

Abstract
Overlapping substrate specificities within the family of matrix metalloproteinases (MMPs), usually caused by their highly conserved structural topology, increase the potential for a substrate to be cleaved by multiple enzymes within this family, which leads to the decrease in the selectivity of MMP substrate-based probes. To resolve this issue, MT1-MMP activatable fluorogenic probes for tumor detection with enhanced specificity were developed by combining a fluorescence resonance energy transfer (FRET) peptide substrate and its specific binding peptide with different lengths of linkers. The specificity of the probes increased profiting from the high affinity of the MT1-MMP specific binding peptide while keeping the ability to amplify the output imaging signals in response to MMP activity with the FRET substrate. Enzyme kinetics analysis clearly demonstrated that the conjugation of P-1 and MT1-AF7p enhanced both the specificity and selectivity of the fluorogenic probes for MT1-MMP, and introducing a linker composed of 12 PEG subunits into these two fragments led to optimized specificity and selectivity of the fluorogenic probe for MT1-MMP. Both in vitro and in vivo results revealed that the imaging probe with the linker composed of 12 PEG subunits based on our designed strategy could be effectively applied for MT1-MMP positive tumor imaging. Since this strategy for enhancing the specificity of protease sensing probes can be applied to other proteases and is not just limited to MT1-MMP, it is an appealing platform to achieve selective tumor imaging.
Introduction
Under the initiative of developing precision medicine, there is an urgent need to visualize diseased cells to enable diagnosis, facilitate surgical resection, and monitor therapeutic response.
The primary challenge for imaging in cancer treatment is finding effective ways to define the boundaries between tumors and healthy surrounding tissues at the cellular level.1 Hence, the specificity of a probe is the basis for precision tumor imaging. Compared to other imaging modalities, optical imaging, which is non-invasive, relatively inexpensive, and simple to perform, has great potential to be utilized in precision medicine for cancer treatment, as exemplified by optical surgical guidance with molecular precision, etc. Therefore, there is an unmet medical need for highly specific and sensitive targeted fluorogenic probes for cancer imaging.
Matrix metalloproteinases (MMPs) have been demonstrated to play key roles in the progression and dissemination of cancer, including the modulation of tumor-associated angiogenesis, rendering them valuable targets of probes designed for tumor diagnosis and therapy.2 MMPs can be principally categorized into two types: secreted soluble type (extracellular MMPs, EC-MMPs) and membrane-tethered type (membrane-type MMPs, MT-MMPs). As active secreted MMPs are known to circulate in the bloodstream,3 the latter subtype with a more fixed localization due to its unique membrane-tethering property is deemed to be a valuable candidate for targeting in precision imaging to provide a direct cellular target for molecular imaging to detect invasive cancer cells. Among MT-MMPs, MT1-MMP is the most prominent member, and it has been demonstrated to play an essential role in cell migration, invasion, cell morphology, metastasis, and angiogenesis in normal physiological and pathological events.4–7 Therefore, the development of an MT1-MMP probe for the in vivo monitoring of the activity of MT1-MMP in cancers is a valuable approach for precision imaging in tumor detection and disease control.
To date, various kinds of MMP imaging probes have been developed.8–13 Based on the primary strategies used to induce tumor-specific probe accumulation, these probes can be principally divided into two categories: affinity-based probes and substrate-based probes.1,13 Affinity-based probes mostly rely on the specific affinity between a peptide/inhibitor and MMPs that are overexpressed in cancer cells or tumor-associated cells, indicating reasonable practicability, but their use might be limited to high background signals due to non-specific molecular recognition.8,14 Substrate-based probes mostly rely on the specific interaction and catalyzation between MMPs and their substrates.13,15 Since MMPs are one of the typical subtypes of proteases in humans, their altered activity in a disease of interest cannot be simply connected to a differential expression of the protein,16 and so the activity based detection of MMPs by using substrate-based probes has became a promising strategy for disease diagnosis. Among the substrate-based probes, the most prominent ones used for imaging MMPs are fluorogenic activatable probes, which are able to amplify output imaging signals in response to the activity of MMPs in real time and provide high-resolution imaging with ultralow background signals. Currently, one activatable cell penetrating peptide-based probe for detecting MMPs’ activity, AVB-620, is undergoing an initial pilot study in patients with breast cancer (NCT02391194). Although these MMP substrate-based probes have achieved success in the area of tumor imaging, they still suffer from limitations preventing the establishment of sufficient imaging contrast at disease sites. For example, overlapping substrate specificities within the family of MMP enzymes due to their highly conserved structural topology adds the potential for a substrate to be cleaved by multiple enzymes within this family,17–19 which leads to a decreasing selectivity of MMP substrate-based probes. Hence, the development of highly sensitive and specific MMP substrate-based probes for cancer imaging still remains a daunting challenge.
Strategies have been developed to improve the specificity of the MMP substrate-based imaging probes, including screening the substrate sequence to improve the specificity for its given enzyme13,19 or introducing an exosite-binding domain adjacent to a protease cleavage site, which improves both the affinity and specificity for its given enzyme.20 Based on these strategies, the designed probes have been successfully used to profile the activity of the MMPs in tumors; however, their specificity and sensitivity for MMP could be further improved, and new strategies are still needed to address this issue.
In this study, we report a novel strategy to enhance the specificity of MT1-MMP activatable fluorogenic probes for tumor detection with the combination of an advanced MMP (fluorescence resonance energy transfer) FRET substrate and its specific binding peptide via different lengths of linkers (Fig. 1). The specificity of the probes was increased profiting from the high affinity of the MT1-MMP specific binding peptide, while keeping the ability to amplify the output imaging signals in response to MMP activity with the FRET peptide substrate.
Fig. 1.

Schematic illustration of the imaging process of the designed MT1-MMP probe.
Both in vitro and in vivo “proof-of-concept” investigations were carried out to confirm the feasibility and success of this innovative strategy for effective tumor-imaging via enhancing the specificity of the MT1-MMP activatable fluorogenic probes.
Experimental
Materials
Isopropyl-β-d-1-thiogalactopyranoside (IPTG), yeast extract, and tryptone were provided by Sigma-Aldrich (Saint Louis, MO). Competent E. coli BL21(DE3)pLysS cells were obtained from CWBIO (Beijing, China). The nickel affinity column was purchased from QIAGEN (Hilden, Germany). PageRuler-prestained protein ladder, RPMI1640 and DMEM medium were provided by Thermo Fisher Scientific (Waltham, MA). Anti-MMP14 antibody was purchased from Abcam (Cambridge, MA). DAPI and the DiI dye were purchased from Beyotime Biotechnology (Shanghai, China). The broad-spectrum MMP inhibitor GM6001 was purchased from Bio-Techne China Co. Ltd (Shanghai, China). All the other chemicals were of the highest grade commercially available.
Cell lines and animals
HT-1080 (human fibrosarcoma), MCF-7 (human breast adenocarcinoma), and HUVEC (human umbilical vein endothelial cell) cell lines were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). BALB/c nude mice (4 weeks old) were obtained from the Chinese Academy of Military Medical Sciences (Beijing, China). All the animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Tianjin Medical University and approved by the Animal Ethics Committee of Tianjin Medical University.
Synthesis of MT1-MMP substrate-based fluorogenic probes
All the designed probes were synthesized by ChinaPeptides Corporation via standard solid-phase Fmoc peptide chemistry, the sequences of which are shown in Tables 1 and 2. The structures of these probes were verified by electrospray ionization-mass spectrometry (ESI-MS), and the purity of these probes (95%) was analyzed by high-performance liquid chromatography.
Table 1.
