

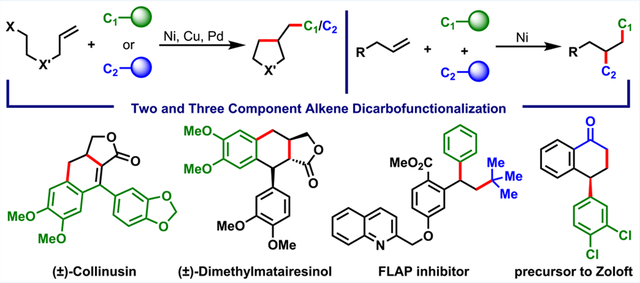
CONSPECTUS:
Recently, alkene dicarbofunctionalization, i.e., the powerful organic synthesis method of alkene difunctionalization with two carbon sources, emerged as a formidable reaction with immense promise to synthesize complex molecules expeditiously from simple chemicals. This reaction is generally achieved with transition metals (TMs) through interception by carbon sources of an alkylmetal [β-H–C(sp3)–[M]] species, a key intermediate prone to undergo rapid β-H elimination. Related prior reports, since Paolo Chiusoli and Catellani’s work in 1982 [Tetrahedron Lett. 1982, 23, 4517], have used bicyclic and disubstituted terminal alkenes, wherein β-H elimination is avoided by geometric restriction or complete lack of β-H’s. With reasoning that β-H–C(sp3)–[M] intermediates could be rendered amenable to interception with the use of first row late TMs and formation of coordination-assisted transient metallacycles, these two strategies were implemented to address the β-H elimination problem in alkene dicarbofunctionalization reactions.
Because first row late TMs catalyze C(sp3)–C(sp3) coupling, Cu and Ni were anticipated to impart sufficient stability to β-H–C(sp3)–[M] intermediates, generated catalytically upon alkene carbometalation, for their subsequent interception by carbon electrophiles/nucleophiles in three-component reactions. Additionally, such an innate property could enable alkene difunctionalization with carbon coupling partners through entropically driven cyclization/coupling reactions. The cyclometalation concept to stabilize intractable β-H–C(sp3)–[M] intermediates was hypothesized when three-component reactions were performed. The idea of cyclometalation to curtail β-H elimination is founded upon Whitesides’s [J. Am. Chem. Soc. 1976, 98, 6521] observation that metallacycles undergo β-H elimination much slower than acyclic alkylmetals.
In this Account, examples of alkene dicarbofunctionalization reactions demonstrate that Cu and Ni catalysts could enable cyclization/coupling of alkenylzinc reagents, alkyl halides, and aryl halides to afford complex carbo- and heterocycles. In addition, forming coordination-assisted transient nickellacycles enabled regioselective performance of three-component dicarbofunctionalization of various alkenyl compounds. In situ reaction of [M]-H with alkenes generated after β-H elimination induced an unprecedented metallacycle contraction process, in which six-membered metal-containing rings shrank to five-membered cycles, allowing creation of new carbon–carbon bonds at allylic (1,3) positions. Applications of these regioselective alkene dicarbofunctionalization reactions are discussed.
Graphical Abstract
1. INTRODUCTION
Transition metal (TM)-catalyzed creation of two carbon–carbon bonds across an alkene, termed dicarbofunctionalization (Scheme 1), is an effective strategy to generate complex molecules from readily available chemicals.5,6 This method combines an alkene with two carbon electrophiles/nucleophiles and generates products with stereocenters. Its catalytic cycle involves four steps—activation of the first carbon source, reaction with the alkene, activation of the second carbon source, and reductive elimination from a diorgano-TM intermediate (4) (Scheme 1). However, sequential execution of the four steps is particularly challenging as there are two mainstream chemistries that function as side reactions. The first is the direct coupling between two carbon sources prior to reaction with the alkene.7 The second is the Heck reaction after the intermediates (3 and 4) undergo β-H elimination.8 Consequently, alkene dicarbofunctionalization reactions require that the rate of the alkene reaction be faster than coupling between the two carbon sources. Also, the rate of activation of the second carbon source and reductive elimination must be faster than β-H elimination. Moreover, controlling the regiochemistry of products is also difficult because the order of addition of the electrophiles and nucleophiles could be reversed. In some cases, intermediate 3 could also undergo a β-H elimination/[M]–H re-insertion step to generate rearranged dicarbofunctionalized products.
Scheme 1.
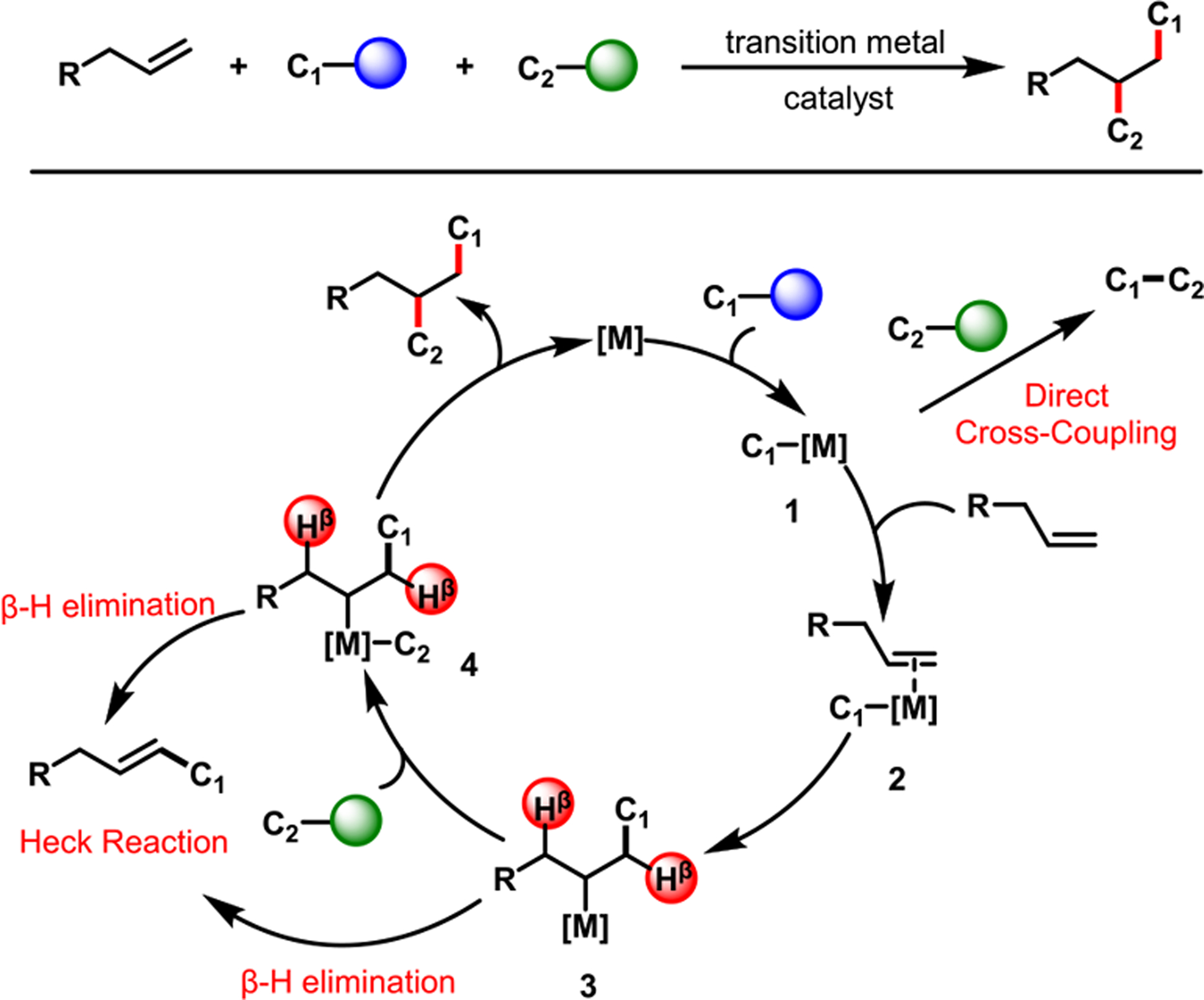
General alkene dicarbofunctionalization reaction and catalytic cycle
Historically, alkene dicarbofunctionalization reactions have been conducted through cross-coupling with organic halides and organometallic reagents using Pd as a catalyst. In these reactions, the predominant tactic has been the use of bicyclic cis-alkenes, since the work of Paolo Chiusoli and Catellani in 1982,9 to promote alkene insertion, and prevent β-H elimination by creating a geometry with β-H trans to Pd(II) (Scheme 2a).10 Conjugated dienes have also served as effective substrates for alkene dicarbofunctionalization with Pd.11–13 Conjugated dienes promote alkene insertion by strong bidentate coordination, generating π-allyl–Pd(II) intermediates to suppress β-H elimination (Scheme 2b). Alternatively, many reactions have been performed with disubstituted terminal alkenes through cyclization (Scheme 2c).14,15 Upon migratory insertion, the disubstituted terminal alkenes generate C(sp3)–Pd(II) intermediates lacking β-H’s. Unfortunately, these substrates were designed to circumvent β-H elimination. Therefore, the scope of the reaction is seriously limited, ultimately restricting its synthetic application. Details of developments and advances of these reactions have recently been reviewed in a number of articles.16–24 In this Account, we present our studies on the development of Ni-, Cu-, and Pd-catalyzed dicarbofunctionalization reactions of alkenes that generate C(sp3)–[M] intermediates containing β-H’s, which eliminate constraints on substrate scope. In these studies, we use cross-coupling for bond formation, in which organic halides and organometallic reagents serve as two carbon sources. Other bond-forming approaches, such as reductive coupling,25,26 radical reaction27–29 and photoredox catalysis,30–34 have also remained successful. We illustrate that the β-H–C(sp3)–[M] intermediates of Ni and Cu are sufficiently stable to enable transmetalation and reductive elimination in cyclization/coupling processes. For more challenging three-component reactions, we implement Ni catalysts in conjunction with a coordination strategy, facilitating the generation of metallacycles to stabilize β-H–C(sp3)–[M] intermediates.
Scheme 2.
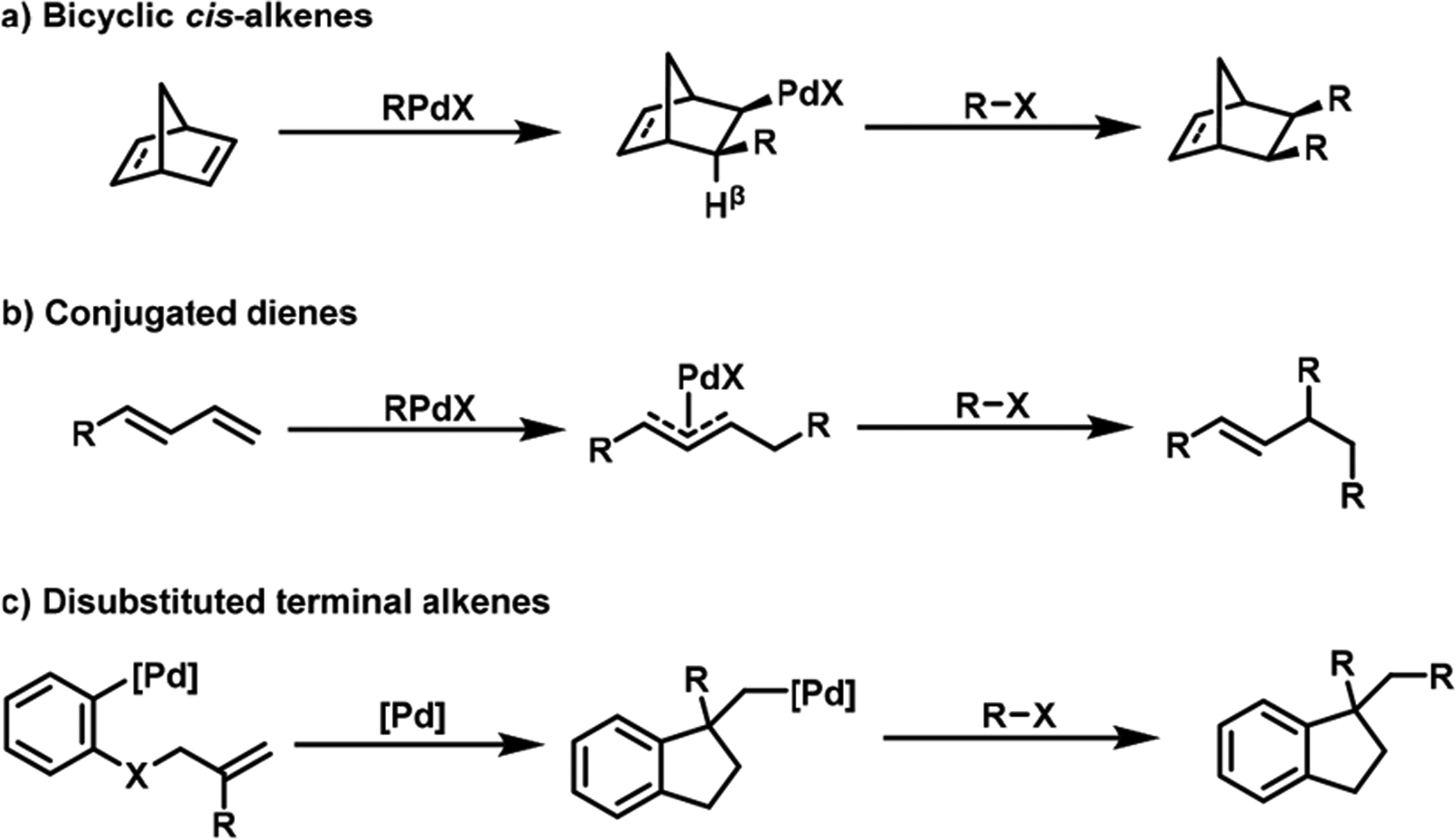
Structural Variations of Alkenes in Early Reactions
2. THREE-COMPONENT ALKENE DICARBOFUNCTIONALIZATION
2.1. Coordination-Controlled Vicinal Dicarbofunctionalization
Recently, we implemented the concept of forming transient β-H–C(sp3)–metallacycles with a removable coordinating group for alkene difunctionalization with organic halides and organometallic reagents (Scheme 3).3 Our approach relied upon Whitesides et al.’s observation35 that β-H–C(sp3)–platinacycles undergo β-H elimination much slower than acyclic β-H–C(sp3)–Pt(II) complexes. In metallacycles, the TM and β-H’s are oriented out of syn-coplanarity due to geometric restriction. We envisioned that alkenyl substrates bearing a heteroatom coordinating group would function as a bidentate ligand. Bidentate coordination would be critical for promoting pseudo-intramolecular migratory insertion of TM-bound alkenes, and for stabilizing postalkene insertion β-H–C(sp3)–[M] intermediates as metallacycles. Previously, Neufeldt and Sanford36 and Toste et al.37 utilized heteroatom coordination for dioxygenation of O-allyloximes and fluoroarylation of 2-vinylarenes with Pd catalysts. In 2009, Larhed et al.38 implemented -NMe2 for a Pd-catalyzed homodiarylation of an alkene with arylboronic acids (Scheme 3a).
Scheme 3.
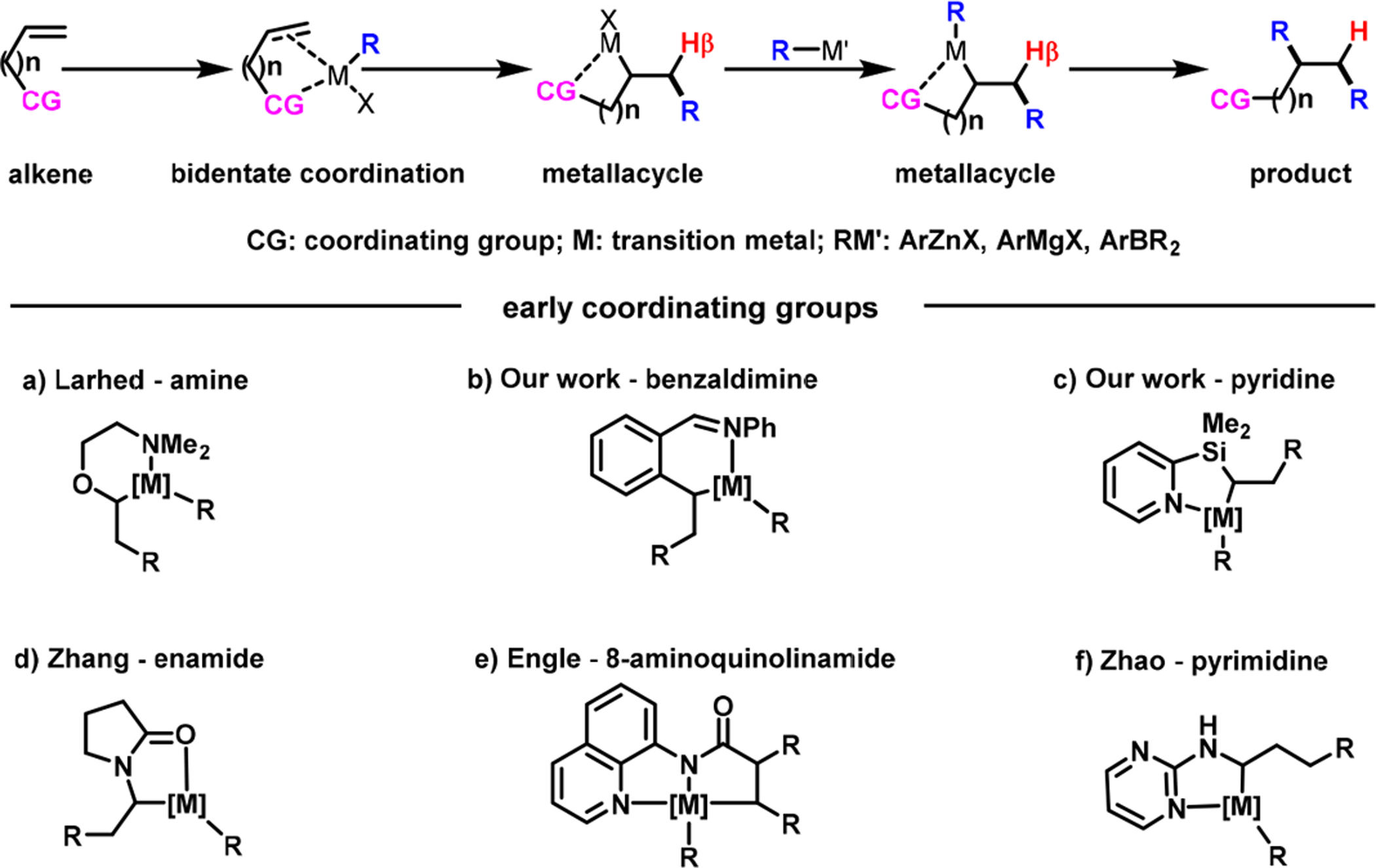
Concept of Forming a Transient Metallacycle for Alkene Dicarbofunctionalization
In 2017, we disclosed a Ni(cod)2-catalyzed diarylation of 2-alkenylbenzaldimines with aryl halides and arylzinc reagents (Scheme 4).3 During our work, Zhang et al.,39,40 Engle et al.41 and recently Koh et al.,42 and Zhao et al.43 also disclosed similar coordination strategies with enamide, bidentate 8-amino-quinolinamide, and pyrimidine, respectively, for alkene dicarbofunctionalization (Scheme 3d–f). Our benzaldimine substrate was designed to orient both the γ,δ-alkene and the imine groups syn to each other. The lateral arene facilitated the formation and stabilization of six-membered nickellacycles (5 and 8) by planarization and rigidification (Scheme 5). This thermodynamic stabilization of transient metallacycles enabled β-H elimination to proceed slower than transmetalation and reductive elimination, as required for alkene difunctionalization.
Scheme 4.
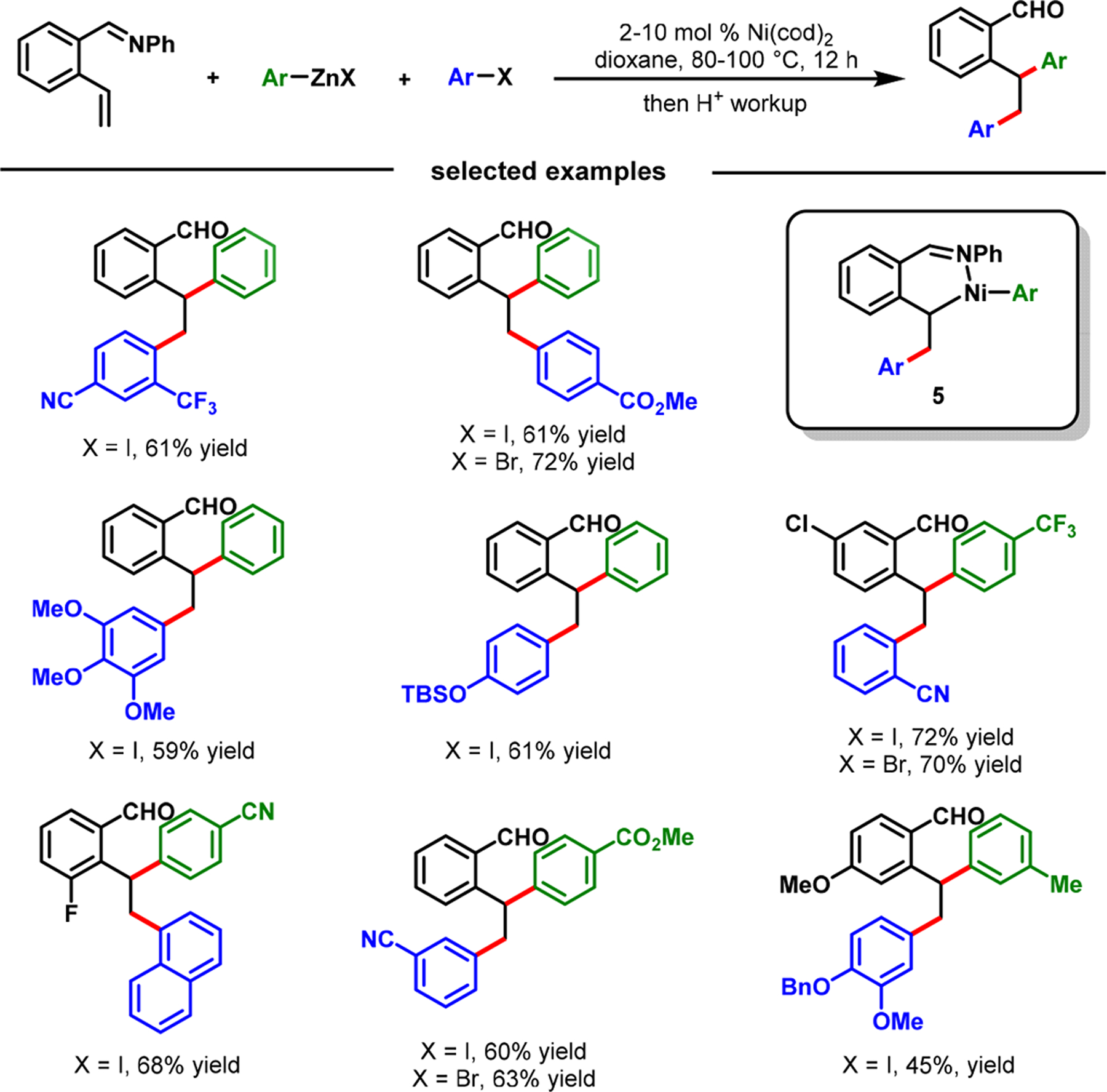
Imine-Assisted Nickel-Catalyzed Diarylation of 2-Alkenylbenzaldehydes
Scheme 5.
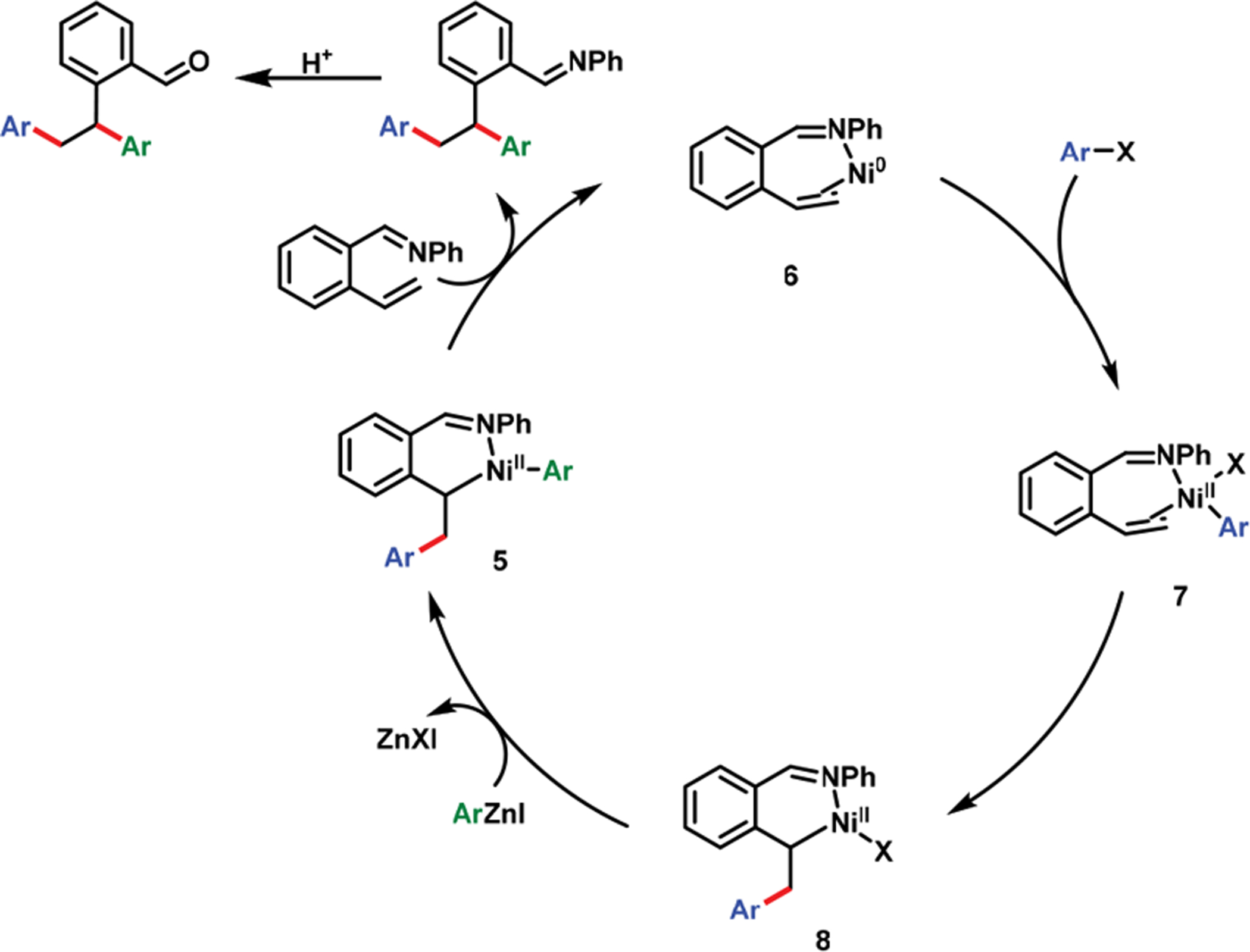
Catalytic Cycle via Nickellacycle Formation
Our imine-assisted reaction demonstrated an excellent scope with aryl iodides, aryl bromides, aryl triflates, arylzinc reagents, and 2-alkenylbenzaldimines. The reaction condition was also amenable to the diarylation of 2-alkenylaniline-derived imines (Scheme 6). This latter reaction proceeded with five-membered nickellacycles (9). Unlike 8-aminoquinolinamide, enamide, and pyrimidine, our imine coordinating group was readily removed instantaneously during a simple acid workup.
