

Abstract
Background:
Huntington’s disease (HD) is caused by an expanded (>35) CAG trinucleotide repeat in huntingtin (HTT). Age-at-onset of motor symptoms is inversely correlated with the size of the inherited CAG repeat, which expands further in brain regions due to somatic repeat instability. Our recent genetic investigation focusing on autosomal SNPs revealed that age-at-onset is also influenced by genetic variation at many loci, the majority of which encode genes involved in DNA maintenance/repair processes and repeat instability.
Objective:
We performed a complementary association analysis to determine whether variants in the X chromosome modify HD.
Methods:
We imputed SNPs on chromosome X for ~9,000 HD subjects of European ancestry and performed an X chromosome-wide association study (XWAS) to test for association with age-at-onset corrected for inherited CAG repeat length.
Results:
In a mixed effects model XWAS analysis of all subjects (males and females), assuming random X-inactivation in females, no genome-wide significant onset modification signal was found. However, suggestive significant association signals were detected at Xq12 (top SNP, rs59098970; p-value, 1.4E-6), near moesin (MSN), in a region devoid of DNA maintenance genes. Additional suggestive signals not involving DNA repair genes were observed in male- and female-only analyses at other locations.
Conclusion:
Although not genome-wide significant, potentially due to small effect size compared to the power of the current study, our data leave open the possibility of modification of HD by a non-DNA repair process. Our XWAS results are publicly available at the updated GEM EURO 9K website hosted at https://www.hdinhd.org/ for browsing, pathway analysis, and data download.
Keywords: Huntington’s disease, genetic modifier, XWAS, residual age-at-onset
INTRODUCTION
Huntington’s disease (HD; OMIM #143100) is a dominantly inherited neurodegenerative disease, caused by an expanded unstable CAG trinucleotide repeat in huntingtin (HTT) [1]. CAG repeats longer than 39 are fully penetrant, resulting in cognitive and behavioral manifestations in addition to a characteristic, diagnostic, movement disorder whose timing is best explained by the length of the inherited CAG repeat, with longer repeats typically leading to earlier onset [1–6]. However, the remaining variance in age-at-onset that is not accounted for by the size of the repeat is heritable, which implicates a role for other genetic factors in influencing HD [7–9] by accelerating or decelerating the rate at which the HTT CAG expansion mutation drives disease onset. Thus, identification of genetic modifiers of HD can inform underlying disease mechanisms of the disease and may point to disease-modifying therapeutic strategies [10].
In order to discover genetic modifiers of HD, we have performed a series of genome-wide association studies (GWAS) [11–13] focusing on residual age-at-onset, which represents age-at-onset normalized by individual CAG repeat size [5, 14]. In our standard GWAS testing SNPs (simple nucleotide polymorphisms) on autosomal chromosomes, significant modification signals were detected at many genomic locations that harbor DNA repair genes involved in CAG repeat instability [13, 14]. These data not only suggest an important role for somatic CAG repeat instability in influencing the onset of HD [12, 13, 15–17] but also support this process as a potential target of disease-delaying therapeutics. Interestingly, although DNA mismatch repair had been proposed as a process important to repeat instability [18–29], the genes implicated by significant age-at-onset modification signals do not completely overlap with canonical mismatch repair genes (https://www.gseamsigdb.org/gsea/msigdb/geneset_page.jsp?geneSetName=GO_MISMATCH_REPAIR). For example, FAN1, which is better known for inter-strand crosslink repair was the most significant modifier in our recent GWAS of ~9,000 HD subjects [13, 30]. Only a subset of canonical mismatch repair genes generated genome-wide significant signals (e.g., MLH1, MSH3), whereas others failed to exhibit even suggestive association signals (e.g., MSH2, MSH6) [13, 14]. In addition, our GWAS also implicated non-DNA repair genes (e.g., CCDC82, TCERG1) as modifiers of HD age-at-onset [13]. These genes might act indirectly on CAG repeat instability processes or might influence HD through a different mechanism [13]. Thus, these human genetic studies indicate that HD can be modified by particular genes involved in DNA repair/maintenance, and also by genes not participating directly in DNA repair processes. Our pursuit of GWAS reflects the potential that identification of either type of modifier gene has implications for understanding the pathogenesis of HD and for the development of disease-delaying therapeutic strategies applicable for this and possibly other DNA expansion disorders [19, 28]. Still, the past GWAS [11, 13] remains incomplete because only autosomes were tested. Therefore, to supplement our recent autosomal GWAS (~9,000 European ancestry) [13], we have now performed an X chromosome-wide association study (XWAS) to determine whether variants in genes on the X chromosome modify the timing of HD onset.
