride and Metoclopramide both significantly reduced NEC severity, as revealed by reduced mucosal Tnf-α expression (P<0.001; Fig. 2, D), improved histology (Fig. 2, E to G) and reduced NEC severity score (P<0.001; Fig. 2, I). On the contrary, the treatment of newborn mice with the anti-kinetic agent loperamide significantly increased NEC severity (P<0.05; Fig. 2, D, H, I). Taken together, these findings suggest that impaired motility could drive NEC development.
Fig. 2. Administration of prokinetic agents reduce the severity of NEC in mice whereas mice lacking enteric glia show increased NEC severity.
(A) a schematic illustration of the experimental protocol for the administration of motility agents in the NEC model in mice. (B) measurement of intestinal motility as determined by the peak fluorescence intensity of FITC-dextran that had been administered orally 30 minutes previously to wild-type mice that had been induced to develop NEC (black) along with the administration of the anti-kinetic agent Loperamide (red), or the pro-kinetic agents Metoclopramide (purple) and Cisapride (green); the anatomic location corelating to the segment of the intestine in which fluorescence is measured is shown on the x-axis (“sto” – stomach; “duo” – duodenum). Arrows show the point of maximum fluorescence (C) quantification of the geometric center corresponding to the FITC-dextran transit time (n≥8 mice/group), (D) qRT-PCR expression of Tnf-α in the indicated group (“Cis” – Cisapride, “Met” – Metoclopramide, “Lop” – loperamide (n≥6 mice/group). (E to H) representative histological images, and (I) NEC severity score (n=6 histological samples scored/group), in mice induced to develop NEC in the absence or presence of pro-kinetic (Cisapride, Metoclopramide) and anti-kinetic (loperamide) agents. (J) measurement of intestinal motility in three separate enteric glial-deficient strains (Sox10 ΔDTR,GfapΔDTR and Plp1ΔDTR) of mice at baseline and after induction of NEC (n≥ 3 enteric glial-deficient and n≥12 mice/group). (K) qRT-PCR expression of Tnf-α in the intestinal mucosa of the indicated mouse strain (n≥5 mice/strain/group) in the absence or presence of NEC as indicated (L to P) representative H&E-stained photomicrographs of the terminal ileum in mice that are either control (L) or induced to develop NEC (M to P) in the indicated strain. (Q) NEC severity scores for the indicated strain (n=6 mice/strain/group). Each dot represents data from individual mice, piglet or human specimens. Statistical significance was determined by one-way ANOVA, followed by Tukey’s multiple comparisons tests using GraphPad prism 9 software. *p<0·05, **p<0·01, ***p<0·001. Scale bar:100μm. Mice experiments were repeated three times with at least 5–6 mice per group in each experiment. For knockout mutant mice experiments, the littermates of wild-type genotypes were used and underwent identical treatments with diphtheria toxin and tamoxifen.
To determine whether the loss of enteric glia could play a causative role in the development of NEC, we generated three separate enteric glial-deficient strains of mice (Sox10ΔDTR, GfapΔDTR, and Plp1ΔDTR) using the breeding schemes described in fig. S5. Each strain showed the anticipated lack of enteric glia (fig. S5) and displayed significant impairment in motility at baseline as compared with wild-type mice (P<0.01; Fig. 2, J). We also note that in the Plp1ΔDTR strain, the female mice had reduced baseline motility as compared with male mice (P<0.05; fig. S6), consistent with previous findings using a similar model (27). We note that there was no evidence of loss of enteric neurons, nor any other intestinal epithelial cell types, in any of the glia-deficient strains that we generated (fig. S7). As shown in Fig. 2, K to Q, NEC severity was significantly greater in each of the glial-deficient mouse strains as compared with wild-type mice, as revealed by increased Tnf-α expression in the intestinal epithelium (P<0.01; Fig. 2, K), greater histologic injury (Fig. 2, L to P) and greater NEC severity scores (P<0.05; Fig. 2, Q). The greater severity of NEC in each of the glial-deficient strains was also associated with greater impairment in motility, as revealed by significantly impaired FITC passage (P<0.01; Fig. 2, J). Taken together, these findings indicate that enteric glia loss worsens NEC-induced inflammation.
Toll-like receptor 4-dependent loss of enteric glia loss is required for the development of NEC in mice
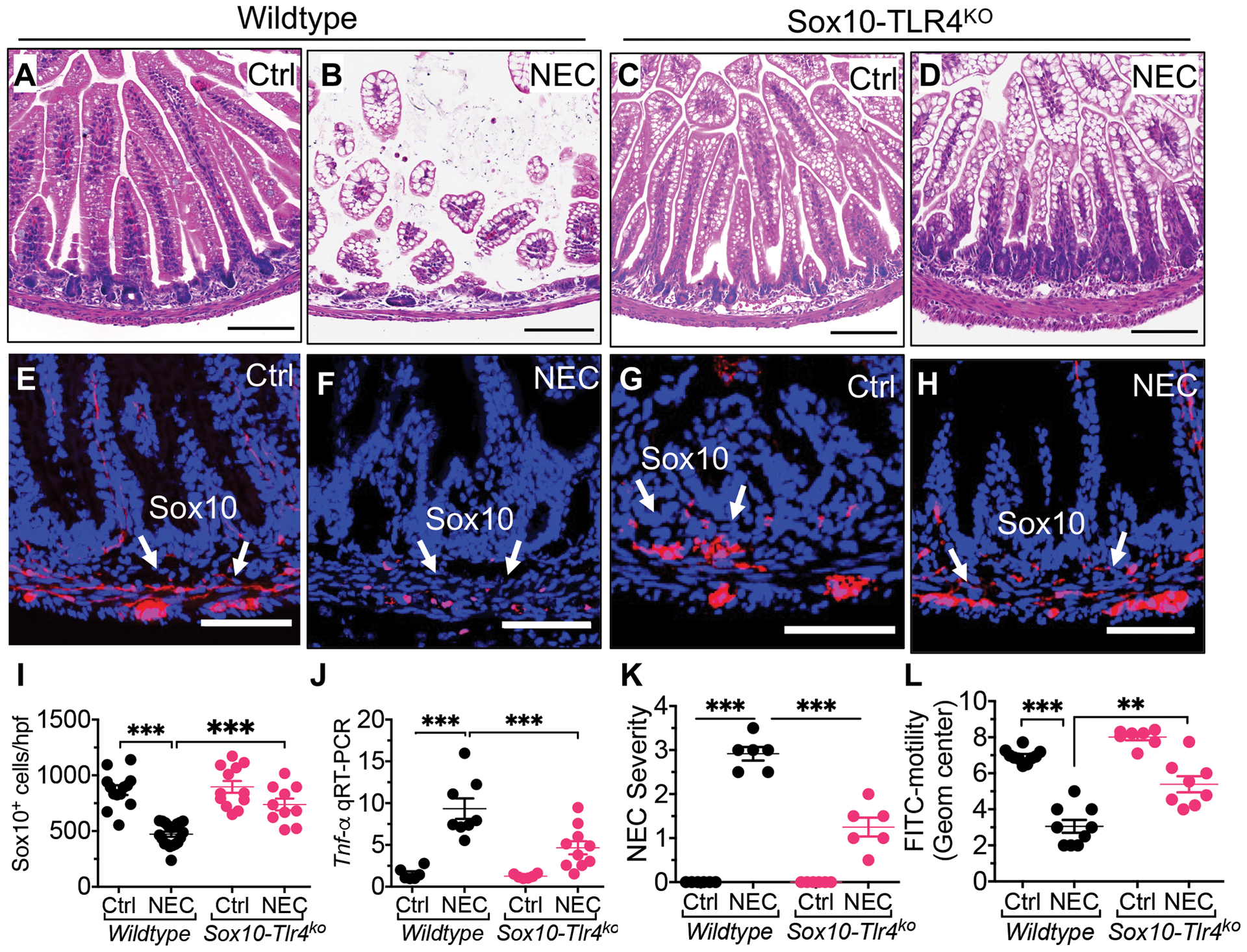
Given that enteric glia express TLR4 (28), we therefore next considered whether TLR4 activation on the enteric glia could lead to enteric glial loss, and if so, whether this loss was required for NEC induction. To evaluate a potential role for TLR4-mediated enteric glia loss in NEC induction, we next generated mice that lack TLR4 on the enteric glia, herein called Sox10-TLR4ko mice as described in Methods and shown in fig. S8. Wild-type mice that were exposed to the NEC model showed intestinal mucosal injury (Fig. 3, A, B), enteric glia loss (Fig. 3, E, F, I to K) and impaired motility (Fig. 3, L) consistent with NEC (Fig. 3, L), as expected. However, Sox10-TLR4ko mice that were exposed to the NEC model retained their enteric glia (Fig. 3, G to H, I) and showed reduced NEC severity (Fig. 3, C, D, J, K). The Sox10-TLR4ko mice also preserved their motility when induced to develop NEC (Fig. 3, L), illustrating the importance of TLR4 signaling on the enteric glia in mediating the dysmotility seen in NEC. These findings indicate that TLR4 signaling on the enteric glia plays a critical role in enteric glia loss, and importantly, that enteric glia loss is required for the development of NEC in this mouse model.