Kinetic constants of the fluorogenic probes catalyzed by MT1-MMP
| Probes | Sequences | Km (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) |
|---|---|---|---|---|
| P-l | (Mca)-PLGL-(Dpa)-AR-NH2 | 2.14 ± 0.15 | 0.97 × 10−3 ± 0.05 × 10−3 | 0.45 × 10−3 ± 0.03 × 10−3 |
| P-3 | (Mca)-PLGL-(Dpa)-AR-(PEG)3-HWKHLHNTKTFL | 0.72 ± 0.03 | 1.20 × 10−3 ± 0.05 × 10−3 | 1.68 × 10−3 ± 0.74 × 10−3 |
| P-4 | (Mca)-PLGL-(Dpa)-AR-(PEG)6-HWKHLHNTKTFL | 0.73 ± 0.06 | 3.21 × 10−3 ± 0.20 × 10−3 | 4.42 × 10−3 ± 0.37 × 10−3 |
| P-5 | (Mca)-PLGL-(Dpa)-AR-(PEG)12-HWKHLHNTKTFL | 0.40 ± 0.03 | 2.44 × 10−3 ± 0.04 × 10−3 | 6.15 × 10−3 ± 0.44 × 10−3 |
| P-6 | (Mca)-PLGL-(Dpa)-AR-(PEG)24-HWKHLHNTKTFL | 1.75 ± 0.14 | 1.94 × 10−3 ± 0.10 × 10−3 | 1.11 × 10−3 ± 0.09 × 10−3 |
Table 2.
Sequence of the MT1-MMP fluorogenic probe for cell imaging
| Probes | Sequences |
|---|---|
| P-7 | (FAM)-PLGLK-(Dabcyl)-AR-(PEG)12-HWKHLHNTKTFL |
| P-8 | (FAM)-PLGLK-(Dabcyl)-AR-NH2 |
| P-9 | (FAM)-PLGLK-(Dabcyl)-AR-HWKHLHNTKTFL |
Expression of the recombinant MT1-MMP
The DNA sequence encoding the catalytic and hinge domains of human MT1-MMP was synthesized by Genewiz Company, and subsequently subcloned into the pET30a vector. The resultant expression construct pET30a-rMT1-MMP-(His)6 was transformed into an E. coli expression strain BL21(DE3)pLysS. Cells were cultured with 2×YT media with 100 mg mL−1 ampicillin at 37 °C. When the optical density at 600 nm reached 0.6–0.8, the cells were induced for 16 h at 15 °C by adding IPTG to a final concentration of 0.5 mM. After induction, the cells were harvested.
Purification and refolding of the recombinant MT1-MMP
The recombinant MT1-MMP was purified and refolded according to the reported method.21 Briefly, a cell pellet was suspended in lysis buffer (50 mM Tris-HCl, 6 M urea, 10 mM imidazole, and 30 mM 2-mercaptoethanol, pH 8.0) at 10 mL per gram wet weight, and then lyzed by sonication. After that, the cell lysate was centrifuged at 12 000 rpm for 1 h and the supernatant was loaded onto Ni-NTA resin pre-equilibrated with lysis buffer. The target protein was eluted with 250 mM imidazole, pH 8.0. To refold the recombinant MT1-MMP, the eluted protein was diluted with buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM CaCl2, 0.5 mM ZnCl2, 150 mM 2-mercaptoethanol, pH 8.0) to 100 μg mL−1 and was gradually dialyzed with a refolding buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM CaCl2, 0.5 mM ZnCl2, pH 8.0). Before harvesting, the protein was dialyzed with a refolding buffer containing 0.05% Brij 35.
Enzyme assay and kinetic experiments
Recombinant human MMP-2 was expressed and purified according to the reported procedure.22 The catalytic activity of recombinant MT1-MMP and MMP-2 was analyzed by a peptide cleavage assay using FRET peptides based on the reported protocols.23 Briefly, fluorescence generated from hydrolysis of the peptide bond was detected at the excitation wavelength of 328 nm and at the emission wavelength of 393 nm. The enzyme reactions were performed in the MT1-MMP assay buffer (20 mM Tris-HCl, 150 mM NaCl, 5 mM CaCl2, 0.1 mM ZnCl2, 0.05% Brij35, pH 8.0) or in the MMP-2 assay buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 10 mM CaCl2, 0.1 mM ZnCl2, and 0.05% Brij 35) in the presence of various concentrations of fluorogenic probes and MT1-MMP or MMP-2 at 37 °C. For the kinetic experiments, 15 nM of the refolded MT1-MMP and different concentrations (0.5–4.5 μM) of the fluorogenic probes were reacted in 200 μL of the MMP assay buffer. The initial rates of the substrate cleavage measured over 28 s were proportional to the substrate concentrations. Km and Vmax were determined from a Michaelis-Menten curve, and kcat was calculated from the Vmax/concentration of the enzyme.
Cell cytotoxicity assay
The cytotoxicity of P-7 was determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay by using the HUVEC cells based on a modified manufacturer’s protocol. Briefly, cells were seeded into 96-well plates at a density of 4 × 103–5 × 103 cells per well and incubated for 12 h. Then, the cells were treated with P-7 at set concentrations (from 10 μM to 100 μM). After an additional 48 h incubation, 5 mg mL−1 MTT in PBS (20 μL) was added to each well and incubated for another 4 h. Finally, the medium was replaced with 150 μL of DMSO to dissolve the purple formazan crystals. The absorbance intensity in each well was detected at 490 nm by a microplate spectrophotometer (SpectraMax M2E (Molecular Device, Inc.)).
Cell imaging
HT-1080 cells were cultured in RPMI 1640 medium and MCF-7 cells were cultured in high glucose DMEM medium, with 10% fetal bovine serum and 100 μg mL−1 penicillin, and 100 μg mL−1 streptomycin under standard cell culture conditions in a well-humidified incubator with 5% CO2 and 95% air at 37 °C. The glass bottom dishes were added in a 24-well plate for cell imaging before the cells were seeded. Then, the cells were transferred to the 24-well plate and cultured for one night before the experiments. After that, the culture medium was replaced with a fresh one. The dishes were further incubated with the probe in a 37 °C incubator for 2 h. After incubation, the cells were quickly washed with PBS three times, then fixed with 4% paraformaldehyde solution for 10 min. Finally, the cells were washed with PBS and stained with DAPI before imaging.
Ex vivo tumor imaging
HT-1080 tumor models were established by subcutaneous injection of the suspensions of HT-1080 cells (1 × 106 cells in PBS buffer) into the right flank of nude mice. When the average tumor volume reached approximately 0.5–1 cm3, 12 mice with similar tumor volumes were selected and divided into four groups (n = 3 per group). The tumor-bearing mice of each group were intravenously injected with 50 nmol of probe. The tumors were collected for cryosection 4.5 h post injection. Then, the slides were stained with DAPI and DiI. The fluorescence was observed with a confocal microscope (Olympus, Japan).
Optical in vivo imaging
As described previously, 12 mice with a similar size of tumor were divided into four experimental groups. Then the tumor-bearing mice were intratumorally injected with 50 nmol of the probe/control, and an IVIS spectrum imaging system (PerkinElmer, Massachusetts, USA) was used to detect the FAM fluorescence (Ex/Em = 492/518 nm). During the imaging, the mice were anesthetized with 2.5% isoflurane gas in an oxygen flow (1.5 L min−1). The recorded images were analyzed using Living Image 4.3.1 software (Xenogen).