MATERIALS AND METHODS
Study subjects and genotype imputation of SNPs on chromosome X
A total of 9,058 HD subjects (CAG 40 to 55) of European ancestry were previously analyzed in our GWA study to test SNPs on autosomes [13]. Details of study approval, genotyping, determination of CAG repeat size, and calculation of residual age-at-onset are described elsewhere [13]. Typed chromosome X SNP data of the same 9,058 subjects were used to impute SNPs on the X chromosome. Details of typed genotype data are described elsewhere [13]. The chromosome X typed GWA data were subjected to initial quality control (QC) to include subjects with X chromosome sample genotyping call rate > 90%, SNP call rate > 95% and SNP minor allele frequency > 1%). Subsequently, X chromosome SNP imputation, including additional QC, was carried out by the Michigan Imputation Server (https://imputationserver.sph.umich.edu/index.html#!!; v.1.1.4) using the Haplotype Reference Consortium (HRC; r1.1) as the reference panel; phasing and imputation was performed using EAGLE (v.2.4), and MINIMAC4, respectively. Then, imputed X SNPs with imputation R square values higher than 0.5 and minor allele frequency (MAF) greater than 0.1% were taken, generating genotypes at 214,728 SNPs in 8,963 HD subjects for the genetic association study. The pseudoautosomal regions (PAR; ChrX: 60001–2699520 and ChrX:154931004–155260560 relative to GRCh37/hg19 coordinates) were not analyzed in this study due to low imputation quality. SNP IDs and genomic coordinates of SNPs were based on dbSNP151 and GRCh37/hg19, respectively.
XWAS using a residual age-at-onset phenotype
Imputed X chromosome SNPs were tested for modification of HD. We used a continuous residual age-at-onset phenotype, which represents the difference between observed and expected age-at-onset based on individual CAG repeat size. For example, positive and negative residual age-at-onset values represent later and earlier age-at-onset compared to expected age-at-onset based on CAG repeat size [5]. For genetic association testing, residual age-at-onset was modeled as a function of the minor allele count of each test SNP in a linear mixed effects model using the GEMMA program (v0.98.1); a set of covariates such as study group, sex, and ancestry characteristics was also included [13]. Familial relationship was corrected in the mixed effects model by including the kinship matrix based on SNPs on autosomes. Using this statistical framework, we performed three separate continuous phenotype XWAS: 1) combined analysis of males and females as the main analysis, 2) male-specific analysis, and 3) female-specific analysis. For male-female combined analysis, we used a genotype coding method that considers X chromosome inactivation in females. For example, the genotype of males who carry a major and a minor allele were respectively coded as 0 and 2; while the genotype of females who were homozygous major allele, heterozygous, and homozygous minor alleles were coded as 0, 1, and 2, respectively. This approach, therefore, assumes the magnitude of the effect of two alleles in females is similar to that of one in males due to the random inactivation of one allele in a female’s cells. In addition, we performed dichotomous phenotype XWAS analysis for the combined data. Details of dichotomous phenotype analysis were also described previously [13]. Briefly, subjects were sorted based on residual age-at-onset values, and the top 30% of subjects were compared to the bottom 30% samples in a logistic regression model for a given test SNP.
Conditional analysis of the MSN region
To determine whether the suggestive significant association signal at Xq12 represented a single independent modifier haplotype, a conditional analysis was performed for SNPs in chrX: 64550000–65550000. For each test SNP in the region, we constructed a mixed effects model involving the same set of covariates that were used in our standard XWAS analysis and the genotype of the top SNP in the region (rs59098970).
Incorporation of XWAS data into the GEM EURO 9K website
The construction of the original website for autosomal GWAS of 9,058 HD subjects with European ancestry [13] was described previously [14]. We updated the original website by incorporating XWAS results. Briefly, we combined association results of SNPs on autosomes and chromosome X generating a single summary data file. This summary data file contains association analysis results of more than 11 million SNPs with minor allele frequencies > 0.1%, and is available for download at the updated website. In addition, our updated website implements the same regional plot function and UCSC custom track using the combined summary data file for user-friendly browsing and annotation. For gene set enrichment analysis functionality, we applied the same methods for SNP-to-gene mapping and gene size correction [14] to XWAS results to generate the data set that allows the updated website to perform the same permutation-based enrichment analysis of a user provided gene set. Autosome-based gene set enrichment analysis of comprehensive pathways annotated in various databases was performed and reported previously [13]. Therefore, it is recommended to refer to our previous report [13] for gene set enrichment analysis results of curated/annotated pathways. However, if a user’s gene set is not represented in the curated pathway databases, and/or a gene set has a significant number of genes on the X chromosome, our updated website provides a means to evaluate their significance as a set. Our updated website (namely, GEM Euro 9K) is available at https://www.hdinhd.org/.
Genomic coordinates
Genomic coordinates are based on Grch37/hg19 genome assembly.
RESULTS AND DISCUSSION
Following genotype imputation and quality control, we analyzed 214,728 SNPs (minor allele frequency > 0.1%) on chromosome X in 8,963 HD subjects (4,345 males and 4,618 females) who carry 40–55 CAG repeats (S. Fig. 1A). Similar to our autosomal GWAS [13], we focused on a phenotype that represents age-at-onset corrected for individual CAG repeat size (i.e., residual age-at-onset). Consistent with our previous analysis of a relatively small number of HD subjects [31], age-at-onset is similar between male and female HD subjects for a given CAG repeat (S. Fig. 1B) indicating no significant effect of the subject’s sex on the timing of onset. As anticipated, residual age-at-onset is also not significantly different between males and females (S. Fig. 1C; t-test p-value, 0.5415).
