

Abstract
Clarithromycin is an improved semi-synthetic analogue of the naturally occurring macrolide, erythromycin. The subtle modification of a methyl group on the C-6 hydroxyl group endows the molecule with improved acid stability and results in a clinically useful antibiotic. Here we show that the effector specificity of the biosensor protein, MphR, can be evolved to selectively recognize clarithromycin, and therefore report on the production of this molecule in vivo. In addition, a crystal structure of the evolved variant illustrates the molecular basis for selectivity and provides a guide for the evolution of new metabolic function using this biosensor.
Keywords: macrolide, polyketide, biosensor, transcription factor, synthetic biology
Graphical Abstract
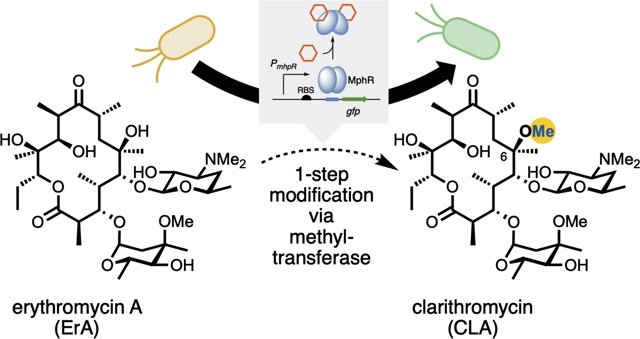
INTRODUCTION
Macrolides are a large class of natural products that display potent biological activities that span many therapeutic areas.1 The core structures of macrolides are biosynthesized by large, modular enzymes known as polyketide synthases (PKSs).2,3 The prototypical macrolide antibiotic, erythromycin A (ErA) while clinically useful, has limitations of acid instability and limited bioavailability.4 Clarithromycin (CLA), a second-generation macrolide antibiotic derived from ErA (Figure 1A), effectively addresses these limitations, while also expanding the scope of activity to other microorganisms not impacted by ErA.4–12 Moreover, CLA has served as a platform for the synthesis and derivatization of ketolide antibiotics including telithromycin and solithromycin (Figure 1B).13–18 CLA and ErA differ at the C6-position of the macrolactone core: the C6-hydroxyl group in ErA is replaced with a methoxy group in CLA. Currently, CLA is produced through semi-synthesis in a six-step sequence from ErA, including protection, methylation and deprotection.19 Despite the fact that ErA is produced by fermentation on massive scale (4,000 metric tons annually),20 semi-synthesis is costly and produces a substantial amount of chemical waste products.21,22 Moreover, given that the essential C6 O-methylation is also a key functional group of many later generation macrolide antibiotics, strategies to improve the efficiency and yield of this modification are highly sought after.
Figure 1.
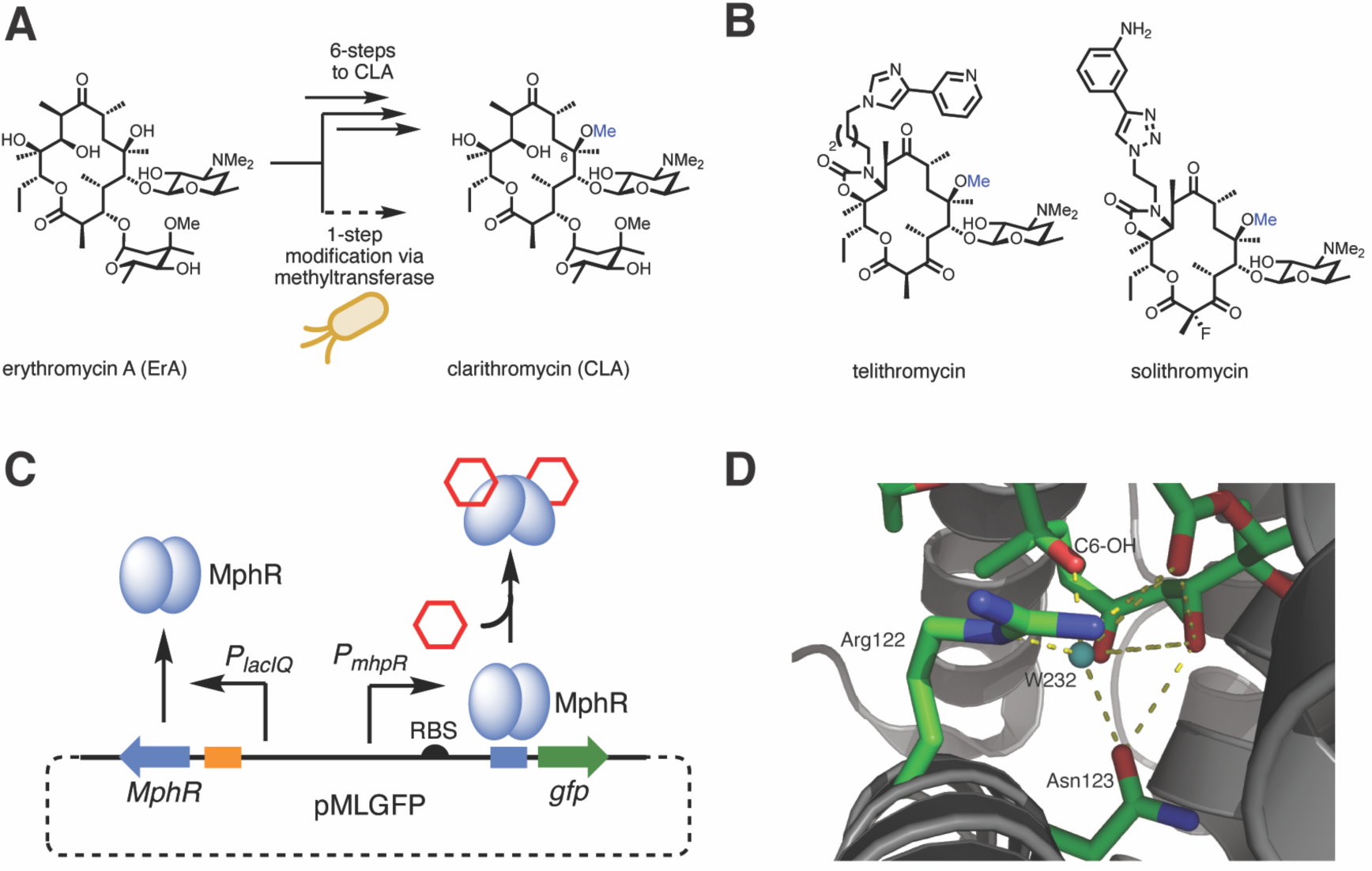
Erythromycin derivatives as antibiotics and their detection by the macrolide sensing transcription factor MphR. (A) Chemical conversion of ErA to CLA. In principle, this could be carried out in a single step with a suitable MT. (B) Structures of semi-synthetic antibiotics derived from CLA. (C) Scheme illustrating the MphR macrolide biosensor. In the presence of a macrolide effector (hexagon) and MphA, MphR undergoes a conformational change and leaves the operator sequence, allowing transcription of the downstream reporter, gfp. For clarity, phosphorylation of ErA via MphA (housed in pJZ12) is omitted from the figure. (D) Ligand binding site of wild-type MphR (monomer A, PDB: 3FRQ) with two key residues and a water molecule (teal sphere) surrounding the C6-OH of ErA shown (green sticks) and labeled.
A potential route to C6-methylated macrolides involves the regiospecific methylation of ErA (Figure 1A), which coupled with ErA biosynthetic machinery23,24 could furnish an engineered microbial strain programmed for the manufacture of CLA or related derivatives. Yet, methyltransferases (MTs) with the desired specificity are not currently known. It is not yet practical to search the massive pool of MTs and to characterize their substrate specificities via traditional low throughput or computational approaches. Similarly, while directed evolution25,26 could address our limited ability to rationally reprogram MT specificity, high-throughput tools are not available to identify improved MT variants while searching through large mutant libraries. Screens or selections for many polyketide biosynthesis steps are not available or are not adaptable to in vivo experiments. This evaluation bottleneck could be eliminated by leveraging a genetically-encoded biosensor that reports the conversion of ErA to CLA in vivo. Biosensors based on transcription factors have been developed for a wide range of small molecules including phenylpropanoids, steroids, and lactams,27 but none are currently available for the selective detection of CLA.
The repressor protein MphR controls the deactivation of ErA by regulating the transcription of the ErA resistance gene mphA, encoding a macrolide 2’-phosphotransferase.28 The protein, a member of the TetR class of repressor proteins, binds to a DNA operator but in the presence of a small molecule effector ligand, releases DNA allowing for transcription to take place. MphR is de-repressed by other macrolides, including CLA, oleandomycin, azithromycin, roxithromycin, and others.29–31 The MphR gene cassette has been redesigned to control a green fluorescence protein (GFP) reporter in E. coli in response to macrolides (Figure 1C).29,32 The sensitivity and specificity of MphR have been tuned by directed evolution.29 Accordingly, it was hypothesized that given the ability of MphR to be efficiently de-repressed by both ErA and CLA, that this inherent promiscuity could be leveraged by directed evolution to provide a MphR variant with specificity towards CLA. A variant biosensor system that is de-repressed in the presence of CLA but not ErA (or vice versa) provides a potential platform for the discovery and directed evolution of a MT for the biocatalytic synthesis of C6-methylated macrolides.
Herein, a genetically-encoded biosensor capable of reporting the conversion of ErA to CLA was engineered. Inspired by rare examples33–35 that engineer transcription factor ligand specificity by combining rational design and directed evolution strategies, a structure-guided mutagenesis followed by random mutagenesis and gene circuit engineering was developed to engineer the MphR-based sensor. This led to the identification of a quadruple MphR mutant dubbed “M9C4” that can discriminate between ErA and CLA. Extensive mutagenesis and structural studies enabled the contribution of each mutation to the specificity of M9C4 to be elucidated, suggesting a rationale for the emergence of exquisite effector selectivity.