Fig. 3. Toll-like receptor 4 dependent loss of enteric glia loss is required for the development of NEC in mice.
(A to D) representative H&E-stained photomicrographs of wild-type and Sox10-Tlr4ko mice that are either breast fed and allowed to stay with the dam (“Ctrl”) or induced to develop NEC (“NEC”). (E to H) Representative confocal images of terminal ileum of wild-type and Sox10-Tlr4ko mice that were either control or induced to develop NEC; sections were stained for DAPI (blue) and enteric glia (Sox10, red). (I) quantification of Sox10 immunofluorescence in wild type and Sox10-TLR4ko intestinal sections corresponding to E to H, reflecting the abundance of enteric glia (n≥10 sections/group). (J) qRT-PCR expression of Tnf-α in the intestinal mucosa of the indicated group (n≥6 mice/group) (K) NEC severity scores corresponding to E to H (n=6 histological sections scored/group). (L) Intestinal motility in wild-type and Sox10-TLR4ko mice with and without NEC as indicated (n≥7 mice/group); Each dot represents data from individual mice. Statistical significance was determined by one-way ANOVA, followed by Tukey’s multiple comparisons tests using GraphPad prism 9 software. ***p<0·001. Scale bars: 100μm on panels A to D, and 50μm on panels E to H. Mouse experiments were repeated three times with at least 3–4 mice per group in each experiment.
The release of BDNF from the enteric glia restrains TLR4 signaling and prevents NEC development
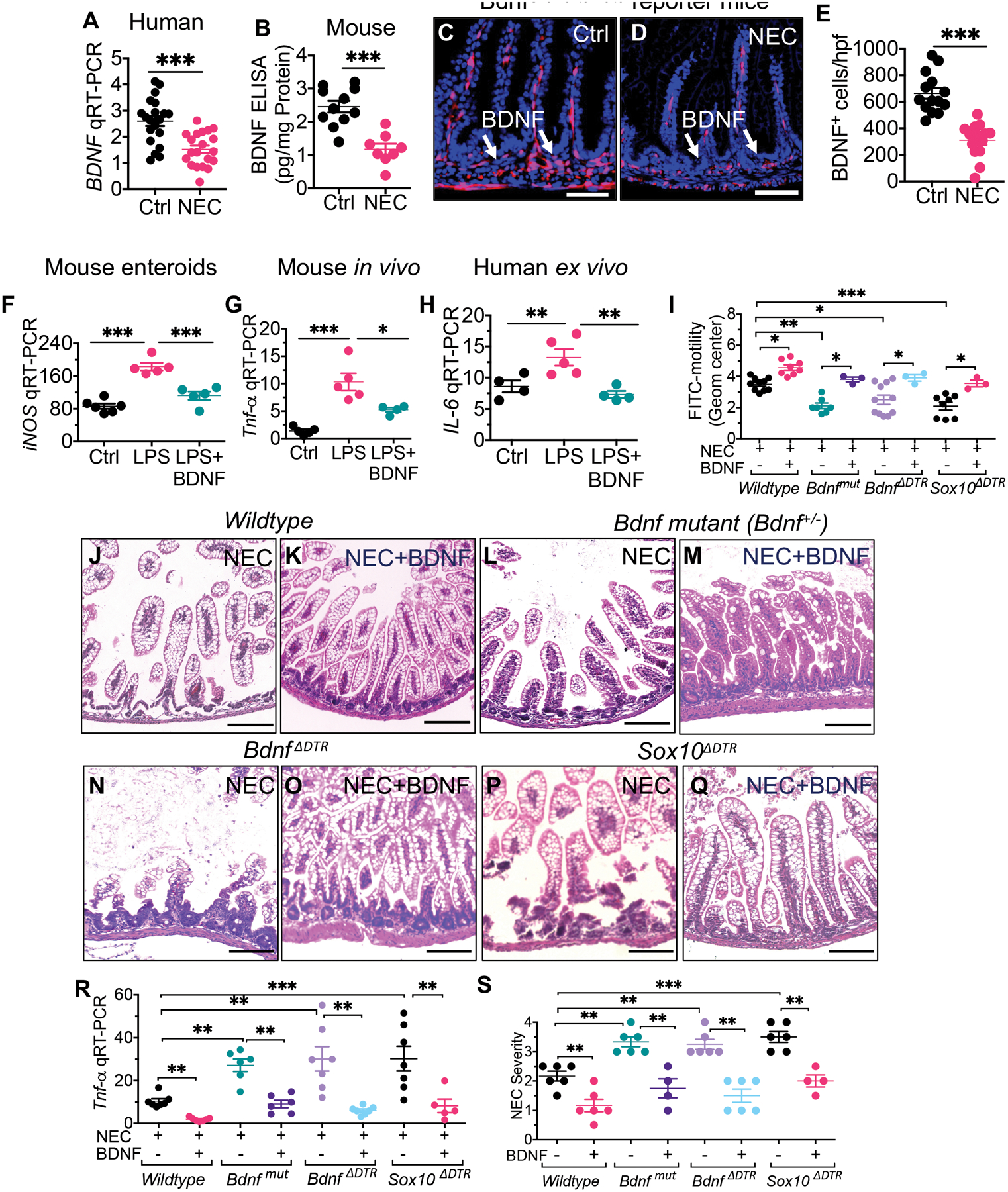
Having shown that the development of NEC requires an initial TLR4-dependent loss of enteric glia, we next explored potential enteric glia-derived factors whose loss could result in NEC development. To do so, we focused on brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family of growth factors that is constitutively released from the enteric glia (29, 30), and which has anti-inflammatory properties (31). The expression of BDNF was reduced in intestinal tissue that was removed from infants with NEC as compared with controls (P<0.001; Fig. 4, A), and in mouse NEC as compared with control tissue as revealed by ELISA (P<0.001; Fig. 4, B). In addition, BDNF+ cells were reduced in a Bdnf-tomato reporter mouse strain with NEC (Fig 4, C to E), consistent with the ELISA findings in patients and mice.
Fig. 4. The release of BDNF from the enteric glia restrains TLR4 signaling and prevents NEC development.
(A) qRT-PCR expression of BDNF expression in the intestine of de-identified, human specimens resected at the time of stoma closure (control) or surgical NEC (n=20 ctrl and n=20 NEC intestinal specimens). (B) Measurement of BDNF in the intestinal mucosa of mice with and without NEC by ELISA (n≥8 mouse ileal tissues/group); (C to E) representative confocal images of sections stained for DAPI (blue) and BDNF (red) in BdnfCreTd-tomato reporter mice that were either controls (C) or were induced to develop NEC (D). (E) quantification of BDNF+ cells in the indicated group (n=14 ileal histological sections/group). (F) qRT-PCR expression of iNOS in small intestinal enteroids from mice that were treated with LPS (50μg/ml, 6h) and recombinant BDNF (1μg/ml, 1h pretreatment) (n≥5 enteroids cultured wells/group). (G) qRT-PCR expression of Il-6 in the intestinal mucosa of C57Bl/6 mice that were treated with LPS (5mg/kg, 6h) and BDNF (5μg/kg, 1h prior) (n≥4 mice/group). (H) qRT-PCR expression of IL-6 in de-identified samples of the small intestine removed at surgery from infants with NEC, then treated ex-vivo with LPS (50μg/ml, 6h) and BDNF (1μg/ml, 1h pre-treatment) (n≥4 specimens/treatment group). (I) FITC-dextran motility assay in wildtype, Bdnf-deficient (“Bdnfmut”), BdnfΔDTR , and enteric glia deficient (“idtr-Sox10ΔDTR”) mouse strains that were either control or were induced to develop NEC in the absence or presence of exogenous BDNF (5μg/kg/d, 3d treatment) as indicated (n≥3 enteric glia deficient mice and n≥8 wildtype mice/group). (J to Q) representative H&E-stained micrographs of the terminal ileum of the indicated strain of mice with NEC in the absence of presence of BDNF treatment as indicated, (R) qRT-PCR expression of Tnf-α (n≥5 mice/group). (S) NEC severity scores in mice of the indicated strain that were induced to develop NEC in the absence or presence of BDNF administration as indicated (n≥5 mice/group). Each dot represents data from individual mice. Statistical significance was determined by one-way ANOVA, followed by Tukey’s multiple comparisons tests or unpaired t-test using GraphPad prism 9 software. ***p<0·001. Scale bars: 25μm on panels C to D and 100μm on panels J to Q. Mouse experiments were repeated 2–3 times with at least 3–4 mice per group for most of the experiments. For knockout mutant mouse experiments, the littermates of wild-type mice were used and underwent identical treatments with diphtheria toxin and tamoxifen.