Next, we performed XWAS using the combined (males plus females) data to evaluate the levels of association between residual age-at-onset and SNPs on chromosome X (excluding pseudoautosomal regions due to low genotype imputation quality) as the primary analysis. For a given test SNP, we constructed a linear mixed effects model to correct for familial relationship, using residual age-at-onset as the continuous phenotype variable and a single SNP coded based on X-chromosome inactivation in females [32, 33]. We also performed separate XWAS for males and females. In addition, we performed a separate XWAS with a dichotomized residual age-at-onset phenotype, comparing subjects with the top 30% and bottom 30% residual age-at-onset values using logistic regression models similar to the dichotomous analysis performed for SNPs on autosomes [13]. The X chromosome contains 19 genes annotated as ‘DNA_Repair’ in gene ontology (GO:0006281) [34] (Table 1), but none of these is specifically associated with gene ontology_Mismatch_Repair (GO:0006298). Overall, neither continuous (top panel) nor dichotomous phenotype (bottom panel) XWAS analysis revealed genome-wide significant (p-value < 5E-8) association signals for SNPs near any of the genes involved in DNA repair (Fig. 1, blue circles; Table 1) or elsewhere on the X chromosome (Fig. 1; grey circles).
Table 1.
Association analysis of DNA repair genes on chromosome X.
| Gene symbol | RefSeq | Number of SNPs | Top SNP | Top SNP p-value |
|---|---|---|---|---|
| FANCB | NM_001018113.3 | 289 | rs139578952 | 0.002315 |
| KLHL15 | NM_030624.3Tetx | 94 | rs138596629 | 0.118225 |
| POLA1 | NM_001330360.2 | 403 | rs185474865 | 0.029276 |
| WAS | NM_000377.3 | 0 | NA | NA |
| SMC1A | NM_006306.4 | 21 | rs12009181 | 1425990.142599 |
| HUWE1 | NM_031407.7 | 50 | rs6638361 | 0.073692 |
| APEX2 | NM_014481.4 | 20 | rs113783082 | 0.223833 |
| USP51 | NM_201286.3 | 32 | rs2473060 | 0.05954 |
| NONO | NM_007363.5 | 0 | NA | NA |
| ATRX | NM_000489.5 | 278 | rs35865732 | 0.016954 |
| RPA4 | NM_013347.4 | 218 | rs188707019 | 0.011041 |
| MORF4L2 | NM_012286.3 | 147 | rs5945700 | 0.072105 |
| RADX | NM_018015.6 | 236 | rs138511267 | 0.013999 |
| UBE2A | NM_003336.4 | 0 | NA | NA |
| RNF113A | NM_006978.3 | 25 | rs144292694 | 0.122693 |
| CUL4B | NM_001079872.1 | 82 | rs150396008 | 0.041952 |
| CETN2 | NM_004344.3 | 0 | NA | NA |
| TREX2 | NM_080701.3 | 0 | NA | NA |
| BRCC3 | NM_001018055.2 | 0 | NA | NA |
A list of genes annotated as “DNA REPAIR” (GO:0006281) was obtained (http://www.gseamsigdb.org/gsea/msigdb/cards/GO_DNA_REPAIR). Among 551 genes in the list, 19 are located on the chromosome X. XWAS analysis results using the continuous residual age-at-onset phenotype were mapped to those 19 genes. SNPs located between the 50KB flanking region of the transcription start site and 50KB flanking region of the transcription end site are identified for each gene; the SNP that showed the smallest p-value in XWAS analysis is shown.
Figure 1. XWAS analysis of continuous or dichotomized residual age-at-onset in HD.