Results and discussion
Design of the new MT1-MMP fluorogenic probes
According to the principle of our designed imaging probes, conjugation of the MMP FRET substrate and its specific binding peptide would lead to enhanced specificity of the probes for MMP detection, where the specificity of the probes was proposed to increase profiting from the high affinity of the MMP specific binding peptide, while keeping the ability of amplify output imaging signals in response to MMP activity with the FRET peptide substrate. To verify the feasibility of our design, a non-substrate MT1-MMP binding peptide (MT1-AF7p, HWKHLHNTKTFL) with high affinity identified by Zhu and co-workers8 was linked to the FRET peptide substrate (Mca)-PLGL-(Dpa)-AR-NH2 (P-1), [Mca: (7-methoxycoumarin-4-yl)acetyl, Dpa: N-3-(2, 4-dinitrophenyl)-l-2,3-diaminopropionyl] to improve the specificity of the FRET probe by taking advantage of the specific interaction between MT1-AF7p and MT1-MMP. P-1 is a FRET peptide substrate that is extensively applied for monitoring MMP activity in biological samples, including crude cell culture supernatants, and for the screening of MMP inhibitors. This fluorogenic peptide indicated a high kcat/Km value, especially for the measurement of the enzymatic activity of gelatinases (MMP-2, MMP-9), and a relatively lower kcat/Km value for MT1-MMP.24 Since kcat/Km value is known as the specificity constant, P-1 with a low specificity for MT1-MMP, is a valuable candidate to evaluate the contribution to the enhanced specificity induced by the non-substrate MT1-MMP binding peptide MT1-AF7p.
As the binding peptide MT1-AF7p was introduced to increase the specificity of the probe, for a start, the effect of its binding on the catalysis of P-1 by MT1-MMP was evaluated first via the enzyme assay by using the recombinant human MT1-MMP. The catalytic domain and hinge region of MT1-MMP (Tyr112-Ile318) fused with His-tag were expressed in E. coli BL21(DE3)pLysS cells, and purified under the denaturing conditions. The purified protein was confirmed by SDS-PAGE and Western blot using anti-MT1-MMP antibody, indicating a molecular weight of 24.2 kDa (Fig. 2a), which corresponds to the calculated molecular weight. The active form of recombinant MT1-MMP was generated by successive autoproteolysis of the N- and C-terminal sites during refolding by a gradient dialysis according to the reported procedures.21 As shown in Fig. 2b, increasing the concentration of the binding peptide from 0.47 μM to 640 μM had no effect on the catalysis of P-1 by the recombinant MT1-MMP, thus indicating the feasibility of the conjugation of P-1 with MT1-AF7p.
Fig. 2.
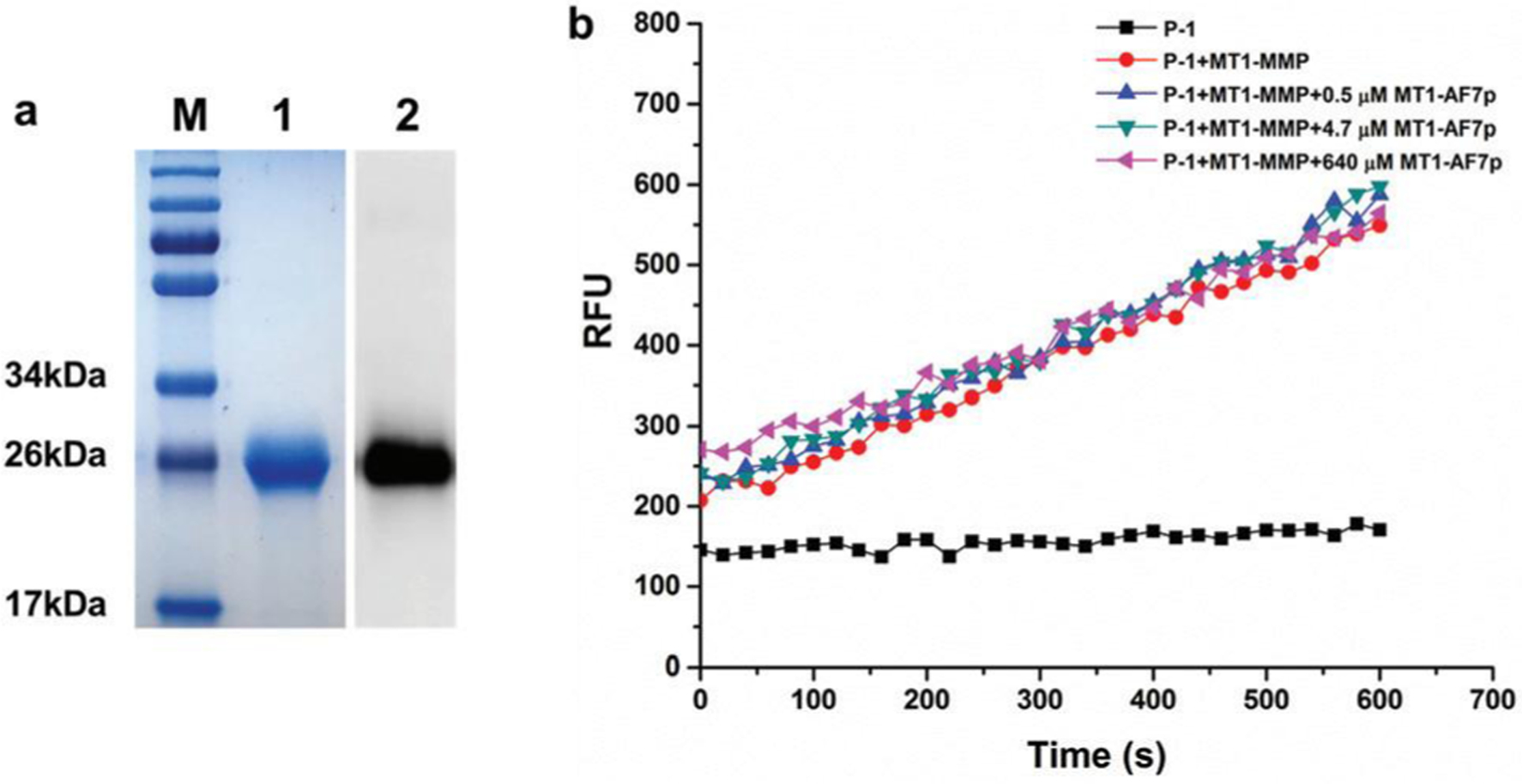
Effect of the binding peptide MT1-AF7p on the catalyzation of P-1 by MT1-MMP was evaluated via the enzyme assay, (a) SDS-PAGE and Western blot assay results for the recombinant MT1-MMP. Lane M: markers of the protein molecular weight standard. Lane 1 represents the protein purified using a Ni-NTA column, whereas lane 2 denotes the Western blot results for the purified recombinant MT1-MMP. (b) Effect of the binding peptide MT1-AF7p on the catalyzation of P-1 by MT1-MMP with increasing the concentration of the binding peptide MT1-AF7p from 0.5 μM to 640 μM pre-incubated with MT1-MMP before applying the cleavage kinetic assay with MT1-MMP.
According to the reported crystal structure of MT1-MMP25 and the binding site of MT1-AF7p with MT1-MMP,8 there was a certain distance between the substrate binding site and the MT1-AF7p binding site, therefore, PEG linkers with different lengths were introduced, in order to modulate for the best fitting between MT1-MMP and the designed fluorogenic probe. Accordingly, five fluorogenic probes were synthesized, among which P-2 was without a linker [(Mca)-PLGL-(Dpa)-AR-HWKHLHNTKTFL], and P-3, P-4, P-5, and P-6 had different lengths of PEG linker (from 3 to 24 PEG subunits) (Table 1).
