

Abstract
Pancreatic Ductal Adenocarcinoma (PDAC) is a lethal aggressive cancer, in part due to elements of the microenvironment (hypoxia, hypoglycemia) that cause metabolic network alterations. The FDA approved anti-helminthic Pyrvinium Pamoate (PP) has been previously shown to cause PDAC cell death, although the mechanism has not been fully determined. We demonstrated that PP effectively inhibited PDAC cell viability with nanomolar IC50s (9-93nM) against a panel of PDAC, patient-derived, and murine organoid cell lines. In vivo, we demonstrated that PP inhibited PDAC xenograft tumor growth with both intraperitoneal (IP; p<0.0001) and oral administration (PO; p=0.0023) of human-grade drug. Metabolomic and phosphoproteomic data identified that PP potently inhibited PDAC mitochondrial pathways including oxidative phosphorylation and fatty acid metabolism. As PP treatment reduced oxidative phosphorylation (p<0.001) leading to an increase in glycolysis (p<0.001), PP was 16.2-fold more effective in hypoglycemic conditions similar to those seen in PDAC tumors. RNA sequencing demonstrated that PP caused a decrease in mitochondrial RNA expression, an effect which was not observed with established mitochondrial inhibitors rotenone and oligomycin. Mechanistically, we determined that PP selectively bound mitochondrial G-quadruplexes and inhibited mitochondrial RNA transcription in a G-quadruplex dependent manner. This subsequently led to a 90% reduction in mitochondrial encoded gene expression. We are preparing to evaluate the efficacy of PP in PDAC in an IRB approved window of opportunity trial (IND:144822).
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a lethal cancer with a five year overall survival rate of 10% (1). Contributing to this high mortality is the resistant nature of PDAC which has been largely attributed to the PDAC tumor-microenvironment (TME). The PDAC TME is nutrient poor, with decreased vascularization leading to both hypoxic and hypoglycemic conditions which drive chemo-resistance(2-4). While hypoxic, the levels of oxygenation displayed in PDAC (1.5% oxygen vs normal 5% oxygen) are still permissive for the effective utilization of the electron transport chain (5,6). Accordingly, PDAC tumors adapt and become more reliant on oxidative phosphorylation as opposed to glycolysis to increase glucose efficiency (36 vs 2 ATP per glucose molecule respectively) (5,7). Increased reliance on mitochondria in PDAC xenografts has been demonstrated by simultaneously measuring oxygenation, glucose uptake and glycolysis spatially throughout tumors (6). Accordingly, multiple mitochondrial inhibitors have shown promise in pre-clinical models for the treatment of PDAC (7-9), thus there is a strong premise to target mitochondria in the uniquely nutrient-deprived TME of PDAC tumors (4).
To target the nutrient deprived PDAC TME (3,5,6), we utilized the FDA-approved drug Pyrvinium Pamoate (PP), which has previously been shown to have multiple potential mechanisms of action including targeting mitochondrial pathways (10-16) and to be particularly effective in low glucose settings.
Materials and Methods
Cell culture
Cell lines (PANC-1, Capan-1, HS766T, MIA-PaCa2, and CFPAC) were purchased from ATCC (Manassas, VA). HuR knockout clones were generated as previously described (17,18). PDX line SPC_145 was a gift from Dr. Talia Golan (Sheba Medical Center) (19). All cell lines were tested monthly for mycoplasma and every 6 months STR validated. Cell lines for all experiments were below passage 20.
Cell growth and survival assays
For IC50 analysis, cells were seeded at 1,000 cells/well in 96-well plates and treated with drugs the next day. Cell viability was assessed after five days utilizing Quant-iT™ Pico Green™ (Invitrogen, St. Louis MS) and a GloMax® Explorer Multimode Microplate Reader (Promega, Madison, WI). IC50s were calculated utilizing non-linear curve fitting in GraphPad Prism 8.0.1. (GraphPad Software, San Diego, CA).
Colony formation was performed by plating 1,000 cells/well of a six-well plate, followed by drug dosing the next day. After ten days, colony growth was assessed by staining as previously described (18).
For experiments comparing differing glucose conditions, cells were pretreated for 12-24 hours with indicated glucose concentration prior to drug treatment.
Organoids were cultured as previously described (17). For organoid IC50s, organoids were broken down to single cells and seeded at 1,500 cells/well in a 96-well plate. The next day, cells were treated with drug. After five days, cell viability was determined via Cell Titer Glo (Promega, Madison, WI) and a GloMax® Explorer Multimode Microplate Reader (Promega, Madison, WI). IC50s were calculated utilizing non-linear curve fitting in GraphPad Prism 8.0.1.
Immunoblot analysis
Westerns were performed using the following antibodies α-Tubulin (B-5-1-2, 1:5000, Thermo Scientific); HuR (3A2, 1:4000, Santa Cruz Biotechnology) COX-2 (H-62, 1:1000, Santa Cruz), Total OXPHOS Human WB Antibody Cocktail (ab110411, 1:1000, Abcam), Anti-TOMM20 antibody (ab56783, 1:1000, Abcam), Beta-actin (13E5 1:10,000, Cell Signaling). Membranes were scanned using Odyssey Infrared Imaging System (LI-COR Biosciences) and analyzed with Image Studio Lite Version 5.2.
Immunofluorescence
Cells were plated at 10,000 cells/well in a 24-well plate containing 8mm coverslips. Cells were treated for 1 hour with PP 0.3μM and/or 0.1μM MitoTracker Green FM (Thermofisher, Waltham, MA) PP was imaged using the Texas Red channel. Coverslips were then removed and imaged live within 10 minutes with a Leica DM4 B microscope (Leica Microsystems, Wetzlar, Germany).
Immunohistochemistry (IHC)
IHC was performed as previously described (20). Patient samples were collected in compliance with IRB 06U.76. Mouse tumors were TUNEL stained using diaminobenzidine (DAB). Five 20X images per group were quantified and compared utilizing a t-test in GraphPad Prism 8.0.1.
Cellular Thermal Shift Assay
PANC-1 cells were plated at 6x106 cells/10cm dish. Cells were treated with PP (10μM), DHTS (10μM) or DMSO for 4 hours at 37°C prior to collection. Cell pellets were suspended in PBS with suitable protease inhibitor. Aliquots of 5x105 cells/100μL were made in separate tubes. On a thermocycler, cells were exposed to different temperatures (37°C-61°C) for 3 mins and then kept directly on dry ice. Cells were lysed by freeze-thaw cycle (dry ice to 25°C three times). Cells were centrifuged at 20,000 x g for 20 mins at 4°C, supernatant was collected and mixed with loading buffer (PBS and SDS loading buffer). Samples were analyzed by immunoblotting.
Phosphoproteomics
HS766T, MIA-PaCa2 and PANC-1 cells were seeded at 500,000 cells/10cm plate and treated with PP at 0.3μM for 48 hours. Cells were collected and run for phosphoproteomic reverse phase microarray analysis (previously described) (21). Phosphoproteomic analysis was performed utilizing Genesis version 1.8.1, and Average Linkage WPMGA Pearson Correlation Hierarchical Clustering.