RESULTS
Structure-guided mutagenesis of MphR to provide a CLA specific biosensor
Previously, it has been demonstrated that wild-type MphR does not discriminate between ErA and CLA.29 The structure of MphR bound with ErA reveals that Arg122 and Asn123 are involved in hydrogen bonding interactions with water molecules within the effector binding pocket and with the ErA C6-OH (Figure 1D).28 It was envisioned that disruption of this H-bonding network by mutagenesis at Arg122 or Asn123 may negatively impact the ability of MphR to bind ErA and concomitantly provide more space for the C6-O-methyl group of CLA. In addition, the sequence of amino acids from Arg133 to Ala140 are part of a turn that connects the two helices that accommodates the macrolide (Figure 1D). Inserting an amino acid into this turn might create flexibility and create space for the additional C6-O-methyl of CLA. To test these hypotheses, saturation mutagenesis of Arg122 and Asn123 was carried out, resulting in 19 possible variants at each residue, and the mutant Gly137insAla was constructed. Unique mutants from the Arg122X and Asn123X libraries were identified by DNA sequencing. The corresponding biosensor strains, consisting of the plasmids pMLGFP and pJZ12 (Figure 1C), were screened at a fixed concentration of ErA and CLA to determine their specificity, along with the Gly137insAla mutant (Supplementary Figure S1). Of the mutations tested, only R122V and R122T displayed a potential change in effector specificity compared to the wild-type MphR biosensor, supporting 2.7- and 2.8-fold increased reporter fluorescence, respectively, with CLA compared to ErA. A possible explanation for this specificity is that substitution of Arg122 with valine or threonine accommodates the increased bulk of the C6-O-methyl group of CLA and disturbs the hydrogen bonding between MphR and the ErA C6-OH. Notably, every substitution at Arg122 tested drastically decreased the sensitivity of MphR with ErA compared to the wild-type, possibly due to a critical salt bridge between Arg122 and Glu109 (Supplementary Figure S2). Interestingly, substitutions at Asn123 were better tolerated, as judged by the fluorescence response of the biosensor strains with ErA and CLA. However, none of the Asn123X library members displayed an improvement in specificity towards CLA, compared to the wild-type biosensor strain.
To further characterize the R122V and R122T biosensor strains, the GFP fluorescence of each mutant in the presence of various concentrations of CLA and ErA was determined. The biosensor performance characteristics were extracted by fitting the dose response data to the Hill equation (Figure 2A–C and Table 1, entries 1–3). The dose-response curves of the two mutants R122V and R122T were very different from that of the wild-type biosensor strain. In fact, for each mutant, the normalized fluorescence is higher with CLA than with ErA at every effector concentration tested, indicating significant changes in effector specificity. Compared to wild-type, the sensitivity (K1/2) and dynamic range of the two mutants towards each macrolide were poorer, perhaps due to disturbance of the salt bridge between Arg122 and Glu109. Additionally, reduced sensitivity towards ErA might be due to disruption of a hydrogen bonding network between the ErA C6-OH, a water molecule, Arg122, and Asn123 (Figure 1D). Nevertheless, the similarity of Hill coefficients between mutants and wild-type indicates that the degree of positive cooperativity has not been affected by the mutations. Notably, the R122T and R122V mutations shift selectivity toward CLA ~2- and ~4-fold, respectively, compared to the wild-type biosensor strain, as judged by the ratio of K1/2’s for each effector. Although these mutants are a good first step towards a CLA-specific biosensor, their relatively small dynamic range and poor selectivity needed to be further addressed.
Figure 2.
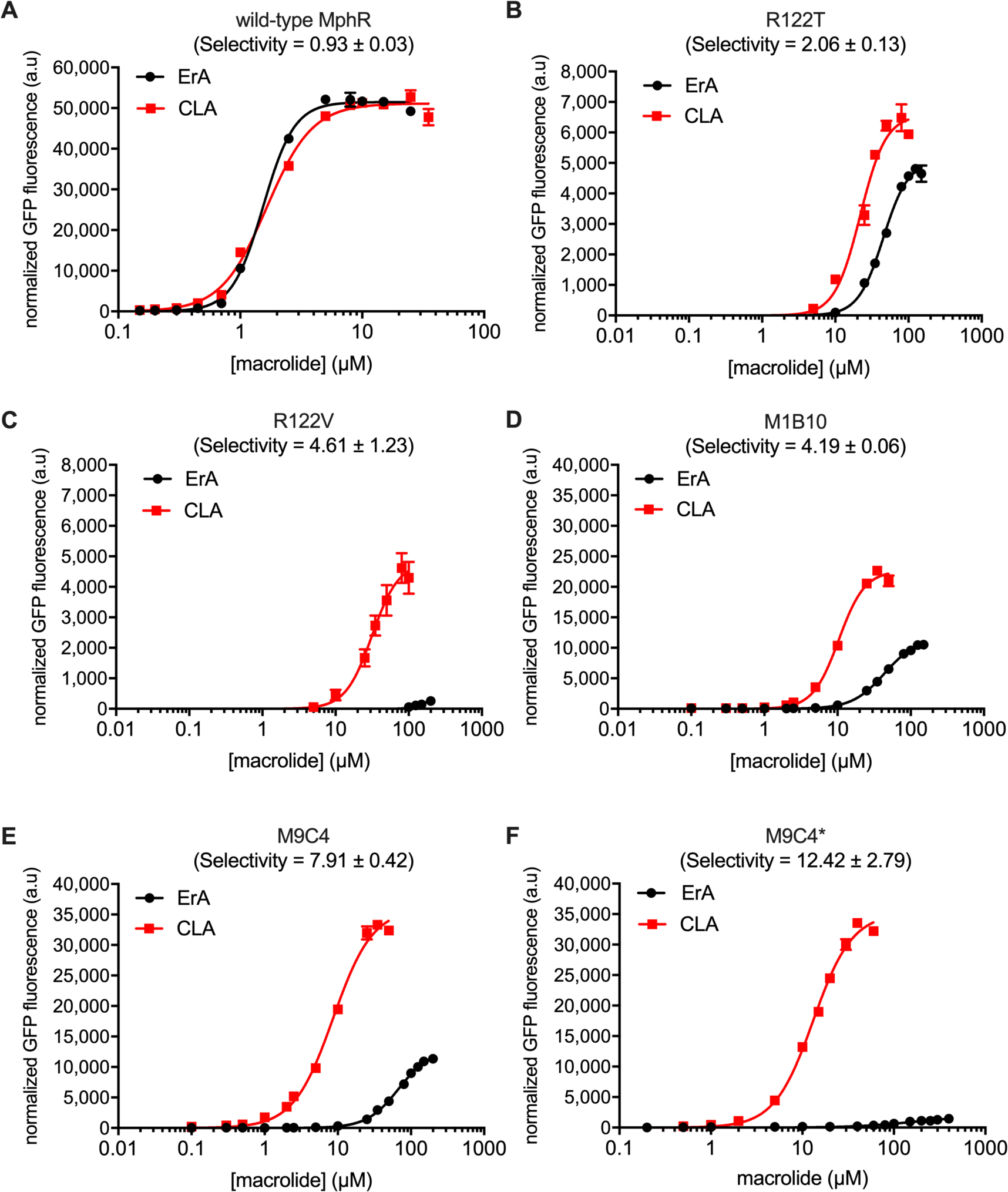
Characterization of MphR biosensor variants with engineered ErA/CLA specificity. (A) Wild-type MphR dose-response curves. (B) MphR variant R122T dose-response curves. (C) MphR variant R122V dose-response curves. (D) MphR variant M1B10 dose-response curves. (E) MphR variant M9C4 dose-response curves. (F) MphR variant M9C4* dose-response curves. Error-bars represent the standard error of the mean (n = 3) and are only visible when larger than the data point symbol.
Table 1.
Performance features of wild-type and mutant MphR biosensor strains.
| entry | biosensor | mutationsa | effector | K1/2 (μM)b | dynamic rangec | n d | selectivitye |
|---|---|---|---|---|---|---|---|
| 1 | wild-typef | - | ErA | 1.54 ± 0.03 | 51,491 | 3.39 ± 0.13 | |
| CLA | 1.65 ± 0.05 | 51,143 | 2.31 ± 0.17 | 0.93 ± 0.03 | |||
| 2 | R122Tf | R122T | ErA | 45.22 ± 1.42 | 5,078 | 2.54 ± 0.17 | |
| CLA | 21.96 ± 1.15 | 6,574 | 2.48 ± 0.28 | 2.06 ± 0.13 | |||
| 3 | R122Vf | R122V | ErA | 145.40 ± 37.82 | 300 | 5.24 ± 4.10 | |
| CLA | 31.56 ± 1.95 | 4,807 | 2.39 ± 0.34 | 4.61 ± 1.23 | |||
| 4 | M9C4f | R122T, K132N, A151T, | ErA | 68.32 ± 1.65 | 12,892 | 2.01 ± 0.06 | |
| H184Q | CLA | 8.64 ± 0.41 | 36,012 | 1.61 ± 0.08 | 7.91 ± 0.42 | ||
| 5 | M1B10g | T49I, L89V, D98N, | ErA | 43.62 ± 0.85 | 11,559 | 1.98 ± 0.06 | |
| E109D | CLA | 10.41 ± 0.33 | 22,643 | 2.45 ± 0.16 | 4.19 ± 0.16 | ||
| 6 | R122T/K132Ng | R122T, K132N | ErA | 25.52 ± 1.01 | 12,007 | 2.18 ± 0.16 | |
| CLA | 14.40 ± 0.44 | 13,923 | 2.15 ± 0.14 | 1.77 ± 0.09 | |||
| 7 | R122T/A151Tg | R122T, A151T | ErA | 71.77 ± 3.43 | 4,126 | 2.61 ± 0.16 | |
| CLA | 12.21 ± 0.48 | 21,985 | 2.18 ± 0.17 | 5.88 ± 0.36 | |||
| 8 | R122T/K132N/A151Tg | R122T, K132N, A151T | ErA | 45.26 ± 1.69 | 13,506 | 2.27 ± 0.12 | |
| CLA | 8.43 ± 0.50 | 38,308 | 1.64 ± 0.13 | 5.37 ± 0.38 | |||
| 9 | M9C4*g | R122I, K132N, A151T, | ErA | 163.70 ± 36.52 | 1,876 | 1.35 ± 0.18 | |
| H184Q | CLA | 13.18 ± 0.36 | 35,175 | 2.04 ± 0.10 | 12.42 ± 2.79 | ||
| 10 | M9C4-A3g | ErA | 37.77 ± 3.18 | 10,984 | 4.41 ± 0.88 | 7.71 ± 0.59 | |
| R122T, K132N, A151T, H184Q + GGAAGGT |
CLA | 6.62 ± 0.44 | 51,252 | 1.26 ± 0.07 | |||
| 11 | M9C4-E7g | R122T, K132N, A151T, | ErA | 37.58 ± 1.57 | 13,418 | 2.26 ± 0.19 | |
| H184Q + AGATGGT | CLA | 5.21 ± 0.37 | 45,331 | 1.69 ± 0.16 | 7.21 ± 0.59 | ||
| 12 | M9C4-H4g | R122T, K132N, A151T, | ErA | 49.38 ± 0.97 | 12,809 | 2.05 ± 0.08 | |
| H184Q + AGAAGGC | CLA | 8.05 ± 0.42 | 48,381 | 1.46 ± 0.08 | 6.13 ± 0.34 | ||
| 13 | M9C4-E7 (2-plasmid strain w/EryG)h | R122T, K132N, A151T, | ErA | 16.30 ± 0.98 | 70,063 | 1.76 ± 0.13 | |
| H184Q + AGATGGT | CLA | 0.95 ± 0.14 | 97,213 | 1.82 ± 0.48 | 17.16 ± 2.73 | ||
| 14 | M9C4-E7 (1-plasmid strain w/EryG)i | R122T, K132N, A151T, | ErA | 14.03 ± 2.11 | 43,501 | 1.96 ± 0.42 | |
| H184Q + AGATGGT | CLA | 0.88 ± 0.09 | 64,900 | 2.69 ± 0.69 | 15.94 ± 2.90 |
For ribosome binding site mutants, the RBS sequence is shown, mutated nucleotides are underlined
Concentration of ligand at half-maximum normalized GFP fluorescence
GFPmax−GFPmin
Hill coefficient, a measure of cooperativity within the MphR biosensor. Values >1 indicate positive cooperativity.