Specificity evaluation of the MT1-MMP fluorogenic probes
In order to evaluate the activation efficiency of our designed fluorogenic probes by MT1-MMP, six fluorogenic probes (P-1 to P-6) were initially examined by detecting the fluorescence produced by MT1-MMP activation at a set time (catalysis velocity), and P-1 was used as a control. As shown in Fig. 3a, all the designed probes could be effectively activated by the recombinant MT1-MMP; however, P-2 induced substrate inhibition of MT1-MMP when its concentration was higher than 1.5 μM (data not included). P-2, which was the conjugate of P-1 with the MT1-MMP specific binding peptide MT1-AF7p, indicated similar activation efficiency by MT1-MMP as P-1, while the other four probes (P-3 to P-6) with PEG linkers exhibited a higher activation efficiency than P-1. This result implied that conjugation of the binding peptide MT1-AF7p to P-1 might lead to enhanced specificity of the probes for MT1-MMP, and it also confirmed the fact that there was a certain distance between the substrate binding site and the MT1-AF7p binding site of MT1-MMP, whereby direct conjugation of P-1 with MT1-AF7p might affect the catalyzation of P-1 by MT1-MMP, and therefore introducing a linker between these two peptides would render increased activation efficiency by MT1-MMP to the probes.
Fig. 3.
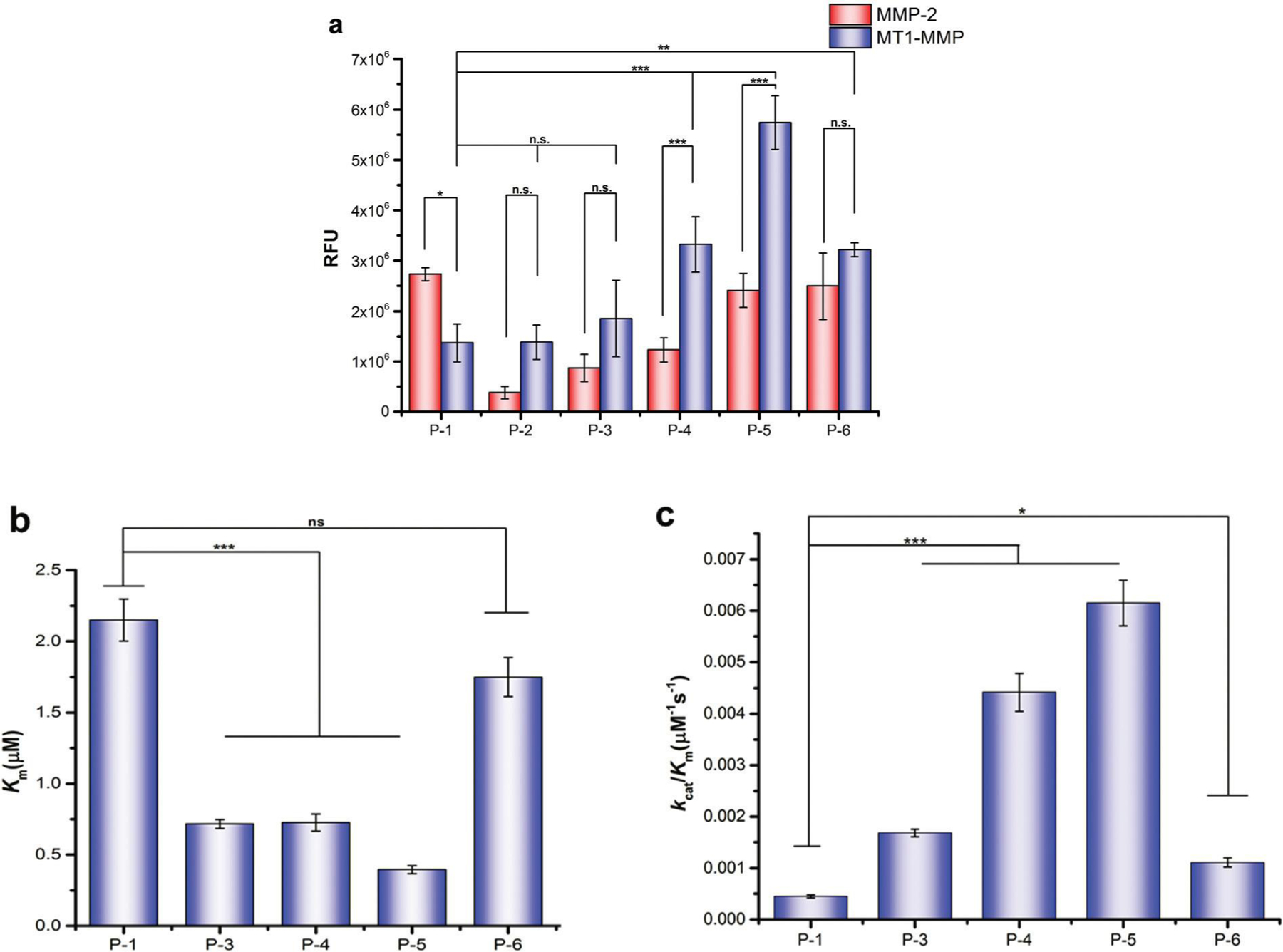
Specificity and selectivity evaluation of the MT1-MMP fluorogenic probes, (a) Fluorescence activation of the fluorogenic probes (1 μM) by the same concentration (15 nM) of MT1-MMP and MMP-2, respectively, following a 7 min incubation at 37 °C. The Km (b) and kcat/Km (c) value comparisons of the five fluorogenic probes with different lengths of PEG linkers. The data are shown as the mean ± SD. n.s., not significant; asterisks (*) represent statistically significant (*p < 0.05, **p < 0.01, and ***p < 0.001).
To thoroughly assess the activation efficiency of our designed fluorogenic probes by MT1-MMP, kinetic constants of the four fluorogenic probes with different lengths of PEG linker (P-3 to P-6) were further characterized via enzyme kinetics measurements using P-1 as the control. It can be seen from the data in Table 1 that, compared with the control probe P-1, the Km values of the four probes (P-3 to P-6) were all decreased, and their kcat values were all increased; these results indicated that the conjugation of P-1 and MT1-AF7p with the PEG linker improved both the affinity and hydrolysis rate of the probe for its given enzyme MT1-MMP. The specificity of probes to MT1-MMP, as characterized by the kcat/Km, value consistently increased as the PEG linker increased, it nevertheless reached the maximum level when the linker was composed of 12 PEG subunits.
It should be noted that probe P-5 with the linker composed of 12 PEG subunits (PEG12) had a 5.4-fold decreased Km value, indicating an enhanced binding affinity to the MT1-MMP (Fig. 3b), and a 2.5-fold enhanced hydrolysis rate (kcat) compared with the control probe P-1. Additionally, P-5 exhibited the highest specificity to MT1-MMP over the other probes analyzed, up to almost 14-fold specificity to MT1-MMP compared with the control probe P1 (Fig. 3c). This result demonstrated that conjugation of the binding peptide MT1-AF7p to P-1 via the PEG linker vastly enhanced the specificity of the probes for MT1-MMP. The selectivity of the designed fluorogenic probes for MT1-MMP was further evaluated by comparing with MMP-2 in the same condition. As shown in Fig. 3a, compared with P-1, the activation efficiencies of probes P-2 to P-6 by MMP-2 all decreased, and these results implied that the conjugation of P-1 and the MT1-MMP binding peptide MT1-AF7p with or without the PEG linker both led to the decreased selectivity of these probes for MMP-2. On the contrary, compared with P-1, the conjugation of P-1 with MT1-AF7p via the PEG linker enhanced the activation efficiencies of the probes P-3 to P-6 by MT1-MMP, where P-5 demonstrated significantly higher activation folds when incubated with MT1-MMP than with MMP-2. This implied that conjugation of the binding peptide MT1-AF7p to P-1 enhanced the selectivity of the probes for MT1-MMP but not for MMP-2, and P-5 can be used as an MT1-MMP specific probe.
