

SUMMARY
Anti-angiogenic therapies, such as anti-VEGF antibodies (AVAs), have shown promise in clinical settings. However, adaptive resistance to such therapies occurs frequently. We use orthotopic ovarian cancer models with AVA-adaptive resistance to investigate the underlying mechanisms. Genomic profiling of AVA-resistant tumors guides us to endothelial p130cas. We find that bevacizumab induces cleavage of VEGFR2 in endothelial cells by caspase-10 and that VEGFR2 fragments internalize into the nucleus and autophagosomes. Nuclear VEGFR2 and p130cas fragments, together with TNKS1BP1 (tankyrase-1-binding protein), initiate endothelial cell death. Blockade of autophagy in AVA-resistant endothelial cells retains VEGFR2 at the membrane with bevacizumab treatment. Targeting host p130cas with RGD (Arg-Gly-Asp)-tagged nanoparticles or genomic ablation of vascular p130cas in p130casflox/floxTie2Cre mice significantly extends the survival of mice with AVA-resistant ovarian tumors. Higher vascular p130cas is associated with shorter survival of individuals with ovarian cancer. Our findings identify opportunities for new strategies to overcome adaptive resistance to AVA therapy.
In brief
The scaffolding protein p130cas is important during integrin-mediated angiogenesis under VEGF-A stimulation. Wen et al. discover that internalization of VEGFR2/p130cas fragments in endothelial cells, followed by TNKS1BP1-mediated cell death, is responsible for angiogenesis inhibition with AVA therapy. Targeting endothelial p130cas defers adaptive resistance to AVA therapy and reduces tumor growth.
Graphical Abstract
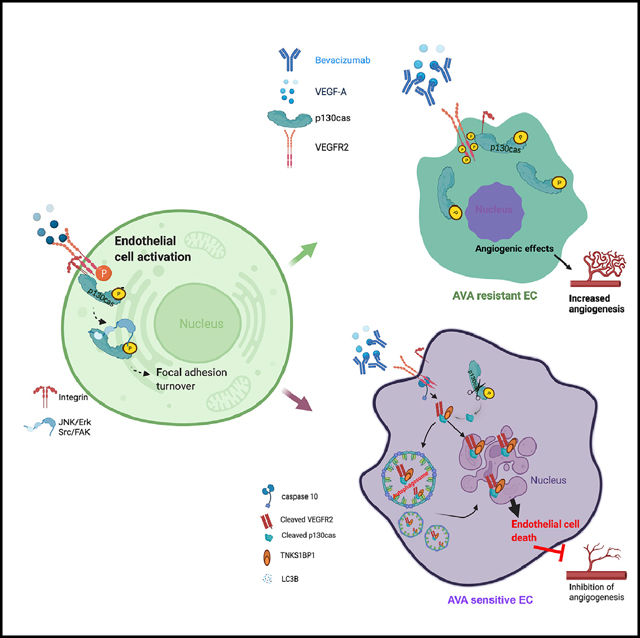
INTRODUCTION
Angiogenesis is a vital part of cancer growth and progression. As a result, a large proportion of targeted therapies aim to reduce tumor-associated vasculature (Folkman, 1990). Anti-angiogenic strategies, such as anti-vascular endothelial growth factor (VEGF) antibody (AVA) therapy, have shown promise in clinical trials among individuals with ovarian cancer, with response rates ranging from 15.9%–21% (Burger et al., 2007; Cannistra et al., 2007). Bevacizumab has been approved by the US Food and Drug Administration for use in individuals with newly diagnosed or relapsed ovarian cancer (Burger et al., 2011). However, the vast majority of tumors rapidly acquire resistance, with rebound tumor growth or revascularization following termination of anti-angiogenic therapies. Our genomic profiling data identified p130cas (Crk-associated substrate) as being significantly upregulated in tumor-associated endothelium in AVA-resistant compared with AVA-sensitive tumors. It is well known that p130cas functions as a central node in many normal and pathologic signaling pathways involved in tumor growth and shaping of the tumor microenvironment (Cabodi et al., 2006; Chodniewicz and Klemke, 2004; Klemke et al., 1998; Sanders and Basson, 2005). Silencing p130cas in ovarian cancer cells results in a robust reduction in tumor growth (Nick et al., 2011). In addition to serving as an adaptor for integrin-focal adhesion kinase (FAK)-Src signaling pathways, p130cas plays a central role in development: p130cas-null mice develop cardiovascular abnormalities resulting in embryonic lethality (Honda et al., 1998). Intriguingly, the fragment of p130cas originating from caspase-dependent cleavage has a role opposite that of the full-length p130cas; the fragment functions as a pro-apoptotic factor and disrupts integrin-FAK-mediated survival signaling in fibroblasts (Jeong et al., 2014; Kim et al., 2004; Kook et al., 2000). Although the role of p130cas is well understood in cancer cells, its role in the tumor microenvironment and in shaping the response to anti-angiogenic therapy is not well understood.
Here we characterized the functional effects of full-length and cleaved p130cas and its binding partners VEGFR2 and TNKS1BP1 (tankyrase-1-binding protein; a factor reportedly involved in p53-mediated cell cycle arrest; Chalabi Hagkarim et al., 2018) in endothelial cells in response to AVA treatment. Using an array of model systems, we found that blocking p130cas in the tumor vasculature reduces adaptive resistance to AVA therapy and results in robust anti-tumor effects.
RESULTS
Role of endothelial 130cas in resistance to AVA therapy
To investigate the mechanisms of adaptive resistance to anti-angiogenic therapy, we compared the genomic profiles of endothelial cells from orthotopic ovarian tumor samples that were resistant or sensitive to an anti-VEGF-A antibody (B20) via cDNA microarray (Figure 1A; Table S2). Three canonical pathways—protein ubiquitination, autophagy (mTOR [mammalian target of rapamycin]/p70 S6K), and integrin signaling—were significantly upregulated in endothelial cells from B20-resistant tumors. We chose to further interrogate p130cas because of its central role in integrin signaling and other pathologic signaling pathways (Chodniewicz and Klemke, 2004). To investigate the biological effects of AVA therapy, we first tested the in vitro effects of VEGF versus VEGF + bevacizumab (Bev) treatment in two non-immortalized human primary endothelial cells. Human primary coronary artery endothelial cells (HCAECs) and human primary pulmonary artery endothelial cells (HPAECs) as well as immortalized RF24 human umbilical vein endothelial cells (HUVECs), were exposed to two different dosages of Bev: 5 μg/mL and 5 μg/mL. First we examined cell proliferation (Figure 1B) and tube formation (Figure 1C) with these two primary endothelial cells. After treatment with VEGF-A (10 ng/mL) + Bev (5 μg/μL) or VEGF-A (10 ng/mL) + Bev (5 μg/mL), both doses of Bev significantly reduced the EdU+ (5-ethynyl-2′-deoxyuridine) proliferative population (Figure 1B) and tube formation (Figure 1C) in comparison with VEGF-A (10 ng/mL) stimulation in all three endothelial cell lines. Therefore, we used VEGF-A (10 ng/mL) + Bev (5 μg/mL) for subsequent studies.
Figure 1. Cellular effects of different doses of AVA therapy in ECs.

(A) Schematic representing Ingenuity Pathway Analysis of a cDNA microarray of ECs isolated from anti-VEGF-A antibody (B20)-sensitive ID8 tumors and B20-resistant ID8 tumors.
(B) Effects of AVA therapy on proliferation of human primary ECs. Human primary pulmonary artery ECs (HPAECs) and human primary coronary artery ECs (HCAECs) were treated with control (CTL), VEGF only, or VEGF in combination with Bev at two different dosages, 5 μg/μL (higher dose) or 5 μg/mL (lower dose), and proliferation was assessed by 5-ethynyl-2-deoxyuridine (5-EdU+) incorporation. Representative flow plots are shown on the left, and graphs are shown on the right. Data are expressed as mean ± SD, determined by two-tailed Student’s t test (n = 3).
(C) Formation of tube nodes in HPAECs, HCAECs, and parental RF24 cells (RF24-par) under the same treatments as in (B) Data are expressed as mean ± SD, two-sided Student’s t test (n = 3).
To investigate the mechanisms underlying AVA resistance, we next established RF24 cells (Imbert et al., 1998) that were resistant to Bev (Narayana et al., 2009). Bev-resistant (RF24-Bev) cells expressed higher levels of pTyr410-p130cas than the Bev-sensitive RF24-parental (par) cells in response to treatment with Bev (5 μg/μL) and recombinant human VEGF (10 ng/mL) for 72 h (Figure S1A; Chan et al., 2009). Next we knocked down p130cas using a short hairpin RNA (shRNA) in RF24-Bev cells to create stable RF24-Bev-shRNA-p130cas cells and re-introduced wild-type (WT) p130cas tagged with green fluorescent protein (GFP) into p130cas-knockdown cells (RF24-Bev-shRNA-p130cas/WT-p130cas-GFP) (Figure S1B). RF24-Bev cells exhibited ~1.7-fold greater tube formation and a ~3.5-fold higher proliferation rate (Figure S1C, top panel). p130cas knockdown reversed the proliferative and angiogenic responses of these cells to the levels of RF24-par cells (Figure S1C, bottom panel). RF24-Bev cells showed ~2.1-fold greater migration capacity than RF24-par cells (Figure S1D). Increased p130cas expression in RF24-par cells (RF24-par-WT-p130cas) led to resistance to AVA therapy, whereas stable knockdown of p130cas in RF24-Bev cells (RF24-Bev-shRNA-p130cas) sensitized them to Bev treatment with decreased cell viability (Figure S1E). Ectopic expression of WT-p130cas-GFP restored resistance to AVA therapy in RF24-Bev-shRNA-p130cas cells (Figure S1F).
Because hypoxia levels increase in response to AVA therapy and can promote vessel growth (Krock et al., 2011), we compared RF24-par and RF24-Bev cells under 20% oxygen (normoxia) and 1% oxygen (hypoxia) conditions and observed that RF24-Bev cells showed increased p130cas levels under hypoxia, which was the opposite of RF24-par cells (Figure S1G). These cell-based assays point to an important role of p130cas in mediating resistance to Bev in RF24 cells.
The role of p130cas and VEGFR2 fragments in endothelial cell autophagy under AVA therapy
Because binding of VEGF to VEGFR2 activates p130cas-mediated angiogenic signaling in endothelial cells (Eliceiri et al., 2002; Mitra and Schlaepfer, 2006), we next measured VEGFR2 expression in RF24-par and RF24-Bev cells. We found that VEGFR2 gene expression was higher in RF24-par cells than in RF24-Bev cells (Figure S2A). Furthermore, VEGFR2 expression remained at the membrane in both types of cells after VEGF monotherapy. However, treatment with VEGF + Bev caused internalization of VEGFR2 into the cytosol and nucleus in RF24-par cells, whereas no change in VEGFR2 localization was noted in RF24-Bev cells (Figure 2A).
Figure 2. p130cas and VEGFR2 are internalized into autophagosomes and the nucleus, followed by caspase-10 cleavage, in ECs treated with Bev.