Metabolomics
MIA-PaCa2 cells were plated in 6-cm dishes 24 hours prior to treatment with fresh medium containing DMSO or 0.3μM PP. After treatment for 6 hours, cells were washed with PBS, and metabolites were extracted with 1ml cold 80% methanol/water on dry ice for subsequent LC-MS/MS analysis (22). For mouse samples mice were treated with 1mg/kg PP IP and blood was collected utilizing heperanized tubes and retro-orbital bleeding at the indicated time points. A hydrophilic interaction chromatography method (HILIC) on an Acquity UPLC BEH Amide column (2.1x150mm i.d., 1.7μm; Waters) was used for compound separation. A Xevo-TQ-S mass spectrometer (Waters) was used to detect all compounds with multiple reaction monitoring (MRM) methods. Peak areas were integrated using Skyline (23) and normalized to cell number. Peak areas were analyzed using MetaboAnalyst 4.0 (http://www.metaboanalyst.ca/MetaboAnalyst/)(24).
ATP Quantification
Cells were plated at 5,000 cells/well in a 96-well plate. Cells were pretreated for 16-24hrs in media without glucose or FBS. Cells were treated with Oligomycin A (10μM), FCCP (1μM), Gemcitabine (1μM), or PP (0.3μM) +/− 25mM Glucose for 1hr and ATP was quantified utilizing Cell Titer Glo (Promega, Madison, WI, catalog# G7570).
Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR)
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using the Seahorse XFp Analyzer (Seahorse Bioscience, Agilent, CA). MIA-PaCa2 cells were plated at 22,000 cells/well on XFp cell plates 24 prior to 12 hours treatment with 0.3μM PP or vehicle. The concentration of FCCP, antimycin A, rotenone and Oligomycin A were 4μM, 1μM, 4μM and 2μM. For acute treatment, PP in PBS at 0.3μM was injected after basal OCR measurement and monitored for 90 min, followed by inhibiting the complex IV and then the electron transport chain. For oxygen consumption quantification, total OCR was defined as the basal OCR rates, non-mitochondrial OCR is defined as the OCR subsequent to antimycin-a and rotenone treatment, and mitochondrial OCR is defined as the difference between the two.
Mitochondrial DNA quantification
MIA-PaCa2 cells were plated at 1x106 cells/10cm plate and treated with PP at 0.3μM for indicated times, at which time cells were collected and DNA extracted (DNeasy blood and tissue kit, Qiagen). Mitochondrial DNA was compared to nuclear DNA using qRT-PCR.
Mitochondrial RNA Quantification
MIA-PaCa2 cells were plated at 1x106 cells/10cm plate and treated with PP at 0.3μM for 1 hour, at which time cells were collected and RNA extracted (RNeasy Mini Kit, Qiagen). Mitochondrial RNA was quantified using qRT-PCR utilizing 18s as a reference.
RNA Sequencing
MIA-PaCa2 cells were plated at 1x106 cells/10cm plate and treated with PP at 0.3μM, Oligomycin at 4μM or rotenone at 0.5 μM for 48 hours at which time cells were collected and RNA extracted (RNeasy Mini Kit, Qiagen). Whole-exome sequencing was performed by Novogene utilizing Illumina Platform PE150. Single sample geneset enrichment analysis was performed using hallmark genesets from the MSigDB Collections at https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp as previously described (25). Integrative metabolomic and RNA sequencing analysis was performed using MetaboAnalyst5.0 (26). Results can be found at (GSE176305).
Synergy
Synergy was determined utilizing BLISS synergy with Combenefit software (27).
Xenograft Studies
We used 6-week-old Athymic female nude mice from Envigo (East Millstone, NJ), all procedures were approved by IACUC. Mice were engrafted subcutaneously with 5x106 MIA-PaCa2 cells/flank, prepared in 200μl solution comprised of 1:1 DPBS:Matrigel (Corning, MA). For the first PP efficacy, mice were randomized with tumors at 100mm3 to either vehicle or 1mg/kg PP 3 times weekly. One PP treated mouse was removed from analysis after utilizing Grubb’s outlier test, and one died 11 days into the study. It is unknown if this was drug related. The control arm is the same control arm in a previously published paper as we tested both drugs in vivo at the same time and shared control animals (18).
For the Kaplan Meier study, mice were randomized when tumor volumes reached 100mm3 into groups: vehicle and PP IP 1mg/kg, with 10 mice per arm. Mice were sacrificed when tumor burden reached 2000mm3.
For analysis of human-grade oral PP treatment, mice were randomized and treatment was initiated the day after engraftment. The arms for this experiment were 5mg/kg, 20mg/kg, 35mg/kg and vehicle. Pyrvin tablets were crushed, resuspended in saline, and dosed orally 5 days/week. There were five mice/arm except for the vehicle arm with ten mice.
Pharmacokinetics
For pharmacokinetic IV work mice were dosed at 1mg/kg PP (100uL injection) by tail vein, and blood was collected utilizing heperanized tubes and retro-orbital bleeding at the indicated time points. For IP and oral dosing mice were dosed either IP at 1mg/kg or orally at 5, 20 or 35 mg/kg and serum and tissue samples were collected by Alliance Pharma, with blood collected utilizing cardiac puncture. All samples were analyzed by Alliance Pharma using HPLC (UPLC, Shimadzu), MS/MS (API 4000, AB Sciex).
G-Quadruplex bound PP fluorescence shift assay
Similar to the method described (28), mitochondrial DNA and control dsDNA fragments were created by mixing oligonucleotides in annealing buffer (10mM TrisHCl pH 7.52,50 mM NaCl and 1 mM EDTA). Annealing sequence using a thermocycler was 2 minutes @ 95°C > cool-down at 0.5°C/minute down to 65°C > 20 minutes @ 65°C > cool-down at 0.5°C/minute down to 55°C > 45 minutes @ 55°C > cool-down at 0.5°C/minute down to 35°C. dsDNA fragments were in buffer (100 mM potassium chloride and 20mM potassium phosphate) for 1 hour to promote and stabilize G-Quadruplex formation. PP was added at a ratio of 1:2 (4μM Pyrvinium and 8μM DNA) and incubated in the dark for 1hr at RT and measured using the Bio-Rad ChemiDoc MP (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with excitation/emission of 556nm/573nm to recognize the spectral and intensity shift previously described by Stockert et al(29). Oligonucletides used can be found as previously described (28,30).
G-Quadruplex bound Stop-PCR assay
Mitochondrial DNA was amplified from PANC-1 DNA using the REPLI-g Mitochondrial DNA Kit (Qiagen). Similar to the method described by (30), PCR reactions were run with increasing concentrations of PP. Briefly, 25μl reactions were performed with 0.625 units Ex-Taq DNA Polymerase (Takara), 0.2mM dNTP , 1 μM of each primer, and 0.375 μl of mtDNA. Reaction mixes were incubated on ice for 15 minutes and then moved to the thermocycler for a sequence of 30 cycles of (10s at 98C > 30s at 55C> 15s at 72). Reaction samples were run on a 0.8% agarose gel and imaged on a Bio-Rad ChemiDoc MP (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Primers used are as follows (target fwd/reverse):
ND1 GGCTACTACAACCCTTCGCT/ACGGGAAGGGTATAACCAACA,
ND2 ATCATAGCAGGCAGTTGAGG/GGGTGGGTTTTGTATGTTCA,
Non-G4 TCGAGTCTCCCTTCACCATT/GCACTCGTAAGGGGTGGAT.
