

Abstract
Background and purpose:
Human papillomavirus (HPV) is known as the main reason for cervical cancer. According to carcinogenic risk, HPV can be located into two classes, counting the low-risk virus, which is the main cause of genital warts and low-grade cervical epithelial lesions. HPV-16 is one of the high-risk HPV subtypes in the spectrum of cervical diseases.
Experimental approach:
The PubChem database was screened in order to identify potential anti-HPV hits followed by ADMET predictions. Then, molecular docking was performed to improve the accuracy of screening and also to find the details of the interactions of the hit compounds with the active site. Finally, molecular dynamic (MD) simulations and free binding energy on top-ranked structures CID_73212812, CID_91059286, CID_69838075, cidofovir, and jaceosidin were carried out with protein to compute the interaction energies and stability of the top-ranked compounds at the active site.
Findings/Results:
Based on molecular docking studies, three compounds including CID_73212812, CID_91059286, and CID_69838075 exhibited the best results among compounds against the E6 protein of HPV-16. Furthermore, RMSD, RMSF, hydrogen binds, Rg, and energy analysis during MD simulation certainly indicated the stable binding of selected compounds with E6 protein of HPV-16 active site.
Conclusion and implications:
Docking and MD results revealed that hydrophobic contacts and optimum hydrogen bonds were determinant factors in the interactions of hits and the E6 protein of HPV-16. In addition, the binding energy portions exposed that Van der Waals and non-polar interactions were fundamental factors in the molecule binding.
Keywords: ADMET, Cervical cancer, HPV, Molecular docking, Virtual screening
INTRODUCTION
Human papillomaviruses (HPVs) are small, circular, and double-stranded DNA viruses from the papillomavirus family with a genome of about 8 kb and mainly infects both external skin and mucosal surfaces (1). HPV infection is one of the most common diseases that spread mostly by sexual transmission and has been related powerfully to cervical cancer (2). DNA of HPV has been discovered in over 90% of cervical dysplasia or cancer cases which are diagnosed by cervical biopsy. In 2018, an estimated 570,000 women were identified with cervical cancer, and about 311,000 women died from this disease (3).
The HPV is divided into two categories including the low-risk HPV (e.g. types 6 and 11) and the high-risk HPV (e.g. types 16 and 18). They are the primary drug targets to discover and design anticancer drugs. The structure of the HPV-16 genome has E6 and E7 proteins that collaborate to transform and immortalize cells (4).
The expression of the E6 and E7 genome was observed in cancer biopsies and cervical cancer cell lines. The E6 and E7 specifically interact with p53 and pRb, respectively (4).
HPV plays a key role in cervical cancer but unfortunately, there are not any approved suitable drugs to treat HPV infections (3). There are three approved vaccines to avoid HPV infection, but these preventive vaccines are not effective in women who have previously been infected with high-risk HPV kinds. Various chemotherapeutic routes are in hand against cervical intraepithelial neoplasia resulting from HPV infections. Cidofovir (an acyclic nucleoside phosphonate) (Fig. 1) unselectively inhibits viral replication by the selective inhibition of a viral DNA polymerase. This compound also is an E6 and E7 HPV inhibitor. Although cidofovir inhibits human polymerases, it is a weaker inhibitor when compared to viral DNA polymerases (5). Photfrin (Fig. 1) has a considerable potential to provide an effective non-surgical treatment for both low- and high-grade HPV-related dysplasia (6). Imiquimod, an immune activator (Fig. 1) does not have any direct antiviral activity, but it activates cytokines which subsequently promotes immunological clearance of the viruses. The topical application of this agent has been used extensively to treat HPV-related genital warts (7). Immunoenhancers such as interferon and imiquimod inhibit HPV replication. Moreover, cytotoxic drugs such as 5-fluorouracil (5-FU) (Fig. 1), radiotherapy, and surgery procedures are other therapeutic approaches for HPV infection treatment (7). It should be noted that these treatments have high costs and incomplete efficacy with many side effects and safety concerns, which greatly restricts their applications (7).
Fig. 1.

Some representative examples of human papillomavirus inhibitors.
Natural products have been recognized as suitable sources to prevent or treat HPV infections and some of them have been submitted to preclinical and clinical trials (Fig. 1). Flavonoids (Fig. 2) containing one of the largest groups of secondary metabolites have been found in plants, including vegetables, fruits, seeds, nuts, and tea. Natural flavonoids have been an important source of medicines for many years (8). Although various classes of flavonoids have different structures, they exhibit important therapeutic and pharmacological properties. It has been reported that flavonoids have been described with a broad spectrum of biological activities such as anti-inflammation, antioxidant, antibacterial, antiviral, anticancer, and neuroprotection (9). The catechin-like flavonoid and epigallocatechin gallate (Fig. 2) increased the level of the p53 protein accompanied by reducing the E6 protein of HPV-16 in HeLa and CaSki cells. Furthermore, the flavonoid luteolin (Fig. 2) and synthetic flavonoid-like compounds inhibited the binding between the E6 protein of HPV-16 and E6AP in vitro and induced an increased expression of p53 and p21 proteins in cervical cancer cells (10). In addition, previous studies proved the activity of a series of flavonoid compounds as HPV inhibitors (11,12). Some representative examples of these compounds have been shown in Fig. 2.
Fig. 2.

(A) Some natural compounds with flavonoid scaffold and (B) flavonoid derivatives prepared by previous studies as human papillomavirus inhibitors.
Jaceosidin (4’,57-trihydroxy-3’,6-dimethoxy- flavone; Fig. 2), isolated from Artemisia argyi as a putative oncogene inhibitor, inhibits binding between oncoprotein E6 of the HPV and the p53 tumor suppressor protein. In addition, it inhibits binding between the E7 oncoprotein and the Rb tumor suppressor protein, and also the function of HPV-16 harboring cervical cancer cells, including SiHa and CaSki. Overall, jaceosidin inhibits the functions of the E6 and E7 oncoproteins of the HPV-16 (13).
Virtual screening (VS) is a computer- assisted protocol comprised of one or more computational methods applied to select the best compounds with the desired biological activities among the compounds in a large molecular database. VS can be defined in two categories, structure-based VS (SBVS) and ligand-based VS (LBVS) (14). The selection of methods especially depends on the presence or absence of information regarding a biological target and the molecules interacting with this target. In summary, SBVS applies docking approaches for searching small molecule databases and ranking them (15) to find appropriate ligands that might be able to make adaptable stereo electronic fitness within the binding site of the desired target. When the receptor structure is not available, the information which the ligands carry sheds light on the drug design and discovery plans. Defining statistical models which describe the dependence of the biological properties to the structural features of the bioactive compounds, quantitative structure-activity relationship (QSAR) studies, are the most known examples of LBVS approaches. Sometimes a combination of both LBVS and SBVS is used for a more successful VS protocol which may start with a similarity search or pharmacophore screening based on a biologically active compound. Then, more expensive computational structure-based methods such as molecular docking are used to narrow down the collection of hits (16). Previous research studies have reported the identification of new E6 protein of HPV-16 inhibitors using VS technique (17,18,19). For instance, VS could be successfully applied to the ZINC database to introduce new E6 protein of HPV-16 protein inhibitors (20,21). In other studies, several natural products were recognized via molecular docking methods as inhibitors of the E6 protein of HPV-16 and 18 (22).
In order to discover novel in silico hit compounds with a potential affinity toward the E6 protein of HPV-16, in the present contribution jaceosidin, was selected (flavonoid scaffold) to search structurally related compounds in available databases. A similarity-based search on the online PubChem database was carried out to extract some compounds’ databases. These compounds were subjected to some filters to select the most desired compounds as E6 protein of HPV-16 inhibitors. These filters were the docking of the compounds using PyRx to select the ones with the highest estimated binding energies, drug- likeness properties, evaluating the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of the selected compounds, selecting the compounds with the lowest binding free energies and the best interactions with the HPV active site residues using Autodock software, the molecular dynamics simulations of the best compounds for further investigations of the interactions and stabling them in E6 protein of the HPV-16 active site and finally, the calculation of binding free energies of the highest-ranked compounds using the molecular mechanics Poisson- Boltzmann surface area (MM-PBSA) technique. The exploited filters led us to select compounds with the best pharmacokinetics and pharmacodynamics features rendering them drug-likeness properties. To be more illustrative, a hierarchical view of the work is depicted in Fig. 3.