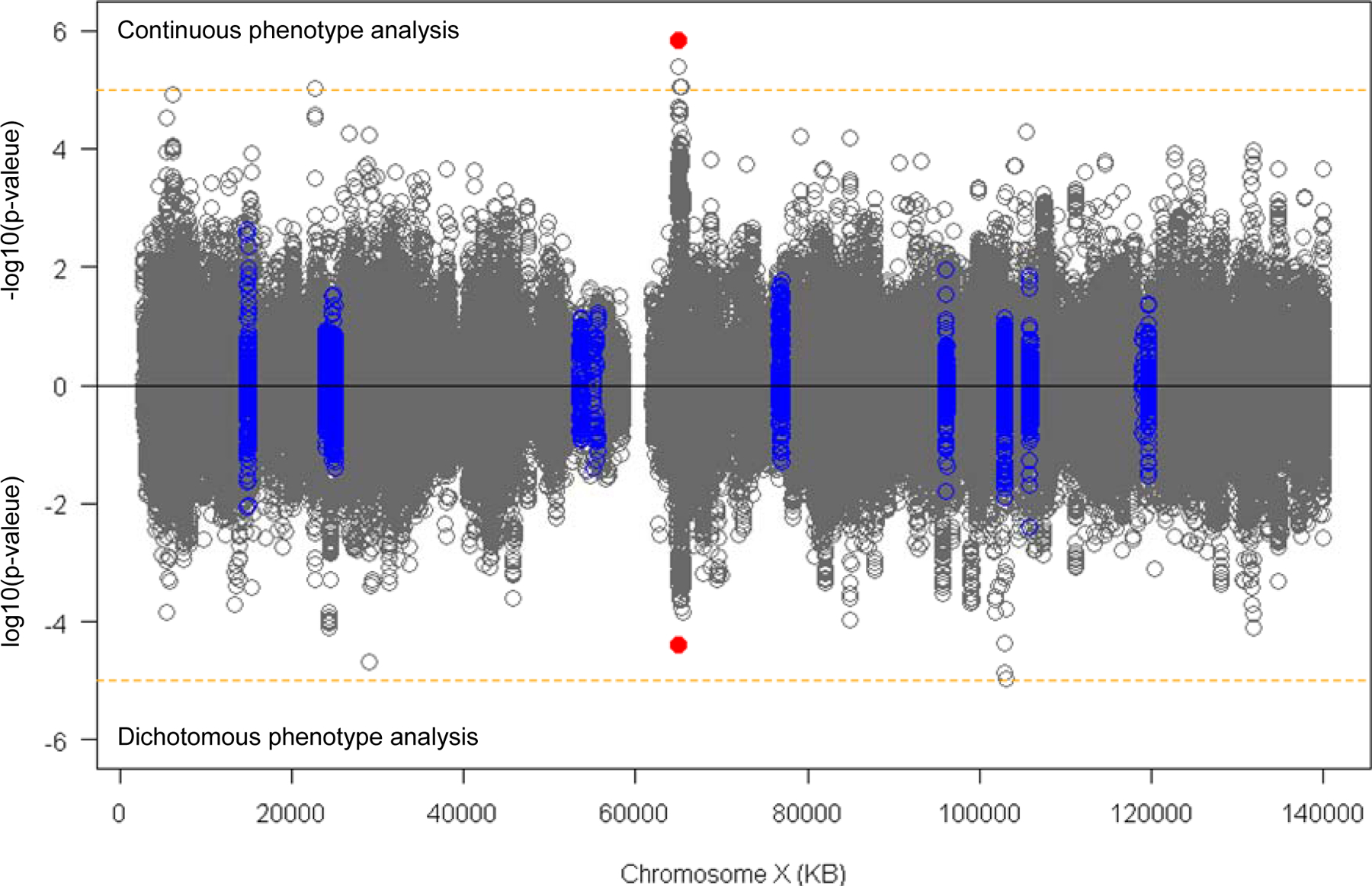
Linear mixed effects model XWAS analysis was performed to evaluate the levels of association between the test SNP and residual age-at-onset in continuous phenotype analysis (top panel) and dichotomous phenotype analysis (bottom panel). The Y-axis of top and bottom panels represent −log10(p-value) in continuous phenotype analysis and log10(p-value) in dichotomous phenotype analysis, respectively. Blue circles represent SNPs located in 50 KB flanking regions of DNA repair genes (see Table 1). Orange horizontal lines indicate suggestive significance (p-value, 0.00001). Red dots indicate the top SNP (rs59098970) at Xq12 region that generated suggestive significant modification signals. PARs (ChrX: 60001–2699520 and ChrX:154931004–155260560) were excluded from our XWAS due to low quality in genotype imputation
Despite the absence of genome-wide significant associations, a number of suggestive (p-value < 1E-5) signals were observed (Table 2). For example, the Xq12 region (chrX:64550000–65550000) exhibited four SNPs near MSN (moesin) and VSIG4 (V-set and immunoglobulin domain-containing protein 4) with suggestive association signals in the continuous phenotype XWAS (Fig. 1; Table 2) that showed contribution from both male and female HD subjects (Table 2; Fig. 2; S. Fig. 2). Patterns of linkage disequilibrium (Fig. 3) and conditional analysis using the top SNP (rs59098970) (S. Fig. 3) revealed two relatively independent signals of suggestive significance in this region. The top SNP (rs59098970) in this region was still suggestive significant (p-value, 2.6E-6) in an association analysis model with additional covariates that included a set of previously discovered autosomal modifier SNPs (tagging modifier haplotype 2AM1, 3AM1, 5AM1–3, 5BM1, 7AM1, 8AM1, 11AM1, 15AM1–4, and 19AM1–3) [13]. A single suggestive signal (rs10284175) was also observed at Xp22.11 in an intron of the non-coding RNA PTCHD1-AS (Patched-domain containing 1 antisense RNA).
Table 2.
Suggestive significant SNPs.
| SNP | Coordinate | P-value | ||
|---|---|---|---|---|
| Males + Females | Males | Females | ||
| rs766033603 | 6525210 | 3.79E-04 | 6.82E-01 | 9.75E-06 |
| rs10284175 | 22746142 | 9.74E-06 | 5.67E-05 | 4.89E-02 |
| rs59098970 | 64985533 | 1.45E-06 | 5.06E-04 | 2.02E-04 |
| rs773999898 | 65015633 | 4.11E-06 | 3.81E-05 | 1.22E-02 |
| rs192170082 | 65231566 | 8.94E-06 | 3.81E-05 | 2.56E-02 |
| rs183103894 | 65251211 | 8.94E-06 | 3.81E-05 | 2.56E-02 |
| rs1853544 | 95232656 | 5.82E-02 | 9.53E-01 | 3.83E-06 |
| rs747527548 | 95238800 | 5.82E-02 | 9.53E-01 | 3.83E-06 |
| rs1951846 | 95247994 | 5.82E-02 | 9.53E-01 | 3.83E-06 |
| rs1781122 | 123312624 | 1.34E-03 | 7.55E-06 | 5.71E-01 |
| rs2133238 | 123314352 | 1.46E-03 | 7.55E-06 | 5.42E-01 |
| rs77800759 | 123316132 | 1.80E-03 | 9.87E-06 | 5.19E-01 |
SNPs generating suggestive significant p-values in either 1) combined analysis (males + females), 2) male-specific analysis, or 3) female-specific analysis using the continuous residual age-at-onset phenotype are listed.