Ratio of K1/2(CLA)/K1/2(ErA)
Two plasmid system consisting of pMLGFP/pJZ12
Two plasmid system consisting of pMLGFPK/pJZ12
Two plasmid system consisting of pSense-MphR-MphA/pCOLADuet-EryG
One plasmid system consisting of pSense-MphR-MphA-EryG
Directed evolution of MphR mutants
It is difficult to predict which additional residues to target for mutagenesis to further improve the effector selectivity of MphR. Accordingly, a directed evolution approach was explored. Three error-prone PCR mutant libraries of MphR were constructed using wild-type, R122T, and R122V MphR as each template. Flow cytometry was first used to enrich the population of mutants that were induced poorly by ErA by collecting ~5% of each library with the lowest fluorescence (Supplementary Figure S3). Next, those clones were screened in microplates either in the tier screening strategy, two mutants were selected for further analysis: ‘M9C4’ from the error-prone library based on R122T and ‘M1B10’ from the error-prone library based on the wild-type MphR.
Subsequently, ErA/CLA dose-response curves of M9C4 and M1B10 were determined (Figure 2D–E and Table 1, entries 4 and 5, respectively). Notably, the K1/2 of M9C4 with ErA and CLA is increased 45- and 5-fold, respectively, compared to the wild-type, resulting in a variant that is ~8-fold more selective for CLA vs. ErA. Additionally, M9C4 displays a 2.8-fold higher dynamic range with CLA than with ErA. Similarly, the K1/2 of M1B10 with ErA and CLA is increased 29- and 6.3-fold, respectively, compared to the wild-type, resulting in a variant that is ~4-fold more selective for CLA vs. ErA. However, M1B10 displays only a 2-fold higher dynamic range with CLA than with ErA. The DNA sequences of each variant revealed four amino acid changes: T49I, L89V, D98N and E109D for M1B10; and R122T, K132N, A151T and H184Q for M9C4. In addition, the sequence of M9C4 also revealed a non-coding mutation between the mphR promoter and its RBS (Supplementary Table S1). Both R122T and K132N are located in the ligand binding domain of MphR while A151T and H184Q are located in the regulatory core that forms the dimerization interface. Given that the selectivity and dynamic range of M9C4 was superior compared to those of M1B10, M9C4 was chosen as the template for mutational analysis.
Mutational analysis of MphR M9C4
To establish which mutations contributed to the CLA selectivity of M9C4, all possible single (4), double (6), and triple (4), mutants were generated by site directed mutagenesis, in addition to a mutant which only contained the non-coding mutation. Each variant biosensor strain was then assayed at fixed concentrations of ErA and CLA (Figure 3). Notably, the specificity of the non-coding MphR variant was indistinguishable from that of the wild-type (‘NCR’, Figure 3), indicating that the nucleotide mutation in the non-coding region is not responsible for the CLA specificity of M9C4.
Figure 3.
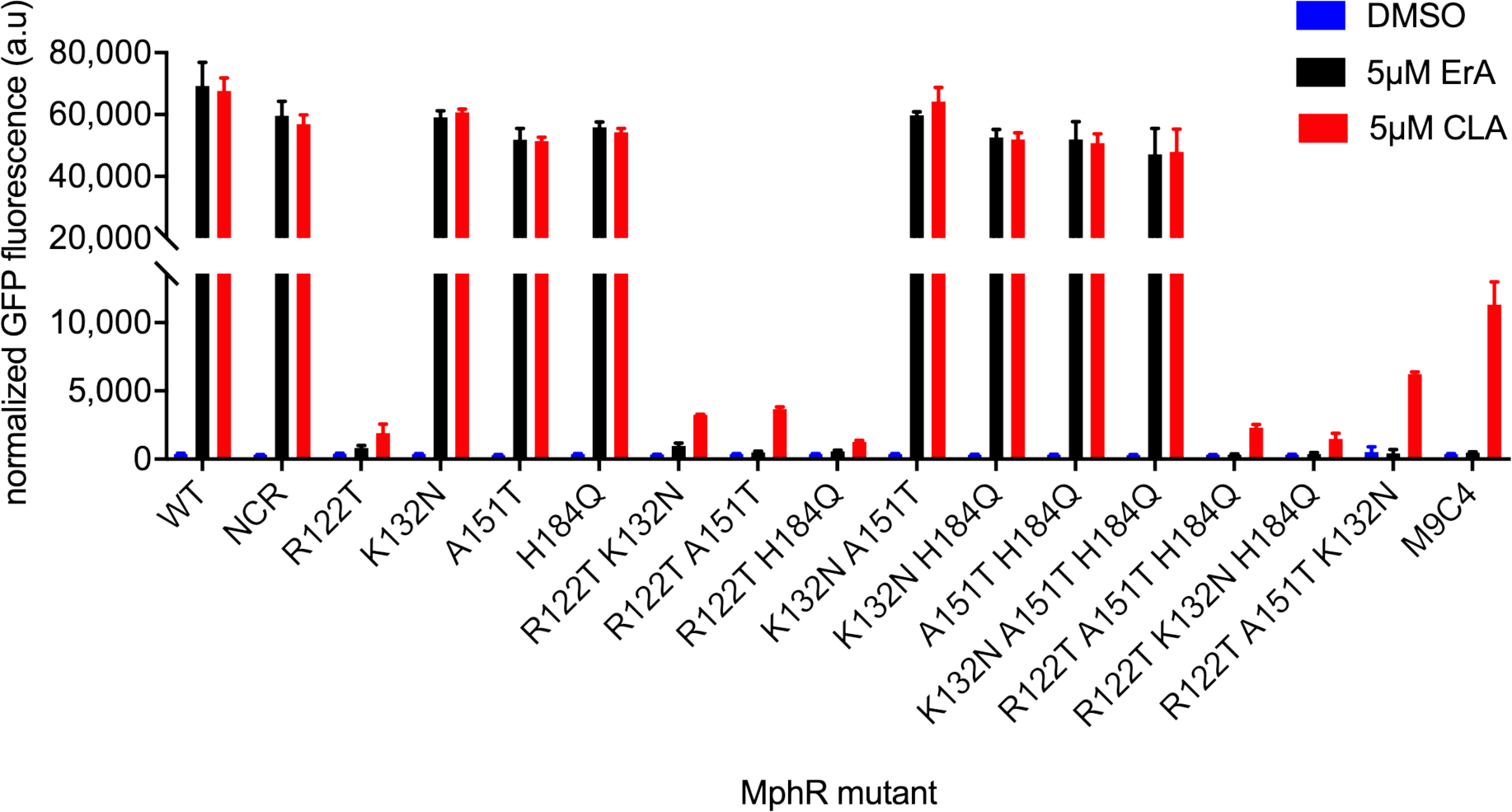
Activities of individual and combined mutations from M9C4 using 5μM ErA and CLA. NCR, non-coding region mutant. Error-bars represent the standard error of the mean (n = 3).
Turning to the amino acid mutation, of the four single mutants, R122T is the only mutation that affects the specificity between CLA and ErA, while K132N, A151T, and H184Q were indistinguishable from the wild-type MphR biosensor. However, the combination of K132N or A151T with R122T (R122T/K132N and R122T/A151T) further shifts specificity towards CLA and suggests that M9C4 may derive most of its selectivity from these three mutations (Figure 3). The combination of R122T and H184Q are indistinguishable in comparison to R122T alone. Furthermore, the double mutants that lack R122T (K132N/A151T, K132N/H18Q, A151T/H184Q) are indistinguishable from wild-type MphR. Together, the single and double mutant data support the importance of the semi-rational R122T mutation in combination with some of those derived from random mutagenesis. Dose-response curves show that R122T in combination with K132N or A151T improve the MphR biosensor properties in different respects (Table 1 and Supplementary Figure S4). For example, in comparison to R122T, the double mutant R122T/K132N appears to improve the dynamic range with CLA ~2-fold. Both the dynamic range with CLA and the CLA/ErA selectivity of the R122T/A151T mutant improves ~3-fold compared to R122T. As anticipated by these results, the combination of R122T, K132N, and A151T into the corresponding triple mutant results in a mutant MphR biosensor strain with the highest dynamic range and selectivity for CLA compared to any of the parent single and double mutants (Figure 3). Interestingly, even though H184Q did not seem to contribute significantly to the CLA selectivity of the double or triple mutants, inclusion in M9C4 does improve selectivity 1.5-fold compared to R122T/K132N/A151T, largely through increasing the K1/2 for ErA (Table 1).