Statistical Significance
For all figures asterisks are designated for p-values (or q-values where appropriate) as follows *<0.05, **<0.01, ***<0.005, ****<0.001 unless otherwise designated.
NCI60 data
Publicly available data for the activity of PP across the NCI60, and expression levels of genes from microarray data were accessed using CellMinerCDB version 1.2 (31).
Results
PP’s has significant in vitro cytotoxic efficacy against PDAC cells
To determine PP efficacy in vitro, we tested PP in a panel of PDAC and PDX-derived cell lines where we found PP to be effective at inhibiting cell viability at the nanomolar range (HS766T, MIA-PaCa2, PANC-1, CFPAC, SPC_145 respective IC50s of 93, 40, 92, 21, 9, 93, 58, and 9 nM). PP effectively inhibited the growth of two PDAC organoid lines KPC and KTC (KTC having an activating KRAS mutation and HuR overexpression and KPC organoids having an activating KRAS mutation and p53 inactivation) (32) with IC50sof 93 and 58 nM respectively. These results indicated that PP was effective at inhibiting cell growth in multiple PDAC models.
PP does not rely on HuR as a target in PDAC
Several mechanisms of action have been proposed for PP, including the inhibition of the RNA binding protein HuR (15). HuR has been recognized as a vital target in PDAC, and to be active in the nutrient-deprived PDAC TME (3,32,33). Utilizing a cellular thermal shift assay and demonstrated that PP did not strongly interact with HuR as compared to the validated HuR inhibitor dihydrotanshinone I (34) (Supplemental Figure 1A-C). To assess if PP was indirectly affecting HuR we tested PP along with several control therapeutics in HuR KO cells. We found HuR knockout lines were more sensitive to multiple clinically utilized therapeutics tested (Supplemental Table 1), however they were not resistant to PP (Supplemental Table 1). We confirmed the lack of resistance to PP using a colony formation assay(Supplemental Figure 1 D-F). We were unable to supplement these in vitro findings with in vivo experimentation, as the HuR KO cells were xenograft lethal (Supplemental Figure 1G) (17). These results demonstrate that PP induces PDAC cell death in an HuR-independent manner.
Screening for the primary target of PP in PDAC
Several other mechanisms of action for PP have been proposed including targeting autophagy, mitochondrial oxidative phosphorylation, WNT response, the unfolded protein response, androgen receptor signaling, and adipogenic differentiation (10-16). To investigate which if any of these mechanisms of action were predominant in PDAC cells in an unbiased manner, we utilized phosphoproteomic reverse-phase microarray (PRPMA). Across HS766T, PANC-1 and MIA-PaCa2 cells we found a number of significantly affected targets were down-regulated, in particular several consistently down regulated genes were regulators of cell-cycle and cellular energetics pathways including S6 ribosomal protein s240/244, RB S780, and mTOR S2448 among others. Of the commonly up-regulated genes many were associated with cell scavenging, inflammation, and cell death including cleaved caspase 3, and cleaved lamin A and AKT S473 (Supplemental Figure 2A-C). Based on these findings, we hypothesized that the broad changes observed from our PRPMA data could result from a large shift in cellular metabolism.
To investigate if/how PP was affecting metabolism in PDAC, we performed a comprehensive and unbiased analysis of metabolites after PP treatment. We observed significant modulations in metabolites involved in the citric acid cycle upon PP treatment (Figure 1A), along with increases in glycolytic metabolites (Supplementary Figure 2D). This may indicate a compensatory reaction of increased glycolysis in response to PP induced inhibition of the TCA cycle/oxidative phosphorylation. There were several relatively small changes among arginine and proline metabolites (Supplemental Figure 2E). Interestingly we observed a large increase in N-Carbamoyl Aspartate indicating alterations in pyrimidine biosynthesis (Figure 1B). We also observed an increase in long chain acylcarnitine levels, but significant decreases in free carnitine along with short and medium acylcarnitine chains (Figure 1C). In particular we observed large increases in acylcarnitine C4-OH and acylcarnitine C5-DC which have been clinically characterized to be markers of mitochondrial disfunction leading to medium/short-chain acyl-CoA dehydrogenase deficiency (M/SCHAD deficiency) and glutaric acidemia type I respectively(35). These data suggest an alteration of multiple mitochondrial pathways upon PP treatment including an inhibition of the citric acid cycle, alteration of fatty acid metabolism, and inhibition of pyrimidine biosynthesis (Figure 1D).
Figure 1. Pyrvinium Pamoate Metabolomic Changes:
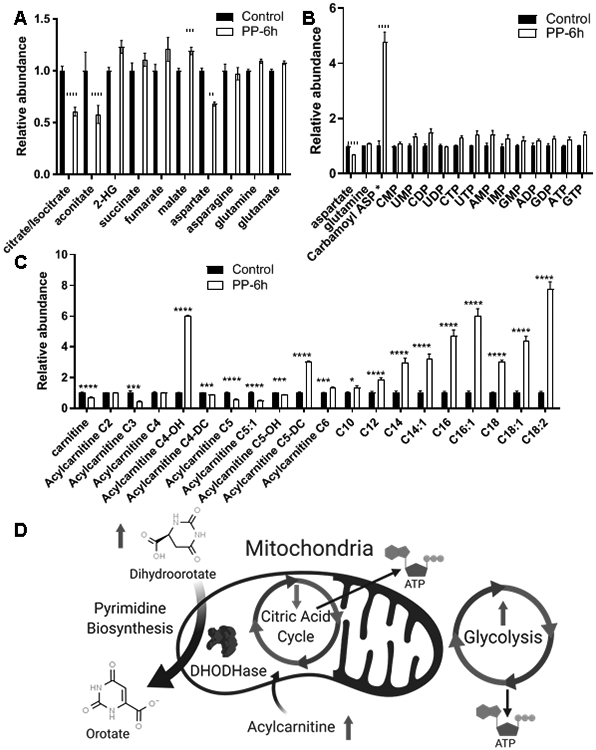
MIA-PaCa2 cells were treated with PP .3uM for 6 hours and metabolite changes were compared to control (n=5) A) Citric acid cycle metabolites B) Purine and Pyrimidine metabolites C) Acylcarnitine metabolites D) These changes may indicate a global alteration of mitochondrial function with a modulation in the Citric Acid Cycle leading to increased reliance on glycolysis for ATP production and increases in Acylcarnitine being caused by a reduction of fatty acid oxidation and thus buildup of acylcarnitine, the mitochondrial DHODHase is a critical member of Pyrimidine biosynthesis and a loss of function of this enzyme could potentially cause a buildup of the precursor N-carbamoyl aspartate
PP inhibits mitochondrial oxidative phosphorylation in PDAC
To assess the effect of PP on mitochondria, we first confirmed previous reports that PP localizes to mitochondria (36). In PDAC cells, PP localized to the mitochondria for prolonged periods of time (Figure 2A, Supplemental Figure 3A,B) (12-14). Chronic treatment (12 hour PP pre-treatment) caused a significant decrease of mitochondrial oxygen consumption (i.e. the electron transport chain), while not significantly effecting non-mitochondrial oxygen consumption (Figure 2B,C). In control cells acute oligomycin treatment caused an increase in glycolysis (1.44 fold increase P<0.0001) (Figure 2D), as determined through measuring the extracellular acidification rate (ECAR). This was expected as oligomycin A inhibits mitochondrial ATP generation, and cells frequently up-regulate glycolytic flux (glycolysis) to compensate. PP pre-treated cells were insensitive to oligomycin A with no increase in glycolysis, consistent with the hypothesis that PP pretreatment inhibited mitochondrial ATP generation (Figure 2D). Conversely, acute PP treatment caused a loss of mitochondrial oxidative phosphorylation along with a concomitant increase in glycolytic function (1.56 fold increase P<0.0001) (Figure 2E,F). These results complemented our metabolomics data where PP treatment resulted in altered glycolytic metabolites indicating a potential increase in glycolytic function. To prevent cells from upregulating glycolysis in response to PP treatment, we pre-treated PDAC cells with no-glucose media. PP treatment in these glucose-starved PDAC cells resulted in a significant decrease in ATP levels, which did not occur with the negative control gemcitabine, and could be rescued with co-treatment with glucose (Figure 2G). We demonstrated similar results with other mitochondrial inhibitors and were also able to replicate these results in PANC1 cells (Supplemental Figure 3C,D).