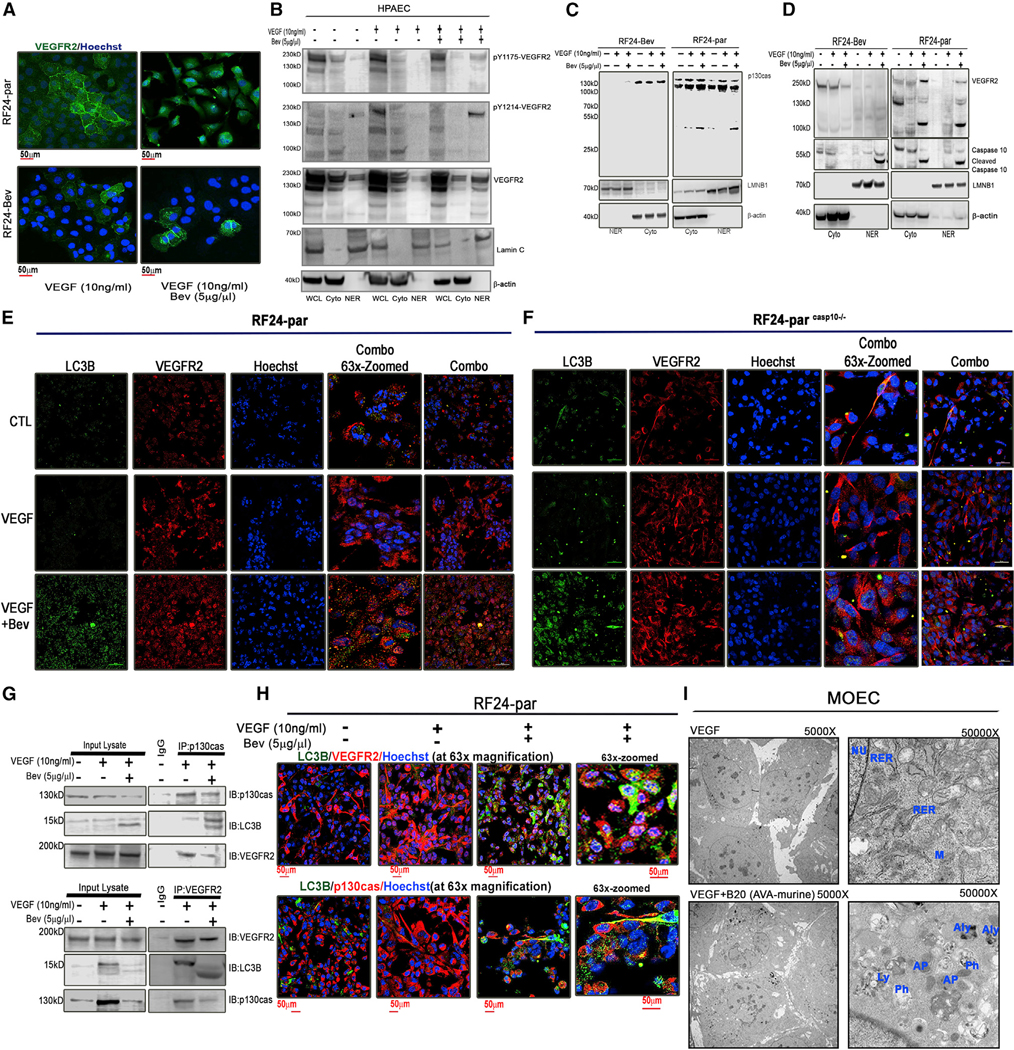
(A) Representative confocal images showing expression of VEGFR2 (green) in Bev-sensitive RF24-par and Bev-resistant RF24-Bev cells treated with VEGF only or VEGF + Bev. Scale bar, 50 μm; n = 3.
(B) Expression of VEGFR2 pY1175 and pY1214 and total VEGFR2 in subcellular fractions of HPAECs. Only under VEGF-A (10 ng/mL) + Bev (5 μg/μL) treatment did the ~100-kD fragment of VEGFR2 appear together with the phosphorylated and total matured VEGFR2 (~220 kD). We used lamin A/C (LMNC) as a marker for the nuclear fraction (NER) and β-actin as a marker for whole-cell lysate (WCL) and cytoplasmic (Cyto) fractions.
(C and D) Expression of Cyto p130cas and its 31-kDa nuclear fragment was observed in subcellular fractions from RF24-par cells but not from RF24-Bev cells (C). We used lamin B1 (LMNB1) as a marker for NER and β-actin as a marker for Cyto. When treated with VEGF + Bev, the 100-kDa nuclear fragment of VEGFR2 was observed only in subcellular fractions of RF24-par cells but not of RF24-Bev cells (D). Activated/cleaved caspase-10 was also observed in the Cyto and NER fractions of RF24-par cells treated with VEGF + Bev.
(E and F) Representative confocal images showing co-localization of LC3B (green) and VEGFR2 (red) in RF24-par (E) or caspase-10-depleted RF24casp10—/— (F) cells in response to treatment with CTL, VEGF only, or VEGF + Bev. Scale bar, 50 μm; n = 3.
(G) Top: co-immunoprecipitation of p130cas and LC3B or VEGFR2 in RF24-par cells treated with CTL, VEGF, or VEGF + Bev. Bottom: reciprocal immunoprecipitates of VEGFR2 and LC3B or p130cas.
(H) Representative confocal images showing co-localization of LC3B and VEGFR2 or p130cas in RF24-par cells treated with CTL, VEGF only, or VEGF + Bev. Scale bar, 50 μm; n = 3.
(I) Representative transmission electron microscopy images of mouse ovarian ECs (MOECs). In comparison with VEGF treatment, in which nuclei (NUs), rough endoplasmic reticulum (RER), and regular mitochondria (Ms) were visible, VEGF + B20 (murine AVA) treatment induced numerous autophagosomes (APs), lysosomes (Ly), and autolysosomes (Aly); abundant phagophores (Ph) were identified in Aly membranes. The substructure was observed under 5,000× and 50,000× magnification (n = 5).
Next we measured VEGFR2 expression in whole-cell lysates, the cytoplasmic fraction, and the nuclear fraction from HPAECs and RF24-par and RF24-Bev cells (Figures 2B–2D). Expression of VEGFR2 in response to VEGF-A stimulation or Bev treatment in HPAECs appeared to be similar to RF24-par cells. We also observed that the ~100-kD fragment of VEGFR2 appeared in the nuclear fraction in HPAECs and RF24-par cells following treatment with VEGF-A (10 ng/mL) + Bev (5μg/μL). In addition, phosphorylated VEGFR2 (pY1175 and pY1214; Takahashi et al., 2001) appeared in the nuclear fraction of HPAECs after VEGF-A + Bev treatment (Figures 2B and 2D). Nuclear distribution of the ~31-kD fragment of p130cas was shown to initiate apoptosis in malignant cells (Kim et al., 2004), but its role in endothelial cells is not known. We found a fragment of p130cas at ~31 kD that appeared in cytoplasmic and nuclear extracts from RF24-par cells, but not RF24-Bev cells, in response to VEGF + Bev treatment (Figure 2C). Moreover, the ~100-kD fragment of VEGFR2 was detected in cytoplasmic and nuclear extracts from RF24-par cells but not from RF24-Bev cells (Figure 2D).
We next tested the effect of VEGFR2 levels on Bev resistance. We knocked out VEGFR2 expression using CRISPR-Cas9 KDR (human VEGFR2) in RF24-par and RF24-Bev cells and confirmed knockout efficacy by western blotting (Figures S2B and S2C). Results from SYTOX flow cytometry indicated that endogenous levels of VEGFR2 are required for sensitivity to Bev treatment in RF24-par cells; knockdown of VEGFR2 in RF24-par cells reversed the inhibitory effect of Bev with regard to decreasing cell viability, and the SYTOX+ population of RF24-par cells was approximately equal after treatment with VEGF-A (10 ng/mL) alone or with Bev (5 μg/μL) (Figure S2D). On the other hand, knockdown of VEGFR2 in RF24-Bev cells did not change their resistance to Bev (Figure S2E), possibly because the endogenous levels of VEGFR2 in RF24-Bev cells were very low in comparison with RF24-par cells at the mRNA (Figure S2A) and protein (Figures S2B and S2C) levels.
Given that VEGFR2 autophosphorylation can contribute to its activity in endothelial cell signaling, we next used pazopanib (a VEGFR tyrosine kinase inhibitor (Merritt et al., 2010)) to test the effects of inhibition of VEGFR2 phosphorylation on suppressing VEGFR2 expression, phosphorylation, and cell viability in RF24-par and RF24-Bev cells. The results from SYTOX flow cytometry analysis (Figures S2F and S2G) and western blotting (Figures S2H and S2I) showed that pazopanib reduced phosphorylation of pY1175 and pY1214 of VEGFR2 in RF24-par and RF24-Bev cells, but it did not affect the cellular effects of Bev in terms of reducing viability of RF24-par cells (Figure S2F) and maintaining Bev resistance in RF24-Bev cells (Figure S2G). It is also note-worthy that, in RF24-par cells but not RF24-Bev cells, the 100-kD VEGFR2 fragment and 31-kD p130cas fragment appeared under VEGF + Bev treatment (Figures S2H and S2I). Pazopanib treatment had minimal effects on pY410 phosphorylation of p130cas in both cell lines (Figures S2H and S2I).
To determine the mechanism of VEGFR2 fragmentation and nuclear internalization, we analyzed its amino acid sequence (NP_002244.1) and found a unique single cleavage site at amino acid (aa) 852 for caspase-10, an initiator caspase and protease (Figure S3A). Indeed, we observed the cleaved (active) form of caspase-10 in nuclear extracts from RF24-par and RF24-Bev cells but only following treatment with VEGF + Bev (Figure 2D). We also observed expression of cleaved caspase-10 in cytoplasmic extract from RF24-par cells but not from RF24-Bev cells (Figure 2D), indicating potential translocation of cleaved caspase-10 into the nucleus in RF24-par cells following AVA treatment. To determine the potential role of caspase-10 in VEGFR2 internalization, we used CRISPR-Cas9 to knock out caspase-10 in RF24-par cells (Figure S3B). In comparison with RF24-par cells treated with a scrambled control, the absence of caspase-10 in the CRISPR-Cas9-edited RF24-par cells led to disappearance of nuclear VEGFR2 with and without Bev treatment (Figure S3C). By comparing the localization of VEGFR2 with that of LC3B (autophagosomes) or Hoechst (nucleus) in RF24-par and RF24-caspase10—/— cells (Figures 2E and 2F), we discovered that absence of caspase-10 led to retention of VEGFR2 at the membrane (Figure 2F).