Given that TLR4 activation on the intestinal epithelium is required for NEC development (5, 7, 9, 32), we next examined whether BDNF release from the enteric glia could reduce TLR4 signaling in the intestinal epithelium. To do so, we first treated enteroids derived from the ileum of wild-type C57-BL/6 mice (as in (24)), with LPS, in the presence or absence of recombinant BDNF. As shown in Fig. 4F, BDNF treatment reduced LPS-induced iNos expression in enteroids (P<0.001). Moreover, recombinant BDNF inhibited TLR4 signaling in vivo, as BDNF injection into newborn C57-BL/6 mice reduced LPS-induced Il-6 expression in the intestinal mucosa (P<0.05; Fig. 4, G). The human relevance of the ability of BDNF to inhibit TLR4 signaling is shown in Fig. 4H, in which BDNF reduced LPS-mediated IL-6 expression in human ileal explants ex vivo.
We next explored whether a reduction in BDNF release could account for the critical role that enteric glia loss exerts in the pathogenesis of NEC. To do so, we first administered BDNF orally to wild-type mice on each day during NEC development in mice. As shown in Fig. 4 (I to K), BDNF treatment significantly improved intestinal motility (P<0.05; Fig. 4, I) and reduced the severity of experimental NEC, as revealed by reduced mucosal injury (Fig. 4, J to K), reduced expression of Tnf-α in the intestinal mucosa (P<0.01; Fig. 4, R), and improved histological severity score (P<0.01; Fig. 4, S), as compared with saline treated mice with NEC.
Next, to assess whether a loss of BDNF could account in part for the role that enteric glia loss (Fig. 1, R to Y) plays in NEC development, we generated two strains of Bdnf-deficient mice as described in Methods, namely “Bdnf-mutant mice” (Bdnf+/−) and BdnfΔDTR mice. BDNF-deficient strains of mice developed worse NEC as revealed by greater mucosal injury (Fig. 4L, N), higher expression of Tnf-α in the intestinal mucosa (P<0.01; Fig. 4, R), and higher NEC severity scores (P<0.01; Fig. 4, S), as compared with wild-type mice with NEC. It is noteworthy that the Bdnf-deficient mice with NEC also showed impaired motility as compared with wild-type mice (P<0.05; Fig. 4, I), confirming the importance of endogenous BDNF in maintaining intestinal motility.
To directly determine whether the loss of BDNF could explain in part the critical role played by enteric glia loss in in NEC development as seen in Fig. 2 to 3, we next administered BDNF to enteric glia-deficient mice in an “add-back experiment”. Replenishing BDNF to the glia-deficient mice restored motility (Fig. 4, I), and reduced the development of NEC, as revealed by intact histology (Fig. 4, P to Q), reduced intestinal expression of Tnf-α (P<0.01; Fig. 4, R), and reduced NEC severity score (P<0.01; Fig. 4, S).
Taken in aggregate, these findings show that a reduction in BDNF release accounts for the critical role that enteric glia loss plays in the pathogenesis of experimental NEC in mice.
The identification of a molecule, J11, induces BDNF release from enteric glia, improves motility, and reduces NEC in mice and human tissue ex vivo
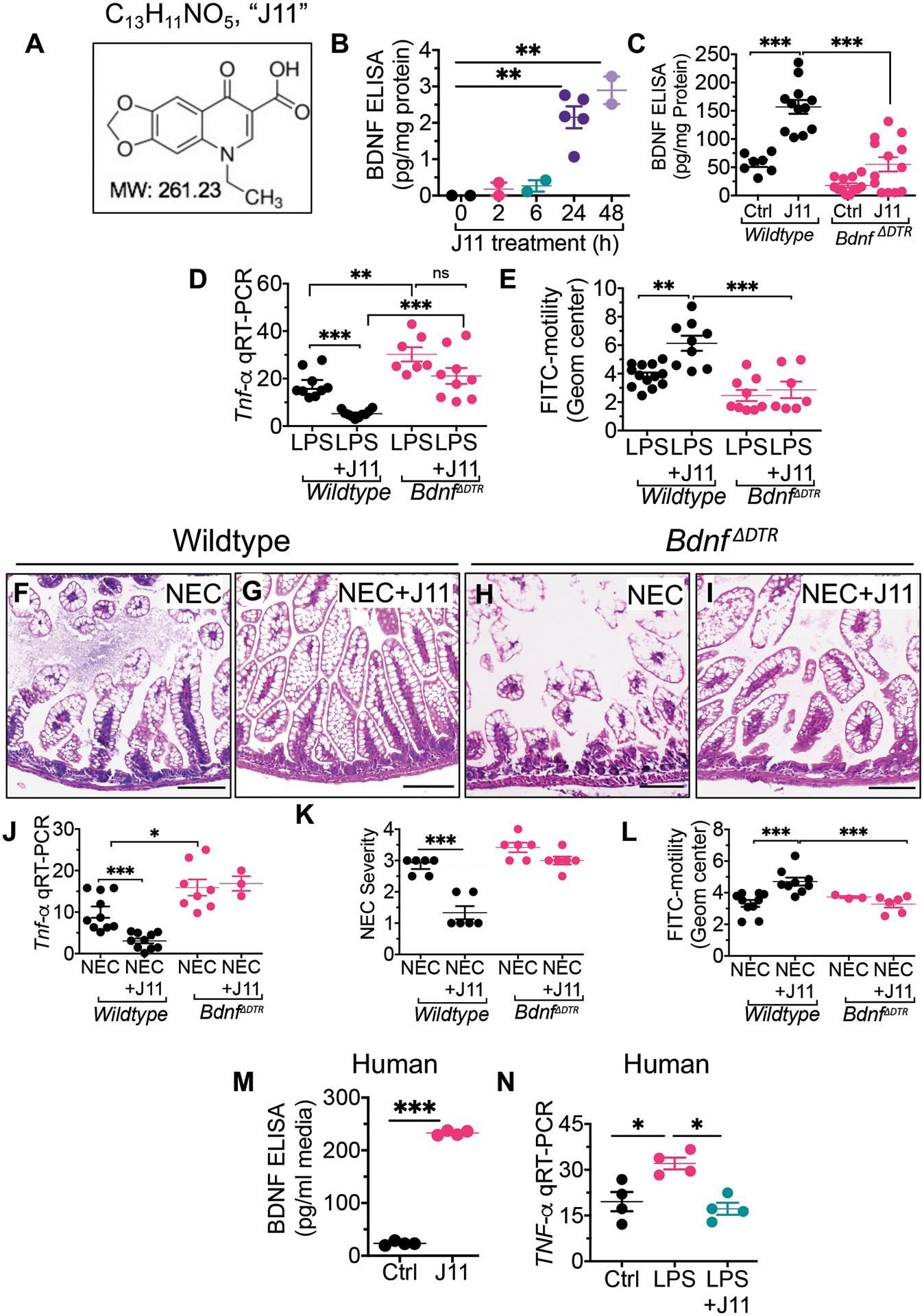
We next embarked on a strategy to identify therapeutic agents for NEC based upon their ability to enhance glial function and thus increase BDNF release. To do so, we screened several libraries of compounds for their ability to stimulate glia in culture and induce BDNF secretion into the media. Hits were then validated by their ability to reduce TLR4 signaling and attenuate NEC severity in mice. Our lead compound (Fig. 5, A) herein called “J11”, has the molecular formula C13H11NO5 and MW 261.23Da, and is oxolinic acid, a molecule that belongs to the quinolone class of antibiotics (33). As shown in Fig. 5B, J11 treatment induced the release of BDNF from cultured enteric glia, and also increased the expression of BDNF in the intestine 6 hours after oral administration to wild-type mice (Fig. 5, C). In control experiments, J11 did not induce BDNF release from BDNF-deficient mice, confirming the specificity of the BDNF measurement assay. (Fig. 5, C).
Fig. 5. Identification of a molecule, J11, able to induce BDNF release from enteric glia, improve motility, and reduce NEC in mice and human tissue ex vivo.
(A) The structural formula of “J11”, a molecular “hit” from a drug screen seeking molecules that induce BDNF from enteric glia. (B) Determination of BDNF concentration by ELISA from the enteric glia cell line EGC/PK060399egfr in which cells were treated with J11 (20μM) for the indicated time (n≥2 wells/time point) (C) Determination of BDNF concentration by ELISA in small intestinal lysates from wildtype or BdnfΔDTR mice at p11 after treatment with J11 (n≥7 mouse ileal tissues/group). (D) qRT-PCR expression of Tnf-α in ileal tissue of premature mice treated with LPS (5mg/kg) and J11 (20mg/kg) as indicated (n≥7 mouse ileal tissues/group). (E) FITC-dextran motility assay in wildtype or BdnfΔDTR mice at p11 treated with LPS (5mg/kg) and J11 (20mg/kg) as indicated (n>7 mice/group). (F to L) wildtype or BdnfΔDTR mice were induced to develop NEC in the presence or absence of J11(20mg/kg); shown at the end of the four-day model are (F to I) representative H&E images, (J) qRT-PCR expression in the intestinal epithelium of Tnf-α (n≥3 Bdnf deficient mice and n≥8 wildtype mice/group) (K) NEC severity scores (n n=6 histological sections scored/group). (L) FITC-dextran motility assay (n≥3 Bdnf deficient mice and n≥8 wildtype mice/group). (M, N) human intestinal tissue was obtained from patients undergoing surgery for NEC, were treated for 5 hours with either saline (“ctrl”) or J11 (50μg/ml) ex vivo in 10% fetal bovine containing Dulbecco’s modified growth media and assessed for (M) release of BDNF in the condition media by ELISA (n≥3 specimens/group) and (N) qRT-PCR expression of TNF-α in the tissue (n≥3 specimens/group). Each dot in dot graphs represents data from individual mice. Statistical significance was determined by one-way ANOVA, followed by Tukey’s multiple comparisons tests using GraphPad prism or unpaired t-test 9 software. *p<0·05, **p<0·01, ***p<0·001. Scale bars: 100μm. Mice experiments were repeated two-three times with at least 3–4 mice per group in each experiment. For knockout mutant mice experiments, the littermates of wild-type genotypes were used and underwent identical treatments with diphtheria toxin. Data of 4–5 de-identified resected bowl specimens at the time of stoma closure (healed NEC).