Figure 2. Pyrvinium Pamoate Inhibits Mitochondrial Oxidative Phosphorylation:
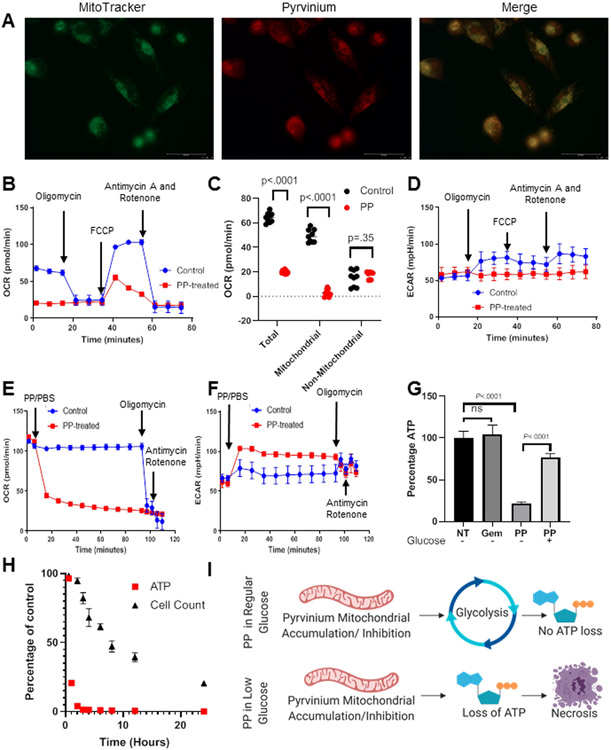
A) Treatment of MIA-PaCa2 cells with mitotracker (green) and PP (red) for one hour displayed co-localization of these two agents B,C,D) Cells were pretreated with either PP .3uM or vehicle for 12 hours prior to analysis of cellular Oxygen Consumption Rate B) which was used to quantify total, mitochondrial and Non-mitochondrial OCR (C) and concomitant ECAR measurements were also taken (D) E,F) Cells were treated with PP .3uM and OCR and ECAR were measured over time (n=1) G) Cells were treated with PP .3uM +/− glucose (25mM), gemcitabine (negative control) (no glucose), or vehicle and then ATP levels were determined (n=3) H) Cell treated with PP (.3μM) without glucose experience a decrease in ATP prior to a decrease in viability (n=3) I) In regular glucose conditions PP inhibits mitochondrial ATP production, however, cells can increase glycolysis leading to a protection from acute ATP loss. Contrastingly, in low glucose cells cannot rescue ATP production leading to necrosis.
As PP effectively inhibited mitochondrial oxidative phosphorylation, we investigated whether PP was more effective under relevant metabolic stress conditions that occur in the PDAC TME (i.e., hypoxia and hypoglycemia). We demonstrated that PP was similarly effective in normoxic and hypoxic conditions, although it was significantly more effective in hypoglycemic conditions (Supplemental Figure 3E). We demonstrated that the increased efficacy of PP in low glucose was due to a rapid reduction in ATP levels in glucose-starved PDAC cells, which was followed by a decrease in cellular viability, an effect which was negated with glucose supplementation (Figure 2H, Supplemental Figure 3F,G).
Overall, we found that acute PP treatment led to mitochondrial dysfunction which, in a glucose-deplete environment, led to a decrease in ATP levels, followed by cell death. These effects could be partially rescued by glucose (Figure 2I). Importantly, we demonstrated that these effects on mitochondria were preferential to cancer cells by comparing MIA-Paca2 sensitivity to MRC5 (immortalized fibroblasts) and HPNE (human pancreatic normal epithelial cells). PP caused less of a decrease in ATP production in MRC5 and HPNE cells as compared to MIA-PaCa2 cells (Supplemental Figure 4A). Accordingly MRC5 and HPNE cells were 111 and 6.6 fold less sensitive to PP induced cell-kill than MIA-PaCa2 cells (Supplemental Figure 4B). These data indicate that PDAC cells are more susceptible to PP treatment than normal cells.
To further augment the ability of PP to sensitize PDAC cells to their hypoglycemic TME (4), we co-treated cells with PP and the competitive inhibitor of glucose, hexokinase 2-deoxy-glucose (2DG) (37). Both agents were more effective in low glucose conditions, and they acted synergistically to inhibit ATP production (Supplemental Figure 4C-F). We demonstrated 93.9% decreased viability at clinically achievable levels of 2DG (.125mM) (37), and at low nanomolar levels of PP (25nM) (Supplemental Figure 4G).
PP decreases mitochondrial RNA
We performed RNA sequencing analysis comparing PP to the complex I inhibitor rotenone and the ATP synthase inhibitor oligomycin A. Geneset enrichment analyses indicated that PP significantly affected oxidative phosphorylation, fatty acid metabolism, glycolysis, and genesets related to cell cycle progression including E2F targets, MYC targets and G2M checkpoint (Figure 3A,B). Using MetaboAnalyst 5.0 we integrated metabolomic and RNA sequencing data (26) and found that PP affected pathways included the citric acid cycle, oxidative phosphorylation, pyrimidine biosynthesis, glycolysis or gluconeogenesis, central carbon in cancer and cell cycle (Figure 3C).