Grey table cells indicate suggestive significance.
Figure 2. XWAS of residual age-at-onset in males and females.
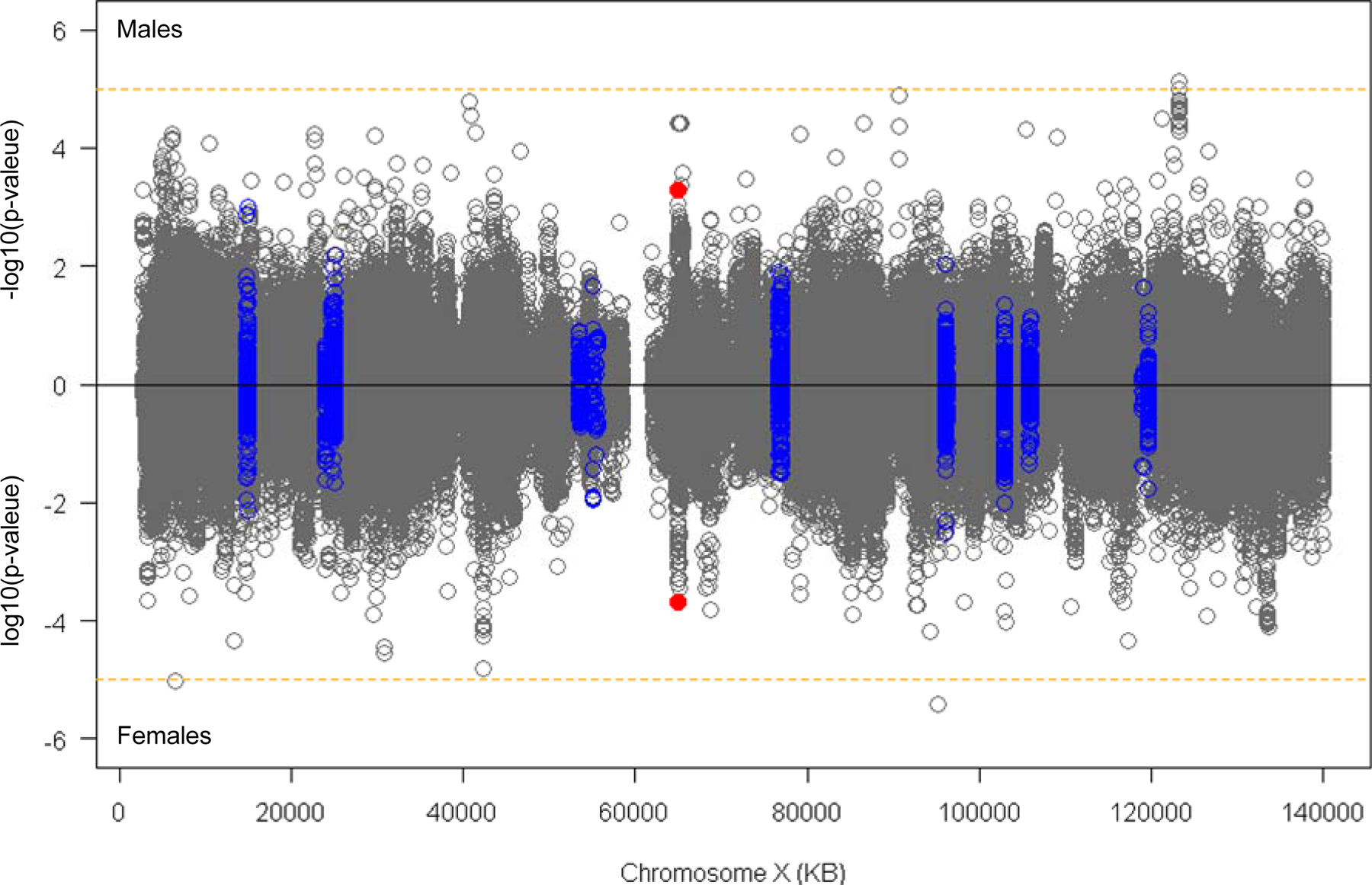
XWAS analysis was performed for males (top panel) and females (bottom panel) separately. Y-axis represent − log10(p-value) for male-specific analysis and log10(p-value) for the female-specific analysis. Orange horizontal lines indicate suggestive significance. Blue circles represent SNPs located in 50 KB flanking regions of DNA repair genes. Orange horizontal lines indicate suggestive significance (p-value, 0.00001). Red dots indicate the top SNP (rs59098970) at Xq12 region.
Figure 3. Suggestive significant association signals at Xq12.