In order to assess whether J11-mediated BDNF release could reduce TLR4 signaling in the intestine, we treated wild-type mice with LPS in the presence of J11. As shown in Fig. 5D, J11 inhibited LPS-mediated induction of Tnf-α in the intestine of wild-type mice but not Bdnf−/− mice, confirming that J11 inhibits TLR4 signaling in vivo by modulating BDNF. It is noteworthy that the BDNF-deficient mice show higher inflammation after LPS treatment as compared with wild-type mice (Fig. 5D), supporting the protective role that endogenous BDNF expression has on limiting TLR4 signaling. J11 treatment improved motility in LPS-treated wild-type but not Bdnf−/− mice (P<0.01; Fig. 5, E), consistent with the fact that J11 exerted its protective effects on motility by increasing BDNF release.
In the next series of experiments, we sought to assess whether J11 could prevent NEC by enhancing BDNF release. To do so, we administered J11 within the infant formula to wild-type and Bdnf−/− mice. As shown in Fig. 5 (F to L), J11 treatment significantly reduced the severity of NEC in wild-type mice, as revealed by a reduction in mucosal injury (Fig. 5, F, G), reduced expression of Tnf-α in the intestinal mucosa (P<0.001; Fig. 5, J), reduced NEC severity score (P<0.001; Fig. 5, K) and improved small intestinal motility (P<0.001; Fig. 5, L) as compared to untreated mice with NEC. J11 did not reduce NEC severity (Fig. 5, H to K) or improve motility (Fig. 5, L) in Bdnf−/− mice, revealing that BDNF was required for the NEC protection by J11.
In the final series of experiments, we sought to determine the potential effects of J11 on reducing inflammation in intestinal tissue from human infants with NEC. To do so, we obtained intestinal samples of infants undergoing surgery for NEC, and treated them with BDNF. J11 induced the release of BDNF from the human intestine ex vivo as measured by ELISA (Fig. 5, M), and also reduced LPS-TLR4 induced TNF-α expression from the human tissue (P<0.05; Fig. 5, N).
Altogether, these findings raise the possibility that J11 may have therapeutic benefit to human infants with NEC by enhancing BDNF release and inhibiting TLR4 in the premature bowel.
Discussion
We now provide five lines of evidence that intestinal dysmotility, as a consequence of TLR4-mediated loss of the enteric glia, is a critical factor in NEC pathogenesis in preclinical models and possibly in humans: [1] Three different strains of mice that are deficient in enteric glia show impaired gastrointestinal motility and greater NEC severity than wild-type mice; [2] mice lacking TLR4 on the enteric glia (Sox10-TLR4ko mice) maintain their enteric glia and do not develop dysmotility when induced to develop NEC and are protected from NEC, indicating that enteric glia loss is required for the development of NEC; [3] The increased severity of NEC in glial-deficient mice can be reversed by the administration of BDNF, revealing that enteric glia-derived BDNF release is a critical factor in the protective role of enteric glia against NEC; [4] BDNF inhibits TLR4 signaling in the intestinal epithelium, providing a molecular explanation for the protective role of BDNF against NEC; [5] the identification of compound “J11”, also known as oxolinic acid, was found to enhance BDNF release from enteric glia, reverse dysmotility, and attenuate NEC severity in wild-type but not BDNF-deficient mice and reduce inflammation in human intestinal tissue ex vivo, thus providing proof-of-concept for the translational potential of the current studies. Taken in aggregate, these findings identify a critical role for impaired gastrointestinal motility via enteric glia loss in the pathogenesis of NEC, and identify opportunities for NEC therapies.
The current study is supported by prior work showing that mice lacking enteric glia develop fulminant jejuno-ileitis (34), that in utero inflammation is associated with enteric glia loss and intestinal inflammation in sheep (35), and that rat and human NEC are each associated with reduced enteric glia (36). Our work showing enteric glia depletion leads to NEC is also supported by earlier studies linking abnormalities in the enteric nervous system to the development of inflammatory bowel diseases (37). However, recent studies have challenged the role played by enteric glia loss in the development of intestinal inflammation, by raising concerns about epithelial toxicity of the virus-mediated enteric glia depletion strategy used (27, 34). Accordingly, in the current studies, we used three different strains of enteric glia-deficient mice, all of which display a similar phenotype of increased NEC severity. We also show that restoring BDNF to enteric glia-deficient mice (which is itself reduced in the intestines of mice and humans with NEC and in the enteric glia-deficient strains) reversed the NEC-phenotype and prevented NEC induction, thus excluding a non-specific epithelial effect of enteric glia depletion, and providing validity to the current approach.
One of the key unanswered questions in the field relates to why premature infants are at risk for developing NEC. In answering this question, previous investigators including ourselves have attributed various features of the premature bowel to NEC development, including the elevated expression of TLR4 in the intestinal mucosa, which leads to an exaggerated pro-inflammatory signaling response to colonizing microbes (11–13, 38, 39), a relative increase in endovascular tone which predisposes to intestinal ischemia (14, 40–42), and disturbed development of the microbiome and of the host immune system in the premature gut, leading to impaired adaptation to colonizing microbes and an overwhelming septic response to bacteria (40, 43, 44). Missing in these potential explanations however is a unifying model to explain how these factors could be linked in a way that leads directly to NEC. Based upon the current findings, we now propose a unifying model by identifying that enteric glia serve to restrain TLR4 signaling in the gut via the release of BDNF. As a corollary, we suggest that the premature gut is at a dual disadvantage when fed non-breast milk formula in the setting of bacterial colonization. On the one hand, the impaired motility makes digestion inefficient and potentially damaging, by preventing the intestinal lumen from efficiently clearing its contents, leading to blooms of dysbiotic bacteria which can then further activate TLR4. In parallel, the loss of enteric glia leads to unrestrained TLR4 signaling via BDNF loss, and a subsequent hyper-inflammatory response to bacteria, leading to mucosal injury and delayed repair (5). Accordingly, strategies that either enhance motility directly (including the prokinetic agents Cisapride and Metoclopramide) or restore the function of lost glia (such as the administration of BDNF), or that enhance glia function (as in the Sox10-Tlr4ko mouse or the administration of J11) were successful in attenuating NEC. As a surprising additional proof-of-concept, it is noteworthy that breast milk contains BDNF (45), which might explain the effects of breast milk on increasing gastrointestinal motility (46) and in preventing NEC (47).
There are several findings of the current work that open the door for further study. First, the fact that TLR4 signaling on enteric glia is necessary for the enteric glia loss that precedes the development of NEC suggests the importance of understanding the transcriptional profile of the bacteria that could induce TLR4 signaling on the enteric glia and lead to their death, in comparison with those bacteria that may be required for the maintenance of enteric glia survival (48, 49). Second, the precise mechanisms by which luminal bacteria activate TLR4 on the enteric glia remain unknown. To this end, enteric glia have been shown to send cellular processes which pierce from the lamina propria into the epithelium, and which might account for how TLR4 ligands on the luminal surface could activate enteric glia that are seemingly in the submucosa (50). Third, the current findings may be of relevance to Hirschsprung’s disease associated enterocolitis, a severe condition of infants in which an atonic bowel leads to intestinal inflammation and overwhelming sepsis (51, 52). Additional studies could shed light on whether administration of prokinetic agents, BDNF, or J11, may play a role in preventing this disease. Finally, the precise mechanisms by which J11 acts to enhance BDNF release and thus prevent NEC remain incompletely understood. J11 has the chemical structure of Oxolinic acid, a quinolone antibiotic that can inhibit dopamine uptake (53), which would be expected to increase gastrointestinal motility (54), suggesting additional studies are necessary to elucidate the precise mechanism of action of J11. Quinolone antibiotics can also influence motility through GABA receptors(55) or Ghrelin receptor signaling (56), and these pathways represent important targets to further investigate how J11 causes BDNF release in the protection from NEC. It is noteworthy that because J11 did not prevent mucosal inflammation or NEC in mice that lack BDNF, its mechanism of protective action for NEC is likely to mainly involve its effects on BDNF release, although these other biochemical effects of J11 could also play secondary or even synergistic effects in NEC reduction.