Figure 3. PP Regulates Mitochondrial RNA Expression Levels:
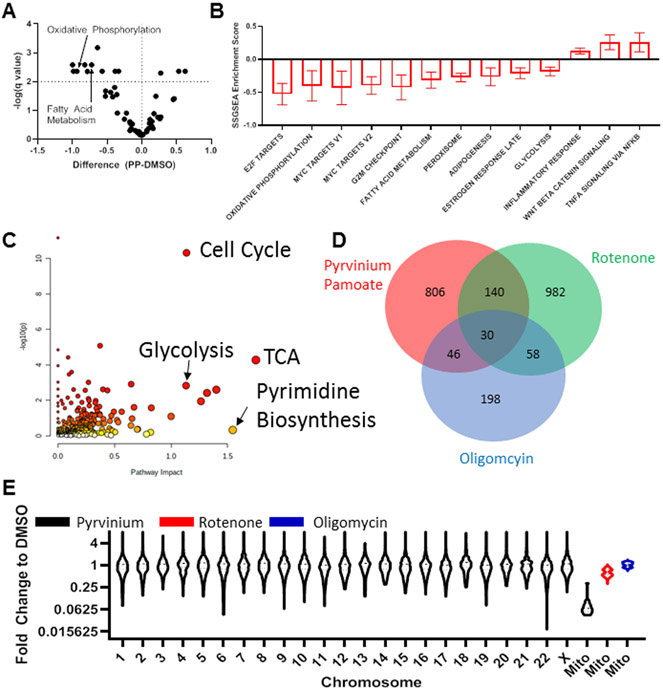
A) Changes induced after PP treatment as assessed using Geneset Enrichment Analysis B) Statistically significant changes from A C) Integrative analysis from RNA sequencing and metabalmics was assessed using MetaboAnalyst 5.0 indicating significant changes in Cell Cycle, Glycolysis, TCA and Pyrimidine biosynthesis among other pathways, for this graph along with x and y positioning larger points indicate increased pathway impact and p value is indicated by the color of points from least (white) to most (red) significantly effected D) Venn diagram demonstrating genes differentially regulated by PP, Rotenone and Oligomycin treatment as compared to DMSO E) PP changes to all genes as marked by chromosome or mitochondrial genes and compared to changes of mitochondrial transcripts as induced by rotenone and oligomycin
Comparing PP to oligomycin and rotenone we found of the 1022 genes that PP significantly altered, only 13.7% and 4.5% were similarly altered by rotenone or oligomycin respectively (Figure 3D). We also observed divergent geneset enrichment results. Interestingly, for genesets that encompassed mitochondrial function (i.e. oxidative phosphorylation, fatty acid metabolism, glycolysis) rotenone caused similar changes as compared to PP. Conversely cell cycle related pathway alterations caused by PP (i.e. E2F targets, G2M checkpoint, MYC Targets) were similarly affected by oligomycin treatment (Supplemental Figure 4H). These data suggest that PP inhibited the mitochondria in manner alternative to specific complex I or ATP synthase inhibition.
The mitochondrial genome is small in comparison to the nuclear genome, and the mitochondria encodes only 37 genes, with 13 of these genes encoding enzymes involved in oxidative phosphorylation. We observed that PP caused a significant reduction in all mitochondrial protein coding genes preferentially to changes induced in nuclear genes (Figure 3E). Furthermore, the down-regulation of mitochondrial genes was not seen with either rotenone or oligomycin A. We validated the decrease in mitochondrial RNA in separate qPCR experiments (Supplemental Figure 5A). We found that PP treatment also caused loss of protein expression of multiple members of the mitochondrial electron transport chain, which was not observed with the mitochondrial inhibitors oligomycin A or rotenone (Figure 4A,B).
Figure 4: PP binds mitochondrial DNA, ablates mitochondrial transcripts, and decreases mitochondrial protein expression:

A) Cells treated for 48 hours with PP.3μM, 4μM oligomycin A, 2μM rotenone, or 10μM MS444 displayed significant changes in protein levels of electron transport chain complex subunits B) PANC-1 and CAPAN-2 cells were treated with PP .3μM for 48 hours and protein levels of electron transport chain members were assessed C) PP and known G-Quadruplex binder Pyridostatin were assessed using a STOP-PCR assay D) RNA sequencing data was analyzed and known nuclear g-quadruplex targets were assessed for changes in response to PP, rotenone and oligomycin treatment along with mitochondrial g-quadruplex containing targets MT-ND1 and MT-ND2
To determine if loss of mitochondrial gene and protein expression could be a precursor or sign of mitophagy, we assessed mitochondrial DNA or mitochondrial mass as markers of mitophagy. Six hours of PP treatment caused a slight but significant decrease in mitochondrial DNA (22.9% P=0.0097) (Supplemental Figure 5B). Longer treatments (48 hours) did not cause significant changes in mitochondrial TOMM20 levels which are indicative of total mitochondrial mass (Supplemental Figure 5C). These data indicated that PP affected mitochondrial gene and protein expression in a manner that was not caused by specific inhibition of complex I or ATP synthase, and was not dependent on mitophagy.
PP binds mitochondrial G-quadruplexes and decreases mitochondrial RNA expression
DNA G-quadruplexes are tertiary structured formed in guanine rich regions of either DNA or RNA. The mitochondrial genome contains significantly more potential G-quadruplex forming regions per kb of DNA than nuclear DNA (38). These structures have roles in normal mitochondrial functions, however some compounds can bind to and stabilize mitochondrial G-quadruplexes thereby disrupting mitochondrial transcription (39). Mitochondrial genes, unlike nuclear genes, are transcribed in a polycistronic fashion, and thus, failing of the transcription bubble can inhibit the transcription of multiple genes (39). We hypothesized that PP was binding to and stabilizing mitochondrial G-Quadruplexes, thereby disrupting mitochondrial transcription. We found that PP weakly bound negative controls and had an increased binding for a known MYC G-Quadruplex and for multiple known and imputed mitochondrial G-Quadruplex target sequences (Supplemental Figure 5D) (28,30). We confirmed these results using stop-PCR analysis, where PP inhibited two mitochondrial targets with imputed G-quadruplexes (39) at concentrations that did not inhibit targets that lacked G-quadruplexes, similar to the known G-quadruplex inhibitor pyridostatin (Figure 4C) (40).
We assessed published nuclear G-quadruplexes, and while PP inhibited mitochondrial targets more than rotenone or oligomycin, it did not appear to have similar inhibitory effects on known nuclear G-quadruplex targets(41) (Figure 4D). Overall, we determined that PP bound to mitochondrial G-Quadruplexes, and led to a decrease in mitochondrial RNA and protein expression.
In vivo evaluation of PP
In a xenograft model of MIA-PaCa2 tumors, PP treatment three times weekly at 1mg/kg IP caused a 3.4-fold reduction in tumor growth while not significantly affecting mouse weights (Figure 5A P<0.0001, Supplemental Figure 5E). Comparing tumors from the vehicle control group to the PP treated group, we found there was a significant increase in TUNEL staining (24.7-fold P<0.0001 Figure 5B, Supplemental Figure 5F). Furthermore, PP treatment led to a 1.5-fold increase in median survival (Figure 5C P=.0036). We treated mice with human grade PP (Pyrvin 100mg tablets, Orion Pharma, Espoo, Finland) at 5, 20 and 35mg/kg. Daily oral treatment of PP led to significant and dose-dependent reductions in tumor growth, although 35mg/kg daily treatment caused a slight but significant decrease in average mouse weight gain (4.6%, p=.0174) (Figure 5D, Supplemental Figure 5G).