Next we performed co-immunoprecipitation using antibodies for p130cas, LC3B, or the 1,200–1,300 aa portion of VEGFR2. We detected associations between p130cas and LC3B and between p130cas and VEGFR2 following VEGF + Bev treatment in RF24-par cells (Figure 2G). Internalization of VEGFR2 relies on formation of large vesicles such as autophagosomes, which also direct cells’ bioenergetic and biosynthetic responses to metabolic stress (Basagiannis et al., 2016; Schaaf et al., 2019). In RF24-par cells, VEGF treatment resulted in membrane-associated expression of VEGFR2 and p130cas; VEGF + Bev treatment led to high levels of cytosolic VEGFR2 or p130cas co-localized with LC3B, a marker specific for autophagosome assembly (Jackson et al., 2005; Figures 2H and S4A). Specifically, we measured the red fluorescence intensity in the nucleus of each cell in each field. The dimensional intensity plots indicate a significant increase in cell populations containing nuclear VEGFR2 (red fluorescence) following VEGF + Bev treatment in comparison with control or VEGF-A treatment, with a similar trend observed for nuclear p130cas expression (Figures S4A and S4B). In addition, we performed immunofluorescence staining for vascular endothelial (VE)-cadherin/VEGFR2/Hoechst in RF24 cells for detection of membrane, cytosolic, and nuclear VEGFR2 expression (Figure S5A), and the same quantification method using Imaris confirmed the increase in cell populations with nuclear VEGFR2 under VEGF + Bev treatment in comparison with control or VEGF-A treatment (Figure S5B). To further confirm this observation of AVA therapy-induced autophagy in endothelial cells, we treated another endothelial cell line (mouse ovarian endothelial cells [MOECs]) with B20, an anti-human/murine VEGF-A antibody, and performed transmission electron microscopy. We observed extensive cytoplasmic autophagic vacuolization, including formation of autophagosomes and autolysosomes, after VEGF-A + B20 treatment (Figure 2I).
To investigate the role of autophagy during internalization of the VEGFR2 complex in response to AVA therapy, we stably transfected RF24-par and RF24-Bev cells with LC3B-GFP (Figure 3A). Accumulation of LC3B loci was increased substantially in RF24-par cells treated with VEGF + Bev (Figure 3A, top panel). However, the number of LC3B loci in RF24-Bev cells was not changed significantly by treatment with the control, VEGF, or VEGF + Bev (Figure 3A, bottom panel). We used CRISPR-Cas9 (Ran et al., 2013) to generate LC3B knockout clones (Figures 3B and 3C) and found that the absence of LC3B in CRISPR-Cas9-edited RF24-par cells diminished VEGFR2 internalization after treatment with VEGF + Bev in comparison with RF24-par-WT cells (Figure 3D). Furthermore, we used hydroxychloroquine (HCQ) to inhibit autophagy and determined the expression of membrane, cytosolic, and nuclear VEGFR2 in RF24 cells in response to VEGF-A (10 ng/mL) + Bev (5 μg/μL) treatment. Immunofluorescence staining of LC3B/Hoechst and VE-cadherin/VEGFR2/Hoechst indicated that HCQ treatment induced enrichment of LC3B loci in the cytosol of RF24 cells (Figure 3E). In addition, the percentage of acidic vesicular organelles induced by VEGF-A (10 ng/mL) + Bev (5 μg/μL) was reduced significantly by HCQ treatment (Figure 3F). On the other hand, the membrane-tethered VEGFR2 was retained at the cell membrane under Bev treatment in RF24 cells that were pre-treated with the autophagy inhibitor HCQ (Figure 3G). This result was consistent with what we observed in LC3B knockout RF24 cells (Figure 3D).
Figure 3. Autophagy is essential for VEGFR2 internalization under AVA therapy.
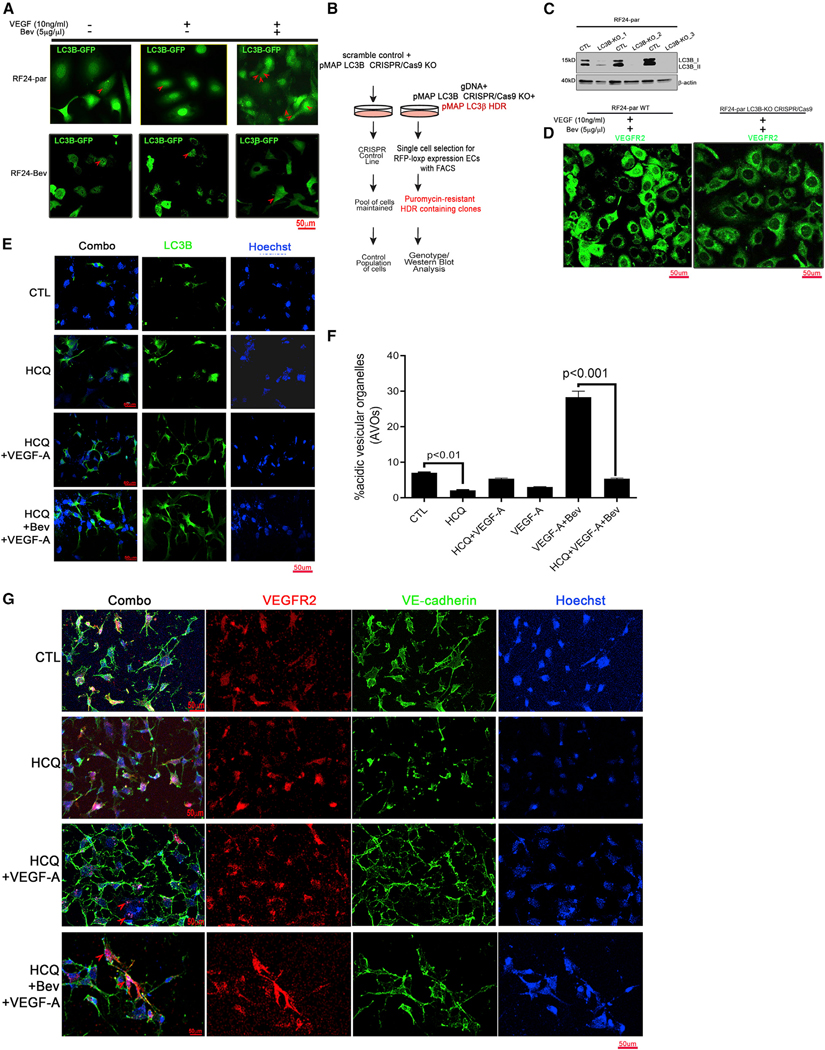
(A) Bevacizumab (Bev) induced enrichment of LC3B loci only in AVA-sensitive RF24-par cells, not in resistant RF24-Bev cells. Shown are representative confocal microscopy images for GFP-LC3B (green) expression in RF24-par or RF24-Bev cells in response to treatment with CTL, VEGF only, or VEGF + Bev. Scale bar, 50 μm; n = 3. Red arrows show the LC3B loci formed in the cells.
(B) In vitro transfection with the pMAP-LC3B CRISPR-Cas9 construct used to knock out LC3B and insertion of pMAP LC3B homology-directed DNA repair (HDR) in RF24-par endothelial cells (ECs).
(C) Expression or absence of LC3B in 3 CRISPR-Cas9 knockout (KO) cells. β-Actin was used as a loading CTL.
(D) Representative confocal microscopy images of VEGFR2 (green) in RF24-par WT cells or LC3B2 CRISPR-Cas9 KO cells treated with VEGF + Bev. Scale bar, 50 μm; n = 3.
(E–G) Autophagy inhibition by hydroxychloroquine (HCQ) reduced VEGFR2 internalization into the LC3B lysosomal compartment and NU in RF24 cells. RF24-par cells pre-treated with 40μM HCQ (24 h) were subjected to VEGF-A alone or Bev + VEGF-A treatment for another 48 h.
(E) LC3B loci in cells were first determined by immunofluorescence staining with an anti-LC3B antibody. Scale bar, 50μm.
(F) The autophagy flux in RF24-par cells with or without HCQ treatment was measured by acridine orange (AO) staining and analyzed with fluorescence-activated cell sorting (FACS). The percentage of acidic vesicle organelles (AVOs) was statistically compared between CTL and HCQ or VEGF-A + Bev and HCQ + VEGF-A + Bev; two-tailed Student’s t test. Data are expressed as mean ± SD (n = 3).
(G) Representative images of confocal microscopy on RF24-par cells treated with HCQ or left untreated and stained with Hoechst (blue)/VE-cadherin(green)/VEGFR2 (red) for each group (CTL, VEGF, and VEGF + Bev). Each experiment was repeated at least three times, and representative images from 15 high-power fields in each condition are shown. Membrane VEGFR2 and VE-cadherin were present in RF24-par cells pretreated with HCQ + VEGF-A + Bev, where the nuclear VEGFR2 was minimal in comparison with the internalized VEGFR2 in Figure 2F and/or 2H. Scale bar, 50 μm; n = 3.
To better understand the dynamics of VEGFR2 translocation, we treated RF24-par cells with Bev for 24 or 72 h and then replaced the medium with fresh VEGF-containing medium. In cells treated for 24 h, VEGFR2 expression was detected only in the cytoplasmic fraction, whereas in cells treated for 72 h, VEGFR2 was detected in the cytoplasm and nucleus (Figure S6A). We also assessed VEGFR2 internalization in two other types of endothelial cells (G1S1 dermal endothelial cells and MOECs). Both of these showed an ~100-kD VEGFR2 fragment in the nucleus following treatment with VEGF + AVA (Figure S6B). Notably, nuclear VEGFR2 was not detected in SKOV3 ovarian cancer cells regardless of Bev treatment (Figure S6C), indicating that internalization of VEGFR2 in response to AVA therapy is an endothelial-cell-specific effect.
TNKS1BP1 chaperones nuclear VEGFR2 fragments in endothelial cells
To identify the downstream effects of nuclear VEGFR2 in endothelial cells, we immunoprecipitated VEGFR2 from the nuclear fraction of RF24-par cells and performed mass spectrometry (Figure S7A). Among the top factors associated with nuclear VEGFR2 (Figure S7B), TNKS1BP1 is a binding partner of tankyrase 1 through poly(ADP-ribosyl)ation and is associated with DNA damage response mechanisms such as double-strand break repair (Smith et al., 1998). In RF24-par cells, TNKS1BP1 was mostly expressed in the cytoplasmic and membrane fractions without Bev treatment. Under untreated control or VEGF treatment conditions, most of the TNKS1BP1 was expressed in the membrane-associated and cytoplasmic fractions, whereas VEGF + Bev treatment significantly elevated the level of TNKS1BP1 in the nuclear fractions of RF24-par cells (Figure 4A). We also examined TNKS1BP1 expression in the tumor vasculature of individuals with ovarian cancer who received Bev-based therapy. TNKS1BP1 expression was substantially higher in the tumor-associated vasculature of individuals whose tumors were responsive to AVA therapy (Bev) than in those whose tumors were resistant to such therapy (Figure 4B). We also found that VEGFR2, TNKS1BP1, and p130cas formed a complex, as shown by their co-immunoprecipitation in nuclear fractions from RF24-par cells treated with Bev (Figure 4C). Co-localization of VEGFR2 and TNKS1BP1 in the nuclei of RF24-par cells treated with Bev was confirmed by confocal microscopy (Figure 4D). When TNKS1BP1 was silenced in RF24-par cells with endoribonuclease-prepared short interfering RNAs (siRNAs) (Figure S7C), we found that the cytotoxicity of VEGF + Bev treatment was decreased (Figure S7D). We further generated stable TNKS1BP1shRNA clones from RF24-par cells (Figure 4E). In RF24-par cells, VEGF + Bev treatment significantly decreased the SYTOX— viable population compared with VEGF alone (p < 0.001; Figures 4F and 4G), whereas knockdown of TNKSBP1 by shRNAs (A or C) reversed the sensitivity of RF24-par cells to Bev treatment (Figures 4F and 4G). We also found that VEGFR2 translocated into the nucleus in RF24-parscramble shRNA cells treated with VEGF + Bev but was restricted to the membrane in RF24-parTNKS1BP1 knockdown cells (Figure 4H). Next we tested the effects of TNSK1BP1 shRNA in RF24-Bev cells in terms of autophagy flux and cell viability. Results from SYTOX staining and acridine orange staining showed that knockdown of TNKS1BP1 did not have a significant effect on AVA resistance in RF24-Bev cells (Figure S8), possibly because the endogenous levels of TNKS1BP1 in AVA-resistant endothelial cells were very low (Figure 4B).
Figure 4. TNKS1BP1 and nuclear VEGFR2 mediate AVA therapy-induced EC death.

(A) Nuclear TNKS1BP1 enrichment in RF24-par cells in response to Bev treatment. MOF, membranous fraction. β-Actin was used as Cyto CTL, p-cadherin as MOF CTL, and LMNB1 as NER CTL.
(B) Expression of endothelial TNKS1BP1, shown by dual immunofluorescence staining for TNKS1BP1 (red) and CD31 (green), in ovarian tumor samples that were sensitive or resistant to AVA therapy (Bev).
(C) Co-immunoprecipitation (coIP) of VEGFR2 and TNKS1BP1 in RF24-par cells under Bev treatment. The anti-VEGFR2 and anti-TNKS1BP1 immunoprecipitates were re-probed with an anti-p130cas antibody.
(D) Representative confocal microscopy images showing TNKS1BP1 (green) and VEGFR2 (red) distributed into the NUs of RF24-par cells in response to Bev treatment (VEGF + Bev); this is distinctly different from the expression patterns in cells treated with CTL or VEGF only. Scale bar, 50 μm; n = 3.
(E) Knockdown of TNKS1BP1 in RF24-par cells with shRNAs (A–D).
(F and G) The graph shows mean numbers of SYTOX— live cells for each treatment group (F; data are expressed as mean ± SD, n = 3, p < 0.001 or not significant [ns], two-tailed Student’s t test. Notably, in cells treated with scramble shRNA, the percentage of SYTOX— viable cells was 36.3% under VEGF + Bev treatment; in cells transfected with shRNA A or C, the percentages of SYTOX— live cells were 72.61% and 84.98%, respectively, under VEGF + Bev treatment. Also shown are representative plots of SYTOX— populations from FACS analysis of RF24-par cells transfected with scramble shRNA, shRNA A, or shRNA C against TNKS1BP1 and treated with CTL, VEGF only, or VEGF + Bev (G).
(H) Representative confocal images showing nuclear TNKS1BP1 and VEGFR2 in RF24-parscramble shRNA cells; VEGFR2 remained at the membrane in RF24-parshRNA-TNKS1BP1—A cells under VEGF + Bev treatment. Scale bar, 50 mm; n = 3.
Targeting vascular p130cas inhibits tumor growth and progression
Next we tested the effects of endothelial p130cas silencing on adaptive resistance to AVA therapy using the murine-specific siRNA mp130cas (selected from a pool of validated siRNAs) incorporated in chitosan nanoparticles tagged with RGD (Arg-Gly-Asp) peptide (CH-NP-RGD) (Figure S9A; Han et al., 2010), which targets integrin on angiogenic vasculature (Eliceiri et al., 2002). Mice bearing orthotopic OVCA432 tumors were randomized into two groups after breakthrough (defined as tumor growth despite AVA therapy): one group received CH-NP-RGD-control siRNA and the other group received CH-NP-RGD-siRNA-mp130cas (Figure 5A). In comparison with the control, treatment with CH-NP-RGD-siRNA-mp130cas decreased tumor weight (p < 0.001; Figure 5B), the number of tumor nodules (p = 0.005; Figure 5C), and the volume of ascites (p = 0.04; Figure 5D). There was no significant difference in body weight between the two groups (Figure 5E). CH-NP-RGD-siRNA-mp130cas also significantly decreased microvessel density (MVD) by 55% (p = 0.003) and increased expression of LC3B in CD31+ cells by 38% (p < 0.05; Figure S9B). As expected, CH-NP-RGD-siRNA-mp130cas effectively reduced p130cas expression in tumor endothelial cells by 85% (p < 0.001; Figure S9C). The membrane-associated expression of VEGFR2 observed in tumor-associated vasculature in mice treated with CH-NP-RGD-control siRNA was instead observed in the perinuclear area in mice treated with CH-NP-RGD-siRNA-mp130cas (Figure S9D). In addition, the TUNEL+ population of CD31+ endothelial cells increased in tumors of mice treated with CH-NP-RGD-siRNA-mp130cas (p < 0.001; Figure S9E).
Figure 5. Ablation of vascular p130cas delays progression of tumors with adaptive resistance to AVA therapy.

(A) Mouse models of adaptive resistance to the anti-VEGF antibody (B20) were established using an orthotopic model of OVCA432 cells labeled with luciferase (OVCA432-luc). Mice with adaptive resistance (e.g., after development of ‘‘breakthrough’’ disease) were randomized and treated with CH-NP-RGD-CTL siRNA or CH-NP-RGD-siRNA-mp130cas.
(B–E) The (B) tumor weight, (C) number of tumor nodules, (D) volume of ascites, and (E) body weight of each mouse were recorded at necropsy. Data are expressed as mean ± SD: (B) p < 0.001, (C) p = 0.005, (D) p = 0.05, and (E) p = 0.262, determined by two-tailed, nonparametric t test.
(F) Schematic of the excision of exon 2 from the p130casflox/flox allele by Cre recombinase expressed in Tie2Cre mice to generate p130casflox/floxTie2Cre mice.
(G) Schematic of the dosing and in vivo bioluminescence imaging regimen used to establish the syngeneic ID8 ovarian tumor mouse model with adaptive resistance to B20 treatment.
(H and J) Representative gross images from C57/BL6 (H, on day 35) or p130casflox/floxTie2Cre (J, on day 98) mice. Syngeneic ID8 tumors, which were implanted surgically into the left ovary of each mouse, are labeled with yellow ovals (H).
(I and K) ID8 tumor burden, demonstrated with bioluminescence in C57/BL6 or p130casflox/floxTie2Cre mice.
(L) Kaplan-Meier survival plot of C57/BL6 and p130casflox/floxTie2Cre mice bearing intra-ovarian ID8 tumors and receiving B20 treatments; p < 0.001, log rank test.
Acknowledging the potential off-target effects of siRNAs (Jackson and Linsley, 2010), we generated endothelial-cell-specific p130cas-deficient (p130casflox/floxTie2Cre) mice by crossing p130casflox/flox mice (Riccomagno et al., 2014) with endothelial-cell-specific Tie2Cre mice (Kisanuki et al., 2001; Figure 5F). Next we established ID8 syngeneic tumors with AVA resistance in age-matched C57/BL6 WT and p130casflox/floxTie2Cre mice using the B20 antibody to further investigate the role of p130cas in AVA adaptive resistance (Figure 5G). Other groups in this experiment included control immunoglobulin G (IgG)-treated animals (WT; p13cas [Riccomagno et al., 2014]; Tie2Cre [in which p130cas was not floxed]; p130casflox/flox Tie2Cre) and B20 antibody-treated animals (WT, p130flox/flox, Tie2Cre, and p130cas flox/flox Tie2Cre) (Figures S10A and S10B). Among them, development and progression of ID8 tumors were only delayed substantially in p130casflox/floxTie2Cre mice compared with C57/BL6 WT mice (Figures 5H–5K). As expected, the levels of vascular p130cas were diminished in tumor samples from p130casflox/floxTie2Cre mice (p < 0.001; Figure S11A). Tumor-bearing p130casflox/floxTie2Cre mice survived significantly longer than C57/BL6 WT mice (p < 0.001; Figure 5L). In addition, p130casflox/floxTie2Cre mice had significantly lower tumor MVD (p = 0.001) and higher expression of LC3B in CD31+ endothelial cells in comparison with their C57/BL6 WT counterparts (3.2-fold, p = 0.03; Figure S11B). Expression of TNKS1BP1 was higher in endothelial cells from p130casflox/floxTie2Cre mice (Figure S11C), and endothelial VEGFR2 was mostly nuclear in p130casflox/floxTie2Cre mice (Figure S11D).
Vascular p130cas expression in human ovarian cancer samples
Next we assessed the clinical outcomes of individuals with high-grade serous ovarian cancer (n = 91) based on their vascular p130cas expression. All tumors were co-stained with p130cas (Figure 6A, top) and CD31 (Figure 6A, bottom). Sixty-nine individuals (76%) had high p130cas expression in the tumor-associated vasculature, and 22 individuals (24%) had low p130cas expression. High expression of vascular p130cas was associated with a higher rate of mortality (p < 0.001; Figure 6B). The association of p130cas and CD31 in ovarian tumors was verified by immunofluorescence staining with CD31 and p130cas (Figure 6C). Correspondingly, we observed higher MVD, reflected by CD31+ endothelial cells, in ovarian tumors than in normal ovaries (Figure 6D). We also assessed p130cas expression in the endothelial cell compartment by qRT-PCR and found that endothelial cells isolated from invasive epithelial serous ovarian carcinomas demonstrated 6.94-fold greater p130cas expression than those from normal ovaries (p < 0.05; Figure 6E). To confirm our findings regarding the differences of subcellular location of VEGFR2 and p130cas in response to AVA therapy, we compared the expression of VEGFR2 and LC3B as well as endothelial p130cas in specimens from individuals with ovarian cancer who had different responses to Bev. IHC (immunohistochemistry) staining showed lower levels of co-localized LC3B and VEGFR2 or CD31 and p130cas in specimens from a responder to Bev (individual C), an intermediate level of co-localized LC3B and VEGFR2 or CD31 and p130cas in specimens from an inconclusive responder to Bev (individual B), and higher levels of VEGFR2 and p130cas in autophagosomes and nuclei in specimens from a non-responder to Bev (individual A) (Figure 6F). Our study revealed a previously unrecognized mechanism by which an axis formed by fragments of p130cas, VEGFR2, and TNKS1BP initiates endothelial cell death in response to AVA therapy. Depletion of vascular p130cas blocks this adaptive resistance to AVA therapy (Figure 6G).
Figure 6. A higher level of vascular p130cas associated with worse survival in individuals with ovarian cancer and its functional mechanism in mediating resistance to AVA therapy in ECs.
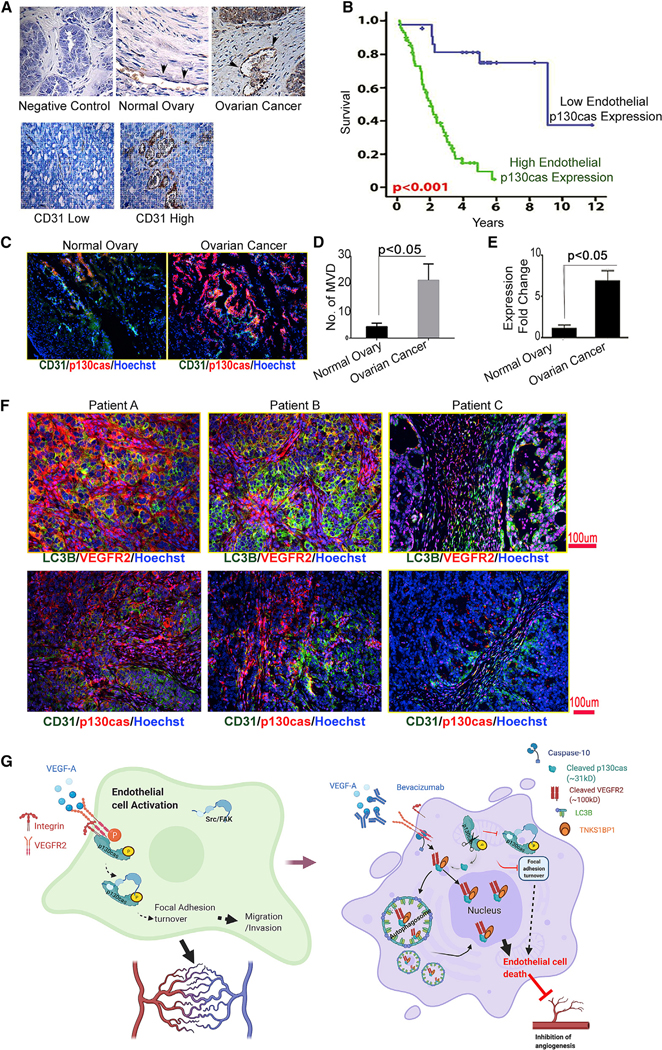
(A) Representative images of immunohistochemical peroxidase staining for p130cas in ECs in normal human ovary or ovarian cancer tissue. Negative CTL represents a sample of ovarian cancer tissue used in the current study, processed for immunohistochemistry with a secondary antibody alone.
(B) Kaplan-Meier curves showing disease-specific mortality estimates for individuals with ovarian cancer based on the degree of p130cas expression in the tumor-associated vasculature; p < 0.001, determined by unpaired two-sided Student’s t test.
(C) Representative images of dual immunofluorescence staining for p130cas (red) and CD31 (green) in the same sets of human ovary or ovarian cancer samples.
(D) Microvessel density (MVD) was calculated by averaging MVD from four random fields per sample. Data are expressed as mean ± SD; p < 0.05, determined by two-sided Student’s t test (n = 3).
(E) p130cas expression in normal and tumor-associated ECs from normal human ovary and ovarian tumors was measured by quantitative real-time PCR. Data are expressed as mean ± SD. p < 0.05, determined by two-sided Student’s t test (n = 3).
(F) Representative images of dual immunofluorescence staining of LC3B (green) and human VEGFR2 (red) (top panel) or CD31 (green) and human p130cas (red) (bottom panel) in advanced-stage human ovarian cancer samples. Individual A had an omentum tumor and responded to Bev-based therapy, individual B hada right ovary tumor as the primary site and inconclusive response to Bev, and individual C had a right ovary tumor and did not respond to Bev-based therapy. Hoechst staining (blue) was used to shown nuclei.
(G) Schematic of internalization of p130cas/VEGFR2 fragments and initiation of EC death through binding with TNKS1BP1 in response to AVA treatment. Left: activated p130cas (phosphorylated) serves as a scaffold protein for activating the downstream FAK-mediated angiogenic processes (focal adhesion turnover, cell survival, and/ or migration/invasion) in ECs when stimulated by VEGF-A. Right: in response to treatment with Bev, membrane-tethered VEGFR2 is cleaved by caspase-10. Together with the ~31-kD p130cas fragment, the VEGFR2 fragment is internalized into LC3B-tagged APs. Next, a complex formed by the VEGFR2 and p130cas fragments and TNKS1BP1 translocates into the nucleus and initiates endothelial cell death, which leads to reduced angiogenesis, followed by inhibition of tumor growth.
DISCUSSION
Clinically, anti-angiogenic therapies such as Bevacizumab are known to be beneficial for individuals with cancer, but the vast majority of tumors develop resistance to such therapy and eventually progress. Prior studies have examined some of the mechanisms underlying adaptive resistance, including stromal factors, macrophages, and some proinflammatory cytokines, such as T helper type 17 and interleukin-17 (Chung et al., 2013). Here we report an unexpected role of p130cas and VEGFR2 in endothelial cells following AVA treatment whereby these proteins are internalized and cell death is initiated by forming a complex with TNKS1BP1.
p130cas is known to be involved in VEGF-stimulated endothelial cell migration by functioning as a scaffold protein in a VEGFR2/FAK-dependent mechanism (Avraham et al., 2003). It plays an important role in recruiting the FAK/Ephrin-B1 complex to induce membrane ruffling in human aortic endothelial cells (Nagashima et al., 2002). However, the effects of anti-angiogenic therapy on p130cas activity in endothelial cells and its role in adaptive resistance were not known previously. We found that a fragment of p130cas translocates to the nucleus to orchestrate cell death in AVA-sensitive endothelial cells in response to anti-VEGF therapy. In AVA-resistant endothelial cells, p130cas levels remained steady, and expression of VGFR2 remained at the membrane, regardless of the presence of Bev. In our genetically engineered mice, we also found that targeting endothelial p130cas significantly reduced the growth of ovarian tumors with adaptive resistance to AVA therapy. Although it is possible that p130cas may also be expressed in cancer-associated fibroblasts and CD36+ macrophages, our results point to a specific and important role of p130cas in tumor endothelial cells during acquisition of resistance to anti-angiogenic therapy. To our knowledge, this is the first study to link degradation of p130cas and VEGFR2 fragments to tumor angiogenesis. We also identified p130cas as an important target for overcoming adaptive resistance to AVA therapy.
VEGF receptors are well known to be important for mediating the pro-angiogenic effects of VEGF. Bev blocks binding of VEGF to its receptors on the endothelial membrane. However, how VEGFR2-mediated signaling pathways are altered in endothelial cells during development of adaptive resistance is not fully understood. Our findings indicate that, upon Bev treatment, membrane-tethered VEGFR2, together with p130cas, is internalized into autophagosomes assembled by LC3-II. Moreover, we discovered that VEGFR2 can be cleaved by caspase-10 (the initiator caspase in death receptor signaling; Wang et al., 2001) and that the resultant VEGFR2 fragment binds to p130cas and TNKS1BP1 (a death factor involved in p53-mediated cell cycle arrest; Chalabi Hagkarim et al., 2018) to initiate cell death in tumor-associated endothelial cells. Knockdown of caspase-10 in AVA-sensitive endothelial cells abolished internalization of VEGFR2 and made them resistant to AVA. Based on these findings, we propose a model of the role of the VEGFR2/p130cas/TNKS1BP1 complex in endothelial cells during development of resistance to AVA therapy (Figure 6F). To date, our understanding of the mechanisms whereby VEGFR2 is internalized and trafficked remains incomplete. Development of resistance to anti-VEGF therapy is complex. We reported previously that Cd11b+Gr1+ myeloid cells play an important role in adaptive resistance to anti-VEGF therapies (Dalton et al., 2017; Lyons et al., 2017). However, the current study has provided unique insights into the varying roles of endothelial VEGFR2 depending on its localization switching from proangiogenic activity when expressed on the membrane to anti-angiogenic activity when expressed in the nucleus. Our findings have important implications for p130cas as a target for overcoming adaptive resistance to anti-angiogenic therapy and for the distinct pools of VEGFR2 in autophagosomes and the nucleus, which may provide opportunities for further anti-angiogenic approaches.
Limitations of the study
The cell-based in vitro experiments using HUVEC-RF24 cells suggest that internalization of VEGFR2 with p130cas fragments into autophagosomes and nuclei following AVA therapy is limited by using RF24 cells, which reportedly have different growth behavior compared with other endothelial cells (ECs) (Fontijn et al., 1995). We included two additional human primary ECs (HPAEC and HCAEC) to validate our findings. However, this study could be enhanced by directly using tumor ECs (Hida et al., 2018). Furthermore, our study could benefit from additional models. Our experimental design for generating EC-specific, p130cas-deficient (p130casflox/floxTie2Cre) mice by crossing p130casflox/flox mice (Riccomagno et al., 2014) with Tie2Cre mice (Kisanuki et al., 2001) does not exclude the possibility that the Tie2Cre promoter might not be exclusively EC specific (Challa et al., 2019). In this complex scenario, future studies using a more endothelium-specific promoter (Kano et al., 2003) could help to further define the specific roles of p130cas and VEGFR2 fragments in response to anti-angiogenic therapy.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yunfei Wen, Ph.D., (ywen2@mdanderson.org).
Materials availability
All published and inaugural reported reagents and mouse lines will be shared upon request within the limits of the respective material transfer agreements.
Data and code availability
This study did not generate code and all data have been presented in figures and supplemental figures. The uncropped western blots related to Figures 2, 3, 4, S1–S3, S6, and S7, and the IPA analysis and graphic are included in Data S1. Other original images will be made available upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human ovarian cancer specimens, endothelial cell isolation, validation, and microarray
After approval by the Institutional Review Board for the protection of human subjects at The University of Texas MD Anderson Cancer Center (protocol No. PA17–0271), archived clinical specimens of epithelial ovarian cancer were obtained. Ninety-one paraffin-embedded tumor specimens were obtained for expression analysis. The median age at diagnosis of the patient cohort was 61 years (range = 36–89 years), and the median follow-up interval was 2.32 years (range = 0.07–13.01 years). Approximately 80% of the examined tumors had serous histology and 87% were high-grade carcinomas. Among the 91 patients (all female), 83% had advanced-stage disease (III or IV) at initial diagnosis and 74% had associated ascites. Clinical outcome data for these patients were obtained by chart review. The following data were obtained from the medical record: patient age, tumor grade, tumor histology, disease stage, presence of ascites, and volume of residual disease at surgical cytoreduction. Time to recurrence and disease-specific survival at the time of chart review were also recorded.
As previously described (Lu et al., 2007), fresh tissue samples (5 normal ovaries and 10 epithelial high-grade, stage III or IV invasive serous ovarian cancers) were obtained from patients undergoing primary surgical exploration at MD Anderson Cancer Center after approval from the Institutional Review Board. The minced tissue was digested with collagenase A, elastase, and DNase 1 at 37°C for 90 min to yield a single-cell suspension. A number of negative selections followed, including removal of platelets and red blood cells by Percoll separation; removal of epithelial cells using M450 beads, which are prebound to BerEP4 antibody; and removal of leukocytes using anti-CD14, anti-CD45, and anti-CD64 beads (Dynal Biotech, Brown Deer, WI). Positive selection was done with P1H12 (CD146) immunobeads (Chemicon, Temecula, CA), and the beads linked to secondary antibodies were from Dynal Biotech. Immunostaining was then done using von Willebrand factor and 4′,6-diamidino-2-phenylindole nuclear staining to confirm the purification of endothelial cells. Next, two rounds of amplification were used to generate sufficient labeled cRNA for microarray analysis from 25 ng of total RNA using the Two-Cycle cDNA Synthesis Kit (Affymetrix, Santa Clara, CA) and oligo-dT24-T7 (5′-GGCCAGTGAATTG TAATACGACTCACTATAGGGAGGCGG-3′) primer according to the manufacturer’s instructions followed by amplification with the MEGA script T7 Kit (Ambion, Inc., Austin, TX). After cleanup of the cRNA with a GeneChip Sample Cleanup Module IVT column (Affymetrix), second-round double-stranded cDNA was amplified using the IVT Labeling Kit (Affymetrix). A 15.0-μg aliquot of labeled product was fragmented by heat and ion-mediated hydrolysis at 94°C for 35 min in 24 μL of H2O and 6 μL of 5× Fragmentation Buffer (Affymetrix). The fragmented cRNA was hybridized for 16 h at 45°C in a Hybridization Oven 640 to a U133 plus 2.0 oligonucleotide array (Affymetrix). Washing and staining of the arrays with phycoerythrin-conjugated streptavidin (Molecular Probes, Eugene, OR) was completed in a Fluidics Station 450 (Affymetrix). The arrays were then scanned using a confocal laser GeneChip Scanner 3000 and GeneChip Operating Software (Affymetrix). Data normalization and filtering and class comparison analysis for cDNA microarray analysis were performed as in a previously described protocol (Lu et al., 2007). Data presented in the sensitive versus resistant groups were analyzed by Ingenuity Pathway Analysis (IPA, Qiagen). The p values associated with pathways in IPA analysis were calculated using the right-tailed Fisher exact test.
Animal models
Female athymic nude mice (4 to 6 weeks old) and female C57/BL6 mice were purchased from Taconic Biosciences and housed in specific-pathogen–free conditions. Animals were cared for in accordance with the guidelines set forth by the American Association for Accreditation for Laboratory Animal Care and the U.S. Public Health Service Policy on Human Care and Use of Laboratory Animals. All studies were first approved and supervised by the MD Anderson Institutional Animal Care and Use Committee (protocols 00001171 and 00001019).
Development and characterization of an orthotopic murine model of advanced ovarian cancer has been previously described by our group (Landen et al., 2005). For in vivo therapeutic experiments, murine-specific p130cas siRNA constructs (Table S1) were incorporated into chitosan nanoparticles (CH; β (1–4)-linked 2-amino-2deoxy-D-glucose [D-glucosamine, D-GlcN] and 2-acet-amido-2-deoxy-D-glucose [N-acetyl-D-glucosamine, D-GlcNAC]). To establish the model of adaptive resistance to AVA therapy, female athymic nude mice (4–6 weeks old) were inoculated intraperitoneally with luciferase-labeled OVCA432 high-grade serous carcinoma cells (1×106 cells per mouse) (Domcke et al., 2013). Each mouse received initial treatments with B20, a murine monoclonal VEGF-A antibody (5 μg/kg intraperitoneally, twice weekly; B20–4, MTA OR-502783; Genentech, Inc., South San Francisco, CA). After ~6 weeks, when tumor burden began to increase from previously stable disease (e.g., breakthrough, an indication of acquired resistance), mice were randomized into the following treatment groups (n = 10 mice/group): control siRNA-CH (5.0 μg/mouse intravenously, twice weekly) and murine-specific p130cas-siRNA-CH (5.0 μg/mouse intravenously, twice weekly). After treatment began, mice were closely monitored for adverse effects, and all mice were sacrificed when the control mice became moribund. Mouse body weight, tumor weight, tumor distribution, number of tumor nodules, and amount of ascites were recorded at the time of necropsy. Portions of tissue specimens were snap frozen for subsequent protein or RNA analysis, fixed in formalin for paraffin embedding, or snap frozen in optimal cutting medium (Miles, Inc., Elkhart, IN) for frozen slide preparation.
To establish the p130casflox/floxTie2Cre model, p130casflox/flox female mice (Riccomagno et al., 2014) were crossed with Tie2Cre male mice (Kisanuki et al., 2001) to selectively deplete the expression of p130cas in the endothelial cells of the offspring. Genomic DNA was isolated from tail biopsies of the mice, and expression of the Tie2-Cre transgene and floxed p130cas allele were confirmed by PCR (primers listed in Table S1). To establish the syngeneic tumor model, murine ID8-luciferase ovarian cancer cells (1 × 106) were injected directly into the left ovarian parenchyma of age-matched female C57/BL6 or p130casflox/floxTie2Cre mice through a surgical incision. After the initial establishment of tumors as measured by IVIS imaging (2×105 photons/second/cm2/sr), each mouse received B20 or control treatment as described above. We recorded the tumor weight, number, and distribution of tumors at the death of each animal. Individuals who performed the necropsies were blinded to the treatment group assignments. Tissue specimens were processed as described above.
Cell lines and culture media
The immortalized human endothelial RF24 cell line was generated from primary human vascular endothelial cells by immortalization through the integration of a single DNA copy of an amphotrophic, replication-deficient retrovirus containing the E6/E7 genes of human papillomavirus (Fontijn et al., 1995). The cell line has been used by us and others as a substitute for human umbilical vein endothelial cells (HUVECs) in multiple studies (Haemmerle et al., 2017; Noh et al., 2017; Previs et al., 2017). We cultured WT parental RF24 cells (RF24-par) in minimum essential medium (MEM) supplemented with 15% fetal bovine serum (FBS), 0.1% gentamicin sulfate, 10 ng/ml basic fibroblast growth factor, 1× NEAA, 1 mM sodium pyruvate, 2 mM L-glutamine, and 1× MEM vitamin solution. The RF24 cells were rendered resistant to bevacizumab (Bev) by selection with Bev (5μg/μl) for 2 weeks and were maintained at a median inhibitory Bev concentration of 1.0 μg/μL in the same culture medium as RF24-par cells; the Bev-resistant cells are referred to here as RF24-Bev.
The non-immortalized primary human coronary artery endothelial cells (ATCC® PCS-100–020™) and primary human pulmonary artery endothelial cells (ATCC® PCS-100–022™) were maintained in Endothelial Cell Growth Kit-BBE supplement (ATCC® PCS-100–040, not containing VEGF). All the in vitro cell-based assays including EdU+ incorporation, tube formation, and protein lysate collection were performed within 3~5 passages of both cell lines. Under these conditions, these two cell lines and murine ovarian endothelial cells (MOECs) were cultured in Dulbecco modified essential medium supplemented with 10% FBS and 0.1% gentamicin sulfate. The OVCA432-luciferase cell lines were cultured in RPMI-1640 medium supplemented with 10% FBS and 0.1% gentamicin sulfate (Gemini Bioproducts, Calabasas, CA). The human epithelial ovarian cancer cell line SKOV3ip1 and the mouse ovarian cancer cell line ID8 were maintained as described previously (Lengyel et al., 2014). All experiments were performed with cells at 60–80% confluence. All cell lines were routinely tested to confirm the absence of mycoplasma with an ATCC (30–1012K) Universal Mycoplasma Detection Kit (https://www.atcc.org/products/all/30–1012K.aspx) and were validated by the MD Anderson Cancer Center Characterized Cell Line Core Facility. For in vivo experiments, OVCA432-luciferase or ID8-luciferase cells were subjected to trypsinization and centrifugation at 1000 rpm for 7 minutes at 4°C, washed twice with Hanks balanced salt solution, and re-suspended at a concentration of 5×106 cells/ml.
siRNA, shRNA, and CRISPR/Cas9 knockout constructs
EGFP-LC3B (Addgene plasmid #11546; http://n2t.net/addgene:11546) was provided by Dr. Karla Kirkegaard. Human-specific p130cas siRNA was purchased from Sigma-Aldrich (St. Louis, MO) and used to silence p130cas expression in ovarian cancer cell lines (target sequence: CAGCATCACGCAGGGCAA). Murine-specific p130cas siRNA was purchased from Sigma-Aldrich and used to silence expression in MOECs (target sequence: TTGACTAATAGTCTACATTA). Control non-targeting (“scramble”) siRNA (target sequence: AATTCTCCGAACGTGTCACGT) was confirmed to have no sequence homology with any known human or murine mRNA by BLAST analysis. Information on sequences is available in Table S1.
METHOD DETAILS
Immunohistochemistry
Formalin-fixed, paraffin-embedded samples were sectioned at 5 μm thickness and stained with hematoxylin and eosin; adjacent sections were used for immunohistochemical staining as described elsewhere (Nick et al., 2011). All sections were reviewed by a board-certified pathologist who was blinded to the clinical outcomes of the patients. p130cas expression in the tumor endothelium was determined semi-quantitatively by assessing the distribution of positive cells and the staining intensity in the tumor-associated endothelial cells by methods previously described by our group (Lin et al., 2007). Each specimen was assigned a score and categorized into one of four groups. An overall score of 0 for negative expression of p130cas was assigned if ≤ 5% of cells stained, regardless of intensity.
Microvessel density (MVD) in each clinical sample was determined by immunohistochemical staining with a mouse anti-human CD31 monoclonal antibody (dilution 1:20; Dako, Carpinteria, CA) on sequentially cut sections of the tumors (Lin et al., 2007). Tumor MVD was calculated as the average CD31-positive vessel count over five sections under ×200 high-power fields. A vessel was defined as an open lumen with one or more CD31-positive cells immediately adjacent to it.
RT-PCR and invasion and migration assays
TRIzol reagent (Invitrogen) was added to the cells and total RNA was extracted using reagents and protocol provided by Zymo Research RNA isolation kit. Complementary DNA was synthesized with 1 μg of total RNA using reagents from a Verso cDNA synthesis kit (Thermo Fisher Scientific). Oligo-dt primers and random hexamers were used in a 1:3 ratio. Quantitative PCR was performed with Sybr Green Master Mix on a 7500 Real-Time PCR machine (Applied Biosystems, Carlsbad, CA, USA).
Cell motility was determined by using modified Boyden Transwell chambers in a membrane invasion culture system containing a polycarbonate filter with 8-μm pores coated with 0.1% gelatin, as previously described. For the invasion assay, the chamber was pre-coated with human defined matrix composed of 50 μg/ml human laminin (L6274; Sigma-Aldrich), 50 μg/ml human type IV collagen (C6745; Sigma-Aldrich) in 10 mM acetic acid, or 2 mg/ml gelatin (G1393; Sigma-Aldrich) (Pecot et al., 2013). RF24-par or RF-Bev cells were transfected with either p130cas or control siRNA for 48 hours, and then were seeded in each upper chamber and allowed to incubate at 37°C and migrate for 6 hours toward serum-free MEM containing 10 ng/ml VEGF in the lower chamber. The number of cells that migrated to the lower side of each membrane was determined by examining five random fields (100×) per condition. Each condition was carried out in triplicate and the results were averaged.
Multiphoton confocal microscopy and quantification of fluorescence density in intracellular compartments
Stained cells were visualized with a Nikon laser scanning multiphoton confocal microscope. Each experiment was repeated at least three times, and representative images from 15 high-power fields are shown. Intracellular localization was quantified using the Cell function in Imaris software. Segmentation algorithm involved nuclei detection using Hoechst channel and cell body detection using the red channel with adjacent cell splitting based on one nucleus per cell and cell body signal intensity. For each cell, the sum of each channel intensity in cytoplasm and nucleus was exported to Microsoft Excel to then calculate the cytoplasm-to-whole cell intensity ratios. Statistical analysis and graphing were performed in GraphPad Prism software.
Transmission electron microscopy
MOECs were fixed for 2 hours with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4), fixed again in 1% OsO4 in the same buffer, and then subjected to electron microscopy analysis as described previously (Klionsky et al., 2008). Ultra-thin sections in representative areas from five fields are shown. Sections were viewed with a Hitachi 7600 electron microscope (Hitachi High Technologies America, Inc., Pleasanton, CA). The substructure was observed under 5000× and 50,000× magnification.
Immunoblotting, subcellular fractionation, and co-immunoprecipitation
For immunoblotting, lysates from cultured endothelial cells (RF24, MOEC, G1S1, etc.) were prepared using modified cell lysis buffer (Cell Signaling Technology, Boston, MA) plus 1 unit of Halt™ Protease and Phosphatase Inhibitor Cocktail and 1 unit of complete, Mini Protease Inhibitor Cocktail (Roche). The protein concentrations were determined by using a BCA Protein Assay Reagent kit (Pierce Biotechnology, Rockford, IL). Lysates were loaded and separated by 8% sodium dodecyl sulfate (SDS)—polyacrylamide gel electrophoresis (PAGE).
For immunoprecipitation and co-immunoprecipitation, the cell lysates were incubated with A/G-conjugated antibody at 4°C for 2 hours. For immunoprecipitation, the beads were washed twice with radioimmunoprecipitation assay buffer, once with 0.5 m LiCl in 0.1 m Tris (pH 8.0), and once with phosphate-buffered saline. Reactions were boiled in sample buffer, and proteins were then subjected to 10% SDS-PAGE and immunoblotting.
For immunoblotting, proteins were transferred to a nitrocellulose membrane by semi-dry electrophoresis (Bio-Rad Laboratories, Hercules, CA) overnight, blocked with 4% bovine serum albumin, and then incubated at 4°C with primary antibody overnight. After washing with a mixture of tris-buffered saline and 0.8% Tween 20 (TBST) solutions, the membranes were incubated with horseradish peroxidase-conjugated horse anti-mouse IgG (1:2000, GE Healthcare, Amersham Place, UK) for 2 h. Horseradish peroxidase was visualized by an enhanced chemiluminescence detection kit (Pierce).
Subcellular fractionation were performed using a Subcellular Protein Fractionation Kit for Tissues (Thermo Fisher Scientific). RF24-Bev and -par cells were treated with control, VEGF only, or VEGF + Bev. Subcellular fractions were subjected to immunoblotting for p130cas, Lamin B1 (LMNB1) or Lamin A/C (Lamin C) as markers for nuclear extracts, and β-actin as a marker for cytoplasmic extracts.
Annexin V and 7-AAD staining with flow cytometry
Apoptosis was evaluated by using the Annexin V–phycoerythrin apoptosis detection kit according to the manufacturer’s instructions (BD Biosciences, San Diego, CA). Each experiment was repeated three times independently.
Tube formation assay
HUVECs (1×106) were plated in 10-cm plates and treated with p130cas and control siRNA. After incubation for 36 hours, cells were subjected to trypsinization and collected for a tube formation assay as previously described (Lu et al., 2007). Untreated, control, siRNA–treated, or p130cas siRNA–treated cells were then transferred to Matrigel-containing wells at a concentration of 2×104 cells/well and allowed to incubate for 6 hours. Cells were examined under a microscope (Axiovert S100; Zeiss, White Plains, NY) to identify formation of capillary-like structures and photographed (×50; Retiga 1300 camera, Teledyne QImaging, Surrey, BC, Canada). Capillary-like structures formed in the gel were quantified by counting node points over four random fields per condition.
Liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS)
Myc-DDK (Flag)–tagged VEGFR2 (CAT#: RC219851; Origene, Rockville, MD) was stably overexpressed in RF24-par cells. These cells were then treated with VEGF (10 ng/ml) + Bev (5μg/μl), and the nuclear extracts were prepared according to the manufacturer’s instructions (Thermo Fisher Scientific). These nuclear extracts were incubated with anti-Flag M2 affinity gel (Sigma-Aldrich) overnight at 4°C, and the pulled-down complex was pH-adjusted by adding 50 mM ammonium bicarbonate and digested by adding 200 ng modified sequencing-grade trypsin (Promega, Madison, WI) for 18 hours at 37°C. The resulting peptides and compound were analyzed by LC-MS/MS on an Orbitrap-Elite mass spectrometer (Thermo Fisher Scientific) as described previously (Chan et al., 2013). The expression of VEGFR2 on the pulled-down complex was validated by applying an anti-VEGFR2 antibody (ab39256; Abcam, Cambridge, MA).
QUANTIFICATION AND STATISTICAL ANALYSIS
For evaluation of clinical samples, the chi-square test was performed to determine the associations between clinicopathological variables, MVD, and p130cas staining in the tumor-associated endothelial cells. The optimal MVD cut-off point had been determined previously on a separate set of tumors (Lin et al., 2007). Kaplan-Meier survival curves were generated and compared using a two-sided log-rank statistic. Patients who were alive at the last follow up were censored at that date. For in vivo experiments, 10 mice were assigned per treatment group in order to achieve 80% power to detect a 50% reduction in tumor weight with 95% confidence. Tumor weight and number, locations of nodules, volume of ascites, and mouse body weight means for each group were compared using the two-tailed Student t-test. Measurements for in vitro assays are depicted as the mean ± standard error of the mean. Nonparametric distributions were compared with the Mann-Whitney rank sum test. All statistical analyses were performed with SPSS software (SPSS, Inc., Chicago, IL). For all analyses, a p value < 0.05 was considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT/ANIMAL | SOURCE/SUPPLIER | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse anti-human p13cas antibody | Lab Vision/Neomarkers, Fremont, CA | Cat# MS-855-P1ABX, RRID:AB_145261 |
| Anti-human CD31 monoclonal antibody | Dako, Garpinteria, CA | Agilent Cat# GA610, RRID:AB_2892053 |
| Anti-VEGF Receptor 2 antibody (ab39256), cytoplasmic portion | Abcam | Abcam Cat# ab39256, RRID:AB_883437 |
| Anti-VEGF Receptor 2 antibody (ab45010), extracellular domain | Abcam | Abcam Cat# ab45010, RRID:AB_883436 |
| Bevacizumab | Genentech Inc | N/A |
| B20, anti-human/murine VEGF A antibody | Genentech Inc | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| EGFP-LG3B | Karla Kirkegaard (Addgene plasmid # 11546) | RRID:Addgene_11546 |
| MAP-LC3β2 GRISPR/Gas9 knockout plasmids (Human) | Santa Cruz Biotechnology | Cat# sc-417828, sc-417828-HDR |
| CASP10 - human gene knockout kit via CRISPR/Cas9 (KN206379) | Origene | Cat# KN2G6379 |
| VEGF Receptor 2 (KDR) (NM_GG2253) Human Tagged ORF Glone | Origene | Cat# NM_GG2253 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Human-specific p130cas siRNA (CAGCATCACGCAGGGCAA) | Sigma-Aldrich (St. Louis, MO) | NM_G14567 |
| Murine-specific p130cas siRNA (TTGACTAATAGTCTACATTA) | Sigma-Aldrich (St. Louis, MO) | NM_GG9954 |
| Control non-targeting siRNA (AATTCTCCGAACGTGTCACGT) | Sigma-Aldrich (St. Louis, MO) | SIGGG1 |
| MISSION® esiRNA human TNKS1BP1 (EHU090681) | Sigma-Aldrich (St. Louis, MO) | Cat# EHUG9G681 |
| TNKS1BP1 - Human shRNA constructs in lentiviral vector (TL300907) | Origene | Cat# TL3GG9G7 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6-p130casflox/flox female and male | Dr. Alex Kolodkin, Solomon H. Snyder Department of Neuroscience, Howard Hughes Medical Institute, The Johns Hopkins School of Medicine | N/A |
| Mouse: C57BL/6- Tie2-Cre+/− female and male | Genetically Engineered Mouse Facility at MD Anderson Cancer Center https://www.mdanderson.org/research/research-resources/core-facilities/genetically-engineered-mouse-facility/mouse-resource-facility.html | N/A |
| Female athymic nude mice (NCr-nu) | Taconic | Model # NGRNU-F |
| Female C57/BL6 mice | Taconic | Model # B6-F |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism 7.0 | GraphPad Software, Inc., San Diego, GA | GraphPad Prism, RRID:SGR_002798 |
| Kaplan-Meier survival curves | SPSS version 12 for Windows statistical software (SPSS, Inc., Ghicago, IL) | survival, RRID:SGR_021137 |
Highlights.
p130cas contributes to endothelial resistance to anti-VEGF antibody (AVA) therapy
Internalization of VEGFR2/p130cas occurs during AVA-induced endothelial cell death
TNKS1BP1 mediates cell death associated with nuclear p130cas/VEGFR2
Targeting vascular p130cas extends survival of AVA-resistant ovarian tumors
ACKNOWLEDGMENTS
Portions of this work were supported by the Department of Defense Ovarian Cancer Research Program (W81XWH-20-1-0335 to Y.W.); the National Institutes of Health Uterine Cancer SPORE P50CA098258 (to Y.W. and A.K.S.); the National Comprehensive Cancer Network (to Y.W.); the Marsha Rivkin Center for Ovarian Cancer Research (to Y.W.); NIH grants P50CA217685 (to A.K.S.), R35CA209904 (to A.K.S.), and P30CA016672 (used the Bioinformatics Shared Resource and the Functional Proteomics RPPA Core Facility); the American Cancer Society Research Professor Award (to A.K.S.); the Frank McGraw Memorial Chair in Cancer Research (to A.K.S.); the Ovarian Cancer Research Alliance, the Ovarian Cancer Moonshot Program, the Dunwoody Fund, the Gordon Fund (to A.K.S.); and the Blanton-Davis Ovarian Cancer Research Program (to Y.W. and A.K.S.). A.C.-R. is supported by funds from Deutsche Forschungsgemeinschaft (CH 1733/1-2). We thank Drs. Malgorzata A. Zal and Tomasz Zal for confocal imaging, Imaris image analysis, and quantification. We thank Dr. Bulent Ozpolat and Kenneth Dunner, Jr., for transmission electron microscopy. We thank Amy Ninetto, Kathryn Hale, and Sunita Patterson (Scientific Publications, Research Medical Library, MD Anderson Cancer Center) for editorial work.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110301.
REFERENCES
- Avraham HK, Lee TH, Koh Y, Kim TA, Jiang S, Sussman M, Samarel AM, and Avraham S. (2003). Vascular endothelial growth factor regulates focal adhesion assembly in human brain microvascular endothelial cells through activation of the focal adhesion kinase and related adhesion focal tyrosine kinase. J. Biol. Chem 278, 36661–36668. [DOI] [PubMed] [Google Scholar]
- Basagiannis D, Zografou S, Murphy C, Fotsis T, Morbidelli L, Ziche M, Bleck C, Mercer J, and Christoforidis S. (2016). VEGF induces signalling and angiogenesis by directing VEGFR2 internalisation through macropinocytosis. J. Cell Sci 129, 4091–4104. [DOI] [PubMed] [Google Scholar]
- Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, Mannel RS, Homesley HD, Fowler J, Greer BE, et al. (2011). Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med 365, 2473–2483. [DOI] [PubMed] [Google Scholar]
- Burger RA, Sill MW, Monk BJ, Greer BE, and Sorosky JI (2007). Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: a Gynecologic Oncology Group Study. J. Clin. Oncol 25, 5165–5171. [DOI] [PubMed] [Google Scholar]
- Cabodi S, Tinnirello A, Di Stefano P, Bisaro B, Ambrosino E, Castellano I, Sapino A, Arisio R, Cavallo F, Forni G, et al. (2006). p130Cas as a new regulator of mammary epithelial cell proliferation, survival, and HER2-neu oncogene-dependent breast tumorigenesis. Cancer Res. 66, 4672–4680. [DOI] [PubMed] [Google Scholar]
- Cannistra SA, Matulonis UA, Penson RT, Hambleton J, Dupont J, Mackey H, Douglas J, Burger RA, Armstrong D, Wenham R, et al. (2007). Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J. Clin. Oncol 25, 5180–5186. [DOI] [PubMed] [Google Scholar]
- Chalabi Hagkarim N, Ryan EL, Byrd PJ, Hollingworth R, Shimwell NJ, Agathanggelou A, Vavasseur M, Kolbe V, Speiseder T, Dobner T, et al. (2018). Degradation of a novel DNA damage response protein, tankyrase 1 binding protein 1, following adenovirus infection. J. Virol 92, e02034–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challa DK, Wang X, Montoyo HP, Velmurugan R, Ober RJ, and Ward ES (2019). Neonatal Fc receptor expression in macrophages is indispensable for IgG homeostasis. MAbs 11, 848–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, Stagg LJ, Lad-bury JE, Cai Z, Xu D, et al. (2013). Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 154, 556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KT, Cortesio CL, and Huttenlocher A. (2009). FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J. Cell Biol 185, 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodniewicz D, and Klemke RL (2004). Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim. Biophys. Acta 1692, 63–76. [DOI] [PubMed] [Google Scholar]
- Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV, et al. (2013). An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat. Med 19, 1114–1123. [DOI] [PubMed] [Google Scholar]
- Dalton HJ, Pradeep S, McGuire M, Hailemichael Y, Ma S, Lyons Y, Armaiz-Pena GN, Previs RA, Hansen JM, Rupaimoole R, et al. (2017). Macrophages facilitate resistance to anti-VEGF therapy by altered VEGFR expression. Clin. Cancer Res 23, 7034–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S, Sinha R, Levine DA, Sander C, and Schultz N. (2013). Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun 4, 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliceiri BP, Puente XS, Hood JD, Stupack DG, Schlaepfer DD, Huang XZ, Sheppard D, and Cheresh DA (2002). Src-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signaling. J. Cell Biol 157, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. (1990). What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst 82, 4–6. [DOI] [PubMed] [Google Scholar]
- Fontijn R, Hop C, Brinkman HJ, Slater R, Westerveld A, van Mourik JA, and Pannekoek H. (1995). Maintenance of vascular endothelial cell-specific properties after immortalization with an amphotrophic replication-deficient retrovirus containing human papilloma virus 16 E6/E7 DNA. Exp. Cell Res 216, 199–207. [DOI] [PubMed] [Google Scholar]
- Haemmerle M, Taylor ML, Gutschner T, Pradeep S, Cho MS, Sheng J, Lyons YM, Nagaraja AS, Dood RL, Wen Y, et al. (2017). Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat. Commun 8, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HD, Mangala LS, Lee JW, Shahzad MM, Kim HS, Shen D, Nam EJ, Mora EM, Stone RL, Lu C, et al. (2010). Targeted gene silencing using RGD-labeled chitosan nanoparticles. Clin. Cancer Res 16, 3910–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hida K, Maishi N, Annan DA, and Hida Y. (2018). Contribution of tumor endothelial cells in cancer progression. Int. J. Mol. Sci 19, 1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda H, Oda H, Nakamoto T, Honda Z, Sakai R, Suzuki T, Saito T, Nakamura K, Nakao K, Ishikawa T, et al. (1998). Cardiovascular anomaly, impaired actin bundling and resistance to Src-induced transformation in mice lacking p130Cas. Nat. Genet 19, 361–365. [DOI] [PubMed] [Google Scholar]
- Imbert E, Poot AA, Figdor CG, and Feijen J. (1998). Different growth behaviour of human umbilical vein endothelial cells and an endothelial cell line seeded on various polymer surfaces. Biomaterials 19, 2285–2290. [DOI] [PubMed] [Google Scholar]
- Jackson AL, and Linsley PS (2010). Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov 9, 57–67. [DOI] [PubMed] [Google Scholar]
- Jackson WT, Giddings TH Jr., Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, and Kirkegaard K. (2005). Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3, e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong DE, Lee EK, Song WK, and Kim W. (2014). The 31-kDa caspase-generated cleavage product of p130Cas antagonizes the action of MyoD during myogenesis. Biochem. Biophys. Res. Commun 444, 509–513. [DOI] [PubMed] [Google Scholar]
- Kano A, Wolfgang MJ, Gao Q, Jacoby J, Chai GX, Hansen W, Iwamoto Y, Pober JS, Flavell RA, and Fu XY (2003). Endothelial cells require STAT3 for protection against endotoxin-induced inflammation. J. Exp. Med 198, 1517–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Kook S, Kim DJ, Teodorof C, and Song WK (2004). The 31-kDa caspase-generated cleavage product of p130cas functions as a transcriptional repressor of E2A in apoptotic cells. J. Biol. Chem 279, 8333–8342. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, and Yanagisawa M. (2001). Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Develop. Biol 230, 230–242. [DOI] [PubMed] [Google Scholar]
- Klemke RL, Leng J, Molander R, Brooks PC, Vuori K, and Cheresh DA (1998). CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J. Cell Biol 140, 961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. (2008). Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4, 151–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kook S, Shim SR, Choi SJ, Ahnn J, Kim JI, Eom SH, Jung YK, Paik SG, and Song WK (2000). Caspase-mediated cleavage of p130cas in etoposide-induced apoptotic Rat-1 cells. Mol. Biol. Cell 11, 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krock BL, Skuli N, and Simon MC (2011). Hypoxia-induced angiogenesis: good and evil. Genes Cancer 2, 1117–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landen CN Jr., Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, and Sood AK (2005). Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 65, 6910–6918. [DOI] [PubMed] [Google Scholar]
- Lengyel E, Burdette JE, Kenny HA, Matei D, Pilrose J, Haluska P, Nephew KP, Hales DB, and Stack MS (2014). Epithelial ovarian cancer experimental models. Oncogene 33, 3619–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YG, Han LY, Kamat AA, Merritt WM, Landen CN, Deavers MT, Fletcher MS, Urbauer DL, Kinch MS, and Sood AK (2007). EphA2 over-expression is associated with angiogenesis in ovarian cancer. Cancer 109, 332–340. [DOI] [PubMed] [Google Scholar]
- Lu C, Bonome T, Li Y, Kamat AA, Han LY, Schmandt R, Coleman RL, Gershenson DM, Jaffe RB, Birrer MJ, et al. (2007). Gene alterations identified by expression profiling in tumor-associated endothelial cells from invasive ovarian carcinoma. Cancer Res. 67, 1757–1768. [DOI] [PubMed] [Google Scholar]
- Lyons YA, Pradeep S, Wu SY, Haemmerle M, Hansen JM, Wagner MJ, Villar-Prados A, Nagaraja AS, Dood RL, Previs RA, et al. (2017). Macrophage depletion through colony stimulating factor 1 receptor pathway blockade overcomes adaptive resistance to anti-VEGF therapy. Oncotarget 8, 96496–96505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt WM, Nick AM, Carroll AR, Lu C, Matsuo K, Dumble M, Jennings N, Zhang S, Lin YG, Spannuth WA, et al. (2010). Bridging the gap between cytotoxic and biologic therapy with metronomic topotecan and pazopanib in ovarian cancer. Mol. Cancer Ther 9, 985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, and Schlaepfer DD (2006). Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cel. Biol 18, 516–523. [DOI] [PubMed] [Google Scholar]
- Nagashima K, Endo A, Ogita H, Kawana A, Yamagishi A, Kitabatake A, Matsuda M, and Mochizuki N. (2002). Adaptor protein Crk is required for ephrin-B1-induced membrane ruffling and focal complex assembly of human aortic endothelial cells. Mol. Biol. Cell 13, 4231–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayana A, Kelly P, Golfinos J, Parker E, Johnson G, Knopp E, Zagzag D, Fischer I, Raza S, Medabalmi P, et al. (2009). Antiangiogenic therapy using bevacizumab in recurrent high-grade glioma: impact on local control and patient survival. J. Neurosurg 110, 173–180. [DOI] [PubMed] [Google Scholar]
- Nick AM, Stone RL, Armaiz-Pena G, Ozpolat B, Tekedereli I, Graybill WS, Landen CN, Villares G, Vivas-Mejia P, Bottsford-Miller J, et al. (2011). Silencing of p130cas in ovarian carcinoma: a novel mechanism for tumor cell death. J. Natl. Cancer Inst 103, 1596–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh K, Mangala LS, Han HD, Zhang N, Pradeep S, Wu SY, Ma S, Mora E, Rupaimoole R, Jiang D, et al. (2017). Differential effects of EGFL6 on tumor versus wound angiogenesis. Cell Rep. 21, 2785–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C, Wu S, Han H-D, Shah MY, Rodriguez-Aguayo C, et al. (2013). Tumour angiogenesis regulation by the miR-200 family. Nat. Commun 4, 2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previs RA, Armaiz-Pena GN, Ivan C, Dalton HJ, Rupaimoole R, Hansen JM, Lyons Y, Huang J, Haemmerle M, Wagner MJ, et al. (2017). Role of YAP1 as a marker of sensitivity to dual AKT and P70S6K inhibition in ovarian and uterine malignancies. J. Natl. Cancer Inst 109, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccomagno MM, Sun LO, Brady CM, Alexandropoulos K, Seo S, Kurokawa M, and Kolodkin AL (2014). Cas adaptor proteins organize the retinal ganglion cell layer downstream of integrin signaling. Neuron 81, 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MA, and Basson MD (2005). p130cas but not paxillin is essential for Caco-2 intestinal epithelial cell spreading and migration on collagen IV. J. Biol. Chem 280, 23516–23522. [DOI] [PubMed] [Google Scholar]
- Schaaf MB, Houbaert D, Meçe O, and Agostinis P. (2019). Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ. 26, 665–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, Giriat I, Schmitt A, and de Lange T. (1998). Tankyrase, a poly(-ADP-ribose) polymerase at human telomeres. Science 282, 1484–1487. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Yamaguchi S, Chida K, and Shibuya M. (2001). A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 20, 2768–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Chun HJ, Wong W, Spencer DM, and Lenardo MJ (2001). Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. U S A 98, 13884–13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate code and all data have been presented in figures and supplemental figures. The uncropped western blots related to Figures 2, 3, 4, S1–S3, S6, and S7, and the IPA analysis and graphic are included in Data S1. Other original images will be made available upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.