Overall, all of these findings confirmed the feasibility of our strategy for enhancing both the specificity and selectivity of the fluorogenic probes for MT1-MMP by conjugating P-1 and MT1-AF7p, and the conjugate with a linker of PEG12 showed the optimized specificity and selectivity of the fluorogenic probes for MT1-MMP.
MT1-MMP fluorogenic probe with enhanced specificity for tumor cell imaging
Since a fluorogenic probe with enhanced specificity for MT1-MMP was screened, its applicability for tumor cell imaging was investigated. To overcome the limited penetration depths using visible FRET pairs, the FRET pair (Mca/Dpa) of probe P-5 was replaced by a new FRET pair (Dabcyl and FAM, Dabcyl: 4-(dimethylaminoazo)benzene-4-carboxylic acid, FAM: Carboxyfluorescein) to produce a new designed probe P-7 (Table 2), which was more suitable for cell and in vivo imaging. Additionally, in the probe P-7, FAM worked as the fluorophore and Dabcyl acted as the quencher. The specificity of P-7 for its given enzyme MT1-MMP was evaluated initially, as shown in Table S1, † where P-7 indicated a high specificity (kcat/Km = 3.56 × 10−3 ± 0.33 × 10−3) for MT1-MMP similar to P-5. Activation of the probe was further evaluated by tumor cells-expressed MT1-MMP. Two cell lines, the HT-1080 and control MCF-7 cells, were used in the experiment. The cell uptake results revealed that the HT-1080 cells with a higher MT1-MMP expression level indicated a much higher cell uptake compared with the control cells MCF-7 (Fig. 4a). Besides the different MT1-MMP expression levels, the different uptake efficiencies might also be caused by the different nature of endocytosis in the two different cell lines. So the cellular uptake experiments were also conducted by HT-1080 cells with or without the treatment of the broad-spectrum MMP-inhibitor GM6001.26,27 As shown in Fig. 4b, the cell uptake of P-7 by HT-1080 cells decreased when the cells were treated with the MMP inhibitor. These results revealed that the activation of P-7 by tumor cells was MMP dependent.
Fig. 4.
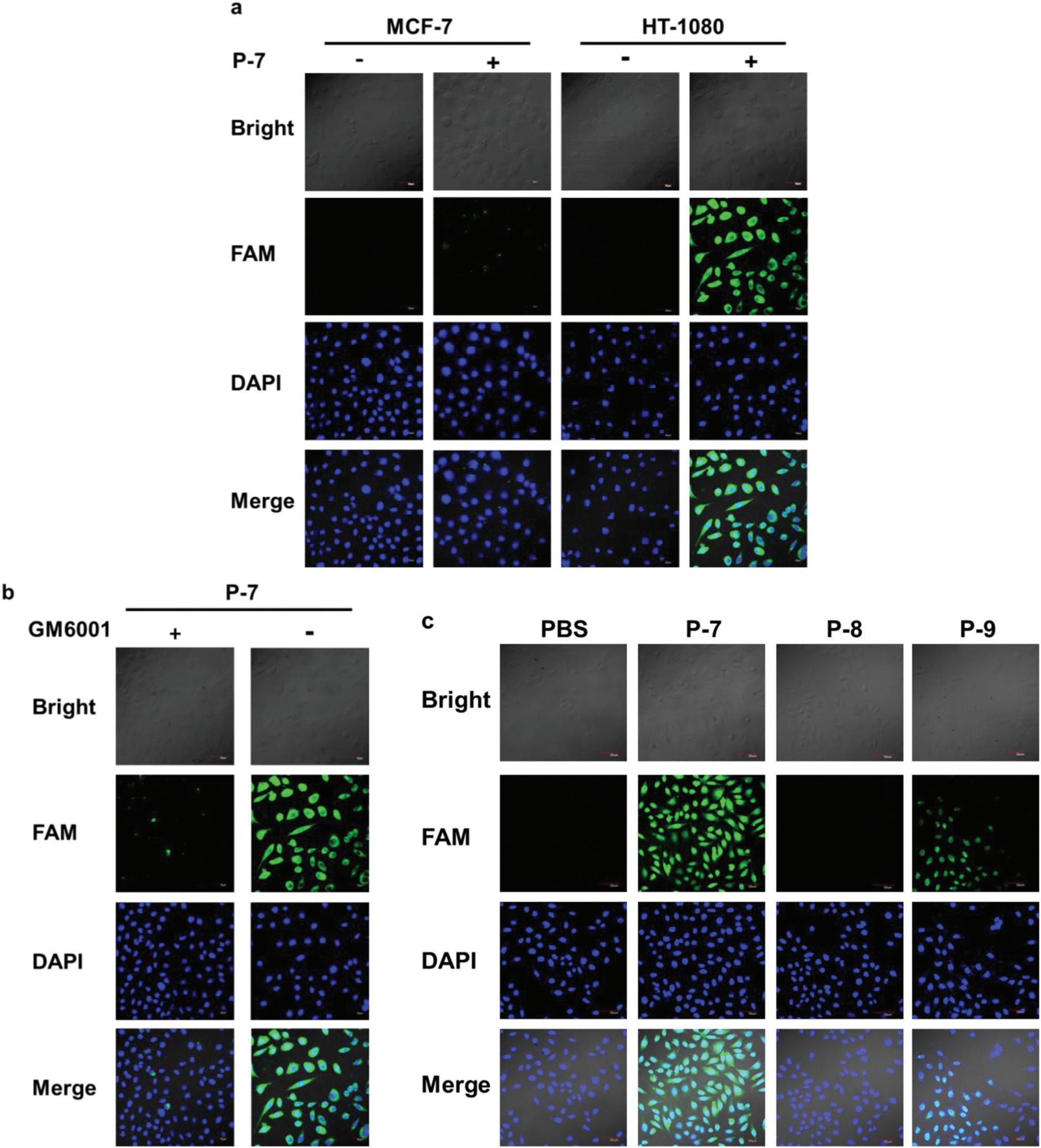
Cell uptake of the MT1-MMP fluorogenic probes, (a) Cell uptake of P-7 by HT-1080 cells and MCF-7 cells, (b) Cellular uptake of P-7 by the HT-1080 cell line in the presence or absence of the broad-spectrum MMP-inhibitor GM6001 (5 μM). (c) Cell uptake of the P-7, P-8, and P-9 probes by HT-1080 cells. The concentration of these probes used in the experiment was 5 μM. The slides were stained for cell nuclei with DAPI (blue), and the fluorescence of FAM is shown in green. Scale bar, 50 μm.