Scheme 6.
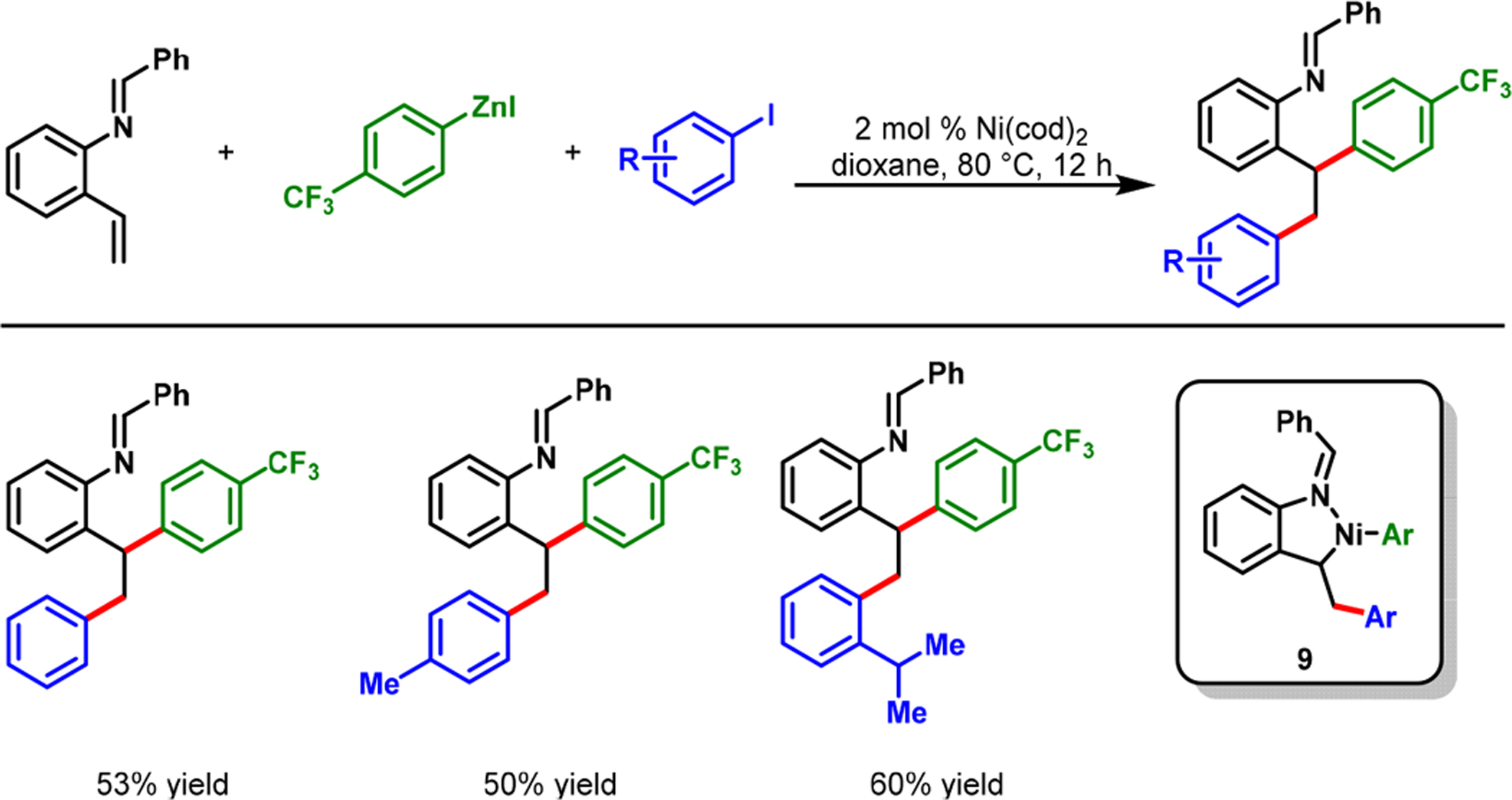
Imine-Assisted Nickel-Catalyzed Diarylation of 2-Alkenylanilines
Catalytic alkene difunctionalization with two C(sp3) sources is more challenging than with C(sp) and C(sp2) sources, as they generate additional β-H–C(sp3)–[M] intermediates (10) during the reaction (Scheme 7). Since the extra β-H–C(sp3)–[M] intermediate makes the reaction more susceptible to β-H elimination, regioselective intermolecular addition of two C(sp3) groups to an alkene remains a serious problem. As such, these reactions are exceptionally rare and inefficient, requiring an excess of reactive C(sp3) sources, high catalyst loadings,44 and vinylboron reagents as an alkene source.45 In 2020, we reported that 2-alkenylbenzaldimines were excellent substrates for regioselective dialkylation with benzyl bromides and alkylzinc reagents using Ni(cod)2 (Scheme 8).46 Concomitantly, Koh et al.47 disclosed a Ni-catalyzed dialkylation of alkenylquinolinamides with redox active esters. Our dialkylation reaction was highly efficient and could be conducted with 500 ppm catalyst, registering up to 2 × 103 catalytic turnover number (TON) and 165 h−1 turnover frequency (TOF) at room temperature. The reaction was also scalable, and proceeded with electronically varied benzyl bromides, alkylzinc reagents, and 2-alkenylbenzaldimines.
Scheme 7.
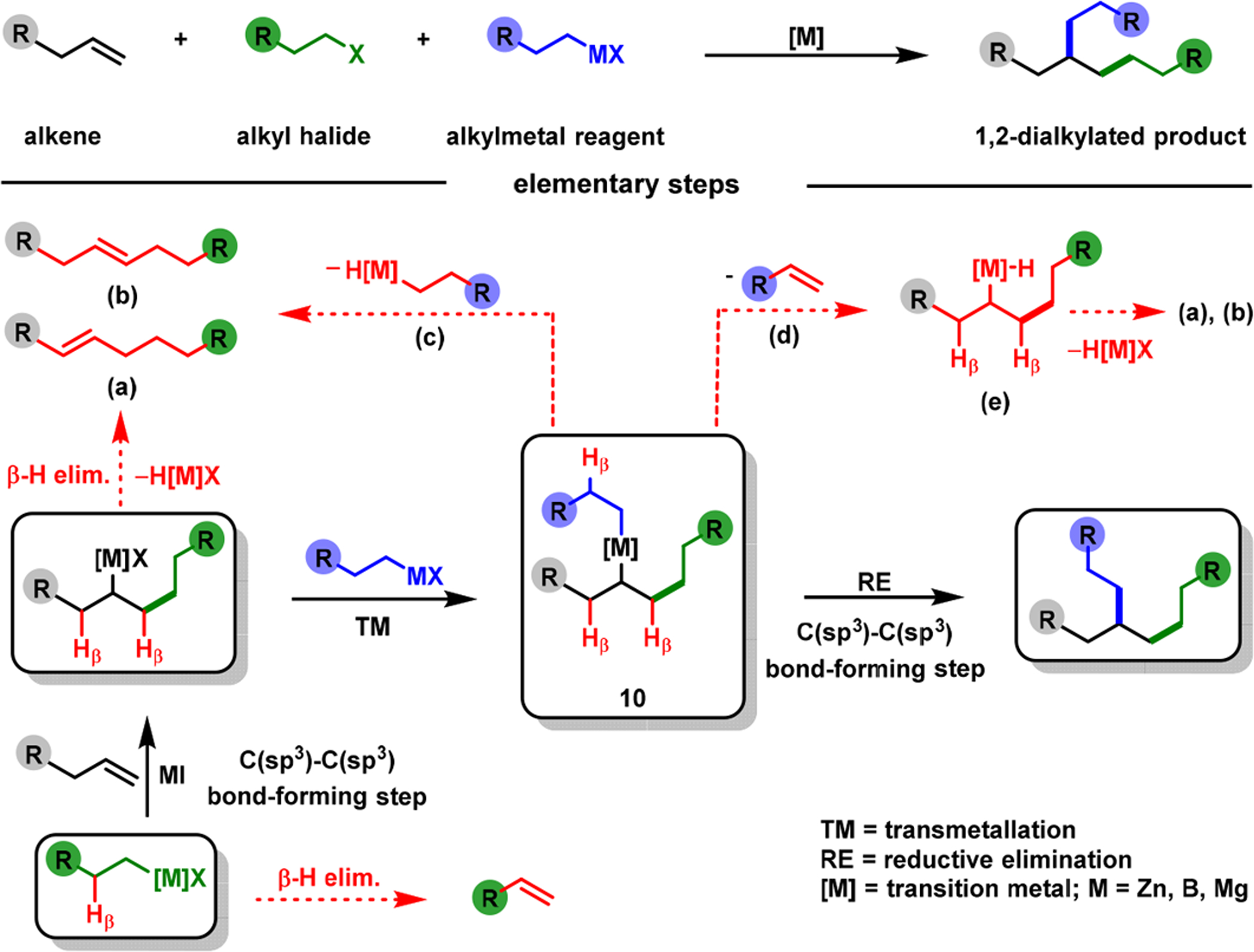
Problems in Metal-Catalyzed Alkene Dialkylation Reactions
Scheme 8.
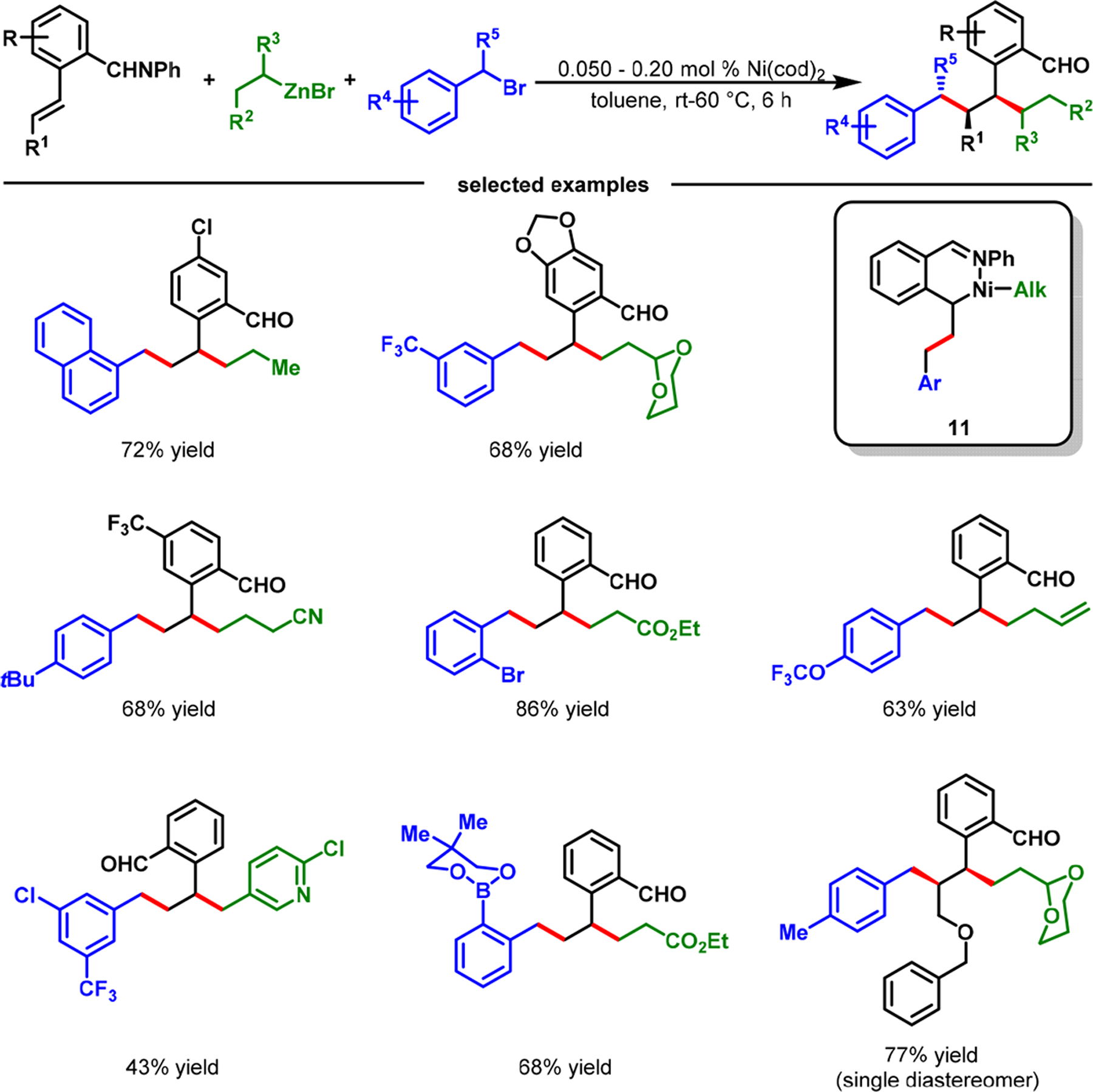
Imine-Assisted Nickel-Catalyzed Dialkylation of 2-Alkenylbenzaldehydes
The alkene dialkylation reaction was also effective for the coupling of secondary benzyl bromides and secondary alkylzinc reagents with internal alkenes, and generating products with three contiguous all-carbon secondary stereocenters (Scheme 9). Difunctionalization of internal alkenes with two discrete secondary C(sp3) reagents is a desired reaction that generates branched carbon frameworks with multiple contiguous stereocenters. However, such a process is fundamentally challenging, since internal alkenes are less polarized and more sterically crowded than terminal alkenes. In addition, these reactions require the formation of C(sp3)–C(sp3) bonds between secondary C(sp3) reagents and secondary C(sp3)–[M] intermediates, which further raises the activation barrier of the transition state due to increased sterics.
Scheme 9.
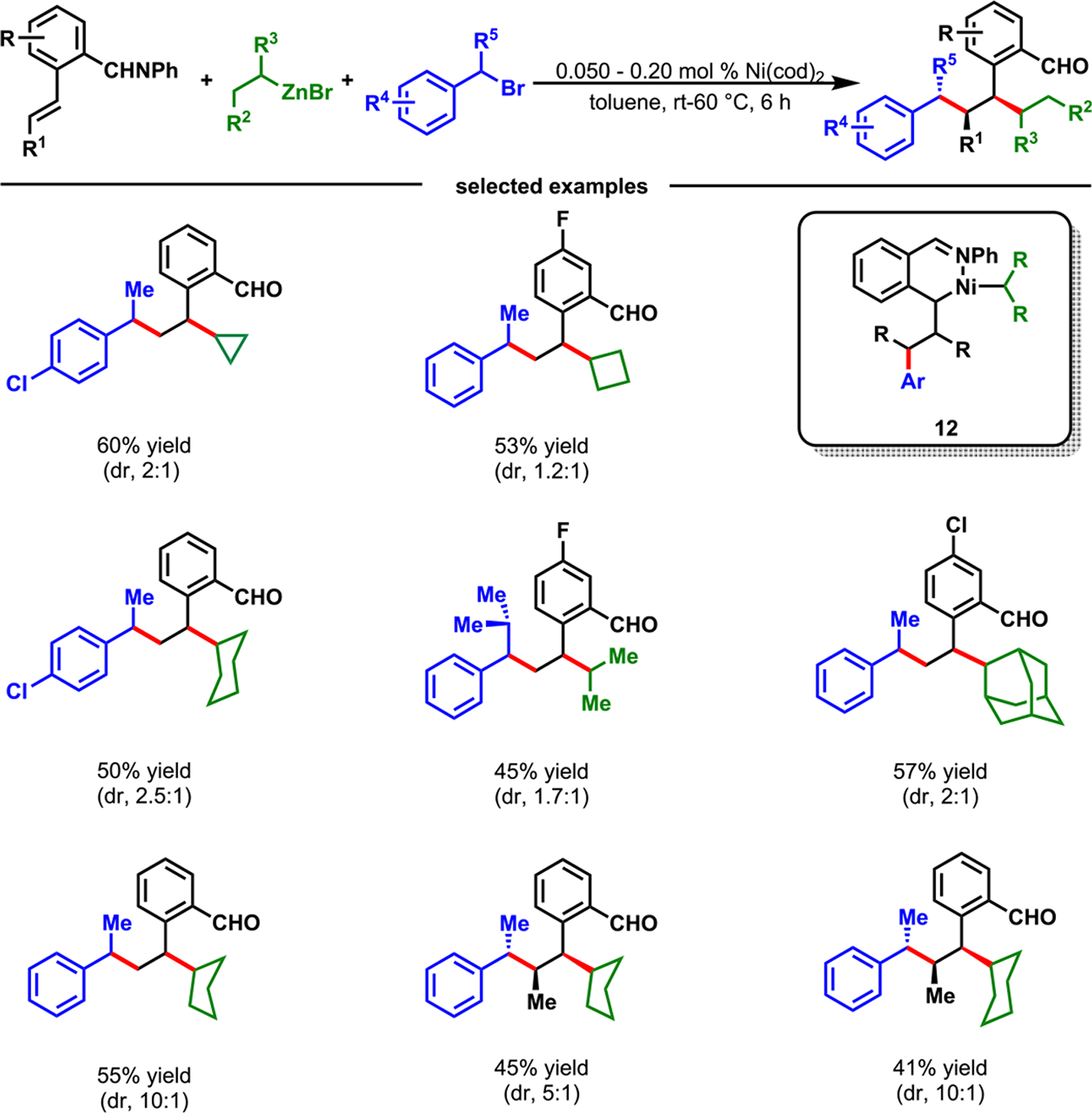
Scope with Internal Alkenes, Secondary Halides, and Alkylzinc Reagents
In our dialkylation reaction, the o-aldehyde remained vital, not only for the generation of an aldimine for coordination, but also as a synthetic handle for further functionalization of the dialkylated products. The dialkylated benzaldimine products could be reduced in situ with NaBH4 to access advanced secondary arylbenzylamines (Scheme 10). In addition, the reaction could be performed with alkylzinc reagents bearing ester, nitrile, and protected aldehyde groups, wherein the carbonyl α-carbons could be condensed with the o-aldehyde to produce tetralenes and benzosuberenes (Scheme 11). Likewise, the use of 2-bromobenzyl bromide and 2-(dioxanyl)ethylzinc bromide enabled us to create an arene-studded bicyclo[4.2.2]-decene (14) by the Heck reaction subsequent to dialkylation and cyclization (Scheme 12).
Scheme 10.
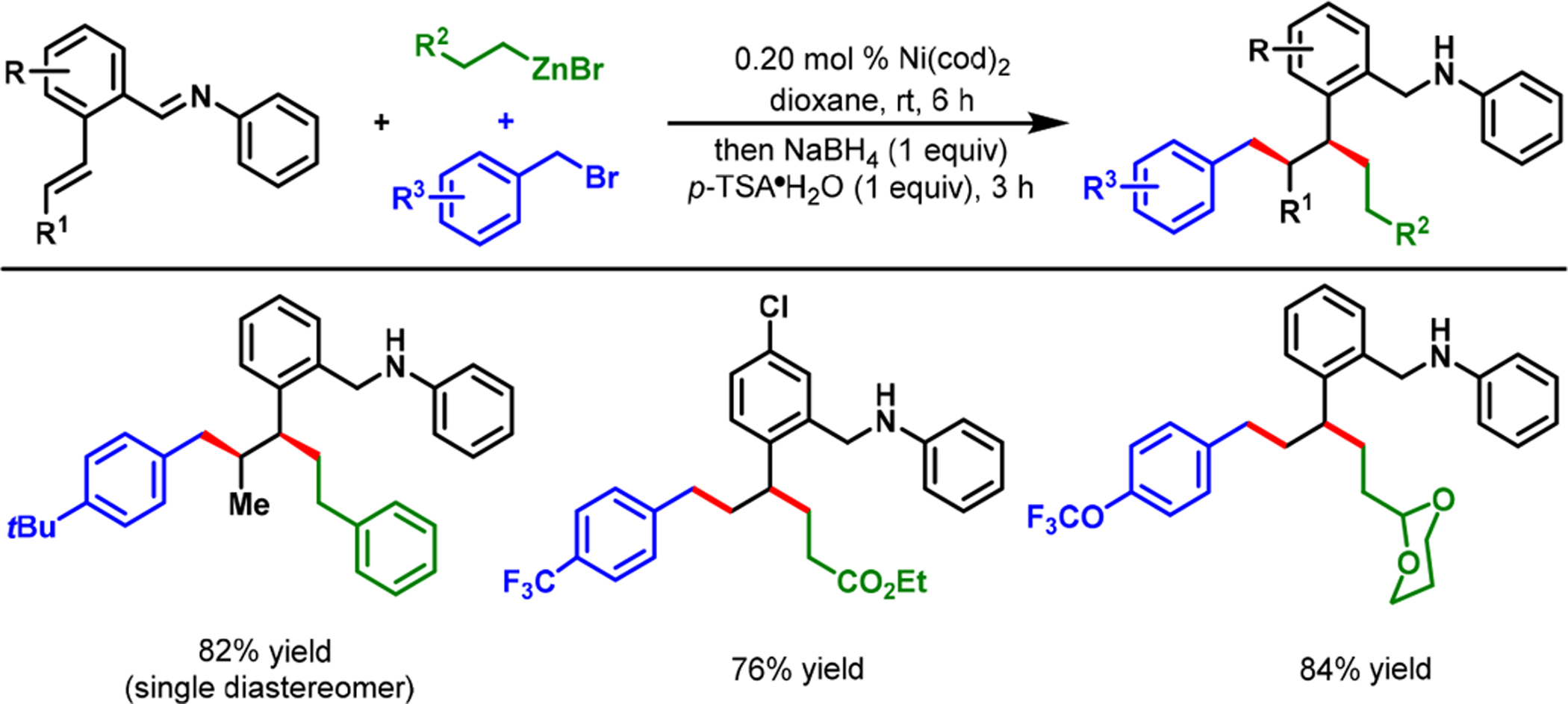
Rapid Access to Complex Secondary Arylbenzylamines
Scheme 11.
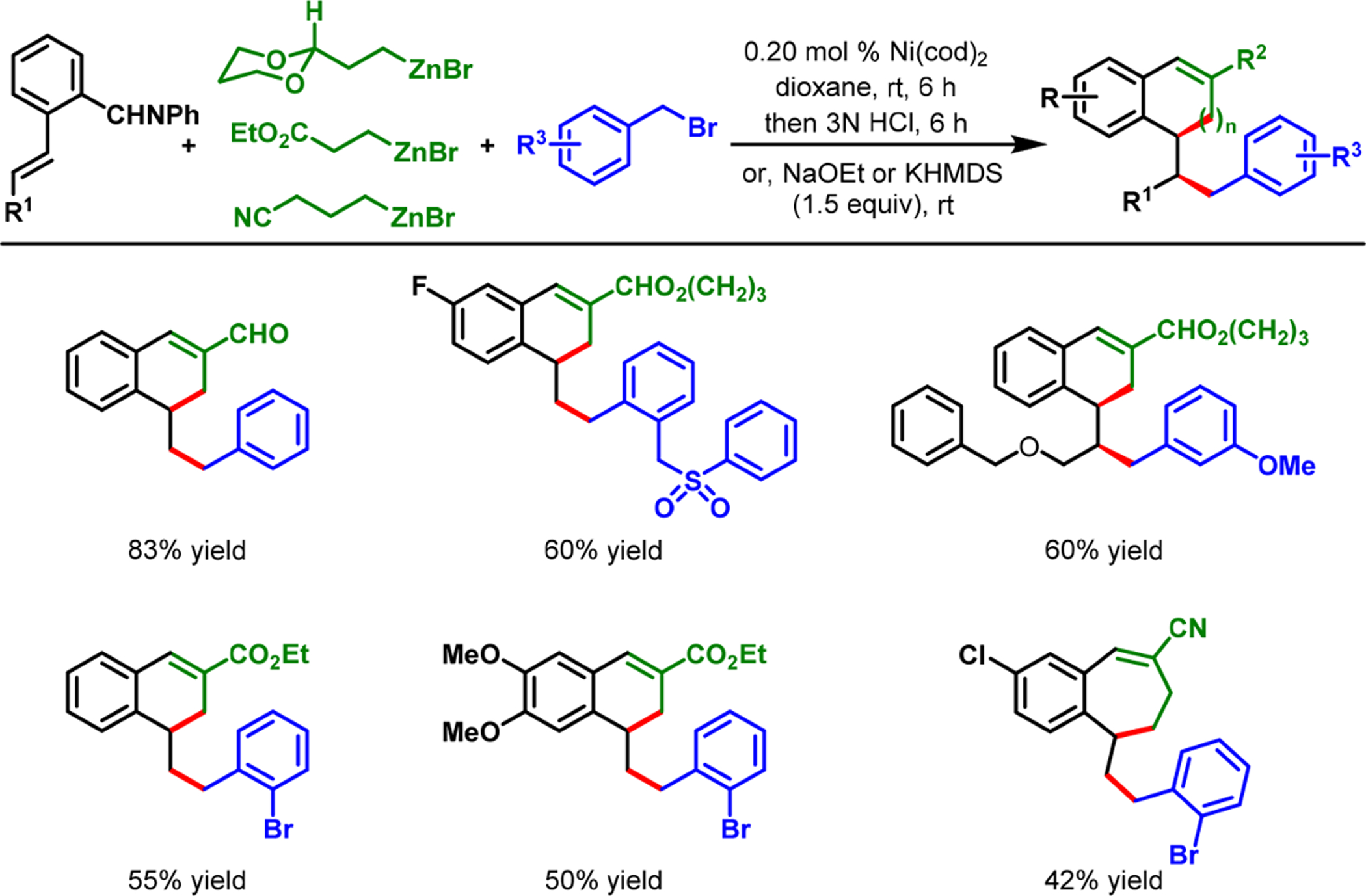
Rapid Access to Complex Tetralene and Benzosuberene Derivatives
Scheme 12.