The importance of R122T in the triple mutant, coupled with the incomplete coverage of the original R122X saturation library (Supplementary Figure S1), prompted another substitution in M9C4 to be tested. Given its similarity to threonine (valine was already tested as a single substitution, entry 3, Table 1), isoleucine was introduced at Arg122 in the quadruple mutant M9C4, generating M9C4* (entry 9, Table 1). Compared to M9C4, the dynamic range of this new quadruple amino acid mutant (R122I/K132N/A151T/H184Q) towards ErA decreased ~7-fold while towards CLA was unchanged. As anticipated, selectivity of this new quadruple amino acid mutant was improved 1.5-fold, compared to M9C4 (Figure 2 and Table 1). Although the selectivity of M9C4* was improved on the basis of the K1/2 for each macrolide, the K1/2(CLA) of M9C4* was 1.5-fold higher than the parent M9C4. Subsequently, the sensitivity of M9C4 at lower concentrations of CLA is better than that of M9C4*, with no de-repression by ErA up to ~10 μM ErA. Thus, M9C4 was chosen to be further investigated.
Validation of the simulated conversion of ErA to CLA via MphR M9C4
The low K1/2 of M9C4 for CLA, it’s lack of de-repression by ErA up to 10 μM, and its large dynamic range could enable a sensitive interrogation of the bioconversion of ErA to CLA in living cells. Initially, in the presence of an active MT, the ErA concentration would be high (e.g. at 0% conversion), while after some time, the concentration of ErA would decrease in proportion to the increase in concentration of CLA. Since an enzyme to effect conversion of ErA to clarithromcyin is not yet available, to mimic the bioconversion, different ratios of ErA and CLA were applied as a mixture to cultures of the wild-type and M9C4 biosensor strains and the GFP outputs were determined (Figure 4A). Gratifyingly, the GFP output of the M9C4 biosensor strain is linear across the entire range of macrolide mixtures tested, while the wild-type response was not strongly correlated. Additionally, M9C4 was able to differentiate as low as 10% conversion of ErA to CLA (equivalent to 1 μM or 0.748 μg/mL CLA) as mimicked by these assay conditions (Figure 4B). This confirms that the M9C4 biosensor strain is an effective reporter to validate the ability of a MT to biosynthesize CLA from ErA, with the caveat that the conversion needs to reach 10%.
Figure 4.
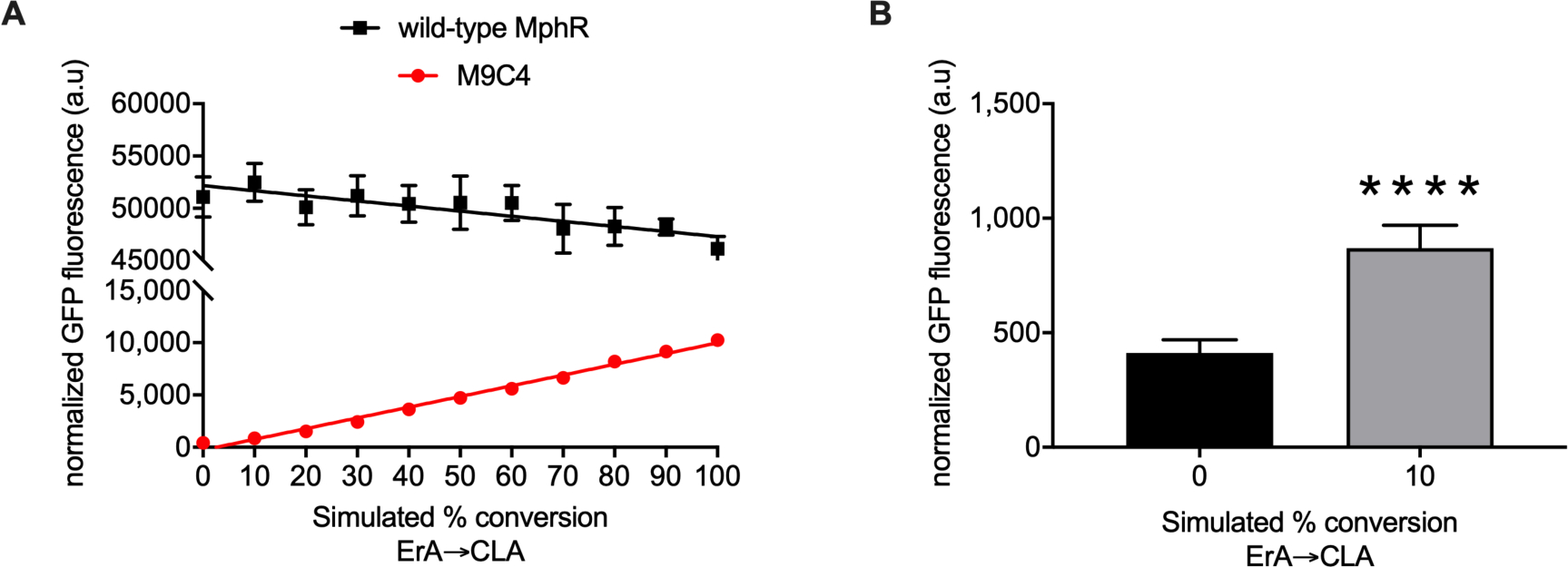
Fluorescence response of the wild-type and M9C4 mutant MphR biosensor strain with mixtures of ErA/CLA simulating their conversion. (A) The total concentration of ErA/CLA is 10 μM such that 0 % conversion represents 10 μM ErA and 100 % conversion represents 10 μM CLA. Error-bars represent the standard error of the mean (n = 7) and are only visible when larger than the data symbol. (B) Student’s unpaired two-tailed t-test indicated p < 0.0001 between 0 and 10% simulated conversion.
Improving the CLA sensitivity of MphR M9C4
Mutagenesis of the ribosome binding site (RBS) region of MphR M9C4 was expected to enhance the sensitivity towards CLA (and ErA) by modulating the expression of the regulatory protein. Three previously described RBS sensitivity mutants29 were introduced into M9C4 (Supplementary Table S2) and their dose-response with ErA and CLA determined (entries 10–12, Table 1; Supplementary Figure S5). Gratifyingly, the K1/2 of all three mutants with both ErA and CLA decreased, in comparison to that of M9C4, demonstrating that incorporation of the RBS mutants into the M9C4 mutant improved sensitivity towards both macrolides. Notably, the CLA sensitivity of M9C4-E7 was the most improved of the three variants (1.7-fold, compared to M9C4) and its dynamic range for CLA was improved 1.3-fold, compared to M9C4. Collectively, this data suggests that M9C4-E7 is more suitable for screening the conversion of ErA to CLA at lower concentrations of macrolides, compared to M9C4.
Prototype in vivo system for screening MT candidates
At this point, the biosensor strain consists of two plasmids, pMLGFP(K) and pJZ12, that both house the biosensor components. As a preliminary step towards construction of a single E. coli strain capable of screening libraries of MT variants, two new plasmids were constructed: one that housed all of the biosensor components in one place (pSense-M9C4-E7-MphA), and another that carried a potential candidate MT gene (pCOLADuet-EryG) (Supplementary Figure S6 and Supplementary Table S2). Given that an MT capable of converting ErA to CLA is not yet known, eryG, responsible for conversion of Erythromycin C (ErC) to ErA in S. erythrae, was used as a proxy to determine the detection characteristics of the prototype biosensor strain.36,37 Importantly, EryG does not methylate ErA or CLA (data not shown), so exogenous mixtures of ErA/CLA will not be modified in the presence of this MT. Notably though, conversion of ErC to ErA was detected by LC-MS analysis of the culture medium after feeding ErC into a biosensor strain containing pCOLADuet-EryG, indicating that this system is competent for MT expression (Supplementary Figure S7). In addition, even with the potential metabolic burden of hosting two plasmids, the two-plasmid strain containing pSense-M9C4-E7-MphA/pCOLADuet-EryG maintained and even enhanced the selectivity of CLA over ErA ~2-fold as well as increased the sensitivity of ErA and CLA ~2.3 and ~5.5 fold, respectively, compared to the parent strain (entry 13 vs. 11, Table 1; Figure 5A). In order to reduce the metabolic load of this two-plasmid system and to improve the transformation efficiency of potential MT libraries into the host as part of the screening procedure, eryG was integrated into the biosensor plasmid to furnish the one-plasmid system, pSense-M9C4-E7-MphA-EryG (Supplementary Figure S6). Notably, the identity of the RBS and promoter sequence driving EryG expression in this one plasmid system proved critical for proper expression of the MT. For example, a calculated RBS sequence and the original promoter from pSense proved insufficient to drive EryG expression, as judged by LC-MS analysis of the conversion of ErC to ErA (pSense-1, Supplementary Figure S8), whereas the RBS sequence from pET28a (AAGGAG) and the strong T7 promoter was able to support 16-fold higher conversion of ErC to ErA than the initial strain (pSense-3, Supplementary Figure S8). The sensitivity and selectivity of the one-plasmid biosensor strain optimized for MT expression (AAGGA RBS and T7 promoter) was indistinguishable from the corresponding 2-plasmid system (entries 13 vs. 14, Table 1; Figure 5B), albeit with a concomitant ~67% reduction in the dynamic range for CLA. The one-plasmid system response retained the linear relationship with the proportion of CLA in the two macrolides combination (Figure 5C). However, the detection capabilities of this system with mixtures of ErA/CLA that simulate the conversion of ErA to CLA now reflect the improved K1/2 for CLA, as low as 2% conversion of ErA to CLA (equivalent to 0.2 μM or 0.15 μg/mL CLA) can be detected (Figure 5D).
Figure 5.