Moreover, according to the literature, HT-1080 cells overexpress not only MT1-MMP but also MMP-2, and therefore to confirm the activation of P-7 exclusively by MT1-MMP, two control probes P-8 and P-9 with the same FRET pair as P-7 (Table 2) were used in the cell imaging experiments. Compared with the structure of P-7, P-8 was a control probe without the PEG12 linker and the MT1-MMP specific binding peptide part (HWKHLHNTKTFL), while P-9 was a control probe without the PEG12 linker. P-8 and P-9 induced the substrate inhibition of MT1-MMP at higher concentrations (data not included), so their activation efficiency by MT1-MMP was characterized via the catalysis velocity as P-2 at the concentration of 1 μM. Both P-8 and P-9 exhibited a higher selectivity for MT1-MMP compared with MMP-2 as P-7, but a lower activation efficiency than P-7 (Fig. S1†). As can be seen from Fig. 4c, P-7 with a higher activation efficiency by MT1-MMP displayed a much higher cell uptake compared with P-8 and P-9. Also, what stands out in the figure is P-9, which exhibited a lower activation efficiency by MT1-MMP than P-8 and presented a higher cell uptake. Since P-9 and P-7 both contained the same peptide fragment (MT1-MMP specific binding peptide MT1-AF7p peptide sequence), the most likely cause of the higher cell uptake might be the contribution from the internalization of the membrane-tethered MT1-MMP, which has been reported to be a major short-term level of enzyme regulation for controlling the net amount of active enzyme present at the plasma membrane.28,29 Thus P-7, which contained the MT1-MMP specific binding peptide MT1-AF7p peptide sequence, might enter the cells by MT1-MMP internalization via the binding peptide of the probe. On the other hand, as shown in Table S1, † P-7 indicated a higher kcat value than P-5 by MT1-MMP, and presented a high specificity for MT1-MMP, so if the activation of P-7 by MT1-MMP is faster than the internalization of MT1-MMP, P-7 is proposed to be activated extracellularly by the membrane-tethered MT1-MMP. Therefore, it is speculated that P-7 might be activated by MT1-MMP by two different pathways: after being incubated with cells, (1) part of the probes entered the cells by MT1-MMP internalization via the binding peptide of the probe, then were activated intracellularly by the internalized MT1-MMP, and (2) part of the probes were activated extracellularly by the membrane-tethered MT1-MMP. Also, as P-9 and P-7 contained the same peptide fragment (MT1-MMP specific binding peptide MT1-AF7p peptide sequence) both exhibited a higher cell uptake than P-8, so pathway (1) was proposed to be the main pathway. Yet, all these hypotheses still need rigorous testing. These results revealed that the MT1-AF7p peptide sequence of the probe induced enhanced cellular uptake of the probes. Besides, since MT1-AF7p was the MT1-MMP specific binding peptide, P-7 was exclusively activated by MT1-MMP and led to a high cell uptake.
Taken together, these results confirmed the feasibility and success of the tumor cell imaging by P-7, and demonstrated that the activation of P-7 by tumor cells was MT1-MMP dependent, showing P-7 with low toxicity (Fig. S2†) can be effectively applied for MT1-MMP positive tumor cell imaging.
MT1-MMP fluorogenic probe with enhanced specificity for in vivo tumor imaging
According to the principle of our designed imaging probe P-7, the fluorescence signal of FAM was quenched by the Dabcyl group, before the cleavable substrate of the designed fluorogenic probes was hydrolyzed by MT1-MMP, which is highly overexpressed in tumor tissues. During application, the fluorogenic probes were retained at the tumor tissues via the high affinity of the MT1-MMP specific binding peptide of the probe. Then, after the FRET substrate was hydrolyzed by MT1-MMP, the fluorophore FAM was released and detained around the tumor tissues.
To verify the feasibility of our designed probe for detecting MT1-MMP activity in tumors, the fluorogenic probes (P-7, P-8, and P-9) with the dose of 50 nmol were implemented with tumor-bearing mice via tail vein injection, and P-8 and P-9 were used as controls. Mice with a similar tumor volume were divided into four groups to investigate the accumulation of the FAM fluorescence in the major organs and tumors with time. Based on the biodistribution studies, P-7 was activated predominantly in the tumor, liver, and kidney over other organs, and the fluorescence signal of P-7 stayed high from 2 h to 6 h post injection at the tumor site (Fig. S3†), and therefore the mice were sacrificed 4.5 h post injection, and the tumor tissues were collected for the cryosection process. The tissue slices were stained for the cell membrane and nuclei and then observed using fluorescence microscopy. As shown in Fig. 5, the imaging probes were activated by the overexpressed MT1-MMP of the tumor, leading to the restoration of FAM fluorescence in the tumor. Compared with the two control probes P-8 and P-9, P-7 with the highest activation efficiency by MT1-MMP displayed the strongest fluorescence intensity in the tumors, while P-8 and P-9 with the lower activation efficiency by MT1-MMP displayed weaker fluorescence intensities. The spatial distribution of fluorescence for the MT1-MMP activatable probe in the frozen tissue slices indicated that after being activated by MT1-MMP, P-7 was located in the cytosol. This finding demonstrated that P-7 presents the potential for the in vivo imaging of MT1-MMP activity in tumor. It should be noted that compared with P-8, P-9 exhibited a higher accumulation at the tumor site. Since P-9 was the conjugate of P-8 with the MT1-MMP specific binding peptide MT1-AF7p, this further confirmed that the specificity and targeting efficiency of the probe increased, profiting from the high affinity of the MT1-MMP specific binding peptide.
Fig. 5.
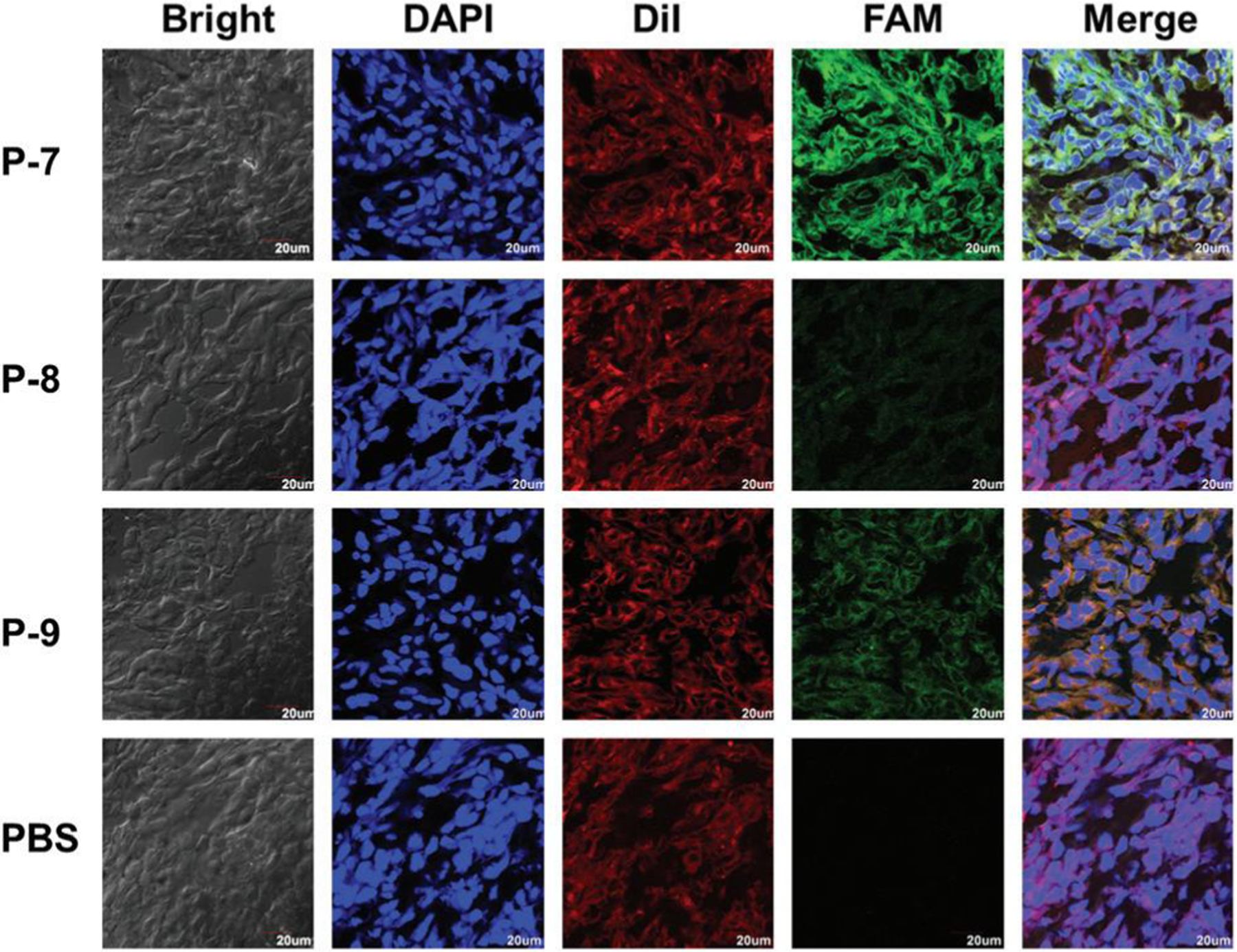
In vivo imaging of HT-1080 tumor-bearing nude mice after intravenous injection of the fluorogenic probe P-7. Spatial distribution of fluorescence for the MT1-MMP activatable probe in frozen tissue slices 4.5 h after injection. The slides were stained for cell membrane and nuclei with DiI (red) and DAPI (blue), respectively. FAM fluorescence is shown in green. Scale bar, 20 μm. Original magnification: 100×.