Fig. 3.
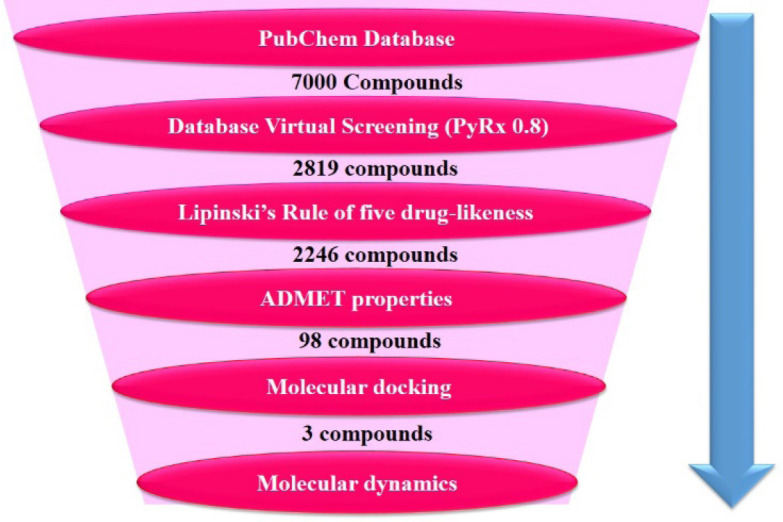
Representation of the overall filtering process in order to reduce false positives and recognize the best possible hits.
MATERIALS AND METHODS
Virtual screening
The crystal structure of the E6 protein of HPV-16 was retrieved from the protein data bank (www.rcsb.org, PDB ID; 4GIZ). Jaceosidin, a bioactive flavone structure from genus Artemisia that inhibits the functions of the E6 and E7 oncoproteins of the HPV, used as a template for a similarity search (70% similarity to the template) in PubChem online database (https://pubchem.ncbi.nlm.nih.gov/). Obtained small molecule structures in the structure data file (SDF) format were converted to pdbqt by file format by PyRx 0.8 software (freely downloaded from http://PyRx. sourceforge.net/downloads) as an input ligand for the VS. Then, the obtained compounds from the similarity search with an SDF format for binding to the E6 protein of HPV-16 active site were docked using Autodock vina in PyRx 0.8. Following that, compounds with the best docking scores were selected for the next step.
Drug-likeness profile
Drug-like molecules should satisfy Lipinski’s rule of five and have a balance between lipophilicity and hydrophilicity. The criteria of these rules include (I) the number of hydrogen bond donors under 5; (II) the number of hydrogen bond acceptors under 10; (III) the molecular weight (MW) less 500 (g/mol); (IV) the partition coefficient LogP (CLogP) less 5 (23). Additionally, to the criteria of this rule, if the topological polar surface area (TPSA) value is < 60 Å2, a drug can be absorbed over 90% (24). The number of rotatable bonds (nRTBs) is also very vital for the absorptive ability of candidate molecules (24). The obtained compounds from the previous step were passed for drug-likeness properties. The drug-likeness properties of the candidate ligands were estimated using Molinspiration online server (http://www.molinspiration.com/). Compounds with appropriate structural features for an orally active E6 protein of the HPV-16 inhibitor were selected. Selected compounds were evaluated for in silico ADMET properties analysis in the next step.
ADME calculation
One of the important characteristics of oral drugs is the quick and total absorption from the gastrointestinal tract and subsequent distribution into its place of action in the body. Metabolism is also very important and the last step involves the proper removal without producing any harm (25). Thus, the therapeutic applications of inhibitors are up to the appropriate ADMET profiles. Such simulation procedure may lead to the selection of relatively safer inhibitors with little or no side effects (26). Good efficiency with a satisfactory ADMET profile is the significant criterion for a drug (26). Therefore, it is important to calculate pharmacokinetic attributes in the hit discovery and identification process.
SwissADME (http://www.swissadme.ch) server, a chemoinformatics-based web server that can predict the most important molecular properties such as absorption, inhibiting enzyme interactions, and toxicity (acute and carcinogenicity) (27) was applied to acquire the ADMET properties of the selected compounds to yield ones with the best properties for further in silico investigations.
Molecular docking study
The molecular modeling calculations of the filtered ligands into the E6 protein of the HPV- 16 active site were made by Autodock 4.2 software. The target protein was the E6 protein of HPV-16. The crystal structure of the E6 protein of HPV-16 (PDB ID; 4GIZ) was retrieved from the PDB (https://www.rcsb.org/). Water molecules inside the crystal structure of the protein were removed by discovery studio visualizer 4 software (Accelrys lnc, San Diego, CA, USA) and polar hydrogens/Kollman charges were added to protein structure using Autodock tools package (MGTools 1.5.6). Then, the Gasteiger charges of energy minimized structure of ligands were calculated by Autodock tools package too. The number of runs for each docking analysis was set to 150 and Lamarckian genetic algorithm was applied. A grid box of 70×70×70 Å3 was made within the X, Y, and Z-axis, so that the grid point spacing was set at 0.375 Å.
The center of the grid box was set to the coordinates of the α-carbon of the catalytic residue Leu50 (28). Thus, the binding modes of cidofovir and jaceosidin, two well-known HPV E6 inhibitors, within the entire protein were explored via a blind-docking procedure to validate the applied docking protocol. When the calculations finished, the ligands were sorted by free binding energy to the target active site. All complex interactions were investigated using Autodock tools, discovery studio visualizer 4 and LigPlus software.
Molecular dynamic simulation study
GROningen machine for chemical simulations (GROMACS) V4.6.5 package was used to preformat the molecular dynamic (MD) simulation. The geometries and topologies parameters for the filtered molecules were retrieved from PRODRG server (29). Water molecules were characterized by the SPC216 model and the Gromos43a1 force field was chosen for MD simulation. The MD simulation was carried out based on the described procedure in the previous article (24). The final step or the production phase of MD simulation was accomplished for 20 ns with a 2 fs time step under the NPT ensemble with Nose-Hoover thermostat, and the position restraints were removed in this step. Three ligands selected in molecular docking studies (the highest score docking) and cidofovir and jaceosidin (two well-known inhibitors of the E6 protein of HPV-16) were subjected to a MDs simulation study.
Binding free energy calculation by MM-PBSA method
The g_mmpbsa tool was applied to calculate the average free binding energies. This tool computes free binding energy components using the MM-PBSA procedure (30). MM-PBSA is extensively performed in drug discovery to evaluate the binding affinity of ligand-enzyme interactions. The appendix equation reveals the free energy of binding:
ΔGbinding=ΔGcomplex-(ΔGprotein+ΔGligand) (1)
The total free binding energy of the E6 protein of HPV-16 hit compound complexes is termed as ΔGcomplex, and ΔGprotein+ ΔGligand are the total free energies of the E6 protein of HPV-16 enzyme and hit compounds in the solvent, respectively.
ΔGbinding=ΔEMM-TΔS+ΔGsol 2)
ΔEMM=ΔEbonded+ΔEele+ΔEvdw (3)
Gsol=Gpol+Gnonpol (4)
Gnonpol=γΔSASA+b (5)
ΔEbonded contains the angle, dihedral angle, and total bond interactions; the Van der Waals energy, the electrostatic energy, electrostatic energy of solvation, and non-electrostatic free energy of solvation are recognized as ΔEvdw, ΔEele, Gpol, and Gnonpol, respectively. T is the temperature, S is the entropy, and TS is the entropic contribution in a vacuum. Since the change of TS period does not progress the predicted results and ΔEbonded is zero in the single trajectory method, they were ignored in the calculation. Polar interactions among the solvent and solute are characterized by the solvation energy (ΔEpolar), also the non-polar solvation energy (ΔESASA) is calculated using the solvent-accessible surface area (SASA) nonpolar model. The γ constant was set at 0.0226778 kJ/mol/Å2. In the present study, the binding free energy of the three selected compounds, cidofovir, and jaceosidin were estimated during the last stable 10 ns period of MD simulation analysis using the g_mmpbsa tool of GROMACS, based on MM-PBSA technique.