There are several limitations to the current work. First, although J11 was found to be effective in the prevention of NEC in the mouse model, we did not evaluate any off-target effects, nor did we perform a full toxicity profile. Along these same lines, the strategy that we employed in order to deplete the enteric glia using the iDTR system could potentially lead to unintended cytotoxic effects – either from the impact of the iDTR transgene on cell loss, or from the administration of tamoxifen, that could compound the increased NEC severity that was observed. The lack of any baseline toxicity in littermate control mice that were subjected to similar administration of tamoxifen and diphtheria toxin is reassuring.
In summary, we now identify a critical role for TLR4-induced enteric glia loss leading to dysmotility and increased inflammation, via the loss of BDNF, in the pathogenesis of experimental NEC. Moreover, our discovery of a compound J11 that can reverse these processes offers the potential for enteric glia-based approaches for the prevention and treatment of this devastating disease.
Materials and Methods
Study design
The objective of the study was to evaluate the mechanisms by which impaired intestinal motility can contribute to the pathogenesis of NEC, and to test the ability of a compound, J11, to enhance motility and prevent NEC.
All experiments and animal protocols were approved by the Johns Hopkins University in accordance with the Guide for the Care and Use of Laboratory Animals (8th Edition, The National Academies Press 2011). The goals of this study were to elucidate the pathophysiology leading to the NEC development. Using a preclinical experimental model that closely resembles the intestinal disease in mice, we correlated our findings with those present in patients and in resected intestinal specimens of infants with NEC. All mice were bred in-house to generate various mutant mouse lines as per protocol approved by the Johns Hopkins University Institutional Animal Care and Use Committee (protocol MO20M276) in accordance with the Guide for the Care and Use of Laboratory Animals and the Public Health Service Policy on Humane Care and Use of Laboratory Animals. In genetically modified mouse experiments, all mice were backcrossed at least 8 times with C57BL/6. Both genders were used in equal ratios in all experiments. For human specimens, de-identified human ileal samples were collected via waiver of consent from the Office of Human Subjects Research Review Boards at Johns Hopkins University (IRB00094036), during surgery for NEC or at the time of stoma closure.
A power analysis was performed for each experiment to detected statistically significant differences at a p value p<0.05, 6–8 animals per group is required. The precise number of animals per group is shown by dots in each figure. All data points were included for evaluation; in models of NEC, all points were included, and outliers were included in the statistical analysis and included as raw data points in each of the Figures. Randomization: pups at the same gestational age were randomized to each group without bias to size; all genders were included. Blinding: All data examination and histologic assessment was performed on samples that were blinded to the study group. Replication: The precise number of repeats varies by experiment and is included in each figure legend.
Cell culture, enteroid isolation, antibodies, and reagents
The enteroglial cell line (EGC/PK060399egfr (ATCC CRL-2690) was obtained from ATCC and maintained in Dulbecco’s Modified Eagle’s Medium (ATCC 30–2002) containing 10% fetal bovine serum (ATCC 30–2020) in a humidified 37°C CO2 incubator. Intestinal enteroids were isolated and maintained in Matrigel (Corning) as described previously (57). The enteroids were digested and passed using TrypLE Express (Gibco) weekly and used between passage 3 and 10 for all experiments.
Reagents were obtained as follows: Cisapride monohydrate (catalog no. C4740, Sigma), Loperamide hydrochloride (catalog no. L4762, Sigma), Metoclopramide hydrochloride (catalog no. M0763, Sigma), LPS from Escherichia coli 0127:B8 (catalog no. L3129, Sigma), Diphtheria Toxin (DT) (catalog no. D0564, Sigma), 4’,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (catalog no. D9542, Sigma), 70kda Fluorescein isothiocyanate–dextran (FITC-dextran) (catalog no. 46945, Sigma), Tamoxifen (catalog no. 1052910, Peprotech), recombinant BDNF (catalog no. 450–02, Peprotech), RNeasy Kit (catalog no. 74106, Qiagen), QuantiTect Reverse Transcription (catalog no. 205313, Qiagen), Anti-GFAP antibody (Catalog no. ab4674, Abcam), Sox10 antibody (catalog no. AF2864, R&D systems), s100b antibody (Catalog no. 612376, BD Bioscience), β-tubulin (Tubb3) antibody (Catalog no. ab78078, Abcam), C-kit antibody (Catalog no. MAB1356, R&D systems), p75NTR antibody (Catolog no. AB8875, Abcam), DuoSet ELISA (catalog no. DY248; R&D systems), Pierce BCA Protein Assay Kit (ThermoFisher scientific), Forward and reverse primers (Table. 1) were custom designed using NCBI Primer-BLAST online program and ordered from Integrated DNA Technologies. RNeasy kit (catalogue no. 74106; Qiagen) and QuantiTect Reverse Transcription (catalogue no. 205313; Qiagen).
Table 1.
primer sequences
| qRT-PCR primers | |
|---|---|
| BDNF (human) | GACTTCGGGAAGGTGTCTGG; ACAGGATGGGCAGAAGGTTG |
| ChgA (mouse) | AAGGTGATGAAGTGCGTCCTGGAA; AGCAGATTCTGGTGTCGCAGGATA |
| cKit (mouse) | CCTCTGGGAGCTCTTCTCCT; TGTTGGCCTTTTCAAGGGGT |
| Il6 (mouse/rat) | CCAATTTCCAATGCTCTCCT; ACCACAGTGAGGAATGTCCA |
| IL6 (human) | TCTCCACAAGCGCCTTCG; CTCAGGGCTGAGATGCCG |
| iNOS (mouse) | CTGCTGGTGGTGACAAGCACATTT; ATGTCATGAGCAAAGGCGCAGAAC |
| Ki67 (mouse) | CCAAGGCCCAAGTTTGATGC; GACTTGGCCCCGAGATGTAG |
| Lgr5 (mouse) | TGAGCGGGACCTTGAAGATTTCCT; TACCAAATAGGTGCTCACAGGGCT |
| Lyz1 (mouse) | AAGCTGGCTGACTGGGTGTGTTTA; CACTGCAATTGATCCCACAGGCAT |
| Muc2 (mouse) | TAGTGGAGATTGTGCCGCTGAAGT; AGAGCCCATCGAAGGTGACAAAGT |
| p75NTR | CCGCTGACAACCTCATTCCT; TGCTTGCAGCTGTTCCATCT |
| Tnf-α (mouse) | TTCCGAATTCACTGGAGCCTCGAA; TGCACCTCAGGGAAGAATCTGGAA |
| Tnf-α (pig) | GCCCTTCCACCAACGTTTTC; TCTGGCAAGGGCTCTTGATG |
| TNF-α (human) | GGCGTGGAGCTGAGAGATAAC; GGTGTGGGTGAGGAGCACAT |
| Tlr4 (k/d rat) | TGTTGGATGGAAAAGCCTTG; GTTCTCACTGGGCCTTAGCC |
| Tlr4 (k/o mouse) | CAGCAAAGTCCCTGATGAC; TCCAGCCACTGAAGTTCTGA |
| Tomato (Td-Tomato reporter mouse) | CACCATCGTGGAACAGTACG; GCGCATGAACTCTTTGATGA |
| Rplp0 | GGCGACCTGGAAGTCCAACT; CCATCAGCACCACAGCCTTC |
| Vil1 (mouse) | AAATTGCAGCCTCGGCGTATCAAG; GGCACAGGCTCCAAGTTGTTCTTT |
| Genotyping primers | |
| Bdnf2lox mouse | TGTGATTGTGTT TCTGGTGAC; GCCTTCATGCAACCGAAGTATG |
| Bdnfmutant mouse | ATGAAAGAAGTAAACGTCCAC; CCAGCAGAAAGA GTAGAGGAG; GGGAACTTCCTGACTAGGGG |
| Cre-transgenic mouse | GTTCGCAAGAACCTGATGGACA; CTAGAGCCTGTTTTGCACGTTC |
| ROSA26 iDTR | CATCAAGGAAACCCTGGACTACTG; AAAGTCGCTCTGAGTTGTTAT; GGAGCGGGAGAAATGGATATG |
| ROSA26 tdTomato | CTGTTCCTGTACGGCATGG; GGCATTAAAGCAGCGTATCC; AAGGGAGCTGCAGTGGAGTA; CCGAAAATCTGTGGGAAGTC |
| Tlr4loxp mouse | CAAGGATCCGATGATGAGTACC; CTGGGATCAGAGGCTGTCTTATAG |
Mouse strains
Mice were obtained from Jackson Laboratory as follows: C57BL/6J (Stock No: 000664), BdnfCre (B6.FVB-Bdnf em1(cre)Zak/J, Stock No: 030189), BdnfKO (B6.129S4-Bdnf tm1Jae/J, Stock No: 002266), Bdnf2lox (Bdnf tm3Jae/J, Stock No: 004339), GfapCre/ERT2 (B6.Cg-Tg(GFAP-cre/ERT2)505Fmv/J, Stock No: 0128490), ROSA26iDTR (C57BL/6-Gt(ROSA)26Sor tm1(HBEGF)Awai/J, Stock No: 007900), ROSA26tdTomato (B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J, Stock No: 007909), Sox10Cre/ERT2 (CBA;B6-Tg(Sox10-icre/ERT2)388Wdr/J, Stock No: 02765). TLR4KO were generated in our lab as described previously (32). All mice were housed in a specific pathogen free environment on a 12-hour-light/12-hour-dark cycle with free access to water and standard rodent chow (Teklad global 18% protein rodent diets, Envigo).
