


Abstract
The Cas9 nuclease from Staphylococcus aureus (SaCas9) holds great potential for use in gene therapy, and variants with increased fidelity have been engineered. However, we find that existing variants have not reached the greatest accuracy to discriminate base mismatches and exhibited much reduced activity when their mutations were grafted onto the KKH mutant of SaCas9 for editing an expanded set of DNA targets. We performed structure-guided combinatorial mutagenesis to re-engineer KKH-SaCas9 with enhanced accuracy. We uncover that introducing a Y239H mutation on KKH-SaCas9’s REC domain substantially reduces off-target edits while retaining high on-target activity when added to a set of mutations on REC and RuvC domains that lessen its interactions with the target DNA strand. The Y239H mutation is modelled to have removed an interaction from the REC domain with the guide RNA backbone in the guide RNA-DNA heteroduplex structure. We further confirmed the greatly improved genome-wide editing accuracy and single-base mismatch discrimination of our engineered variants, named KKH-SaCas9-SAV1 and SAV2, in human cells. In addition to generating broadly useful KKH-SaCas9 variants with unprecedented accuracy, our findings demonstrate the feasibility for multi-domain combinatorial mutagenesis on SaCas9’s DNA- and guide RNA- interacting residues to optimize its editing fidelity.
INTRODUCTION
Translating genome editing technologies for therapeutic applications requires tools that are highly specific, robust, as well as being able to be delivered in vivo, and applicable for targeting a wide range of disease-relevant loci. CRISPR (i.e. clustered regularly interspaced short palindromic repeats), a programmable gene-targeting system capable of knocking out genes in human cells, has become a widely used tool for genome editing and hold great potentials for gene therapy applications (1,2). The CRISPR system has two components: (i) the Cas9 nuclease and (ii) a single guide RNA (sgRNA) that provides DNA sequence-targeting accuracy. The targeting of the Cas9–sgRNA complex is mediated by the protospacer adjacent motif (PAM) located at the DNA for Cas9 recognition and the homology between the ∼20-nucleotide recognition sequence encoded in the sgRNA and the genomic DNA target. The targeted protein-coding gene can be knocked out after the Cas9–sgRNA complex finds and cleaves the exonic region of the gene to generate frameshift mutations. The more commonly used CRISPR enzyme for genome editing, SpCas9, is derived from the bacteria strain Streptococcus pyogenes, and it finds DNA targets carrying an “NGG” PAM site. SpCas9 variants with a specific combination of mutations were engineered to minimize its off-target editing (3–9). However, fewer studies have been conducted on SaCas9, although SaCas9 holds an important advantage of being smaller than SpCas9 that enables its efficient packaging using adeno-associated virus vectors for in vivo gene editing and gene therapy applications.
SaCas9 was reported to edit the human genome with similar efficiency as with SpCas9 (10). Using SaCas9 for genome editing requires its target site to contain a longer PAM site (i.e. “NNGRRT”). To overcome this limitation, several mutational studies on SaCas9 were carried out to broaden its PAM recognition (11–13), and KKH-SaCas9 is one of the identified variants that recognizes an “NNNRRT” PAM site. This variant is useful for therapeutic genome editing because it can edit sites with PAM that other small-sized Cas orthologs such as Cas9 from Campylobacter jejuni (14) and Neisseria meningitidis (15), as well as Cas12a (16) and CasΦ (17) cannot recognize. In terms of editing fidelity, SaCas9 variants (including SaCas9-HF (18) and eSaCas9 (3)) carrying a specific combination of mutations at its amino acid residues that interact with the targeting or non-targeting DNA strand and the sgRNA were shown to exhibit reduced off-target activity. Comparison between SaCas9-HF with eSaCas9 revealed that they have comparable on-target activity while SaCas9-HF has a lower genome-wide off-target activity (18). However, grafting the mutations (i.e. R245A/N413A/N419A/R654A) from SaCas9-HF onto KKH saCas9 greatly reduced its on-target activity in targeting many of the tested gene targets (18). There is no SaCas9 variant with a broad targeting range (such as KKH-SaCas9) being engineered with both high efficiency and proven genome-wide accuracy, which is needed for therapeutic applications. Here we engineered and characterized a panel of combination mutants for KKH-SaCas9 and identified new variants that are super-accurate and efficient.
MATERIALS AND METHODS
Construction of DNA vectors
The vector constructs used in this study (Supplementary Table S1) were generated using standard molecular cloning techniques, including PCR, restriction enzyme digestion, ligation, and Gibson assembly. Custom oligonucleotides were purchased from Genewiz. To create the expression vector encoding KKH-SaCas9-HF, KKH-efSaCas9 and KKH-eSaCas9, the SaCas9 sequences were amplified/mutated from Addgene #61591 and #117552 by PCR and cloned into the pFUGW lentiviral vector backbone. To construct the expression vector containing U6 promoter-driven expression of a sgRNA that targeted a specific locus, oligo pairs with the gRNA target sequences were synthesized, annealed, and cloned in the pFUGW-based vector. The gRNA spacer sequences are listed in Supplementary Table S2. The constructs were transformed into E. coli strain DH5α, and 50 μg/ml of carbenicillin/ampicillin was used to isolate colonies harboring the constructs. DNA was extracted and purified using Plasmid Mini (Takara) or Midi (Qiagen) kits. Sequences of the vector constructs were verified with Sanger sequencing.
A library of KKH-SaCas9 variants with combinations of substitution mutations was constructed. Based on predictions from protein structure models, we focused on 12 amino acid residues that were predicted to make contacts with or be in close proximity to the DNA and sgRNA backbones, and modified them to harbor specified substitutions (Supplementary Table S3). Some of those mutations are present in SaCas9-HF (18) and eSaCas9 (3). We hypothesized that specific combinations of these mutations in KKH-SaCas9 could reduce its undesirable off-target activity, while maximizing its on-target editing efficiency. To assemble the KKH-SaCas9 variants with combinations of substitution mutations, the KKH-SaCas9 sequence is modularized into four parts (P1 to P4). The modularized parts with specific mutations were generated by PCR or synthesis, and each of them is flanked by a pair of type IIS restriction enzyme cut sites on their two ends. The variants within each part were pooled together. Type IIS restriction enzymes were used to iteratively digest and ligate to the subsequent pool of DNA parts in a lentiviral vector to generate higher-order combination mutants. Since digestion with type IIS restriction enzymes generates compatible overhangs that are originated from the protein-coding sequence, no fusion scar is formed in the ligation reactions. A set of 27 variants (i.e. v3.1-20, v3.22-25 and v3.27-29) were randomly sampled from the combination mutant library of KKH-SaCas9 and their editing activities were individually characterized using multiple sgRNA reporter lines.
Human cell culture
HEK293T and SK-N-MC cells were obtained from American Type Culture Collection (ATCC). MHCC97L cells were gifts from S. Ma (School of Biomedical Sciences, The University of Hong Kong). OVCAR8-ADR cells were gifts from T. Ochiya (Japanese National Cancer Center Research Institute, Japan), and the identity of the OVCAR8-ADR cells was confirmed by a cell line authentication test (Genetica DNA Laboratories). OVCAR8-ADR cells were transduced with lentiviruses encoding RFP and GFP genes expressed from UBC and CMV promoters, respectively, and a tandem U6 promoter-driven expression cassette of sgRNA targeting GFP site. ON1 and ON2 lines harbor sgRNA’s spacer that matches completely with the target sites on GFP, while OFF1, OFF2 and OFF3 lines harbor single-base mismatches to the targets site. To generate cell lines stably expressing SaCas9 protein, cells were infected with a lentiviral expression vector encoding KKH-SaCas9, KKH-SaCas9- SAV1 and SAV2, followed by P2A-BFP. These cells were sorted using a Becton Dickinson BD Influx cell sorter. HEK293T, SK-N-MC and MHCC97L cells were cultured in Dulbecco's Modified Eagle Medium (DMEM). supplemented with 10% heat-inactivated FBS and 1× antibiotic-antimycotic (Thermo Scientific) at 37°C with 5% CO2. OVCAR8-ADR cells were cultured in RPMI supplemented with 10% heat-inactivated FBS and 1× antibiotic-antimycotic (Thermo Scientific) at 37°C with 5% CO2. Cells were regularly tested for mycoplasma contamination and were confirmed to be negative. Lentivirus production and transduction were carried out as previously described (9).
Fluorescent protein disruption assay
Fluorescent protein disruption assays were performed to evaluate DNA cleavage and indel-mediated disruption at the target site of the fluorescent protein (i.e. GFP) brought by SaCas9 and gRNA expressions, which results in loss of cell fluorescence. Cells harboring an integrated GFP and RFP reporter gene and together with SaCas9 and sgRNA were washed and resuspended with 1× PBS supplemented with 2% heat-inactivated FBS, and assayed with a Becton Dickinson LSR Fortessa Analyzer or ACEA NovoCyte Quanteon. Cells were gated on forward and side scatter. At least 1 × 104 cells were recorded per sample in each data set.
Immunoblot analysis
Immunoblotting experiments were carried out as previously described (9). Primary antibodies used were: anti-SaCas9 (1:1,000, Cell Signaling #85687) and anti-GAPDH (1:5000, Cell Signaling #2118). Secondary antibody used was HRP-linked anti-mouse IgG (1:10 000, Cell Signaling #7076) and HRP-linked anti-rabbit IgG (1:20 000, Cell Signaling #7074).
T7 Endonuclease I assay
T7 endonuclease I assay was carried out as previously described (9). Amplicons harboring the targeted loci were generated by PCR. The PCR primer sequences are listed in Supplementary Table S4. Quantification was based on relative band intensities measured using ImageJ. Editing efficiency was estimated by the formula, 100 × (1 − (1 − (b + c)/(a + b + c))1/2) as previously described (19), where a is the integrated intensity of the uncleaved PCR product, and b and c are the integrated intensities of each cleavage product. Normalized editing efficiency brought by the KKH-SaCas9 variants to those by wild-type are calculated for each sgRNA.
GUIDE-seq
Genome-wide off-targets were accessed using the GUIDE-seq method (20). Experimental procedures for preparing sequencing libraries were carried out as previously described (9). For each GUIDE-seq sample, 1.6 million MHCC97L cells infected with SaCas9 variants and sgRNAs (EMX1-sg2, EMX1-sg7, VEGFA-sg3, AAVS1-sg4 and CCR5-sg2) were electroporated with 1100 pmol freshly annealed GUIDE-seq end-protected dsODN using 100 μl Neon tips (ThermoFisher Scientific) according to the manufacturer's protocol. The dsODN oligonucleotides used for annealing were 5′-P-G*T*TTAATTGAGTTGTCATATGTTAATAACGGT*A*T-3′ and 5′-P-A*T*ACCGTTATTAACATATGACAACTCAATTAA*A*C-3′, where P represents 5′ phosphorylation and asterisks indicate a phosphorothioate linkage. The electroporation parameters used were 1100 volts, 20 pulse width, and pulse 3. Similarly, 1.5 million OVCAR8-ADR cells infected with SaCas9 variants and the VEGFA-sg8 sgRNA were electroporated with the dsODN. Sequencing libraries were sequenced on Illumina NextSeq System and analysed using the GUIDE-seq software (21). Our updated GUIDE-seq software is based on tsailabSJ/guideseq with the following modifications: (i) changes to make it compatible with python 3.8; (ii) configurable UMI length and sample index length; (iii) configurable PAM sequence; (iv) tox automated testing for python 3.8 to test against alignment data generated by bwa-0.7.17.
Deep sequencing
Deep sequencing was carried out as previously described (22). OVCAR8-ADR cells were infected with SaCas9 variants and the sgRNAs bearing perfectly matched or single-base-pair-mismatched protospacer sequences. Amplicons harboring the targeted loci were generated by PCR. ∼1 million reads per sample on average were used to evaluate the editing consequences of >10 000 cells. Indel quantification around the protospacer regions was conducted using CRISPresso2 (23).
Reverse transcription quantitative PCR (RT-qPCR)
OVCAR8-ADR cells were transduced by BFP-tagged KKH-dSaCas9-KRAB variants and then by GFP-marked sgRNA lentiviral vectors 3 days after. Co-infected cells were sorted by BD FACSAria SORP based on the fluorescent signals 7 days post-sgRNA infection. Total RNA was extracted from the sorted cells and reverse transcription were done by using MiniBEST Universal RNA Extraction Kit and PrimeScript™ RT Reagent Kit (TaKaRa), respectively, according to the manufacturer's instructions. qPCR was performed using TB Green Premix Ex Taq (TaKaRa), with the standard PCR protocol. Relative gene expressions were determined relative to GAPDH using standard ΔΔCt method (2–ΔΔCt). qPCR primers used are listed in Supplementary Table S4.
Molecular modelling
Molecular dynamic simulations were conducted on the variants using DynaMut (24). The variants mutations were singly inputted into the webserver, and the structural outputs were then aligned with the crystal structure of SaCas9 (PDB: 5CZZ) on PyMol. The predicted rotamer of the mutations as indicated by DynaMut was then used to replace the amino acid positions on the SaCas9 crystal structure. The predicted interactions determined by DynaMut and Pymol were then drawn on the crystal structure to provide a putative representation of the SaCas9 variants. Chimera v.1.4 was used for intermolecular contacts estimation, atom-atom distance calculation, and visualization of the protein model.
RESULTS
Identification of KKH-SaCas9 SAV1 and SAV2 variants with enhanced accuracy
We have previously described over 50% reduction of KKH-SaCas9’s activity on editing three out of five endogenous loci when mutations found on SaCas9-HF (i.e. R245A/N413A/N419A/R654A) was directly grafted onto it (18). We have confirmed this observation by evaluating the editing efficiency of KKH-SaCas9-HF against additional endogenous loci and detected 88% of reduction (averaged from nine sgRNAs) in its on-target activity when compared to KKH-SaCas9 (Figure 1A; Supplementary Figure S1). Here we attempted to re-engineer KKH-SaCas9 with low off-target and high on-target editing activities. It is plausible that specific mutations found on SaCas9-HF may be detrimental to KKH-SaCas9’s overall activity, and alternative residues and substitutions could be exploited to more optimally engineer the enzyme. Based on protein structure analyses, multiple residues of SaCas9, including those scattered over its REC and RuvC domains, are predicted to interact with the DNA and sgRNA backbones (Supplementary Table S3). To gain insights on which of those residues play more important roles in affecting KKH-SaCas9’s activity, we started off to assemble and randomly-sample variants with combinations of 12 substitution mutations that are located at the different regions of the protein (see Materials and Methods) and measure their on- and off-target editing activities of variants. An initial set of 27 variants (i.e. v3.1-20, v3.22-25 and v3.27-29) carrying different sets of these substitution mutations were individually constructed and characterized using two sgRNAs targeting a GFP reporter. Among these variants analyzed, we noted that there was a stark contrast of on-target activities between variants with and without R245A. Variants harboring R245A showed >60% of reduction (and in most cases >80% reduction) in at least one out of the two tested sgRNAs at day 15 post-transduction in the green fluorescent protein (GFP) disruption assays, which is similar to that detected for the R245A-containing KKH-SaCas9-HF (Figure 1B). Based on molecular modelling, R245 is predicted to make multiple contacts with the DNA backbone (Figure 1C). Losing too many interactions with the DNA may explain the incompatibility of adding R245A to N413A/N419A/R654A mutations that were found on SaCas9-HF in the KKH-SaCas9 setting. Indeed, grafting only three mutations N413A/N419A/R654A but not including R245A completely restored the on-target activities of KKH-SaCas9 (Figure 1B). However, there was also a minimal reduction of its off-target activities (Figure 1B), indicating the need for alternative mutations to improve KKH-SaCas9’s editing accuracy.
Figure 1.

Identification of KKH-SaCas9- SAV1 and SAV2 variants with enhanced accuracy. (A) KKH-SaCas9-HF exhibits low on-target activity assessed by T7E1 assay. Activity is normalized to those of KKH-SaCas9, and mean and standard deviation are shown for sgRNAs targeting 9 loci. The full dataset is presented in Supplementary Figure S1. (B–D) KKH-SaCas9 variants carrying different mutation combinations were characterized using GFP disruption assays. Editing efficiency of KKH-SaCas9 variants was measured as percentage of cells with depleted GFP fluorescence and compared to efficiency for KKH-SaCas9. The percentages of off-target GFP disruption are presented in Supplementary Figure S2. Data in D shown are mean and standard deviation obtained from three biological replicates. Molecular models of R245A mutation in SaCas9 are shown in panel C.
Maintaining the core stability of the Cas9 protein and the intricate balance of contacts between sgRNA and DNA seems to be crucial for retaining the on-target activities while reducing off-targeting (8,9). Promising KKH-SaCas9 variants with high activity and targeting accuracy were identified among the variants analyzed. Eight of the variants (i.e. v3.18, v3.8, v3.22, v3.24, v3.19, v3.16, v3.10, v3.2) showed high on-target activities (with >60% of KKH-SaCas9 activity at day 15 post-transduction, averaged from two sgRNAs), and 7 of them exhibited greatly reduced off-target activities (decreased by >90%; being characterized using 3 individual sgRNAs each bearing a single-base-pair-mismatched protospacer sequence) (Figure 1B). In particular, the variant v3.16 harboring Y239H/N419D/R499A/Q500A/Y651H mutations generated the fewest off-target edits after 15 days post-transduction (reduced by >95%) and resulted in an average of ∼70% of on-target activity, when compared to KKH-SaCas9 (Figure 1B; Supplementary Figure S2). This variant v3.16 was able to better discriminate most of the single-base-pair mismatches between the DNA target and the tested sgRNA, which span over the entire protospacer sequence (Figure 1D; Supplementary Figure S3). We designated this super-accurate variant as KKH-SaCas9-SAV1 (hereinafter also referred to as “SAV1”). Furthermore, we identified another variant v3.10 (hereinafter referred to as KKH-SaCas9-SAV2 or “SAV2”) harboring Y239H/N419D/R654A/G655A mutations that showed very few off-target edits at day 7 post-transduction similar to SAV1, exhibiting comparable on-target activity (an average of ∼80%) to KKH-SaCas9 (Figure 1B, D; Supplementary Figure S2; S3). Variants harboring additional substitution(s) over the quadruple mutant SAV2 exhibited lower on-target activities (Figure 1B; 2A). Variant v3.1, another tested variant which is like SAV2 but carries R245A instead of Y239H, also resulted in less on-target edits than SAV2 (Figure 1B). The R245-containing v3.1 when added with Y239H (as well as other tested variants containing both Y239H and R245A) indeed showed reduced editing more consistently across the two on-target sites tested (Figure 1B), which suggests that Y239H and R245A co-mutation is particularly detrimental to the enzyme's activity. SAV1 and SAV2 were thus selected for further characterization.
We attempted to gain structural insights on why KKH-SaCas9-SAV1 and SAV2 exhibit low off-target and high on-target activities, and our results revealed a pivotal role of the previously unreported Y239H substitution in determining target accuracy while maintaining the activity of KKH-SaCas9. We found that SAV2 lacking Y239H (i.e. variant v3.2) generated substantially more off-target edits (Figure 1B; Supplementary Figure S2). In other words, the triple mutant combination (N419D/R654A/G655A) alone is insufficient in minimizing off-target editing. Replacing Y239H with R245A in SAV2 also increased off-target edits, and such variant exhibited reduced on-target activity (Figure 1B). Based on molecular modelling, mutating Y239 into histidine could weaken the enzyme's bonding with the sgRNA backbone to reduce off-target editing, while preserving the π–π interactions with its F418 (Figure 2B). We also mutated Y239 into arginine that could maintain its bonding with the sgRNA backbone but form a cation–π interaction with F418 (Figure 2B). The variant bearing Y239R instead of Y239H indeed generated less overall activity with a more similar on-to-off targeting ratio as KKH-SaCas9 (Figure 2A). This observation supports the possible involvement of the π-π interaction in maintaining the enzyme's structural stability and activity. We also observed that adding substitutions including N394T (i.e. v3.24 in Figure 1B) or T392A/N394T (Figure 2A) to SAV2 decreased its activity. N394T is modelled to reduce interaction with the sgRNA backbone at the side opposite to where Y239 interacts (Supplementary Figure S4). Losing multiple interactions with the sgRNA backbone may account for the drop of SAV2’s activity when these mutations were added. The result showing that SAV2 with Y239R instead of its original Y239H mutation was slightly less susceptible to the drop of activity brought by T392A/N394T addition could be due to the prediction that Y239R does not lose its interaction with the sgRNA backbone. Thus, tuning the enzyme's interaction with the sgRNA backbone to reduce off-target editing, while maintaining high on-target activity, requires optimal engineering at specific site(s). In addition, we tested the replacement of R245A with Y239H in KKH-SaCas9-HF to improve its activity and target accuracy. We found that such replacement resulted in fewer off-target edits while partially restoring the enzyme's on-target activity (Figure 1B; Supplementary Figure S2). Nonetheless, this variant still exhibited far lower on-target activity and greater off-target activity than SAV2, suggesting that Y239H orchestras with the also off-target-reducing N419D/R654A/G655A mutations (i.e. variant v3.2, Figure 1B) to achieve optimal editing performance for SAV2.
Figure 2.

Y239H is important for KKH-SaCas9-SAV2’s editing specificity and activity.(A) KKH-SaCas9-SAV2 carrying a Y239R substitution and/or additional mutations were individually constructed and characterized using GFP disruption assays. OVCAR8-ADR cells harboring reporter constructs with two on-target sgRNAs and three off-target sgRNAs were infected with lentiviruses encoding the individual KKH-SaCas9 mutants. After 7- and 15- day post-infection, the editing efficiency of the KKH-SaCas9 variants was measured as the percentage of cells with depleted GFP fluorescence using flow cytometry. Mean and standard deviation obtained from at least two biological replicates are shown. (B) Molecular modelling of Y239H/R mutations in SaCas9 depicts their differential interactions with F418 of SaCas9 and the sgRNA backbone.
Comparison of on- and off- target activities of KKH-SaCas9 variants
We compared the on- and off-target activities of SAV1 and SAV2 with existing/candidate high-fidelity variants of KKH-SaCas9. Our results from GFP disruption assays showed that SAV1 and SAV2 exhibited higher on-target editing activity than KKH-SaCas9-HF, (i.e. ∼70%, ∼85% and ∼40% of KKH-SaCas9 activity for SAV1, SAV2 and KKH-SaCas9-HF, respectively), and generated much less off-target edits (i.e. reduced by >98%, ∼95% and ∼60% for SAV1, SAV2 and KKH-SaCas9-HF, respectively, when compared to KKH-SaCas9) (Figure 3A). A candidate N260D substitution was more recently reported to reduce the off-target activity of SaCas9 (25). We grafted this mutation onto KKH-SaCas9 to generate KKH-efSaCas9. Despite this variant exhibited comparable on-target activity to KKH-SaCas9 and SAV2, it generated a high frequency of off-target edits (i.e. reduced by only ∼40% for KKH-efSaCas9 versus ∼95% for SAV2, when compared to KKH-SaCas9) (Figure 3A). Adding N260D to mutations on SAV1 and SAV2 greatly reduced their on-target activities (Supplementary Figure S5). Another variant with mutations grafted from eSaCas9 (3) onto KKH-SaCas9 generated off-target edits fewer than those by wild-type but more than those by SAV2, while it produced more on-target edits with the two tested sgRNAs (Figure 3B). The differences in the editing efficiencies observed were not due to the discrepancy in protein expression levels of the variants (Supplementary Figure S6). These results indicated that SAV1 and SAV2, along with KKH-eSaCas9, showed superior fidelity and efficiency to other existing variants (Figure 3C).
Figure 3.
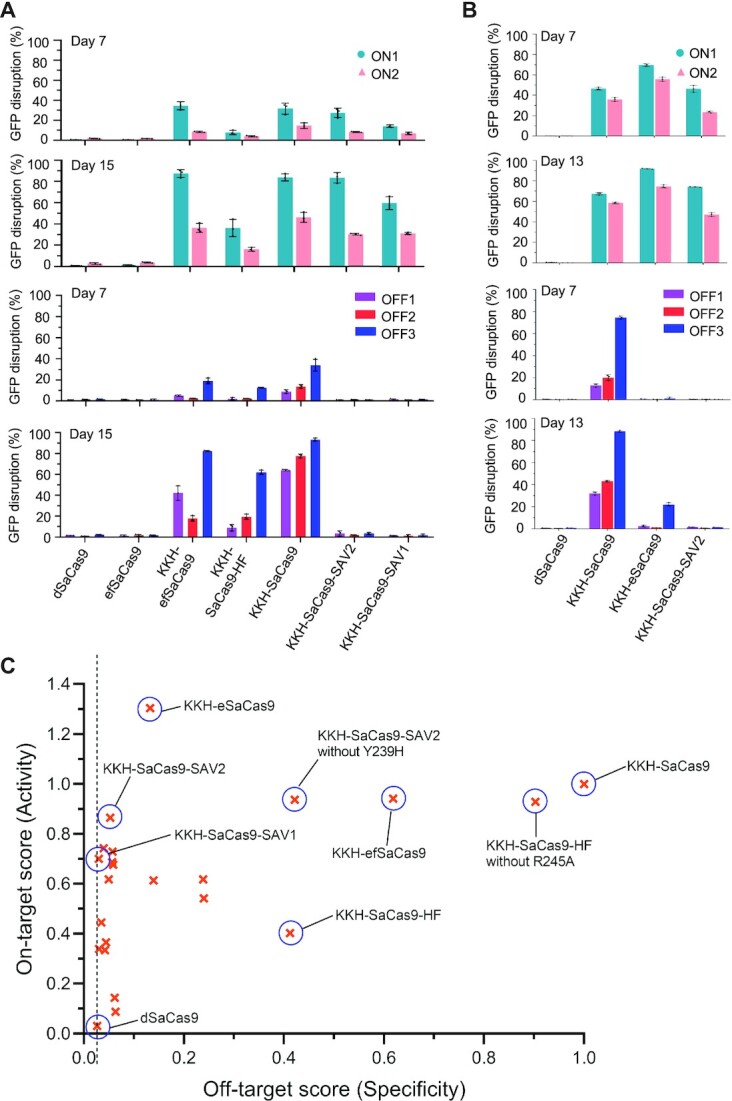
Comparison of on- and off- target activities of KKH-SaCas9 variants. (A, B) KKH-SaCas9 variants carrying different mutation combinations were constructed and characterized using GFP disruption assays. The editing efficiency of the KKH-SaCas9 variants was measured as the percentage of cells with depleted GFP fluorescence and compared to the efficiency for KKH-SaCas9. Mean and standard deviation obtained from three biological replicates are shown. (C) Specificity and activity scores for tested variants.
We further characterized the performance of SAV1, SAV2 and KKH-eSaCas9 in editing endogenous genomic loci, we performed T7 Endonuclease I mismatch detection assay, Genome-wide unbiased identification of double-strand breaks enabled by sequencing (GUIDE-seq), and deep sequencing to evaluate their on- and off-target activities in three cell lines. By assaying multiple endogenous loci that we and others have previously studied (11,18), we showed that SAV1, SAV2 and KKH-eSaCas9 exhibited a median editing efficiency of 51%, 72% and 87% of KKH-SaCas9’s activity, respectively (Figure 4A; Supplementary Figure S7) in OVCAR8-ADR cells, which is much higher than KKH-SaCas9-HF (Figure 1A). Comparison of the variants’ on-target activity was extended to SK-N-MC and MHCC97L cells, and our results showed a similar trend. The normalized median editing efficiency for each variant was: 43% for SAV1, 65% for SAV2 and 111% for KKH-eSaCas9 in SK-N-MC cells, while 65% for SAV1, 81% for SAV2 and 110% for KKH-eSaCas9 in MHCC97L cells (Figure 4A). GUIDE-seq results indicated that SAV1 and SAV2 resulted in higher on- to off-targeting ratio than wild-type in all tested loci (Figure 4B; Supplementary Figure S8). We further evaluated the variants’ ability to discriminate target sequences with single-base mismatches. Deep sequencing analysis was performed using a panel of sgRNAs that are perfectly matched or carry a single-base mismatch to the target sequences (i.e. VEGFA and FANCF). Compared with wild-type and KKH-eSaCas9, SAV1 and SAV2 much better discriminated the single-base mismatches, including those located distal to the PAM region, between the endogenous target and the sgRNAs (Figure 5). These results corroborate the on- and off-target activities observed for these variants in our experiments using GFP disruption assays and confirmed the high fidelity of KKH-SaCas9-SAV1 and KKH-SaCas9-SAV2 efficiently generated more accurate genomic edits against sequences with a single-base mismatch.
Figure 4.

Evaluation of endogenous on-target and genome-wide off-target activities of KKH-SaCas9 variants. (A) Assessment of KKH-SaCas9 variants’ on-target editing with sgRNAs targeting endogenous loci. The percentage of sites with indels was measured using a T7 endonuclease I (T7E1) assay. The ratio of the on-target activity of KKH-SaCas9-SAV1, SAV2 and KKH-eSaCas9 to the activity of KKH-SaCas9 was determined, and the median and interquartile range for the normalized percentage of indel formation are shown for the 5-6 loci tested in three cell lines. Each locus was measured twice or three times; the full dataset is presented in Supplementary Figure S7. (B) GUIDE-seq genome-wide specificity profiles for the KKH-SaCas9 variants paired with the indicated sgRNAs in MHCC97L (for EMX1-sg7 and AAVS1-sg4) and OVCAR8-ADR (for VEGFA-sg8) cells. Mismatched positions in off-target sites are colored, and GUIDE-seq read counts were used as a measure of the cleavage efficiency at a given site.
Figure 5.

KKH-SaCas9- SAV1 and SAV2 showed improved single-base mismatch discrimination. Assessment of the specificity of the KKH-SaCas9 variants when there were single-base mismatches between the sgRNA and the target endogenous loci. The percentage of indels at the target loci was measured using deep sequencing assay.
DISCUSSION
In summary, through combinatorial mutagenesis, we successfully identified KKH-SaCas9-SAV1 and KKH-SaCas9-SAV2, which harbor new sets of mutations that confer KKH-SaCas9 with high editing accuracy and efficiency. Our work addresses the unmet need for highly specific and efficient variants of KHH-SaCas9 that can make edits across a broad range of genomic targets (i.e. with “NNNRRT” PAM), including sites harboring “NHHRRT” PAM that could not be targeted by other high-fidelity SpCas9 variants that recognize “NGG” PAM. We further reveal that SAV1 and SAV2 have an enhanced ability to distinguish targets with single-nucleotide differences including those located distantly from the PAM. Current strategies to target mutant allele using SaCas9 requires the pathogenic single-nucleotide polymorphism (SNP) or mutation to be located within the seed region of the sgRNA or using an SNP-derived PAM to achieve SNP-specific targeting without cleaving the wild-type allele. However, these do not apply to SNPs that are located outside of the seed region or those that do not generate a new PAM for SaCas9 targeting. The unique ability of SAV1 and SAV2 in distinguishing a broader range of single-nucleotide mismatches could expand the scope and capabilities of genome editing at loci with SNPs and mutations located further away from the PAM, which has not been previously achieved. When compared to wild-type KKH-SaCas9, we observed that some of the endogenous target loci showed a greater reduction in editing efficiency when SAV1 and SAV2 were used. Such variability of the relative editing efficiency among loci was also previously reported for other high-fidelity SpCas9 variants (5,26). This could be due to the sgRNA/target sequence dependencies for each variant, because each variant was engineered with mutations that interact with different regions of the DNA and/or sgRNA backbone(s). We anticipate that a comprehensive investigation using a large repertoire of sgRNAs paired with their endogenous target sequences will help delineate the relationship between sgRNA sequence features and activity for individual variants in future experiments.
Screening combinatorial mutations have been technically challenging due to the huge combinatorial space to search in, and only a limited number of mutants could be characterized in practice. For example, performing a saturated mutagenesis screen on 12 amino acid residues requires 2012 (i.e. 4 × 1015) variants to be screened, which is practically infeasible. In this work, we applied a structure-guided approach to rationally select mutations for our engineering and testing. Our results demonstrate the feasibility of engineering highly accurate KKH-SaCas9 enzyme via mutating multiple DNA- and sgRNA- interacting residues that span over the different parts of the protein. Of note, we observed that grafting SAV1 and SAV2 mutations onto the nuclease-dead version of KKH-SaCas9 showed comparable gene knockdown efficiency and specificity to their wild type and KKH-eSaCas9 counterparts (Supplementary Figure S9). This suggests that the enhanced editing specificity of SAV1 and SAV2 nucleases is more likely dictated at the DNA cleavage level, rather than the DNA binding level. The mutations, including Y239H, identified in this work are thus useful building blocks for further engineering of KKH-SaCas9 nucleases. With new methods being developed for combinatorial mutagenesis en masse (9) and protein engineering using machine learning (27,28), we anticipate a more systematic follow-up work to be carried out to exploit the large combinatorial mutational landscape for identifying other optimal sets of mutations that confer the enzyme with ultimate accuracy and higher efficiency for KKH-SaCas9, as well as wild-type SaCas9 and other engineered SaCas9 variants with altered PAM (12,13).
DATA AVAILABILITY
The sequencing data generated during this study are available. GUIDE-seq datasets are available from Sequence Read Archive (SRA) under accession SUB10793397.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Wong lab for helpful discussions. We thank the Centre for PanorOmic Sciences at the LKS Faculty of Medicine, The University of Hong Kong, for providing and maintaining the equipment needed for flow cytometry analysis and cell sorting.
Contributor Information
Chaya T L Yuen, Laboratory of Combinatorial Genetics and Synthetic Biology, School of Biomedical Sciences, The University of Hong Kong, Pokfulam, Hong Kong SAR, China.
Dawn G L Thean, Laboratory of Combinatorial Genetics and Synthetic Biology, School of Biomedical Sciences, The University of Hong Kong, Pokfulam, Hong Kong SAR, China.
Becky K C Chan, Laboratory of Combinatorial Genetics and Synthetic Biology, School of Biomedical Sciences, The University of Hong Kong, Pokfulam, Hong Kong SAR, China; Centre for Oncology and Immunology, Hong Kong Science Park, Hong Kong SAR, China.
Peng Zhou, Laboratory of Combinatorial Genetics and Synthetic Biology, School of Biomedical Sciences, The University of Hong Kong, Pokfulam, Hong Kong SAR, China.
Cynthia C S Kwok, Laboratory of Combinatorial Genetics and Synthetic Biology, School of Biomedical Sciences, The University of Hong Kong, Pokfulam, Hong Kong SAR, China.
Hoi Yee Chu, Laboratory of Combinatorial Genetics and Synthetic Biology, School of Biomedical Sciences, The University of Hong Kong, Pokfulam, Hong Kong SAR, China; Centre for Oncology and Immunology, Hong Kong Science Park, Hong Kong SAR, China.
Maggie S H Cheung, Laboratory of Combinatorial Genetics and Synthetic Biology, School of Biomedical Sciences, The University of Hong Kong, Pokfulam, Hong Kong SAR, China.
Bei Wang, Laboratory of Combinatorial Genetics and Synthetic Biology, School of Biomedical Sciences, The University of Hong Kong, Pokfulam, Hong Kong SAR, China.
Yee Man Chan, Ming Wai Lau Centre for Reparative Medicine, Karolinska Institutet, Hong Kong SAR, China.
Silvia Y L Mak, Ming Wai Lau Centre for Reparative Medicine, Karolinska Institutet, Hong Kong SAR, China.
Anskar Y Leung, Centre for Oncology and Immunology, Hong Kong Science Park, Hong Kong SAR, China; Division of Haematology, Department of Medicine, LKS Faculty of Medicine, The University of Hong Kong, Pokfulam, Hong Kong SAR, China; The Jockey Club Centre for Clinical Innovation and Discovery, LKS Faculty of Medicine, The University of Hong Kong, Pokfulam, Hong Kong SAR, China.
Gigi C G Choi, Laboratory of Combinatorial Genetics and Synthetic Biology, School of Biomedical Sciences, The University of Hong Kong, Pokfulam, Hong Kong SAR, China; Centre for Oncology and Immunology, Hong Kong Science Park, Hong Kong SAR, China.
Zongli Zheng, Ming Wai Lau Centre for Reparative Medicine, Karolinska Institutet, Hong Kong SAR, China; Department of Biomedical Sciences, City University of Hong Kong, Hong Kong SAR, China; Biotechnology and Health Centre, City University of Hong Kong Shenzhen Research Institute, Shenzhen, China.
Alan S L Wong, Laboratory of Combinatorial Genetics and Synthetic Biology, School of Biomedical Sciences, The University of Hong Kong, Pokfulam, Hong Kong SAR, China; Centre for Oncology and Immunology, Hong Kong Science Park, Hong Kong SAR, China; Department of Electrical and Electronic Engineering, The University of Hong Kong, Pokfulam, Hong Kong SAR, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Centre for Oncology and Immunology under the Health@InnoHK Program launched by the Innovation and Technology Commission, the Government of Hong Kong Special Administrative Region of the People's Republic of China; Hong Kong Research Grants Council [TRS-T12-702/20-N]; National Natural Science Foundation of China Excellent Young Scientists Fund [32022089]; A.S.L.W. is a Ming Wai Lau Centre for Reparative Medicine (MWLC) Associate Member; Ming Wai Lau Centre for Reparative Medicine Associate Member Programme (in part). Funding for open access charge: Centre for Oncology and Immunology under the Health@InnoHK Program launched by the Innovation and Technology Commission, the Government of Hong Kong Special Administrative Region of the People's Republic of China; Hong Kong Research Grants Council [TRS-T12-702/20-N]; National Natural Science Foundation of China Excellent Young Scientists Fund [32022089]; A.S.L.W. is a Ming Wai Lau Centre for Reparative Medicine (MWLC) Associate Member; Ming Wai Lau Centre for Reparative Medicine Associate Member Programme (in part).
Conflict of interest statement. The authors have filed a patent application based on this work.
REFERENCES
- 1. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A.et al.. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M.. RNA-guided human genome engineering via Cas9. Science. 2013; 339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F.. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016; 351:84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K.. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016; 529:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen J.S., Dagdas Y.S., Kleinstiver B.P., Welch M.M., Sousa A.A., Harrington L.B., Sternberg S.H., Joung J.K., Yildiz A., Doudna J.A.. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017; 550:407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Casini A., Olivieri M., Petris G., Montagna C., Reginato G., Maule G., Lorenzin F., Prandi D., Romanel A., Demichelis F.et al.. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018; 36:265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee J.K., Jeong E., Lee J., Jung M., Shin E., Kim Y.H., Lee K., Jung I., Kim D., Kim S.et al.. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat Commun. 2018; 9:3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J.et al.. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018; 24:1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choi G.C.G., Zhou P., Yuen C.T.L., Chan B.K.C., Xu F., Bao S., Chu H.Y., Thean D., Tan K., Wong K.H.et al.. Combinatorial mutagenesis en masse optimizes the genome editing activities of SpCas9. Nat. Methods. 2019; 16:722–730. [DOI] [PubMed] [Google Scholar]
- 10. Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S.et al.. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015; 520:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kleinstiver B.P., Prew M.S., Tsai S.Q., Nguyen N.T., Topkar V.V., Zheng Z., Joung J.K.. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 2015; 33:1293–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma D., Xu Z., Zhang Z., Chen X., Zeng X., Zhang Y., Deng T., Ren M., Sun Z., Jiang R.et al.. Engineer chimeric Cas9 to expand PAM recognition based on evolutionary information. Nat. Commun. 2019; 10:560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luan B., Xu G., Feng M., Cong L., Zhou R.. Combined computational-experimental approach to explore the molecular mechanism of SaCas9 with a broadened DNA targeting range. J Am Chem Soc. 2019; 141:6545–6552. [DOI] [PubMed] [Google Scholar]
- 14. Kim E., Koo T., Park S.W., Kim D., Kim K., Cho H.Y., Song D.W., Lee K.J., Jung M.H., Kim S.et al.. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun. 2017; 8:14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Edraki A., Mir A., Ibraheim R., Gainetdinov I., Yoon Y., Song C.Q., Cao Y., Gallant J., Xue W., Rivera-Perez J.A.et al.. A compact, high-accuracy Cas9 with a dinucleotide PAM for in vivo genome editing. Mol Cell. 2019; 73:714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A.et al.. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015; 163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pausch P., Al-Shayeb B., Bisom-Rapp E., Tsuchida C.A., Li Z., Cress B.F., Knott G.J., Jacobsen S.E., Banfield J.F., Doudna J.A.. CRISPR-CasPhi from huge phages is a hypercompact genome editor. Science. 2020; 369:333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tan Y., Chu A.H.Y., Bao S., Hoang D.A., Kebede F.T., Xiong W., Ji M., Shi J., Zheng Z.. Rationally engineered Staphylococcus aureus Cas9 nucleases with high genome-wide specificity. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:20969–20976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guschin D.Y., Waite A.J., Katibah G.E., Miller J.C., Holmes M.C., Rebar E.J.. A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 2010; 649:247–256. [DOI] [PubMed] [Google Scholar]
- 20. Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P.et al.. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015; 33:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsai S.Q., Topkar V.V., Joung J.K., Aryee M.J.. Open-source guideseq software for analysis of GUIDE-seq data. Nat. Biotechnol. 2016; 34:483. [DOI] [PubMed] [Google Scholar]
- 22. Wong A.S., Choi G.C., Cui C.H., Pregernig G., Milani P., Adam M., Perli S.D., Kazer S.W., Gaillard A., Hermann M.et al.. Multiplexed barcoded CRISPR-Cas9 screening enabled by CombiGEM. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:2544–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E.et al.. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019; 37:224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rodrigues C.H., Pires D.E., Ascher D.B.. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018; 46:W350–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xie H., Ge X., Yang F., Wang B., Li S., Duan J., Lv X., Cheng C., Song Z., Liu C.et al.. High-fidelity SaCas9 identified by directional screening in human cells. PLoS Biol. 2020; 18:e3000747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kulcsar P.I., Talas A., Toth E., Nyeste A., Ligeti Z., Welker Z., Welker E.. Blackjack mutations improve the on-target activities of increased fidelity variants of SpCas9 with 5'G-extended sgRNAs. Nat. Commun. 2020; 11:1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Biswas S., Khimulya G., Alley E.C., Esvelt K.M., Church G.M.. Low-N protein engineering with data-efficient deep learning. Nat. Methods. 2021; 18:389–396. [DOI] [PubMed] [Google Scholar]
- 28. Wu Z., Kan S.B.J., Lewis R.D., Wittmann B.J., Arnold F.H.. Machine learning-assisted directed protein evolution with combinatorial libraries. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:8852–8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data generated during this study are available. GUIDE-seq datasets are available from Sequence Read Archive (SRA) under accession SUB10793397.