Although preliminary proof-of-concept in vivo studies have been carried out to assess the feasibility of the P-7 probe in tumor imaging, the application needs to go through a cryosection process. The relatively low tumor targeting efficiency of P-7 might be due to the relatively short biological half-life of its peptide backbone, which is the common feature of peptide-based probes.30 Besides, the bulky structures of the FRET pair (FAM and Dabcyl) might lead to the decreased binding affinity and less effective cleavage of P-7 for MT1-MMP.9 The much higher Km value of P-7 than P-5 implied the weaker binding affinity of P-7 to MT1-MMP based on the enzyme kinetics study (Table S1†). Therefore, the in vivo tumor-activated fluorescent imaging of P-7 was further performed to confirm this via intratumoral injection to improve the concentration of the probes accumulated in the tumor. Briefly, tumor-bearing mice were subjected to the IVIS imaging system following intratumoral injection of the three probes (P-7, P-8, and P-9) with the dose of 50 nmol, and the fluorescent images were acquired 0.5, 1, 2, 4.5, and 6 h post injection. Twelve mice with a similar tumor volume were divided into four groups (n = 3 per group) and administrated with P-7, P-8, and P-9, respectively. As shown in Fig. 6, while the fluorescent intensity of P-7 consistently increased over the incubation time, it nevertheless reached the maximum level after about 1 h of incubation, and the fluorescent intensity stayed high from 0.5 h to 4.5 h post injection. This result suggested that P-7 continuously accumulated at the tumor region in a time-dependent manner and was then rapidly activated by the overexpressed MT1-MMP of the tumor. Similar to the results from the ex vivo tumor imaging, the fluorescence of P-8 and P-9 was hardly detected in the tumor site. These findings further confirmed the in vivo feasibility of our designed probe P-7 for tumor-activated fluorescent imaging, and suggested the fundamental plausibility of our strategy to enhance the specificity of the MT1-MMP activatable fluorogenic probe for tumor imaging.
Fig. 6.
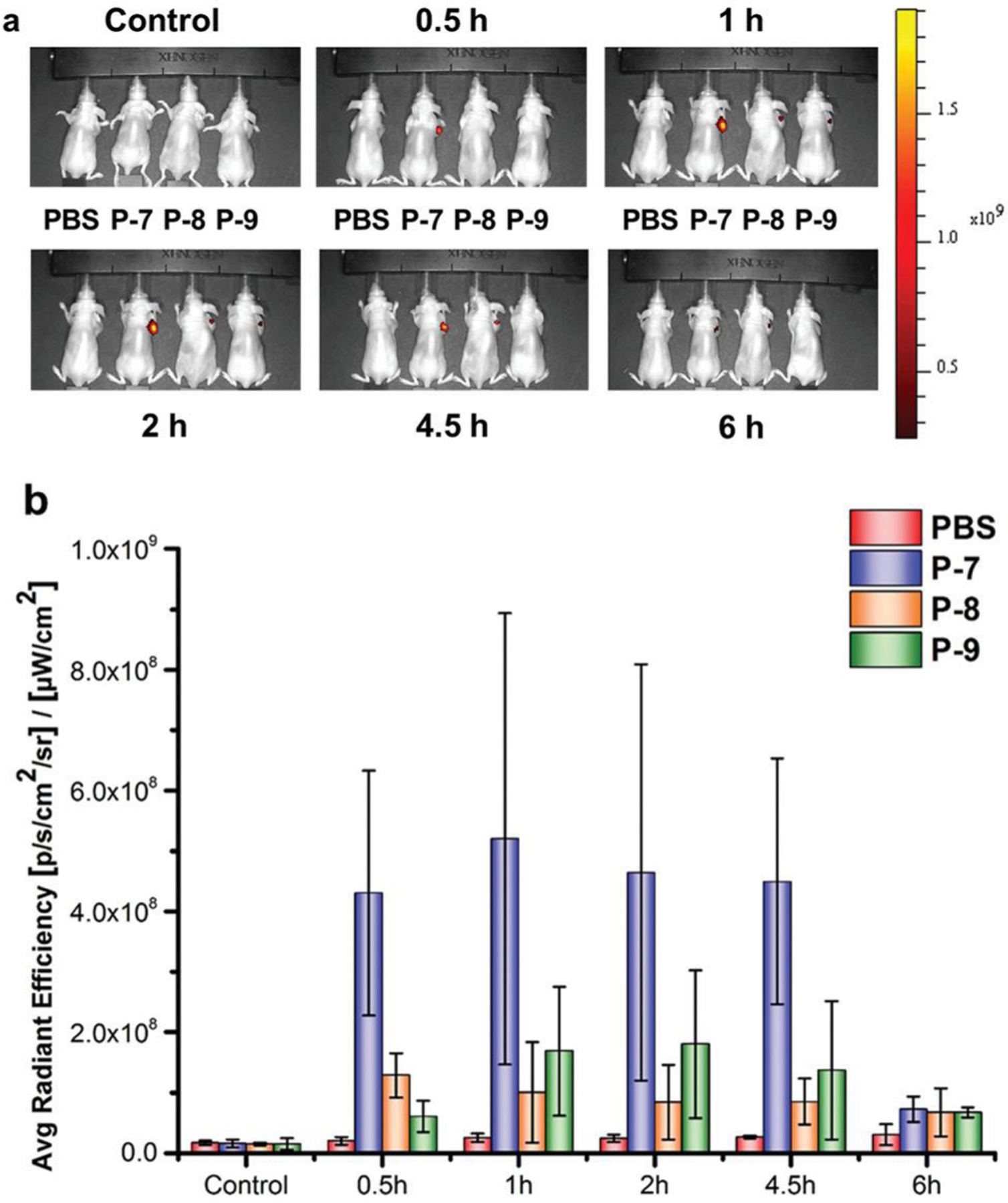
In vivo imaging of HT-1080 tumor-bearing nude mice after intratumoral injection of the probes, (a) Fluorescence images at various time points after intratumoral injection. Fluorescence images were acquired 0.5, 1, 2, 4.5, and 6 h post injection, (b) Radiant efficiency at the tumor sites.