Figure 5: PP efficacy in-vivo:
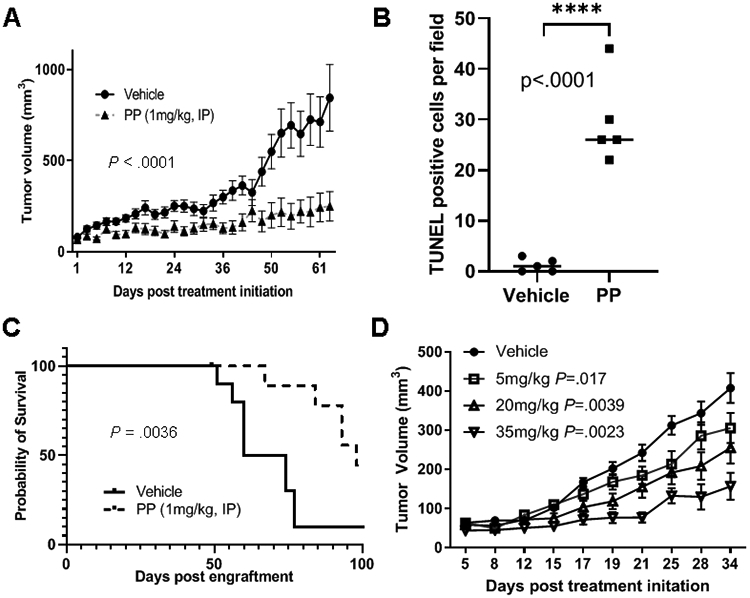
A) Five mice per arm were treated with either vehicle or 1mg/kg PP IP after tumors reached 100mm3 B) Tumors from A were TUNEL stained indicating an increase in apoptosis in PP treated tumors C) Ten mice per arm were treated with IP 1mg/kg PP after tumors reached 150mm3, mice were sacrificed when tumors reached 2000mm3 D) Mice were treated PO (orally) with crushed Pyrvin (human-grade PP) at 5, 20 and 35mg/kg
We performed pharmacokinetic profiling of PP to determine its bioavailability. We initially dosed animals IV at 1mg/kg and determined plasma half-life was 0.54 hours (Supplemental Figure 6A). We evaluated PP accumulation in tissues by treating mice with single doses of PP at 1mg/kg IP, or with PO dosing of 5, 20, or 35 mg/kg PP and collected plasma, muscle, fat and pancreas tissues (Supplemental Tables 2A,B). We determined across all groups that while there was little to no detectable serum accumulation, PP appeared to accumulate at the pancreas and in fat tissue (Supplemental Figure 6B-E). Interestingly, we observed significant accumulation of PP at target organ sites even at concentrations where PP was undetectable in the plasma (Supplemental Figure 6F,G). This may explain the discrepancy where PP has previously been observed to have low oral bioavailability in plasma (42), but has anti-tumor effects when dosed orally (43,44).
In vivo plasma metabolomics results from mice treated with PP at 1mg/kg largely corroborated the hypothesis that PP affected mitochondria systemically in vivo. In particular, we observed an increase in plasma levels of N-carbamoyl aspartate along with a concomitant decrease in orotate, and a trend towards a decrease in all un-modified acylcarnitines tested (Supplemental Figure 6H,I). These data were highly similar to the metabolomic results we observed in vitro indicating similar systemic alteration in mitochondrial pathways including de novo pyrimidine biosynthesis and fatty acid metabolism.
PP as a potential therapeutic in other cancer types
Beyond PDAC, we explored whether PP was effective in other cancers utilizing publicly available data from the NCI60. We observed that PP appeared to be particularly effective in colon, leukemia and lung cell lines, with renal cell lines displaying the least sensitivity (Supplemental Figure 6J). PP efficacy correlated with the expression of mitochondrial related genes MDH2, NDUFB11, HARS2, and inversely correlated with the drug transporter ABCB1 (Supplemental Figure 6K). These data support the hypothesis that PP works through targeting the mitochondria, and may be resisted through the up-regulation of drug-efflux pathways.
Discussion
In this study, we demonstrated that PP is effective for inhibiting PDAC cell growth and proliferation in-vitro and in vivo (Figure 5). We determined through phosphoproteomic, metabolomic, and RNA sequencing approaches (Figure 1,3 Supplemental Figure 2) that PP was acting predominantly through altering cellular metabolism, and validated that PP inhibited mitochondrial oxidative phosphorylation (Figure 2).
Mitochondrial depleted cells are resistant to PP confirming PP’s action through targeting mitochondria(13), however how PP inhibits mitochondria is less well understood. Previous work proposed that PP inhibits fumarate reductase, a critical member in an alternative mitochondrial pathway proposed to function in cancer cells in hypoxic conditions whereby mitochondria can generate ATP in an oxygen independent fashion(12). As PP effectively inhibits cancer cell mitochondria in non-hypoxic settings, specific targeting of this pathway is unlikely. PP has also previously been shown to inhibit complex I in the mitochondria (13). However it has previously been demonstrated that specifically inhibiting complex I causes an increase in beta oxidation as indicated by slight increases in free carnitine and long-chain acyl carnitines (45). We demonstrated that PP causes a larger increase in long-chain acyl carnitines along with increases in Acylcarnitine C4-OH and Acylcarnitine 5-DC, and decreases in short chain acylcarnitines and free carnitine (Figure 1). Taken together, this is indicative of an inhibition of beta oxidation, rather than the increase of beta oxidation that would be expected if PP was solely inhibiting complex I (35,45). Similarly, the increase in N-Carbamoyl aspartate and decrease in orotate observed with PP treatment (Figure 1, Supplemental Figure 6) suggests an inhibition of DHODH, which can be caused through the specific inhibition of complex III but not complex I (46). The inhibition of DHODH may further augment PP’s efficacy in PDAC as KRAS driven tumors such as PDAC are uniquely sensitive to DHODH inhibition (47). Furthermore, PP treatment caused transcriptomic changes which were largely distinct from those caused by the complex I inhibitor rotenone (Figure 3). Overall, our data indicated that PP was not acting through the specific inhibition of Complex I, or fumarate reductase, but rather a more global inhibition of mitochondrial function.
We further identified a novel mechanism whereby PP caused ablation of mitochondrial RNA expression (Figure 3) followed by downregulation of mitochondrial protein expression (Figure 4). Our results indicate that this ablation of mitochondrial RNA is through PP binding mitochondrial DNA at regions of mitochondrial G-quadruplexes, and thus inhibiting transcription (Figure 4). This is supported by previous work which demonstrated that the G-quadruplex stabilizer RHPS4 bound mitochondrial G-quadruplexes and caused a depletion in mitochondrial RNA (39). This novel mechanism underlies the multifaceted mitochondrial effects PP treatment causes through affecting multiple mitochondrial pathways simultaneously.
We demonstrated that PP is more effective at inhibiting ATP levels and cell viability in PDAC cells as compared to non-cancerous cells. Beyond this increased sensitivity, the hypoglycemic TME characteristic of PDAC may further sensitize PDAC cancers to PP in comparison to normal tissues (4). Importantly for a predominantly metastatic disease, cancer stem cells which are critical for metastatic spread are critically reliant on oxidative phosphorylation (48). This reliance of primary PDAC tumors and metastases on mitochondrial pathways may be why targeting the mitochondria in PDAC models has shown such promising results (7-9).