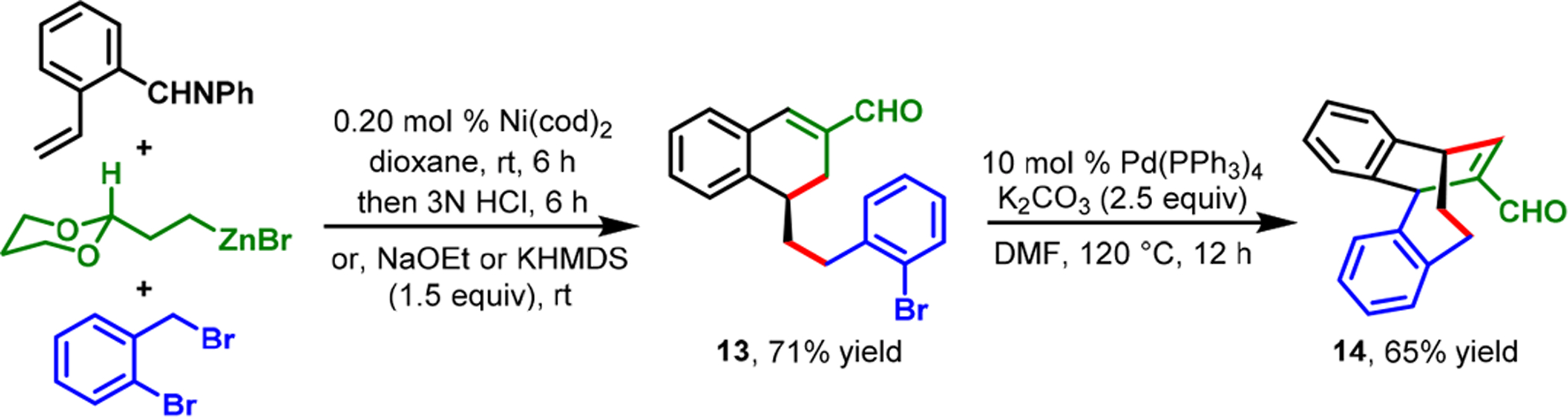
Rapid Access to Arene-Studded Bicyclo[4.2.2]decene Structures
Our Ni-catalyzed alkene difunctionalization method was also amenable to other coordinating groups, in addition to aldimines. In 2018, we disclosed that pyridine could also function as a coordinating group for regioselective diarylation of alkenes in alkenylpyridylsilanes with aryl halides and arylzinc reagents (Scheme 13).48 The reaction required NiBr2 as a catalyst. However, on the basis of regiochemical outcomes, the reaction is catalyzed by Ni(0) formed upon in situ reduction of NiBr2 and proceeds via a pyridine-stabilized five-membered nickellacycle 15. The reaction afforded 1,2-diarylethylsilanes, which could be oxidized to 1,2-diarylethanols.
Scheme 13.
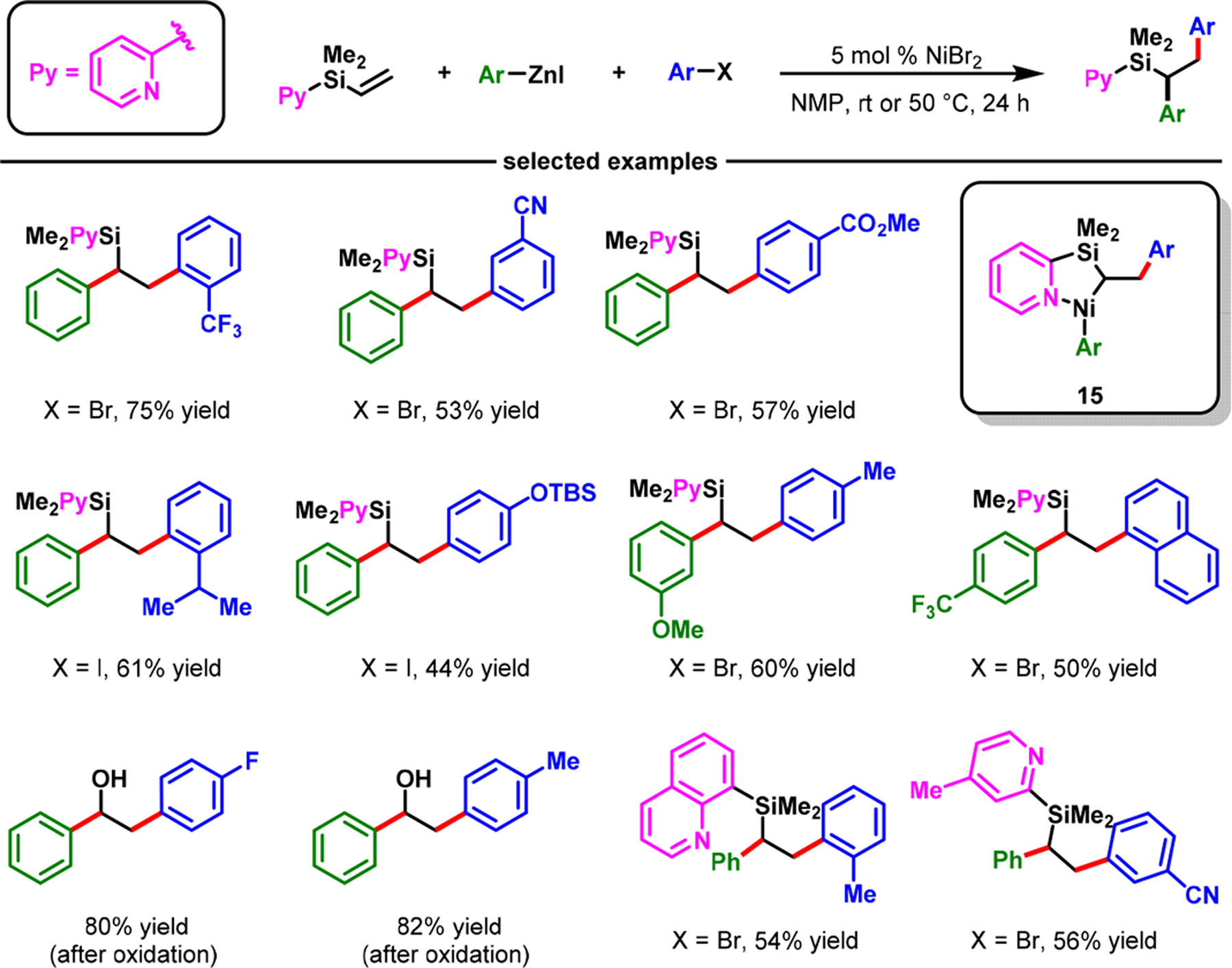
Pyridine-Assisted Nickel-Catalyzed Diarylation of Vinylpyridylsilanes
Our studies established reaction parameters for the dicarbofunctionalization of 2-alkenylbenzaldimines involving six-membered transient nickellacycles. However, the translation of the design principle with 2-alkenylaldimines to linear γ,δ-alkenylimines was infeasible on account of C(sp3) hybridization on the substrate backbone. The six-membered metallacycles (16 and 17) would remain in chair conformation, as a fluxional structure, prompting both the imine and alkene to disengage from TMs, imparting low stabilization to the metallacycles (Scheme 14). In addition, fluxionality would permit β-H’s to attain syn-coplanarity with TMs, causing β-H elimination. Therefore, we hypothesized that difunctionalization of linear γ,δ-alkenylimines would require tinkering with relative kinetic energetics of the migratory insertion, β-H elimination, transmetalation, and reductive elimination steps. Literature reports have indicated that Ag salts can abstract halides from ArPdX to generate cationic [ArPd(II)]+ species and promote both alkene migratory insertion49 and transmetalation.50 The fast kinetics of migratory insertion and transmetalation are critical to overcome the competing cross-coupling and β-H elimination reactions.
Scheme 14.
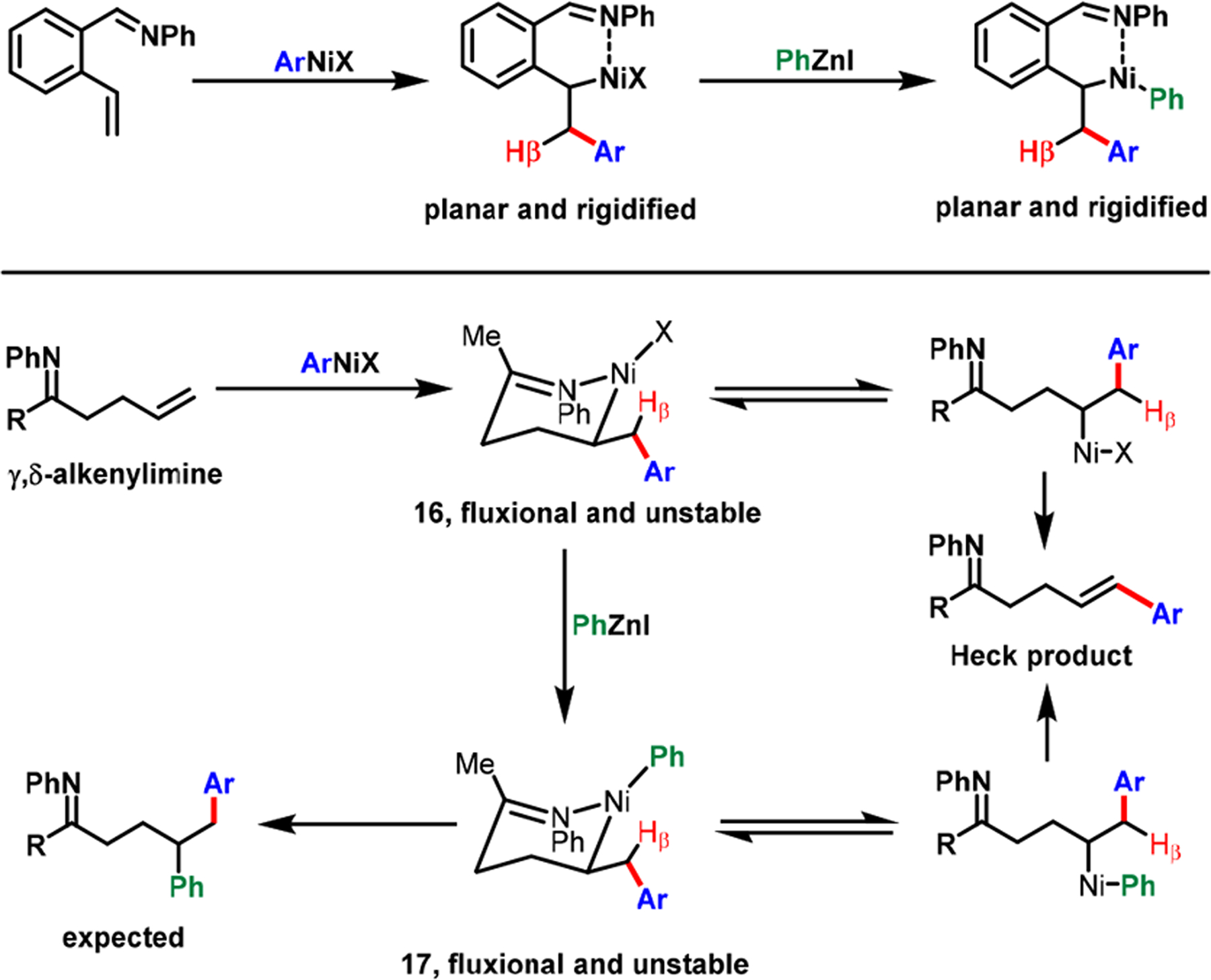
Stability of Planar and Fluxional Six-Membered Metallacycles
In 2018, we disclosed that catalytic amounts of AgBF4 or CuX (X = I, BF4) promoted Ni(cod)2-catalyzed regioselective γ,δ-diarylation of γ,δ-alkenylketimines with aryl halides and arylzinc reagents (Scheme 15).51 The reaction worked well with a variety of γ,δ-alkenylketimines, aryl halides, and arylzinc reagents and tolerated a range of functional groups. α-Substituted products were formed as a single diastereomer (racemic), underscoring the potential use of the reaction for stereoselective processes. Such a high degree of stereocontrol could be explained by the formation of a tight pseudochair conformation with the cationic Ni(II) during migratory insertion.
Scheme 15.
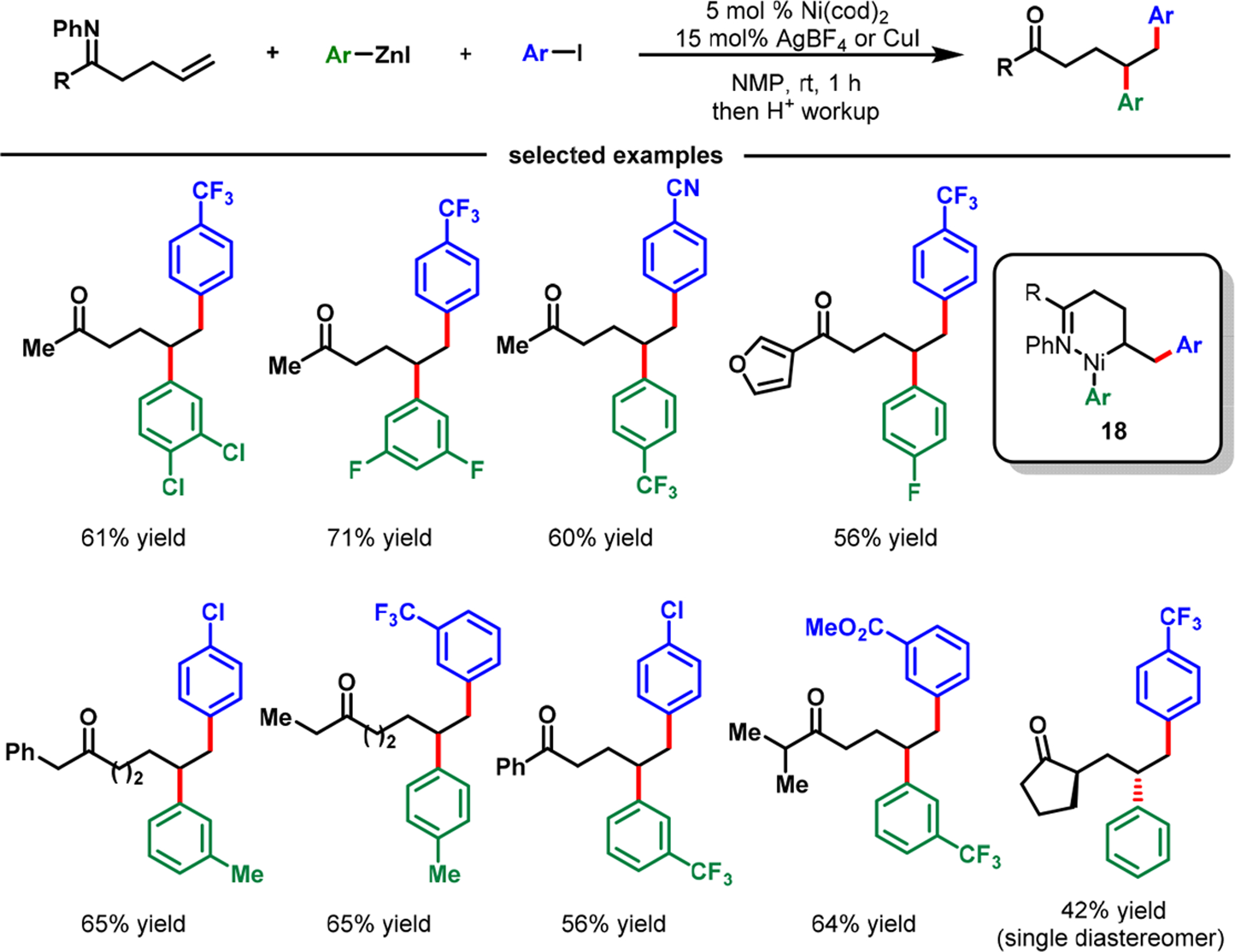
Imine-Assisted Nickel-Catalyzed γ,δ-Diarylation of γ,δ-Alkenylketones
Real-time monitoring of the reaction progress by 19F NMR indicated that both AgBF4 and CuX dramatically increased the rate of alkene diarylation (Figure 1a). In the absence of AgBF4 or CuX, the reactions generated diarylated products only in 15–30% yields, before terminating in 3 h. Stoichiometric reactions between 4-FC6H4ZnI and CuX, and between 4-FC6H4ZnI and AgBF4 showed that [Ar–Ag] and [Ar–Cu] complexes did not form, suggesting that AgBF4 and CuX did not act as cotransmetalating reagents. Cross-coupling and Heck products were also suppressed in the presence of AgBF4 and CuX (Figure 1b), indicating that the cationic Ni(II) catalyst promoted migratory insertion faster than cross-coupling and transmetalation faster than β-H elimination.
Figure 1.
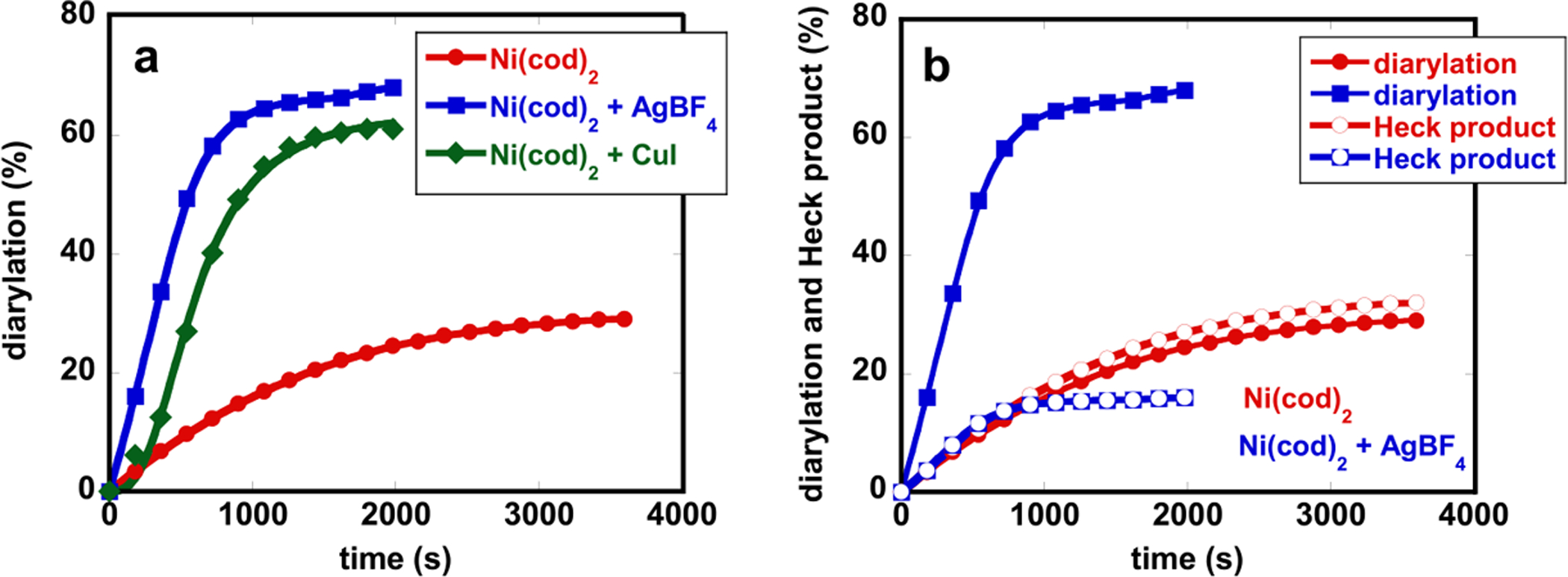
(a) Effects of AgBF4 and CuI cocatalyst on alkene diarylation. (b) Effect of AgBF4 on diarylation and the Heck product.
2.2. Coordination-Controlled Homovicinal Dicarbofunctionalization
In alkene difunctionalization, β-H elimination is generally considered a nuisance, and, as such, many strategies such as those outlined in Section 2.1 are employed to overcome this problem. However, recent reports have shown that the process of β-H elimination can also be exploited, to our benefit, to create new bonds at sites that are not generally considered for bond formation.20 Harnessing β-H elimination for alkene difunctionalization strictly depends on the ability of TMs to undergo reversible β-H elimination/alkene hydrometalation.52 During hydrometalation, TMs could reside on geminal, allylic, or remote carbons relative to the original position of alkenes, and generate products with two new bonds at 1,1-, 1,3-, or 1,n-positions (Scheme 16).20 Among these possibilities, dicarbofunctionalization has been mostly observed for 1,1-13,53–55 along with limited examples for 1,3-43,56,57 and 1,n-positions.57–59
Scheme 16.
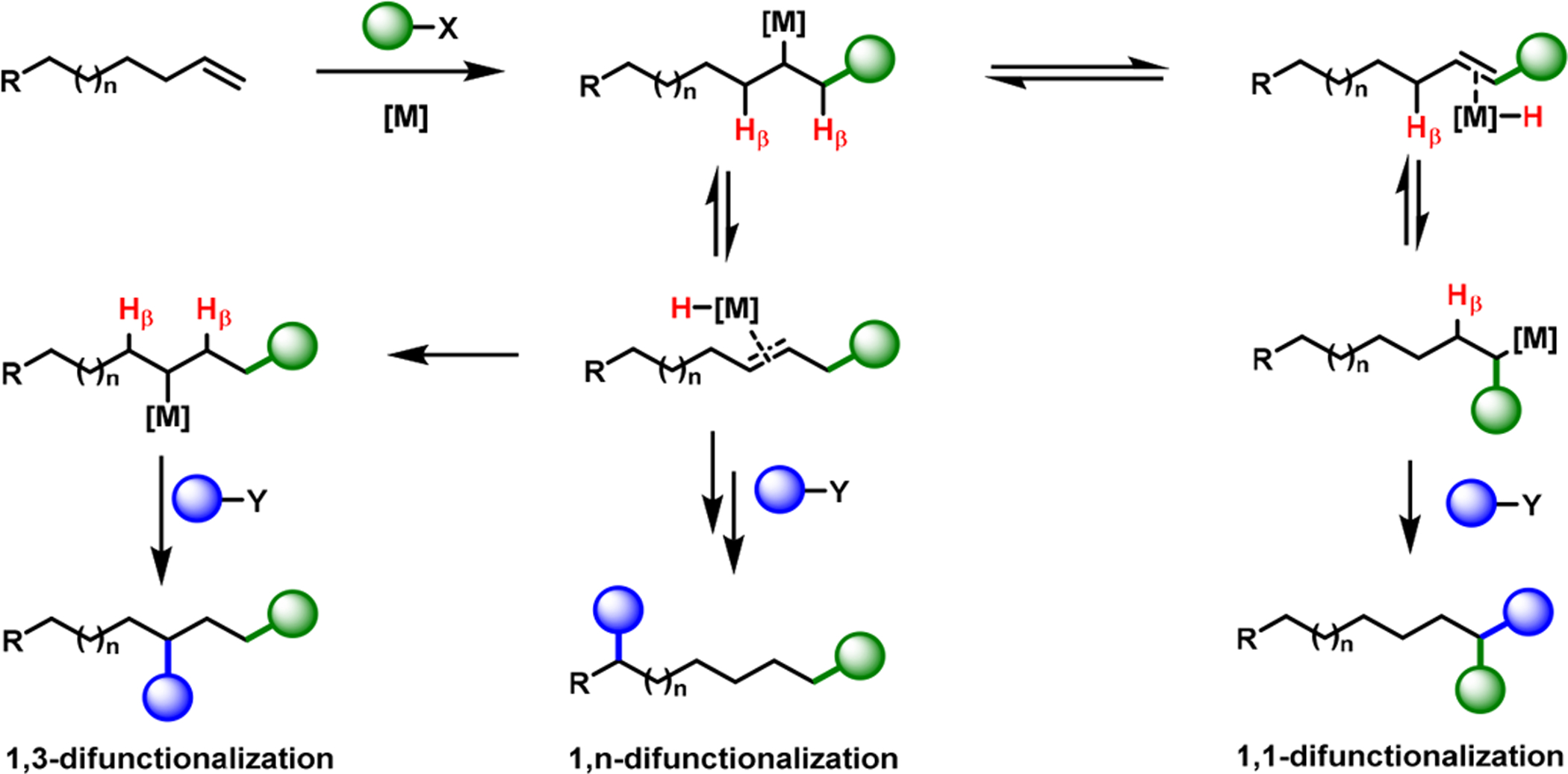
1,1-, 1,3-, or 1,n-Difunctionalizations via β-H Elimination/Hydrometallation
In our efforts to expand the imine-assisted Ni-catalyzed alkene dicarbofunctionalization to γ,δ-alkenylketimines, we observed that a Heck product was consistently generated.56 We reasoned that the fluxional six-membered nickellacycle 16 or 17 (Scheme 14) was unable to retain its integrity due to facile β-H elimination. Therefore, the resultant [Ni]–H species was incapable of performing hydronickellation on the alkene intermediate. Upon examining a series of phosphine ligands, we discovered, in 2018, that (PhO)3P promoted hydronickellation of the resultant alkene intermediate, leading to the formation of γ,δ-diarylation products (Scheme 17).56 (PhO)3P stabilized a thermodynamically stable five-membered nickellacycle 24, generated during the reaction after ring contraction of the initial six-membered nickellacycle 22, via a sequential β-H elimination and alkene hydronickellation process (Scheme 18).