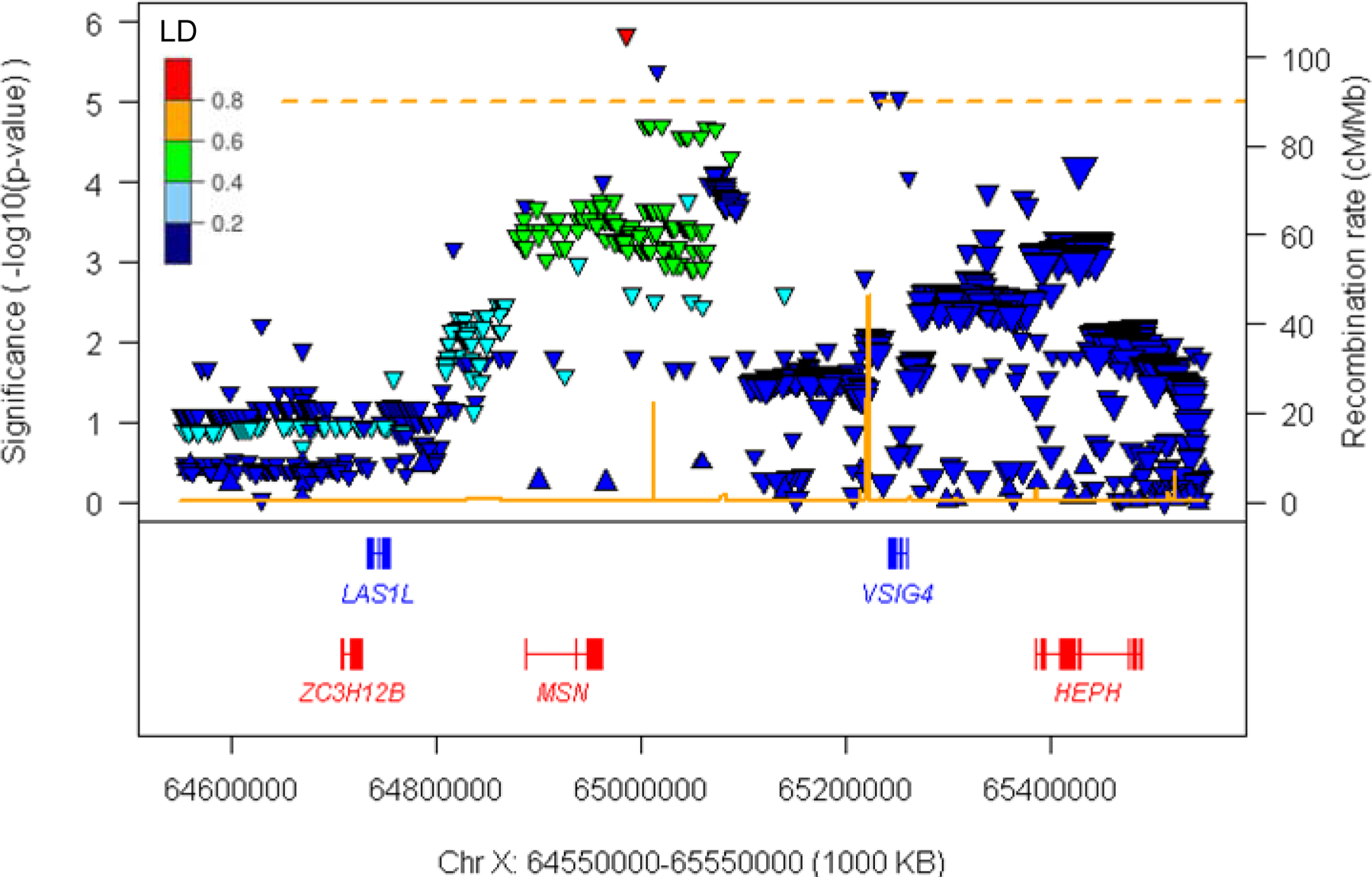
Suggestive significant association signals at Xq12 are displayed. The Y-axis and X-axis represent the levels of association significance (−log10(p-value)) and genomic coordinate, respectively. The secondary Y-axis represents the recombination rate based on HapMap data. Upward and downward triangles represent SNPs whose minor alleles are associated with delayed and hastened age-at-onset, respectively. SNPs are colored based on linkage disequilibrium with the top SNP in this region (i.e., rs59098970). The sizes of triangles are proportional to MAF. Genes in red and blue represent RefSeq Select transcripts on the plus and minus strand, respectively.
Several different suggestive signals were observed in the sex-specific XWAS. Three Xq25 SNPs with common minor alleles in GRIA3, which encodes subunit 3 of the AMPA type ionotropic glutamate receptor, achieved suggestive significance in males but were not even nominally significant (P<5E-2) in females (Table 2). By contrast, three SNPs with low frequency minor alleles in Xq21.22 and one in Xp22.31, both regions representing large intergenic segments, all reached suggestive significance only in females (Table 2).
Any of these suggestive association signals may represent genuine modifier effects that will become genome-wide significant with larger XWAS sample sizes or be revealed by complementary evidence. Alternatively, they may be due to chance. If the former, they would more likely be acting through the mechanism that causes neuronal toxicity in HD rather than through the CAG repeat expansion that leads to triggering of that toxicity mechanism when a threshold CAG length is reached through somatic expansion of the repeat in vulnerable neurons [14]. None of the association signals is near any genes reported to be involved in DNA repair, being instead near genes that could conceivably be participating in causing or protecting the cell from toxic damage. For example, excitotoxicity via glutamate receptor activation has long been postulated to play a role in HD making GRIA3 an interesting candidate [35]. Similarly, the level of expression of moesin (‘membrane-organizing extension spike protein’), which links integral membrane proteins to the cytoskeleton and regulates signaling from surface receptors [36] has recently been implicated in neuronal viability [37]. Whether these or other genes in the vicinity of the suggestive signals are modifiers of HD will require additional evidence that we hope to gain from future genetic studies or from other experimental approaches.