Conclusions
Due to the overlapping substrate specificities among the family of MMP enzymes, most reported peptide substrates show sensitivity to multiple MMPs, making it therefore challenging to develop highly selective substrate-based fluorogenic probes for specific MMPs. To resolve this issue, we developed a probe design strategy taking advantage of the combination of an advanced MMP FRET substrate and an MMP specific binding peptide for MMP detection in tumors. According to the principle of our designed imaging probe, conjugation of the MMP FRET substrate and its specific binding peptide led to enhanced specificity of the probe for MMP detection, and the specificity of probe was proposed to increase profiting from the high affinity of the MMP specific binding peptide, while maintaining the ability to amplify the output imaging signals in response to MMP activity with the FRET peptide substrate. A series of probes was fabricated using a reported MT1-MMP specific binding peptide and an MMP peptide substrate with different lengths of PEG linkers between these two peptides. Both the in vitro and in vivo results revealed that the probe with a linker composed of 12 PEG subunits based on our designed strategy could be effectively applied for MT1-MMP positive tumor imaging. Since this strategy that enhances the specificity of protease sensing probes can be applied to other proteases and not just limited to MT1-MMP, it is an appealing platform to achieve selective tumor imaging. Moreover, as MMPs have been investigated as robust tumor microenvironmental stimuli for “smart” MMP-responsive drug delivery and tumor targeting systems, which have shown great potential in cancer diagnosis and therapy,31–33 this work is also expected to shed light on the design of an MMP-responsive system that is sensitive only to a specific MMP family member, and so its impact could therefore be significant, far-reaching, and widespread.
Supplementary Material
Acknowledgements
This work was supported by School of Pharmacy, Tianjin Medical University & the Open Project Program of Tianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnostics (Theranostics) (CTD2018-04). This research was also partially sponsored by National Key Research and Development Plan (2016YFE0119200).
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm02007a
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1.Garland M, Yim JJ and Bogyo M, Cell Chem. Biol, 2016, 23,122–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kessenbrock K, Plaks V and Werb Z, Cell, 2010, 141, 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isaacson KJ, Martin Jensen M, Subrahmanyam NB and Ghandehari H, J. Controlled Release, 2017, 259, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seiki M, Cancer Lett, 2003, 194, 1–11. [DOI] [PubMed] [Google Scholar]
- 5.Itoh Y and Seiki M, J. Cell. Physiol, 2006, 206, 1–8. [DOI] [PubMed] [Google Scholar]
- 6.Holmbeck K, Bianco P and Birkedal-Hansen H, Cancer Cell, 2003, 4, 83–84. [DOI] [PubMed] [Google Scholar]
- 7.Marco M, Fortin C and Fulop T, J. Leukocyte Biol, 2013, 94, 237–246. [DOI] [PubMed] [Google Scholar]
- 8.Zhu L, Wang H, Wang L, Wang Y, Jiang K, Li C, Ma Q, Gao S, Wang L, Li W, Cai M, Wang H, Niu G, Lee S, Yang W, Fang X and Chen X, J. Controlled Release, 2011, 150, 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park J, Yang J, Lim EK, Kim E, Choi J, Ryu JK, Kim NH, Suh JS, Yook JI, Huh YM and Haam S, Angew. Chem., Int. Ed. Engl, 2012, 51, 945–948. [DOI] [PubMed] [Google Scholar]
- 10.Chung EY, Ochs CJ, Wang Y, Lei L, Qin Q, Smith AM, Strongin AY, Kamm R, Qi YX, Lu S and Wang Y, Nano Lett, 2015, 15, 5025–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L, Xie S, Qi J, Liu E, Liu D, Liu Q, Chen S, He H and Yang VC, ACS Appl Mater. Interfaces, 2017, 9, 39209–39222. [DOI] [PubMed] [Google Scholar]
- 12.Bazeli R, Coutard M, Duport BD, Lancelot E, Corot C, Laissy JP, Letourneur D, Michel JB and Serfaty JM, Invest. Radiol, 2010, 45, 662–668. [DOI] [PubMed] [Google Scholar]
- 13.Knapinska A and Fields GB, ChemBioChem, 2012, 13, 2002–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner S, Breyholz HJ, Law MP, Faust A, Holtke C, Schroer S, Haufe G, Levkau B, Schober O, Schafers M and Kopka K, J. Med. Chem, 2007, 50, 5752–5764. [DOI] [PubMed] [Google Scholar]
- 15.Lee S, Xie J and Chen X, Chem. Rev, 2010, 110, 3087–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turk B, Nat. Rev. Drug Discovery, 2006, 5, 785–799. [DOI] [PubMed] [Google Scholar]
- 17.Kukreja M, Shiryaev SA, Cieplak P, Muranaka N, Routenberg DA, Chernov AV, Kumar S, Remacle AG, Smith JW, Kozlov IA and Strongin AY, Chem. Biol, 2015, 22, 1122–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ratnikov BI, Cieplak P, Gramatikoff K, Pierce J, Eroshkin A, Igarashi Y, Kazanov M, Sun Q, Godzik A, Osterman A, Stec B, Strongin A and Smith JW, Proc. Natl. Acad. Sci U. S. A, 2014, 111, E4148–E4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryu JH, Lee A, Na JH, Lee S, Ahn HJ, Park JW, Ahn CH, Kim BS, Kwon IC, Choi K, Youn I and Kim K, Amino Acids, 2011, 41,1113–1122. [DOI] [PubMed] [Google Scholar]
- 20.Crisp JL, Savariar EN, Glasgow HL, Ellies LG, Whitney MA and Tsien RY, Mol. Cancer Ther, 2014, 13, 1514–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koo HM, Kim JH, Hwang IK, Lee SJ, Kim TH, Rhee KH and Lee ST, Mol. Cells, 2002, 13, 118–124. [PubMed] [Google Scholar]
- 22.Goncalves AN, Meschiari CA, Stetler-Stevenson WG, Nonato MC, Alves CP, Espreafico EM and Gerlach RF, J. Biotechnol, 2012, 157, 20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carmona AK, Schwager SL, Juliano MA, Juliano L and Sturrock ED, Nat. Protoc, 2006, 1, 1971–1976. [DOI] [PubMed] [Google Scholar]
- 24.Neumann U, Kubota H, Frei K, Ganu V and Leppert D, Anal Biochem, 2004, 328, 166–173. [DOI] [PubMed] [Google Scholar]
- 25.Fernandez-Catalan C, Bode W, Huber R, Turk D, Calvete JJ, Lichte A, Tschesche H and Maskos K, EMBO J, 1998, 17, 5238–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grobelny D, Poncz L and Galardy RE, Biochemistry, 1992, 31, 7152–7154. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto M, Tsujishita H, Hori N, Ohishi Y, Inoue S, Ikeda S and Okada Y, J. Med. Chem, 1998, 41,1209–1217. [DOI] [PubMed] [Google Scholar]
- 28.Remacle A, Murphy G and Roghi C, J. Cell Sci, 2003, 116, 3905–3916. [DOI] [PubMed] [Google Scholar]
- 29.Pahwa S, Stawikowski MJ and Fields GB, Cancers, 2014, 6, 416–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun X, Li Y, Liu T, Li Z, Zhang X and Chen X, Adv. Drug Delivery Rev, 2017, 110–111, 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao Q, Kou L, Tu Y and Zhu L, Trends Pharmacol. Sci, 2018, 39, 766–781. [DOI] [PubMed] [Google Scholar]
- 32.Wang H, Huang Q, Chang H, Xiao J and Cheng Y, Biomater. Sci, 2016, 4, 375–390. [DOI] [PubMed] [Google Scholar]
- 33.Wang Q, Jiang N, Fu B, Huang F and Liu J, Biomater. Sci, 2019, 7, 4888–4911. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.