Scheme 17.
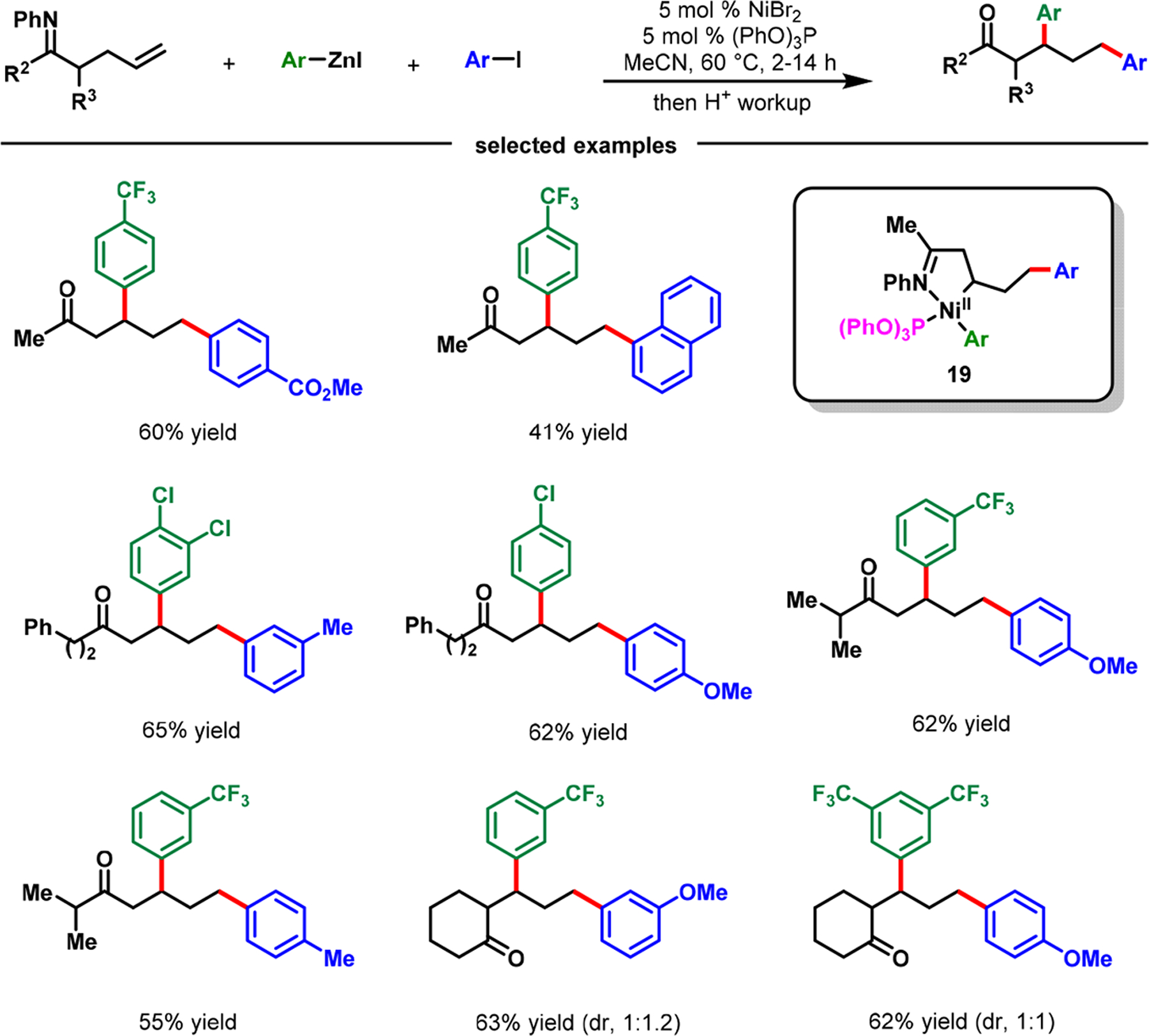
Imine-Assisted Nickel-Catalyzed β,δ-Diarylation of γ,δ-Alkenylketones via the Contraction of Transient Nickellacycles
Scheme 18.
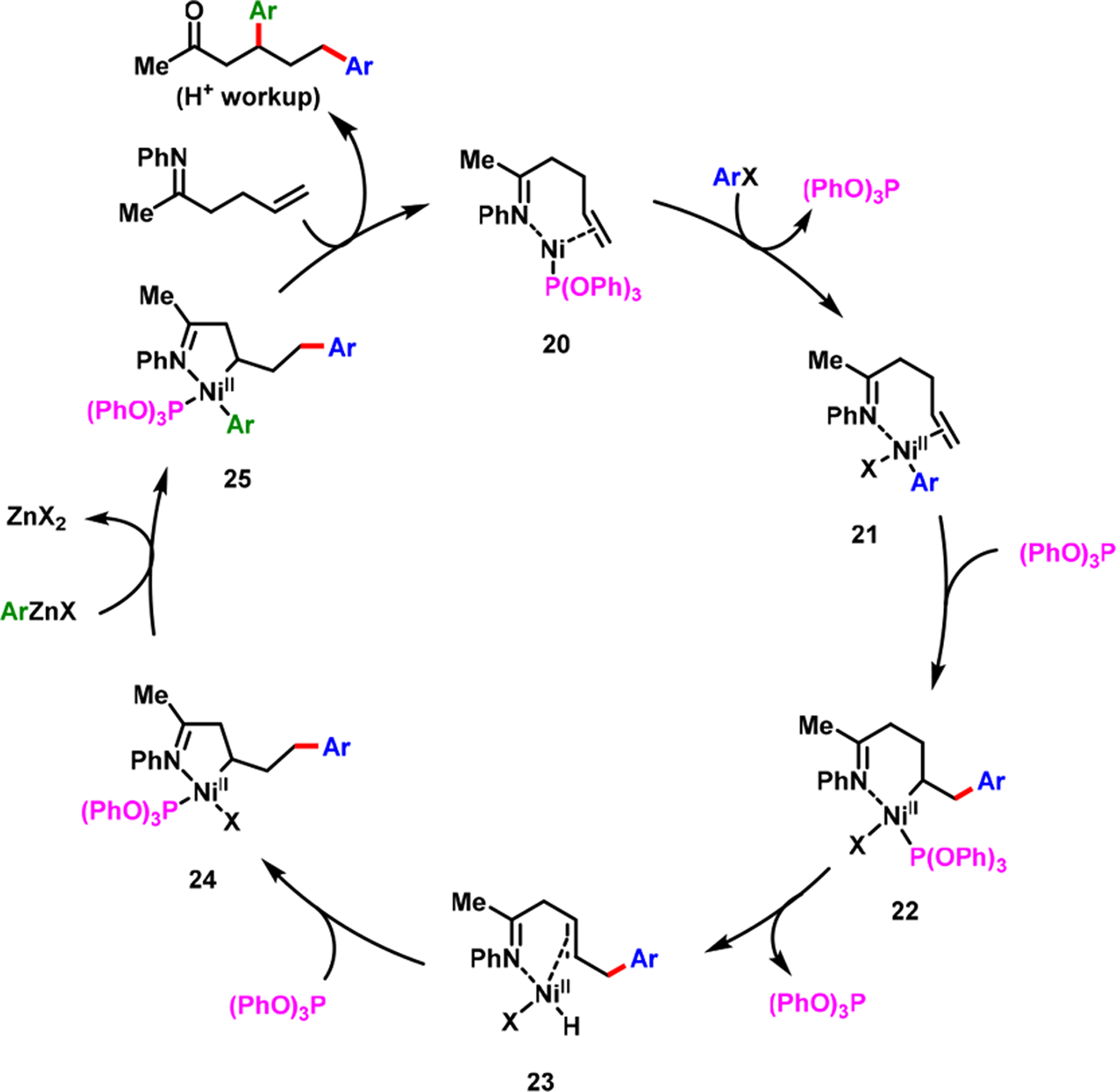
Catalytic Cycle for Alkene Diarylation by the Nickellacyle Contraction Process
Deuterium labeling at the allylic position of the substrate and a crossover experiment with a putative Heck product indicated that contraction of the six-membered nickellacycle to a five-membered ring proceeded via β-H elimination and alkene hydronickellation and that the [Ni]–H species remained bound to the alkene intermediate during ring contraction (Scheme 19). Metallacycle contraction has been previously proposed for CO deinsertion60 and propylene polymerization.61 Therefore, this unique process has the potential to be further developed to create new bonds at nonclassical alkene sites. Concurrent to our work, Zhao et al.43 also observed a similar phenomenon during difunctionalization of N-allyl-2-aminopyrimidine with aryl iodides and arylboronic acids. Our reaction worked with a wide range of γ,δ-alkenylketimines in the presence of electron-deficient triarylphosphite and tris(pyrrolyl)phosphine ligands, and tolerated various functional groups.
Scheme 19.
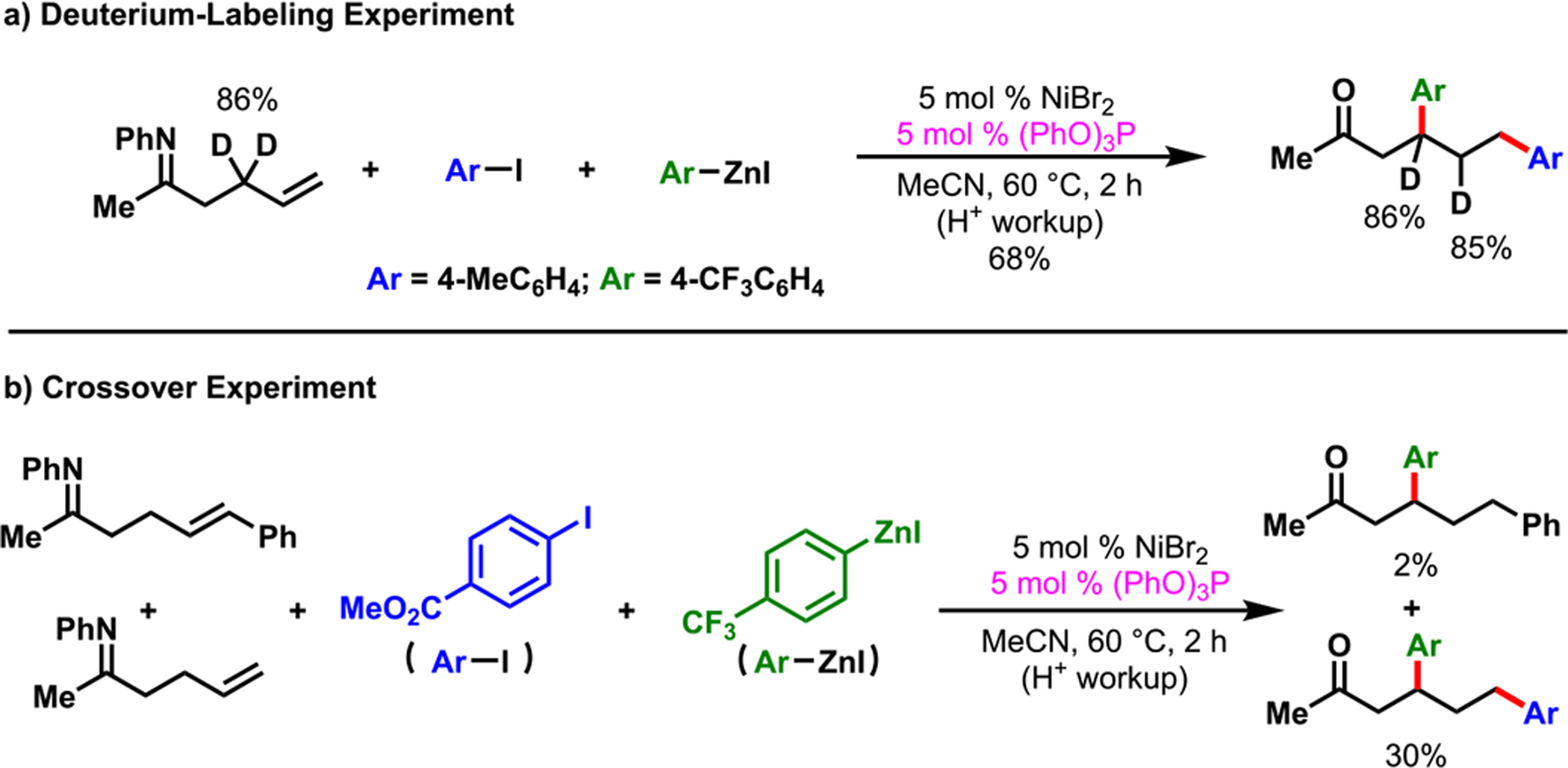
Deuterium Labeling and Crossover Experiments
The discovery above indicated that strong coordinating groups, such as imine and pyrimidine, could be crucial to trigger ring contraction and stabilize five-membered nickellacycles. However, we hypothesized that metallacycle contraction could be more readily executed by weak coordination, in contrast to strong binding; since the fluxional six-membered metallacycle is more susceptible to undergo β-H elimination due to fast ligand dissociation. This possibility would open a completely new scope for metallacycle contraction because any simple functional group could participate in the process. On the basis of this idea, we disclosed a NiCl2-catalyzed β,δ-alkenylarylation of γ,δ-alkenyl-α-cyanocarboxylic esters with alkenyl triflates and arylzinc reagents (Scheme 20).56 In this reaction, a simple cyanoester group promoted nickellacycle contraction and stabilized five-membered nickellacycles. Our reaction proceeded with terminal and internal alkenes and furnished complex aliphatic α-cyanoesters. The α-cyanoester products could be selectively hydrolyzed to access α-cyanocarboxylic acids, dicarboxylic acids, dicarboxylic acid monoamides, monocarboxylic acids, nitriles, and spirolactones, further attesting to the reaction’s wide synthetic applicability (Scheme 21).
Scheme 20.
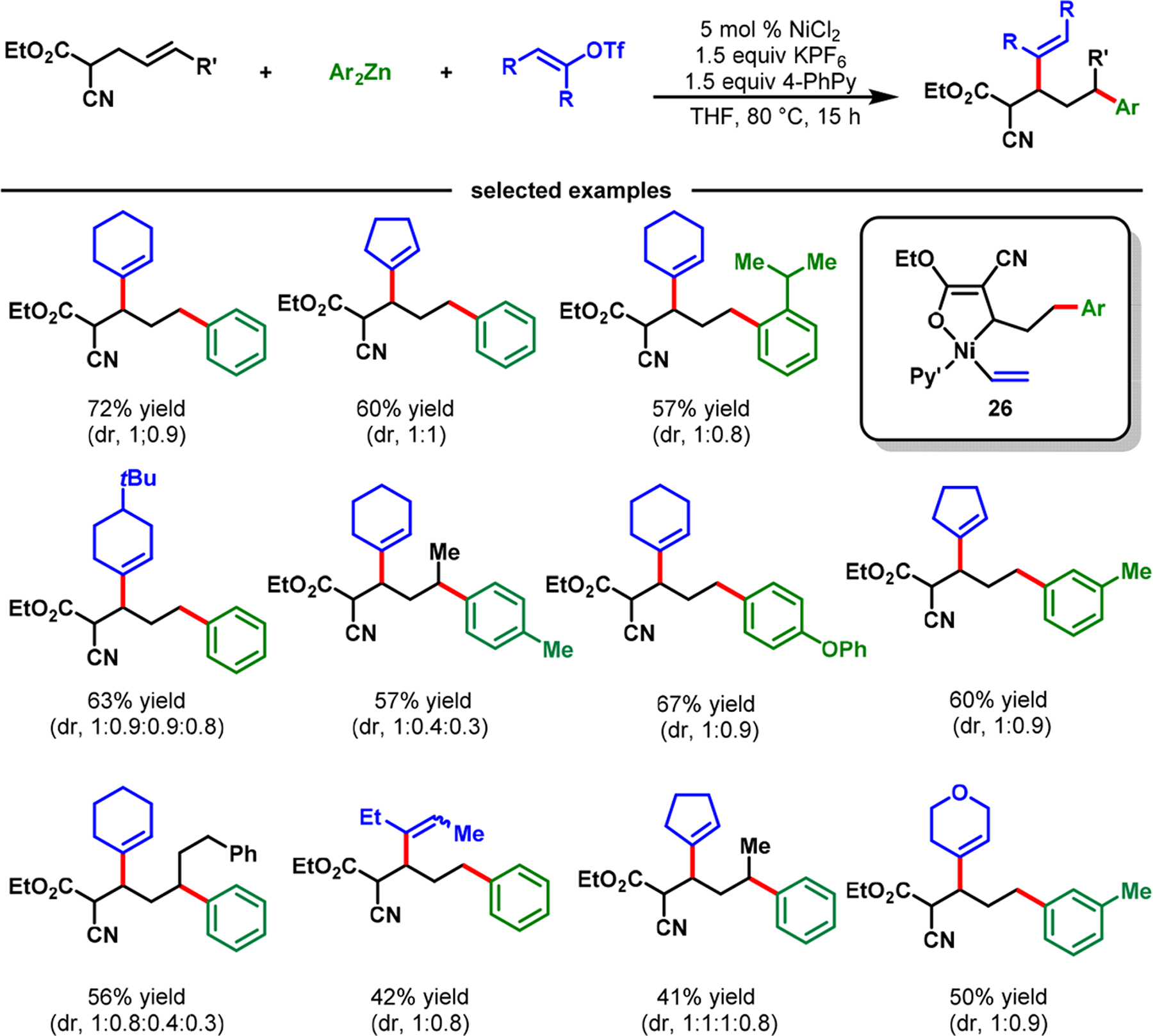
Carbonyl-Assisted Nickel-Catalyzed β,δ-Alkenylarylation of γ,δ-Alkenyl-α-cyanoesters via the Contraction of Transient Nickellacycles
Scheme 21.
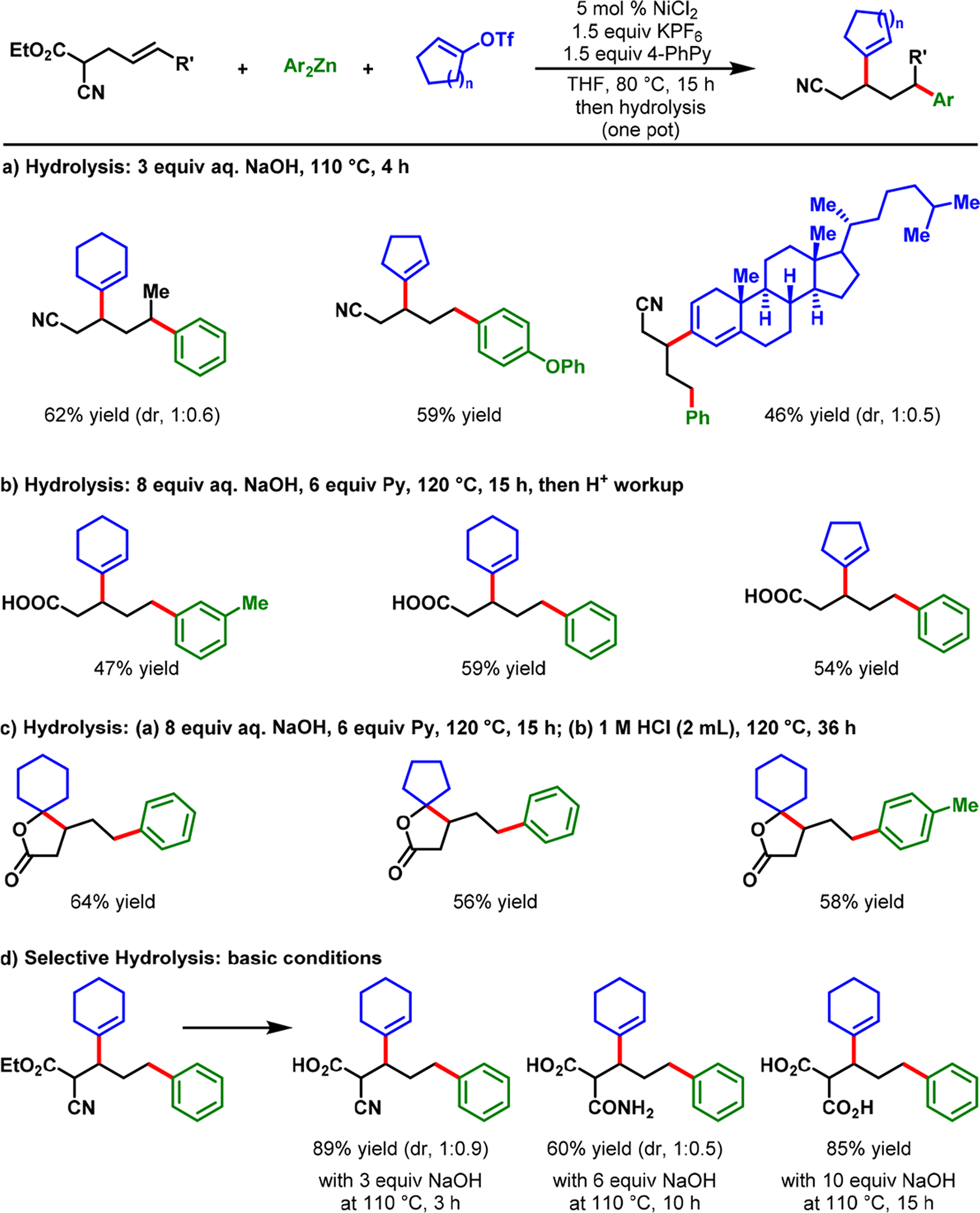
Rapid Access to Synthetically Appealing Precursors
The reaction was unique because the products were formed with regioreversal for the addition of alkenyl triflates and arylzinc reagents. Therefore, this reaction was transmetalation-initiated, contrary to oxidative addition-initiated, which is the most common reaction initiation pathway. On the basis of the product regiochemistry, and additional control, deuterium-labeling, and crossover experiments, we proposed that the reaction proceeds via a Ni(I)-catalyzed metallacycle contraction process (Scheme 22). This catalytic cycle ensues with a combination of cationic, neutral, and anionic speciation and with a high fidelity of the alkene intermediate binding to the [Ni]–H species during ring contraction. The requirement for a weak organic base (4-PhPy) and a cationizing agent (KPF6) supports a possible reversible step to generate a cationic Ni(I) species 28 to initiate transmetalation with an arylzinc reagent, and further stabilize the β-H–C(sp3)–[Ni] intermediate 32 as an enolate-bound metallacycle.
Scheme 22.
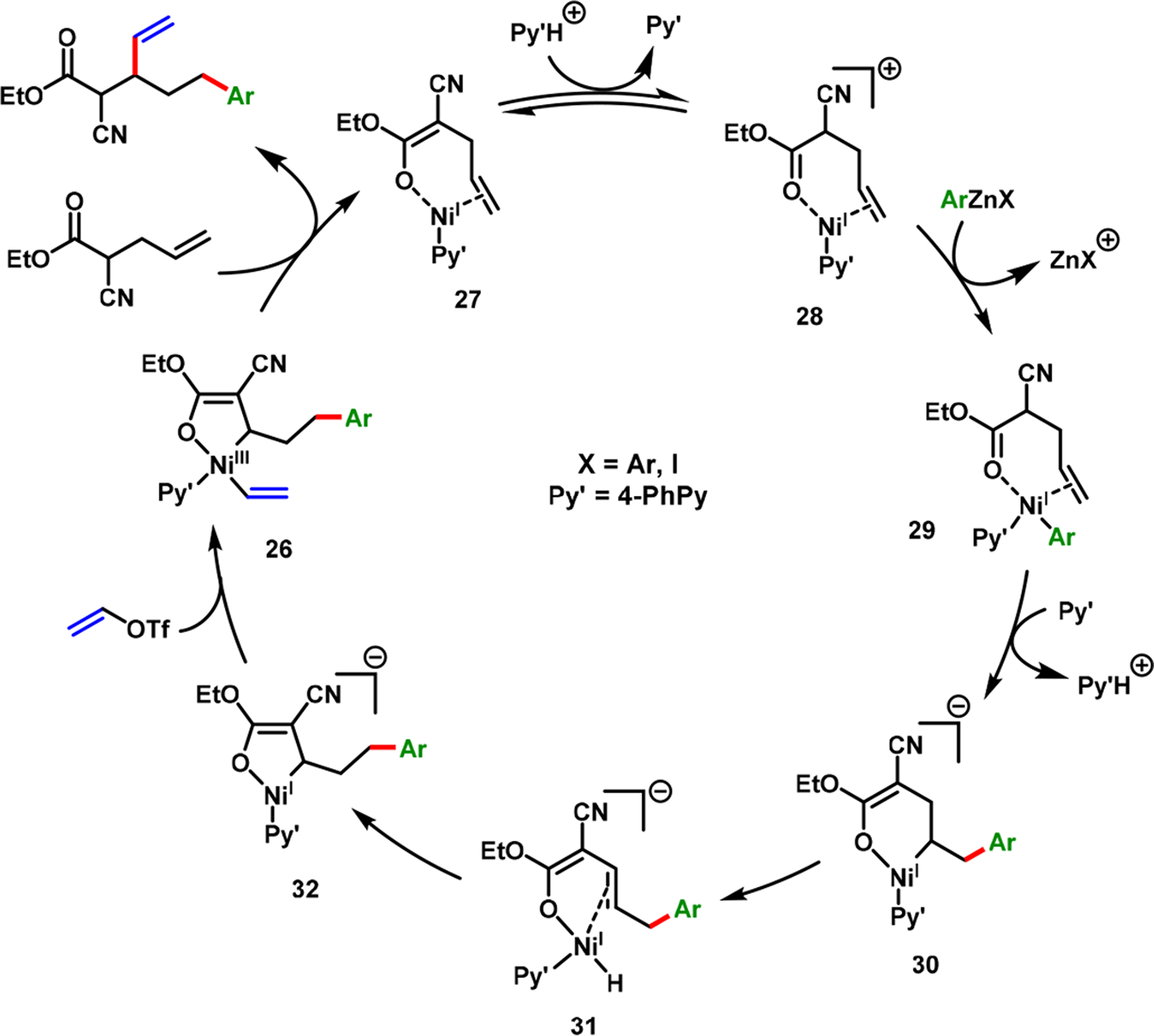
Catalytic Cycle for Alkene Alkenylarylation by the Nickellacyle Contraction Process
2.3. Electronically-Controlled Vicinal Dicarbofunctionalization
Three-component alkene dicarbofunctionalization without heteroatom coordination suffers seriously from the β-H elimination issue. When alkenyl-OTf is used, the resultant β-H elimination products proceed with hydropalladation to generate π-allylpalladium(II) intermediates and eventually furnish 1,1-difunctionalized products.13 Since Takai’s Cr-mediated work in 1998,62 stabilization of alkylmetal species as π-allylmetal intermediates has served as fodder for developing vicinal dicarbofunctionalization in conjugated dienes.11–13 An analogous reaction scenario can be anticipated with alkenylarenes, in which the electronically polarized alkene can enhance reactivity and regiocontrol, and the native arene can stabilize the resultant migratory insertion intermediate as a π-benzylmetal species.63,64 Consequently, alkenylarenes have always been at the forefront for alkene dicarbofunctionalization since Kambe’s Cp2TiCl2-catalyzed dialkylation in 1998;64 although, the general scope has so far been surprisingly limited.