Of note, previous work performed in the 1970’s indicated that there was poor systemic bioavailability of PP after oral treatment (42). We have demonstrated in this work that PP has therapeutic efficacy for PDAC when dosed orally in mice (Figure 5D), and this is supported by other studies where oral administration of PP has demonstrated significant systemic efficacy (43,44). In fact, the doses used in this study span the standard daily dosing of PP in humans (5mg/kg) to the maximum dose utilized in humans (35mg/kg)(49). Indeed, 35mg/kg does not represent a true maximum tolerated dose as higher doses have not been tested, and rhesus monkeys have been treated up to 80mg/kg daily of PP(50). In pre-clinical characterization studies of PP, we demonstrated that PP accumulated at target tissues after oral administration (Supplemental Figure 6) while still being undetectable in plasma. This may explain why PP bioavailability appears to be poor based on drug accumulation in the plasma, yet PP demonstrates in vivo efficacy against tumors. We believe this work supports repurposing PP for the treatment of PDAC. Based on this work, we have received IND approval for the treatment of PDAC patients with PP (IND 144822) and have recently received IRB approval to further explore PP as an anti-PDAC agent in human patients.
Supplementary Material
Financial Information:
R01CA212600-01 (J.R. Brody), R01CA243445 (D.A. Dixon), R01 CA163649 (P.K. Singh), R01CA216853 (P.K. Singh), P01CA217798 (P.K. Singh), P50CA127297 (P.K. Singh) NCI-CCR Intramural support Z01 006150 (Y. Pommier), T32 training grant NIH/NIGMS T32GM008562 (T. Dhir), the Mary Halinski Pancreatic Cancer Research Fund (J.R. Brody and A. Nevler). Dr. Brody was supported in part by the NCI of the NIH Award Number P30CA056036 SKCC Core Grant (Thomas Jefferson University); OHSU Knight Cancer Institute P30 CA069533 (Druker); in part, by U01CA224012 (NCI, NIH).
Abbreviations:
- ATP
Adenosine triphosphate
- DHODH
Dihydroorotate dehydrogenase
- ECAR
Extracellular Acidification Rate
- KO
Knockout
- OCR
Oxygen Consumption Rate
- PDAC
Pancreatic Ductal Adenocarcinoma
- PP
Pyrvinium
- TME
Tumor Microenvironment
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Footnotes
Disclosures: The authors have declared that no conflict of interest exists.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68(1):7–30 doi 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys 2000;48(4):919–22 doi 10.1016/s0360-3016(00)00803-8. [DOI] [PubMed] [Google Scholar]
- 3.Blanco FF, Jimbo M, Wulfkuhle J, Gallagher I, Deng J, Enyenihi L, et al. The mRNA-binding protein HuR promotes hypoxia-induced chemoresistance through posttranscriptional regulation of the proto-oncogene PIM1 in pancreatic cancer cells. Oncogene 2016;35(19):2529–41 doi 10.1038/onc.2015.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 2015;75(3):544–53 doi 10.1158/0008-5472.CAN-14-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaziri-Gohar A, Zarei M, Brody JR, Winter JM. Metabolic Dependencies in Pancreatic Cancer. Front Oncol 2018;8:617 doi 10.3389/fonc.2018.00617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto K, Brender JR, Seki T, Kishimoto S, Oshima N, Choudhuri R, et al. Molecular Imaging of the Tumor Microenvironment Reveals the Relationship between Tumor Oxygenation, Glucose Uptake, and Glycolysis in Pancreatic Ductal Adenocarcinoma. Cancer Res 2020. doi 10.1158/0008-5472.CAN-19-0928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Q, Li M, Gan Y, Jiang S, Qiao J, Zhang W, et al. Mitochondrial Protein UQCRC1 is Oncogenic and a Potential Therapeutic Target for Pancreatic Cancer. Theranostics 2020;10(5):2141–57 doi 10.7150/thno.38704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu M, Nguyen ND, Huang Y, Lin D, Fujimoto TN, Molkentine JM, et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 2019;5 doi 10.1172/jci.insight.126915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alistar A, Morris BB, Desnoyer R, Klepin HD, Hosseinzadeh K, Clark C, et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol 2017;18(6):770–8 doi 10.1016/S1470-2045(17)30314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones JO, Bolton EC, Huang Y, Feau C, Guy RK, Yamamoto KR, et al. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc Natl Acad Sci U S A 2009;106(17):7233–8 doi 10.1073/pnas.0807282106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu DH, Macdonald J, Liu G, Lee AS, Ly M, Davis T, et al. Pyrvinium targets the unfolded protein response to hypoglycemia and its anti-tumor activity is enhanced by combination therapy. PLoS One 2008;3(12):e3951 doi 10.1371/journal.pone.0003951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomitsuka E, Kita K, Esumi H. An anticancer agent, pyrvinium pamoate inhibits the NADH-fumarate reductase system--a unique mitochondrial energy metabolism in tumour microenvironments. J Biochem 2012;152(2):171–83 doi 10.1093/jb/mvs041. [DOI] [PubMed] [Google Scholar]
- 13.Harada Y, Ishii I, Hatake K, Kasahara T. Pyrvinium pamoate inhibits proliferation of myeloma/erythroleukemia cells by suppressing mitochondrial respiratory complex I and STAT3. Cancer Lett 2012;319(1):83–8 doi 10.1016/j.canlet.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 14.Deng L, Lei Y, Liu R, Li J, Yuan K, Li Y, et al. Pyrvinium targets autophagy addiction to promote cancer cell death. Cell Death Dis 2013;4:e614 doi 10.1038/cddis.2013.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo J, Lv J, Chang S, Chen Z, Lu W, Xu C, et al. Inhibiting cytoplasmic accumulation of HuR synergizes genotoxic agents in urothelial carcinoma of the bladder. Oncotarget 2016;7(29):45249–62 doi 10.18632/oncotarget.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Dai Z, Luo Z, Zuo C. Identification of Pyrvinium, an Anthelmintic Drug, as a Novel Anti-Adipogenic Compound Based on the Gene Expression Microarray and Connectivity Map. Molecules 2019;24(13) doi 10.3390/molecules24132391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lal S, Cheung EC, Zarei M, Preet R, Chand SN, Mambelli-Lisboa NC, et al. CRISPR Knockout of the HuR Gene Causes a Xenograft Lethal Phenotype. Mol Cancer Res 2017;15(6):696–707 doi 10.1158/1541-7786.MCR-16-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhir T, Schultz CW, Jain A, Brown SZ, Haber A, Goetz A, et al. Abemaciclib Is Effective Against Pancreatic Cancer Cells and Synergizes with HuR and YAP1 Inhibition. Mol Cancer Res 2019. doi 10.1158/1541-7786.MCR-19-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golan T, Stossel C, Schvimer M, Atias D, Halperin S, Buzhor E, et al. Pancreatic cancer ascites xenograft-an expeditious model mirroring advanced therapeutic resistant disease. Oncotarget 2017;8(25):40778–90 doi 10.18632/oncotarget.17253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang W, Dulaimi E, Devarajan K, Parsons T, Wang Q, Liao L, et al. Immunohistochemistry Successfully Uncovers Intratumoral Heterogeneity and Widespread Co-Losses of Chromatin Regulators in Clear Cell Renal Cell Carcinoma. PLoS One 2016;11(10):e0164554 doi 10.1371/journal.pone.0164554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wulfkuhle JD, Aquino JA, Calvert VS, Fishman DA, Coukos G, Liotta LA, et al. Signal pathway profiling of ovarian cancer from human tissue specimens using reverse-phase protein microarrays. Proteomics 2003;3(11):2085–90 doi 10.1002/pmic.200300591. [DOI] [PubMed] [Google Scholar]