To facilitate engagement of the research community with our autosomal GWAS data [13], we previously created a web site that allows interested investigators to browse the association data to view SNP location and identity, minor allele frequency, association p-value and effect size by SNP or by gene, as well as providing a link to a custom track in the UCSC Genome Browser to permit direct integration with the multitude of other regional annotations available [14]. The GWAS web site also allows download of the full autosomal data set. For additional useful functionality, the web site provides the capacity to test user-defined gene sets for significant association with HD age-at-onset [14]. Consequently, to ensure that investigators using complementary approaches can capitalize on the information provided by this XWAS and potentially develop evidence that assists in defining autosomal or X-linked modifiers, we have updated our GEM Euro 9K web site by combining the autosomal and X-chromosome data, with all of the functionality described above. Our updated ‘GEM Euro 9K’ website is available at https://www.hdinhd.org/.
Our autosomal GWAS identified multiple loci involved in DNA repair processes that implicated somatic expansion of the CAG repeat as the rate driver leading to disease onset and other loci that might be indirectly involved in this process or act later, on the toxicity mechanism. On the assumption that the toxicity mechanism in human HD involves the expression of huntingtin protein with an expanded polyglutamine segment, which remains uncertain, huntingtin lowering strategies are now being tested as possible therapeutic interventions to slow the worsening of HD symptoms. The identification of multiple DNA maintenance genes as modifiers of onset has subsequently led to preclinical efforts to interfere with somatic CAG expansion as a potential treatment. Identification of additional candidate modifiers of the toxicity driver mechanism might similarly help to define the nature of that mechanism and to provide additional routes to treatments validated directly by human data. Although the current XWAS did not reveal genome-wide significant modification signals or add additional loci to the DNA maintenance category of modifiers, our data remain consistent with the possibility that age-at-onset in HD can be modified by non-DNA repair mechanisms in some X chromosome genes. As revealed by our GWAS, identification of functionally-related sets of modifier genes and thereby informing the underlying mechanisms of modification can significantly contribute to the development of effective disease modifying treatments for HD. Consequently, we are currently in the process of further expanding the GWAS and XWAS datasets with the goal of further dissecting the rate driving process and delineating the mechanism(s) involved in causing and modifying the toxic damage.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank research participants and their families. This research was supported by the CHDI Foundation Inc., the U.S. National Institutes of Health (NS082079, NS091161, NS016367, NS049206, NS105709, NS119471), the Medical Research Council (UK MR/L010305/1and fellowship MR/P001629/1), and a Cardiff University School of Medicine studentship.
CONFLICT OF INTEREST
J.F.G. is a Scientific Advisory Board member and has a financial interest in Triplet Therapeutics, Inc. His NIH-funded project is using genetic and genomic approaches to uncover other genes that significantly influence when diagnosable symptoms emerge and how rapidly they worsen in Huntington disease. The company is developing new therapeutic approaches to address triplet repeat disorders such Huntington’s disease, myotonic dystrophy and spinocerebellar ataxias. His interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. J.F.G. has also been a consultant for Wave Life Sciences USA, Inc. J.M.L serves in the scientific advisory board of GenEdit, Inc. Within the last five years D.G.M. has been a scientific consultant and/or received an honoraria/stock options from AMO Pharma, Charles River, LoQus23, Small Molecule RNA, Triplet Therapeutics and Vertex Pharmaceuticals and held research contracts with AMO Pharma and Vertex Pharmaceuticals. J.D.L. is a paid Advisory Board member for F. Hoffmann-La Roche Ltd and uniQure biopharma B.V., and he is a paid consultant for Vaccinex Inc, Wave Life Sciences USA Inc, Genentech Inc, Triplet Inc, and PTC Therapeutics Inc. L.J. is a member of the scientific advisory boards of LoQus23 Therapeutics and Triplet Therapeutics. V.C.W. is a Scientific Advisory Board member of Triplet Therapeutics, Inc., a company developing new therapeutic approaches to address triplet repeat disorders such Huntington’s disease and Myotonic Dystrophy. Her financial interests in Triplet Therapeutics were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict of interest policies. She is a scientific advisory board member of LoQus23 Therapeutics and has provided paid consulting services to Alnylam and Acadia Pharmaceuticals.