In 2018, our laboratory developed a Ni-catalyzed three-component alkylarylation of alkenylarenes with alkyl halides and arylzinc reagents, which generated 1,1-diarylalkane products (Scheme 23).2 The reaction was achieved via the formation of two C(sp3)–C(sp3) and C(sp3)–C(sp2) bonds. Concurrently, a similar Ni-catalyzed diarylation of alkenylarenes with aryl halides and arylboronic esters was disclosed by Brown et al.,65 which created two C(sp3)–C(sp2) bonds. Our reaction tolerated various functional groups on alkenylarenes, alkyl halides, and arylzinc reagents. The reaction could be conducted with primary, secondary, and even tertiary alkyl halides. The coupling of primary and secondary alkyl halides was performed with NiBr2 as a catalyst, while that of tertiary alkyl halides required (Ph3P)2NiCl2.
Scheme 23.
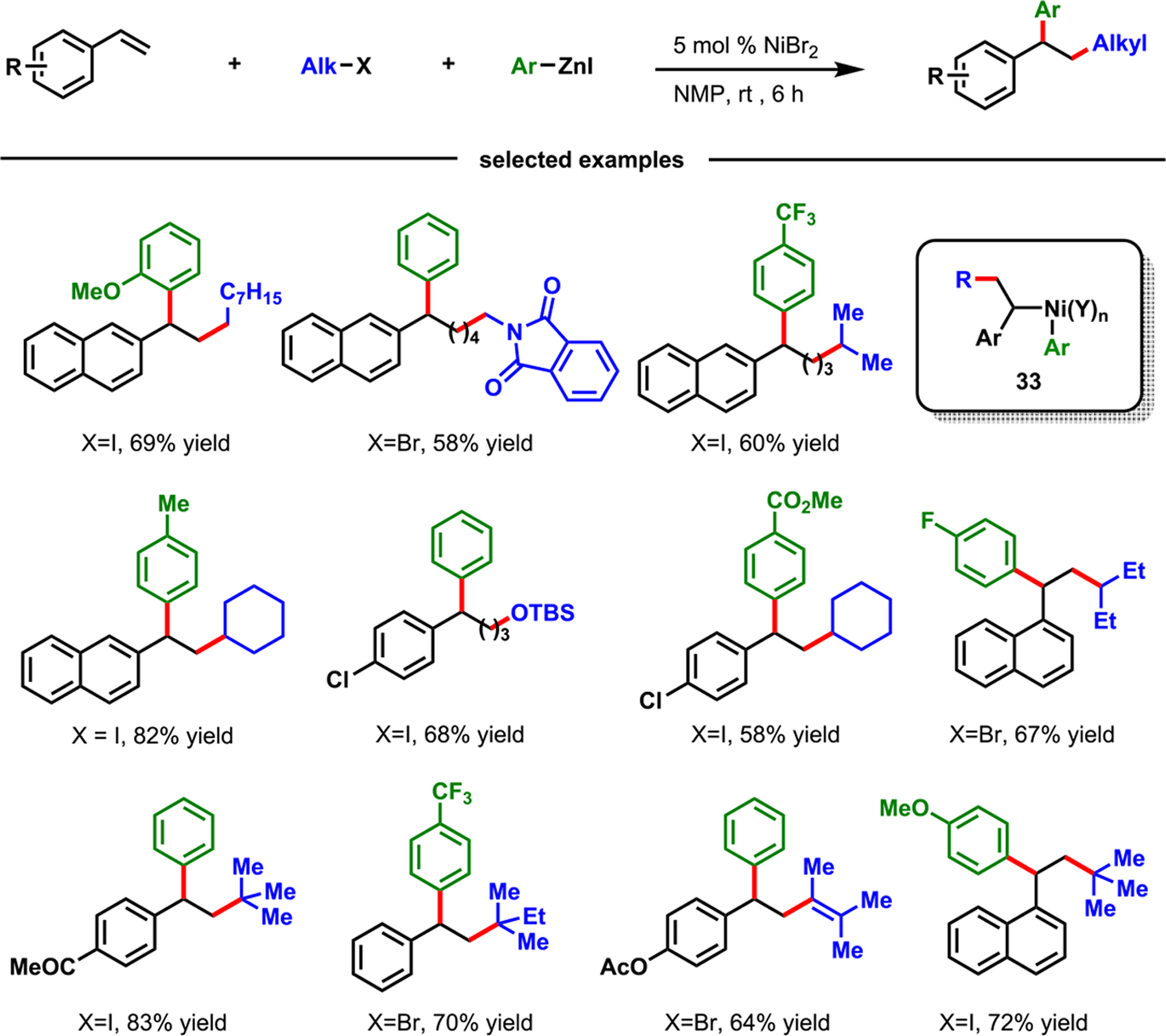
Nickel-Catalyzed Alkylarylation of 2-Alkenylarenes
Real-time monitoring of the reaction between 4-fluorophenylzinc bromide and NiBr2·DME by 19F NMR suggested that Ni(II) was reduced in situ to Ni(0) and that the latter was the catalytically active species. We also performed competition studies between primary, secondary, and tertiary alkyl bromides (3° > 2° > 1°) and between alkyl iodide, bromide, and chloride (I > Br > Cl) (Scheme 24). The outcomes indicated that the reaction was initiated by a rate-limiting halogen-atom abstraction via inner-sphere SET from Ni(0) to alkyl halides. The presence of carbon-centered radicals (34 and 35) was also supported by a radical clock experiment and the isolation of a dimeric product 37 from a catalytic reaction. The rate-limiting alkyl halide activation was also supported by a competition study between electronically biased arylzinc reagents that showed no product selectivity, and our further quantitative kinetic studies examining rate dependence and reaction order. Overall, the complete catalytic cycle involved Ni(0), Ni(I), and Ni(II) speciation (Scheme 25) and proceeded via a rate-limiting halogen-atom abstraction by inner-sphere SET, followed by carbon-centered radical addition to alkene, recombination of benzylic radical with Ni(I), transmetalation, and, finally, reductive elimination.
Scheme 24.
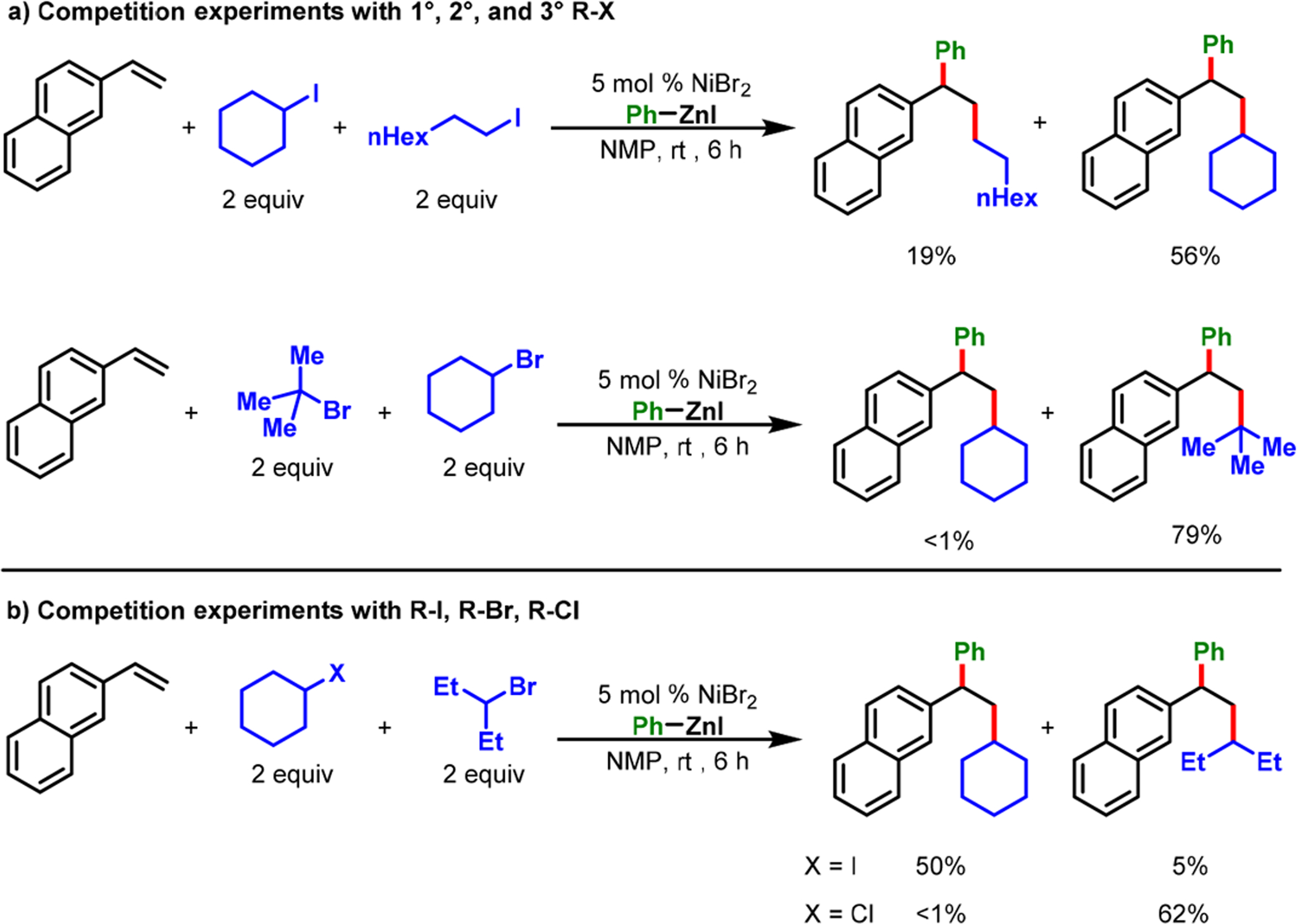
Competition Experiments
Scheme 25.
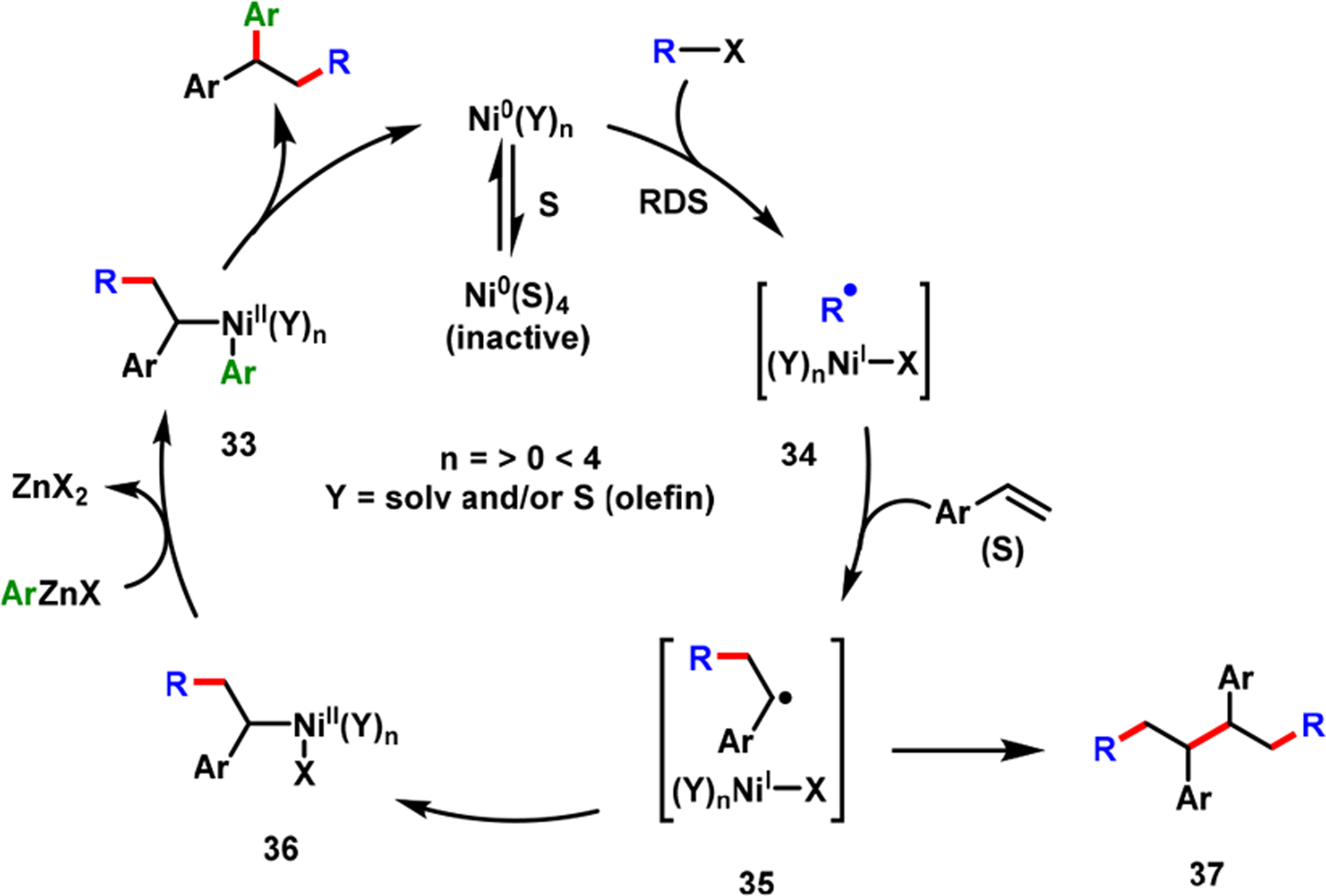
Catalytic Cycle for Nickel-Catalyzed Alkene Alkylarylation
In 2020, we reported that alkyl halides could be replaced with α-halocarbonyl compounds as C(sp3) sources to difunctionalize alkenylarenes to access γ,γ-diarylcarbonyl compounds (Scheme 26).66 Although this reaction could be conducted under otherwise identical conditions developed for alkylarylation using NiBr2 as the catalyst, Ni(cod)2 proved to be more competent, furnishing products in higher yields. Examination of the reaction scope revealed that variously substituted alkenylarenes could be difunctionalized with primary, secondary, and tertiary α-halocarboxylic esters, and electronically varied aryl/heteroarylzinc reagents. α-Halolactones and α-haloketones also participated in the reaction, although the scope was somewhat limited.
Scheme 26.
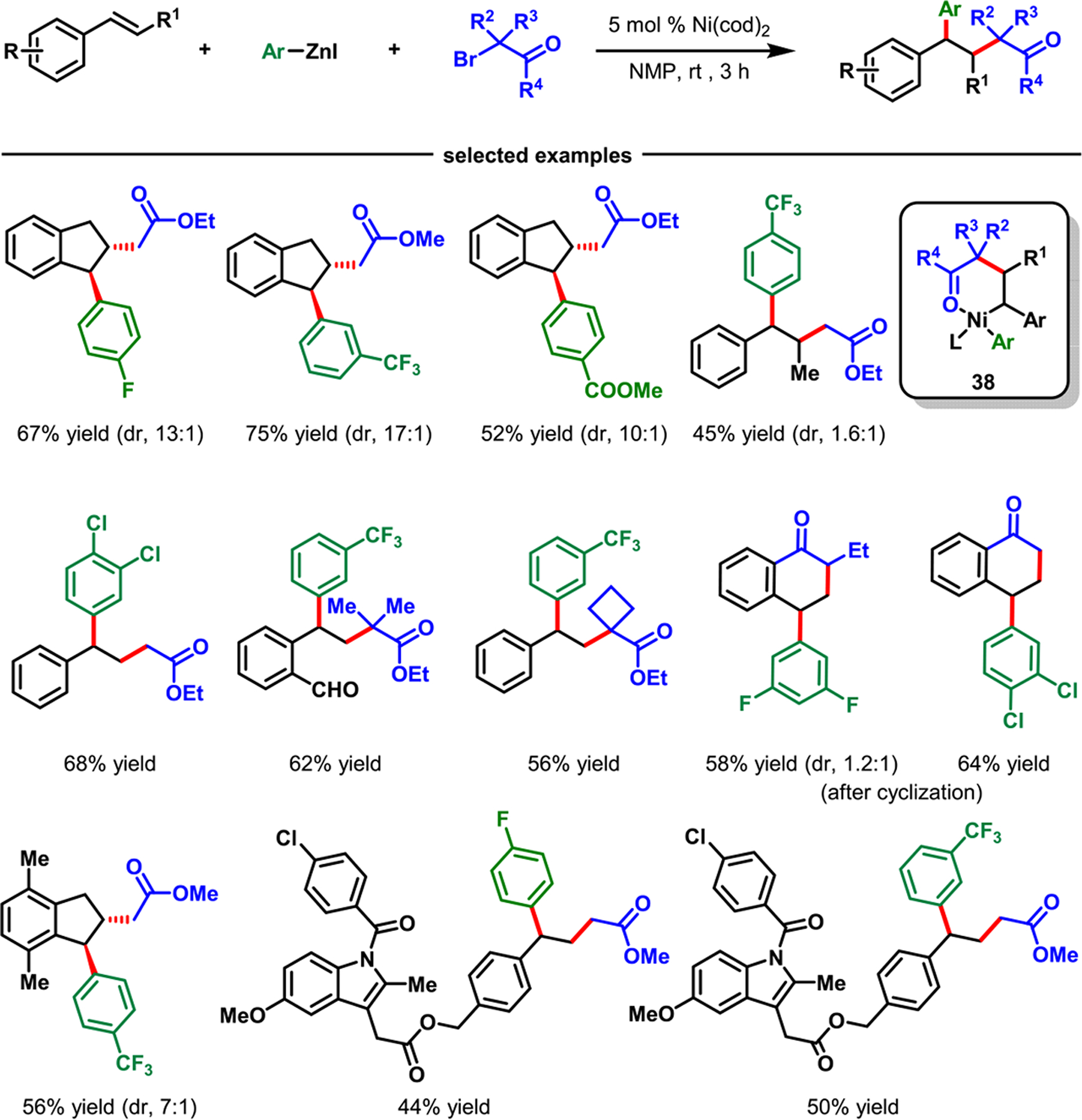
Nickel-Catalyzed α-Carbonylalkylarylation of 2-Alkenylarenes
The reaction also tolerated internal alkenes, which were difunctionalized with absolute regioselectivity and good diastereoselectivity. Cyclic internal alkenes, such as indenes, proceeded with high diastereoselectivity (>10:1) affording trans-stereoisomers as the major products. Preliminary mechanistic studies with a radical clock and TEMPO, as a radical trap, indicated that the reaction generated a stable α-carbon radical. This radical could be intercepted with TEMPO and electronically polarized intermolecular alkenes in alkenylarenes but was unable to undergo radical cyclization onto an electronically unbiased tethered alkene. On the basis of these studies, we presume that the catalytic cycle is analogous to that for alkylarylation described in Scheme 25.
3. TWO-COMPONENT ALKENE DICARBOFUNCTIONALIZATION
Two-component alkene dicarbofunctionalization reactions are conducted through cyclization/coupling in which an alkene is tethered to an organic electrophile or nucleophile. In these reactions, the alkylmetal species generated upon cyclization is intercepted by a second carbon source; creating five- and six-membered carbo- and heterocycles. Cyclization/coupling is entropically favored for migratory insertion, which, along with intramolecular alkene activation, helps to overcome cross-coupling.
Pd-catalyzed cyclization/coupling is the most studied alkene dicarbofunctionalization reaction. However, β-H elimination still dominates the outcome of these reactions. Consequently, the reactions were compelled to employ disubstituted terminal alkenes,14,15 which generated C(sp3)–Pd(II) intermediates lacking β-H’s. Tour and Negishi67 and Suzuki et al.28 independently demonstrated that the use of alkenes predestined for β-H elimination was also possible for cyclization/coupling. Recently, Alexanian et al. observed analogous reactivity with a Mn2(CO)10 catalyst.29 However, such reactions required a combination of photolytic conditions amenable to the formation of carbon-centered radicals and capture by CO. Last year, Glorius et al. disclosed that a photoexcited Pd catalyst could also accomplish cyclization/coupling reactions of alkenes prone to cause β-H elimination via carbon-centered radicals.27
In 2017, we attempted Pd-catalyzed cyclization/coupling of alkenyl carboxamides with intramolecular enolates as nucleophiles (Scheme 27).68 To our surprise, our studies showed that Pd(dba)2 could catalyze such reactions to generate 1,3,4-trisubstituted pyrrolidinones, products that could arise from the interception of β-H–C(sp3)–Pd(II) species with ArI. In 1987, Balme et al. also developed a similar Pd-catalyzed cyclization/coupling of alkenyl dicarbonyl compounds bearing an enolizable α-hydrogen (DMSO, pKa ~ 13).69 Our reaction could employ alkenyl carboxamides bearing an enolizable α-hydrogen with a much higher pKa (DMSO, ~27), and various electronically modified ArI’s. Recently, Newhouse et al. also disclosed a similar cyclization/coupling of alkenyl carbonyl compounds with aryl electrophiles using a Ni catalyst.70 Likewise, Wolfe et al. reported a (BrettPhos)Pd-catalyzed cyclization/coupling of alkene-tethered ArX and alkenyl-OTf in which malonates functioned as nucleophiles.71
Scheme 27.
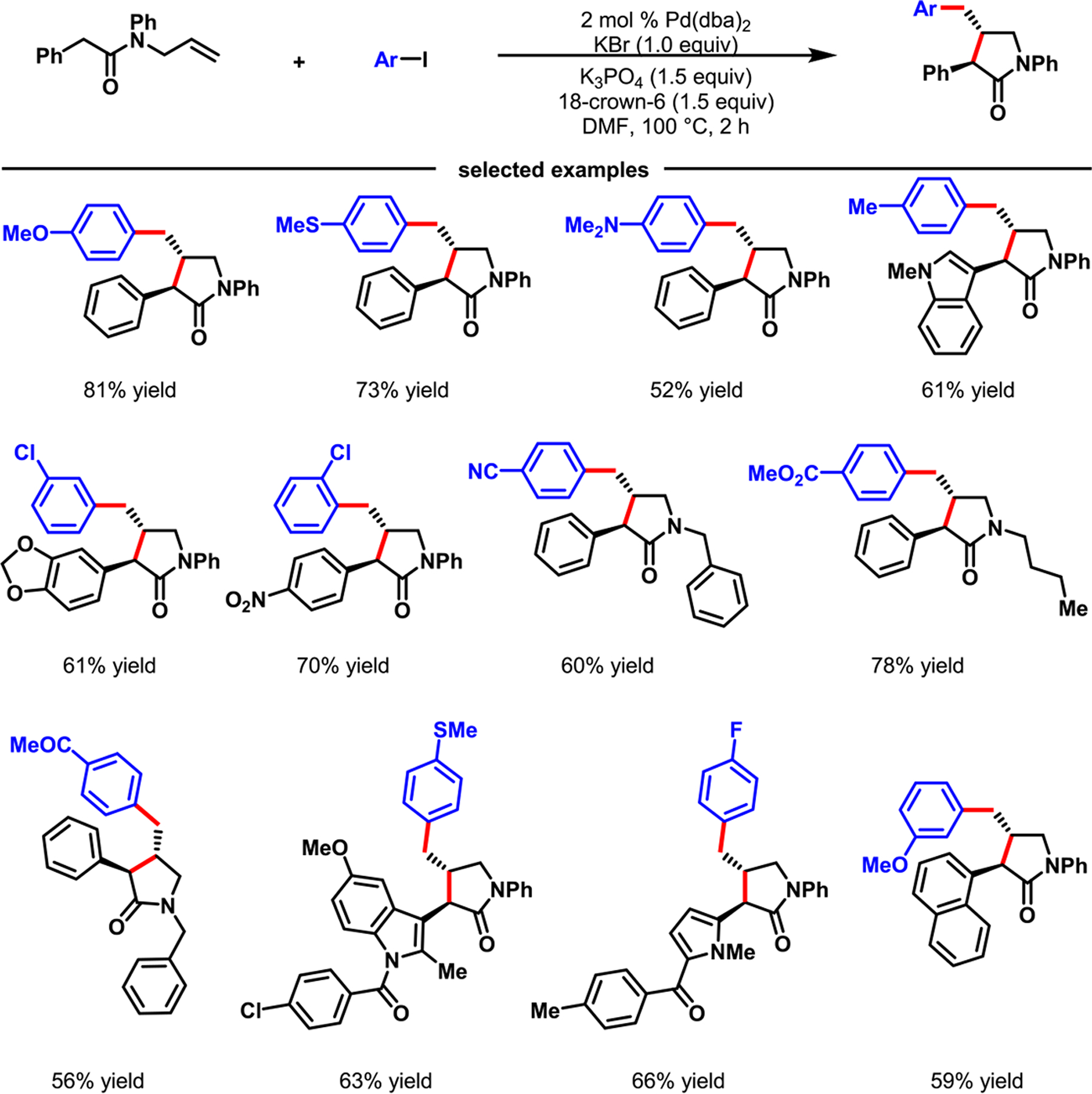
Palladium-Catalyzed Cyclization/Coupling of N-Allylcarboxamides
Puzzled by the observation of zero to minute amounts of Heck products, we monitored the progress of the reaction over time (Scheme 28). The experiment immediately revealed that β-H–C(sp3)–Pd(II) intermediates, generated upon migratory insertion of alkenes to ArPd(II), indeed underwent β-H elimination to form Heck products as reaction intermediates. In the presence of K3PO4, the styryl group in the Heck product 40 would undergo in situ base-promoted conjugate addition with the internal enolate to furnish final products.