RESULTS
Virtual screening
In the areas of drug design and discovery, VS can be effectively used to select privileged potentially bioactive small molecules from a library of predefined structures with the aim of binding to target proteins or enzymes. The online database of PubChem was searched for compounds with 70% similarity to jaceosidin (flavonoid scaffold and the E6 protein of HPV inhibitor). PubChem database keeps more than 35 million diverse compounds (natural, synthetic, and commercial types). This database is free of charge and provides a great opportunity for researchers to do the VS procedure (31). The result of this similarity search was a library of 7000 compounds that were subjected to the following pharmacokinetics and pharmacodynamics filters.
The VS of collected compounds was carried out by the Autodock vina in PyRx software. Then, the molecules were ranked on the basis of evaluated affinity energies in the active site of the protein. In this stage, 2819 molecules were selected with the affinity energies equal to or higher than -8.60 kcal/mol for the next step (equal and higher than evaluated affinity energy of jaceosidin, ΔGbinding= -8.60 kcal/mol).
Drug-likeness profile
One of the vital factors to discover and progress the bioactive ligands as an oral drug is their high oral bioavailability. The significant forecasters of good oral bioavailability are rotatable bonds of molecules which became known under molecular flexibility, a good gastrointestinal absorption, and a low polar surface area (total of acceptor and donor hydrogen bonds) (32). TPSA of a molecule is defined as the surface sum over all polar atoms or molecules, primarily oxygen and nitrogen, also including their attached hydrogen atoms. Additionally, Lipinski et al. reported drug-likeness features such as MW, hydrogen bond donor and acceptor values, LogP (partition coefficient) under the ‘Rule of Five’ term. This rule helps to make an easier selection of molecules with better pharmacokinetic properties in the human body for oral formulations. In this step, physicochemical parameters (e.g. CLogP, TPSA, MW, hydrogen bond donor and acceptor, TPSA, and rotatable bonds) of the approved compounds from the previous step were evaluated. Finally, 2246 out of 2819 compounds were fitted with these properties and revealed a satisfied drug-likeness demonstrating perhaps a good permeability through the biological membranes.
ADME calculation
The calculation of the pharmacological properties e.g. ADME for a molecule(s) is significant in their primary identification as a chemical lead and makes a benchmark against which synthesized compounds were assessed during lead optimization (33). The use of ADMET prediction of ligands is to eliminate the weak drug candidates and focus on the compounds with more likely successful drug properties. In silico key physicochemical and pharmacokinetic properties of selected compounds were calculated by swissADME, a free web tool. These parameters include lipophilicity value, permeability, blood-brain barrier (BBB) penetration, solubility, absorption, plasma protein binding, and metabolism in addition to the prediction of the influence of ligands on liver enzymes e.g cytochrome P450 and being p-glycoprotein inhibitor or substrate (27). SwissADME server was used to predict the ADMET properties of 2246 compounds that passed the last step. In this stage, 55 compounds with the best ADMET properties were selected. Drug-likeness and pharmacokinetic results of the 55 compounds are summarized in Tables 1 and 2, respectively.
Table 1.
Drug-likeness calculations of filtered molecules by Molinspiration server.
| Compounds | CLogP | TPSA | nAtoms | MW | HBAs | HBDs | nRotb | Volume (Å3) |
|---|---|---|---|---|---|---|---|---|
| CID_78385623 | 2.18 | 90.89 | 20 | 270.24 | 5 | 3 | 1 | 224.05 |
| CID_85044918 | 2.15 | 86.99 | 21 | 284.27 | 15 | 3 | 1 | 240.85 |
| CID_76319859 | 2.41 | 131.35 | 24 | 328.28 | 7 | 5 | 2 | 267.50 |
| CID_73197708 | 1.70 | 111.12 | 21 | 286.24 | 6 | 4 | 1 | 232.07 |
| CID_73212812 | 1.63 | 113.29 | 25 | 342.30 | 7 | 3 | 3 | 285.38 |
| CID_73318750 | 1.27 | 86.99 | 20 | 272.26 | 5 | 3 | 1 | 230.29 |
| CID_101382379 | 1.02 | 107.22 | 21 | 288.25 | 6 | 4 | 1 | 238.31 |
| CID_129646445 | 1.69 | 128.19 | 23 | 314.25 | 7 | 4 | 2 | 251.05 |
| CID_5322048 | 2.18 | 90.89 | 20 | 270.24 | 5 | 3 | 1 | 224.05 |
| CID_11313032 | 2.59 | 128.19 | 23 | 314.25 | 7 | 4 | 2 | 251.05 |
| CID_16743457 | 1.27 | 86.99 | 20 | 272.26 | 5 | 3 | 1 | 230.29 |
| CID_21500651 | 2.66 | 107.97 | 20 | 272.21 | 6 | 3 | 1 | 215.62 |
| CID_22298206 | 1.73 | 96.22 | 22 | 302.28 | 6 | 3 | 2 | 255.83 |
| CID_23626869 | 2.38 | 107.22 | 23 | 314.29 | 6 | 4 | 2 | 265.70 |
| CID_89490339 | 2.40 | 111.12 | 22 | 302.28 | 6 | 4 | 3 | 259.78 |
| CID_90666180 | 1.63 | 128.19 | 23 | 314.25 | 7 | 4 | 2 | 251.05 |
| CID_91059286 | 1.89 | 107.22 | 22 | 300.27 | 6 | 4 | 1 | 248.87 |
| CID_91413267 | 2.52 | 124.29 | 21 | 290.23 | 7 | 4 | 4 | 234.47 |
| CID_101342513 | 3.93 | 120.36 | 25 | 344.32 | 7 | 4 | 3 | 290.68 |
| CID_101342516 | 3.93 | 120.36 | 25 | 344.32 | 7 | 4 | 4 | 291.56 |
| CID_101584198 | 2.57 | 131.35 | 25 | 346.33 | 7 | 5 | 3 | 295.92 |
| CID_101593135 | 1.75 | 100.13 | 22 | 300.27 | 6 | 3 | 2 | 249.59 |
| CID_101782169 | 1.78 | 100.13 | 22 | 300.27 | 6 | 3 | 2 | 249.59 |
| CID_122182396 | 2.57 | 96.22 | 23 | 314.29 | 6 | 3 | 2 | 266.40 |
| CID_102292606 | 1.49 | 137.43 | 25 | 344.27 | 8 | 4 | 3 | 276.60 |
| CID_101566521 | 1.49 | 137.43 | 25 | 344.27 | 8 | 4 | 3 | 276.60 |
| CID_76330740 | 2.67 | 111.12 | 23 | 312.28 | 6 | 4 | 2 | 259.48 |
| CID_71514077 | 1.03 | 104.06 | 21 | 286.24 | 6 | 3 | 1 | 232.44 |
| CID_71330931 | 2.46 | 111.53 | 25 | 348.31 | 8 | 2 | 7 | 295.08 |
| CID_70544322 | 1.40 | 128.19 | 24 | 328.28 | 7 | 4 | 3 | 267.88 |
| CID_313107 | 2.64 | 87.74 | 21 | 282.25 | 5 | 2 | 2 | 253.01 |
| CID_193980 | 3.02 | 100.90 | 24 | 326.30 | 6 | 2 | 7 | 281.64 |
| CID_122718 | 1.01 | 117.97 | 25 | 344.32 | 7 | 2 | 2 | 290.31 |
| CID_90443 | 2.15 | 86.99 | 20 | 272.26 | 5 | 3 | 1 | 230.26 |
| CID_73200 | 1.66 | 82.07 | 23 | 316.31 | 6 | 1 | 3 | 273.69 |
| CID_67111 | 1.76 | 90.89 | 20 | 270.24 | 5 | 3 | 1 | 224.05 |
| CID_40586 | 1.12 | 100.90 | 22 | 302.28 | 6 | 2 | 2 | 255.57 |
| CID_17366 | 2.80 | 122.53 | 24 | 334.28 | 8 | 3 | 6 | 277.55 |
| CID_12548 | 2.75 | 115.05 | 21 | 286.24 | 6 | 4 | 0 | 231.21 |
| CID_10169 | 2.13 | 132.12 | 22 | 300.22 | 7 | 4 | 1 | 233.63 |
| CID_69840182 | 2.35 | 111.12 | 22 | 300.27 | 6 | 4 | 1 | 248.63 |
| CID_69839295 | 2.41 | 90.89 | 21 | 284.27 | 5 | 3 | 1 | 240.61 |
| CID_69838991 | 1.91 | 111.12 | 21 | 286.24 | 6 | 4 | 1 | 232.07 |
| CID_69838075 | 1.95 | 111.12 | 22 | 300.27 | 6 | 4 | 1 | 248.63 |
| CID_67182664 | 2.15 | 96.22 | 22 | 302.28 | 6 | 3 | 2 | 255.81 |
| CID_56975176 | 2.52 | 124.29 | 21 | 290.23 | 7 | 4 | 4 | 234.47 |
| CID_9879020 | 1.41 | 93.07 | 21 | 288.25 | 6 | 2 | 1 | 238.79 |
| CID_5281295 | 1.76 | 90.89 | 20 | 270.24 | 5 | 3 | 1 | 224.05 |
| CID_24885046 | 1.03 | 104.06 | 21 | 286.24 | 6 | 3 | 1 | 232.44 |
| CID_24885048 | 1.05 | 104.06 | 21 | 286.24 | 6 | 3 | 1 | 232.44 |
| CID_42636438 | 1.58 | 120.36 | 21 | 290.23 | 7 | 4 | 1 | 230.19 |
| CID_44187799 | 1.02 | 107.22 | 21 | 288.25 | 6 | 4 | 1 | 238.31 |
| CID_44355975 | 2.02 | 100.13 | 22 | 300.27 | 6 | 3 | 2 | 249.59 |
| CID_45257240 | 2.70 | 111.12 | 23 | 312.28 | 6 | 4 | 2 | 259.48 |
| CID_46881083 | 2.15 | 96.22 | 22 | 302.28 | 6 | 3 | 2 | 255.81 |
CLogP, Calculated LogP; TPSA, topological polar surface area; nAtoms, number of atoms; MW, molecular weight; HBAs, number of hydrogen bond donors; HBDs, number of hydrogen bond acceptors; nRotb, number of rotatable bonds.