REFERENCES
- 1.The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–83. [DOI] [PubMed] [Google Scholar]
- 2.Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet. 1993;4:398–403. [DOI] [PubMed] [Google Scholar]
- 3.Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet. 1993;4:387–92. [DOI] [PubMed] [Google Scholar]
- 4.Bates GP. History of genetic disease: the molecular genetics of Huntington disease - a history. Nature Reviews Genetics. 2005;6:766–73. [DOI] [PubMed] [Google Scholar]
- 5.Lee JM, Ramos EM, Lee JH, Gillis T, Mysore JS, Hayden MR, et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012;78:690–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nature Reviews Disease Primers. 2015:15005. [DOI] [PubMed] [Google Scholar]
- 7.Djousse L, Knowlton B, Hayden M, Almqvist EW, Brinkman R, Ross C, et al. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. Am J Med Genet A. 2003;119A:279–82. [DOI] [PubMed] [Google Scholar]
- 8.Li JL, Hayden MR, Almqvist EW, Brinkman RR, Durr A, Dode C, et al. A genome scan for modifiers of age at onset in Huntington disease: The HD MAPS study. Am J Hum Genet. 2003;73:682–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci U S A. 2004;101:3498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gusella JF, MacDonald ME, Lee JM. Genetic modifiers of Huntington’s disease. Mov Disord. 2014;29:1359–65. [DOI] [PubMed] [Google Scholar]
- 11.GeM-HD Consortium. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell. 2015;162:516–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JM, Chao MJ, Harold D, Abu Elneel K, Gillis T, Holmans P, et al. A modifier of Huntington’s disease onset at the MLH1 locus. Hum Mol Genet. 2017;26:3859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.GeM-HD Consortium. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell. 2019;178:887–900 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong EP, MacDonald ME, Wheeler VC, Jones L, Holmans P, Orth M, et al. Huntington’s Disease Pathogenesis: Two Sequential Components. J Huntingtons Dis. 2021;10:35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ciosi M, Maxwell A, Cumming SA, Hensman Moss DJ, Alshammari AM, Flower MD, et al. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine. 2019;48:568–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monckton DG. The Contribution of Somatic Expansion of the CAG Repeat to Symptomatic Development in Huntington’s Disease: A Historical Perspective. J Huntingtons Dis. 2021;10:7–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swami M, Hendricks AE, Gillis T, Massood T, Mysore J, Myers RH, et al. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. 2009;18:3039–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massey TH, Jones L. The central role of DNA damage and repair in CAG repeat diseases. Dis Model Mech. 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bettencourt C, Hensman-Moss D, Flower M, Wiethoff S, Brice A, Goizet C, et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann Neurol. 2016;79:983–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morales F, Vasquez M, Santamaria C, Cuenca P, Corrales E, Monckton DG. A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair (Amst). 2016;40:57–66. [DOI] [PubMed] [Google Scholar]
- 21.Pinto RM, Dragileva E, Kirby A, Lloret A, Lopez E, St Claire J, et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet. 2013;9:e1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dragileva E, Hendricks A, Teed A, Gillis T, Lopez ET, Friedberg EC, et al. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis. 2009;33:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kovtun IV, McMurray CT. Features of trinucleotide repeat instability in vivo. Cell Res. 2008;18:198–213. [DOI] [PubMed] [Google Scholar]
- 24.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–42. [DOI] [PubMed] [Google Scholar]
- 25.Owen BA, Yang Z, Lai M, Gajec M, Badger JD 2nd, Hayes JJ, et al. (CAG)(n)-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat Struct Mol Biol. 2005;12:663–70. [DOI] [PubMed] [Google Scholar]
- 26.Gomes-Pereira M, Fortune MT, Ingram L, McAbney JP, Monckton DG. Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion. Hum Mol Genet. 2004;13:1815–25. [DOI] [PubMed] [Google Scholar]
- 27.Wheeler VC, Lebel LA, Vrbanac V, Teed A, te Riele H, MacDonald ME. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum Mol Genet. 2003;12:273–81. [DOI] [PubMed] [Google Scholar]
- 28.Flower M, Lomeikaite V, Ciosi M, Cumming S, Morales F, Lo K, et al. MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tome S, Manley K, Simard JP, Clark GW, Slean MM, Swami M, et al. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet. 2013;9:e1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim KH, Hong EP, Shin JW, Chao MJ, Loupe J, Gillis T, et al. Genetic and Functional Analyses Point to FAN1 as the Source of Multiple Huntington Disease Modifier Effects. Am J Hum Genet. 2020;107:96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keum JW, Shin A, Gillis T, Mysore JS, Abu Elneel K, Lucente D, et al. The HTT CAG-Expansion Mutation Determines Age at Death but Not Disease Duration in Huntington Disease. Am J Hum Genet. 2016;98:287–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozbek U, Lin HM, Lin Y, Weeks DE, Chen W, Shaffer JR, et al. Statistics for X-chromosome associations. Genet Epidemiol. 2018;42:539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science. 2004;303:644–9. [DOI] [PubMed] [Google Scholar]
- 34.Gene Ontology C Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wan J, Savas JN, Roth AF, Sanders SS, Singaraja RR, Hayden MR, et al. Tracking brain palmitoylation change: predominance of glial change in a mouse model of Huntington’s disease. Chem Biol. 2013;20:1421–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faure S, Salazar-Fontana LI, Semichon M, Tybulewicz VL, Bismuth G, Trautmann A, et al. ERM proteins regulate cytoskeleton relaxation promoting T cell-APC conjugation. Nat Immunol. 2004;5:272–9. [DOI] [PubMed] [Google Scholar]
- 37.Luo T, Ou JN, Cao LF, Peng XQ, Li YM, Tian YQ. The Autism-Related lncRNA MSNP1AS Regulates Moesin Protein to Influence the RhoA, Rac1, and PI3K/Akt Pathways and Regulate the Structure and Survival of Neurons. Autism Res. 2020;13:2073–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.