Scheme 28.
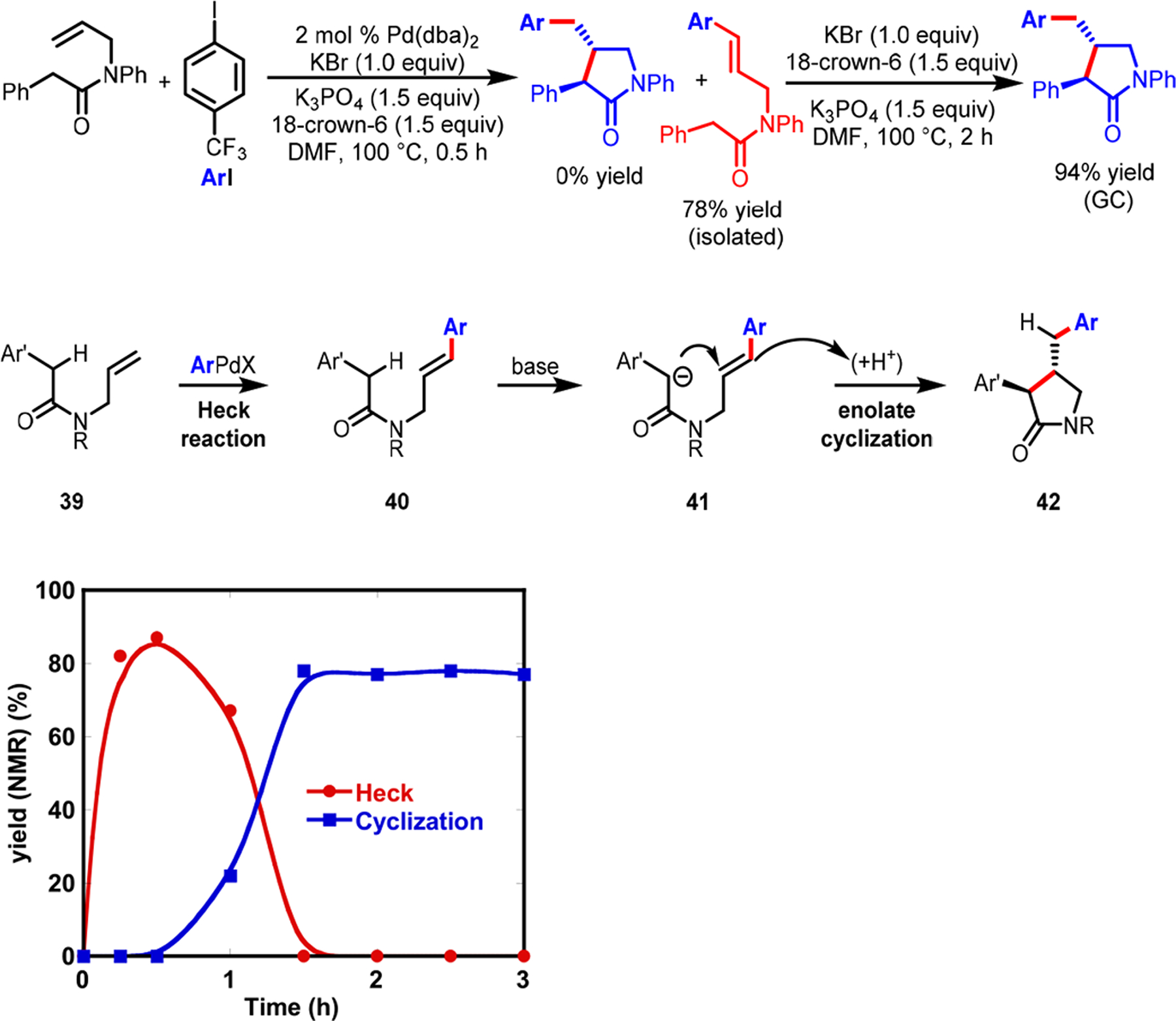
Reaction Kinetic Profile and Mechanism
Reports of catalytic reactions7 have shown that β-H–C(sp3)–[M] intermediates of first row late TMs are slower to undergo β-H elimination than β-H–C(sp3)–Pd(II) species. In 1994, Delgado et al. demonstrated that a stoichiometric amount of Ni(cod)2 could catalyze cyclization/coupling of alkene-tethered vinyl bromides amenable to generate β-H–C(sp3)–[Ni] intermediates.72 Although, their stabilization was attributed to intramolecular nitrogen coordination. In 2001, Oshima et al. demonstrated that (dppe)CoCl2 was an effective catalyst to perform cyclization/coupling of alkene-tethered alkyl halides with ArMgX.73 In 2007, Cárdenas et al. disclosed that (pybox)NiCl2 could overcome even more challenging cyclization/coupling of alkene-tethered alkyl halides with alkylzinc reagents.74 The overall observation of successful examples unequivocally substantiates the notion that the first row TMs are great catalysts for alkene dicarbofunctionalization involving β-H–C(sp3)–[M] intermediates.
In 2017, we demonstrated that Cu(I) salts could catalyze cyclization/coupling of alkene-tethered organozinc reagents with aryl/heteroaryl iodides (Scheme 29).4 The reaction furnished a wide range of complex carbocyclic and N,O-heterocyclic products containing cyclopentyl, pyrrodinyl, furanyl, dihydrofuranyl, indanyl, and indolinyl cores in excellent diastereoselectivity. Prior to our work, Cong and Fu75 and You and Brown,76 in 2014, also disclosed similar cyclization/coupling of alkenyl-aryl-9-BBN.
Scheme 29.
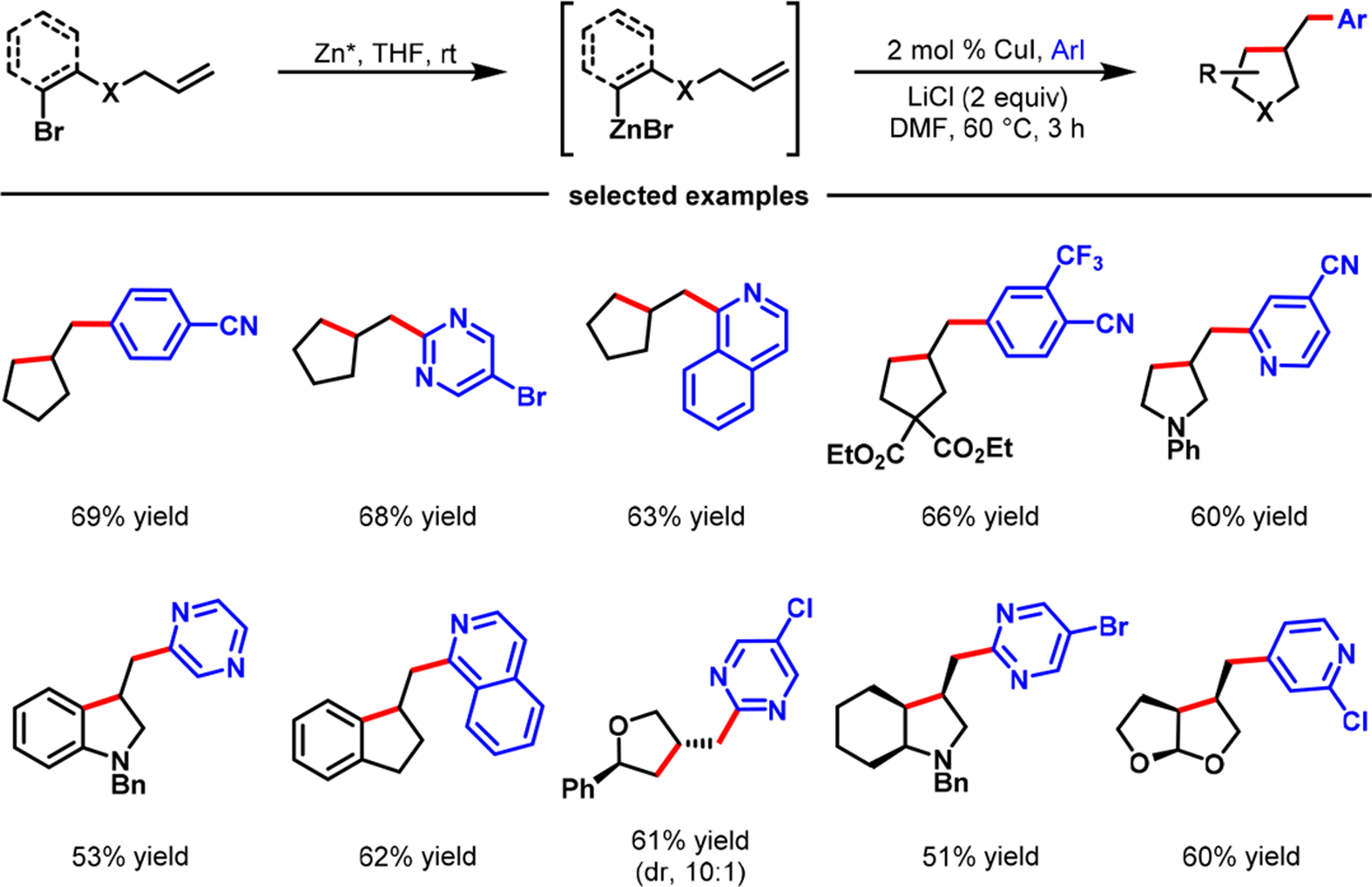
Copper-Catalyzed Cyclization/Coupling of Alkenyl Organozinc Reagents
Analysis of the products generated and the stereoselectivity observed during organozinc synthesis, under radical conditions, highlighted different scenarios for cyclization of alkenes tethered to alkyl and arylzinc reagents. In alkenylalkylzinc reagents, the radical cyclization ensued in situ during alkylzinc preparation, and the resultant cyclized alkylzinc species underwent transmetalation with Cu(I) during the catalytic reaction to generate β-H–C(sp3)–[Cu] intermediates (Scheme 30). In contrast, alkenylarylzinc reagents remained mostly intact during arylzinc synthesis and participated in radical cyclization during the Cu-catalyzed reaction (Scheme 31). This resulted in the formation of β-H–C(sp3)–[Cu] intermediates for subsequent reaction with aryl halides.
Scheme 30.
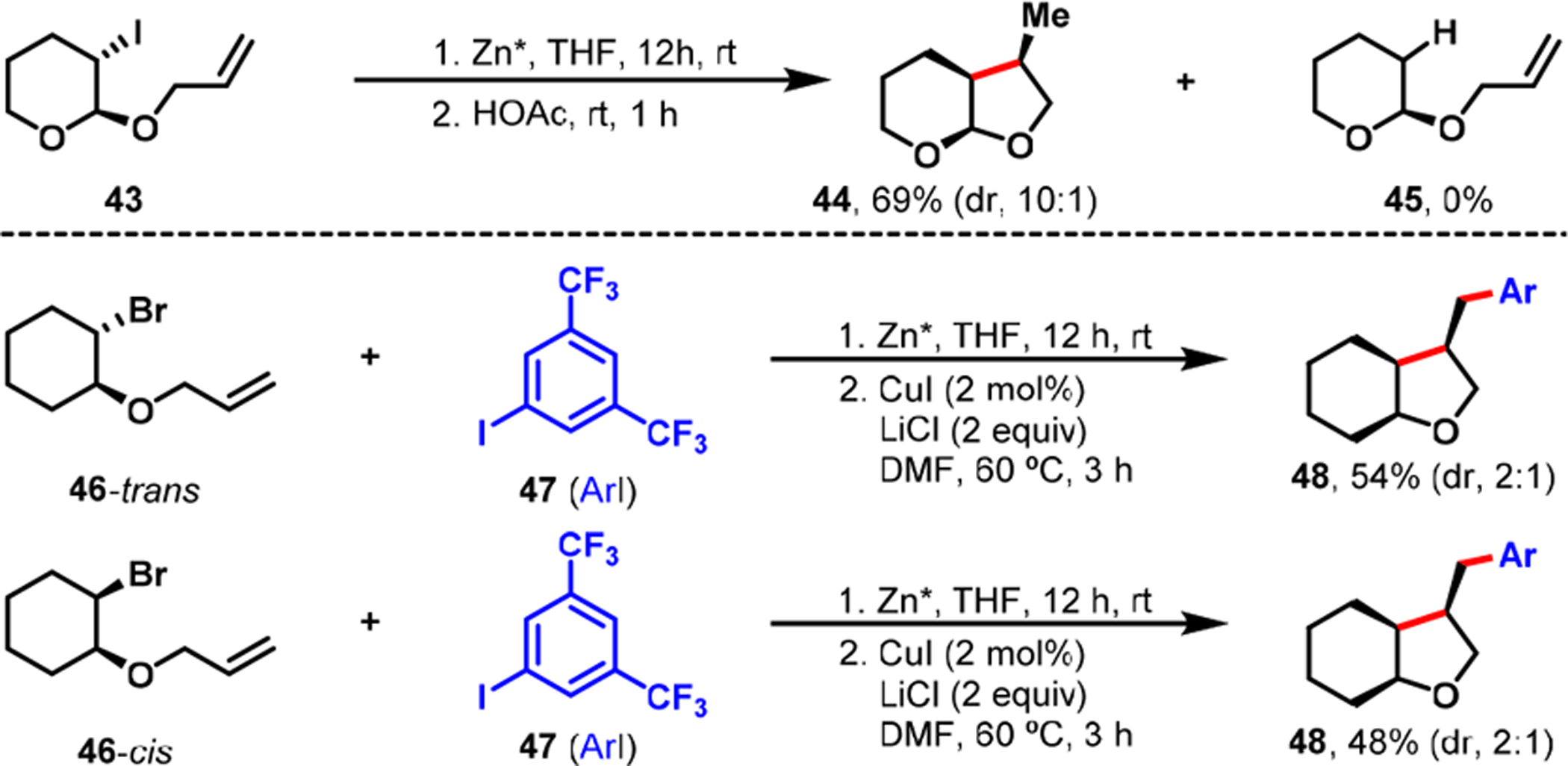
Radical Cyclization during the Preparation of Alkenyl Alkylzinc Reagents
Scheme 31.
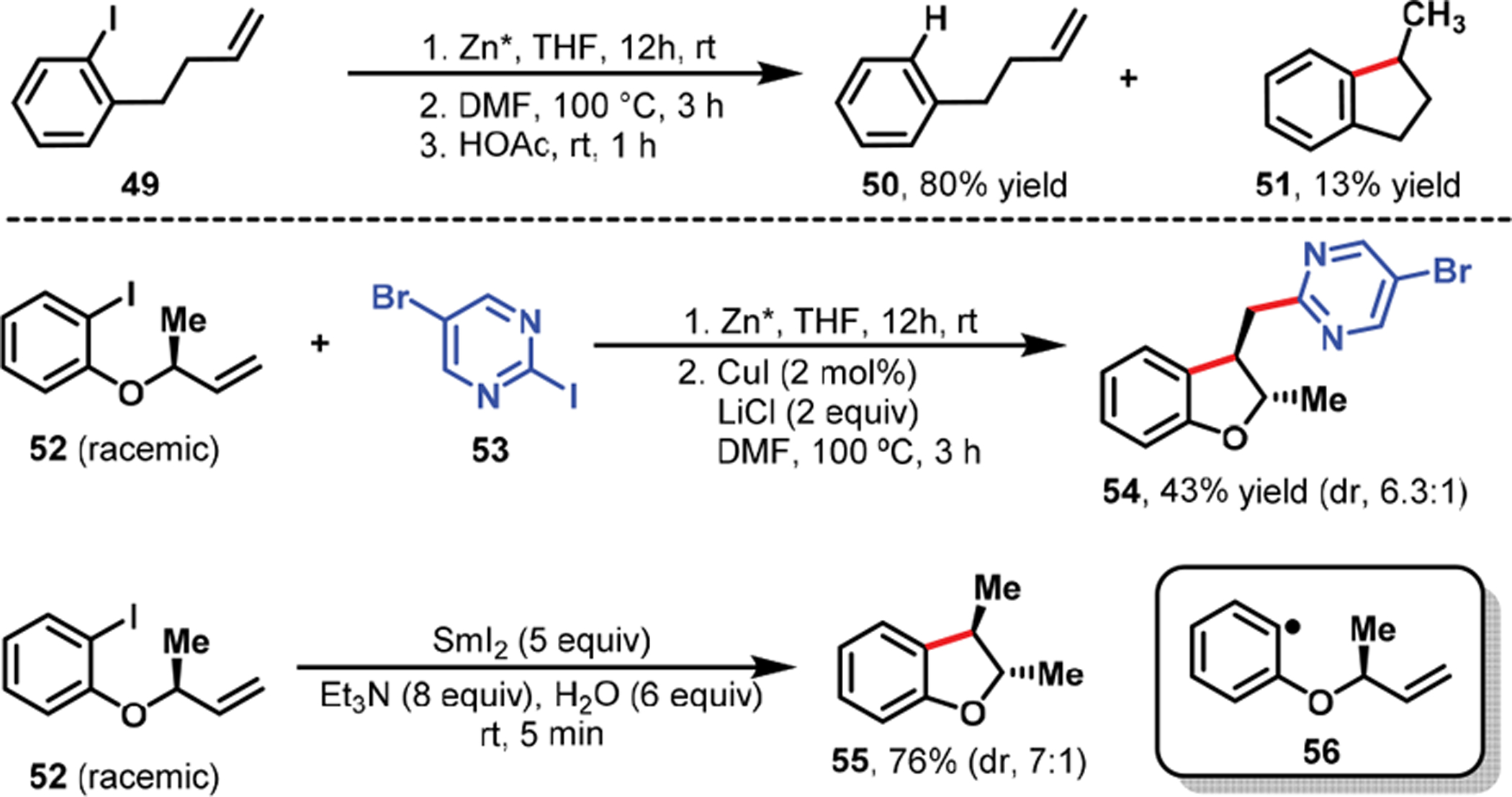
Radical Cyclization during CuI- and SmI2-Catalyzed Reactions
The Cu-catalyzed reaction illustrated the synthetic aptitude of cyclization/coupling to generate rapidly complex carbo- and heterocycles. The reaction was also amenable for cyclization/coupling with heterocyclic arenes. However, the reaction scope remained largely limited to electron-deficient and heteroaryl iodides. In order to surmount this shortcoming, we switched the tethering of alkenes from alkylzinc reagents to alkyl halides and developed a NiBr2-catalyzed cyclization/coupling in which arylzinc reagents supplied the arene component (Scheme 32).77 The reaction required terpyridine as a ligand. The (terpy)Ni-catalyzed cyclization/coupling proceeded in high diastereoselectivity and tolerated a number of sensitive functional groups and molecules with racemizable stereocenters. One of the appealing outcomes of the Ni-catalyzed reaction, that is complementary to the Cu-catalyzed process, is the access to (arylmethyl)carbo- and heterocyclic scaffolds containing electron-rich arenes, structural cores that are ubiquitous in various lignan natural products.78 The reaction proceeded via a SET process involving carbon centered radicals for cyclization (Scheme 33).21
Scheme 32.
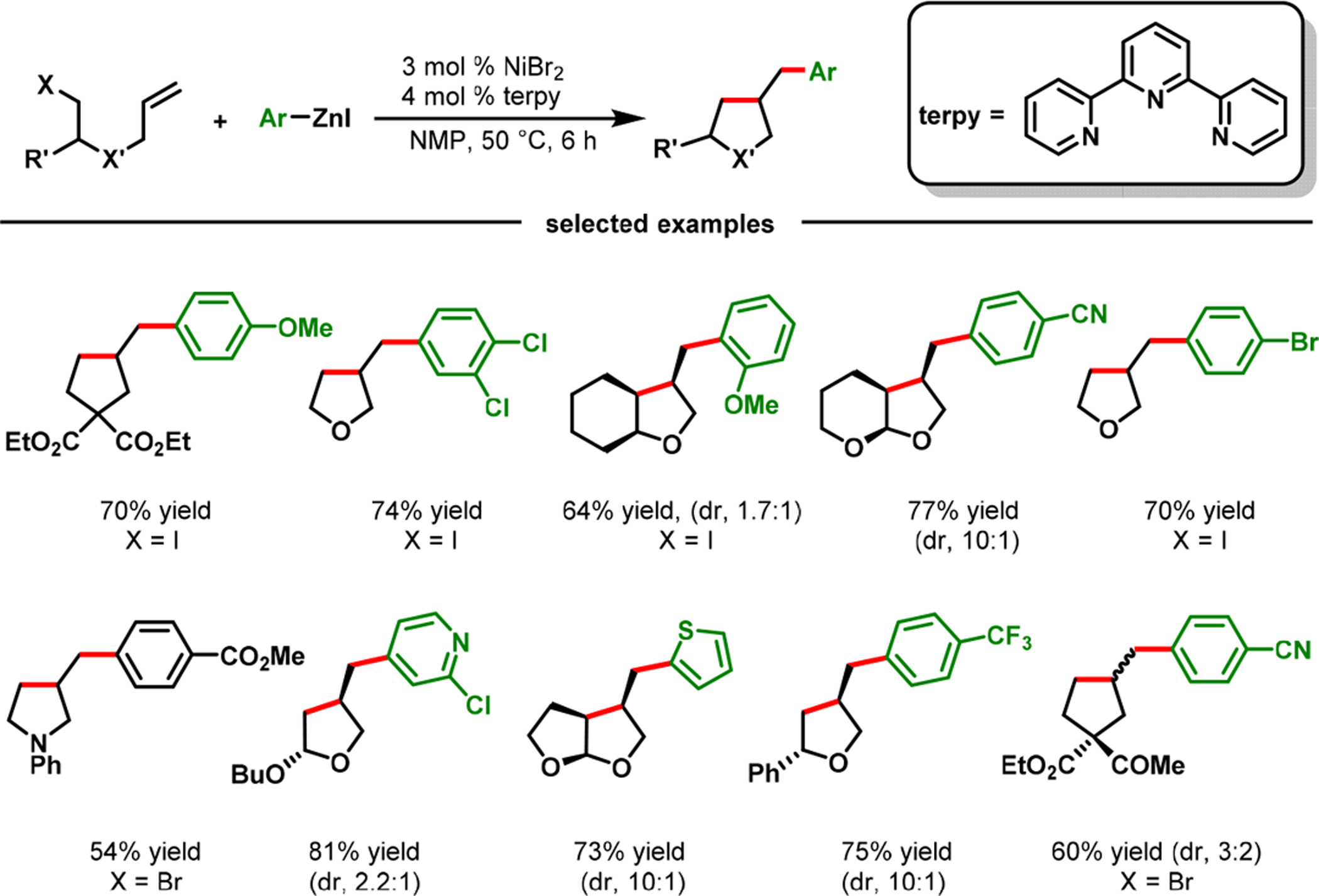
(Terpy)Ni-Catalyzed Cyclization/Coupling of Alkenyl Alkyl Halides
Scheme 33.