Table 2.
Pharmacokinetic properties of filtered molecules explored using Swiss ADME server.
| Compounds | AS | GIA | BBBP | PGPS | CYP 1A2 inhibitor | CYP2C19 inhibitors | CYP2C9 inhibitors | CYP2D6 inhibitors | CYP3A4 inhibitors | Lead Likeness |
|---|---|---|---|---|---|---|---|---|---|---|
| CID_78385623 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_85044918 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_76319859 | Soluble | High | No | No | Yes | No | No | Yes | No | Yes |
| CID_73197708 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_73212812 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_73318750 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_101382379 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_129646445 | Soluble | High | No | No | No | No | No | Yes | Yes | Yes |
| CID_5322048 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_11313032 | Soluble | High | No | No | No | No | No | Yes | No | Yes |
| CID_16743457 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_21500651 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_22298206 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_23626869 | Soluble | High | No | No | Yes | No | No | No | No | Yes |
| CID_89490339 | Soluble | High | No | No | No | No | Yes | No | No | Yes |
| CID_90666180 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_91059286 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_91413267 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_101342513 | Soluble | High | No | No | Yes | No | Yes | No | No | Yes |
| CID_101342516 | Soluble | High | No | No | Yes | No | Yes | No | No | Yes |
| CID_101584198 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_101593135 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_101782169 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_122182396 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_102292606 | Soluble | High | No | No | Yes | No | No | No | Yes | Yes |
| CID_101566521 | Soluble | High | No | No | Yes | No | No | No | Yes | Yes |
| CID_76330740 | Soluble | High | No | No | No | No | No | Yes | No | Yes |
| CID_71514077 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_71330931 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_70544322 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_313107 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_193980 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_122718 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_90443 | Soluble | High | No | No | Yes | No | No | No | Yes | Yes |
| CID_73200 | Soluble | High | No | No | Yes | No | No | No | No | Yes |
| CID_67111 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_40586 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_17366 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_12548 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_10169 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_69840182 | Soluble | High | No | No | Yes | No | No | No | Yes | Yes |
| CID_69839295 | Soluble | High | No | No | Yes | No | No | No | Yes | Yes |
| CID_69838991 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_69838075 | Soluble | High | No | No | Yes | No | No | No | Yes | Yes |
| CID_67182664 | Soluble | High | No | No | Yes | No | No | No | Yes | Yes |
| CID_56975176 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_9879020 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_5281295 | Soluble | High | No | No | No | No | No | No | Yes | Yes |
| CID_24885046 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_24885048 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_42636438 | Soluble | High | No | No | Yes | No | No | No | No | Yes |
| CID_44187799 | Soluble | High | No | No | No | No | No | No | No | Yes |
| CID_44355975 | Soluble | High | No | No | Yes | No | No | No | No | Yes |
| CID_45257240 | Soluble | High | No | No | Yes | No | No | Yes | No | Yes |
| CID_46881083 | Soluble | High | No | No | Yes | No | No | No | Yes | Yes |
AS, Aqueous solubility; GIA, gastrointestinal absorption; BBBP: blood-brain barrier permeant; PGPS: P-glycoprotein substrate.
Molecular docking study
The molecular docking simulation was carried out to study the binding mode of the selected compounds inside the active site. The analysis of the docking results assisted the rationalization of the E6 protein of HPV-16 inhibition predictions made in the previous steps. At the first step of docking, numerous PDB structures were subjected and chosen based on their crystallographic resolutions. Using conformation population in the top-ranked cluster of the AutoDock output file, the 4GIZ was selected as the most appropriate crystallographic structure for further modeling studies. In addition, most of the previous research studies have been used this PDB code (4,17,13,34,35). The crystal structure of the E6-E6AP complex (PDB ID: 4GIZ) was retrieved from the PDB.
Molecular docking study
The molecular docking simulation was carried out to study the binding mode of the selected compounds inside the active site. The analysis of the docking results assisted the rationalization of the E6 protein of HPV-16 inhibition predictions made in the previous steps. At the first step of docking, numerous PDB structures were subjected and chosen based on their crystallographic resolutions. Using conformation population in the top- ranked cluster of the AutoDock output file, the 4GIZ was selected as the most appropriate crystallographic structure for further modeling studies. In addition, most of the previous research studies have been used this PDB code (4,17,13,34,35). The crystal structure of the E6- E6AP complex (PDB ID: 4GIZ) was retrieved from the PDB.
The docking scores (estimated free binding energy ΔGbinding)) and the interactions with the key amino acids of each ligand were explored to find the best conformation and orientation of the ligand within the active site of the enzyme. The final docking results were sorted according to the docking scores. The runs with the best docking scores were regarded as the most stable conformations and orientations.
From the previous step, 55 molecules were subjected to molecular docking studies. These compounds were docked into the active site of the E6 protein of HPV-16. Then, they were sorted based on free binding energy, and 11 molecules with the highest free binding energies were selected (Table 3 and Fig. 4). Accomplished docked poses were assessed to find the best binding mode of the molecule in the active site. Three compounds; viz. CID_73212812, CID_91059286, and CID_69838075 exhibited the highest docking score and the best interactions with binding residues when compared to the other compounds and jaceosidin and cidofovir in the E6 protein of HPV-16 (Table 3).
Table 3.