Discovery of enteric glia agonist “J11”
For the discovery of agents that could prevent or treat NEC, we screened the Johns Hopkins University Drug Library, containing a series of approved compounds, for their ability to stimulate cultured glia (cell line EGC/PK060399, ATCC) and induce BDNF release. Hits were secondarily validated by their ability to reduce TLR4 signaling and NEC in mice. Our lead compound has the chemical formula C13H11NO5, MW 261.23Da, also known as oxolinic acid, and is herein called “J11”.
Induction of necrotizing enterocolitis in mice
Experimental NEC was induced in ~7-day old (3–3.5g body weight) mice as we have previously described and validated (32, 57), by gavage feeding (40μl/g body weight, 5 times daily from 7am–7pm) infant formula containing Similac Advance infant formula (Abbott Nutrition): Esbilac (PetAg) canine milk replacer, 2:1 ratio, which was supplemented with enteric bacteria made from a stock created from a specimen obtained from an infant with surgical NEC five times per day. Additionally, the mice were subjected to hypoxia (5% O2-95% N2) for 10min in a hypoxia chamber (Billups-Rothenberg) twice daily for 4 days. Age-matched breast milk-fed mouse pups were used as healthy controls. Evaluation of ileal histology and expression of pro-inflammatory cytokines by qRT-PCR at a fixed point in the terminal ileum 2cm proximal to the cecum, was used to determine the disease severity.
Induction of NEC in piglets
To induce NEC in piglets, timed-pregnant White Yorkshire (Yorkshire × Landrace) sows were obtained from Oak Hill Genetics, and piglets were delivered prematurely via cesarean section at ~95% gestation as described (57). In brief, NEC was induced in piglets by gavage formula at 15ml/kg every 3h (120ml/kg per day) for 4 days of the following (per liter): Pepdite Junior (93.9g; Nutricia), MCT Oil (38.3g, USP grade; Now Foods), whey protein isolate (56g, Now Foods), and 837g water, which was supplemented with enteric bacteria made from a specimen obtained from an infant with surgical NEC.
Human ileal sample collection and explant culture
For human explant intestinal cultures, fresh ileum samples were obtained at the time of surgical resection in the treatment of NEC, of during re-anastomosis. Freshly obtained intestinal specimens were washed with sterile phosphate-buffered saline containing gentamycin (5μg/ml), minced into 2- to 4-mm diameter pieces, and then cultured in Dulbecco’s modified Eagle growth medium supplemented with 10% fetal bovine serum, 4μg/mL human recombinant insulin and 100μg/mL Primocin. Human ilea explant cultures were then pre-treated with 10μM J11 or 1μg/ml BNDF for 1h before treatment with LPS (50μg/mL) for 6h, then processed for total RNA isolation followed by qRT-PCR.
Intestinal transit time and intestinal motility studies
For measurement of intestinal motility, mice were fasted for 2h before being studied as described previously (58) with modifications. A solution containing 70kda fluorescein-dextran (50μl; 10mg/ml in 2% methylcellulose; or Similac/puppy milk formula without bacteria) was administered to each mouse by gavage using a 24-French angiocatheter. Animals were killed 30min after gavage; the whole intestine from the stomach up to colon was then divided into precise 1cm sections. Each piece of tissue was then transferred to bead homogenizing tubes containing 100μl Dulbecco’s Phosphate Buffered Saline (DPBS). The samples were either stored in −20°C until processed for later study, or immediately homogenized using a bead homogenizer and centrifuged at 12000×g for 3 minutes to obtain clear supernatants. 10μl of the clear supernatants was then diluted with 100μL DPBS and was used to measure FITC-fluorescence (λ excitation: 492nm, emission 518nm) using the SpectraMax M3 spectrophotometer (Molecular Devices). The small intestinal transit time was then derived from the position of the geometric (Geom) center of FITC-dextran in the small bowel (59).
Treatment of mice with LPS, BDNF, and J11
Endotoxemia in mice was induced in mice by administering 5mg/kg lipopolysaccharides (LPS) via intraperitoneal injection, and ileal samples were harvested 6h after LPS treatments. Recombinant BDNF (5μg/kg), and J11 (20mg/kg) by intraperitoneal injections as pretreatment or co-treatments wherever indicated.
ELISA assay
To measure the concentration of BDNF in cells or tissue lysate, we performed the DuoSet ELSIA (R&D systems) Sandwich ELISA per manufacturer instructions. Briefly, freshy harvested cells or tissue were either immediately processed or cryopreserved at −80°C until use. Cell or tissues were homogenized using a bead homogenizer in ice cold DPBS, centrifuged at 13000rpm 4°C for 10 min, and clear supernatant was separated for total protein and ELISA assay. Total protein was measured using Pierce BCA Protein Assay Kit. Amount of BDNF is calculated as picogram BDNF/mg protein.
Isolation of the longitudinal muscle myenteric plexus
The longitudinal muscle myenteric plexus was isolated from the small intestine of weanling Td-tomato-Sox10creER2 or Td-tomato-TLR4-Sox10creER2 CKO mice, and used to isolate total RNA to verify the successful glial-specific TLR4-CKO mice as described recently (60).
Histologic evaluation and NEC severity assessment
Freshly harvested intestinal tissues were fixed with 4% Paraformaldehyde (PFA), and processed for paraffin embedding. 5μm tissue sections were cut using a CUT 6062 microtome (SLEE Medical GmbH, D-55129), deparaffinized using heating vacuum heating incubator at 56°C, followed by a series of Xylene changes and rehydration ethanol gradients, and H&E stained. NEC severity was determined based upon a validated scoring system as described previously (61): 0 (no injury), 1 (minor-submucosal, lamina propria separation), 2 (moderate separation of submucosa, lamina propria and oedema in submucosal and muscular layers) and 3 (severe separation of submucosa, lamina propria, severe oedema and villous sloughing or loss of villi). In cases in which there is total destruction of the intestinal epithelium, a score of 4 is assigned.
Immunofluorescence staining
Freshly harvested intestinal tissues were fixed overnight fixed with 4% Paraformaldehyde (PFA), and either processed for paraffin embedding or imbibed in 30% sucrose solution and mounted in Tissue Freezing Medium (Electron Microscopy Sciences). Subsequently, 5μm tissue sections were cut from either paraffin blocks using a CUT 6062 microtome (SLEE Medical GmbH, D-55129 Mainz, Germany), or frozen blocks using a CryoStar NX50 (Thermo Scientific). Paraffin-embedded sections were deparaffinized, rehydrated, heated in 10mmol/L citric acid buffer for antigen retrieval. Cryosections were rehydrated and not subjected to antigen retrieval. Sections were permeabilized with 0.1% Tween-20, blocked with 5% bovine serum albumin (BSA) for no-specific binding, probed with primary antibodies overnight at 4°C, washed and probed with secondary antibody containing DAPI solution for 1 hour at room temperature. Finally, the sections were mounted using Gelvatol mount media, air dried, and imaged using the Nikon Eclipse Ti microscope confocal laser microscope system (Nikon Instruments Inc.). TUNEL staining was performed using manufacturer’s instructions. Confocal images were processed using Image J software for fluorescent intensity quantification or quantification of cells.
Quantitative real-time PCR
Total RNA was isolated using the RNeasy mini kit (Qiagen) and complementary DNA (cDNA) was synthesized from 0.5μg total RNA using QuantiTect Reverse Transcription kit (Qiagen) following the manufacturer’s instructions. The quantitative reverse transcription Polymerase chain reaction (qRT-PCR) was performed on the Bio-Rad CFX96 Real-Time System (Bio-Rad) using iTaq universal SYBR Green mix (Bio-Rad) using gene specific primers as listed in table S1. The mRNA expression relative to the housekeeping gene ribosomal protein large P0 (Rplp0) was calculated using the 2−ΔΔCt method as described (61).
Statistics
All data were analyzed using Graph Pad Prism 9 (GraphPad Software). Data were analyzed for statistical significance by ordinary one-way ANOVA followed by Tukey’s multiple comparison test. Two-tailed unpaired t test with Welch’s correction was used to compare data from experiments involving 2 treatment groups. Biologic data from the experimental data groups in the newborn mice was normally distributed. A p value of less than 0.05 (95% confidence level) was considered statistically significant, and data are presented as mean ± SEM. All experiments were performed with at least three biological replicates and at least three animals per group. Graphs show individual animals or patient samples as dots for each mouse, piglet or human specimens.