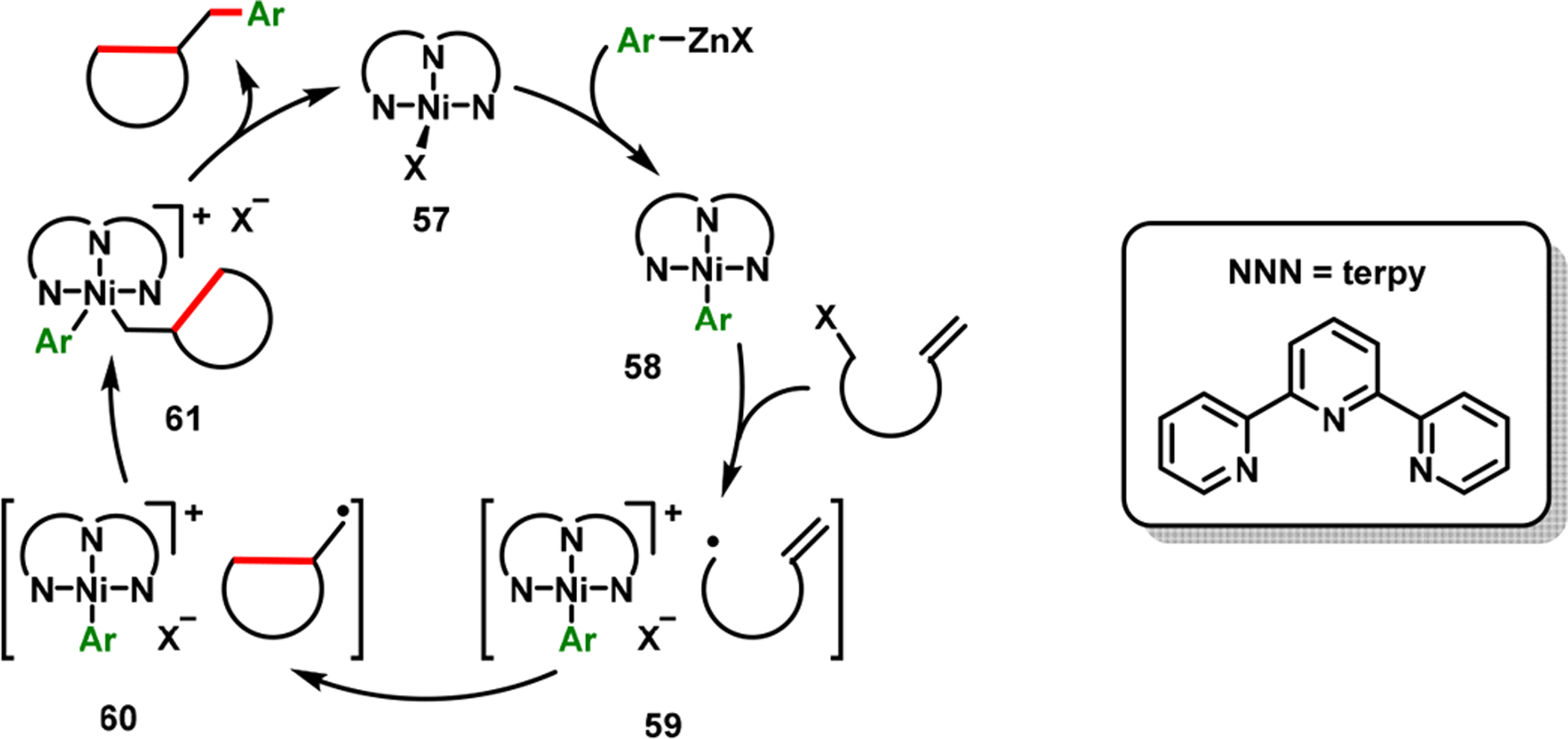
Catalytic Cycle for the (terpy)Ni-Catalyzed Cyclization/Coupling Reaction
4. APPLICATIONS
The robustness of a chemical method is measured against its potential in the synthesis of complex natural products and bioactive molecules.79 Since the development of metal-catalyzed alkene dicarbofunctionalization reactions has been relatively dormant for almost four decades, their vetting against long-established synthetic protocols has not been thoroughly investigated. Yet, the potential of alkene dicarbofunctionalization through cyclization/coupling was realized early on in the rapid synthesis of complex natural products. In 1994, Balme and Bouyssi described the total synthesis of a sesquiterpene, (±)-D9(12) capnellene,80 through cyclization/coupling of α-allyl-dicarboxylic esters with organic halides.69
In 2018, we scrutinized our Ni-catalyzed cyclization/coupling for the concise synthesis of six lignan natural products with three different structural frameworks.77 In 2014, Cong and Fu also synthesized a dihydrobenzofuran core of fasiglifam via a Ni-catalyzed cyclization/coupling of alkenylaryl-9-BBN.75 Recently, Diao et al. utilized Ni-catalyzed reductive cyclization/coupling to prepare a key intermediate toward the synthesis of an epoxide hydrolase inhibitor.26 Our target lignan natural products contain a di(arylmethyl)butyrolactone core and display a wide range of biological activity.78 We accessed the 3-arylmethylbutyrolactone intermediates (63 and 64) required for (±)-kusunokinin (65), (±)-dimethylmatairesinol (66), (±)-bursehernin (67), and (±)-yatein (68), through cyclization/coupling of 1-(1-(allyloxy)-2-iodoethoxy)butane (62) with corresponding arylzinc reagents, followed by the Jones oxidation (Scheme 34). The butyrolactone derivatives could be generated in a one-pot, two-step process in gram-scale quantities. These butyrolactone intermediates were subsequently converted to the natural products upon reaction with corresponding benzyl bromides.
Scheme 34.
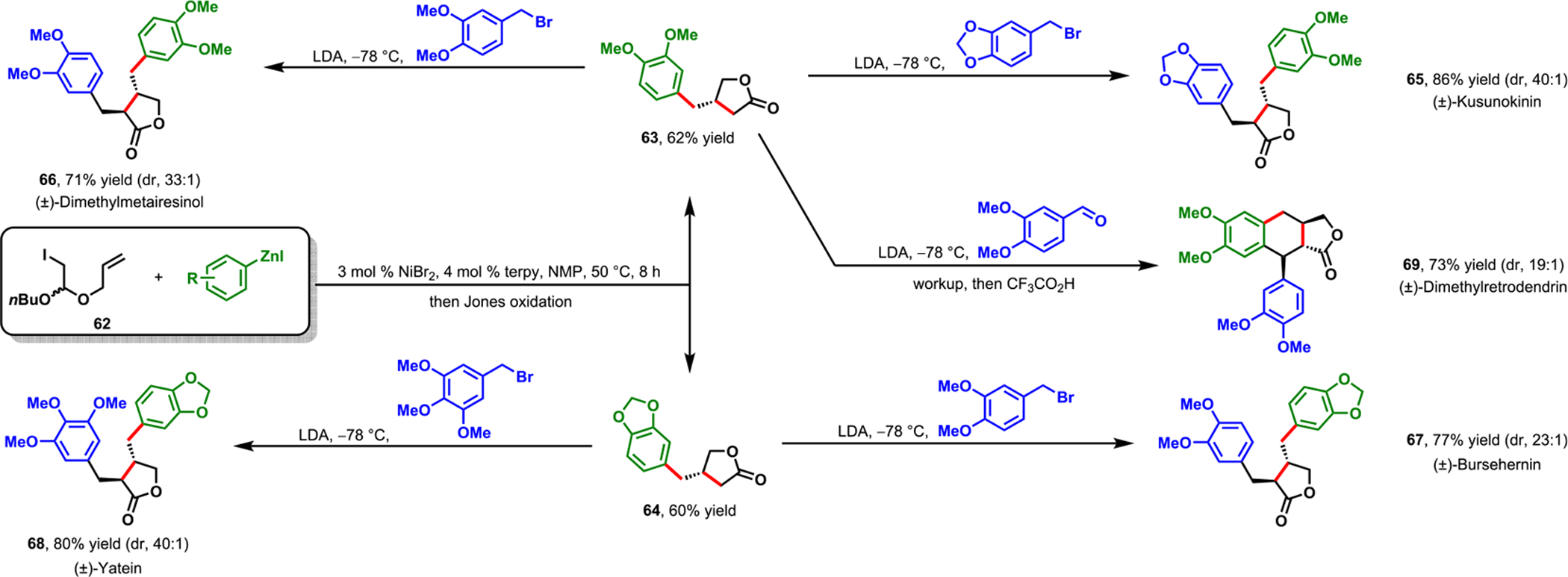
Concise Synthesis of (±)-Kusunokinin, (±)-Dimethylmatairesinol, (±)-Bursehernin, (±)-Dimethylretrodendrin, and (±)-Yatein via Nickel-Catalyzed Cyclization/Coupling
The 3-arylmethylbutyrolactone derivatives also serve as intermediates for more complex lignan natural products, including (±)-dimethylretrodendrin (69) and (±)-collinusin (73). For example, 3-(3,4-dimethoxybenzyl)butyrolactone 63 could be condensed with 3,4-dimethoxybenzaldehyde followed by Friedel–Crafts cyclization to synthesize (±)-dimethylretrodendrin (69) in high yield and excellent diastereoselectivity (Scheme 34). The carbonylbutyrolactone 72 necessary for the synthesis of (±)-collinusin (73) was also accessed in a concise manner following the same one-pot, two-step process beginning with a (2-aryoylaryl)zinc reagent 71 (Scheme 35). Through this synthesis, we demonstrated the astonishing efficiency and practicality of our new method, as the synthesis of similar lignan products required several steps to construct the dihydronaphthofuranone core.78
Scheme 35.
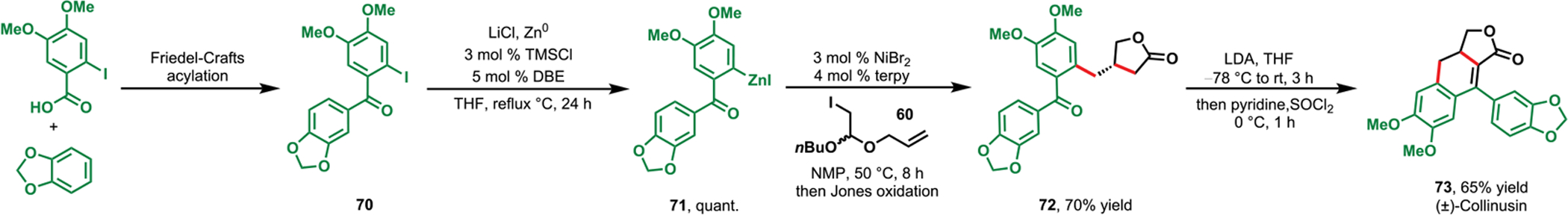
Concise Synthesis of (±)-Collinusin via Nickel-Catalyzed Cyclization/Coupling
The application of three-component alkene dicarbofunctionalization in the synthesis of natural products/bioactive molecules remains extremely scarce. We recently applied our Ni-catalyzed alkylarylation of alkenylarenes to synthesize a potential 5-lipoxygenase activating protein (FLAP) inhibitor 75 along with a few of its analogs (Scheme 36) and a regioisomer (Scheme 37).81 The bioactive compound is constructed on a 1,1-diarylalkane framework, which has broadly shown bio-activity against FLAP, lung cancer, breast cancer, and brain cancer.82–84 In particular, in 2012, the Merck Research Laboratory reported the FLAP inhibitory activity of compound 75, which they synthesized in 12 steps.82 In contrast, we were able to successfully synthesize the same potential FLAP inhibitor 75 in just five steps by using our Ni-catalyzed alkylarylation reaction. Concomitantly, Brown et al. also synthesized rac-lasofoxifene through a Ni-catalyzed alkenylarene diarylation reaction.65
Scheme 36.
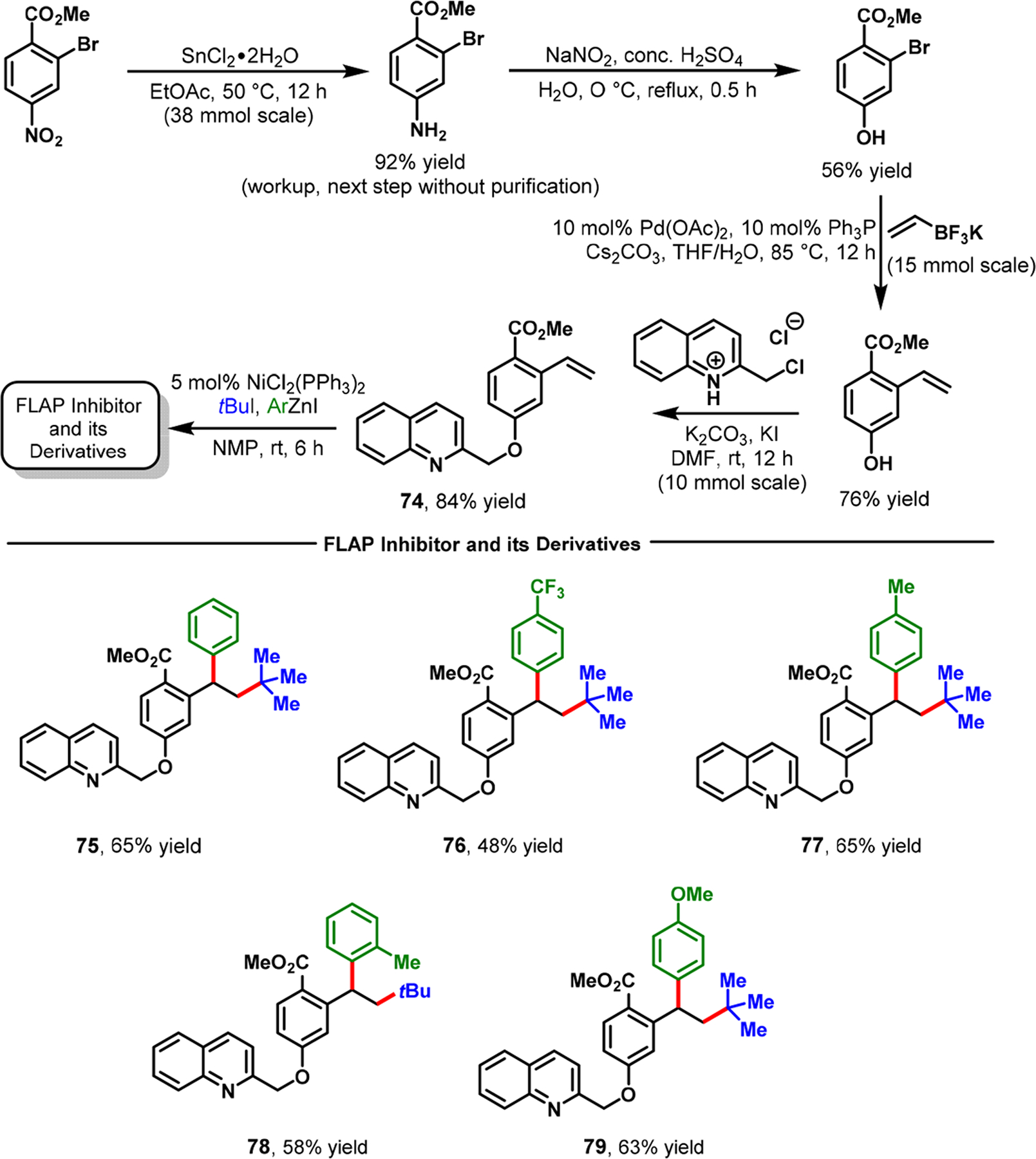
Synthesis of a Potential FLAP Inhibitor and Its Derivatives via Nickel-Catalyzed Alkene Alkylarylation
Scheme 37.
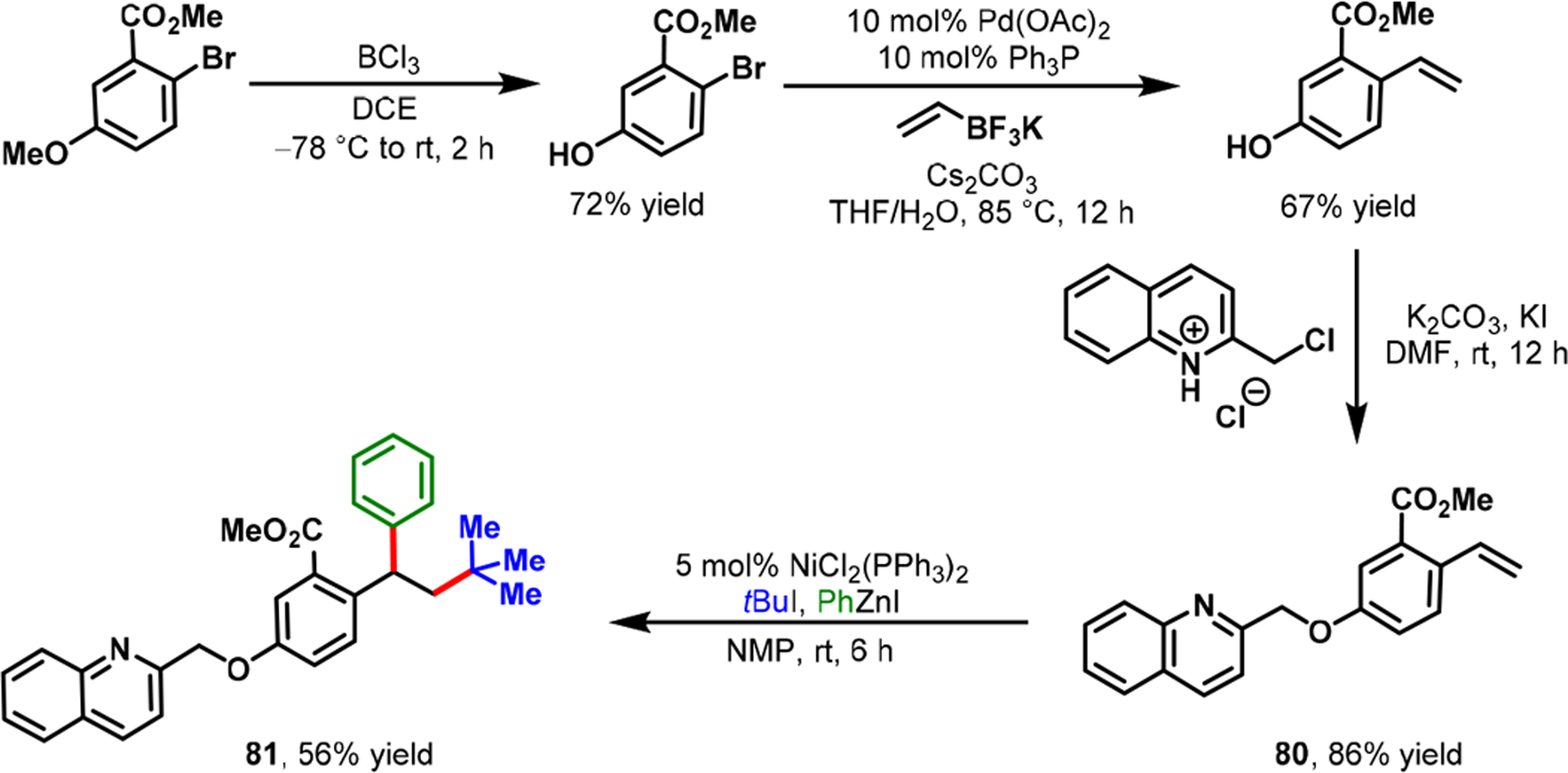
Synthesis of a Regioisomer of the Potential FLAP Inhibitor via Nickel-Catalyzed Alkene Alkylarylation
The required alkenylarene 74 was conveniently synthesized through four steps from commercially available 2-bromo-4-nitrobenzoic acid methyl ester. The alkenylarene 74 was subjected to Ni-catalyzed alkylarylation with tBuI and PhZnI to yield the potential FLAP inhibitor 75 in 65% yield. Since our Ni-catalyzed alkylarylation was used at the last step in the synthetic sequence, we were also able to synthesize additional analogs of the potential FLAP inhibitor (76–79) with the same alkenylarene intermediate 74. A transposition of the O-quinolinyl group on the central arene also enabled us to synthesize the FLAP inhibitor’s regioisomer 81 in just four steps from the commercially available 2-bromo-5-methoxybenzoic acid methyl ester (Scheme 37). Similarly, we applied our Ni-catalyzed regioselective α-carbonylalkylarylation reaction to the rapid synthesis of different aryltetralone derivatives.66 This ability enabled us to synthesize dichlorophenyltetralone 82, a known synthetic precursor to a commercial antidepressant drug, sertraline·HCl (Zoloft) 83 (Scheme 38).85
Scheme 38.
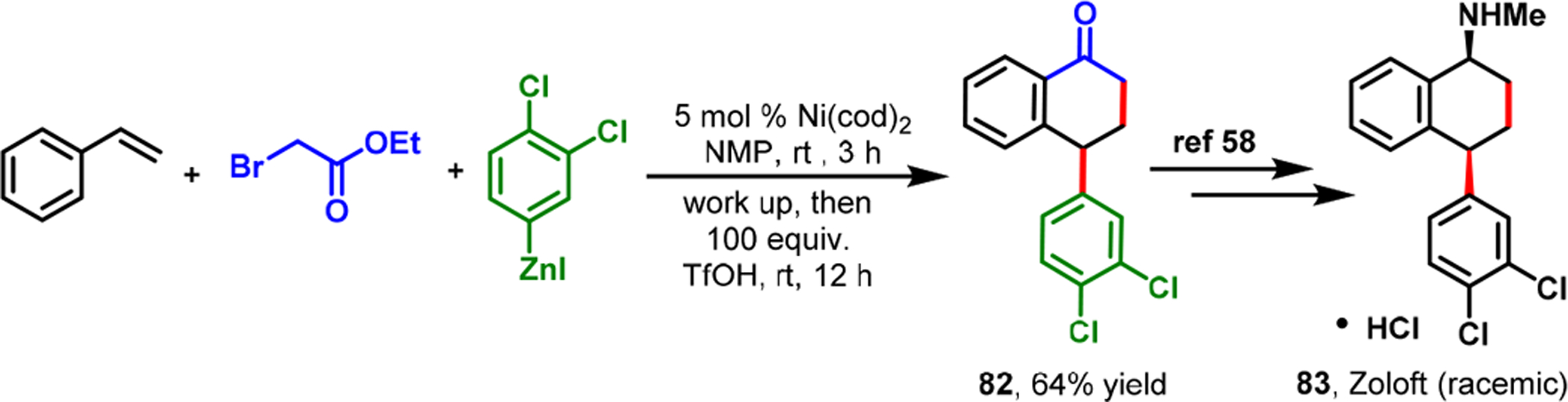
Synthesis of Zoloft Precursor via Nickel-Catalyzed Alkene α-Carbonylalkylarylation
In another synthetic application, we applied our Ni(cod)2-catalyzed diarylation of 2-alkenylbenzaldimines with aryl halides and arylzinc reagents to synthesize 9-arylmethylanthracene derivatives (Scheme 39).86 We found that the o-aldimine group was a synthetic bonus in this reaction, since it could be condensed with the proximal arene by acid-catalyzed in situ deaminative aromatization to generate functionalized anthracenes. This method illustrated a new three-component approach to synthesize 9-arylmethylanthracene derivatives.
Scheme 39.
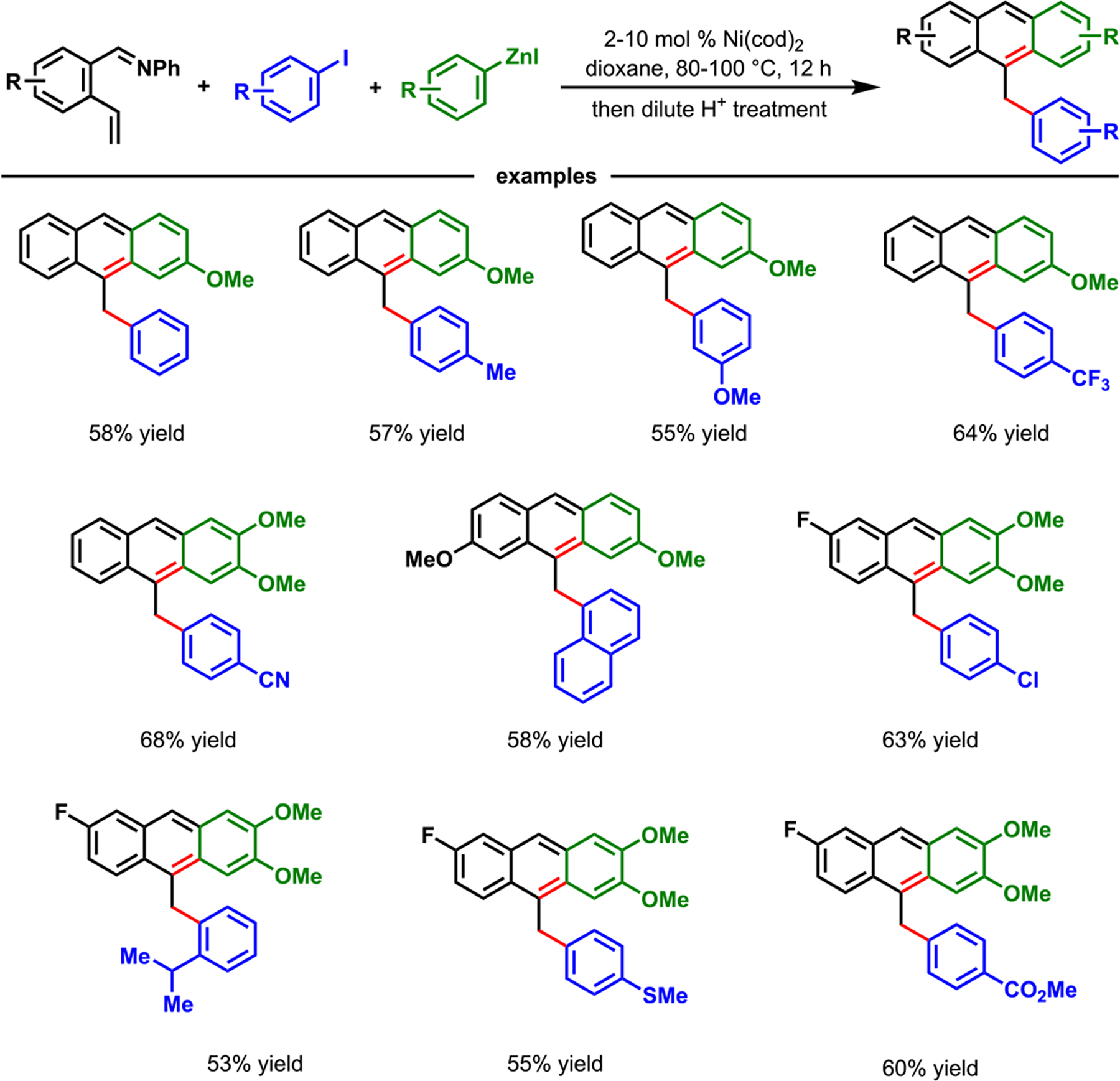
Synthesis of 9-Arylmethylanthracene Derivatives via a Nickel-Catalyzed Three-Component Alkene Diarylation Reaction
5. CONCLUSION AND OUTLOOK
As described in this Account, alkene dicarbofunctionalization with organic halides and organometallic reagents is emerging as a formidable method to construct complex molecules rapidly from readily available chemicals. Early discoveries were limited, as the methods relied on bicyclic and disubstituted terminal alkenes to circumvent the β-H elimination issue from the inevitable C(sp3)–[M] intermediates generated during the reaction. Recent studies have shown that alkenes that generate C(sp3)–[M] intermediates bearing β-H’s can now be difunctionalized without suffering from β-H elimination. The immense success and the current rise in alkene dicarbofunctionalization lends credit to the fundamental understanding of the nature of β-H–C(sp3)–[M] intermediates and their susceptibility toward β-H elimination. In particular, studies have shown that β-H–C(sp3)–[M] species of first row late metals are more resistant to β-H elimination than those of Pd and that these species can be intercepted as coordination-assisted transient metallacycles to bestow further stability to them. These two factors, collectively or individually, have contributed to the tremendous success of alkene dicarbofunctionalization through both entropically driven cyclization/coupling and more intractable three-component reactions in some of the most common organic molecules, including, aryl and alkyl halides, organometallics, aromatics, and carbonyl compounds. However, general chemical space is yet to be fully revealed, and a number of challenges, including an expanding scope to simple alkenes without coordinating groups, solving regioselectivity issues, and developing enantioselective variations, are still to be met. New catalytic systems and ligand discoveries will likely address these challenges in the future. In addition, the rapid rise in the method development has not parlayed into the synthetic output of complex natural products and bioactive molecules, which could serve as a touchstone for the method’s promise. Some optimism is on the horizon with the synthesis of a few natural products, pharmaceuticals, and bioactive molecules, from us and others. We anticipate that this field will soon grow exponentially and live up to synthetic chemists’ expectations to assemble complex molecules expeditiously.