Hydrophobic, hydrogen, and cation-π stacking interactions for screened molecules in the E6 protein of HPV-16 active site.
| Compounds | ΔGbinding (Kcal/mol) | Hydrophobic | hydrogen bonds | Cation-π |
|---|---|---|---|---|
| CID_76319859 | -9.06 | Pro335, Leu373, Arg383, Gln376, Cys51, Gly380, Ser71, Ser74 | Gln336, Glu381, Arg10, Glu377 | - |
| Val53, Ile52, Arg8, Pro9, Glu7, Pro5, Tyr32, | ||||
| CID_73212812 | -11.17 | Leu50, Val62, Cys64, Leu67, Tyr70, Arg102, | Tyr54, Glu377, Arg10 | - |
| Ile128, Arg131, Cys51, Ser71, Ser74 | ||||
| CID_23626869 | -9.61 | Val53, Ile52, Glu377, Gln376, Glu381, Arg102, Val62, Leu67, Lys65, Tyr32, Tyr70 | Ile52, Arg10, Arg131, Gly380, Arg383 | - |
| CID_90666180 | -9.52 | Pro9, Tyr54, Glu377, Gly380, Val53, Gln376, Val53, Tyr32, Leu50 | Ile52, Arg10, Glu381, Arg8 | - |
| Val53, Arg102, Val62, Lys180, Ile179, Thr374, | ||||
| CID_91059286 | -10.43 | Leu50, Asp64, Leu67, Arg10, Lys65, Tyr32, | Ile179, Lys34 | - |
| Tyr70, Ser74, Leu373, Cys51 | ||||
| CID_102292606 | -9.71 | Pro9, Glu7, Pro5, Tyr54, Val53, Gly380, Tyr32, Leu50, Arg131 | Arg10, Ile52, Glu377, Arg8 | - |
| CID_76330740 | -9.24 | Ile52, Val53, Cys51, Arg8, Pro5, Arg10, Gln6, Tyr70, Leu50, Tyr32 | Glu7, Glu377, Tyr54 | Arg8 |
| CID_69840182 | -10.04 | Arg8, Tyr54, Pro9, Cys51, Pro5, Glu377, Leu50, Gly380, Val53, Leu67, Tyr70, Tyr32 | Arg10, Ile52, Glu381, Gln376 | - |
| CID_69839295 | -9.35 | Pro5, Arg10, Ile52, Arg8, Cys51, Val53, Leu67, Tyr70, Tyr32, Leu50 | Glu382, Glu377, Tyr54 | Arg10 |
| CID_69838075 | -10.87 | Val53, Val62, Pro5, Ser74, Glu75, Ser71, Tyr70, Leu67, Ile52, Arg10, Cys51, Asp64, Leu50, Ile104, Arg131, Tyr32 | Glu381, Glu377, Arg8, Tyr54, Ile52 | Arg10 |
| CID_45257240 | -9.29 | Val53, Tyr54, Glu377, Gly380, Arg10, Gln376, Glu381 | Arg131, Ile52, Gly380 | - |
| Cidofovir | -9.12 | Gln376, Glu381, Arg102, Val62, Tyr54, Lys65, Tyr32, Leu50 | Arg10, Glu377, Val53, Ile52 | - |
| Jaceosidin | -8.60 | Val53, Cys51, Gln376, Arg10, Ile104, Tyr70, Leu50 | Arg8, Ile52, Leu67 | - |
The obtained results proposed that CID_73212812, CID_91059286, and CID_69838075 were possibly more potent in the E6 protein of the HPV-16 active site. CID_73212812 had a docking score of -11.17 kcal/mol. Similarly, CID_91059286 and CID_69838075 exhibited docking scores equal to -10.43 and -10.87 kcal/mol, respectively. Free binding energies values and different interactions of molecules in the E6 protein of HPV-16 active site are summarized in Table 3.
MD simulation studies
MD simulations were performed on the best- docked conformations of the three selected compounds (CID_73212812, CID_91059286, and CID_69838075) and cidofovir and jaceosidin as the references with the best docking scores and orientations in the active site. The results of MD would be suitable to confirm the docking outputs since both protein and ligand are flexible.
Root mean square deviations
To explore the stability of the relevant ligand-protein complexes during the simulation, the root means square deviations (RMSD) of the protein backbone atoms was plotted vs primary crystal protein as a function of time (36). Throughout MD simulation, the RMSD of the system tends to be converged demonstrating that the system is stable and well equilibrated. The analysis of the backbone RMSDs plot exhibited that after a minor fluctuation from the primary conformation, the complex retained its stability throughout MD simulation. The RMSD plots of all complexes vs simulation time frames are shown in Fig. 5. The produced RMSD graph lines indicated growing trends with increasing RMSD values from 0.10 to 0.30 nm during 0-1100 ps. At About 5400 ps the CID_73212812 complex decreased a little while CID_91059286 and CID_69838075 displayed growing trends. Finally, all compounds reached equilibration and stability of about 11000 ps.
Fig. 5.

Root mean square deviations (RMSD) backbone for complexes of the E6 protein of HPV-16 with CID_73212812, CID_91059286, CID_69838075, cidofovir, and jaceosidin during molecular dynamic simulations. Five color graph lines characterized the docked complexes.
Root mean squared fluctuation
Root mean square fluctuation (RMSF) was performed for the top-ranked molecules and control drugs to unravel the effect of ligand binding on the flexible structure of protein and the behavior of key amino acids as well. Higher RMSF values indicate more flexibility, whereas lower RMSF values display more rigidity throughout the simulation. The RMSF of all complexes was estimated to be similar to each other and was depicted in Fig. 6. According to the RMSF values, the fluctuations of some residues in some complexes were more than that of other complexes, while residues fluctuations in the active site and in particular in Asp66, Val53, Tyr32, Leu50, Arg10, Arg8, Cys51, Tyr70, and Tyr54 were very low.
Fig. 6.

Root mean square fluctuation (RMSF) graphs for CID_73212812, CID_91059286, CID_69838075, cidofovir, and jaceosidin.
Radius of gyration
The radius of gyration (Rg) study was carried out to understand the compactness measure of the protein. The lower Rg value of an enzyme is indicative of its compactness. The stability and proper folding of the enzyme structure are shown by stable and lower Rg value while the structural flexibility and lack of appropriate folding of the protein introduces a highly fluctuating Rg. The average Rg values of CID_73212812, CID_69838075, CID_91059286, cidofovir, and jaceosidin complexes were found to be 2.55 nm, 2.54 nm, 2.62 nm, 2.55 nm, and 2.54 nm, respectively. The Rg value of CID_73212812 increased after 12 ns while the Rg value of CID_69838075 decreased during the MD simulations. In general, the average Rg of all complexes was approximately similar throughout the MD simulations (Fig. 7).
Fig. 7.

The radius of gyration (Rg) for backbone atoms of protein throughout the simulation, in the existence of CID_73212812, CID_69838075, CID_91059286, cidofovir, and jaceosidin.
Hydrogen bonds analysis
A strong inhibitor appears to form hydrogen bonds with its target protein. In examining the ligand-protein complexes, hydrogen bonds have a critical role. Therefore, the stability measure of the ligand-protein complex, to some extent depended on the number of intermolecular hydrogen bonds. The hydrogen bonds were analyzed to describe the stability of top bound molecules and cidofovir and jaceosidin as the standard compounds (Fig. 8). in the case of cid_73212812 and cid_69838075, the maximum of four hydrogen bond interactions could be recorded while two or three hydrogen bonds were observed at most of the simulation time. The number of hydrogen bonds reached uptofive some time during the CID_91059286 simulation.
Fig. 8.

Hydrogen bonds numbers made between (A) CID_73212812, (B) CID_91059286, (C) CID_69838075, (D) cidofovir, and (E) jaceosidin the E6 protein of HPV-16 active site residues during MD simulations.
Binding free energy analysis
Binding free energies were calculated using the MM-PBSA technique. According to Table 4, the free binding energy of cidofovir and jaceosidin were -168.152 kJ/mol and -135.476 kJ/mol, respectively. Compared to the standard ligands, the selected compounds are associated with higher free binding energies. The energy components including polar, electrostatic, Van der Waals, and SASA are listed in Table 4.
Table 4.
