

Summary
Gene silencing by heterochromatin plays a crucial role in cell identity. Here, we characterize the localization, the biogenesis, and the function of an atypical heterochromatin, which is simultaneously enriched in the typical H3K9me3 mark and in H3K36me3, a histone mark usually associated with gene expression. We identified thousands of dual regions in mouse embryonic stem (ES) cells that rely on the histone methyltransferases SET domain bifurcated 1 (SETDB1) and nuclear set domain (NSD)-containing proteins to generate H3K9me3 and H3K36me3, respectively. Upon SETDB1 removal, dual domains lose both marks, gain signatures of active enhancers, and come into contact with upregulated genes, suggesting that it might be an important pathway by which genes are controlled by heterochromatin. In differentiated tissues, a subset of these dual domains is destabilized and becomes enriched in active enhancer marks, providing a mechanistic insight into the involvement of heterochromatin in the maintenance of cell identity.
Keywords: heterochromatin, gene expression, enhancer, cellular identity
Graphical abstract
Highlights
-
•
H3K9me3 and H3K36me3 dual domains form on thousands of regions in ES cells
-
•
Dual domains depend on SETDB1 and NSD enzymes
-
•
Most upregulated genes in Setdb1 KO cells are not normally heterochromatinized
-
•
Dual domains become enhancers for these genes upon Setdb1 loss
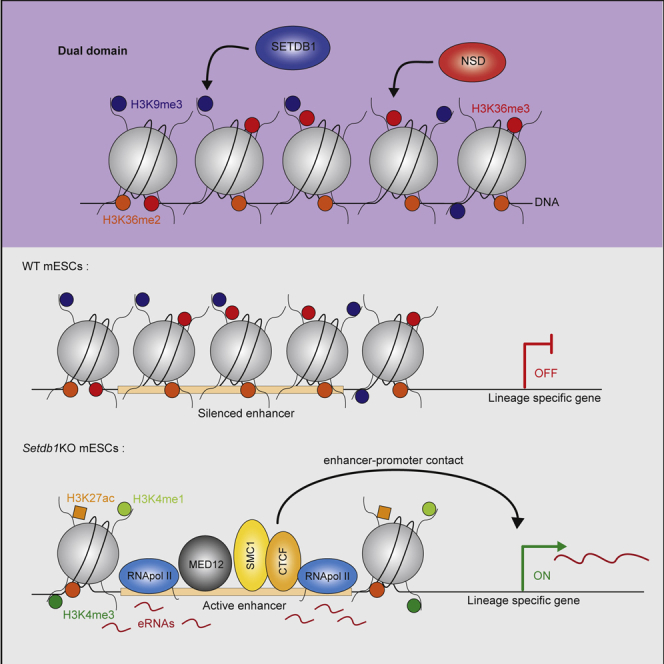
H3K9me3-marked heterochromatin is important to maintain cellular identity, but the mechanism is unknown. Barral et al. discover that in mouse ES cells, heterochromatin can control gene expression by concealing a subset of enhancers.
Introduction
Cellular identity is conferred by specific gene expression profiles that are established and maintained by tissue-specific transcription factors and chromatin regulation. Combinations of specific histone modifications are characteristic signatures of gene activity. Active genes typically harbor an open chromatin structure at their promoters, which allows access to the transcription machinery. They also display an enrichment in histone H3 lysine 4 trimethylation (H3K4me3) at their promoters, a mark deposited by mixed lineage leukemia (MLL) complexes (Dou et al., 2006; Steward et al., 2006; Wang et al., 2009), whereas gene bodies are enriched in histone H3 lysine 36 trimethylation (H3K36me3). In mammals, H3K36me3 is deposited by SET domain 2 (SETD2) (Edmunds et al., 2008; Yuan et al., 2009b), an enzyme that interacts with the RNA polymerase II (RNA Pol II) complex during transcriptional elongation. Enrichment of both H3K4me3 on the promoter and of H3K36me3 on the gene body represents a universal signature of gene activity (Huang and Zhu, 2018; Hyun et al., 2017; McDaniel and Strahl, 2017; Ruthenburg et al., 2007; Zhang et al., 2015).
Epigenomic signatures characteristic of gene silencing are more complex and diverse. A large subset of developmentally silenced genes is targeted by proteins of the Polycomb group and usually harbors an enrichment in promoter-bound H3 lysine 27 trimethylation (H3K27me3) (Simon and Kingston, 2009). Other silent genes are not controlled by the Polycomb pathway but either harbor no typical histone modification or an enrichment in H3K9 trimethylation (H3K9me3) at their promoter (Ernst and Kellis, 2010). H3K9me3 can be synthesized by three histone methyltransferases: SET domain bifurcated-1 (SETDB1) and suppressor of variegation 3–9 homologs 1 and 2 (SUV39H1/2) (O’Carroll et al., 2000; Peters et al., 2001; Schultz et al., 2002; Wang et al., 2003). During lineage specification and terminal differentiation, H3K9me3 is deposited and removed in a cell-type-specific fashion, suggesting an involvement in development (Hawkins et al., 2010; Nicetto et al., 2019). During this process, the loss of H3K9me3 by abrogating SETDB1 and SUV39H leads to the acquisition of aberrant gene expression profiles, suggesting that H3K9me3-dependent activities are crucial to control cellular identity. The bulk of H3K9me3 is found on DNA repeats, such as transposable elements and satellite DNA, which usually assemble into heterochromatin (Saksouk et al., 2015). Gene silencing by heterochromatin is thought to essentially occur either via a direct assembly on the promoter or via the spreading of heterochromatin from a repeat region into a neighboring promoter, which restricts transcription, a phenomenon called the position effect (Allshire and Madhani, 2018; Elgin and Reuter, 2013; Nicetto and Zaret, 2019; Timms et al., 2016).
In cells that have not yet committed to a specific developmental fate, like embryonic stem cells, H3K9me3 does not spread much from DNA repeats to flanking regions (Rebollo et al., 2011). This mark is also found on several thousand genes (Bilodeau et al., 2009). SETDB1 installs H3K9me3 at these genes and at LTR retrotransposon insertions (Bilodeau et al., 2009; Matsui et al., 2010), while SUV39H1 and SUV39H2 enzymes essentially target pericentromeric regions (Bulut-Karslioglu et al., 2014; Peters et al., 2001). Knocking out Setdb1 results in early embryonic lethality, likely because embryonic stem cells acquire an aberrant gene expression profile (Bilodeau et al., 2009; Lohmann et al., 2010; Yuan et al., 2009a). The loss of SETDB1 in more differentiated tissues can also lead to the activation of transcriptional programs typical of other tissues (Jiang et al., 2017; Koide et al., 2016; Nicetto et al., 2019; Tan et al., 2012; Wu et al., 2020), suggesting that SETDB1 controls tissue-specific gene expression profiles. However, most upregulated genes in Setdb1 KO cells are not marked by H3K9me3 on their promoters in a normal context, suggesting that gene control does not generally occur through direct heterochromatinization (Karimi et al., 2011). Recently, the loss of SETDB1 in neurons was shown to affect the protocadherin gene cluster and higher-order organization by allowing aberrant CTCF redistribution (Jiang et al., 2017), suggesting that SETDB1 might also control long-range genomic interactions in neurons. Hence, currently available data indicate that SETDB1-dependent activities are crucial for the maintenance of cellular identity, but tissue-specific gene silencing by SETDB1 appears largely indirect.
Several studies have previously uncovered an atypical type of SETDB1-regulated chromatin harboring two marks of seemingly opposing functions at the 3′ end of a subset of zinc finger genes (Blahnik et al., 2011; Valle-García et al., 2016). These regions have both H3K9me3 and H3K36me3, most likely on the same nucleosome, and they are also found on weakly transcribed regions and in a subset of weak putative enhancers (Mauser et al., 2017). We also found this atypical combination on telomeres in ES cells, which are bound by SETDB1 and transcribed (Gauchier et al., 2019). Whether the co-occurrence of both marks is a mere consequence of a pre-existing H3K9me3 domain inside a transcribed region or represents a signature for a specific regulatory function is unknown.
Here, we used mouse embryonic stem cells to characterize H3K9me3/H3K36me3 domains. These ∼4-kb domains are SETDB1, as well as nuclear set domain (NSD) containing enzymes dependent for H3K9me3 and H3K36me3, respectively. Unexpectedly, however, they are mostly SETD2 independent for H3K36me3. They are functionally distinct from other H3K9me3 regions since they form on enhancers to restrict their gene-activating function. We propose that the maintenance of cell identity mediated by SETDB1 operates, at least in part, through the direct control of these dual domains.
Results
H3K9me3/H3K36me3 domains
To explore the distribution of H3K9me3 and H3K36me3 in ES cells, we used chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq). We used ChIP-grade H3K9me3- and H3K36me3-specific antibodies, which we independently validated (Figures S1A–S1C). As expected, the majority of H3K9me3 is found outside genes (67% of the 55,770 called peaks that could be merged into 31,424 genomic regions; Figure 1A), often (70%) colocalizing with annotated retrotransposon insertions (Figure S1D), while H3K36me3 is essentially found within genes (93% of the 110,986 called peaks that were merged into 25,436 regions; Figure 1A). While generally not correlating, they co-occurred at 3,015 regions (4,744 peaks; Figures 1B–1D, see experimental procedures) that have an average size of 4 kb. The number of observed overlapping peaks is significantly higher than simulated overlaps (Figure S1E), suggesting a concerted process at these regions. The H3K36me3 signal colocalizing with H3K9me3 is generally comparable to that found on moderately transcribed genes (data not shown). We confirmed the presence of both marks on a subset of targets by ChIP-qPCR (Figure S1F). The majority (65%) of these domains were located inside genes, mostly in introns (Figure 1C) and had a lower content of retrotransposons than other H3K9me3 regions (48% versus 70%, Figure S1D). We categorized two types of H3K9me3/H3K36me3 regions (Figure 1E): 62% that we termed “colocalized” because both H3K9me3 and H3K36me3 profiles were similar (1,866 regions/3,207 peaks where more than 60% of an H3K36me3 region is also fully covered by H3K9me3 or when an H3K9me3 region is only partly covered by H3K36me3). This category is essentially found in intergenic regions and within poorly expressed or silent genes (Figures S1G and S1H). The second type consists of “embedded” domains (38%), corresponding to H3K9me3 islands within broader H3K36me3 domains (1,149 regions/1,537 peaks). Embedded domains occur inside active genes and result from the presence of H3K9me3 islands within an active gene body (Figures 1E, S1G, and S1H).
Figure 1.

H3K9me3 and H3K36me3 colocalize at thousands of sites in mouse ES cells
(A) Left: H3K9me3 distribution and tracks for intergenic and intragenic regions (blue). Right: H3K36me3 distribution and tracks for intergenic and intragenic regions (red). Reference sequence (RefSeq) track with annotated genes from mm10 genome. Endogenous retrovirus (ERV) track contains all annotated LTR transposable elements. Rmsk, RepeatMasker, track contains all annotated DNA repeat sequences.
(B) Venn diagram showing the overlap between H3K9me3 and H3K36me3.
(C) Genome-wide distribution of H3K9me3/H3K36me3 colocalized domains and genome browser tracks of H3K9me3 and H3K36me3. Purple bars show H3K9me3/H3K36me3 domains based on peak calling.
(D) H3K9me3 and H3K36me3 distributions on H3K9me3/H3K36me3 regions, ranked by H3K9me3 enrichment.
(E) Distribution of H3K9me3/H3K36me3 regions according to percentage of overlap between H3K9me3 domains and H3K36me3 domains allowing to categorize “colocalized” and “embedded” domains (STAR Methods for details). Right: tracks of H3K9me3 and H3K36me3 at “colocalized” H3K9me3/H3K36me3 regions (top) and “embedded” H3K9me3/H3K36me3 regions (bottom).
(F) Enrichment of H3K9me3 (blue), H3K36me3 (red), and CIDOP (purple) chromatin on H3K9me3/H3K36me3 regions or on other H3K9- or H3K36me3 regions.
(G) Tracks of H3K9me3, H3K36me3, and CIDOP on “colocalized” H3K9me3/H3K36me3 regions (left) and “embedded” H3K9me3/H3K36me3 regions (right).
(H) ChIP-reChIP for H3K9me3 (blue) and H3K36me3 (red) fold-enrichment after input normalization in positive dual regions (In.), or in flanking regions (Flk.), intracisternal A particle (IAPs, used as a H3K9me3 control) or Bzw1 transcribed gene body (used as H3K36me3 control) in WT cells (∗, Student’s t test p value < 0.05). Also see Figure S1 and Table S1.
The overlap of the two marks could occur on the same fiber, could result from allelic differences in the epigenome, or to some admixture of distinct cell populations locally bearing only H3K9me3 or H3K36me3, as ES cells are notoriously heterogeneous. To address this, we used chromatin interacting domain precipitation (CIDOP; Figure S1I) that exploits an artificial adapter protein (PWWP-MPP8) made of the DNA methyl transferase 3A PWWP domain, which binds to H3K36me2/3, and of the MPP8 chromodomain, which binds to H3K9me3. The presence of both marks on the same chromatin fiber stabilizes binding of the dual adapter, thereby allowing specific capture, while either mark alone is not sufficient (Mauser et al., 2017). We used this protein to capture mononucleosomal chromatin and confirmed enrichment of both H3K9me3 and H3K36me3 in these preparations (Figures S1J and S1K). When combined with sequencing, CIDOP showed a robust enrichment of the H3K9me3/H3K36me3 regions defined by ChIP-seq analyses, while other H3K9me3 or H3K36me3 regions were weakly or not significantly enriched (Figures 1F, 1G, S1L, and S1M). This was validated by qPCR (Figure S1N). As the adapter used in CIDOP is able to also capture H3K9me3/H3K36me2 chromatin, we mapped H3K36me2 by ChIP-seq. H3K9me3/H3K36me3 doubly marked regions were strongly enriched for H3K36me2 (Figure S1O), while other H3K9me3 regions have a weak enrichment for H3K36me2, which might explain the very weak CIDOP enrichments occasionally observed on these regions (Figures 1F and S1P). Regions highly enriched for H3K36me2 or for H3K9me3 were not detected by the H3K36me3 antibody in ChIP-seq (Figure S1Q), arguing against the possibility that the H3K36me3 antibody cross-reacted with H3K36me2 or H3K9me3 in our assays. In support, we performed ChIP-reChIP assays on mononucleosomal chromatin (Figure 1H). H3K9me3 and H3K36me3 signals are coenriched on recaptured mononucleosomes from H3K9me3/H3K36me3 regions. Thus, these chromatin regions are made of nucleosomes doubly modified for H3K9me3 and H3K36me3.
H3K9/K36me3 domains are not bivalent
Both H3K4me3 and H3K27me3 are enriched in a subset of poised gene promoters in ES cells. Gene-specific activation or repression during differentiation correlates with the corresponding loss of one of the two marks, resolving into a monovalent domain. Hence, the term bivalent was coined to describe the apparent antagonism that exists locally between H3K4me3- and H3K27me3-dependent activities. We wanted to test if the signature described here follows this rule by perturbing either H3K9me3 or H3K36me3 and measuring the other mark. Most of these domains are strongly bound by SETDB1 (Figures S2A and S2B), with a generally stronger SETDB1 binding than on other H3K9me3 regions. Hence, we used an ES cell line harboring a tamoxifen-inducible Setdb1 knockout (Matsui et al., 2010) to perturb H3K9me3 deposition. Setdb1 KO induced a loss of H3K9me3, both at the H3K9me3 and H3K36me3 domains and also on other H3K9me3 regions (Figures 2A–2C, S2C, and S2D). There was also a ∼50% reduction in SETD2 protein levels, which led to a corresponding reduction in global H3K36me3 levels (Figure 2A). Consequently, H3K36me3 levels at gene bodies were also affected (Figure S2C). Intriguingly, the reduction of H3K36me3 was comparatively more drastic at the domains bearing both H3K9me3 and H3K36me3 (FC = −0,487 at dual domains and −0,296 at other H3K36me3 regions, Figures 2B, 2C, and S2D), while H3K36me2 was still strongly enriched upon SETDB1 loss (Figure S2E). Thus, both H3K9me3 and H3K36me3 seem to rely on SETDB1 in these regions, while H3K36me2 enrichment remains largely independent. We also analyzed inducible Setdb1 KO cells re-expressing a SETDB1 catalytic dead mutant, called Rescue-C1243A/T (Figure S2F). In these cells, the SETDB1 loss led to a loss of both H3K9me3 and H3K36me3 on dual domains (Figure S2G), indicating that both marks are dependent on SETDB1 catalytic activity. Since classical bivalent domains rely on distinct machineries, the domains we describe here do not seem to have this feature, and henceforth, we use the term “dual” to define them.
Figure 2.

Both H3K9me3 and H3K36me3 depend on SETDB1
(A) SETDB1, H3K9me3, SETD2, H3K36me3, and PCNA immunoblots performed on nuclear extracts from WT, Setdb1 KO, Setd2 KO, and double KO cells.
(B) Enrichment of H3K9me3 and H3K36me3 on H3K9me3/H3K36me3 regions and on other H3K9me3 or H3K36me3 regions in WT and Setdb1 KO cells (Wilcoxson signed rank test: ∗p value = 2.2 × 10−16; 2.2 × 10−16; and 2.2 × 10−16, respectively).
(C) Tracks of H3K9me3 and H3K36me3 on H3K9me3/H3K36me3 regions compared with other H3K9me3 or H3K36me3 regions in WT and Setdb1 KO cells. Also see Figure S2 and Table S1.
H3K36me3 in dual domains essentially relies on NSD1/2/3 proteins
Since SETD2 is the major trimethylase for H3K36 (Edmunds et al., 2008; Yuan et al., 2009b), we also generated a Setd2 KO by CRISPR to be able to analyze the impact of the loss of either or both H3K9me3 and H3K36me3. The Setd2 KO resulted in a loss of both SETD2 proteins and mRNA (Figures 2A and S3A). It also led to the anticipated H3K36me3 abrogation in gene bodies (Figure S3B) and had no effect on SETDB1 expression, H3K9me3 deposition, or ES cell pluripotency (Figures 2A, 3A, S3C, and S3D and data not shown). More unexpected was the modest H3K36me3 reduction on dual domains (Figures 3A, S3C, and S3D). Moreover, the dynamics of H3K36me2 and H3K36me1 were also distinct: both increased on active gene bodies in the Setd2 KO (Figures 3B and S3B), as described (Edmunds et al., 2008; Zhou et al., 2018), but were globally unaffected on the dual domains, suggesting different regulations. In more detail, we found that the loss of SETD2 had no major effect on H3K36me3 levels in the “colocalized” domains (5% reduction; Figure 3C), while H3K36me3 levels were reduced by 2-fold (but not abrogated) in “embedded” domains. These observations argue against the interpretation that dual domains are the mere result of H3K9me3 regions located inside SETD2-regulated regions. This was also unexpected because SETD2 is thought to be responsible for all H3K36 trimethylation. Consistently, H3K36me3 signals measured by immunoblots were strongly reduced (4–8 times less) in the Setd2 KO cells, but not abrogated (Figure S3E). To confirm that there is a pool of H3K36me3 that is SETD2 independent, we used quantitative mass spectrometry on acid-extracted histones to measure this mark specifically. H3K36me3 was reduced, but not abrogated in the Setd2 KO cells (Figure S3F), in line with previous findings (Baubec et al., 2015). Our reanalysis of H3K36me3 ChIP-seq data from WT and Setd2KO mesenchymal stem cells (Weinberg et al., 2019) indicated that those cells also have a pool of H3K36me3, which is insensitive to Setd2 KO (Figure S3G) and also enriched for H3K9me3 and bound by SETDB1 (Figure S3H). Because dual domains were also strongly enriched in H3K36me2 (Figure S1O), we hypothesized that other H3K36 methylases normally involved in H3K36 mono- and di-methylations, like NSD-containing proteins 1, 2, or 3 (NSD1/2/3) (Kuo et al., 2011; Li et al., 2009b), might also be involved in H3K36 trimethylation activity in a SETDB1-dependent manner. Recent analyses (Villaseñor et al., 2020), and our unpublished locus-specific proteomics results, indicated that both NSD1 and NSD2 were found enriched with SETDB1 on H3K9me3 regions. We thus knocked out each NSD by CRISPR (Figure S3I) and found that NSD1, NSD2, and NSD3 were involved in H3K36me3 enrichment in the dual domains, as measured by ChIP-qPCR (Figure 3D); while there was an increase (albeit not significant) in local H3K9me3. We also reanalyzed H3K36me3 ChIP-seq profiles in Nsd1 KO ESCs (Weinberg et al., 2019) and found that there was a drop of H3K36me3, more pronounced in “colocalized” domains than in “embedded” domains (Figures 3E and 3F). H3K36me3 reductions were observed particularly on colocalized domains in Nsd1;Nsd2 double KO cells (Figure S3J). These data suggest that NSD proteins act together downstream of SETDB1 to deposit the H3K36me3 mark on dual domains, that SETD2 is minimally involved in this process, and that NSD proteins might counteract SETDB1 on dual domains.
Figure 3.

NSD proteins trimethylate H3K36 on dual domains
(A) Enrichment of H3K9me3 and H3K36me3 on dual domains and on other H3K9me3 or H3K36me3 regions in WT, Setd2KO, Setdb1 KO, and double KO cells (Wilcoxson signed rank test: ∗p value = 2.2 × 10−16; 2.2 × 10−16; 2.2 × 10−16, respectively).
(B) H3K36me3 (red), H3K36me2 (orange) and H3K36me1 (yellow) enrichment on dual domains and other H3K36me3 regions in WT and Setd2 KO cells (Wilcoxson signed rank test: ∗p value = 2.2 × 10−16; 2.2 × 10−16 for H3K36me3; p value = 6.83 × 10−3; 2.4 × 10−4 for H3K36me2; p value = 6.83 × 10−3; 2.4 × 10−4 for H3K36me1 on dual domains and other H3K36me3 regions, respectively).
(C) Left: H3K36me3 enrichment on “colocalized” and on “embedded” dual domains in WT and Setd2 KO cells (Wilcoxson signed rank test: ∗p value = 2.2 × 10−16; 2.2 × 10−16 on colocalized dual domains and embedded dual domains, respectively). Right: tracks of H3K9me3 and of H3K36me3 on colocalized and embedded dual domains in WT and Setd2 KO cells.
(D) ChIP-qPCR showing the relative H3K36me3 fold-enrichment (left) and H3K9me3 fold-enrichment (right) after input normalization inside positive H3K9me3/H3K36me3 regions (In.), or in Flanking regions (Flk), on intracisternal A particle (IAP, used as a H3K9me3 domain control) or on the Csrnp1 transcribed gene body (used as a H3K36me3 domain control) in WT, Nsd1 KO, Nsd2 KO, and Nsd3 KO cells (∗: Student’s t test p value < 0.05).
(E) Left: H3K36me3 on transcribed gene body regions and on embedded dual domains in WT and in Nsd1 KO cells. Right: tracks of H3K36me3 on a gene body and on an “embedded” dual domain in WT and Nsd1 KO cells.
(F) Left: H3K36me3 enrichment on “colocalized” dual domains in WT and Nsd1 KO cells. Right: Tracks of H3K36me3 on a “colocalized” dual domain in WT and Nsd1 KO cells. Also see Figure S3 and Table S1.
Dual domains do not generally control host genes expression
We performed RNA-seq experiments in Setdb1 KO ES cells (Figure S4A) and also analyzed data from other Setbd1 KO tissues (Jiang et al., 2017; Nicetto et al., 2019) but found no general correlation between promoter H3K9me3 changes and changes in gene expression profiles (Figures S4B–S4D), arguing against a direct gene promoter repression. In ES cells, only 7% (61/908) of upregulated genes had H3K9me3 on their promoters, 5% (43/908) had a neighboring (<5 kb) retrotransposon insertion, and 11% (103/908) hosted a dual domain (Figure 4A). Therefore, the bulk of upregulated genes (77%, 701/908) are not direct SETDB1 targets, in line with previous observations (Karimi et al., 2011). Most genes that host a dual domain (1,130/1,233) did not change their expression either, indicating that a dual domain generally does not control the expression of its associated host gene (Figure 4B). To explore host gene regulation further, we also analyzed other marks and players involved in transcription. We noticed a very small enrichment of H3K4me3, RNA Pol II, and its initiating phosphorylated form on Serine 5 (Ser5-P) at intragenic and intergenic dual domains, which increased in Setdb1 KO cells, while these markers were unchanged at the promoter (Figures 4C, 4D, S4E, and S4F). It is unlikely that intragenic dual domains represent cryptic promoters because the stranded enrichment profiles did not display the classic biphasic shape observed at active transcription start sites (Guenther et al., 2007; Howe et al., 2017) (Figure S4G), and their enrichment levels were much lower (19.83- and 8.37-fold lower for H3K4me3 and RNA Pol II, respectively). We also checked if intragenic dual domains were involved in modulating splicing, as both H3K9me3 and H3K36me3 marks are known to regulate this process (Luco et al., 2010; Pradeepa et al., 2012; Saint-André et al., 2011). Exon read enrichment along genes, or exon junctions, as well as unusual splicing junctions, was comparable between host genes and a set of random genes upon Sedtb1 KO (Figure S4H), suggesting no major effect on splicing modulation. However, we observed local gains in RNA matching dual domains on both strands, regardless of whether they were intragenic or intergenic (Figures 4E, 4F, and S4I). To confirm these results, we performed a northern blot using a probe able to detect transcripts from the Etv5 gene, which is active and hosts a dual domain. SETDB1 removal had no effect on the steady-state level of the Etv5 mRNA, and there was also no appearance of alternative transcripts. However, when we made a probe specific to the dual domain, we noticed the appearance of RNAs of about 1,000 nt in length (Figure S4J) upon SEDTB1 removal, consistent with the sequencing data. Hence, dual domains might inhibit local transcription.
Figure 4.

Dual domain might repress local bidirectional transcription
(A) Number of upregulated genes in Setdb1 KO cells and their status in WT cells.
(B) Log2 fold change in expression for a random set of genes (white), for genes hosting a dual domain (purple) and for upregulated genes in Setdb1 KO (gray) (Wilcoxson signed rank test: × p value = 2.2.10−16).
(C) H3K4me3, RNA Pol II and Serine 5 (Ser-5) on dual domains and on other H3K9- or H3K36me3 regions in WT and Setdb1 KO cells (Wilcoxson signed rank test: ∗p value = n.s; 0.042; 0.021 for H3K4me3, p value = 1.10 × 10−9; 2.2 × 10−16; 5.538 × 10−4 for RNA Pol II, and p value = 2.2 × 10−16; 2.2 × 10−16; n.s. for Ser5-P on dual domains, other H3K9me3 regions and other H3K36me3 regions, respectively).
(D) H3K4me3, RNA Pol II and Serine 5 (Ser5-P) levels on intragenic (left) and on intergenic (right) dual domains in WT, Setdb1 KO cells.
(E) Expression levels, log2(Fpkm+1), of intragenic (left) and intergenic (right) dual domains in WT and Setb1 KO cells (Wilcoxson signed rank test: ∗p value = 8.9 × 10−3; ∗∗p value = 5.2 × 10−12).
(F) Sense (left) and anti-sense (right) transcription levels, log2(Tpm+1), on dual domains in WT and Setdb1 KO cells (Wilcoxson signed rank test: ∗p value = 3.8 × 10−6; ∗∗p value < 2.2 × 10−16). Also see Figure S4 and Table S1.
Dual domains restrict enhancer elements
The above findings prompted us to explore the possibility that dual domains might conceal putative enhancers. We thus analyzed known enhancer marks such as H3K27 acetylation (H3K27ac) and H3K4 monomethylation (H3K4me1) (Creyghton et al., 2010; Heintzman et al., 2009; Rada-Iglesias et al., 2011; Whyte et al., 2013). There was a small enrichment of both marks on dual domains in wild-type cells, which increased upon SETDB1 removal, a feature that was not generally observed for other H3K9me3 or H3K36me3 domains (Figures 5A and S5A). We also measured mediator complex recruitment by analyzing the binding of one of its subunits (Med12), as it is known to mark active enhancers (Kagey et al., 2010). We detected Med12 enrichment on 9% (287/3,015 regions; 391/4,744 peaks) of dual domains, which increased in both signal intensity and number to 30% (904/3,015 regions; 1,281/4,744 peaks) upon SETDB1 removal, a feature uncommon for other H3K9me3- or H3K36me3-marked regions (Figures 5B and S5A). Finally, we measured local DNA accessibility by performing ATAC-seq (Corces et al., 2017). Dual domains are essentially inaccessible but become broadly open upon SETDB1 removal (Figures 5C, S5B, and S5C). A much weaker gain in DNA accessibility was also measured on other H3K9me3 regions, likely due to specific LTR reactivation in the absence of SETDB1. While other putative enhancers did not seem affected upon SETDB1 removal, dual domains systematically gained enhancer chromatin hallmarks to a similar or even higher extent (Figures S5D and S5E). We also noted that the two types of dual domain, colocalized and embedded, gained both H3K27ac and DNA accessibility (Figure S5F) and other enhancer chromatin marks (H3K4me1, H3K4me3, RNA Pol II, and MED12, data not shown) to similar extents, suggesting that they are not functionally distinct. Thus, dual domains restrict putative enhancers. To test the functionality of these putative enhancers, we cloned 7 dual domains that we placed downstream of a luciferase reporter gene on a plasmid (Figure 5D). All but one domain tested in transient transfections (6/7) had a stimulatory effect on luciferase, comparable to the SV40 enhancer used as a positive control, while sequences from other H3K9me3 or H3K36me3 domains seemed to have a silencing effect (Figure 5D). We conclude that the sequences forming dual domains can act as functional enhancers.
Figure 5.

Dual domains repress functional enhancers
(A) H3K27ac (left) and H3K4me1 (right) enrichment on dual domains, on other H3K9me3 regions or other H3K36me3 regions in WT and Setdb1 KO cells (Wilcoxson signed rank test: ∗: p value = 2.3 × 10−6; 1.85 × 10−15; n.s. for H3K27ac and p value = 1.18 × 10−7; n.s.; 1.36 × 10−10 for H3K4me1 on dual domains, other H3K9me3 regions and other H3K36me3 regions, respectively).
(B) MED12 enrichment on dual domains and on other H3K9me3 regions or other H3K36me3 regions at in WT and Setdb1 KO cells. Heatmaps ranked according to the Med12 signal (Wilcoxson signed rank test: ∗p value = 2.05 × 10−7 for dual domains and n.s for other H3K9me3 and H3K36me3 regions).
(C) ATAC-seq enrichment on dual domains and on other H3K9me3 regions, or other H3K36me3 regions in WT and Setdb1 KO cells (Wilcoxson signed rank test: p value < 2.2 × 10−16).
(D) Top: plasmids used in the enhancer assay. Bottom: normalized luciferase activity after transfection of constructs containing a minimal SV40 promoter only, with an SV40 enhancer, with an H3K9me3 “only” region, with an H3K36me3 “only” region, or 7 selected dual domains. Also see Figure S5 and Table S1.
The enhancer function of dual domains is confined within TADs
Enhancers are distal cis-regulatory elements able to stimulate gene expression by forming enhancer/promoter contacts. In contrast to other H3K9me3 regions, dual domain distances relative to genes are comparable to that of enhancers (Figure S6A). Upon SETDB1 depletion, proteins involved in enhancer/promoter contacts like SMC1 and CTCF (Merkenschlager and Nora, 2016) increased (20%–45% and 16%–20% for SMC1 and CTCF, respectively) on the dual domains (Figures S6B and S6C). Since dual domains losing SETDB1 showed enhancer features, we tested whether they might contact genes that are significantly upregulated in the absence of SETDB1 by performing in situ Hi-C in wild-type and Setdb1 KO cells. First, we assessed the distribution across topologically associated domains (TADs) of genes deregulated upon disruption of dual domains. We observed a clear separation of upregulated and downregulated genes in distinct TADs (Figure 6A). Importantly, a significant portion of TADs harboring an upregulated gene also harbor a dual element (357/545, p value = 3.2 × 10−12; Figure 6B). This effect is further accentuated when we only considered the dual domains that gain a strong enrichment in H3K27ac (high-H3K27ac domains, p value = 2.3 × 10−13) compared with dual domains that had a weak gain of H3K27ac (low-H3K27ac domains, p value = 3.1 × 10−8) upon Setdb1 KO (Figures 6A, 6B, and S6D). On the other hand, downregulated genes showed a much lower association with dual elements within the same TAD, suggesting that downregulated genes are mostly the result of indirect effects (Figure 6B). In order to quantify the global effect of Setdb1 KO on genome-wide chromatin architecture, we stratified all identified TADs based on their epigenetic profiles and quantified the average interaction probabilities between them (Figure S6E). We observed no significant change in the overall TAD interaction patterns between WT and Setdb1 KO ES cells. In addition, the overall TAD organization of the two cell populations appears to be largely conserved (Figure S6F). Interestingly, TADs harboring both deregulated genes and dual domains also showed no change in their contact patterns (Figures S6G–S6I), suggesting that loss of H3K9/K36me3 from dual domains does not change general TAD organization. To further investigate the interaction dynamics of dual domains, we performed paired-end spatial chromatin analysis (PE-SCAn) and observed enhanced looping events with active genes in the Setdb1 KO cells (Figure 6C). This gain of preferential interactions appears to be specific to the “high H3K27ac domains” (Figure 6C, second panel). We next assessed direct interactions between dual domains and upregulated genes and observed a significant gain in interaction probability only between upregulated genes and dual domains, again accentuated on high-H3K27ac domains (Figures 6D and S6J), while other H3K9me3 regions did not generally increase their interaction frequencies with active genes or upregulated genes upon SETDB1 loss (Figures 6C and S6K). These results suggest that gene de-repression in Setdb1KO cells is mainly due to the destabilization of H3K9me3/H3K36me3 dual heterochromatin and not due to the loss of H3K9me3 genome-wide. Indeed, dual domains gained interaction with upregulated genes but not with other genes located within the same TAD (Figures 6E, S6L, and S6M). These data confirmed that in the absence of SETDB1, the reactivated enhancer contacted targets to increase their expression while other genes in the same area were unaffected (Figure S6N). An example of this phenomenon is seen in the Ccny gene region (RNA-seq Log2 FC = 4.02; Figures 6F and 6G). While no obvious changes are seen at the multi-Mb scale in the contact pattern in the region surrounding the locus and in its TAD borders (Figure 6F, top and center), a strong contact is formed in Setdb1 KO cells between the region immediately upstream to the Ccny TSS and two upstream regions, both of them dual domains (Figures 6F [bottom] and 6G). A similar change was also observed for other upregulated genes, e.g., Cd200 and Slc18b1 (Figure 6H). Overall, these data suggest that the action of dual domains is restricted by TAD boundaries, and they show increased contact probabilities with upregulated genes upon Setdb1 KO. We used CRISPR/Cas9 to delete some dual domains contacting upregulated genes upon SETDB1 loss (Figure S6O). In the absence of dual domains, gene upregulation was consistently lower in Setdb1KO cells (Figure 6I), confirming that gene upregulation results directly from the action of dual domains on their target promoter. All these data suggest that dual domains behave like functionally poised enhancers in ES cells.
Figure 6.

Dual domains contact promoters of upregulated genes upon SETDB1 depletion
(A) Overlap of TADs harboring upregulated and downregulated genes (left), or upregulated genes and dual domains with high (right) and low (bottom) levels of H3K27ac (hypergeometric test: p value = 0.925; 2.27 × 10−13; 3.13 × 10−8).
(B) Significance of the overlap between misregulated genes and dual domain classes (hypergeometric test values are shown in each square).
(C) PeSCANs of active genes (FPKM > 10) with all dual domains, dual domains with high H3K27ac and low H3K27ac and other H3K9me3 regions, respectively. The maximum distance allowed for interaction scoring is 3 Mb, and the minimum distance is 100 kb. The window sizes span 50 kb upstream and downstream the interaction centers at a resolution of 1 kb.
(D) Empirical distribution functions illustrating the interaction strength dynamics upon SETDB1 depletion between upregulated genes and dual domains (top), high H3K27ac domains (middle), and low H3K27ac domains (bottom) (KS one-sided test: p = 1.4 × 10−3; p = 1.4 × 10−4; n.s. for all dual domains, high- and low-H3K27ac domains, respectively).
(E) Estimation of intra-TAD target gene specificity of high H3K27a dual domains versus upregulated genes (left) and all the other genes in the same TAD (right) (KS one-sided test: p = 5.7 × 10−3; n.s. upregulated gene and all other genes, respectively).
(F) Multiscale representation of the Hi-C results (from 20 Mb to 240 kb). Top: A large genomic region of chromosome 18 is represented based on observed values (log2). TAD calls are highlighted in yellow. TADs harboring both upregulated genes and dual domains are highlighted in blue. Secondary interactions between them are also highlighted as blue boxes. The close-up region represented in the next plot is highlighted by a gray box. Middle: example of a TAD containing an upregulated gene and 4 dual domains. Potential interactions between them are highlighted by the black squares. TAD boundaries are highlighted in blue. Bin-free, distance normalized, interaction scores (shaman) are shown. Bottom: representation of the Ccny locus interaction patterns (score). Interactions between the promoter of Ccny and dual domains are highlighted with black squares on the Hi-C plot.
(G) Left: virtual 4C plots using the Ccny promoter, the dual domain 1 and the dual domain 2 as viewpoints, respectively. Right: tracks showing H3K9me3, H3K27ac and RNA-seq level on the Ccny locus in WT and Setdb1 KO cells.
(H) Top: virtual 4C plots using the Cd200 (left) or Slc18b1 (right) promoter and the dual domain as viewpoint, respectively. Bottom: tracks showing H3K9me3, H3K27ac, and RNA-seq level on the Cd200 (left) and Slc18b1 (right) locus in WT and Setdb1 KO cells.
(I) RT-qPCR to measure the relative mRNA level from target genes, Cd200, Dock9, Mtr, and Ccny, respectively, of after normalization in WT, Setdb1KO, deleted dual domain clones and Setdb1KO-deleted dual domain clones (∗: Student’s t test p value < 0.05). Also see Figure S6 and Table S1.
Dual domains become enhancers in differentiated cells
We analyzed whether dual domains had consensus sequences for transcription factors that would potentially be involved in enhancer function in vivo. We identified binding sites for pioneer transcription factors and a large set of transcription factors involved in differentiation into a broad set of distinct tissues (Figure S7A). These findings prompted us to explore whether dual domains eventually behave as enhancers in specific tissues. Using embryonic or adult H3K27ac ChIP-seq data from ENCODE, we predicted putative enhancers based on H3K27ac peaks in nonpromoter regions. We found that in contrast to all other H3K9me3 regions, where only 12% eventually acquire H3K27ac, 27% (827) of dual domains eventually acquire H3K27ac in a tissue-specific manner (Figure 7A). As shown in Figures S7B–S7D, the number of observed enhancers was often significantly higher than expected by chance for dual domains, whereas it was significantly lower for all other H3K9me3 regions. For instance, it was highly significant in the liver and small intestine for dual domains while it was anticorrelated for all other H3K9me3 regions (Figures S7C and S7D), suggesting that only H3K9me3 is not sufficient to call dormant enhancers. We also analyzed the conditional enhancer formation on dual domains in distinct adult and embryonic tissues by using k-means clustering based on H3K27ac enrichment. By using k-mean clustering from the 3,015 dual domains, we found that 536 dual domains eventually became enriched in H3K27ac in a tissue-specific manner in adult tissues (Figures 7B and 7C). We observed a similar sign of activation for 342 dual domains in embryonic tissues (Figures 7D and 7E). In these specific tissues, those dual domains also showed increased DNA accessibility and a loss of both H3K9me3 and H3K36me3 marks (Figure 7F), suggesting tissue-specific enhancer unmasking during differentiation. Finally, we analyzed the expression levels of genes surrounding dual domains that become putative enhancers in differentiated tissues. As shown in Figure 7G, these genes had a higher expression level than randomly selected genes in these specific tissues. According to Gene Ontology (data not shown), these genes are involved in the regulation of immune response and lipid transport in the placenta (p value = 3.4 × 10−2 and 3.6 × 10−2, respectively), and those in the forebrain are involved in the control of neurogenesis and in the G receptor protein signaling pathway (p value = 5.0.10−3 and 8.5.10−3, respectively). This suggests that dual domains might stimulate the expression of genes that are important for tissue function.
Figure 7.

Dual domains become enhancers in embryonic and adult tissues
(A) Overlap between enhancers or tissue-specific enhancers and dual domains or all other H3K9me3 regions in adult tissues (top) and in embryonic tissues (bottom).
(B) H3K27Ac enrichment on 342 dual domains identified after k-mean clustering (see experimental procedure) in adult tissues.
(C) Top: tracks showing H3K9me3, H3K36me3 and H3K27ac distributions on a dual domain in WT and Setdb1 KO cells. Bottom: tracks of H3K27ac distribution on a dual domain in adult tissues.
(D) H3K27ac enrichment on 342 dual domains identified after k-mean clustering (see experimental procedure) in embryonic tissues.
(E) Top: tracks showing H3K9me3, H3K36me3, H3K27ac distributions on a dual domain in WT and Setdb1 KO cells. Bottom: tracks of H3K27ac distributions on dual domains in embryonic tissues.
(F) H3K27ac, ATAC-seq, H3K9me3, and H3K36me3 on clustered dual domains (based on k-mean clustering) in forebrain tissue (left) and in liver tissue (right).
(G) Transcription levels, log2(fpkm+1), of randomly selected genes (gray) and nearby genes of clustered dual domains (purple) in adult tissues (Wilcoxson signed rank test: ∗: p value = 0.04; 4.3.10−4) and in embryonic tissues (Wilcoxson signed rank test: ∗: p value = 0.036; 1.12.10−6, respectively). Also see Figure S7.
Discussion
We describe here a dual heterochromatin characterized by an enrichment in both H3K9me3 and H3K36me3. These two seemingly opposing histone post-translational modifications cooperate to bookmark enhancers that will eventually become active later in development. It remains unclear how SETDB1 and NSD proteins are recruited to these regions to form dual heterochromatin. While SETDB1 recruitment usually relies on KRAB Zinc finger proteins (Schultz et al., 2002), NSD recruitment has been proposed to rely on interactions with histones. Since H3K36me2 remains enriched at dual domains in the absence of SETDB1 (Figure S2E), NSD enzymes are likely recruited in a SETDB1-independent fashion. Yet, the H3K36 trimethylation activity only occurs if SETDB1 is also present, suggesting that in this particular context, NSD enzymes become capable of trimethylation. Like in the case of other canonical H3K9me3 heterochromatin regions, this dual heterochromatin restricts DNA access and prevents local transcription, a feature that can, therefore, be assigned to H3K9me3-related functions. In contrast with the more canonical H3K9me3 heterochromatin, dual heterochromatin shows significantly more active signatures (as evidenced by the weak recruitment of enhancer markers), suggesting it might be due to H3K36me2/3-related activities. Yet, a functional enhancer is only revealed upon SEDTB1 removal. Thus, H3K36me2/3 is likely involved in bookmarking the enhancer that H3K9me3-linked activities prevent from unwanted activation. Hence, upon differentiation, dual regions that no longer recruit SETDB1 are rapidly commissioned to act as functional enhancers depending on transcription factor availability. H3K36me2/3 marks are known to repulse Polycomb, which is known to interfere with enhancer function (Lhoumaud et al., 2019; Streubel et al., 2018). Thus, NSD activities might protect dual domains from Polycomb silencing action. An alternative, non-mutually exclusive possibility is that the presence of both H3K9me3 and H3K36me2/3 marks provides a specific binding surface for an unknown dual chromatin effector involved in enhancer bookmarking.
Cell-type-specific and locus-specific H3K9me3 domain formation or dissolution are strongly suggestive of a dynamic role for heterochromatin in development. Position effect is the prevalent route proposed to explain how heterochromatin mediates gene silencing. If gene silencing by heterochromatin or by a neighboring heterochromatic region (e.g., a transposable element) was the major means by which heterochromatin controls gene expression, such genes should have H3K9me3 enrichment. While that is certainly the case for some, the mechanism at play for most other genes is different. We describe here a mechanism where heterochromatin adopts a dual signature to operate from a distance by bookmarking and poising enhancers to control gene expression.
Limitations of this study
The majority of upregulated genes in Setdb1 KO cells seem to be targeted by dual domains, while a minority seems directly repressed by local H3K9me3-heterochromatin: our study does not allow to appreciate which regulatory mode is critical to maintain cell identity. We could not find any experimental condition in which H3K36me3 would be maintained in the absence of H3K9me3 at dual domains, and hence, the role of H3K36me3 on these regions remains unknown. Importantly, as dual domains are also highly enriched in H3K36me2, which methylation state for H3K36 is biologically relevant for dual domain function also remains unknown.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Histone H3 (acetyl K27) - ChIP Grade | Abcam | Cat#ab4729; RRID:AB_2118291 |
| Anti-Histone H3 (di methyl K36) - ChIP Grade | Abcam | Cat#ab9049; RRID:AB_1280939 |
| Anti-Histone H3 (mono methyl K36) - ChIP Grade | Abcam | Cat#ab9048; RRID:AB_306964 |
| Anti-Histone H3 (mono methyl K4) - ChIP Grade | Abcam | Cat#ab8895; RRID:AB_306847 |
| Anti-Histone H3 (tri methyl K36) - ChIP Grade | Abcam | Cat#ab9050; RRID:AB_306966 |
| Anti-Histone H3 (tri methyl K4) - ChIP Grade | Abcam | Cat#ab8580; RRID:AB_306649 |
| Anti-Histone H3 (tri methyl K9) - ChIP Grade | Abcam | Cat#ab8898; RRID:AB_306848 |
| Anti-KMT1E / SETDB1 antibody | Abcam | Cat#ab12317; RRID:AB_299005 |
| Rabbit IgG, polyclonal – Isotype Control Grade | Abcam | Cat#ab171870; RRID:AB_2687657 |
| Rabbit anti-MED12 Ab, Affinity Purified | Bethyl Laboratories | Cat#A300-774A; RRID:AB_669756 |
| Rabbit anti-SMC1 Ab, Affinity Purified | Bethyl Laboratories | Cat#A300-055A; RRID:AB_2192467 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling | Cat#7076; RRID:AB_330924 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling | Cat#7074; RRID:AB_2099233 |
| CTCF (D31H2) XP Rabbit mAb | Cell Signaling | Cat#3418S; RRID:AB_2086791 |
| SETD2 Polyclonal Antibody | Life Technologies | Cat#PA5-43071; RRID:AB_2605635 |
| SETDB1 Antibody | ProteinTech | Cat#11231-1-AP; RRID:AB_2186069 |
| Rabbit anti-goat IgG-HRP | SantaCruz | Cat#sc-2768; RRID:AB_656964 |
| Pol II Antibody (F-12) | SantaCruz | Cat#sc-55492; RRID:AB_630203 |
| Anti-phospho RNA Pol II (Ser5), clone 1H4B6 Antibody | Sigma | Cat#MABE954 |
| Monoclonal Anti-PCNA | Sigma | Cat#P8825; RRID:AB_477413 |
| Chemicals, peptides, and recombinant proteins | ||
| Histone H3K36Ac Peptide – biotinylated | Active Motif | Cat#81054 |
| Histone H3K36me2 Peptide – biotinylated | Active Motif | Cat#81056 |
| Histone H3K36me3 Peptide – biotinylated | Active Motif | Cat#81057 |
| Histone H3K9Ac Peptide – biotinylated | Active Motif | Cat#81044 |
| Histone H3K9me2 Peptide – biotinylated | Active Motif | Cat#81046 |
| Histone H3K9me3 Peptide – biotinylated | Active Motif | Cat#81047 |
| Spike-in Antibody | Active Motif | Cat#61686 |
| Spike-in Chromatin | Active Motif | Cat#53083 |
| AMPure XP | Beckman | Cat#A63881 |
| RNAClean XP | Beckman | Cat#A63987 |
| Certified Low Range Ultra Agarose | BioRad | Cat#1613107 |
| Glutathione Sepharose 4B | GE Healthcare | Cat#17-0756-01 |
| Amersham Hybond-XL | GE Healthcare | Cat#RPN203S |
| Dynabeads™ Protein G for Immunoprecipitation | Life Technologies | Cat#10004D |
| Dynabeads™ Protein A for Immunoprecipitation | Life Technologies | Cat#10001D |
| ERCC RNA Spike-In Mix | Life Technologies | Cat#4456740 |
| UltraPure™ Agarose | Life Technologies | Cat#16500100 |
| 4-Hydroxytamoxifen analytical standard, (E) and (Z) isomers (50:50) | Sigma | Cat#T176 |
| IPTG | Sigma | Cat#I6758 |
| L-Glutathione reduced | Sigma | Cat#G4251 |
| PerfectHyb™ Plus Hybridization Buffer | Sigma | Cat#H7033 |
| Critical commercial assays | ||
| ChIP-IT High sensitivity Kit | Active Motif | Cat#53040 |
| Chromatin IP DNA Purification Kit | Active Motif | Cat#58002 |
| Agilent High Sensitivity DNA Kit | Agilent | Cat#5067-4626 |
| Agilent RNA 6000 Pico Kit | Agilent | Cat#5067-4626 |
| TruSeq Chip Librarry Preparation Kit | Illumina | Cat#IP-202-1012 |
| TruSeq Stranded Total RNA | Illumina | Cat#20020596 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat#FC-131-1024 |
| Luciferase Assay System | Promega | Cat#E1500 |
| Prime-a-Gene® Labeling System | Promega | Cat#U1100 |
| EpiTect Hi-C Kit | Qiagen | Cat#59971 |
| MinElute PCR Purification Kit | Qiagen | Cat#28004 |
| DNA Clean & Concentrator-5 w/ Zymo-Spin IC Columns (Capped) | Zymo Research | Cat#ZD4013 |
| Deposited data | ||
| H3K9me3 ChIP-seq in WT cells | This study | GEO: GSE171749 |
| H3K9me3 ChIP-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| H3K9me3 ChIP-seq in Setd2KO cells | This study | GEO: GSE171749 |
| H3K9me3 ChIP-seq in double KO cells | This study | GEO: GSE171749 |
| H3K36me3 ChIP-seq in WT cells | This study | GEO: GSE171749 |
| H3K36me3 ChIP-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| H3K36me3 ChIP-seq in Setd2 KO cells | This study | GEO: GSE171749 |
| H3K36me3 ChIP-seq in double KO | This study | GEO: GSE171749 |
| H3K36me2 ChIP-seq in WT cells | This study | GEO: GSE171749 |
| H3K36me2 ChIP-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| H3K36me2 ChIP-seq in Setd2 KO cells | This study | GEO: GSE171749 |
| H3K36me2 ChIP-seq in double KO cells | This study | GEO: GSE171749 |
| H3K36me1 ChIP-seq in WT cells | This study | GEO: GSE171749 |
| H3K36me1 ChIP-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| H3K36me1 ChIP-seq in Setd2 KO cells | This study | GEO: GSE171749 |
| H3K36me1 ChIP-seq in double KO cells | This study | GEO: GSE171749 |
| H3K4me3 ChIP-seq in WT cells | This study | GEO: GSE171749 |
| H3K4me3 ChIP-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| RNApol II ChIP-seq in WT cells | This study | GEO: GSE171749 |
| RNApol II ChIP-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| Ser5-P ChIP-seq in WT cells | This study | GEO: GSE171749 |
| Ser5-P ChIP-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| H3K27ac ChIP-seq in Setd2 KO cells | This study | GEO: GSE171749 |
| H3K27ac ChIP-seq in double KO cells | This study | GEO: GSE171749 |
| H3K4me1 ChIP-seq in WT cells | This study | GEO: GSE171749 |
| H3K4me1 ChIP-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| MED12 ChIP-seq in WT cells | This study | GEO: GSE171749 |
| MED12 ChIP-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| CTCF ChIP-seq in WT cells | This study | GEO: GSE171749 |
| CTCF ChIP-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| Input ChIP-seq | This study | GEO: GSE171749 |
| PWWP-MPP8 CIDOP-seq in WT cells | This study | GEO: GSE171749 |
| Input CIDOP-seq | This study | GEO: GSE171749 |
| ATAC-seq in WT cells | This study | GEO: GSE171749 |
| ATAC-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| Total RNA-seq in WT cells | This study | GEO: GSE171749 |
| Total RNA-seq in Setdb1 KO cells | This study | GEO: GSE171749 |
| Total RNA-seq in Setd2 KO cells | This study | GEO: GSE171749 |
| HiC in WT cells | This study | GEO: GSE171749 |
| HiC in Setdb1KO cells | This study | GEO: GSE171749 |
| H3K36me3 ChIP-seq embryonic 15.5-day forebrain | ENCODE | ENCSR437SFX |
| H3K36me3 ChIP-seq embryonic 15.5-day liver | ENCODE | ENCSR510CGB |
| H3K9me3 ChIP-seq embryonic 15.5-day forebrain | ENCODE | ENCSR668BBX |
| H3K9me3 ChIP-seq embryonic 15.5-day liver | ENCODE | ENCSR855NKG |
| H3K27ac ChIP-seq embryonic 15.5-day forebrain | ENCODE | ENCSR691NQH |
| H3K27ac ChIP-seq embryonic 15.5-day heart | ENCODE | ENCSR574VME |
| H3K27ac ChIP-seq embryonic 15.5-day hindbrain | ENCODE | ENCSR066XFL |
| H3K27ac ChIP-seq embryonic 15.5-day liver | ENCODE | ENCSR479LFP |
| H3K27ac ChIP-seq embryonic 15.5-day midbrain | ENCODE | ENCSR428GHF |
| H3K27ac ChIP-seq embryonic 15.5-day limb | ENCODE | ENCSR988BRP |
| H3K27ac ChIP-seq embryonic 15.5-day neural tube | ENCODE | ENCSR241BSK |
| H3K27ac ChIP-seq embryonic 15.5-day intestine | ENCODE | ENCSR599GVS |
| H3K27ac ChIP-seq embryonic 15.5-day kidney | ENCODE | ENCSR711SVB |
| H3K27ac ChIP-seq embryonic 15.5-day lung | ENCODE | ENCSR895BMP |
| H3K27ac ChIP-seq embryonic 15.5-day stomach | ENCODE | ENCSR929SEW |
| H3K27Ac ChIP-seq adult 8 weeks bone marrow | ENCODE | ENCSR000CCL |
| H3K27Ac ChIP-seq adult 8 weeks cortical plate | ENCODE | ENCSR000CDD |
| H3K27Ac ChIP-seq adult 8 weeks olfactory bulb | ENCODE | ENCSR000CCE |
| H3K27Ac ChIP-seq adult 8 weeks small intestine | ENCODE | ENCSR000CCQ |
| H3K27Ac ChIP-seq adult 8 weeks spleen | ENCODE | ENCSR000CDJ |
| H3K27Ac ChIP-seq adult 8 weeks placenta | ENCODE | ENCSR000CDO |
| H3K27Ac ChIP-seq adult 8 weeks testis | ENCODE | ENCSR000CCU |
| H3K27Ac ChIP-seq adult 8 weeks thymus | ENCODE | ENCSR000CCH |
| H3K27Ac ChIP-seq adult 8 weeks brown adipose tissue | ENCODE | ENCSR000CEZ |
| ATAC-seq embryonic 15.5-day forebrain | ENCODE | ENCSR976LWP |
| ATAC-seq embryonic 15.5-day liver | ENCODE | ENCSR465PYP |
| PolyA plus RNA-seq embryonic 15.5 days forebrain | ENCODE | ENCS752RGN |
| PolyA plus RNA-seq embryonic 15.5 days liver | ENCODE | ENCSR611PTP |
| SETDB1 ChIP-seq in WT MS cells | (Matsumura et al., 2015) | GEO: GSE73434 |
| H3K9me3 ChIP-seq in WT MS cells | (Matsumura et al., 2015) | GEO: GSE73434 |
| H3K36me3 ChIP-seq in WT ES cells | (Weinberg et al., 2019) | GEO: GSE118785 |
| H3K36me3 ChIP-seq in Nsd1KO ES cells | (Weinberg et al., 2019) | GEO: GSE118785 |
| H3K36me3 ChIP-seq in WT MS cells | (Weinberg et al., 2019) | GEO: GSE118785 |
| H3K36me3 ChIP-seq in WT Setd2KO cells | (Weinberg et al., 2019) | GEO: GSE118785 |
| H3K36me3 ChIP-seq in WT Nsd1;Nsd2KO cells | (Weinberg et al., 2019) | GEO: GSE118785 |
| Experimental models: Cell lines | ||
| CKO mESC | (Matsui et al., 2010) | N/A |
| Setd2KO-CKO mESC | This study | N/A |
| Nsd1KO-CKO mESC | This study | N/A |
| Nsd2KO-CKO mESC | This study | N/A |
| Nsd3KO-CKO mESC | This study | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Feng Zhang | Addgene Plasmid #62988; RRID : Addgene_62988 |
| MPP8PWWP-pGEX-6p-2 | Jeltsch’s lab | N/A |
| pGL3-Enhancer Vectors | Promega | Cat#E1771 |
| pGL3-Promoter Vectors | Promega | Cat#E1761 |
| Software and algorithms | ||
| ATACseqQC (1.14.4) | (Ou et al., 2018) | N/A |
| BedTools (2.92.2) | (Quinlan and Hall, 2010) | N/A |
| Bowtie2 (2.3.5.1) | (Langmead and Salzberg, 2012) | N/A |
| Chipseeker (1.26.0) | (Yu et al., 2015) | N/A |
| Cufflinks (2.2.1) | (Trapnell et al., 2012) | N/A |
| Cutadapt (3.0) | (Martin, 2011) | N/A |
| Deeptools (3.3.0) | (Ramírez et al., 2016) | N/A |
| Deseq2 (1.28.1) | (Love et al., 2014) | N/A |
| Diffbind (3.0.13) | (Ross-Innes et al., 2012) | N/A |
| FastQC (0.11.9) | (de Sena Brandine and Smith, 2019) | N/A |
| Hi-C-Pro (3.001) | (Servant et al., 2015) | N/A |
| IGV (2.3.97) | (Robinson et al., 2011) | N/A |
| MACS2 (v 2.1.2) | (Zhang et al., 2008) | N/A |
| MEME suite (5.0.5) | (Bailey et al., 2009) | N/A |
| Samtools (v 1.9) | (Li et al., 2009a) | N/A |
| STAR (v 2.7.5a) | (Dobin et al., 2013) | N/A |
| scHiC | (Nagano et al., 2017) | N/A |
| TopDom (0.02) | (Shin et al., 2016) | N/A |
| Whippet (0.11) | (Sterne-Weiler et al., 2018) | N/A |
| Other | ||
| Mendeley Data | This study | https://doi.org/10.17632/wf2b5ry478.1 |
| Oligonucleotides used | ||
| Please see Table S1 | This study | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Jérôme Déjardin (jerome.dejardin@igh.cnrs.fr).
Materials availability
Cell lines generated in this study will be available from the lead contact upon request.
Experimental model and subject details
Cell lines
Mouse embryonic stem cells and derivates were grown on gelatin coated dishes in DMEM containing 15% (v/v) fetal bovine serum, Penicillin streptomycin (10 000U/mL), 2mM L-Glutamine, 1X MEM Non-Essential Amino Acids, 10mM 2-Mercaptoethanol and 2mL of homemade Leukemia Inhibitory Factor (LIF) at 37°C with 5% C02. Setdb1 was knocked out by treating cells for 5 days with 800 nM 4-hydroxytamoxifen (4-OHT) in culture medium.
Generation of CRISPR/Cas9 cell line(s)
Setd2KO, Nsd1/2/3KO, CRISPR/Cas9 deleted dual domain mES cell line(s) were generated by CRISPR/Cas9 genome editing. The gRNA sequences were designed using the ATUM gRNA design tool (https://www.atum.bio/eCommerce/cas9/input), and were cloned into the pSpCas9(BB)-2A-Puro V2.0 plasmid (PX459) from Addgene. For Setd2KO and Nsd1/2/3KO cells, at least 50 clones were screened by the T7E1 enzyme. Positive clones were further characterized to confirm gene inactivation by Western-Blot and or RT-qPCR (Setd2) or RT-qPCR only (for Nsd1,2 and 3). For CRISPR/Cas9 deleted dual domain cells, at least 100 clones were screened by PCR.
Methods details
Nuclear protein extraction, Western-Blotting, Silver and colloidal blue stainings
Cells were collected into pre-chilled 2mL Eppendorf tubes, centrifuged twice at 1200g for 3min at 4°C in PBS. 2.107 cells were resuspended into 680μL hypotonic Buffer A (10mM HEPES pH=7,9, 1mM MgCl2, 10mM KCL, 0,5mM DTT, fresh PMSF + protease inhibitor cocktail;) and quickly spun @16,000g for 10sec at 4°C. The pellet was resuspended in 1,5mL Buffer A, incubated for 10min on ice and mixed by vortexing for 10sec. The suspension was spun at 16000g for 10sec at 4°C. The pellet (nuclei) was resuspended into the same volume of extraction buffer (25% Glycerol, 10mM HEPES pH=7.9, 420mM NaCl, 1mM MgCl2, 0,2mM EDTA, 0,5mM DTT, fresh PMSF + 1X PIC). 0,5μL Benzonase was added/ 50μL suspension, incubated 15min on ice and spun at 16,000g for 15min at 4°C. The supernatant (total nuclear extract) was mixed with denaturing sample buffer, boiled 5 min and run onto 4-12% NuPAGE Bis-Tris gels. The Bis-Tris gel were transfer on PVDF membrane at 0.3A for 1h. PDVF is blocked in TBS-T-Milk for 1h at RT, then O/N at 4°C with primary antibody and 2h at RT with secondary antibody. The SilverQuest Silver Stanning Kit from Invitrogen was used for silver staining. The colloidal blue staining kit from Invitrogen was used on acid extracted histones.
Peptide Assays
2μg histone peptides (Active Motif) were boiled in sample buffer (pierce) for 5min at 95°C then chilled on ice, loaded on a 4-12% NuPAGE Bis-Tris gel in 1X MOPS buffer, and transferred to a PVDF membrane. The membranes were probed with primary antibodies against specific histone modifications (1/2000) and revealed by ECL.
Antibody specificity assays
Each antibody (0.5μg) was mixed with a vast excess of modified histone peptides (50μg) and used to probe nuclear extracts (2μg).
Dot blot assays
PVDF membrane was pre-wet with TBS-T in then air-dried for a few minutes. 10μL of different histone peptide concentrations were blotted onto membrane. Membranes were air-dried before blocking in TBS-T-Milk for 1h at RT. Membranes were incubated with primary antibody for O/N at 4°C then 2h at RT with secondary antibody.
Acid histone extraction
106 cells were used to prepare nuclei as above. Nuclei were resuspended in 1ml of NaCl-RIPA buffer (50mM Tris-HCl pH=7,5, 1% NP-40, 0,5% Sodium deoxycholate, 0,1% SDS, 300mM NaCl) and incubated on ice for 10min. The preparation was spun at 1,000g for 10min at 4°C to pellet chromatin. 400μL H2SO4 (55,55μL H2SO4 36 N + 4,95mL water) were added to the pellets and incubated at 4°C for 1h on rotating wheel. Tubes were spun at 16,000g for 10min at 4°C, the supernatant was saved and slowly mixed with 80μl TCA 20% and incubated on ice for 30 mins. The histone pellet was obtained after centrifugation (16,000g, 10 min), washed twice with ice cold acetone and resuspended in 50μl of sample buffer. Histones were resolved on a 12% BIS tris Nupage gels and gel lices were cut out for quantitative LC-MS/MS analysis (performed by the ZfP proteomics platform, Munich).
RNA extractions, qRT-PCR and Northern-Blots
RNAs were extracted with Trizol following the manufacturer’s recommendations. 5μg RNAs were DNAsed with TURBO DNase from Invitrogen and reverse transcribed using an OligodT and Superscript III (Invitrogen). q-PCR was performed using a Light Cycler and SYBR Green Master mix.
1-5 μg RNAs were also analysed by Northern-Blotting using standard procedures and probed using dsDNA radioactively labelled using the prime a gene system (Promega).
Luciferase Assays
H3K9me3 ‘only’ or H3K36me3 ‘only’ or dual domains, were amplified by Q5 DNA polymerase from genomic DNA and cloned into the AfeI site of the pGL3-promoter luciferase reporter plasmid. Primers for amplification were localized upstream and downstream of the domains observed on the IGV browser. For the luciferase assay, ES cells at 30 to 40% of confluence were transfected with 1ug plasmid DNA using Lipofectamine 3000 (Invitrogen). 48h after transfection, the luciferase activity was measured using the Luciferase assay system (Promega).
Chromatin ImmunoPrecipitation: ChIP-qPCR
Two 15cm-dishes with 2.10ˆ7 cells in 20mL of culture media were used for each ChIP experiment. Cells were crosslinked with 1% Formaldehyde for 10 minutes at room temperature on an orbital shaker (60rpm). Formaldehyde was quenched by adding glycine to 75mM, incubated at RT for 5min, and rinsed twice with PBS. Cells were scrapped and transferred into 15mL-Falcon tubes, spin 300g for 5min at 4°C, resuspended with 15mL Buffer A (20mM HEPES pH=7,4, 10mM EDTA, 0,5mM EGTA, 0,25% Triton X-100), incubated at 4°C for 5min on a rotating wheel. Cells were centrifuged 300g for 5min at 4°C then resuspended with 15mL Buffer B (20mM HEPES pH=7,4, 150mM NaCl, 10mM EDTA, 0,5mM EGTA) and incubate at 4°C for 5min on a rotating wheel. Cells were centrifuged 300g for 5min at 4°C and resuspended in 1mL of Buffer C (20mM HEPES pH=7,4, 10mM EDTA, 0,5mM EGTA, 0,1% SDS, 1XPIC). After 10min on ice, cells were transferred into 15-mL sonication tube. Sonication was performed with a Bioruptor: 30sec ON – 30sec OFF, 10 cycles, at high power for a total of 20 cycles with break a 10min pause on ice every 10 cycles. Sonicated chromatin was centrifuged at full speed for 10min at 4°C. To check sonication-efficiency, 10μL of sample was reverse-crosslinked by adding 40μL ChIP elution buffer (10mM Tris pH8, 300mM NaCl, 5mM EDTA, 0,5% SDS). DNA was incubated at 65°C for 1h to 6h 10000 rpm shaking and 1μL RNase A (10mg/mL) was added to sonicated DNA and incubated at 37°C for 1h. Finally, 5μL Proteinase K (20mg/mL) were added and incubate at 55°C for 2h. An 1,5% agarose gel was run to check DNA size distribution.
10μL Protein A Dynabeads and 10μL Protein G Dynabeads (20μL in total) per IP for rabbit antibody or 40μL anti-mouse IgG M-280 Dynabeads per IP for mouse antibody were washed twice in 750μL 1X Incubation buffer (10mM Tris pH=8, 150mM NaCl, 1mM EDTA, 0,5mM EGTA, 0,15% SDS, 1% Triton X-100, 0,1% BSA, 1XPIC). Beads were resuspended beads in 25μL/IP 1X Incubation buffer and incubated O/N at 4°C on a rotating wheel. For ChIP reaction, 20μg chromatin + 2μg antibody for histones were used and 30μg chromatin + 4μg antibody for none-histone proteins were used. For a 500uL reaction, we added x μL chromatin + y μL antibody and 390-(x+y) μL H2O + 100μL 5X incubation buffer (50mM Tris pH=8, 750mM NaCl, 5mM EDTA, 2,5mM EGTA, 0,75% SDS, 5% Triton X-100) + 5μL 10% BSA + 5μL PIC. 1% was saved for input. ChIP reactions were incubated O/N at 4°C on a rotating wheel. On the next day, 25μL washed beads were added per IP then incubated at 4°C for 6h on rotating wheel. IP reactions were washed with 1mL wash buffer 1 (10mM Tris pH 8, 150mM NaCl, 1mM EDTA, 0,5mM EGTA, 0,1% SDS, 0,1% DOC, 1% Triton x-100); with 1mL wash buffer 2 (10mM Tris pH 8, 500mM NaCl, 1mM EDTA, 0,5mM EGTA, 0,1% SDS, 0,1% DOC, 1% Triton x-100); with 1mL wash buffer 3 (10mM Tris pH 8, 250mM LiCl, 1mM EDTA, 0,5mM EGTA, 0,5% DOC, 0,5% NP-40); with 1mL wash buffer 4 (10mM Tris pH=8, 300mM NaCl, 5mM EDTA, 0,5% SDS). IP reactions were resuspended into 100μL ChIP elution buffer then incubated at 65°C for 15min 1000 rpm shaking. The supernatant were transferred into fresh tubes. Elution step was repeated one time then supernatants were pooled for a final volume of 200μL. 1% Input was resuspended into 200μL of ChIP elution buffer. The eluted ChIP-DNA and inputs were incubated at 65°C O/N 1000 rpm shaking then 2μL RNase A (10mg/mL) was added and incubated at 37°C for 1h. 8μL of Proteinase K (20mg/mL) were added and incubated at 55°C for 2h with interval mix, 30 sec ON 500 rpm shaking and 8min OFF. ChIP-DNA was extracted with Phenol/Chloroform following the manufacturer’s recommendations then ethanol precipitated and resuspended into 100μL of water. q-PCR was performed using a Light Cycler and SYBR Green Master mix.
Mononucleosome ChIP-re ChIP
20.106 cells were used for sequential ChIP as above. Cells were resuspend in 5mL of Lysis Buffer (10mM Tris-HCl pH=7.4, 2mM MgCl2, PMSF, 1XPIC) and add 150μL NP-40 20%. Cell were incubated for 5min on ice then centrifuged at 10000g for 10min at 4°C. Cells were washed with 5mL of Lysis Buffer then 10mM Tris-HCl pH=7.4, 2mM MgCl2, 0.5mM PMSF + PIC. Cells were resuspend in 200 to 600μL of Lysis Buffer. For MNAse digestion, 64U of MNAse from NEB per μg of DNA was used with 1mM CaCl2. MNAse reaction was placed directly at 37°C and incubated for 7min to obtain mononucleosomes. Reactions were stopped by adding 2mM EDTA + 30mM NaCl then samples were centrifuged at 13000g for 10min at 4°C. For first IP reaction, 20μg pre-cleared mononucleosome-DNA was used with 2μg of first antibody in 500μL IP Buffer (16.7mM Tris-HCl pH=8, 167mM NaCl, 1.1% TritonX-100, 1.2mM EDTA) and incubated O/N at 4°C on a rotating wheel. Next day, 25μL washed beads were added per IP then incubated at 4°C for 2h on rotating wheel. IP reactions were washed with 1mL wash buffer 1 20mM Tris-HCl pH8, 150mM NaCl, 1%TritonX-100, 2mM EDTA, 0.1%SDS); with 1mL wash buffer 2 (20mM Tris-HCl pH8, 500mM NaCl, 1%TritonX-100, 2mM EDTA, 0.1%SDS); with 1mL wash buffer 3 (10mM Tris-HCl pH8, 250mM LiCl, 1%NP-40, 1mM EDTA, 1% Sodium-deoxycholate); with 1mL wash buffer 4 (10mM Tris-HCl pH8, 1mM EDTA). IP reactions were resuspended in 75μL TE + 10mM DTT then incubated at 37°C for 30min. Eluted DNA is transfer into fresh tube and diluted in 1.5mL of IP Buffer. 2μg of secondary antibody is added into the IP reaction. IP reaction is incubated O/N at 4°C on a rotating wheel. Next day, 25μL washed beads were added per IP then incubated at 4°C for 2h on rotating wheel. After washed steps, beads were resuspended into 100μL of Elution Buffer (50mM Tris-HCl pH8, 50mM NaCl, 1mM EDTA, 1%SDS) + 3μL NEB Proteinase K then incubated at RT for 45min on a rotating wheel then at 55°C for 1h. DNA was purified as described in Chromatin IP DNA purification Kit from Active Motif and IP-DNA was eluted in 100uL DNA purification Elution Buffer for qPCR. q-PCR was performed using a Light Cycler and SYBR Green Master mix.
Chromatin Interacting DOmain Precipitation: CIDOP
MPP8-PWWP adapter plasmid was from the Jeltsch’s lab, the adapter protein was produced into BL21 Competent E.Coli bacteria. At OD600 =0.6, the adapter protein was induced O/N at 22°C in LB medium containing 1M IPTG. Cells were pelleted at 3000g for 20min at 4°C then resuspended in 5mL Resuspension Buffer (20mM HEPES (pH=7,5), 500mM KCl, 0,2mM DTT, 1mM EDTA, 10%Glycerol + PMSF) and incubated 30min on ice. Cells were sonicated with a Bioblock – Vibra Cell 72405, 15sec pulse for 2min then centrifuged 10000g for 10min at 4°C. 1mL of slurry of Glutathione Sepharose 4B beads were washed 3 times in 5mL Resuspension Buffer and spin 2000g for 1min at 4°C. Supernatant was applied on washed beads and incubate at 4°C for 1h on a rotating wheel. Beads were centrifuged at 2000g for 1min at 4°C then washed 3 times with 5mL Resuspension Bacteria Buffer. Beads were resuspended into 500μL Elution Buffer (50mM Tris-HCl, 40mM L-Glutathione reduced) per 1mL of slurry and incubated at 4°C for 10min on a rotating wheel. Beads were centrifuged at 2000g for 1min at 4°C and supernatant was transferred into fresh tube. Elution step was repeated two times for total of 3 elutions and 1,5mL of final elution volume. Elution containing adapter protein was injected adapter into Slide-A-Lyzer Dialysis Cassette then incubated 4°C for 2h against Dialysis Buffer 1 (20mM HEPES pH = 7,5, 200mM KCl, 0.2 mM DTT, 1mM EDTA, 10% Glycerol + PMSF) and O/N on Dialysis Buffer 2 (20mM HEPES pH = 7,5, 200mM KCl, 0.2 mM DTT, 1mM EDTA, 60% Glycerol + PMSF). Colloidal blue staining was performed to control the correct size of the adapter.
CIDOP chromatin capture reaction was performed as described (Mauser et al., 2017) with a few adaptations. For MNAse digestion, 64U of MNAse from NEB per μg of DNA was used with 1mM CaCl2. MNAse reaction was placed directly at 37°C and incubated for 7min to obtain mononucleosomes for CIDOP-qPCR and mononucleosome immunoprecipitation or incubated 5min to obtain 10% of dinucleosomes and 90% of mononucleosomes for CIDOP-seq and library preparation. Reaction were stopped by adding 2mM EDTA + 30mM NaCl then samples were centrifuged at 13000g for 10min at 4°C. For CIDOP reaction, 60μg pre-cleared mononucleosome-DNA was used with 0,5μM HiMID recombinant peptides in 500μL DP Buffer (16.7mM Tris-HCl pH=8, 167mM NaCl, 1.1% TritonX-100, 1.2mM EDTA) and incubated O/N at 4°C on a rotating wheel. Next day, 40μL Glutathione Sepharose 4B beads was added per IP then incubated at 4°C for 2h on a rotating wheel. IP reactions were washed as described in the original protocol. For the elution step followed by mononucleosome immunoprecipitation, 50μL Elution Buffer (50mM Tris-HCl pH8, 50mM NaCl, 1mM EDTA, 1%SDS) was added and mixed with denaturing sample buffer, boiled for 5 min. For the elution step followed by qPCR or library preparation, 100μL Elution Buffer and 3μL NEB Proteinase K were added and incubated at RT for 45min on a rotating wheel then at 55°C for 1h. DNA was purified as described in Chromatin IP DNA purification Kit from Active Motif and CIDOP-DNA was eluted in 100uL DNA purification Elution Buffer for CIDOP qPCR. q-PCR was performed using a Light Cycler and SYBR Green Master mix. For CIDOP-seq, CIDOP-DNA was eluted in 36μL with pre-warmed Elution Buffer then quantified with Qubit dsDNA HS Assay Kit. 1μL was also used to run Agilent High Sensitivity DNA electrophoresis to check DNA fragment size with majority of fragment at 150 nucleotides. 10ng of CIDOP-DNA was used for library preparation.
Chromatin ImmunoPrecipitation Sequencing: ChIP-seq
Two 15cm-dishes with 2.107 cells in 20mL of culture media were used for each ChIP-seq experiment. ChIP-seq was performed as described in ChIP-IT High sensitivity Kit from Active Motif with a few adaptations. Chromatin was sonicated by Bioruptor: 30sec ON – 30sec OFF, 10 cycles, high power. Sonicate for a total of 40 cycles for histones and of 30 cycles for TFs with 10min pause on ice every 10 cycles. For ChIP reactions, 20μg chromatin was used with 4μg antibody for histone and 30μg chromatin + 4μg antibody for none-histone proteins. For quantitative ChIP reaction Drosophila spike-in from Active Motif was used. For histone methylation of lysine 36, 25μg chromatin was used with 50ng Spike-in Chromatin + 4μg antibody + 2ug Spike-in Antibody. Spike-in antibody is required as a blocking step. ChIP-DNA was eluted in 36μL with pre-warmed Elution Buffer then ChIP-DNA concentration was quantified with the Qubit dsDNA HS Assay Kit. 2,5ng to 10ng of ChIP-DNA were used for library preparation.
TruSeq ChIP and CIDOP sample preparation
2.5ng to 10ng ChIP-DNA and 10ng CIDOP-DNA were used for library preparation. ChIP-seq and CIDOP-seq library preparation was performed as described in TruSeq ChIP Sample Preparation kit from Illumina with few adaptations. The Enrich DNA Fragment step was performed before the Purify Ligation Products step. AMPure XP beads were air-dried 7min. 1μL library was diluted 1:10 in H20 to quantify library concentration by Qubit DNA HS assay quantification. Agilent High Sensitivity DNA Electrophoresis was run to check DNA fragment size. Before sequencing, ChIP-seq and CIDOP-seq libraries were quantified by qPCR. Libraries were sequenced 50bp or 100bp single-reads with a depth of 50 million reads on HiSeq 2000 Illumina or NovaSeq 6000 Illumina machine at the MGX platform.
Total RNA-sequencing: Total RNA-seq
RNAs were extracted with Trizol following the manufacturer’s recommendations. Denaturing agarose gel and Agilent High Sensitivity RNA electrophoresis were run to control RNA quality. An RNA integrity number (RIN) of 9 was obtained. 1μg RNA and ERCC RNA Spike-In Mix were used to performed libraries. RNA-seq library preparation was performed as described in TruSeq Stranded Total RNA Sample Preparation kit from Illumina with few adaptations. RNA Clean XP beads and AMPureXP beads were air-dried for 7min. 1uL of libraries were used to determine concentration by Qubit DNA HS assay kit and to run Agilent High Sensitivity DNA Electrophoresis to check DNA fragment size. Before sequencing, RNA-seq libraries were quantified by qPCR. Libraries were sequenced 100bp single-reads with 30 million reads of depth on NovaSeq 6000 Illumina machine at the MGX platform.
Optimized Assay for Transposase-Accessible Chromatin-sequencing: Omni-ATAC-seq
1.10ˆ6 cells were used for each Omni-ATAC-seq experiment. Cells were incubated with DNAse I at 37°C for 30min then cells were collected at 1200g for 3min at 4°C and washed 3 times with 1mL ice-cold PBS. Cells were resuspended into 1mL ice-cold PBS, 50,000 cells (50μL) were transferred into fresh 1.5mL Eppendorf tube. Omni-ATAC-seq was performed as described (Corces et al., 2017) and Omni-ATAC-seq library preparation was performed as described with few adaptations (Buenrostro et al., 2013). The Transposase was from Netera XT DNA library Preparation Kit and Illumina sequencing primer sequences were from (Buenrostro et al., 2013). To determine the number of additional cycles required, Ct corresponding to plateau was identified and ¼ of Ct + 1 defined the number of additional cycles. Then Omni-ATAC-seq libraries were cleaned up with AMPure XP beads to remove DNA fragment smaller than 100bp and longer than 1,5Kb. 30μL H2O was added to libraries to obtain a final volume of 50μL then 25μL AMPure XP beads were added and incubate at RT for 15min. AMPure XP beads were placed on magnetic stand for 5min at RT. Supernatants were transferred into fresh tube and 65μL AMPure XP beads were added to samples then incubated at RT for 15min and were placed on magnetic stand for 5min at RT. Supernatant were discarded. AMPure XP beads were washed with 200μL fresh 80%-EtOH twice. AMPure XP beads were air-dried for 7min at RT. 22μL Buffer EB was added to resuspend AMPure XP beads then incubated at RT for 2min and placed on magnetic stand for 5min at RT. 20μL ATAC-library samples were transferred into fresh 1,5mL Eppendorf tube. 1μL of library was diluted 1:100 to perform qPCR quality controls and to run Agilent High Sensitivity DNA Electrophoresis to check DNA fragment size. Before sequencing, ATAC-seq libraries were quantified by qPCR. Libraries were sequenced 75bp paired-end with 50 million reads of depth on HiSeq 2000 Illumina machine at the MGX platform.
High-throughput Chromatin Conformation Capture-sequencing: Hi-C
Hi-C-seq was performed as described in EpiTect Hi-C Kit from Qiagen. Hi-C sequencing libraries were cleaned up with AMPure XP beads to remove remaining free Illumina adapter. 25μL Buffer EB were added to Hi-C sequencing library to obtain a final volume of 50μL. 0,8V AMPure XP beads was added to Hi-C sequencing library, incubated at RT for 15min and placed tube on a magnetic stand for 5min at RT. Supernatant were discarded and AMPure XP beads were washed with 200μL fresh 80%-EtOH twice. AMPure XP beads were air-dried for 7min at RT. 27μL Buffer EB were added to resuspend AMPure XP beads, incubated at RT for 2min and placed on magnetic stand for 5min at RT. Hi-C libraries were transferred into fresh tubes. 1μL of library was used to run Agilent High Sensitivity DNA electrophoresis for checking DNA fragment size. Before sequencing, Hi-C libraries were quantified by qPCR. Libraries were sequenced 150bp paired-end with 1,2 billion reads of depth on NovaSeq 6000 Illumina machine at the MGX platform.
Quantification and statistical analysis
For ChIP-seq, ATAC-seq, CIDOP-seq and Hi-C, biological duplicates were generated. Biological triplicates were generated for RNA-seq. A spearman correlation score was calculated to ensure reproducibility between samples.
ChIP-seq analyses
Quality controls of ChIP-seq Fastq files were performed by FastQC. ChIP-seq Fastq read files are mapped to the mm10 reference genome using Bowtie2 with default parameters. To determine for ChIP-seq peaks with a narrow profile (CTCF, SMC1 and H3K4me3), datasets were processed using MACS2 with default parameters and --g mm option. For profiles characterized by enrichments in broad regions (H3K9me3, H3K36me3, H3K36me2, H3K36me1, H3K27ac, H3K4me1, SETDB1, MED12, RNApol II and Ser5-P), datasets were processed using MACS2 with --broad and --g mm options. BigWig files were generated using Deeptools bamCoverage with --bs 50 and --normalizeUsing RPKM options. H3K36me3, H3K36me2 and H3K36me1 bigWig files were normalized using drosophila spike-ins, which were mapped to dm6 reference genome using Bowtie2 with default parameters. The normalization factor was determined as described in manufacturr’s protocol (Spike-in Normalization from Active Motif). For each sample, normalized BigWig files were generated using Deeptools bamCoverage with --bs 50, --normalizeUsing RPKM and --scaleFactor options, where scaleFactor is the normalization factor specific to each sample.
CIDOP-seq analyses
Quality controls of CIDOP-seq Fastq files were performed by FastQC. CIDOP-seq Fastq file reads were mapped to the mm10 reference genome using Bowtie2 with default parameters. To determine peaks, CIDOP-seq datasets were processed using MACS2 with --broad and --g mm options. BigWig files were generated using Deeptools bamCoverage with --bs 50 and --normalizeUsing RPKM options.
ATAC-seq analyses
Quality controls of ATAC-seq Fastq files were performed by FastQC. ATAC-seq adaptors and ATAC-seq reads were removed or trimmed using Cutadapt. Paire-end reads of ATAC-seq Fastq files were mapped to the mm10 reference genome using Bowtie2 with default parameters. Quality controls of ATAC-seq alignments were performed with ATACseqQC. To determine peaks for ATAC-seq, datasets were processed using MACS2 with default parameters and --g mm and –B options. BigWig files were generated using Deeptools bamCoverage with --bs 50 and --normalizeUsing RPKM options.
Identification of regions/domains
To identify a region, we merged peaks using bedtools merge with -d option. -d value was specific to each histone marks or transcription factors. We used a -d value of 2,000 for H3K9me3; of 3,000 for H3K36me3, MED12, RNApol II, Ser5-P and ATAC-seq peaks; of 4,000 for H3K4me3 and CIDOP peaks; of 5,000 for H3K27ac and H3K4me1; of 8000 for H3K36me2 and H3K36me1.
Dual domain definition
Dual domains were defined as genomic regions where H3K9me3 peaks and H3K36me3 peaks reciprocally overlap with a minimun of 80%. For identification, we used bedtools intersect firstly with –f 0.8 option then with –F 0.8 option. The two bed files were concatenated and merged using bedtool merge allowing identification of 4,744 dual peaks, corresponding to 3,015 regions across the genome of mESCs.
Categorization of colocalized vs embedded dual domains
We used bedtools intersect to select H3K9me3 regions and H3K36me3 regions overlapping within 3,015 dual regions. The size of each merged dual region was divided by the size of it associated H3K9me3 region to obtain a percentage (%) of H3K9me3 overlap. The size of each dual region was also divided by the size of it associated H3K36me3 region to obtain a percentage (%) of H3K36me3 overlap. Embedded domains were defined as dual regions having a % of H3K9me3 overlap higher than 80% and having a % of H3K36me3 overlap lower than 60% (corresponding to 1151 dual domains). Colocalized domains were defined as merged dual domains having a % of H3K36me3 overlap higher than 60% and a % of H3K9me3 overlap between 0 to 100 (corresponding to 1864 dual domains).
Genome-wide distribution
Distribution of histone marks (H3K9me3 and H3K36me3) and dual domains across the mouse genome were obtained using ChIPseeker.
Definition of regulatory regions and others regions
‘Promoters’ were defined as regions spanning ± 2 kilobase pairs from the transcription start site from RefSeq genes.
‘Enhancers’ were defined as H3K27ac peaks excluded from promoters using bedtools intersect -v. Tissue specific enhancers were defined by excluding enhancers that were found in others tissue using bedtools intersect -v.
Random regions were generated using bedtools random with –n 4744 and –l 420 options.
Random other H3K9me3 peaks and other H3K36me3 peaks were generated using Shuf with -n 4744 option on H3K9me3 peaks and H3K36me3 peaks respectively. Random enhancers and random H3K4me3 peaks were generated from enhancer files and H3K4me3 peak file, respectively, following the same rationale. Random promoters and random genes were generated using Shuf on the promoter file and Refseq genes, respectively.
The transcribed genes in Figure S3B were defined as Refseq genes containing at least one H3K36me3 peak. The H3K9me3 promoters in Figure 4A were defined as promoter, ± 500bp from the transcription start site of RefSeq genes, overlapping with an H3K9me3 peak using bedtools intersect with -f 1E-9 option.
Distance to promoters
Distances between promoters and candidate regions were measured using bedtools closest using -d option.
Kmeans Clustering
The kmeans clustering analysis was performed on the dual domains that are excluded from promoters (4614 peaks). ComputeMatrix from deepTools was used to generate a matrix containing H3K27Ac levels from diverse tissues on 4614 peaks (embryonic and adult tissues were processed separately), then kmeans clustering was performed using --kmeans option of plotHeatmap from deepTools. Clusters, and their associated dual domains, with low H3K27Ac were excluded and a new ComputeMatrix and kmeans clustering were again performed. For kmeans clustering based on H3K27ac level in embryonic tissues 342 dual domains (522 peaks) were clustered into 7 clusters and for kmeans clustering based on H3K27ac level in adult tissues 536 dual domains (849 peaks) were clustered into 8 clusters. The matrix used for cluster calculation were the same as the one used for heatmap visualization.
To identify high and low H3K27ac dual domains, ComputeMatrix from deepTools was used to rank dual domains according H3K27ac intensity in Setdb1KO cells with --sortRegions descend option. The ranked dual domains were used to generate matrix containing H3K27ac levels from WT and Setdb1KO cells using ComputeMatrix from deepTools. Then kmeans clustering was performed using --kmeans 3 option of plotHeatmap from deepTools. The dual domains of cluster 1 and 2 were defined as High H3K27ac dual domains and the dual domains of cluster 3 were defined as Low H3K27Ac dual domains.
Observed and simulated overlaps
For the observed overlap, we analyzed H3K27ac ChIP-seq datasets of embryonic tissues (15.5 days) and of adult tissues (8 weeks) from ENCODE and we identified enhancers and tissue-specific enhancers. Bedtools intersect was used to quantify the overlap between enhancers or tissue specific enhancers and dual domains (4,774 peaks) in each tissue.
For simulated overlaps, we randomly generated 4744 regions of an average size of 420 bp using bedtools random with –n 4744 and –l 420 options. The overlap with enhancers or tissues specific enhancers is computed using bedtools intersect. These steps were iterated 5000 times to obtain a mean of simulated overlaps. The mean of simulated overlaps follows a binomial law B (n;p) where n= 4744 and p is the probability of overlap between a random peaks and enhancers.
A Z-score, quantifying the distance between the observed overlaps and the simulated overlap considering for the variance, was then calculated using the formula:
Sequence analyses
DNA sequences corresponding to dual domains were extracted from the mm10 reference genome by using bedtools getfasta. MEME-ChIP tool from MEME suite was used to detect DNA motifs and transcription factor motifs were obtained from TOMTOM and Jaspar databases.
RNA-seq analysis
Quality controls of RNA-seq Fastq files were performed by FastQC. RNA-seq reads were mapped to the mm10 reference genome using STAR with --library-type fr-firststrand option. BigWig files are generated using Deeptools bamCoverage with --bs 50 and --normalizeUsing RPKM options. To obtain strand specific bigwig files, the --filterRNAstrand option was used.
Differential expression analysis
Fastq reads were mapped to Refseq genes using STAR with --library-type fr-firststrand and --quantMode Transcriptome SAM GeneCounts options to obtain read count files. This was then input processed by deseq2. An adjusted p-value < 0.01 and a base mean > 2 were used as cut-off values for significant differential expression.
Gene expression levels
RNA-seq reads were mapped to Refseq genes using STAR with --library-type fr-firststrand to obtain SAM files. This was processed by Cufflinks. Cufflinks was run with -g and -library-type fr-firsttrand options.
Dual domains and ERVs expression levels
Dual domain and ERVs bed files were converted into gtf using STAR with --runMode genomeGenerate and –sjdbOverhand 95 options. Reads of RNA-seq Fastq were mapped to Refseq genes using STAR with --library-type fr-firststrand to obtain SAM file. This was then processed by Cuffdiff that was use with default parameters.
Identification of sense and anti-sense transcription on dual domains
Dual domain bed files were converted into gtf using STAR with --runMode genomeGenerate and –sjdbOverhand 95 options. Reads were mapped to dual domains using STAR with --library-type fr-firststrand and --quantMode Transcriptome SAM GeneCounts options to obtain read count files. In the read count files, ‘first-stranded’ values show sense transcription and ‘second-stranded’ value corresponds to anti-sense transcription. A dual domain has + (sense) strand when ‘first-stranded’ value > ‘second-stranded’ value and has - (anti-sense) strand when ‘first-stranded’ value < ‘second-stranded’ value.
Identification of alternative-spliced, aberrantly spliced transcripts
Modulation of splicing on hosting genes and randomly selected genes was analysed using Whippet. To be able to detect unannotated spliced variants, an annotation file was created from RNAseq reads using whippet-index. Then RNA-seq reads were quantified using whippet-quant allowing identification of constitutive annotated junctions (CON_ANNO), annotated alternative junctions (ALT_ANNO) and unannotated alternative junctions (ALT_UNIQ) in WT and mutant cells.
Hi-C analysis
Raw Hi-C sequencing data were processed using the “scHiC2” pipeline. Construction of expected models, Hi-C contact scoring, and Hi-C aggregate plots (peSCANs) were performed using the “shaman” R package, v2.0 (https://tanaylab.bitbucket.io/shaman/index.html and https://anaconda.org/bioconda/r-shaman). Hi-C map visualization, segmented interaction frequencies, and downstream analyses were conducted using in-house R scripts using the “misha” package (https://tanaylab.github.io/misha/index.html and https://anaconda.org/bioconda/r-misha, v4.01) to store and access the data. Iced normalized tracks for 25-, 100- and 250-kb resolutions were computed with the Hi-C–Pro pipeline using the ice_norm flag on observed data matrices exported from misha tracks. Hi-C-Pro’s hicpro2juicebox utility was used to convert the transformed adjacency output of the scHiC2 pipeline into Juicebox format (.hic) at fragment resolution. Topologically associated domain (TAD) identification was performed using TopDom on the 100kb resolution iced maps using a window of 5 (ws=5). To optimize resolution and avoid over-segmentation TAD boundaries called on the 100kb bin maps were shifted to the closest boundary called on the 25kb bin maps using a window of 20 (ws=20). TADs overlap significance was calculated using R’s phyper function as follows: n_A=length of TADs overlapping with set1, n_B= length of TADs overlapping with set2, n_C= length of set1, n_A_B=length of TADs overlapping both set1 and set2. The p-value was then calculated as: 1-phyper(n_A_B, n_B, n_C-n_B, n_A). All analyses were performed under an anaconda environment (v3.0) using R version 3.6.3.
Principal component analysis (PCA)
PCA was performed using R based on fold change calculated by DiffBind.
Genome references
The mm10 mouse and the dm6 Drosophila reference genomes were downloaded from UCSC genome data (http://hgdownload.soe.ucsc.edu/downloads.html). The Refseq genes was downloaded from Ensembl (https://www.ensembl.org/index.html). The Repeatmasker annotated region (Rmsk 4.0.5) was downloaded for RepeatMasker (http://www.repeatmasker.org/species/mm.html). This was then input was divided into 4 family repeats: ERVs, LINE, SINE and Simple repeats. ERVs contains all LTR transposable elements.
Figures and IGV browser
Metaplots and hetamaps were generated using deepTools. Bed files used for heatmaps were ranked, using --sortRegions descend, by H3K9me3 intensity except for heatmaps in Figures 5B and S6B which were ranked by MED12 intensity and CTCF or SMC1 intensity, respectively.
Venn Diagrams were generated using VennDis.
Box-plot and Scatter-plots were generated using Matplotlib or ggplot2.
Browser screenshots were generated using IGV viewer. Reference Sequence (Refseq) track contains annotated genes for mm10 reference genome. Endogenous RetroVirus (ERVs) track contains annotated retrotransposons and RepeatMasker (Rmsk) track contains annotated DNA repeat sequences.
Statistical analyses
Statistical analyses of ChIP-seq data were carried out using DiffBind.
The correlation score was calculated using the Spearman’s rank correlation coefficient.
Statistical tests and p-values were calculated using Wilcoxson signed rank test, Student’s T-test, KS one-sided test or hypergeometric test.
Acknowledgments
We would like to thank Y. Shinkai for the gift of the inducible Setdb1 KO ES cell line and derivatives and A. Jeltsch for the gift of the vectors encoding the CIDOP adapter proteins. I. Forne and A. Imhof are thanked for their help in histone MS analyses. A.B. is supported by Labex Epigenmed, the Fondation pour la Recherche Médicale, and an ERC CoG METACHROM PhD fellowship. Work in the lab of J.D. is supported by the ERC CoG METACHROM (724835) and ARC.
Author contributions
A.B. and S.Z. conducted experiments. G.P., A.J.O., and L.D. performed the bioinformatic analyses. G.P. and G.C. performed Hi-C data analyses. A.B. and J.D. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: January 25, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2021.12.037.
Supplemental information
Data and Code availability
Original immunoblot, electrophoresis and slot-blot images reported in this study have been deposited at Mendeley and are publicly available. The DOI is listed in the Key resources table.
This study does not report original code. It uses publicly available software to analyze/re-analyze publicly available NGS data. DOIs and GEO accessions are listed in the Key resources table.
Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.
References
- Allshire R.C., Madhani H.D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2018;19:229–244. doi: 10.1038/nrm.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubec T., Colombo D.F., Wirbelauer C., Schmidt J., Burger L., Krebs A.R., Akalin A., Schübeler D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 2015;520:243–247. doi: 10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- Bilodeau S., Kagey M.H., Frampton G.M., Rahl P.B., Young R.A. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 2009;23:2484–2489. doi: 10.1101/gad.1837309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blahnik K.R., Dou L., Echipare L., Iyengar S., O’Geen H., Sanchez E., Zhao Y., Marra M.A., Hirst M., Costello J.F., et al. Characterization of the contradictory chromatin signatures at the 3′ exons of zinc finger genes. PLoS One. 2011;6 doi: 10.1371/journal.pone.0017121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulut-Karslioglu A., De La Rosa-Velázquez I.A., Ramirez F., Barenboim M., Onishi-Seebacher M., Arand J., Galán C., Winter G.E., Engist B., Gerle B., et al. Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol. Cell. 2014;55:277–290. doi: 10.1016/j.molcel.2014.05.029. [DOI] [PubMed] [Google Scholar]
- Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B., et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A., et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sena Brandine G., Smith A.D. Falco: high-speed FastQC emulation for quality control of sequencing data. F1000Res. 2019;8:1874. doi: 10.12688/f1000research.21142.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y., Milne T.A., Ruthenburg A.J., Lee S., Lee J.W., Verdine G.L., Allis C.D., Roeder R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- Edmunds J.W., Mahadevan L.C., Clayton A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008;27:406–420. doi: 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgin S.C., Reuter G. Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harb. Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a017780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauchier M., Kan S., Barral A., Sauzet S., Agirre E., Bonnell E., Saksouk N., Barth T.K., Ide S., Urbach S., et al. SETDB1-dependent heterochromatin stimulates alternative lengthening of telomeres. Sci. Adv. 2019;5 doi: 10.1126/sciadv.aav3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther M.G., Levine S.S., Boyer L.A., Jaenisch R., Young R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins R.D., Hon G.C., Lee L.K., Ngo Q., Lister R., Pelizzola M., Edsall L.E., Kuan S., Luu Y., Klugman S., et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe F.S., Fischl H., Murray S.C., Mellor J. Is H3K4me3 instructive for transcription activation? BioEssays. 2017;39:1–12. doi: 10.1002/bies.201600095. [DOI] [PubMed] [Google Scholar]
- Huang C., Zhu B. Roles of H3K36-specific histone methyltransferases in transcription: antagonizing silencing and safeguarding transcription fidelity. Biophys. Rep. 2018;4:170–177. doi: 10.1007/s41048-018-0063-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun K., Jeon J., Park K., Kim J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017;49:e324. doi: 10.1038/emm.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Loh Y.E., Rajarajan P., Hirayama T., Liao W., Kassim B.S., Javidfar B., Hartley B.J., Kleofas L., Park R.B., et al. The methyltransferase SETDB1 regulates a large neuron-specific topological chromatin domain. Nat. Genet. 2017;49:1239–1250. doi: 10.1038/ng.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey M.H., Newman J.J., Bilodeau S., Zhan Y., Orlando D.A., van Berkum N.L., Ebmeier C.C., Goossens J., Rahl P.B., Levine S.S., et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M.M., Goyal P., Maksakova I.A., Bilenky M., Leung D., Tang J.X., Shinkai Y., Mager D.L., Jones S., Hirst M., Lorincz M.C. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011;8:676–687. doi: 10.1016/j.stem.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide S., Oshima M., Takubo K., Yamazaki S., Nitta E., Saraya A., Aoyama K., Kato Y., Miyagi S., Nakajima-Takagi Y., et al. Setdb1 maintains hematopoietic stem and progenitor cells by restricting the ectopic activation of nonhematopoietic genes. Blood. 2016;128:638–649. doi: 10.1182/blood-2016-01-694810. [DOI] [PubMed] [Google Scholar]
- Kuo A.J., Cheung P., Chen K., Zee B.M., Kioi M., Lauring J., Xi Y., Park B.H., Shi X., Garcia B.A., et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell. 2011;44:609–620. doi: 10.1016/j.molcel.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhoumaud P., Badri S., Rodriguez-Hernaez J., Sakellaropoulos T., Sethia G., Kloetgen A., Cornwell M., Bhattacharyya S., Ay F., Bonneau R., et al. NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nat. Commun. 2019;10:4843. doi: 10.1038/s41467-019-12811-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Trojer P., Xu C.F., Cheung P., Kuo A., Drury W.J., 3rd, Qiao Q., Neubert T.A., Xu R.M., Gozani O., Reinberg D. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 2009;284:34283–34295. doi: 10.1074/jbc.M109.034462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann F., Loureiro J., Su H., Fang Q., Lei H., Lewis T., Yang Y., Labow M., Li E., Chen T., Kadam S. KMT1E mediated H3K9 methylation is required for the maintenance of embryonic stem cells by repressing trophectoderm differentiation. Stem Cells. 2010;28:201–212. doi: 10.1002/stem.278. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco R.F., Pan Q., Tominaga K., Blencowe B.J., Pereira-Smith O.M., Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. [Google Scholar]
- Matsui T., Leung D., Miyashita H., Maksakova I.A., Miyachi H., Kimura H., Tachibana M., Lorincz M.C., Shinkai Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. 2010;464:927–931. doi: 10.1038/nature08858. [DOI] [PubMed] [Google Scholar]
- Matsumura Y., Nakaki R., Inagaki T., Yoshida A., Kano Y., Kimura H., Tanaka T., Tsutsumi S., Nakao M., Doi T., et al. H3K4/H3K9me3 bivalent chromatin domains targeted by lineage-specific DNA methylation pauses adipocyte differentiation. Mol Cell. 2015;60:584–596. doi: 10.1016/j.molcel.2015.10.025. [DOI] [PubMed] [Google Scholar]
- Mauser R., Kungulovski G., Keup C., Reinhardt R., Jeltsch A. Application of dual reading domains as novel reagents in chromatin biology reveals a new H3K9me3 and H3K36me2/3 bivalent chromatin state. Epigenetics Chromatin. 2017;10:45. doi: 10.1186/s13072-017-0153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel S.L., Strahl B.D. Shaping the cellular landscape with Set2/SETD2 methylation. Cell. Mol. Life Sci. 2017;74:3317–3334. doi: 10.1007/s00018-017-2517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkenschlager M., Nora E.P. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu. Rev. Genomics Hum. Genet. 2016;17:17–43. doi: 10.1146/annurev-genom-083115-022339. [DOI] [PubMed] [Google Scholar]
- Nagano T., Lubling Y., Várnai C., Dudley C., Leung W., Baran Y., Mendelson Cohen N., Wingett S., Fraser P., Tanay A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 2017;547:61–67. doi: 10.1038/nature23001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicetto D., Donahue G., Jain T., Peng T., Sidoli S., Sheng L., Montavon T., Becker J.S., Grindheim J.M., Blahnik K., et al. H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science. 2019;363:294–297. doi: 10.1126/science.aau0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicetto D., Zaret K.S. Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr. Opin. Genet. Dev. 2019;55:1–10. doi: 10.1016/j.gde.2019.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Carroll D., Scherthan H., Peters A.H., Opravil S., Haynes A.R., Laible G., Rea S., Schmid M., Lebersorger A., Jerratsch M., et al. Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol. Cell. Biol. 2000;20:9423–9433. doi: 10.1128/mcb.20.24.9423-9433.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou J., Liu H., Yu J., Kelliher M.A., Castilla L.H., Lawson N.D., Zhu L.J. ATACseqQC: a Bioconductor package for post-alignment quality assessment of ATAC-seq data. BMC Genomics. 2018;19:169. doi: 10.1186/s12864-018-4559-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A.H., O’Carroll D., Scherthan H., Mechtler K., Sauer S., Schöfer C., Weipoltshammer K., Pagani M., Lachner M., Kohlmaier A., et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–337. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Pradeepa M.M., Sutherland H.G., Ule J., Grimes G.R., Bickmore W.A. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S.A., Flynn R.A., Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo R., Karimi M.M., Bilenky M., Gagnier L., Miceli-Royer K., Zhang Y., Goyal P., Keane T.M., Jones S., Hirst M., et al. Retrotransposon-induced heterochromatin spreading in the mouse revealed by insertional polymorphisms. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1002301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes C.S., Stark R., Teschendorff A.E., Holmes K.A., Ali H.R., Dunning M.J., Brown G.D., Gojis O., Ellis I.O., Green A.R., et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–393. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg A.J., Allis C.D., Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol. Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Saint-André V., Batsché E., Rachez C., Muchardt C. Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat. Struct. Mol. Biol. 2011;18:337–344. doi: 10.1038/nsmb.1995. [DOI] [PubMed] [Google Scholar]
- Saksouk N., Simboeck E., Déjardin J. Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin. 2015;8:3. doi: 10.1186/1756-8935-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D.C., Ayyanathan K., Negorev D., Maul G.G., Rauscher F.J., 3rd. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.J., Vert J.P., Heard E., Dekker J., Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H., Shi Y., Dai C., Tjong H., Gong K., Alber F., Zhou X.J. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2016;44:e70. doi: 10.1093/nar/gkv1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J.A., Kingston R.E. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- Sterne-Weiler T., Weatheritt R.J., Best A.J., Ha K.C.H., Blencowe B.J. Efficient and accurate quantitative profiling of alternative splicing patterns of any complexity on a laptop. Mol. Cell. 2018;72:187–200.e6. doi: 10.1016/j.molcel.2018.08.018. [DOI] [PubMed] [Google Scholar]
- Steward M.M., Lee J.S., O’Donovan A., Wyatt M., Bernstein B.E., Shilatifard A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat. Struct. Mol. Biol. 2006;13:852–854. doi: 10.1038/nsmb1131. [DOI] [PubMed] [Google Scholar]
- Streubel G., Watson A., Jammula S.G., Scelfo A., Fitzpatrick D.J., Oliviero G., McCole R., Conway E., Glancy E., Negri G.L., et al. The H3K36me2 methyltransferase Nsd1 demarcates PRC2-mediated H3K27me2 and H3K27me3 domains in embryonic stem cells. Mol. Cell. 2018;70:371–379.e5. doi: 10.1016/j.molcel.2018.02.027. [DOI] [PubMed] [Google Scholar]
- Tan S.L., Nishi M., Ohtsuka T., Matsui T., Takemoto K., Kamio-Miura A., Aburatani H., Shinkai Y., Kageyama R. Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development. 2012;139:3806–3816. doi: 10.1242/dev.082198. [DOI] [PubMed] [Google Scholar]
- Timms R.T., Tchasovnikarova I.A., Lehner P.J. Position-effect variegation revisited: HUSHing up heterochromatin in human cells. BioEssays. 2016;38:333–343. doi: 10.1002/bies.201500184. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle-García D., Qadeer Z.A., McHugh D.S., Ghiraldini F.G., Chowdhury A.H., Hasson D., Dyer M.A., Recillas-Targa F., Bernstein E. ATRX binds to atypical chromatin domains at the 3′ exons of zinc finger genes to preserve H3K9me3 enrichment. Epigenetics. 2016;11:398–414. doi: 10.1080/15592294.2016.1169351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villaseñor R., Pfaendler R., Ambrosi C., Butz S., Giuliani S., Bryan E., Sheahan T.W., Gable A.L., Schmolka N., Manzo M., et al. ChromID identifies the protein interactome at chromatin marks. Nat. Biotechnol. 2020;38:728–736. doi: 10.1038/s41587-020-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., An W., Cao R., Xia L., Erdjument-Bromage H., Chatton B., Tempst P., Roeder R.G., Zhang Y. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol. Cell. 2003;12:475–487. doi: 10.1016/j.molcel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Wang P., Lin C., Smith E.R., Guo H., Sanderson B.W., Wu M., Gogol M., Alexander T., Seidel C., Wiedemann L.M., et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 2009;29:6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg D.N., Papillon-Cavanagh S., Chen H., Yue Y., Chen X., Rajagopalan K.N., Horth C., McGuire J.T., Xu X., Nikbakht H., et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature. 2019;573:281–286. doi: 10.1038/s41586-019-1534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K., Liu H., Wang Y., He J., Xu S., Chen Y., Kuang J., Liu J., Guo L., Li D., et al. SETDB1-mediated cell fate transition between 2C-like and pluripotent states. Cell Rep. 2020;30:25–36.e6. doi: 10.1016/j.celrep.2019.12.010. [DOI] [PubMed] [Google Scholar]
- Yu G., Wang L.G., He Q.Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- Yuan P., Han J., Guo G., Orlov Y.L., Huss M., Loh Y.H., Yaw L.P., Robson P., Lim B., Ng H.H. Eset partners with Oct4 to restrict extraembryonic trophoblast lineage potential in embryonic stem cells. Genes Dev. 2009;23:2507–2520. doi: 10.1101/gad.1831909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W., Xie J., Long C., Erdjument-Bromage H., Ding X., Zheng Y., Tempst P., Chen S., Zhu B., Reinberg D. Heterogeneous nuclear ribonucleoprotein L Is a subunit of human KMT3a/Set2 complex required for H3 Lys-36 trimethylation activity in vivo. J. Biol. Chem. 2009;284:15701–15707. doi: 10.1074/jbc.M808431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Cooper S., Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep. 2015;16:1467–1481. doi: 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Yan X., Feng X., Bu J., Dong Y., Lin P., Hayashi Y., Huang R., Olsson A., Andreassen P.R., et al. Setd2 regulates quiescence and differentiation of adult hematopoietic stem cells by restricting RNA polymerase II elongation. Haematologica. 2018;103:1110–1123. doi: 10.3324/haematol.2018.187708. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original immunoblot, electrophoresis and slot-blot images reported in this study have been deposited at Mendeley and are publicly available. The DOI is listed in the Key resources table.
This study does not report original code. It uses publicly available software to analyze/re-analyze publicly available NGS data. DOIs and GEO accessions are listed in the Key resources table.
Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.