ACKNOWLEDGMENTS
We gratefully acknowledge the NIH NIGMS (Grant R35GM133438), the NSF (Grants CHE-1554299 and CHE-2102394), and The Pennsylvania State University for support of this work.
Biographies
Laura M. Wickham is a Ph.D. student in the Giri group at The Pennsylvania State University. She received her B.S. in Chemistry at Marist College in Poughkeepsie, NY in 2017.
Ramesh Giri is a Weinreb Early Career Professor at the Pennsylvania State University. He earned his M.Phil. in 2003 from Cambridge University, U.K., and Ph.D. in 2009 from the Scripps Research Institute, La Jolla. CA In 2012; he joined the faculty at the University of New Mexico where he was promoted to an Associate Professor before moving to the Pennsylvania State University in 2019.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.accounts.1c00329
The authors declare no competing financial interest.
Contributor Information
Laura M. Wickham, Department of Chemistry, The Pennsylvania State University, University Park, Pennsylvania 16802, United States
Ramesh Giri, Department of Chemistry, The Pennsylvania State University, University Park, Pennsylvania 16802, United States.
REFERENCES
- (1).Basnet P; Dhungana RK; Thapa S; Shrestha B; Kc S; Sears JM; Giri R Ni-Catalyzed Regioselective β,δ-Diarylation of Unactivated Olefins in Ketimines via Ligand-Enabled Contraction of Transient Nickellacycles: Rapid Access to Remotely Diarylated Ketones. J. Am. Chem. Soc 2018, 140 (25), 7782–7786. [DOI] [PubMed] [Google Scholar]; This work demonstrates regioselective β,δ-diarylation of unactivated alkenes in ketimines with various aryl halides and arylzinc reagents.
- (2).Kc S; Dhungana RK; Shrestha B; Thapa S; Khanal N; Basnet P; Lebrun RW; Giri R Ni-Catalyzed Regioselective Alkylarylation of Vinylarenes via C(sp3)-C(sp3)/C(sp3)-C(sp2) Bond Formation and Mechanistic Studies. J. Am. Chem. Soc 2018, 140 (31), 9801–9805. [DOI] [PubMed] [Google Scholar]; This work displays regioselective alkylarylation of vinylarenes with primary, secondary, and tertiary alkyl halides, and electronically diverse arylzinc reagents to produce 1,1-diarylalkanes.
- (3).Shrestha B; Basnet P; Dhungana RK; Kc S; Thapa S; Sears JM; Giri R Ni-Catalyzed Regioselective 1,2-Dicarbofunctionalization of Olefins by Intercepting Heck Intermediates as Imine-Stabilized Transient Metallacycles. J. Am. Chem. Soc 2017, 139 (31), 10653–10656. [DOI] [PubMed] [Google Scholar]; This work describes the use of a readily removable coordinating group to promote Heck carbometalation and stabilize the Heck C(sp3)-NiX intermediates as metallacycles to suppress β-H elimination and facilitate transmetalation/reductive elimination steps to yield regioselective 1,2-dicarbofunctionalized products.
- (4).Thapa S; Basnet P; Giri R Copper-Catalyzed Dicarbofunctionalization of Unactivated Olefins by Tandem Cyclization/Cross-Coupling. J. Am. Chem. Soc 2017, 139 (16), 5700–5703. [DOI] [PubMed] [Google Scholar]; This work illustrates 1,2-difunctionalization of unactivated olefins utilizing alkyl/arylzinc reagents derived from olefin-tethered alkyl/aryl halides that undergo radical cyclization to generate C(sp3)–Cu complexes in situ, which are then intercepted by aryl and heteroaryl iodides to form the difunctionalized product.
- (5).Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents. Science 2016, 352, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhang L; Lovinger GJ; Edelstein EK; Szymaniak AA; Chierchia MP; Morken JP Catalytic conjunctive cross-coupling enabled by metal-induced metallate rearrangement. Science 2016, 351 (6268), 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jana R; Pathak TP; Sigman MS Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners. Chem. Rev 2011, 111 (3), 1417–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Beletskaya IP; Cheprakov AV The Heck Reaction as a Sharpening Stone of Palladium Catalysis. Chem. Rev 2000, 100 (8), 3009–3066. [DOI] [PubMed] [Google Scholar]
- (9).Catellani M; Paolo Chiusoli G One-pot palladium-catalyzed synthesis of 2,3-disubstituted bicyclo[2.2.1]heptanes and bicyclo[2.2.1]hept-5-enes. Tetrahedron Lett 1982, 23 (43), 4517–4520. [Google Scholar]
- (10).Kadam AA; Metz TL; Qian Y; Stanley LM Ni-Catalyzed Three-Component Alkene Carboacylation Initiated by Amide C-N Bond Activation. ACS Catal 2019, 9 (6), 5651–5656. [Google Scholar]
- (11).Mizutani K; Shinokubo H; Oshima K Cobalt-Catalyzed Three-Component Coupling Reaction of Alkyl Halides, 1,3-Dienes, and Trimethylsilylmethylmagnesium Chloride. Org. Lett 2003, 5 (21), 3959–3961. [DOI] [PubMed] [Google Scholar]
- (12).Terao J; Nii S; Chowdhury FA; Nakamura A; Kambe N Nickel-Catalyzed Regioselective Three Component Coupling Reaction of Alkyl Halides, Butadienes, and Ar-M (M = MgX, ZnX). Adv. Synth. Catal 2004, 346 (8), 905–908. [Google Scholar]
- (13).Liao L; Jana R; Urkalan KB; Sigman MS A Palladium-Catalyzed Three-Component Cross-Coupling of Conjugated Dienes or Terminal Alkenes with Vinyl Triflates and Boronic Acids. J. Am. Chem. Soc 2011, 133 (15), 5784–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Grigg R; Sansano J; Santhakumar V; Sridharan V; Thangavelanthum R; Thornton-Pett M; Wilson D Palladium catalysed tandem cyclisation-anion capture processes. Part 3. Organoboron anion transfer agents. Tetrahedron 1997, 53 (34), 11803–11826. [Google Scholar]
- (15).Zhou L; Li S; Xu B; Ji D; Wu L; Liu Y; Zhang Z-M; Zhang J Enantioselective Difunctionalization of Alkenes by a Palladium-Catalyzed Heck/Sonogashira Sequence. Angew. Chem., Int. Ed 2020, 59 (7), 2769–2775. [DOI] [PubMed] [Google Scholar]
- (16).Dhungana RK; Kc S; Basnet P; Giri R Transition Metal-Catalyzed Dicarbofunctionalization of Unactivated Olefins. Chem. Rec 2018, 18 (9), 1314–1340. [DOI] [PubMed] [Google Scholar]
- (17).Giri R; Kc S Strategies toward Dicarbofunctionalization of Unactivated Olefins by Combined Heck Carbometalation and Cross-Coupling. J. Org. Chem 2018, 83 (6), 3013–3022. [DOI] [PubMed] [Google Scholar]
- (18).Derosa J; Apolinar O; Kang T; Tran VT; Engle KM Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes. Chem. Sci 2020, 11 (17), 4287–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Badir SO; Molander GA Developments in Photoredox/Nickel Dual-Catalyzed 1,2-Difunctionalizations. Chem 2020, 6 (6), 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Dhungana RK; Sapkota RR; Niroula D; Giri R Walking metals: catalytic difunctionalization of alkenes at nonclassical sites. Chem. Sci 2020, 11 (36), 9757–9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Diccianni J; Lin Q; Diao T Mechanisms of Nickel-Catalyzed Coupling Reactions and Applications in Alkene Functionalization. Acc. Chem. Res 2020, 53 (4), 906–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Li Y; Wu D; Cheng H-G; Yin G Difunctionalization of Alkenes Involving Metal Migration. Angew. Chem., Int. Ed 2020, 59 (21), 7990–8003. [DOI] [PubMed] [Google Scholar]
- (23).Luo Y-C; Xu C; Zhang X Nickel-Catalyzed Dicarbofunctionalization of Alkenes†. Chin. J. Chem 2020, 38 (11), 1371–1394. [Google Scholar]
- (24).Tu H-Y; Zhu S; Qing F-L; Chu L Recent Advances in Nickel-Catalyzed Three-Component Difunctionalization of Unactivated Alkenes. Synthesis 2020, 52 (09), 1346–1356. [Google Scholar]
- (25).García-Domínguez A; Li Z; Nevado C Nickel-Catalyzed Reductive Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 2017, 139 (20), 6835–6838. [DOI] [PubMed] [Google Scholar]
- (26).Kuang Y; Wang X; Anthony D; Diao T Ni-catalyzed two-component reductive dicarbofunctionalization of alkenes via radical cyclization. Chem. Commun 2018, 54 (20), 2558–2561. [DOI] [PubMed] [Google Scholar]
- (27).Koy M; Bellotti P; Katzenburg F; Daniliuc CG; Glorius F Synthesis of All-Carbon Quaternary Centers by Palladium-Catalyzed Olefin Dicarbofunctionalization. Angew. Chem., Int. Ed 2020, 59 (6), 2375–2379. [DOI] [PubMed] [Google Scholar]
- (28).Ishiyama T; Miyaura N; Suzuki A Palladium-catalyzed carbonylative cross-coupling reaction of iodoalkanes with 9-alkyl-9-BBN derivatives. A direct and selective synthesis of ketones. Tetrahedron Lett 1991, 32 (47), 6923–6926. [Google Scholar]
- (29).McMahon CM; Renn MS; Alexanian EJ Manganese-Catalyzed Carboacylations of Alkenes with Alkyl Iodides. Org. Lett 2016, 18 (16), 4148–4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Campbell MW; Compton JS; Kelly CB; Molander GA Three-Component Olefin Dicarbofunctionalization Enabled by Nickel/Photoredox Dual Catalysis. J. Am. Chem. Soc 2019, 141 (51), 20069–20078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Klauck FJR; Yoon H; James MJ; Lautens M; Glorius F Visible-Light-Mediated Deaminative Three-Component Dicarbofunctionalization of Styrenes with Benzylic Radicals. ACS Catal 2019, 9 (1), 236–241. [Google Scholar]
- (32).Guo L; Yuan M; Zhang Y; Wang F; Zhu S; Gutierrez O; Chu L General Method for Enantioselective Three-Component Carboarylation of Alkenes Enabled by Visible-Light Dual Photoredox/Nickel Catalysis. J. Am. Chem. Soc 2020, 142 (48), 20390–20399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sun S-Z; Duan Y; Mega RS; Somerville RJ; Martin R Site-Selective 1,2-Dicarbofunctionalization of Vinyl Boronates through Dual Catalysis. Angew. Chem., Int. Ed 2020, 59 (11), 4370–4374. [DOI] [PubMed] [Google Scholar]
- (34).García-Domínguez A; Mondal R; Nevado C Dual Photoredox/Nickel-Catalyzed Three-Component Carbofunctionalization of Alkenes. Angew. Chem., Int. Ed 2019, 58 (35), 12286–12290. [DOI] [PubMed] [Google Scholar]
- (35).McDermott JX; White JF; Whitesides GM Thermal decomposition of bis(phosphine)platinum(II) metallocycles. J. Am. Chem. Soc 1976, 98 (21), 6521–6528. [Google Scholar]
- (36).Neufeldt SR; Sanford MS Asymmetric Chiral Ligand-Directed Alkene Dioxygenation. Org. Lett 2013, 15 (1), 46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Talbot EPA; Fernandes T. d. A.; McKenna JM; Toste FD Asymmetric Palladium-Catalyzed Directed Intermolecular Fluoroarylation of Styrenes. J. Am. Chem. Soc 2014, 136 (11), 4101–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Trejos A; Fardost A; Yahiaoui S; Larhed M Palladium(ii)-catalyzed coupling reactions with a chelating vinyl ether and arylboronic acids: a new Heck/Suzuki domino diarylation reaction. Chem. Commun 2009, No. 48, 7587–7589. [DOI] [PubMed] [Google Scholar]
- (39).Gu J-W; Min Q-Q; Yu L-C; Zhang X Tandem Difluoroalkylation-Arylation of Enamides Catalyzed by Nickel. Angew. Chem., Int. Ed 2016, 55 (40), 12270–12274. [DOI] [PubMed] [Google Scholar]
- (40).Xu C; Yang Z-F; An L; Zhang X Nickel-Catalyzed Difluoroalkylation-Alkylation of Enamides. ACS Catal 2019, 9 (9), 8224–8229. [Google Scholar]
- (41).Derosa J; Tran VT; Boulous MN; Chen JS; Engle KM Nickel-Catalyzed β,γ-Dicarbofunctionalization of Alkenyl Carbonyl Compounds via Conjunctive Cross-Coupling. J. Am. Chem. Soc 2017, 139 (31), 10657–10660. [DOI] [PubMed] [Google Scholar]
- (42).Yang T; Chen X; Rao W; Koh MJ Broadly Applicable Directed Catalytic Reductive Difunctionalization of Alkenyl Carbonyl Compounds. Chem 2020, 6 (3), 738–751. [Google Scholar]
- (43).Li W; Boon JK; Zhao Y Nickel-catalyzed difunctionalization of allyl moieties using organoboronic acids and halides with divergent regioselectivities. Chem. Sci 2018, 9 (3), 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Derosa J; van der Puyl VA; Tran VT; Liu M; Engle KM Directed nickel-catalyzed 1,2-dialkylation of alkenyl carbonyl compounds. Chem. Sci 2018, 9 (23), 5278–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Chierchia M; Xu P; Lovinger GJ; Morken JP Enantioselective Radical Addition/Cross-Coupling of Organozinc Reagents, Alkyl Iodides, and Alkenyl Boron Reagents. Angew. Chem., Int. Ed 2019, 58 (40), 14245–14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Dhungana RK; Sapkota RR; Wickham LM; Niroula D; Giri R Ni-Catalyzed Regioselective 1,2-Dialkylation of Alkenes Enabled by the Formation of Two C(sp3)-C(sp3) Bonds. J. Am. Chem. Soc 2020, 142 (50), 20930–20936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Yang T; Jiang Y; Luo Y; Lim JJH; Lan Y; Koh MJ Chemoselective Union of Olefins, Organohalides, and Redox-Active Esters Enables Regioselective Alkene Dialkylation. J. Am. Chem. Soc 2020, 142 (51), 21410–21419. [DOI] [PubMed] [Google Scholar]
- (48).Thapa S; Dhungana RK; Magar RT; Shrestha B; Kc S; Giri R Ni-catalysed regioselective 1,2-diarylation of unactivated olefins by stabilizing Heck intermediates as pyridylsilyl-coordinated transient metallacycles. Chem. Sci 2018, 9 (4), 904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Overman LE; Poon DJ Asymmetric Heck Reactions via Neutral Intermediates: Enhanced Enantioselectivity with Halide Additives Gives Mechanistic Insights. Angew. Chem., Int. Ed. Engl 1997, 36 (5), 518–521. [Google Scholar]
- (50).Chen L; Sanchez DR; Zhang B; Carrow BP Cationic” Suzuki-Miyaura Coupling with Acutely Base-Sensitive Boronic Acids. J. Am. Chem. Soc 2017, 139 (36), 12418–12421. [DOI] [PubMed] [Google Scholar]
- (51).Basnet P; Kc S; Dhungana RK; Shrestha B; Boyle TJ; Giri R Synergistic Bimetallic Ni/Ag and Ni/Cu Catalysis for Regioselective γ,δ-Diarylation of Alkenyl Ketimines: Addressing β-H Elimination by in Situ Generation of Cationic Ni(II) Catalysts. J. Am. Chem. Soc 2018, 140 (46), 15586–15590. [DOI] [PubMed] [Google Scholar]
- (52).Sommer H; Juliá-Hernández F; Martin R; Marek I Walking Metals for Remote Functionalization. ACS Cent. Sci 2018, 4 (2), 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Jeon J; Ryu H; Lee C; Cho D; Baik M-H; Hong S Site-Selective 1,1-Difunctionalization of Unactivated Alkenes Enabled by Cationic Palladium Catalysis. J. Am. Chem. Soc 2019, 141 (25), 10048–10059. [DOI] [PubMed] [Google Scholar]
- (54).Li Y; Wei H; Wu D; Li Z; Wang W; Yin G Nickel-Catalyzed Chemodivergent 1,1-Difunctionalization of Unactivated α-Olefins with Alkynyl Electrophiles and B2pin2. ACS Catal 2020, 10 (9), 4888–4894. [Google Scholar]
- (55).Li Z; Wu D; Ding C; Yin G Modular Synthesis of Diarylalkanes by Nickel-Catalyzed 1,1-Diarylation of Unactivated Terminal Alkenes. CCS Chem 2021, 3 (1), 576–582. [Google Scholar]
- (56).Dhungana RK; Kc S; Basnet P; Aryal V; Chesley LJ; Giri R Ni(I)-Catalyzed β,δ-Vinylarylation of γ,δ-Alkenyl α-Cyanocarboxylic Esters via Contraction of Transient Nickellacycles. ACS Catal 2019, 9 (12), 10887–10893. [PMC free article] [PubMed] [Google Scholar]
- (57).Larock RC; Lu YD; Bain AC; Russell CE Palladium-catalyzed coupling of aryl iodides, nonconjugated dienes and carbon nucleophiles by palladium migration. J. Org. Chem 1991, 56 (15), 4589–4590. [Google Scholar]
- (58).Shu W; Merino E; Nevado C Visible Light Mediated, Redox Neutral Remote 1,6-Difunctionalizations of Alkenes. ACS Catal 2018, 8 (7), 6401–6406. [Google Scholar]
- (59).Huang H-M; Bellotti P; Pflüger PM; Schwarz JL; Heidrich B; Glorius F Three-Component, Interrupted Radical Heck/Allylic Substitution Cascade Involving Unactivated Alkyl Bromides. J. Am. Chem. Soc 2020, 142 (22), 10173–10183. [DOI] [PubMed] [Google Scholar]
- (60).Yamane S; Hinoue T; Usuki Y; Itazaki M; Nakazawa H; Hayashi Y; Kawauchi S; Miura M; Satoh T Iridium-Catalyzed Aerobic Coupling of Salicylaldehydes with Alkynes: A Remarkable Switch of Oxacyclic Product. Chem. - Eur. J 2018, 24 (31), 7852–7855. [DOI] [PubMed] [Google Scholar]
- (61).McLain SJ; Sancho J; Schrock RR Metallacyclopentane to metallacyclobutane ring contraction. J. Am. Chem. Soc 1979, 101 (18), 5451–5453. [Google Scholar]
- (62).Takai K; Matsukawa N; Takahashi A; Fujii T Three-Component Coupling Reactions of Alkyl Iodides, 1,3-Dienes, and Carbonyl Compounds by Sequential Generation of Radical and Anionic Species with CrCl2. Angew. Chem., Int. Ed 1998, 37 (1–2), 152–155. [Google Scholar]
- (63).Kuang Z; Yang K; Song Q Pd-Catalyzed Regioselective 1,2-Difunctionalization of Vinylarenes with Alkenyl Triflates and Aryl Boronic Acids at Ambient Temperature. Org. Lett 2017, 19 (10), 2702–2705. [DOI] [PubMed] [Google Scholar]
- (64).Terao J; Saito K; Nii S; Kambe N; Sonoda N Regioselective Double Alkylation of Styrenes with Alkyl Halides Using a Titanocene Catalyst. J. Am. Chem. Soc 1998, 120 (45), 11822–11823. [Google Scholar]
- (65).Gao P; Chen L-A; Brown MK Nickel-Catalyzed Stereoselective Diarylation of Alkenylarenes. J. Am. Chem. Soc 2018, 140 (34), 10653–10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Kc S; Dhungana RK; Khanal N; Giri R Nickel-Catalyzed α-Carbonylalkylarylation of Vinylarenes: Expedient Access to γ,γ-Diarylcarbonyl and Aryltetralone Derivatives. Angew. Chem., Int. Ed 2020, 59 (21), 8047–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Tour JM; Negishi E Metal promoted cyclization. 9. Controlled and catalytic acylpalladation. A novel route to cyclo-pentenone and cyclohexenone derivatives. J. Am. Chem. Soc 1985, 107 (26), 8289–8291. [Google Scholar]
- (68).Dhungana RK; Shrestha B; Thapa-Magar R; Basnet P; Giri R Pd-Catalyzed Regioselective 1,2-Dicarbofunctionalization of Unactivated Olefins by a Heck Reaction/Enolate Cyclization Cascade. Org. Lett 2017, 19 (8), 2154–2157. [DOI] [PubMed] [Google Scholar]
- (69).Fournet G; Balme G; Gore J Intramolecular nucleophilic attack of organopalladium compounds resulting from carbopalladation of alkylidenecyclopropanes. Tetrahedron Lett 1987, 28 (39), 4533–6. [Google Scholar]
- (70).Huang D; Olivieri D; Sun Y; Zhang P; Newhouse TR Nickel-Catalyzed Difunctionalization of Unactivated Alkenes Initiated by Unstabilized Enolates. J. Am. Chem. Soc 2019, 141 (41), 16249–16254. [DOI] [PubMed] [Google Scholar]
- (71).White DR; Hinds EM; Bornowski EC; Wolfe JP Pd-Catalyzed Alkene Difunctionalization Reactions of Malonate Nucleophiles: Synthesis of Substituted Cyclopentanes via Alkene Aryl-Alkylation and Akenyl-Alkylation. Org. Lett 2019, 21 (10), 3813–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Sole D; Cancho Y; Llebaria A; Moreto JM; Delgado A Intramolecular Nitrogen Assistance in the Nickel-Promoted Tandem Cyclization-Capture of Amino-Tethered Vinyl Bromides and Alkenes. J. Am. Chem. Soc 1994, 116 (26), 12133–12134. [Google Scholar]
- (73).Wakabayashi K; Yorimitsu H; Oshima K Cobalt-Catalyzed Tandem Radical Cyclization and Cross-Coupling Reaction: Its Application to Benzyl-Substituted Heterocycles. J. Am. Chem. Soc 2001, 123 (22), 5374–5375. [DOI] [PubMed] [Google Scholar]
- (74).Phapale VB; Buñuel E; García-Iglesias M; Cárdenas DJ Ni-Catalyzed Cascade Formation of C(sp3)-C(sp3) Bonds by Cyclization and Cross-Coupling Reactions of Iodoalkanes with Alkyl Zinc Halides. Angew. Chem., Int. Ed 2007, 46 (46), 8790–8795. [DOI] [PubMed] [Google Scholar]
- (75).Cong H; Fu GC Catalytic Enantioselective Cyclization/Cross-Coupling with Alkyl Electrophiles. J. Am. Chem. Soc 2014, 136 (10), 3788–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).You W; Brown MK Diarylation of Alkenes by a Cu-Catalyzed Migratory Insertion/Cross-Coupling Cascade. J. Am. Chem. Soc 2014, 136 (42), 14730–14733. [DOI] [PubMed] [Google Scholar]
- (77).Kc S; Basnet P; Thapa S; Shrestha B; Giri R Ni-Catalyzed Regioselective Dicarbofunctionalization of Unactivated Olefins by Tandem Cyclization/Cross-Coupling and Application to the Concise Synthesis of Lignan Natural Products. J. Org. Chem 2018, 83 (5), 2920–2936. [DOI] [PubMed] [Google Scholar]
- (78).Fang X; Hu X Advances in the Synthesis of Lignan Natural Products. Molecules 2018, 23 (12), 3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Nicolaou KC; Bulger PG; Sarlah D Palladium-Catalyzed Cross-Coupling Reactions in Total Synthesis. Angew. Chem., Int. Ed 2005, 44 (29), 4442–4489. [DOI] [PubMed] [Google Scholar]
- (80).Balme G; Bouyssi D Total synthesis of the triquinane marine sesquiterpene (±)Δ9(12) capnellene using a palladium-catalyzed biscyclization step. Tetrahedron 1994, 50 (2), 403–414. [Google Scholar]
- (81).Kc S; Dhungana RK; Aryal V; Giri R Concise Synthesis of a Potential 5-Lipoxygenase Activating Protein (FLAP) Inhibitor and Its Analogs through Late-Stage Alkene Dicarbofunctionalization. Org. Process Res. Dev 2019, 23 (8), 1686–1694. [Google Scholar]
- (82).Chu L; Armstrong HM; Chang LL; Cheng AF; Colwell L; Cui J; Evans J; Galka A; Goulet MT; Hayes N; Lo J; Menke J; Ok HO; Ondeyka DL; Patel M; Quaker GM; Sings H; Witkin SL; Zhao A; Ujjainwalla F Evaluation of endo- and exo-aryl-substitutions and central scaffold modifications on diphenyl substituted alkanes as 5-lipoxygenase activating protein inhibitors. Bioorg. Med. Chem. Lett 2012, 22 (12), 4133–4138. [DOI] [PubMed] [Google Scholar]
- (83).Sanchez LA; Olmedo D; Lopez-Perez JL; Williams TD; Gupta MP Two new alkylresorcinols from Homalomena wendlandii and their cytotoxic activity. Nat. Prod. Commun 2012, 7 (8), 1043–1046. [PubMed] [Google Scholar]
- (84).Yonova IM; Johnson AG; Osborne CA; Moore CE; Morrissette NS; Jarvo ER Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Alkyl Grignard Reagents and Identification of Selective Anti-Breast-Cancer Agents. Angew. Chem., Int. Ed 2014, 53 (9), 2422–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Quallich GJ Development of the commercial process for Zoloft®/sertraline. Chirality 2005, 17 (S1), S120–S126. [DOI] [PubMed] [Google Scholar]
- (86).Niroula D; Sapkota RR; Dhungana RK; Shrestha B; Giri R An Expedient Route to 9-Arylmethylanthracene Derivatives via Tandem Ni-Catalyzed Alkene Dicarbofunctionalization and Acid-Promoted Cyclization-Aromatization. Isr. J. Chem 2020, 60 (3–4), 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]