Free binding energy (kJ/mol) components between candidate compounds and the E6 protein of HPV-16.
| Complex | VWE | EE | PSE | SASA energy | BE |
|---|---|---|---|---|---|
| CID_73212812 | -174.926 ± 1.022 | -176.284 ± 2.006 | 174.833 ± 2.982 | -13.700 ± 0.089 | -190.403 ± 2.679 |
| CID_91059286 | -222.609 ± 1.529 | -3.206 ± 0.928 | 72.493 ± 2.015 | -16.283 ± 0.108 | -169.836 ± 1.880 |
| CID_6983807 | -195.940 ± 1.296 | -53.952 ± 1.301 | 86.917 ± 1.446 | -15.296 ± 0.093 | -178.005 ± 1.978 |
| Cidofovir | -200.595 ± 2.127 | -28.208 ± 1.561 | 76.826 ± 1.943 | -16.254 ± 0.100 | -168.152 ± 2.492 |
| Jaceosidin | -168.033 ± 1.876 | -66.463 ± 1.823 | 114.437 ± 2.081 | -15.393 ± 0.196 | -135.476 ± 2.375 |
VWE, Van der Waals energy; EE, electrostatic energy; PSE, polar solvation energy; SASA, solvent-accessible surface area; BE, binding energy.
DISCUSSION
The highest free binding energy (-11.17 Kcal/mol) for the interaction of CID_73212812 with the E6 protein of HPV-16 active site may be interpreted as follows (Fig. 9): There were two hydrogen bonds between the hydroxyl group of chromene ring and Tyr54 and Glu377 residues. A hydrogen bond was formed between the carboxyl oxygen group of chromene rings and Arg10 residue. Moreover, two hydrogen bonds were detected for NH groups of Arg10 and the carbonyl group of ester moiety at the chromene ring.
Fig. 9.

2D form (right) and 3D form (left) of the best binding poses and interactions of CID_73212812 in the active site of the E6 protein of HPV-16.
Hydrophobic contacts of ligand were formed with Val53, Cys51, Ile52, Arg8, Pro9, Glu7, Pro5, Tyr32, Leu50, Val62, Cys64, Leu67, Tyr70, Arg102, Ile128, Arg131, Cys51, Ser71, and Ser74. In addition, this ligand did not exhibit any π-π or cation-π interactions.
Compound CID_69838075 as the second-ranked binder was found to be associated with the following interactions (Fig. 10): there were four hydrogen bonds between hydroxyl groups of the phenyl ring and Glu377, Ile52, Tyr54, and Arg8 residues. The hydroxyl groups of benzofuran ring participated in two hydrogen bonds with Glu381. This compound formed a cation-π stacking interaction with Arg10 (Fig. 11). Tyr54, Ile52, Arg8, Glu377, Arg10, Pro5, Cys51, Val53, and Glu381 residues formed hydrophobic pockets interacting with CID_69838075 by hydrophobic contacts.
Fig. 10.

2D form (right) and 3D form (left) of best binding poses and interactions of CID_69838075 in the active site of the E6 protein of HPV-16.
Fig. 11.
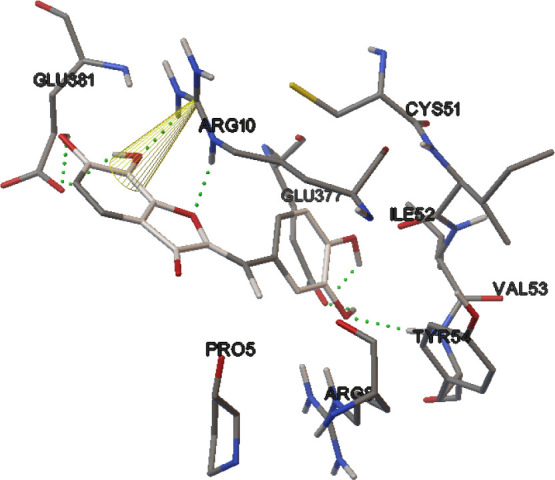
Orientations of benzofuran ring in CID_69838075 with the amine group of Arg10 to cation-π stacking interaction.
The binding mode of CID_91059286 at the E6 protein of HPV-16 active site is shown in Fig. 12. It formed three hydrogen bonds with active site amino acids: a carbonyl group of dihydrobenzo[b]oxepine ring with NH2 group of Lys34 and two hydroxyl groups of phenyl ring with the carbonyl group of Ile179. CID_91059286 displayed hydrophobic interactions with Lys34, Ile179, Val31, Tyr32, Ala371, Arg55, Ala370, Lys180, Thr374, and Leu373 amino acids while no π-π or cation-π interactions could be detected.
Fig. 12.

2D form (right) and 3D form (left) of best binding poses and interactions of CID_91059286 in the active site of the E6 protein of HPV-16.
Previous researches reported Tyr32, Arg10, Arg8, Cys51, Tyr54, Tyr70, Ser71, Phe45, Val53, Ser74, Arg131, Leu67, and Lue50 as key residues for hydrogen bonds and hydrophobic interactions with the reported compounds. In addition, most of the compounds formed hydrogen bonds with Tyr32 and Cys51 (17). It was generally observed that Leu50 and Cys51 are necessary for higher binding affinity (27). CID_76319859, CID_76330740, CID_69840182, and CID_69839295 also showed hydrophobic interactions with Cys51 residue similar to top rank compounds. In addition, CID_90666180, CID_91059286, CID_102292606, CID_76330740, CID_69840182, and CID_69839295 made hydrophobic interactions with Cys50 residue.
Arg10 formed a hydrogen bond with CID_73212812, while it made hydrophobic interactions with CID_69838075 and CID_91059286. Additionally, CID_69838075 displayed cation-π staking interaction with Arg10. CID_76319859, CID_23626869, CID_90666180, CID_102292606, and CID_69840182 made hydrogen bonds with Arg10 residue. CID_69839295 and CID_69838075 showed cation-π staking interaction with Arg10, while CID_76330740 exhibited this interaction with Arg8.
CID_91059286 formed a hydrogen bond with Arg8 but two other compounds showed hydrophobic interactions with it. Also, CID_90666180 and CID_102292606 showed a hydrogen bond with Arg8.
Cidofovir showed a hydrogen bond with Arg10. All compounds showed a hydrogen bond with this amino acid, while CID_91059286, CID_76330740, CID_69839295, CID_69838075, CID_45257240, and jaceosidin formed hydrophobic interaction with this amino acid. Jaceosidin exhibited a hydrogen bond with Arg8, another key amino acid, whereas this interaction did not form with compounds CID_45257240, CID_69839295, CID_69840182, CID_76330740, CID_91059286, CID_23626869, and cidofovir. In addition, cidofovir and jaceosidin formed a hydrogen bond with Ile52 which was formed in most of the compounds.
Jaceosidin and cidofovir did not display any cation-π interaction. This interaction was observed to CID_69838075 and CID_69839295 with Arg10 and CID_76330740 with Arg8. Other compounds also did not show cation-π with active site amino acids.
The obtained docking investigations indicated that Val53, Tyr32, Leu50, Ile52, Arg10, Arg8, Tyr70, Arg131, Ser74, Cys51, and Tyr54 were significant amino acids in keeping the binding modes of the molecules into the E6 protein of HPV-16 because they are involved in a hydrogen bond, hydrophobic, and cation-π interactions with most of the molecules particularly the compounds CID_73212812, CID_91059286, and CID_69838075. These results had a good correlation with the results reported by previous researches (18,13,27). Additionally, in the ligand-enzyme interactions, optimum hydrogen bonds, and hydrophobic interactions displayed a vital and significant role. However, cation-π and π-π interactions did not have an important role in affinity.
All complexes (CID_73212812, CID_91059286, and CID_69838075) exhibited steady stable graph lines associated with very slight fluctuations from 5700 ps to 20000 ps. It is noteworthy that the CID_73212812-protein was relatively more stable than complexes CID_91059286- and CID_69838075-protein. The RMSD values of complexes CID_91059286- and CID_69838075-protein are higher than complex CID_73212812- protein. The RMSD diagram demonstrated that complex CID_73212812-protein is more stable than other complexes and jaceosidin during simulation time but has similar stability to cidofovir (Fig. 5).
These results proposed that in dynamic equilibriums, the stabilities of the complexes were acceptable. Moreover, the stability of the systems confirmed the validity of the docking results.
The residues fluctuations in the active site and in particular in Asp66, Val53, Tyr32, Leu50, Arg10, Arg8, Cys51, Tyr70, and Tyr54 were very low. This suggests that in the E6 protein of HPV-16, when an inhibitor is bound to the enzyme, some amino acids could move far away from their primary positions, but the active site amino acids are more rigid when an inhibitor was bound to them. For example, in CID_69838075, Asp66, Val53, Tyr32, Leu50, Arg10, Arg8, Cys51, Tyr70, and Tyr54 had maximum RMSFs of 0.61, 0.66, 0.53, 0.64, 0.67, 0.52, 0.51, and 0.51 Å, respectively (Fig. 6). The selected compounds were able to make strong interactions with active site amino acids throughout the MD simulations. This could be further proved due to the slight range of RMSFs of the amino acids for each compound.