- 22.Gunda V, Yu F, Singh PK. Validation of Metabolic Alterations in Microscale Cell Culture Lysates Using Hydrophilic Interaction Liquid Chromatography (HILIC)-Tandem Mass Spectrometry-Based Metabolomics. PLoS One 2016;11(4):e0154416 doi 10.1371/journal.pone.0154416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010;26(7):966–8 doi 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, et al. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 2018;46(W1):W486–W94 doi 10.1093/nar/gky310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7 doi 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chong J, Wishart DS, Xia J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr Protoc Bioinformatics 2019;68(1):e86 doi 10.1002/cpbi.86. [DOI] [PubMed] [Google Scholar]
- 27.Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016;32(18):2866–8 doi 10.1093/bioinformatics/btw230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karg B, Funke A, Ficht A, Sievers-Engler A, Lammerhofer M, Weisz K. Molecular Recognition and Visual Detection of G-Quadruplexes by a Dicarbocyanine Dye. Chemistry 2015;21(39):13802–11 doi 10.1002/chem.201502118. [DOI] [PubMed] [Google Scholar]
- 29.Stockert JC, Trigoso CI, Llorente AR, Del Castillo P. DNA fluorescence induced by polymethine cation pyrvinium binding. Histochem J 1991;23(11-12):548–52 doi 10.1007/bf01041182. [DOI] [PubMed] [Google Scholar]
- 30.Huang WC, Tseng TY, Chen YT, Chang CC, Wang ZF, Wang CL, et al. Direct evidence of mitochondrial G-quadruplex DNA by using fluorescent anti-cancer agents. Nucleic Acids Res 2015;43(21):10102–13 doi 10.1093/nar/gkv1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rajapakse VN, Luna A, Yamade M, Loman L, Varma S, Sunshine M, et al. CellMinerCDB for Integrative Cross-Database Genomics and Pharmacogenomics Analyses of Cancer Cell Lines. iScience 2018;10:247–64 doi 10.1016/j.isci.2018.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng W, Furuuchi N, Aslanukova L, Huang YH, Brown SZ, Jiang W, et al. Elevated HuR in Pancreas Promotes a Pancreatitis-Like Inflammatory Microenvironment That Facilitates Tumor Development. Mol Cell Biol 2018;38(3) doi 10.1128/MCB.00427-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schultz CW, Preet R, Dhir T, Dixon DA, Brody JR. Understanding and targeting the disease-related RNA binding protein human antigen R (HuR). Wiley Interdiscip Rev RNA 2020;11(3):e1581 doi 10.1002/wrna.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lal P, Cerofolini L, D'Agostino VG, Zucal C, Fuccio C, Bonomo I, et al. Regulation of HuR structure and function by dihydrotanshinone-I. Nucleic acids research 2017;45(16):9514–27 doi 10.1093/nar/gkx623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rinaldo P, Cowan TM, Matern D. Acylcarnitine profile analysis. Genet Med 2008;10(2):151–6 doi 10.1097/GIM.0b013e3181614289. [DOI] [PubMed] [Google Scholar]
- 36.Xiang W, Cheong JK, Ang SH, Teo B, Xu P, Asari K, et al. Pyrvinium selectively targets blast phase-chronic myeloid leukemia through inhibition of mitochondrial respiration. Oncotarget 2015;6(32):33769–80 doi 10.18632/oncotarget.5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol 2013;71(2):523–30 doi 10.1007/s00280-012-2045-1. [DOI] [PubMed] [Google Scholar]
- 38.Falabella M, Fernandez RJ, Johnson FB, Kaufman BA. Potential Roles for G-Quadruplexes in Mitochondria. Curr Med Chem 2019;26(16):2918–32 doi 10.2174/0929867325666180228165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Falabella M, Kolesar JE, Wallace C, de Jesus D, Sun L, Taguchi YV, et al. G-quadruplex dynamics contribute to regulation of mitochondrial gene expression. Sci Rep 2019;9(1):5605 doi 10.1038/s41598-019-41464-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moruno-Manchon JF, Koellhoffer EC, Gopakumar J, Hambarde S, Kim N, McCullough LD, et al. The G-quadruplex DNA stabilizing drug pyridostatin promotes DNA damage and downregulates transcription of Brca1 in neurons. Aging (Albany NY) 2017;9(9):1957–70 doi 10.18632/aging.101282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Asamitsu S, Obata S, Yu Z, Bando T, Sugiyama H. Recent Progress of Targeted G-Quadruplex-Preferred Ligands Toward Cancer Therapy. Molecules 2019;24(3) doi 10.3390/molecules24030429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith TC, Kinkel AW, Gryczko CM, Goulet JR. Absorption of pyrvinium pamoate. Clin Pharmacol Ther 1976;19(6):802–6 doi 10.1002/cpt1976196802. [DOI] [PubMed] [Google Scholar]
- 43.Esumi H, Lu J, Kurashima Y, Hanaoka T. Antitumor activity of pyrvinium pamoate, 6-(dimethylamino)-2-[2-(2,5-dimethyl-1-phenyl-1H-pyrrol-3-yl)ethenyl]-1-methyl-qu inolinium pamoate salt, showing preferential cytotoxicity during glucose starvation. Cancer Sci 2004;95(8):685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li B, Flaveny CA, Giambelli C, Fei DL, Han L, Hang BI, et al. Repurposing the FDA-approved pinworm drug pyrvinium as a novel chemotherapeutic agent for intestinal polyposis. PLoS One 2014;9(7):e101969 doi 10.1371/journal.pone.0101969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Worth AJ, Basu SS, Snyder NW, Mesaros C, Blair IA. Inhibition of neuronal cell mitochondrial complex I with rotenone increases lipid beta-oxidation, supporting acetyl-coenzyme A levels. J Biol Chem 2014;289(39):26895–903 doi 10.1074/jbc.M114.591354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khutornenko AA, Roudko VV, Chernyak BV, Vartapetian AB, Chumakov PM, Evstafieva AG. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc Natl Acad Sci U S A 2010;107(29):12828–33 doi 10.1073/pnas.0910885107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koundinya M, Sudhalter J, Courjaud A, Lionne B, Touyer G, Bonnet L, et al. Dependence on the Pyrimidine Biosynthetic Enzyme DHODH Is a Synthetic Lethal Vulnerability in Mutant KRAS-Driven Cancers. Cell Chem Biol 2018;25(6):705–17 e11 doi 10.1016/j.chembiol.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 48.Snyder V, Reed-Newman TC, Arnold L, Thomas SM, Anant S. Cancer Stem Cell Metabolism and Potential Therapeutic Targets. Front Oncol 2018;8:203 doi 10.3389/fonc.2018.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wagner ED. Pyrvinium pamoate in the treatment of strongyloidiasis. Am J Trop Med Hyg 1963;12:60–1 doi 10.4269/ajtmh.1963.12.60. [DOI] [PubMed] [Google Scholar]
- 50.Thompson PE, Worley DE, Meisenhelder JE. Anthelmintic studies on pyrvinium pamoate (Povan) and other drugs in rodents, dogs, and monkeys. Am J Trop Med Hyg 1962;11:89–95 doi 10.4269/ajtmh.1962.11.89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.