Supplementary Material
Fig S1. Induction of experimental NEC in mouse, piglets and human
Fig S2. Enteric glia loss in the intestinal epithelium of mice, piglets, and human NEC
Fig. S3. Preservation of enteric neurons and interstitial cells of Cajal in the intestinal epithelium of mice, piglets, and human with NEC.
Fig. S4. Assessment of myenteric and submucosal enteric glial loss in mice, piglets, and human with NEC.
Fig. S5. Generation of enteric glia-deficient mice.
Fig. S6. Assessment of motility in male versus female Plp1ΔDTR-glia deficient mice.
Fig. S7. Phenotypic characterization of enteric glia-deficient mice.
Fig S8. Generation of mice lacking TLR4 on the enteric glia.
Table S1. qRT-PCR gene and primer list.
Acknowledgements
Funding:
This work was funded by RO1 DK117186 to DJH, RO1DK121824 to DJH, and R35GM141956 to DJH and T32D007713 to MLK from the National Institutes of Health.
Footnotes
Competing interests: There are no consultant arrangements associated with the present work. DH, PL, and CPS are co-inventors on a pending PCT application, application number PCT/US2020/012848, filed on 1/9/2020, that has published online as WO 2020/146573. The title is “Compounds and Treatments That Enhance Enteric Nervous System Function”.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials.
References
- 1.Alganabi M, Lee C, Bindi E, Li B, Pierro A, Recent advances in understanding necrotizing enterocolitis. F1000Res 8, 1–8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel RM, Kandefer S, Walsh MC, Bell EF, Carlo WA, Laptook AR, Sanchez PJ, Shankaran S, Van Meurs KP, Ball MB, Hale EC, Newman NS, Das A, Higgins RD, Stoll BJ, Eunice H Kennedy Shriver National Institute of Child, N. Human Development Neonatal Research, Causes and timing of death in extremely premature infants from 2000 through 2011. N Engl J Med 372, 331–340 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharif S, Meader N, Oddie SJ, Rojas-Reyes MX, McGuire W, Probiotics to prevent necrotising enterocolitis in very preterm or very low birth weight infants. Cochrane Database Syst Rev 10, Cd005496 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nino DF, Sodhi CP, Hackam DJ, Necrotizing enterocolitis: new insights into pathogenesis and mechanisms. Nat Rev Gastroenterol Hepatol 13, 590–600 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hackam DJ, Sodhi CP, Toll-Like Receptor-Mediated Intestinal Inflammatory Imbalance in the Pathogenesis of Necrotizing Enterocolitis. Cell Mol Gastroenterol Hepatol 6, 229–238 e221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kovler ML, Sodhi CP, Hackam DJ, Precision-based modeling approaches for necrotizing enterocolitis. Dis Model Mech 13, dmm044388 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Afrazi A, Branca MF, Sodhi CP, Good M, Yamaguchi Y, Egan CE, Lu P, Jia H, Shaffiey S, Lin J, Ma C, Vincent G, Prindle T Jr., Weyandt S, Neal MD, Ozolek JA, Wiersch J, Tschurtschenthaler M, Shiota C, Gittes GK, Billiar TR, Mollen K, Kaser A, Blumberg R, Hackam DJ, Toll-like receptor 4-mediated endoplasmic reticulum stress in intestinal crypts induces necrotizing enterocolitis. J Biol Chem 289, 9584–9599 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sodhi CP, Shi XH, Richardson WM, Grant ZS, Shapiro RA, Prindle T Jr., Branca M, Russo A, Gribar SC, Ma C, Hackam DJ, Toll-like receptor-4 inhibits enterocyte proliferation via impaired beta-catenin signaling in necrotizing enterocolitis. Gastroenterology 138, 185–196 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leaphart CL, Cavallo J, Gribar SC, Cetin S, Li J, Branca MF, Dubowski TD, Sodhi CP, Hackam DJ, A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol 179, 4808–4820 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Jilling T, Simon D, Lu J, Meng FJ, Li D, Schy R, Thomson RB, Soliman A, Arditi M, Caplan MS, The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol 177, 3273–3282 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egan CE, Sodhi CP, Good M, Lin J, Jia H, Yamaguchi Y, Lu P, Ma C, Branca MF, Weyandt S, Fulton WB, Nino DF, Prindle T Jr., Ozolek JA, Hackam DJ, Toll-like receptor 4-mediated lymphocyte influx induces neonatal necrotizing enterocolitis. J Clin Invest 126, 495–508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MohanKumar K, Namachivayam K, Chapalamadugu KC, Garzon SA, Premkumar MH, Tipparaju SM, Maheshwari A, Smad7 interrupts TGF-beta signaling in intestinal macrophages and promotes inflammatory activation of these cells during necrotizing enterocolitis. Pediatr Res 79, 951–961 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng D, Zhu W, Shi HN, Lu L, Wijendran V, Xu W, Walker WA, Toll-like receptor-4 in human and mouse colonic epithelium is developmentally regulated: a possible role in necrotizing enterocolitis. Pediatr Res 77, 416–424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yazji I, Sodhi CP, Lee EK, Good M, Egan CE, Afrazi A, Neal MD, Jia H, Lin J, Ma C, Branca MF, Prindle T, Richardson WM, Ozolek J, Billiar TR, Binion DG, Gladwin MT, Hackam DJ, Endothelial TLR4 activation impairs intestinal microcirculatory perfusion in necrotizing enterocolitis via eNOS-NO-nitrite signaling. Proc Natl Acad Sci U S A 110, 9451–9456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma R, Hudak ML, Tepas JJ 3rd, Wludyka PS, Marvin WJ, Bradshaw JA, Pieper P, Impact of gestational age on the clinical presentation and surgical outcome of necrotizing enterocolitis. J Perinatol 26, 342–347 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Kliegman RM, Walsh MC, Neonatal necrotizing enterocolitis: pathogenesis, classification, and spectrum of illness. Curr Probl Pediatr 17, 213–288 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furness JB, The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol 9, 286–294 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Savidge TC, Newman P, Pothoulakis C, Ruhl A, Neunlist M, Bourreille A, Hurst R, Sofroniew MV, Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology 132, 1344–1358 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Capoccia E, Cirillo C, Gigli S, Pesce M, D’Alessandro A, Cuomo R, Sarnelli G, Steardo L, Esposito G, Enteric glia: A new player in inflammatory bowel diseases. Int J Immunopathol Pharmacol 28, 443–451 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Hoff S, Zeller F, von Weyhern CW, Wegner M, Schemann M, Michel K, Ruhl A, Quantitative assessment of glial cells in the human and guinea pig enteric nervous system with an anti-Sox8/9/10 antibody. J Comp Neurol 509, 356–371 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Ferri GL, Probert L, Cocchia D, Michetti F, Marangos PJ, Polak JM, Evidence for the presence of S-100 protein in the glial component of the human enteric nervous system. Nature 297, 409–410 (1982). [DOI] [PubMed] [Google Scholar]
- 22.Bell MJ, Ternberg JL, Feigin RD, Keating JP, Marshall R, Barton L, Brotherton T, Neonatal necrotizing enterocolitis. Therapeutic decisions based upon clinical staging. Ann Surg 187, 1–7 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma R, Hudak ML, A clinical perspective of necrotizing enterocolitis: past, present, and future. Clin Perinatol 40, 27–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Werts AD, Fulton WB, Ladd MR, Saad-Eldin A, Chen YX, Kovler ML, Jia H, Banfield EC, Buck RH, Goehring K, Prindle T Jr., Wang S, Zhou Q, Lu P, Yamaguchi Y, Sodhi CP, Hackam DJ, A Novel Role for Necroptosis in the Pathogenesis of Necrotizing Enterocolitis. Cell Mol Gastroenterol Hepatol 9, 403–423 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Margolis KG, Gershon MD, Enteric Neuronal Regulation of Intestinal Inflammation. Trends Neurosci 39, 614–624 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scarpignato C, Pharmacological stimulation of gastrointestinal motility: where we are and where are we going? Dig Dis 15 Suppl 1, 112–136 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Rao M, Rastelli D, Dong L, Chiu S, Setlik W, Gershon MD, Corfas G, Enteric Glia Regulate Gastrointestinal Motility but Are Not Required for Maintenance of the Epithelium in Mice. Gastroenterology 153, 1068–1081 e1067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turco F, Sarnelli G, Cirillo C, Palumbo I, De Giorgi F, D’Alessandro A, Cammarota M, Giuliano M, Cuomo R, Enteroglial-derived S100B protein integrates bacteria-induced Toll-like receptor signalling in human enteric glial cells. Gut 63, 105–115 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Hoehner JC, Wester T, Pahlman S, Olsen L, Localization of neurotrophins and their high-affinity receptors during human enteric nervous system development. Gastroenterology 110, 756–767 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Hansebout CR, Su C, Reddy K, Zhang D, Jiang C, Rathbone MP, Jiang S, Enteric glia mediate neuronal outgrowth through release of neurotrophic factors. Neural Regen Res 7, 2165–2175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reinshagen M, Rohm H, Steinkamp M, Lieb K, Geerling I, Von Herbay A, Flamig G, Eysselein VE, Adler G, Protective role of neurotrophins in experimental inflammation of the rat gut. Gastroenterology 119, 368–376 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Sodhi CP, Neal MD, Siggers R, Sho S, Ma C, Branca MF, Prindle T Jr., Russo AM, Afrazi A, Good M, Brower-Sinning R, Firek B, Morowitz MJ, Ozolek JA, Gittes GK, Billiar TR, Hackam DJ, Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 143, 708–718 e705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaminsky D, Meltzer RI, Quinolone antibacterial agents. Oxolinic acid and related compounds. J Med Chem 11, 160–163 (1968). [DOI] [PubMed] [Google Scholar]