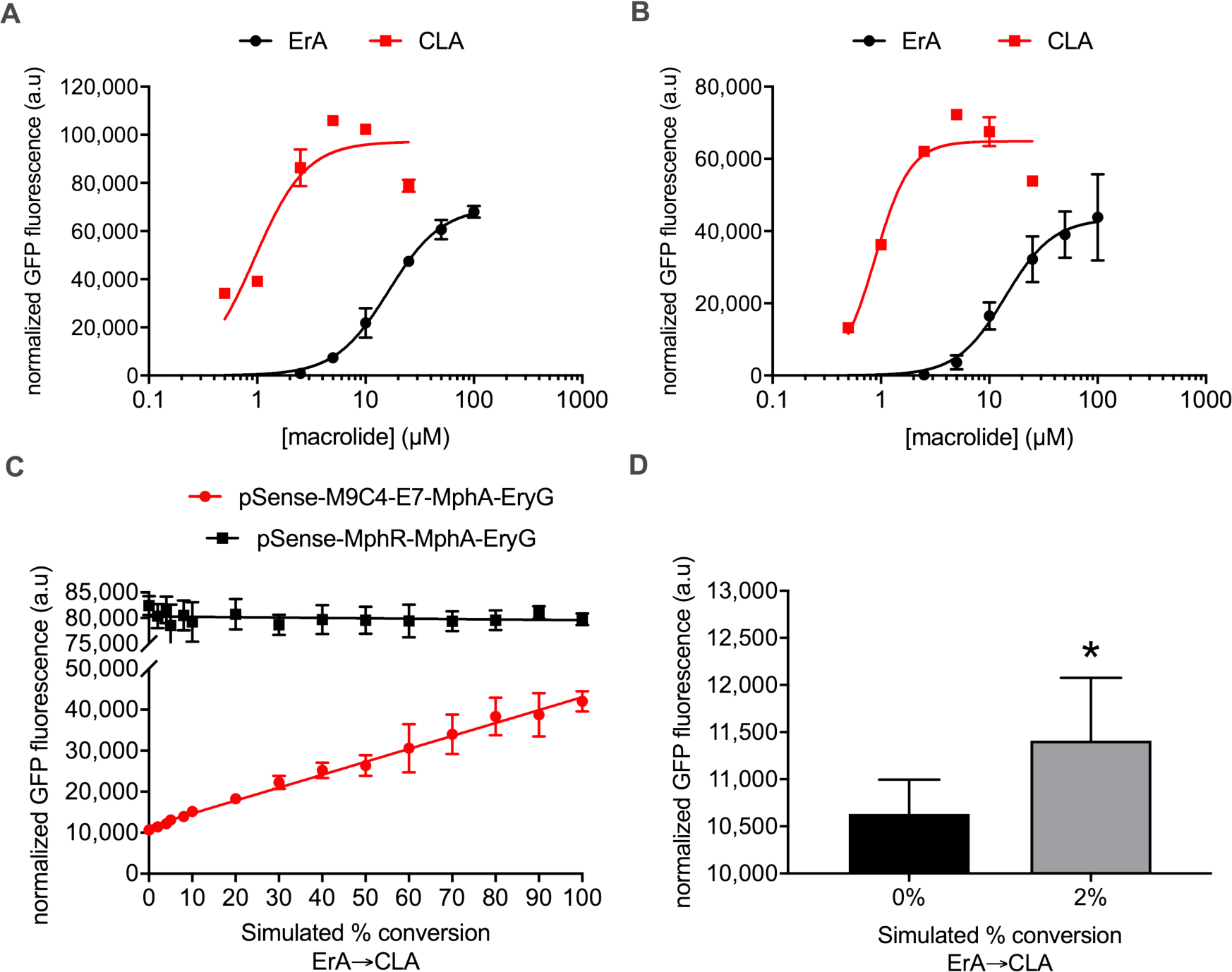
Fluorescence response of various two and one-plasmid MphR biosensor strains with mixtures of ErA/CLA simulating their conversion. (A) Two plasmid system pSense-M9C4-E7-MphA/pCOLADuet-EryG dose-response curves. Error-bars represent the standard error of the mean (n = 3) and are only visible when larger than the data point symbol. (B) One plasmid system pSense-M9C4-E7-MphA-EryG dose-response curves. Error-bars represent the standard error of the mean (n = 3) and are only visible when larger than the data point symbol. (C) pSense-M9C4-E7-MphA-EryG (red) and pSense-MphR-MphA-EryG (black) were screened with different combinations of ErA and CLA. Error-bars represent the standard error of the mean (n = 7) and are only visible when larger than the data point symbol. (D) Student’s unpaired two-tailed t-test indicated p < 0.05 between 0 and 2 % simulated conversion. Error-bars represent the standard error of the mean (n = 7) and are only visible when larger than the data symbol.
Structure of MphR M9C4 bound with CLA
To understand the molecular basis of CLA selectivity in M9C4, we expressed and purified the variant as a hexa-histidine tag fusion protein for crystallographic structure determination. The protein was purified to homogeneity through a sequential process that yielded the protein without tags and containing a glycine residue in the N-terminal position (Supplementary Figure S9). The purified protein was then mixed with CLA and subjected to high throughput screening of crystallization conditions. Several conditions produced crystals with the best diffracting to 2 Å resolution. The structure validated the early hypothesis that R122T is an important mutation that contributes to MphR specificity. Replacement of Arg122 (Figure 6A) in the wild-type MphR with threonine in the M9C4 structure (Figure 6B) shifts the location of a water molecule that originally hydrogen bonded to ErA C6-OH in the wild-type MphR structure. It also leaves the carboxylate of Glu109 (which is paired with the guanidine group of Arg122 in wild-type MphR) unliganded in the mutant structure—buried in the protein and potentially destabilizing to the overall structure unless protonated, as it may be at the pH of the crystal. Notably, the A151T mutation is in proximity to the structural changes that accompany the R122T mutation, and the A151T substitution may play a role in facilitating the reorganization of water that appears to favor binding of CLA by M9C4. Consistent with this, the R122T/A151T double mutant is the most CLA selective of those tested (Figure 3). Other than these subtle changes, the M9C4 structure is overall closely similar to that of the wild-type MphR structure bound to ErA. This suggests that very small structural and possibly dynamic changes can result in significant ligand binding preferences.
Figure 6.
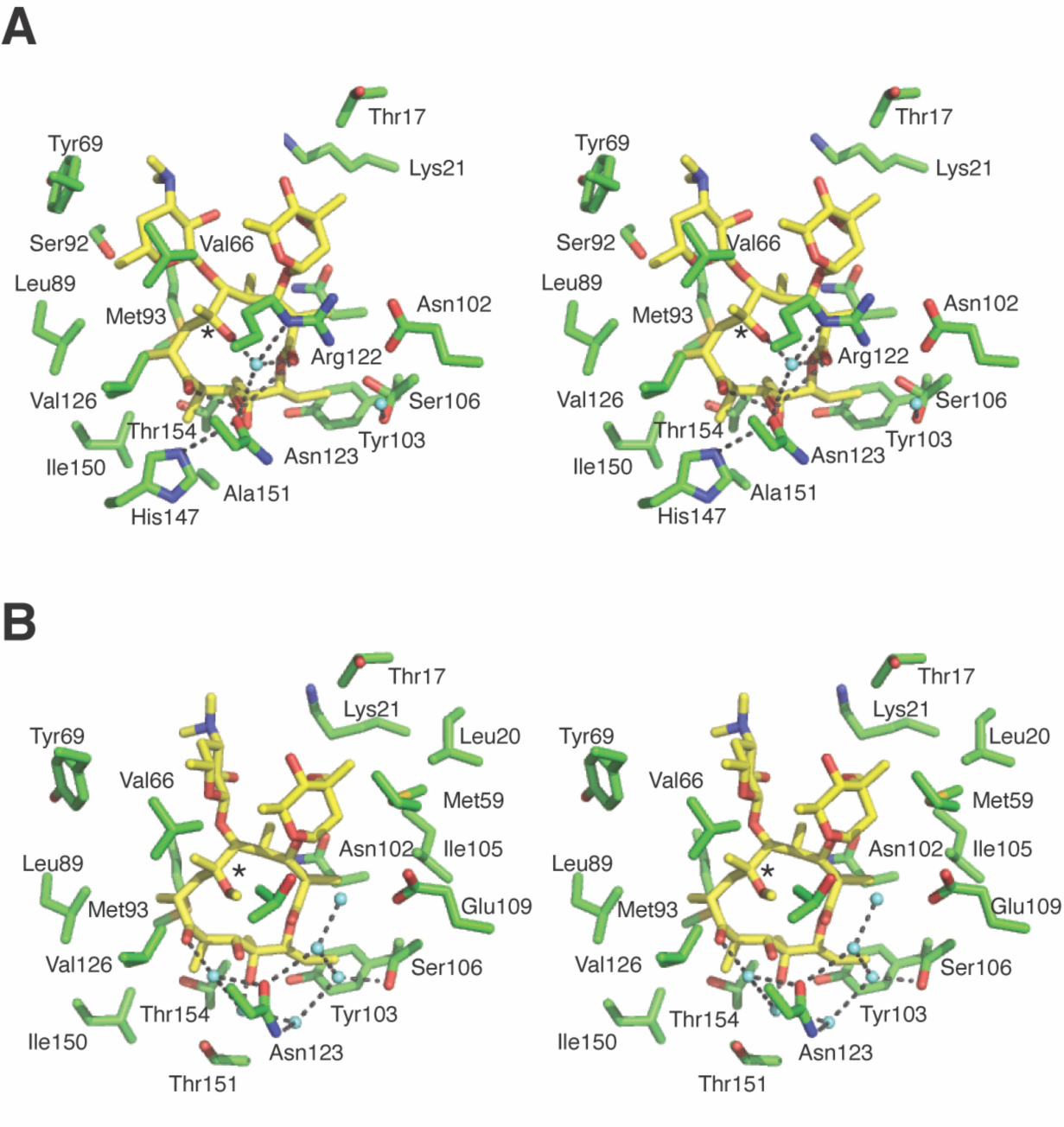
Interactions of MphR and the macrolide effector. (A) The effector binding site of wild-type MphR with bound ErA (yellow sticks). Select water molecules are shown (cyan spheres). Polar contacts within 4 Å of ErA are shown as dotted lines. Hydrogen bonding between the C6-OH (asterisk) of ErA and Arg122 in wild-type MphR is mediated by a water molecule. (B) The effector binding site of M9C4 with bound CLA (yellow sticks). Select water molecules are shown (cyan spheres). Polar contacts within 4 Å of CLA are shown as dotted lines. The CLA C6-OMe is highlighted with an asterisk.
DISCUSSION
Genetically encoded biosensors based on transcription factors offer enormous potential as high-throughput screening tools to report a variety of natural product biosynthetic transformations.38,39 Yet, transcription factors with the required effector specificity are often not available. For example, to accurately detect a post-PKS tailoring step, a biosensor needs to distinguish the tailored natural product from the non-modified intermediate. Here, the effector promiscuity of the macrolide sensing MphR was leveraged as a platform to construct an MphR variant that can detect the methylation of ErA, thereby providing a biosensor for screening the activities of MTs that can convert ErA to CLA. The wild-type MphR is de-repressed by both ErA and CLA, providing a platform for creating a CLA selective variant that is poorly de-repressed by ErA yet retains the ability to be induced by CLA at concentrations that are likely relevant for screening libraries of MT variants. This was accomplished via the combined application of structure-guided mutagenesis and directed evolution approaches to shift the specificity of MphR towards CLA (Figure 7). The full characterization of each variant at each stage enabled a complete evaluation of the suitability of each newly identified mutation and helped inform the overall engineering process. Saturation mutagenesis at Arg122 yielded two variants, R122T and R122V, which were sufficiently improved to be used to parent libraries of random mutants via error-prone PCR, along with the wild-type MphR, with the goal of identifying additional mutations that could improve the selectivity and dynamic range in an additive or synergistic manner. Gratifyingly, this afforded two CLA-selective variants, M9C4 (derived from R122T) and M1B10 (derived from wild-type MphR), each with four amino acid mutations compared to wild-type. The selectivity and dynamic range of M9C4 was superior compared to those of M1B10, suggesting that the initial structure-guided mutations might be critical for providing a minimum level of selectivity and dynamic range identified from the two-tiered flow cytometry-microplate screening strategy (Figure 7). Furthermore, R122T was likely a better starting point than R122V, possibly due to its improved dynamic range, given that improved variants were not identified from the mutant library based on R122V. Remarkably, all four mutations in M9C4 are necessary for the maximum CLA/ErA selectivity (Figure 3), even though three of the four mutations were introduced in a single step from the error-prone PCR mutagenesis. As dictated by the structure-based redesign, the R122T mutation is the major contributor to altering selectivity, while K132N improves the dynamic range and A151T and H184Q improve selectivity to some extent.
Figure 7.
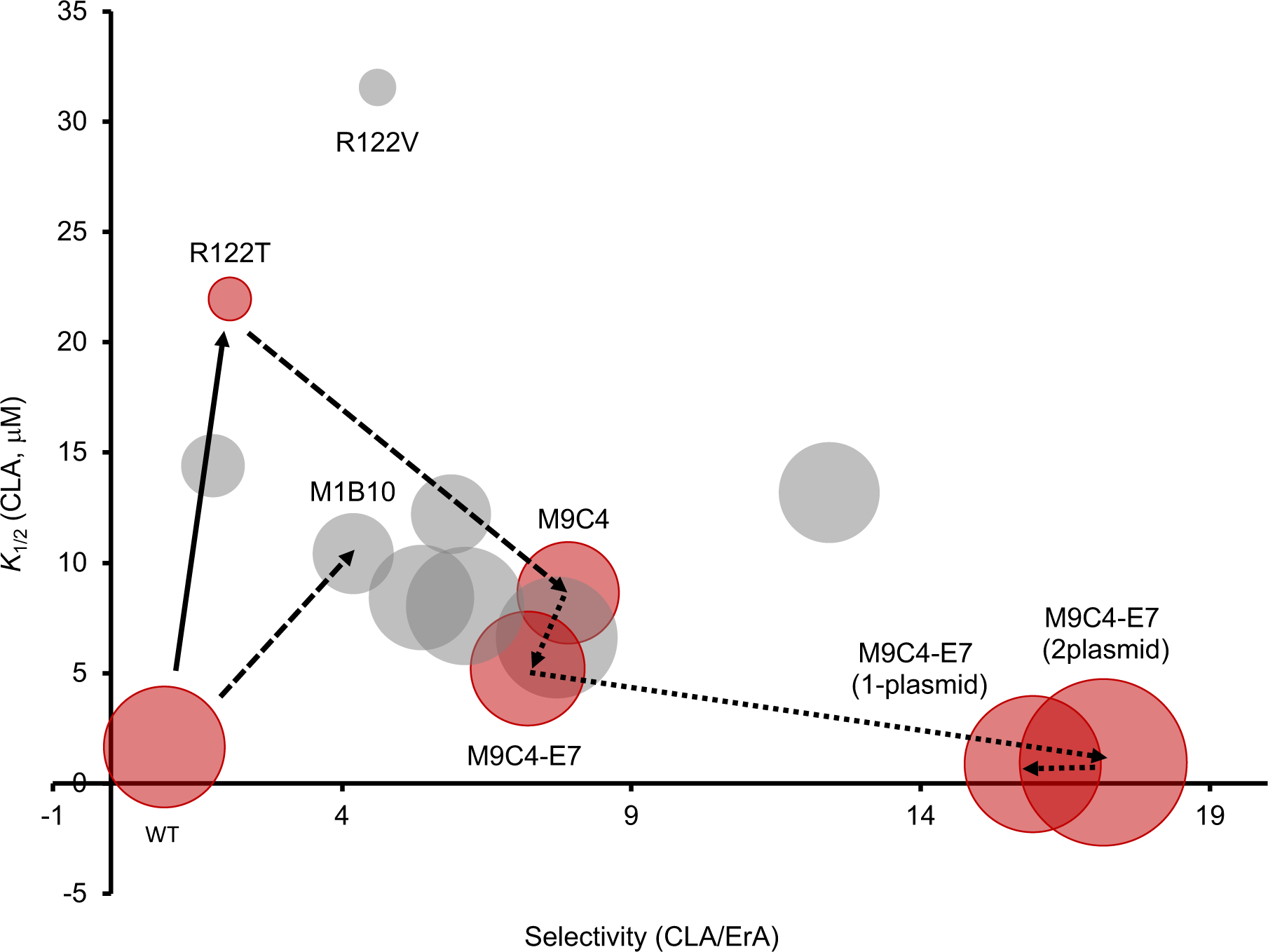
Trajectory of MphR engineering towards a CLA-selective biosensor. Relative size of bubble, dynamic range (GFPmax−GFPmin). Red bubbles, variants that were used in subsequent rounds. Grey bubble, variants that were not used in subsequent rounds. Solid arrows, saturation mutagenesis. Dashed arrows, error-prone PCR. Dotted arrows, non-coding mutagenesis. Selectivity (CLA/ErA), eRatio of K1/2(CLA)/K1/2(ErA).
The crystal structure of M9C4 with bound CLA verifies the critical role of R122T (Figure 6). The substitution of Thr at Arg122 clearly reorganizes the effector binding site of MphR and likely creates space for the C6-methoxy group of CLA to be accommodated. In addition, A151T potentially also plays a role in this structural reorganization, providing an explanation for its appearance in the quadruple mutant, M9C4. However, the structure alone cannot provide a complete explanation for the ability of M9C4 to discriminate CLA and ErA. The mutations in M9C4 may also modulate the complex transmission of information between binding the effector and release of the DNA operator sequence by MphR. Although the random mutant library generated from the wild-type MphR may not have been screened exhaustively, it is notable that this library did not result in rediscovery of the R122T or R122V mutations. Together, these results emphasize that structure-guided mutagenesis and random mutagenesis were both significant in engineering the MphR selectivity (Figure 7) and this combination might be essential to create selective transcription factor-based biosensors.
Aside from mutagenesis of the MphR transcription factor, the biosensor gene circuit itself was also engineered to improve its ability to discriminate CLA/ErA at low concentrations (Figure 7), thereby approximating conditions expected during initial stages of MT directed evolution. This was accomplished by first replacing the RBS driving MphR expression with RBS variants identified in a previous study,29 and then by consolidating the biosensor components into a single plasmid. A second plasmid was constructed that carried EryG as a mock MT to test the ability of the biosensor to discriminate CLA from ErA in the presence of the additional metabolic burden. Finally, both the biosensor components and the MT gene were combined into a single plasmid, pSENSE-M9C4-E7-MphA-EryG-GFP. To simulate the enzymatic conversion of CLA from ErA, different combinations of ErA and CLA were applied to the M9C4 single plasmid biosensor strain. This strain provided the ability to distinguish the equivalent of 2% conversion of ErA to CLA while providing a linear range of detection up to 100% conversion under the conditions tested. Notably, the wild-type sensor strain could not distinguish the simulated conversion across the entire ErA/CLA range tested. Presumably, reconfiguring the biosensor and MT plasmid constructs impacts the expression of the MphR transcription factor relative to the number of operator binding sites and effector molecules, thereby modulating the performance characteristics. Further engineering of the sensor system might be necessary to drive the identification of suitable biocatalysts at a higher load of ErA starting material. In this case, additional rounds of error-prone PCR and screening are likely required to minimize the de-repression of MphR at high ErA concentrations (e.g., >10μM) so that the sensor maintains its CLA selectivity.
In summary, the combination of structure-guided and random mutagenesis followed by plasmid refactoring enabled the rapid development of a mutant MphR biosensor highly optimized for selective CLA detection. This is the first example of combining rational design and directed evolution to engineer repressor protein specificity towards different macrolides. The MphR variant M9C4 was confirmed to be an effective reporter for screening the potential ability of MTs to produce CLA from ErA. Indeed, this system is now poised to be coupled with libraries of MT variants to identify biocatalysts that can support the biosynthesis of CLA from ErA. For example, we anticipate that libraries of error-prone PCR variants can be generated from MTs involved in macrolide biosynthesis and cloned into the sensor strain. Such libraries can now be easily screened for CLA production by feeding in ErA. Similarly, the sensor strain could be leveraged to screen MTs from metagenomic libraries or other gene cluster collections. Potentially, this approach could result in the identification of an MT for the one-step, single-pot biosynthesis of CLA from ErA. Along with a recent example of transcription factor engineering for reporting MTs,40 our work sets the stage for a novel approach to engineering enzymes involved natural product tailoring. In the broader context of biosensor engineering, this study represents a rare demonstration of the combination of semi-rational and directed evolution approaches to provide remarkably selective transcription factor-based biosensors.
MATERIALS AND METHODS
Bacterial Strains, Plasmids, and Materials
The strains and plasmids used in this study are listed in Supplementary Table S2. E. coli strains 10G (Lucigen) TOP10 (Invitrogen) were used for cloning and biosensor expression, respectively. Bacteria were grown in Luria Broth (Fisher Scientific) supplemented with ampicillin (Fisher Scientific) and tetracycline (Fisher Scientific) as appropriate. ErA and CLA were obtained from Sigma Aldrich. Each macrolide was prepared in dimethyl sulfoxide (DMSO) to a stock concentration of 0.5 μM, 5 μM, 50 μM, or 500 μM. DMSO and 96-deepwell plates were purchased from Fisher Scientific. All other chemicals were purchased from Sigma-Aldrich unless stated otherwise. PCR products were extracted with a Bio Basic Gel Extraction Kit. All enzymes for DNA manipulations were purchased from New England Biolabs. Plasmids were isolated using a plasmid miniprep kit from Bio Basic. All oligonucleotides were purchased from Integrated DNA Technologies. Clear and opaque flat-bottom 96-well plates were purchased from Greiner Bio-One. All other chemicals were reagent grade or better.
Site-directed Mutagenesis and Saturation Mutagenesis of MphR
Site-directed mutagenesis for construction of each MphR variant was carried out using the QuikChange Mutagenesis Kit (Agilent) in a total volume of 40 μL containing 100 ng of pMLGFPK 200 μM dNTPs, 0.8 μL Phire Hot Start II DNA Polymerase (Thermo Scientific), and 0.5 mM each forward and reverse primer (entries 1–20, Supplemental Table S3). Standard Phire thermocycling protocols were used with the site-directed mutagenesis protocol as described by the manufacturer instructions. The reaction product was digested with DpnI for 1 h at 37 °C and transformed into chemical competent TOP10 cells. An aliquot of the overnight culture was plated onto LB agar plates supplemented with 100 μg/mL ampicillin. The DNA sequences of plasmids from individual transformants confirmed that the correct mutation was obtained and that there were no spurious mutations except for M9C4 which had a non-coding mutation between the mphR promoter and its RBS (Supplementary Table S1). Purified plasmids were transformed into TOP10 competent cells that carried other plasmids as necessary (e.g. pJZ12) and grown overnight in LB media supplemented with 3 μg/mL tetracycline and 67 μg/mL ampicillin. Glycerol stocks were prepared. For construction of the single, double, and triple mutants derived from M9C4, the template pMLGFPK-R122T was used along with a suitable combination of primers (entries 21−26, see Supplemental Table S3).
General Procedure for Microplate Screening of MphR Variants
Single colonies from mutant libraries in E. coli TOP10 were picked from LB agar plates and used to inoculate 500 μL of LB media containing 67 μg/mL ampicillin and 3 μg/mL tetracycline in wells of a deep-well 96-well microplate. The cultures were incubated for 5 h at 37 °C and with shaking at 350 rpm. Then, 10 μL of each culture was added to 490 μL of LB media containing 67 μg/mL ampicillin, 3 μg/mL tetracycline, and either DMSO, 5 μM ErA, or 5 μM CLA. Plates were incubated overnight at 37 °C and with shaking at 350 rpm. The cultures were centrifuged at 5,000 g for 5 min and the cell pellet was resuspended in 1 mL phosphate-buffered saline (PBS). Then, 100 μL of the cell suspension was used for analyzing the optical density at 600 nm and fluorescence (ex 488 nm/em 515 nm) using a BioTek Hybrid Synergy 4. The fluorescence intensity was divided by the OD600 to yield a normalized GFP fluorescence value.
Dose-Response Analysis of Wild-Type and Variant MphR
Single colonies of the wild-type biosensor or variant MphR biosensor in E. coli TOP10 (that also included pJZ12 if necessary) were picked from LB agar plates and used to inoculate 500 μL of LB media containing 67 μg/mL ampicillin and 3 μg/mL tetracycline in wells of a deep-well 96-well microplate. The cultures were incubated for 5 h at 37 °C and with shaking at 350 rpm. Then, 10 μL of each culture was added to 490 μL of LB media containing 67 μg/mL ampicillin, 3 μg/mL tetracycline, and either ErA or CLA at concentrations between 0 and 200 μM. Plates were incubated overnight at 37 °C and with shaking at 350 rpm. The cultures were centrifuged at 5,000 g for 5 min and the cell pellet was resuspended in 1 mL PBS. Next, 100 μL of the cell suspension was used for analyzing the optical density at 600 nm and fluorescence (ex 488 nm/em 515 nm) using a BioTek Hybrid Synergy 4. The fluorescence intensity was divided by the OD600 to yield a normalized GFP fluorescence value.
The data was fit to the Hill equation to derive the biosensor performance parameters:
where GFP0 is normalized GFP expression in the absence of inducer (i.e., GFPmin), GFPmax is the maximum observed normalized GFP fluorescence, [I] is the effector concentration, K1/2 is the effector concentration resulting in half-maximal induction, and n is the Hill coefficient describing biosensor cooperativity.
Error-prone PCR Library Construction
A KpnI restriction site was inserted into pMLGFP by site directed mutagenesis to generate pMLGFPK using the primers 29–30 (Supplementary Table S3). Error-prone PCR was performed using a GeneMorph II Random Mutagenesis Kit (Agilent Technologies) in a total volume of 50 μL using 2 ng pMLGFPK or pMLGFPK-R122T, 0.8 mM dNTPs, 1μL Mutazyme II DNA polymerase, 1.5 μL of 10 mM forward primer, and 2.1 μL of 10 mM reverse primer (entries 31–32, Supplementary Table S3). Thermocycling conditions were those recommended by the manufacturer’s instructions. The PCR product was a gel purified and double digested using XbaI and KpnI following standard protocols. The double digested product was ligated at 16 °C for 18 h into similarly treated pMLGFPK. Ligation product was ethanol precipitated and electroporated into E.cloni 10G electrocompetent cells and incubated overnight according to the manufacturer’s instructions. An aliquot of the overnight culture was plated onto LB agar plates supplemented with 100 μg/mL ampicillin. Randomly selected colonies were used to prepare plasmids for DNA sequencing, revealing on average 5.3 nucleotide mutations per MphR gene. The rest of the original overnight culture was grown as an overnight liquid culture in LB supplemented with 100 μg/mL ampicillin. A copy of the library was made by purification and storage of the plasmid DNA from an aliquot of the overnight culture. The rest of the overnight culture was stored as glycerol stocks.
Fluorescence Activated Cell Sorting of Biosensor Libraries
The error-prone pMLGFPK plasmid libraries were transformed into TOP10 cells with plasmid pJZ12 and grown overnight in LB media supplemented with 67 μg/mL ampicillin and 3 μg/mL tetracycline. The cell culture was diluted 10-fold into fresh LB media and filtrated through a 30 μm disposable Celtric filter (Partec, Germany) and sorted on a Beckman Coulter MoFlo XDP cell sorter equipped with a 100 μM nozzle operating at 22 psi using a 488 nm excitation laser is and measuring emission at 525 nm. Cells were collected in 1 mL LB broth and incubated at 37 °C for 5 h with shaking at 270 rpm. A portion of the collected population was plated on LB agar supplemented with 67 μg/mL ampicillin and 3 μg/mL tetracycline and the remaining population was grown overnight in LB media with 67 μg/mL ampicillin and 3 μg/mL tetracycline and glycerol stocks were prepared. Putative hit variants from the error-prone PCR libraries were screened in triplicate in microplates according to the method described above.
Preparation of MphR Protein for Crystallization
The MphR M9C4 gene was amplified from pMLGFPK using primers CL2056 and CL2057 (entries 33–34, Supplementary Table S3), which added a NcoI site, 6x-His tag and a Tobacco Etch Protease (TEV) recognition site on the N-terminus, and an EcoRI restriction site on the C-terminus. Gibson Assembly was used to assemble the DNA fragment with the DNA pET28 vector fragment created using the primers CL2062 and CL2063 (entries 35–36, Supplementary Table S3). Protein expression was performed in E. coli BL21 (DE3) cells, which were grown to OD600 = 0.8 and then induced with a final concentration of 1 mM IPTG, for 4 h at 37 °C in 2xYT media. Cells were harvested at 4,000 g, 10 min at 4 °C. Media was removed and pelleted cells were washed in 30 mL nanopure water, then transferred to 50 mL centrifuge tubes. Cells were centrifuged again to remove the water and frozen at −80 °C until ready to purify. Two 1 L frozen cell pellets were defrosted and resuspended in 30 mL lysis buffer (50 mM Tris, 300 mM NaCl, 10 mM imidazole, pH 8) and 1 mM PMSF was added to each. Cells were lysed using lysozyme (1 mg/mL) and incubated on ice for 30 min and then sonicated for 3 × 1.5 min. The cell debris was pelleted and the lysate poured onto HisLink Ni resin and washed using 50 mM Tris, 300 mM NaCl, 80 mM imidazole, pH 8. The N-terminal His-TEV tagged MphR(A) was then eluted using 50 mM Tris, 300 mM NaCl, 500 mM imidazole, 10% glycerol, pH 8. 1 mM EDTA and 1 mM DTT was added to each fraction. The protein was maintained in the same buffer, including the 10% glycerol for protein stability. Purified TEV (2 mL) at 5.675×10−5 M was added to the combined His-TEV-MphR(A) fractions. The protein mixture was digested and dialyzed (Fisherbrand, 6,000–8,000 MWCO tubing) into TEV Working Buffer: 50 mM Tris, 100 mM NaCl, 1 mM DTT, 1 mM EDTA, 10 % glycerol, pH 8.0 at 4 °C for 1 h. In the last exchange, the buffer was switched to TEV Working Buffer minus the DTT and EDTA and allowed to digest and dialyze for 24 h. The final purified protein was obtained after flowing the protein mixture over a new HisLink Ni resin and collecting the flow-through (capturing the TEV and undigested protein).
Protein Crystallization, Data, and Refinement
Purified mutant MphR (~ 9.4 mg/ml) in 10mM Tris, pH 8.0, 50mM NaCl, 1mM DTT was screened in sitting drops (200 nL) for crystallization against 5 commercial screens. CLA was added to a final concentration of 10 mM. The Wizard3 screen (Rigaku) gave a large crystal within 2 weeks at 20 °C with an equal volume of precipitant [20% PEG 3350, 0.2M ammonium citrate dibasic, pH 5.5]. The crystal was cryoprotected briefly (15 sec) in 30% PEG 3350, 0.13M ammonium citrate dibasic and immediately mounted in a liquid nitrogen vapor stream for data collection. Data were collected on an in-house Rigaku Micromax-007 diffractometer with a Dectris Eiger R 4M detector. Intensities to 2Å resolution were processed using CrysAlisPro41 and indexed and scaled in space group P212121 with Pointless42 and Aimless43 in the CCP4 suite.44 The structure was solved by molecular replacement using the structure of the MphR-ErA dimer complex (3frq) as search model and Phaser45 as implemented in Phenix.46 Rigid body refinement of the MphR-CLA model was carried out followed by maximum likelihood parameter refinement in Phenix Refine with recursive rebuilding and solvent addition in Coot.47 Some geometry restraints were applied to the CLA moiety. Supplementary Table S4 summarizes the data collection and refinement statistics. The coordinates for this structure have been deposited to the Protein Data Bank with accession code 6U18.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported in part by the National Institutes of Health grant GM124112 (G.J.W. & T.A.C) and the Thomas Lord Distinguished Professorship Endowment (G.J.W). Crystallography facilities were funded by National Institutes of Health grant OD021756. M.R. acknowledges generous financial support from an Altria Graduate Research Fellowship. We are grateful to Dr. Faik Musayev of the Institute for Structural Biology, Drug Discovery and Development for valuable assistance with crystallization and data collection.
Footnotes
SUPPORTING INFORMATION
Includes supplementary tables, supplemental figures, and detailed methods that describe the construction of various plasmids, in vivo assay of EryG activity, and LC-HRMS analysis of ErA production.
REFERENCES
- 1.Zin PPK, Williams G & Fourches D SIME: synthetic insight-based macrolide enumerator to generate the V1B library of 1 billion macrolides. J Cheminformatics 12, 23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drufva EE, Hix EG & Bailey CB Site directed mutagenesis as a precision tool to enable synthetic biology with engineered modular polyketide synthases. Synthetic Syst Biotechnology 5, 62–80 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hwang S, Lee N, Cho S, Palsson B & Cho B-K Repurposing modular polyketide synthases and non-ribosomal peptide synthetases for novel chemical biosynthesis. Frontiers Mol Biosci 7, 87 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaneko T, Dougherty TJ & Magee TV Comprehensive Medicinal Chemistry II. Anti Bact 519–566 (2007) doi: 10.1016/b0-08-045044-x/00219-4. [DOI] [Google Scholar]
- 5.Dinos GP The macrolide antibiotic renaissance. Brit J Pharmacol 174, 2967–2983 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adachi T & Morimoto S Macrolide Antibiotics. 53–72 (2002) doi: 10.1007/978-3-0348-8105-0_5. [DOI] [Google Scholar]
- 7.Chu SY, Deaton R & Cavanaugh J Absolute bioavailability of clarithromycin after oral administration in humans. Antimicrob Agents Ch 36, 1147–1150 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters DH & Clissold SP Clarithromycin. Drugs 44, 117–164 (1992). [DOI] [PubMed] [Google Scholar]
- 9.Alvarez-Elcoro S & Enzler MJ The Macrolides: Erythromycin, Clarithromycin, and Azithromycin. Mayo Clin Proc 74, 613–634 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Hardy DJ et al. Enhancement of the in vitro and in vivo activities of clarithromycin against Haemophilus influenzae by 14-hydroxy-clarithromycin, its major metabolite in humans. Antimicrob Agents Ch 34, 1407–1413 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heifets LB Clarithromycin against Mycobacterium avium complex infections. Tubercle Lung Dis 77, 19–26 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Ulmer H, Beckerling A & Gatz G Recent Use of Proton Pump Inhibitor-Based Triple Therapies for the Eradication of H. pylori: A Broad Data Review. Helicobacter 8, 95–104 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Fernandes P, Martens E, Bertrand D & Pereira D The solithromycin journey—It is all in the chemistry. Bioorgan Med Chem 24, 1–9 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Agouridas C et al. Synthesis and Antibacterial Activity of Ketolides (6- O –Methyl-3-oxoerythromycin Derivatives): A New Class of Antibacterials Highly Potent Against Macrolide-Resistant and -Susceptible Respiratory Pathogens †. J Med Chem 41, 4080–4100 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Bonnefoy A, Guitton M, Delachaume C, Priol PL & Girard AM In Vivo Efficacy of the New Ketolide Telithromycin (HMR 3647) in Murine Infection Models. Antimicrob Agents Ch 45, 1688–1692 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bryskier A Ketolides—telithromycin, an example of a new class of antibacterial agents. Clin Microbiol Infec 6, 661–669 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Tran MP Telithromycin: A Novel Agent for the Treatment of Community-Acquired Upper Respiratory Infections. Bayl Univ Medical Cent Proc 17, 475–479 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodgers W, Frazier AD & Champney WS Solithromycin Inhibition of Protein Synthesis and Ribosome Biogenesis in Staphylococcus aureus, Streptococcus pneumoniae, and Haemophilus influenzae. Antimicrob Agents Ch 57, 1632–1637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunet E et al. Preparation of clarithromycin. Selective 6-O-methylation of the novel erythromycin A 9-O-(2-pyrimidyl)oxime. Tetrahedron Lett 48, 1321–1324 (2007). [Google Scholar]
- 20.Minas W Microbial Processes and Products. 65–90 (2005) doi: 10.1385/1-59259-847-1:065. [DOI] [Google Scholar]
- 21.Morimoto S et al. CHEMICAL MODIFICATION OF ERYTHROMYCINS. J Antibiotics 43, 286–294 (1990). [DOI] [PubMed] [Google Scholar]
- 22.Morimoto S, Takahashi Y, WATANABE Y & Omura S CHEMICAL MODIFICATION OF ERYTHROMYCINS. J Antibiotics 37, 187–189 (1984). [DOI] [PubMed] [Google Scholar]
- 23.Fang L, Guell M, Church GM & Pfeifer BA Heterologous erythromycin production across strain and plasmid construction. Biotechnol Progr 81, 175–6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Wang Y, Wu J, Skalina K & Pfeifer BA Complete Biosynthesis of Erythromycin A and Designed Analogs Using E. coli as a Heterologous Host. Chem Biol 17, 1232–1240 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Davis AM, Plowright AT & Valeur E Directing evolution: the next revolution in drug discovery? Nat Rev Drug Discov 9, 1–18 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Fryszkowska A & Devine PN Biocatalysis in drug discovery and development. Curr Opin Chem Biol 55, 151–160 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Mitchler MM, Garcia JM, Montero NE & Williams GJ Transcription factor-based biosensors: a molecular-guided approach for natural product engineering. Curr Opin Biotech 69, 172–181 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng J et al. Structure and Function of the Macrolide Biosensor Protein, MphR(A), with and without Erythromycin. J Mol Biol 387, 1250–1260 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Kasey CM, Zerrad M, Li Y, Cropp TA & Williams GJ Development of Transcription Factor-Based Designer Macrolide Biosensors for Metabolic Engineering and Synthetic Biology. Acs Synth Biol 7, 227–239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng Y et al. Comparative study between macrolide regulatory proteins MphR(A) and MphR(E) in ligand identification and DNA binding based on the rapid in vitro detection system. Anal Bioanal Chem 408, 1623–1631 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Möhrle V, Stadler M & Eberz G Biosensor-guided screening for macrolides. Anal Bioanal Chem 388, 1117–1125 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Gardner L, Zou Y, Mara A, Cropp TA & Deiters A Photochemical control of bacterial signal processing using a light-activated erythromycin. Mol Biosyst 7, 2554 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Tang S-Y, Fazelinia H & Cirino PC AraC Regulatory Protein Mutants with Altered Effector Specificity. J Am Chem Soc 130, 5267–5271 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Jha RK, Chakraborti S, Kern TL, Fox DT & Strauss CEM Rosetta comparative modeling for library design: Engineering alternative inducer specificity in a transcription factor. Proteins Struct Funct Bioinform 83, 1327–1340 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Taylor ND et al. Engineering an allosteric transcription factor to respond to new ligands. Nat Methods 13, 177–183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paulus TJ et al. Mutation and cloning of eryG, the structural gene for erythromycin O-methyltransferase from Saccharopolyspora erythraea, and expression of eryG in Escherichia coli. J Bacteriol 172, 2541–2546 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corcoran JW [33] S-adenosylmethionine:Erythromycin C O-methyltransferase. Methods Enzymol 43, 487–498 (1975). [DOI] [PubMed] [Google Scholar]
- 38.Hossain GS, Saini M, Miyake R, Ling H & Chang MW Genetic Biosensor Design for Natural Product Biosynthesis in Microorganisms. Trends Biotechnol (2020) doi: 10.1016/j.tibtech.2020.03.013. [DOI] [PubMed] [Google Scholar]
- 39.Wu Y, Du G, Chen J & Liu L Engineering of Microbial Biosynthetic Pathways. 53–73 (2020) doi: 10.1007/978-981-15-2604-6_4. [DOI] [Google Scholar]
- 40.Kunjapur AM & Prather KLJ Development of a Vanillate Biosensor for the Vanillin Biosynthesis Pathway in E. coli. Acs Synth Biol 8, 1958–1967 (2019). [DOI] [PubMed] [Google Scholar]
- 41.OD R CrysAlisPro 40.64.42a. (2015).
- 42.Evans PR An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr Sect D Biological Crystallogr 67, 282–292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Evans PR & Murshudov GN How good are my data and what is the resolution? Acta Crystallogr Sect D Biological Crystallogr 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr Sect D Biological Crystallogr 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCoy AJ et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr Sect D 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr Sect D Biological Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.