In the presence of selected molecules, the Rg backbone atoms of the E6 protein of HPV-16 slightly decreased during the simulation time. In summary, although molecules binding affected the flexibility of the active site amino acids, the flexibility of the enzyme domain did not significantly decrease, and the protein compactness remained unchanged. In addition, the protein compactness reveals a suitable folding and stability of the protein structure. The Rg is represented in Fig. 7.
CID_73212812 and CID_69838075 showed the maximum of four hydrogen bond interactions and the number of hydrogen bonds reached five for the CID_91059286. For cidofovir and jaceosidin, the number of hydrogen bond interactions during the simulations was similar to CID_73212812 and CID_69838075. It is noticeable that jaceosidin showed fewer hydrogen bonds than cidofovir during the MD simulations (Fig. 8).
According to the lower free binding energy of compound CID_91059286 when compared to the other complexes, it interacted poorly with key active site amino acids. The negative values for free binding energies showed thermodynamic stability and higher binding affinity while the positive values showed less thermodynamic stability. Compound CID_73212812 interacted strongly with the vital active site amino acids and exhibited the highest free binding energy among the modeled compounds. This result confirmed docking binding energy results. The electrostatic energy for CID_73212812, CID_91059286, CID_69838075, cidofovir and jaceosidin complexes are -176.284, -3.206, -53.952, - 28.208, and -66.463 kJ/mol, respectively. Among the complexes, the CID_73212812 complex showed highly negative electrostatic energy and the satisfactory contribution of polar energy.
In summary, analyzing energies revealed that the interaction among the E6 protein of HPV-16 and candidate molecules was mostly determined by appropriate nonpolar interactions, whereas polar interactions are unfavorable to ligand binding.
CONCLUSION
In summary, SBVS as a rational approach could be successfully applied to recognize the potential E6 protein of the HPV-16 inhibitors. A chemical library was developed from compounds with structural similarity to Jaceosidin within the PubChem database. The compounds were retrieved, further analyzed, and screened on the criteria of binding energy, drug-likeness properties, and ADME parameters via computational techniques such as molecular docking and MD simulations. Good physicochemical properties and high affinity toward target could be recorded for the described compounds. The molecular docking was carried out to make qualitative and quantitative analyses on the interactions of the candidate molecules inside the active site. Among all compounds, CID_73212812, CID_91059286, and CID_69838075 were selected and introduced as in silico hit compounds. The MD simulations of CID_73212812, CID_91059286, CID_69838075, cidofovir, and jaceosidin into the E6 protein of HPV-16 active site were performed. Finally, the MD simulations analyses were performed including RMSD, RMSF, hydrogen bond, and Rg and it was revealed that selected complexes retained their stability in the E6 protein of HPV-16 active site during MD time. The docking and MD analysis proposed that optimum hydrogen bonds and hydrophobic contacts were vital in binding interactions of in silico hits.
Conflict of interest statement
The authors declared no conflict of interest in this study.
Authors’ contribution
S. Mirzayi, V. Adhami, and K. Mahnam carried out the experimental studies; N. Razzaghi-Asl analyzed the data and contributed to editing the manuscript; S. Sepehri designed and supervised the project, analyzed the data, and prepared the manuscript. The final version of the manuscript was approved by all authors.
Acknowledgments
This work was financially supported by Ardabil University of Medical Sciences through Grant No. IR.ARUMS.REC.1397.090.
REFERENCES
- 1.Archambault J, Melendy T. Targeting human papillomavirus genome replication for antiviral drug discovery. Antivir Ther. 2013;18(3):271–283. doi: 10.3851/IMP2612. DOI: 10.3851/IMP2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin J, Chen L, Qiu X, Zhang N, Guo Q, Wang Y, et al. Traditional chinese medicine for human papillomavirus (HPV) infections: a systematic review. Biosci Trends. 2017;11(3):267–273. doi: 10.5582/bst.2017.01056. DOI: 10.5582/bst.2017.01056. [DOI] [PubMed] [Google Scholar]
- 3.Beadle JR, Valiaeva N, Yang G, Yu JH, Broker TR, Aldern KA, et al. Synthesis and antiviral evaluation of octadecyloxyethyl benzyl 9-[(2- Phosphonomethoxy)ethyl]guanine (ODE-Bn- PMEG), a potent inhibitor of transient HPV DNA amplification. J Med Chem. 2016;59(23):10470–10478. doi: 10.1021/acs.jmedchem.6b00659. DOI: 10.1021/acs.jmedchem.6b00659. [DOI] [PubMed] [Google Scholar]
- 4.Motavalli Khiavi F, Arashkia A, Nasimi M, Mahdavi M, Golkar M, Roohvand F, et al. Immunization of mice by a multimeric L2-based linear epitope (17-36) from HPV type 16/18 induced cross reactive neutralizing antibodies. Res Pharm Sci. 2017;12(4):265–273. doi: 10.4103/1735-5362.212043. DOI: 10.4103/1735-5362.212043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kachaeva MV, Pilyo SG, Kornienko AM, Prokopenko VM, Zhirnov VV, Prichard MN, et al. In vitro activity of novel 1,3-oxazole derivatives against human papillomavirus. Ibnosina J Med Biomed Sci. 2017;9:111–118. DOI: 10.4103/ijmbs.ijmbs_9_17. [Google Scholar]
- 6.Hillemanns P, Garcia F, Petry KU, Dvorak V, Sadovsky O, Iversen OE, et al. A randomized study of hexaminolevulinate photodynamic therapy in patients with cervical intraepithelial neoplasia 1/2. Am J Obstet Gynecol. 2015;212(4):465. doi: 10.1016/j.ajog.2014.10.1107. e1-e7. DOI: 10.1016/j.ajog.2014.10.1107. [DOI] [PubMed] [Google Scholar]
- 7.Hampson L, Martin-Hirsch P, Hampson IN. An overview of early investigational drugs for the treatment of human papilloma virus infection and associated dysplasia. Expert Opin Investig Drugs. 2015;24(12):1529–1537. doi: 10.1517/13543784.2015.1099628. DOI: 10.1517/13543784.2015.1099628. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Song J, Liu A, Xiao B, Li S, Wen Z, et al. Research progress of the antiviral bioactivities of natural flavonoids. Nat Prod Bioprospect. 2020;10(5):271–283. doi: 10.1007/s13659-020-00257-x. DOI: 10.1007/s13659-020-00257-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zakaryan H, Arabyan E, Oo A, Zandi K. Flavonoids: promising natural compounds against viral infections. Arch Virol. 2017;162:2539–2551. doi: 10.1007/s00705-017-3417-y. DOI 10.1007/s00705-017-3417-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clemente-Soto AF, Salas-Vidal E, Pacheco CM, Carranza JNS, Zaragoza OP, Maya LG. Quercetin induces G2 phase arrest and apoptosis with the activation of p53 in an E6 expression independent manner in HPV positive human cervical cancer derived cells. Mol Med Rep. 2019;19(3):2097–2106. doi: 10.3892/mmr.2019.9850. DOI: 10.3892/mmr.2019.9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cherry JJ, Rietz A, Malinkevich A, Liu Y, Xie M, Bartolowits M, et al. Structure based identification and characterization of flavonoids that disrupt human papillomavirus-16 E6 function jonathan. PloS One. 2013;8(12)(e84506):1–21. doi: 10.1371/journal.pone.0084506. DOI: 10.1371/journal.pone.0084506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar A, Rathi E, Kini SG. E-pharmacophore modelling, virtual screening, molecular dynamics simulations and in-silico ADME analysis for identification of potential E6 inhibitors against cervical cancer. J Mol Struct. 2019;1189:299–306. DOI: 10.1016/j.molstruc.2019.04.023. [Google Scholar]
- 13.Lee HG, Yu KA, Oh WK, Baeg TW, Oh HC, Ahn JS, et al. Inhibitory effect of jaceosidin isolated from Artemisia argyi on the function of E6 and E7 oncoproteins of HPV 16. J Ethnopharmacol. 2005;98(3):339–343. doi: 10.1016/j.jep.2005.01.054. DOI: 10.1016/j.jep.2005.01.054. [DOI] [PubMed] [Google Scholar]
- 14.Kalhor H, Sadeghi S, Marashiyan M, Kalhor R, Gharehbolagh SA, Akbari Eidgahi MR, et al. Identification of new DNA gyrase inhibitors based on bioactive compounds from streptomyces: structure- based virtual screening and molecular dynamics simulations approaches. J Biomol Struct Dyn. 2020;38(3):791–806. doi: 10.1080/07391102.2019.1588784. DOI: 10.1080/07391102.2019.1588784. [DOI] [PubMed] [Google Scholar]
- 15.Kumar A, Rathi E, Kini SG. Exploration of small- molecule entry disruptors for chikungunya virus by targeting matrix remodelling associated protein. Res Pharm Sci. 2020;15(3):300–311. doi: 10.4103/1735-5362.288437. DOI: 10.4103/1735-5362.288437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sepehri S, Saghaie L, Fassihi A. Anti-HIV-1 activity prediction of novel gp41 inhibitors using structure- based virtual screening and molecular dynamics simulation. Mol Inform. 2017;36(3):1600060. doi: 10.1002/minf.201600060. DOI: 10.1002/minf.20. [DOI] [PubMed] [Google Scholar]
- 17.Celegato M, Messa L, Goracci L, Mercorelli B, Bertagnin C, Spyrakis F, et al. A novel small- molecule inhibitor of the human papillomavirus E6-p53 interaction that reactivates p53 function and blocks cancer cells growth. Cancer Lett. 2020;470:115–125. doi: 10.1016/j.canlet.2019.10.046. DOI: 10.1016/j.canlet.2019.10.046. [DOI] [PubMed] [Google Scholar]
- 18.Kumar A, Rathi E, Kini SG. Drug repurposing approach for the identification and designing of potential E6 inhibitors against cervical cancer: an in silico investigation. Struct Chem. 2020;31:141–153. DOI: 10.1007/s11224-019-01378-x. [Google Scholar]
- 19.Kumar S, Jena L, Sahoo M, Nayak T, Mohod K, Daf S, et al. The in silico approach to identify a unique plant-derived inhibitor against E6 and E7 oncogenic proteins of high-risk human papillomavirus 16 and 18. Avicenna J Med Biochem. 2016;4(1)(e33958):1–5. DOI: 10.17795/ajmb-33958. [Google Scholar]
- 20.Kumar S, Jena L, Mohod K, Daf S, Varma AK. Virtual screening for potential inhibitors of high-risk human papillomavirus 16 E6 protein. Interdiscip Sci Comput Life Sci. 2015;7:136–142. doi: 10.1007/s12539-015-0008-z. DOI: 10.1007/s12539-013-0213-6. [DOI] [PubMed] [Google Scholar]
- 21.Ricci-Lopez J, Vidal-Limon A, Zunñiga M, Jimènez VA, Alderete JB, Brizuela CA, et al. Molecular modeling simulation studies reveal new potential inhibitors against HPV E6 protein. PloS One. 2019;14(3)(e0213028):1–22. doi: 10.1371/journal.pone.0213028. DOI: 10.1371/journal.pone.0213028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar S, Jena L, Sahoo M, Kakde M, Daf S, Varma AK. In silico docking to explicate interface between plant-originated inhibitors and E6 oncogenic protein of highly threatening human papillomavirus 18. Genomics Inform. 2015;13(2):60–67. doi: 10.5808/GI.2015.13.2.60. DOI: 10.5808/GI.2015.13.2.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Sessions RB, Prime SS, Shoemark DK, Allen SJ, Hong W, et al. Identification of novel small molecule TGF-β antagonists using structure-based drug design. J Comput Aided Mol Des. 2013;27(4):365–372. doi: 10.1007/s10822-013-9651-9. DOI: 10.1007/s10822-013-9651-9. [DOI] [PubMed] [Google Scholar]
- 24.Razzaghi-Asl N, Mirzayi S, Mahnam K, Sepehri S. Identification of COX-2 inhibitors via structure- based virtual screening and molecular dynamics simulation. J Mol Graph Model. 2018;83:138–152. doi: 10.1016/j.jmgm.2018.05.010. DOI: 10.1016/j.jmgm.2018.05.010. [DOI] [PubMed] [Google Scholar]
- 25.Zolek T, Maciejewska D. Theoretical evaluation of ADMET properties for coumarin derivatives as compounds with therapeutic potential. Eur J Pharm Sci. 2017;109:486–502. doi: 10.1016/j.ejps.2017.08.036. DOI: 10.1016/j.ejps.2017.08.036. [DOI] [PubMed] [Google Scholar]
- 26.Singh S, Das T, Awasthi M, Pandey VP, Pandey B, Dwivedi UN. DNA topoisomerase directed anti- cancerous alkaloids: ADMET based screening, molecular docking and dynamics simulation. Biotechnol Appl Biochem. 2016;63(1):125–137. doi: 10.1002/bab.1346. DOI: 10.1002/bab.1346. [DOI] [PubMed] [Google Scholar]
- 27.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(42717):1–13. doi: 10.1038/srep42717. DOI: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zanier K, Charbonnier S, Sidi AOMO, McEwen AG, Ferrario MG, Poussin-Courmontagne P, et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science. 2013;339(6120):694–698. doi: 10.1126/science.1229934. DOI: 10.1126/science. 1229934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yazdani M, Khezri J, Hadizadeh N, Zamani J, Zakaria A, Naderi M, et al. Depinar, a drug that potentially inhibits the binding and entry of COVID- 19 into host cells based on computer-aided studies. Res Pharm Sci. 2021;16(3):315–325. doi: 10.4103/1735-5362.314830. DOI: 10.4103/1735-5362.314830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun H, Li Y, Tian S, Xu L, Hou T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys Chem Chem Phys. 2014;16(31):16719–16729. doi: 10.1039/c4cp01388c. DOI: 10.1039/C4CP01388C. [DOI] [PubMed] [Google Scholar]
- 31.Kumar A, Shanthi V, Ramanathan K. Discovery of potential ALK inhibitors by virtual screening approach. 3 Biotech. 2016;6(1):21–32. doi: 10.1007/s13205-015-0336-z. DOI: 10.1007/s13205-015-0336-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lone IH, Khan KZ, Fozdar BI. Synthesis, physicochemical properties, antimicrobial and antioxidant studies of pyrazoline derivatives bearing a pyridyl moiety. Med Chem Res. 2014;23(1):363–369. DOI: 10.1007/s00044-013-0643-z. [Google Scholar]
- 33.Krejsa CM, Horvath D, Rogalski SL, Penzotti JE, Mao B, Barbosa F, et al. Predicting ADME properties and side effects: the BioPrint approach. Curr Opin Drug Discov Devel. 2003;6(4):470–480. PMID: 12951810. [PubMed] [Google Scholar]
- 34.Kolluru S, Momoh R, Lin L, Mallareddy JR, Krstenansky JL. Identification of potential binding pocket on viral oncoprotein HPV16 E6: a promising anti-cancer target for small molecule drug discovery. BMC Mol Cell Biol. 2019;20:30–40. doi: 10.1186/s12860-019-0214-3. DOI: 10.1186/s12860-019-0214-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mamgain S, Sharma P, Pathak RK, Baunthiya M. Computer aided screening of natural compounds targeting the E6 protein of HPV using molecular docking. Bioinformation. 2015;11(5):236–242. doi: 10.6026/97320630011236. DOI: 10.6026/97320630011236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahimi H, Negahdari B, Shokrgozar MA, Madadkar- Sobhani A, Mahdian R, Foroumadi A, et al. A structural model of the anaphase promoting complex co-activator (Cdh1) and in silico design of inhibitory compounds. Res Pharm Sci. 2015;10(1):59–67. PMID: 26430458. [PMC free article] [PubMed] [Google Scholar]