- 34.Bush TG, Savidge TC, Freeman TC, Cox HJ, Campbell EA, Mucke L, Johnson MH, Sofroniew MV, Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell 93, 189–201 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Heymans C, de Lange IH, Lenaerts K, Kessels L, Hadfoune M, Rademakers G, Melotte V, Boesmans W, Kramer BW, Jobe AH, Saito M, Kemp MW, van Gemert WG, Wolfs T, Chorioamnionitis induces enteric nervous system injury: effects of timing and inflammation in the ovine fetus. Mol Med 26, 82 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Yang J, Watkins DJ, Boomer LA, Matthews MA, Su Y, Besner GE, Enteric nervous system abnormalities are present in human necrotizing enterocolitis: potential neurotransplantation therapy. Stem Cell Res Ther 4, 157 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villanacci V, Bassotti G, Nascimbeni R, Antonelli E, Cadei M, Fisogni S, Salerni B, Geboes K, Enteric nervous system abnormalities in inflammatory bowel diseases. Neurogastroenterol Motil 20, 1009–1016 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Gribar SC, Sodhi CP, Richardson WM, Anand RJ, Gittes GK, Branca MF, Jakub A, Shi XH, Shah S, Ozolek JA, Hackam DJ, Reciprocal expression and signaling of TLR4 and TLR9 in the pathogenesis and treatment of necrotizing enterocolitis. J Immunol 182, 636–646 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho SX, Rudloff I, Lao JC, Pang MA, Goldberg R, Bui CB, McLean CA, Stock M, Klassert TE, Slevogt H, Mangan NE, Cheng W, Fischer D, Gfroerer S, Sandhu MK, Ngo D, Bujotzek A, Lariviere L, Schumacher F, Tiefenthaler G, Beker F, Collins C, Kamlin COF, Konig K, Malhotra A, Tan K, Theda C, Veldman A, Ellisdon AM, Whisstock JC, Berger PJ, Nold-Petry CA, Nold MF, Characterization of the pathoimmunology of necrotizing enterocolitis reveals novel therapeutic opportunities. Nat Commun 11, 5794 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y, Koike Y, Chi L, Ahmed A, Miyake H, Li B, Lee C, Delgado-Olguin P, Pierro A, Formula feeding and immature gut microcirculation promote intestinal hypoxia, leading to necrotizing enterocolitis. Dis Model Mech 12, dmm040998 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu X, Radulescu A, Zorko N, Besner GE, Heparin-binding EGF-like growth factor increases intestinal microvascular blood flow in necrotizing enterocolitis. Gastroenterology 137, 221–230 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan X, Managlia E, Tan XD, De Plaen IG, Prenatal inflammation impairs intestinal microvascular development through a TNF-dependent mechanism and predisposes newborn mice to necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 317, G57–G66 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sampah MES, Hackam DJ. (Front Immunol, 2020), vol. 11, pp. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elgin TG, Kern SL, McElroy SJ, Development of the Neonatal Intestinal Microbiome and Its Association With Necrotizing Enterocolitis. Clin Ther 38, 706–715 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Nyarady K, Turai R, Funke S, Gyorgyi E, Makai A, Premusz V, Bodis J, Sulyok E, Effects of perinatal factors on sirtuin 3, 8-hydroxy-2’- deoxyguanosine, brain-derived neurotrophic factor and serotonin in cord blood and early breast milk: an observational study. Int Breastfeed J 15, 57 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perrella SL, Hepworth AR, Simmer KN, Geddes DT, Influences of breast milk composition on gastric emptying in preterm infants. J Pediatr Gastroenterol Nutr 60, 264–271 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Herrmann K, Carroll K, An exclusively human milk diet reduces necrotizing enterocolitis. Breastfeed Med 9, 184–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kabouridis PS, Pachnis V, Emerging roles of gut microbiota and the immune system in the development of the enteric nervous system. J Clin Invest 125, 956–964 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anitha M, Vijay-Kumar M, Sitaraman SV, Gewirtz AT, Srinivasan S, Gut microbial products regulate murine gastrointestinal motility via Toll-like receptor 4 signaling. Gastroenterology 143, 1006–1016 e1004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bohorquez DV, Samsa LA, Roholt A, Medicetty S, Chandra R, Liddle RA, An enteroendocrine cell-enteric glia connection revealed by 3D electron microscopy. PLoS One 9, e89881 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pruitt LCC, Skarda DE, Rollins MD, Bucher BT, Hirschsprung-associated enterocolitis in children treated at US children’s hospitals. J Pediatr Surg 55, 535–540 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hackam DJ, Filler RM, Pearl RH, in Journal of Pediatric Surgery. (J Pediatr Surg, 1998), vol. 33, pp. 830–833. [DOI] [PubMed] [Google Scholar]
- 53.Garcia De Mateos-Verchere J, Vaugeois JM, Naudin B, Costentin J, Behavioural and neurochemical evidence that the antimicrobial agent oxolinic acid is a dopamine uptake inhibitor. European Neuropsychopharmacology 8, 255–259 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Marzio L, Neri M, Pieramico O, Delle Donne M, Peeters TL, Cuccurullo F, Dopamine interrupts gastrointestinal fed motility pattern in humans. Effect on motilin and somatostatin blood levels. Dig Dis Sci 35, 327–332 (1990). [DOI] [PubMed] [Google Scholar]
- 55.Di Nucci A, Candura SM, Tagliani M, D’Agostino G, Spelta V, Fiori E, Ricotti P, Tonini M, Fluoroquinolone-induced motor changes in the guinea-pig isolated ileum. Pharmacol Toxicol 83, 263–269 (1998). [DOI] [PubMed] [Google Scholar]
- 56.Torres-Fuentes C, Pastor-Cavada E, Cano R, Kandil D, Shanahan R, Juan R, Shaban H, McGlacken GP, Schellekens H, Quinolones Modulate Ghrelin Receptor Signaling: Potential for a Novel Small Molecule Scaffold in the Treatment of Cachexia. Int J Mol Sci 19, 1605 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sodhi CP, Wipf P, Yamaguchi Y, Fulton WB, Kovler M, Nino DF, Zhou Q, Banfield E, Werts AD, Ladd MR, Buck RH, Goehring KC, Prindle T Jr., Wang S, Jia H, Lu P, Hackam DJ, The human milk oligosaccharides 2’-fucosyllactose and 6’-sialyllactose protect against the development of necrotizing enterocolitis by inhibiting toll-like receptor 4 signaling. Pediatr Res 89, 91–101 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Z, Chalazonitis A, Huang YY, Mann JJ, Margolis KG, Yang QM, Kim DO, Cote F, Mallet J, Gershon MD, Essential roles of enteric neuronal serotonin in gastrointestinal motility and the development/survival of enteric dopaminergic neurons. J Neurosci 31, 8998–9009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miller MS, Galligan JJ, Burks TF, Accurate measurement of intestinal transit in the rat. J Pharmacol Methods 6, 211–217 (1981). [DOI] [PubMed] [Google Scholar]
- 60.Costa DVS, Bon-Frauches AC, Silva A, Lima-Junior RCP, Martins CS, Leitao RFC, Freitas GB, Castelucci P, Bolick DT, Guerrant RL, Warren CA, Moura-Neto V, Brito GAC, 5-Fluorouracil Induces Enteric Neuron Death and Glial Activation During Intestinal Mucositis via a S100B-RAGE-NFkappaB-Dependent Pathway. Sci Rep 9, 665 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sodhi CP, Fulton WB, Good M, Vurma M, Das T, Lai CS, Jia H, Yamaguchi Y, Lu P, Prindle T, Ozolek JA, Hackam DJ, Fat composition in infant formula contributes to the severity of necrotising enterocolitis. Br J Nutr 120, 665–680 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Induction of experimental NEC in mouse, piglets and human
Fig S2. Enteric glia loss in the intestinal epithelium of mice, piglets, and human NEC
Fig. S3. Preservation of enteric neurons and interstitial cells of Cajal in the intestinal epithelium of mice, piglets, and human with NEC.
Fig. S4. Assessment of myenteric and submucosal enteric glial loss in mice, piglets, and human with NEC.
Fig. S5. Generation of enteric glia-deficient mice.
Fig. S6. Assessment of motility in male versus female Plp1ΔDTR-glia deficient mice.
Fig. S7. Phenotypic characterization of enteric glia-deficient mice.
Fig S8. Generation of mice lacking TLR4 on the enteric glia.
Table S1. qRT-PCR gene and primer list.
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials.