


Abstract
Type III CRISPR-Cas systems have a unique mode of interference, involving crRNA-guided recognition of nascent RNA and leading to DNA and RNA degradation. How type III systems acquire new CRISPR spacers is currently not well understood. Here, we characterize CRISPR spacer uptake by a type III-A system within its native host, Streptococcus thermophilus. Adaptation by the type II-A system in the same host provided a basis for comparison. Cas1 and Cas2 proteins were critical for type III adaptation but deletion of genes responsible for crRNA biogenesis or interference did not detectably change spacer uptake patterns, except those related to host counter-selection. Unlike the type II-A system, type III spacers are acquired in a PAM- and orientation-independent manner. Interestingly, certain regions of plasmids and the host genome were particularly well-sampled during type III-A, but not type II-A, spacer uptake. These regions included the single-stranded origins of rolling-circle replicating plasmids, rRNA and tRNA encoding gene clusters, promoter regions of expressed genes and 5′ UTR regions involved in transcription attenuation. These features share the potential to form DNA secondary structures, suggesting a preferred substrate for type III adaptation. Lastly, the type III-A system adapted to and protected host cells from lytic phage infection.
Graphical Abstract
Graphical Abstract.
The type III-A CRISPR-Cas system in Streptococcus thermophilus is adaptation active, and preferentially captures new spacers from DNA that forms transient hairpins. These new spacers are integrated in either orientation.
INTRODUCTION
CRISPR-Cas systems are diverse, adaptive and heritable immune systems that protect many bacteria and archaea against potentially harmful viruses, plasmids and other invasive mobile genetic elements (1–5). CRISPR-Cas immune memories are short fragments of DNA that match sequences within a target mobile genetic element. These DNA fragments (termed spacers) are stored in the repeat array that gave CRISPR its name: Clustered Regularly Interspaced Short Palindromic Repeats. New spacers are acquired through adaptation, a process during which a short fragment of foreign DNA (the protospacer) is captured and integrated into the CRISPR locus (6–8). The CRISPR array is transcribed and the resulting RNA is processed to generate a pool of individual CRISPR RNAs (crRNAs) that each have the potential to direct Cas effector nucleases to destroy complementary nucleic acids (2,9–12). Efficient destruction of the target nucleic acids (termed interference) often requires a short activating sequence immediately adjacent to the protospacer (PAM or protospacer adjacent motif) (13). Successful interference can protect a cell from phage infection or block plasmid uptake.
Of the six broad types of CRISPR-Cas systems identified (I-VI), types I, II and V systems recognize and destroy invasive DNAs, while type VI systems target RNA (5,10). Type III systems are unique in that they recognize and destroy target RNA and are also capable of degrading the associated transcriptionally active DNA (Supplementary Figure S1) (3,4,14–22).
Progress has been made in understanding the molecular mechanisms governing the ability of CRISPR-Cas systems to recognize, excise, process and accurately integrate spacers into CRISPR arrays. Much of the work to date has focused on how type I and II (DNA targeting) systems operate. Available evidence indicates a key and universal role for Cas1-Cas2 integrase complexes in capturing spacer DNA and catalyzing their integration at CRISPR loci (23–28). Spacer integration almost always occurs at one end of the array, at the repeat adjacent to an element called the leader. The leader contains the promoter necessary for crRNA expression (29,30), and in type I and type II systems, it was also shown to harbor sequences that are critical for directing spacer integration to the leader-proximal repeats (23,24,31–34). These key leader sequences have been shown to function either by providing recognition sites for direct binding of Cas1-Cas2 complexes (23,31,34) or for binding of non-Cas factors (such as integration host factor [IHF] for type I-E and I-F systems) that are needed to recruit Cas1-Cas2 complexes to the leader-proximal CRISPR repeat (35,36). The polarized addition of new spacers at the leader end of the CRISPR array can generate a chronological record of past invasions (37).
Biochemical and structural studies have revealed that spacer DNA integration proceeds via a two-step transesterification reaction whereby the 3′ hydroxyl groups of the incoming spacer DNA each attack the terminal nucleotides of the CRISPR repeat element, followed by host cell repair of the gapped DNA intermediates (38,39). While all investigated systems were found to rely on Cas1 and Cas2, the various steps of spacer generation and integration have been shown to require additional Cas (e.g. Cas4, Cas9, Csn2) (26,27,40) and non-Cas factors (e.g. IHF, RecBCD, DNA polymerase) (35,36,41,42) in different systems and in different organisms. Indeed, in some systems but not others, efficient spacer acquisition by the adaptation machinery (including Cas1, Cas2 and any additional adaptation factors) depends upon the presence of the effector crRNP (crRNA-guided Cas protein complex) interference machinery (26,27,43,44).
We are beginning to gain insights into mechanisms directing adaptation by type III systems. Type III systems are among the most abundant and widespread CRISPR systems (second only to type I systems) and occur in both bacteria and archaea (5). Type III systems have been classified into six distinct subtypes (type III-A to III-F) based on the properties of the interference proteins including Cas10, the signature protein of type III systems, and other features (5). Interestingly, one distinguishing feature among different type III systems is the presence or absence of associated cas1 and cas2 genes. Most type III-A systems include cas1, cas2, and the crRNA processing gene cas6 within the locus. In contrast, a majority of other type III systems (III-B, C, D, E and F) lack these genes and so are predicted to be unable to independently adapt (5,45). Instead, it appears that these type III subtypes depend on spacers from other systems since they co-occur with type I systems and use their processed crRNAs (46,47). This finding supports a model wherein most type III systems serve as ‘back-up’ interference modules that enhance the capabilities of existing CRISPR loci by adding the ability to target invaders at the RNA level and to resist phage escape (46,47). A notable exception to this trend is the rare reverse-transcriptase (RT)-Cas1 fusion proteins that enable some type III systems to adapt independently of other systems (48–50). While all other reported systems can only capture DNA as CRISPR spacers, the type III systems with RT activity can use both RNA and DNA as the substrate, and adaptation against RNA is dependent on the RT (48–50). Work to understand how adaptation of type III systems lacking RT domains has just begun. A type III-A system from Thermus thermophilus was recently shown to be active against a phage, with new spacers primarily arising from a region of the phage predicted to harbor genes that are expressed early in infection (51). Further work is necessary to better understand how type III-A systems recognize and integrate spacer sequences into their affiliated CRISPR arrays.
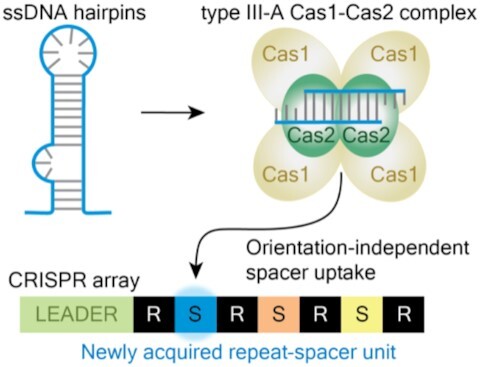
Since interference in type III systems differs from that of type I and II systems in several key ways, adaptation could likewise be expected to have unique attributes. For example, DNA targeting systems require a short (2–5 bp) PAM (protospacer adjacent motif) for efficient interference, and the PAM must be located on the correct side of the protospacer with respect to crRNA binding. PAMs play a key role in self- vs. non-self-recognition since the target protospacer, but not the spacer within the CRISPR array, contains a PAM (13,52). During adaption, protospacers with an adjacent PAM are strongly preferred and new spacers are integrated into the array in the functional orientation (Supplementary Figure S1) (7,45,53). In contrast, type III systems do not require a PAM for interference (54), and it is unclear whether any sequence motifs guide adaptation. Regarding spacer orientation, since type III crRNAs bind transcribed RNA (3,17), newly acquired spacers must be integrated in one of two possible orientations if they are to give rise to functional crRNAs that will be complementary to the transcribed RNA (Supplementary Figure S1). Analyses of native type III-A CRISPR arrays reveal a major bias for spacers inserted in the functional orientation relative to their viral or plasmid RNA targets (55). However, it is not yet known whether type III-A systems utilize a novel (PAM-independent) mechanism to ensure capture of spacers in a particular orientation or whether, as early work suggests (51), spacers are initially acquired in both orientations and downstream processes ultimately preserve spacers that impart a selective advantage to the organism and purge those that either do not confer immunity or induce autoimmunity and host cell death. It is also unclear if the Cas1 and Cas2 proteins are sufficient for executing adaptation in type III systems or if (as with other type I and II systems (26,27,43,44)) the interference components including crRNA, Csm1-6 proteins are needed as well. Given that type III systems ultimately recognize target RNAs from expressed genes, an important question is whether mechanisms governing spacer choice have evolved to enhance the probability of capturing spacers from transcribed DNA and whether there is a bias of these spacers being derived from mobile genetic elements versus the host genome.
In this study, we demonstrate that the native type III-A system of Streptococcus thermophilus JIM 8232 strain is active for adaptation against plasmid and phage invaders. We demonstrate that the type III-A adaptation machinery, composed of Cas1 and Cas2, is required to integrate a relatively broad size range of new spacers into CRISPR loci in a PAM- and orientation-independent fashion. Interestingly, our findings reveal that the type III adaption machinery preferentially acquires spacers from DNA predicted to form hairpins and other DNA secondary structures. We discuss how these observed unique type III spacer selection properties may have evolved to enrich for CRISPR capture of DNA fragments from the expressed genes of mobile genetic elements, capable of being recognized by the acquired crRNAs of RNA-targeting type III-A systems.
MATERIALS AND METHODS
Strain and plasmid manipulation
The JIM8232 strain of Streptococcus thermophilus (Sth) (WT) was kindly provided by Pierre Renault (AgroParisTech, France). Sth DGCC7710 and phage 2972 were kindly provided by Sylvain Moineau (Laval University, Canada). Sth strains were inoculated in M17 medium supplemented with 0.5% lactose (LM17) (Oxoid or HiMedia), and the cultures were incubated at 37°C overnight, or at 42°C during the day. Escherichia coli Top10 was used for plasmid construction and maintenance. E. coli Stellar (dcm-/dam-) was used to generate unmethylated plasmids for the target interference assay. pWAR, pTRK882, pNT1 and pNZ123, pIB184, pG + Off, and pRSNPed plasmids were kindly provided by Michael Federle (University of Illinois, USA), Todd Klaenhammer (North Carolina State University, USA), Sylvain Moineau (Laval University, Canada), Indranil Biswas (University of Kansas Medical Center, USA), Marie-Frédérique Lartigue (University of Tours, France) and John Renye (United States Department of Agriculture, Agricultural Research Service, Temple University, UDSA). M13mp18 single-stranded DNA (ssM13) and M13mp18 RF DNA (dsM13) were purchased from New England BioLabs. Chloramphenicol was supplemented at 2 μg/ml in LM17 liquid broth and at 5 μg/ml in LM17 plates (with 1% agar) for Sth; erythromycin and kanamycin were supplemented at 15 and 150 μg/ml for Sth, respectively.
The construction of Sth mutant and cas deletion strains was achieved by an established natural transformation procedure (56). The primers used for PCR amplification of the recombination templates are listed in Supplementary Table S1. To construct the type II introduced strain, a fragment containing the type II leader and four repeat-spacer units of the array from the DGCC7710 strain was transplanted into JIM8232 at the PTS locus (26).
Target interference assay
Sth strains were inoculated in 5 ml of LM17, and the cultures were incubated at 37°C overnight. About 100 ml of fresh LM17 was inoculated with 1 mL of an overnight culture, and incubated at 42°C until an OD600 value of 0.5 was reached. The culture was then placed on ice for 15 min and then centrifuged for 10 min at 5000 RCF at 4°C. The supernatant was decanted and the pellet was resuspended in 2 ml ice-cold wash solution (10% glycerol + 0.4 M sorbitol). The resuspended cells were washed three times by centrifugation for 1 min at 15 000 RPM at 4°C, followed by resuspension in the wash solution. After the final wash, the electroporation-competent cells were resuspended in 500 μl wash solution and aliquoted. About 1 μg of unmethylated target or control plasmid DNA (generated in the E. coli Stellar strain) was mixed with 40 μl of electrocompetent cells, and electroporation was achieved using a Gene Pulser (BioRad) at 25 μF, 200 Ω and 1.8 KV. The transformants were incubated in 1 ml of recuperation solution (LM17 + 0.4 M sorbitol + 20 mM MgCl2 + 2 mM CaCl2) for 2 h at 42°C and then plated on to an LM17 plate (1% agar) with the appropriate antibiotic.
Adaptation assay
The plasmids were introduced into the Sth cells by an established natural transformation procedure (56). Twenty to thirty colonies of the transformed Sth strains were inoculated into 10 ml of LM17 with appropriate antibiotics, and the cultures were incubated at 37°C overnight. Total DNA of the overnight cultures was extracted by Quick-DNA Fungal/Bacterial Miniprep kit (Zymo Research).
To monitor adaptation, the leader-proximal end of the CRISPR array was amplified by CAPTURE PCR (57) or multiple-round PCR from agarose gel extracted DNAs (Figure 1B). PCR primers are listed in Supplementary Table S1. For type III array amplicon and the introduced type II array amplicon, the forward primer targeted the leader and the reverse primer targeted the first leader-adjacent existing spacer. For the native type II array, we also amplified from the leader to the second leader-adjacent existing spacer. Expanded amplicons from first round PCRs were separated from unexpanded products by agarose gel electrophoresis, bands of the correct size were cut, and DNAs were isolated by a gel recovery kit (Zymo Research). When expanded amplicons were too faint to visualize, the region of a gel lane corresponding to amplicons in the expanded size range was cut. Illumina high-throughput sequencing (HTS) overhangs and Illumina HTS index barcodes were added to the expanded array amplicons by PCR. Purified PCR products were ranked by PCR intensity and then pooled, concentrated by ethanol precipitation, quantified and diluted to a suitable concentration for Illumina platform sequencing.
Figure 1.

Adaptation by type III-A and type II-A CRISPR-Cas systems. (A) The schematic of CRISPR-Cas systems of Sth JIM8232. The cas genes are represented by colored arrows, ‘L’ indicates the leader sequences in green, and the repeats of the CRISPR array are shown in black. (B) Schematic showing steps in the adaptation assay. After isolation of genomic DNA, PCR is used to amplify the leader-adjacent end of the CRISPR array. Expanded PCR products (Ex) contain a new leader-adjacent repeat-spacer unit and so are approximately 70 bp longer; unexpanded PCR products (Un) reflect the original, WT CRISPR array. (C) Length distribution of the new spacers acquired into the type II-A system (cyan) and the type III-A system (peach). (D) Analysis of protospacer adjacent sequences (PFS). Sequences upstream and downstream from aligned new spacers were extracted and used to create weblogos to visualize sequence motifs. (E) Proportion of new spacers that aligned to the plasmid (blue) and genome (green). Pooled data of at least three independent experiments are presented in (C–E).
Array libraries were sequenced on an Illumina MiSeq, set to yield 250 by 50 paired end reads; the 250 base read 1 sequences were used in this study. After sequencing, samples were de-multiplexed by index, and the sequence corresponding to a new (expanded) spacer was extracted from each read. New spacers were aligned to reference sequences (bacterial chromosome and appropriate plasmids) using Bowtie (58) to identify the protospacer sequence. Protospacer sequences were then characterized with respect to length, PAM, and position on the genome or plasmid. To detect PAMs, unique protospacer adjacent upstream and downstream sequences were extracted using bedtools (59) and a consensus sequence logo was made using weblogo (60).
RNA-seq assay
To evaluate RNA expression patterns, RNA sequencing was done on cultures grown to either exponential or stationary phase. Briefly, cultures were pelleted and decanted, then frozen at -80°C. Pellets were thawed and resuspended directly in lysis buffer, and RNA was isolated using the PowerBiofilm RNA Isolation kit (Qiagen). Stranded, total RNA libraries (without rRNA or tRNA depletion) were prepared using the Illumina TruSeq kit and were sequenced on an Illumina NextSeq instrument, generating paired 2 by 150 bp reads. Reads were demultiplexed by index, adapter trimmed and aligned to the appropriate reference sequences (bacterial chromosome, plasmid) by bowtie2 (61).
RNA-seq read and protospacer density analysis
For both the adaptation and RNAseq assays, alignment outputs were processed (59,62) and custom genome browser tracks were generated using tools available from the University of California Santa Cruz Genome Browser (https://genome.ucsc.edu). Tracks for both ‘total’ and ‘unique’ protospacer density were created; for the total tracks, all reads supporting a particular protospacer are counted. For unique tracks, all reads corresponding to a given protospacer that is unique with respect to alignment coordinates and strand were collapsed to a single count. We noted that the distribution of protospacers across the genome was not even, so regions with significant protospacer enrichment were identified using the findPeaks software in the HOMER analysis package (63). For spacers that aligned within the boundaries of annotated protein-coding genes, we determined the percent that matched the coding versus template strand of those annotated genes using a custom python script.
To determine whether spacer uptake was influenced by RNA expression, we divided the genome into 3860 windows, each 500 bp long, and then determined the cumulative RNAseq read coverage for those windows by combining data from exponential and stationary phase cultures, three replicates each. From the cumulative RNAseq coverage, we classified each 500 bp window as either ‘unexpressed’ with zero RNAseq reads, ‘expressed’ with approximately 16 or more aligned RNA fragments (forward and reverse paired reads with intervening space) or ‘marginally expressed’ which had between 0 and 16 aligned RNA fragments. Marginally expressed windows were not considered further for this analysis. We then examined the spacer density (total and strand-specific) for these genome windows by determining the cumulative protospacer coverage for both WT and Δcsm1-6 strains (protospacers from 24 and 18 replicates, respectively, were pooled for this analysis).
Phage infection and BIM isolation and analyses
The Sth strains were inoculated in 5 mL LM17 and the cultures were incubated at 37°C overnight. Five milliliters of fresh LM17 with 10 mM CaCl2 was inoculated with 50 μl of an overnight culture and incubated at 42°C until OD600 value reached 0.3. Phage infection was performed with phage 2972 at multiplicity of infection (M.O.I.) of 0.1, 1 and 10, separately. After phage addition, cells were incubated at 42°C for 60, 90 min or overnight. At the end of each of the three time points, 100 μl of each culture was plated on to an LM17 plate supplemented with 10 mM CaCl2. The plates were incubated at 37°C overnight, and then individual colonies were randomly picked and evaluated for CRISPR array expansion by colony PCR, using the primers listed in Supplementary Table S1. The remaining volume of each of the cultures was subjected to total DNA extraction, CAPTURE PCR and HTS to evaluate adaptation as described above.
RESULTS
The type III-A system of Streptococcus thermophilus actively acquires new spacers at the CRISPR locus
Sth JIM8232 has an intact type III-A CRISPR-Cas system in its genome, containing 17 pre-existing spacers, as well as a type II-A system containing 41 spacers (Figure 1A) (64). An additional type II CRISPR array is present but is devoid of adjacent cas genes and likely to be inactive. Interestingly, the sequences of spacers 3–8 of the type III-A system are identical to those of spacers 9–14, suggesting occurrence of a spacer-repeat duplication event within the CRISPR array.
To test the potential for adaptation by the type III-A CRISPR-Cas system, we transformed cells with pWAR plasmid (26) as heterologous DNA, and then monitored the leader proximal end of the CRISPR array by PCR to look for evidence of array expansion (new spacer uptake). PCR was followed by high-throughput sequencing (HTS) to characterize new spacers (Figure 1B). As a point of comparison, we also examined adaptation by the native type II-A system in the same host, in parallel. We captured about 300 new type II-A spacers and found that they had a downstream NNAGAAW PAM and averaged 30 bp in length (Supplementary Figure S2B, C ‘Native’). Although it was active, spacer uptake for the type II system of Sth JIM8232 strain appeared relatively inefficient and we found that almost all new spacers were integrated between the leader-adjacent spacer and the next downstream spacer rather than between the leader and leader-adjacent spacer. We believe that the limited spacer uptake and the atypical location for integration were due to sequence changes within the leader, as compared to the homologous type II-A system of another well-studied Sth strain, DGCC7710, which has been shown to be active in adaptation (1,26,33) (Supplementary Figure S2A). The rarity of spacer uptake events in the native type II-A array (about 1 read per 1000) limited the resolution of our adaptation characterizations, so we used the DGCC7710 array to improve efficiency. We inserted part of the DGCC7710 type II array (the leader sequence and the following 4 repeat-spacer units) into the JIM8232 genome (65). The inserted type II DGCC7710 array had the same features of adaptation as the native type II JIM8232 array (Supplementary Figure S2B and C) and mediated defense against the tested target plasmid (Supplementary Figure S2D) but was easier to monitor and so data from this strain were used for all subsequent type II-A characterizations. Type III-A characterizations were done in the wild-type JIM8232 strain (labeled ‘WT’, does not have the introduced type II-A array).
Pre-existing spacers of the type III-A CRISPR locus ranged from 35 to 39 bp, with 36 bp being the most common length. Among newly acquired spacers, about 99% of new unique spacers fell within the range of 32 to 42 bp, with 36 bp at the peak of the distribution (Figure 1C). For comparison, >80% of the new spacers acquired by the type II-A system were 30 bp, with most others being 29 or 31 bp (Figure 1C).
For well-studied type I and type II systems, protospacer selection from foreign DNAs is guided by system-specific PAMs, and the resulting pre-spacers are inserted at the leader proximal end of the CRISPR array in a PAM-directed orientation (13,45,52,53). In contrast, adaptation by the tested type III systems appear to be PAM-independent (48–51,54) (Supplementary Figure S1). We examined the upstream and downstream protospacer flanking sequences (PFSs) of the newly acquired type III-A spacers and observed no consensus sequence motifs (Figure 1D), which was expected since target interference by many type III systems tolerate a broad range of PFSs (18,54). Next, we sought to rule out the possibility of survivor bias: since the type III CRISPR-Cas system could carry out interference against newly acquired spacers, immunity itself might eliminate a subset of new spacers and thereby diminish a potential PAM signal. We examined PFSs in a Δcsm1-6 interference-null strain and as in the wild-type strain (Figure 1D) there was no sequence motif or enrichment of any base within 8 bases upstream or downstream of the protospacer (51,655 spacers from 18 replicates). In contrast, the type II-A system in the same host, selected protospacers with a downstream NNAGAAW PAM (Figure 1D and Supplementary Figure S2B). For both systems, a majority of new spacers were derived from the host genome, which is consistent with its greater size (about 464-fold larger than the plasmid) (Figure 1E).
Cas1 and Cas2 are the only two Cas proteins required for adaptation by the type III system
To understand the involvement of Cas proteins in type III-A adaptation, we examined new spacer acquisition in strains with some or all of the crRNA biogenesis (cas6) or interference (csm1-6) genes deleted. Expanded PCR amplicons corresponding to new spacers were apparent after four rounds of PCR for the wild-type (WT) strain, the Δcas6 strain, the Δcsm1-5 strain, and the Δcsm1-6 strain, but not for the Δcas1 or Δcas2 strains (Figure 2A). This indicated that Cas1 and Cas2 were essential for adaptation by the type III-A system and sufficient among the Cas proteins. Deletion of either cas1 or cas2 genes did not affect the ability of the strain to carry out plasmid interference but loss of the csm1-6 genes abolished this activity as expected (Figure 2B). While not essential for adaptation, Csm1-6 interference proteins could potentially influence new spacer characteristics, so the Δcsm1-6 strain was studied alongside the wild-type strain to detect any such differences.
Figure 2.

Cas requirements for adaptation and repeat-spacer duplications. (A) Gel images show bands resulting from four rounds of PCR targeting the leader-adjacent end of the CRISPR array; expanded (Ex) and unexpanded (Un) products are indicated. At least three independent experiments were done; images shown here are representative. Gene deletions were designed to disrupt adaptation (Δcas1 and Δcas2 strains), crRNA maturation (Δcas6) or interference (Δcsm1-5 and Δcsm1-6 strains). (B) Transformation efficiencies for a non-target plasmid (pNo-target), a transcribed target plasmid (pTarget), and a non-transcribed target plasmid (pTarget-NT) are shown for four strains (n = 3). (C) Genome-wide view of aligned new type III spacers. The y-axis indicates the cumulative density of aligned spacers counting either all supporting reads (total) or only unique reads, with respect to alignment coordinates (unique). Spacers aligned to the top strand are colored blue; bottom strand are red. The positions of the native type II and type III arrays are indicated in orange; rRNA and tRNA gene positions are show in green. Black bars above the unique spacer track show regions where significant spacer enrichment was detected. (D) Schematic of PCR products corresponding to an unexpanded array, a novel spacer, or duplication of existing spacers in the array. (E and F) Genome browser tracks show the cumulative density of aligned new spacers across the introduced type II-A array (E) and the native type III-A array in WT and Δcas1 strains (F); new spacers that align to existing spacers correspond to spacer duplications. Spacer duplications were observed for all strains examined; representative genome browser tracks for the Δcas1 and WT strains are shown here (F). At least three independent experiments were conducted for each strain and pooled data were used to determine the percent of all expanded reads that corresponded to spacer duplications (“Dup”, rather than novel spacers); that percent is indicated with along with the total number of aligned spacers and replicates.
Adaptation-independent duplication of repeat-spacer units occurs in both type III and type II systems
We used genome browser tracks to visualize the density of aligned new type III-A spacers across the host chromosome and the introduced plasmid. The distribution of aligned spacers was not even (Figure 2C), suggesting that certain regions of the chromosome and plasmid are more highly sampled during adaptation. A strikingly large cluster of aligned spacers was noted over the type III-A array (Figure 2C, close up in Figure 2F). Since these aligned new spacers were identical to existing spacers from the array and the corresponding protospacers do not exist anywhere else in the chromosome or plasmid, we believe they represent spacer duplications, as illustrated in Figure 2D. Although the first leader-adjacent spacer was the most extensively duplicated, all of the pre-existing spacers in the array were detected at the leader proximal end of the CRISPR array (Figure 2F). The duplications always maintained the same spacer orientation (i.e. pre-existing spacers were not integrated into the leader-adjacent position in the reverse orientation) and, interestingly, the duplications were also observed in the Δcas1 and Δcas2 strains, even though these strains were apparently not active for adaptation (see Figure 2E for Δcas1 duplications; Δcas2 results were similar with 99.96% of aligned new spacers corresponding to duplications, n = 5103 reads, 1 replicate). This suggested that the spacer-repeat duplications were caused by a mechanism that is independent of adaptation, like homologous recombination or template slippage events during DNA replication (66). Repeat-spacer duplications were also detected in the type II CRISPR-Cas locus (Figure 2E), indicating that the adaptation-independent duplication of the repeat-spacer units happens in both Class 1 (type III) and Class 2 (type II) CRISPR-Cas systems.
Type III spacers are integrated in both possible orientations
Interference by type III CRISPR-Cas systems requires target transcription, as the DNase and RNase activity of the crRNP (carried out by Csm1 and Csm3, respectively) and the RNase activity of Csm6 (Csx1 in other systems) are activated by base pairing between the guiding crRNA and the newly transcribed RNA (14,18,21,22,67–73). As a consequence, spacers that correspond to untranscribed regions or spacers that correspond to the coding strand (rather than the template strand) would not be competent for interference since they cannot base pair with transcribed RNA products. Double-stranded DNA pre-spacers could potentially be integrated into the array in two orientations, with only one leading to a functional crRNA (Figure 3A). We examined pre-existing spacers in type III-A arrays from all available sequenced Sth strains, and for those spacers that could be confidently aligned to a phage genome, we determined whether the spacer corresponded to the template or coding strand of the targeted DNA. Nearly all pre-existing spacers corresponded to the template strand and so would generate crRNAs capable of binding nascent target RNA (Figure 3B, first column). The same was true for the Sth strain used in this study, JIM8232 (Figure 3B, second column).
Figure 3.

New type III-A spacers aligned to coding and template strands of expressed genes. (A) Diagram illustrating the outcomes for type III target interference given two possible orientations for spacer integration. When a type III-A spacer corresponds to (aligns to) the coding strand of a gene, the expressed type III-A crRNA will not be able to perform defense (left). In contrast, when a spacer corresponds to the template strand, the expressed type III crRNA can bind nascent RNA and initiate defense (right). (B) Proportions of type III-A spacers derived from the coding strand (blue) and template strand (pink) are shown for existing array spacers and newly acquired, leader-adjacent spacers identified in this work. Existing spacers in type III-A arrays from 63 sequenced S. thermophilus genomes were aligned to a database of phage genomes and for those spacers that aligned, strand was noted (identical spacers were counted once). Sth JIM8232 (WT) existing spacers were aligned similarly. For newly acquired spacers, pooled data of at least three independent experiments are presented. (C–F) Genome browser tracks show cumulative spacer density and RNAseq read density across selected genome and plasmid regions for WT and Δcsm1-6 strains. Spacers aligning to the top strand are colored blue; spacers aligning to the bottom strand are pink. For RNAseq data, RNA reads aligning to the top strand are dark purple and reads aligning to the bottom strand are dark yellow. The y-axis indicates the depth of aligned sequences (spacers or RNAseq reads). For spacers, data from at least three independent experiments was combined on a single track. For RNAseq, aligned data from a single representative replicate is shown.
Among the newly acquired spacers detected in our adaptation assay, a different trend emerged. Approximately 85% of new spacers aligned to the coding strand of the plasmid or host chromosome, implying that a majority of newly captured spacers are non-functional (Figure 3B, third column). However, in the interference-null (Δcsm1-6) strain, there was no bias, with roughly half of the spacers aligning to the coding strand and half to the template strand (53% and 47%, respectively) (Figure 3B, fourth column). This strand bias was most obvious in regions where transcribed ORFs (verified by RNAseq data) with opposing orientations are found side by side; spacers on the coding strand are clearly favored in the WT but not Δcsm1-6 strain (Figure 3C–E). We interpret these findings as evidence of counter-selection. In the WT strain, functional spacers against the genome could result in cell death, effectively removing those cells from liquid culture and eliminating their spacers from the assay. In the Δcsm1-6 strain, the absence of interference meant no counter-selection could occur, and so cells with functional spacers could remain and be detected. In addition, plasmids were selected via antibiotic resistance and we likewise observed a strand bias in the WT but not Δcsm1-6 strain (Figure 3F). Since interference in the type III system requires nascent RNA, we expect that spacers in unexpressed regions of the genome would not be subject to counter-selection. While most of the genome is expressed at some level, we did not find evidence that intergenic spacers show strand bias (Supplementary Figure S3).
Selective targeting of partially palindromic DNA sequences by the type III adaptation machinery
While examining spacer distributions across the pWAR plasmid, we noted that a narrow region around the single-stranded origin (sso) was particularly well-sampled by the type III adaptation machinery (Figure 3F). This plasmid replicates by rolling-circle replication (RCR, Figure 4A) (74,75). Its encoded Rep protein recognizes and generates a nick at the double-stranded origin (dso) of the plasmid. The 5′ end of the plus strand is displaced and unidirectional replication proceeds continuously after initiation at the cognate 3′-OH end of the nicked parental plus strand. The plus strand is re-ligated after the replication. A hallmark feature of plasmids that replicate by the RCR mechanism is the presence of a plus strand, ssDNA intermediate. An extensive hairpin structure forms at the single-stranded origin (sso), which is partially palindromic (Figure 4A), and triggers replication of the minus strand by recruiting host RNA polymerase (or primase in some systems) to synthesize a short RNA primer (Figure 4A) (76,77). The loop distal region of the stem is termed the recombination site (RSB) and is required for RNA polymerase (or primase) binding, together with promoters within the ssos (78–80). There is usually a conserved 6 nucleotide sequence (CS-6) within the loop of a sso, which terminates RNA primer synthesis and is important for the minus DNA strand replication (80–82). New spacers aligned around the pWAR sso in all strains and replicates tested; in particular, we observed extraordinary targeting of one end of the sso in the Δcsm1-6 strain (Figure 4B). Since the sso forms a hairpin during its ssDNA stage (76,77,80), we reasoned that this imperfect dsDNA structure may represent a good substrate for type III adaptation.
Figure 4.

Type III-A spacer uptake patterns across RCR plasmids. (A) Schematic of the steps of rolling-circle replication. The parental plus strand (nicked during the replication) is illustrated in blue, the parental minus strand is illustrated in red, and the newly replicated strands are illustrated with dashed lines. Browser tracks show distributions of aligned type III spacers along the three RCR plasmids pWAR (B), pTRK882 (C) and pNT1 (D); the y-axis indicates the cumulative depth of aligned spacers. Abbreviations: dso = double-stranded origin, sso = single-stranded origin, oriT = origin of transfer, RSB = conserved recombination site, CS-6 = consensus sequence of 6 nt.
To further test this idea, we evaluated adaptation by both type III-A and type II-A CRISPR systems against two other RCR plasmids (pTRK882 (83) and pNT1 (84)), three theta replicating plasmids (pIB184 (85), pRSNPed (86) and pG-off (87)), and one non-replicating plasmid (ssM13 and dsM13), separately. Single stranded origins are diverse in sequence, and are classified into five types according to their secondary structures and consensus motifs. These five types, named ssoA, ssoL, ssoT, ssoU and ssoW (80), all have a predicted hairpin. Among the three RCR plasmids, we examined adaptation against two type sso-Ws (pWAR and pTRK882) and one type sso-A (pNT1). For the type III-A system, all the ssos of RCR plasmids were recognized by the adaptation machinery and covered by new spacer peaks (Figure 4C and D; Supplementary Figure S4). The replication origins (oriA-theta) of the theta replicating plasmids were not as heavily sampled during adaptation (Supplementary Figure S5). pNT1 contains a mob (mobilization) gene for conjugation, as well as a putative oriT (origin of transfer) with a RSA recombination site, inverted repeats and nick site (88). Interestingly, both the sso and oriT of pNT1 were highly sampled by the type III-A system (Figure 4D). Unlike the other plasmids, M13 is not able to replicate in Sth cells and does not contain a selective marker, however, five unique spacers were detected from dsM13 DNA, as well as three unique spacers from ssM13 DNA by six independent experiments (Supplementary Table S2). Notably, all the protospacers of ssM13 were within the lacI gene, which is a hairpin structure enriched region (89), and they were located at partially palindromic sequences. Collectively, these findings indicate that the stem-loop structures formed by a ssDNA may serve as substrates for adaptation by the type III-A CRISPR system. For the type II-A system, a small number of spacers aligned in the vicinity of the the sso, dso, and oriT sequences, but these areas were not particularly preferred over other regions of the plasmid (Supplementary Figure S4), suggesting that a preference for hairpin-forming DNA is unique to type III-A adaptation.
As noted above, the origin of replication for the theta replicating plasmid pIB184 was not enriched for type III spacers relative to upstream and downstream DNA (Supplementary Figure S5). Elsewhere along the plasmid, we did observe regions that were highly sampled, in particular the area between the cop and RepD genes (Figure 5D). This region contains architecture for a transcriptional attenuation mechanism, which controls expression of the RepD protein (90,91) (Figure 5A). One promoter drives expression of a long leader RNA continuous with the RepD mRNA. This leader RNA has multiple inverted repeats that form RNA secondary structures, including a hairpin that creates a rho-independent terminator. A second promoter drives expression of an anti-sense RNA called RNAIII. RNAIII forms its own secondary structure and can interact with the leader RNA structures. In the absence of RNAIII, the leader RNA assumes a conformation that eliminates the terminator and permits RepD transcription. In the presence of RNAIII, an alternative conformation is preferred, the terminator hairpin forms, and RepD transcription is terminated (Figure 5A). Strikingly, hotspots of type III spacer uptake overlap with several of the inverted repeats that make up this riboswitch transcriptional attenuation control region (Figure 5E). The two other theta-replicating plasmids, pRSNPed and pG-off, also contain predicted riboswitch transcriptional attenuation mechanisms between their cop and Rep genes which are likewise targeted by type III adaptation (Supplementary Figure S5). Enrichment at these same locations was not observed for the type II spacers (Supplementary Figure S5).
Figure 5.

Type III-A spacer uptake at regions encoding transcriptional regulators. (A–C) Diagrams illustrating the mechanisms of transcription attenuation for a Rep protein gene (A), the L10 ribosomal protein gene (B) and for a T-box controlled amino acid synthesis operon (C). (A) The Rep gene, which controls plasmid replication at the downstream origin, has a long leader RNA capable of folding into two complex structures. The same region also codes for an anti-sense RNA (RNAIII). When expressed, RNAIII binds the Rep gene leader and promotes formation of a terminator hairpin, which leads to transcription attenuation and therefore loss of Rep expression. (B) L10 ribosomal protein gene likewise contains a long leader RNA which folds into one of two forms mutually exclusive forms, binding of the L10 protein promotes the terminator form, leading to transcriptional attenuation. The T-box mechanism (C) functions similarly, with binding of charged tRNAs promoting attenuation through terminator hairpin formation and binding of uncharged tRNAs promoting the anti-termination conformation. (D–I) Genome browser tracks show cumulative spacer and RNAseq read densities in the vicinity of the three transcription attenuation modules described in (A–C). (D) Spacer and RNAseq density across the full length of plasmid pIB184. Annotated genes, inverted repeats, and terminator/antiterminator structures are shown in gray, spacers are shown in blue (top strand) and pink (bottom strand); RNAseq reads are shown in purple (top strand) and yellow (bottom strand). Black bars show areas of statistically significant spacer enrichment identified by peak calling software. (E) Higher resolution views of spacer and RNAseq density between cop and Rep. (F) Wide view and close view (G) of spacer and RNAseq density around the L10 ribosomal protein gene. (H) Wide view and close view (I) of spacer and RNAseq density overlapping a T-box controlling an operon involved in synthesis of cysteine. Abbreviations: IR = inverted repeat, T = terminator, AT = antiterminator, AAT = anti-antiterminator, TSS = transcription start site.
Riboswitch transcriptional attenuation modules are not restricted to plasmids; these modules are also found in the Sth JIM8232 genome. Upon examining aligned spacer hotspots across the host chromosome, it was apparent that these modules, together with the rRNA and tRNA gene clusters (Figure 2C and Supplementary Figure S6), are the most heavily sampled sites for type III adaptation. Both riboswitch leader RNAs and rRNA and tRNAs are enriched for potential DNA secondary structures as they produce highly structured non-coding RNAs (Figure 3D). Four riboswitches are currently annotated in Sth JIM8232. To identify additional, unannotated riboswitches, we used data from Streptococcus agalactiae as a guide (92). Transcription attenuators were detected in leader regions of 39 genes or operons in the genome of S. agalactiae (92). We found the corresponding genes or operons for 32 of the 39 genes in Sth JIM8232 and examined their upstream regions. Of these 32 genes, 18 had clear clusters of type III aligned spacers in the upstream region or ORF that were significantly enriched above the background spacer density (Supplementary Table S3 and Figure S7). One example lies upstream from the L10 ribosomal protein gene (Figure 5B, F, G). A long leader upstream from the ORF contains three imperfect RNA stem loop structures which can act as a rho-independent transcriptional terminator (T), an anti-terminator (AT) and an anti-antiterminator (AAT). When L10 is present in sufficient quantities, it binds to the anti-antiterminator and favors formation of the terminator stem loop, leading to transcriptional termination (Figure 5B) (93). Type III aligned spacers overlap with the predicted AAT, AT, and T hairpins (Figure 5F,G). We also observed an abrupt drop in RNAseq coverage near the predicted hairpins, which supports the prediction that this leader RNA contains a functional transcriptional attenuation module which is in the termination conformation under the growth conditions herein (Figure 5F,G).
A particularly well-defined cluster of aligned spacers was observed upstream from the PLP-dependent cysteine synthase gene, cysK (Figure 5H). This gene is part of an operon that also contains the ORFs for cystathionine γ-synthase and serine acetyltransferase. These three genes are involved in the production of the amino acid cysteine (94). Although this particular operon was not among the identified transcription attenuators for S. agalactiae, leader sequences of amino acid synthesis genes often contain transcriptional attenuation modules, called T-boxes, which can shut down transcription upon binding of a charged tRNA (Figure 5C) (94). In Bacillis subtilis, serine acetyltransferase (cysE) expression is regulated by transcription termination at a cysteine specific T-box (95). Here we also find a predicted cysteine specific T-box upstream of the operon, although cysK precedes cysE in this case. As with the L10 leader, we observed an abrupt drop in RNAseq coverage near the predicted terminator hairpin and aligned spacers overlapped with the predicted terminator and/or anti-terminator hairpins (Figure 5H,I).
Impact of transcription on protospacer selection
Aside from rRNA/tRNA gene clusters and transcription attenuation modules, we also noted that type III aligned spacers were often enriched around the beginnings of transcribed genes (Figure 6). For example, the second most heavily sampled region of the pIB184 plasmid was just downstream from a promoter (p23) (Figure 5D, close-up in Figure 6B, top panel). Similar spacer clusters were observed near the start codon of many, but not all, protein-coding genes in the host chromosome (e.g. Figure 6A,C). To test whether spacer enrichment was truly linked to the p23 promoter, we inverted it and again assayed type III spacer uptake. For the inverted p23 plasmid, a new dominant peak of aligned spacers appeared on the opposite side of the promoter, again just downstream from where transcription would be predicted to start (Figure 6B, middle panel). Type II spacers sometimes aligned within this same area but were not enriched (Figure 6B, bottom panel).
Figure 6.

Type III-A spacer uptake patterns near gene promoters. (A and C) Genome browser tracks show cumulative spacer and RNAseq densities across two example genome regions with expressed genes. Spacers are shown in blue (top strand) and pink (bottom strand); RNAseq reads are shown in purple (top strand) and yellow (bottom strand). (B) Genome browser tracks show a high resolution view of type III spacer and RNAseq densities around the p23 promoter of plasmid pIB184 in either a forward orientation (top panel) or a reverse orientation (middle panel). For comparison, type II new spacers are shown in the bottom panel. (D) Meta-plot of cumulative spacer density (blue) and RNAseq density (yellow) across all annotated protein-coding genes in the Sth JIM8232 genome. (E-F) Cumulative capture of protospacers acquired by the type III-A system (E) and the type II-A system (F) is plotted in black across annotated, protein-coding Sth JIM8232 genes (ORFs) which have been sorted by expression levels. Genes are ranked by total RNAseq density, with the highest density genes on the left side of the plot. Cumulative capture of protospacers within 200 bp genome windows, rather than annotated protein-coding genes, is plotted in red. These windows were tiled across the entire genome and include ncRNA genes (like rRNAs and tRNAs) and intergenic space. Pooled data of at least three independent experiments are presented in (D–F).
The inverted p23 findings supported an association between upstream regions of expressed genes and spacer uptake, so we next set out to determine how widespread the correlation was. For this, we created a meta-plot of aligned spacer density for all annotated protein-coding genes in the Sth JIM8232 genome. Available annotations for JIM8232 include predicted ORFs but do not provide transcription start sites; however a majority of 5′ UTRs in related species Streptococcus suis, S. agalactiae and Streptococcus pyogenes are <35 bp (92,96,97), so we used start codons as a general proxy for the region of transcription initiation. Bioinformatically, all ORFs were centered according to the first base of the start codon (e.g. A of the ATG) and then the cumulative density of spacers upstream and downstream from the start codons was determined. This cumulative density was then normalized against the average spacer density across the genome to yield a generalized spacer density profile for the meta-ORF. Cumulative normalized RNAseq depth was also plotted the same way. As expected, RNAseq depth for the meta-ORF was generally lower at the 5′ end of the gene then rose across the start codon and stayed relatively high through the body of the ORF (Figure 6D) (98). The profile for type III spacers was different; a peak in spacer enrichment was found approximately 10–50 nucleotides downstream from the start codon (Figure 6D). This enrichment near the start codon was consistent with our p23 results and with the individual peaks observed for certain host genes (Figure 6A,C), suggesting that features or events downstream from gene promoters and/or translation start sites may enhance type III spacer uptake.
Since we observed abundant spacer uptake in the highly expressed rRNA and tRNA genes (Figure 3D and Supplementary Figure S5) and since spacer uptake was also enriched around promoters (Figure 6), we considered the possibility that RNA abundance could drive adaptation. In the type III-B system from Marinomonas mediterranea, Cas1 is fused to a reverse transcriptase domain and RNA can act directly as a pre-spacer (48). A clear relationship between transcript abundance and spacer uptake was observed with this RT-Cas1 fusion protein system (48). In the Sth JIM8232 type III system, Cas1 lacks an RT domain (and there is no predicted Sth RT) and is not expected to use RNA as a substrate, but it is possible that RNA expression influences adaptation indirectly. We assessed the relationship between RNA expression and spacer uptake by calculating the percent of all spacers sampled from protein-coding genes (Figure 6E, black line). This analysis excluded expression from unannotated genes and non-coding genes like rRNA and tRNAs. Next, we did the analysis using 200 bp windows tiled across the entire genome (Figure 6E, red line). When rRNAs and tRNAs were included, the curve rose steeply and then flattened, suggesting that highly expressed non-coding genes are responsible for 30% or more of new spacers. However, when looking only at protein-coding genes, the curve was nearly flat, indicating that expression levels alone are not the primary drivers of spacer uptake (Figure 6E). The same trend was observed for type II spacers, except there was no sharp rise associated with highly expressed rRNA and tRNA non-coding genes (Figure 6F). These results indicate that type III adaptation is linked to events of transcription but not directly responding to the total amount of RNA present.
Type III systems mediate adaptive defense against a lytic phage
From our findings, it appeared that the type III-A system from Sth JIM8232 was fully functional for adaptation and interference and thus could act as a bona fide immune system. However, we had not yet determined whether the type III system could protect against a natural pathogen. To date, no lytic phages capable of infecting Sth JIM8232 strain have been isolated, but phage 2972 can infect the related strain, Sth DGCC7710 (99). To test whether the type III-A CRISPR-Cas system is able to mediate defense against a lytic phage, we replaced the native type III-A CRISPR-Cas system of DGCC7710 with that of JIM8232, and co-cultivated the new strain with phage 2972. We then isolated two type III-A bacteriophage insensitive mutants (BIMs). We amplified the leader-adjacent end of each of their CRISPR-Cas loci to look for evidence of new spacers and found that both BIMs had acquired a new spacer from the template strand of the phage into their type III CRISPR locus and did not acquire any other leader-adjacent spacers in other resident CRISPR loci (Figure 7A). Growth curves and plaque assay results further support that both BIMs are resistant to phage 2972 (Figure 7B,C). Thus, newly acquired spacers in the Sth type III-A system provide protection against lytic phage infection.
Figure 7.

Type III-A spacer uptake after lytic phage challenge. Two Sth DGCC7710 clones resistant to phage 2972 (BIM1 and BIM2) were isolated and genotyped. (A) Gel images show PCR products from the leader-adjacent end of all four CRISPR loci in Sth DGCC7710. Expanded bands (indicated by stars) correspond to a new leader-adjacent spacer in the type III-A array. (B) Growth of WT Sth DGCC7710, type III-A BIM1 and type III-A BIM2 in liquid culture was inferred by measuring changes in optical density over time; cell lysis results in clearing. (C) Plaque assay results for WT Sth DGCC7710, type III-A BIM1 and type III-A BIM2. Serial ten-fold dilutions of phage lysate containing infectious virus were plated onto lawns of each of the three strains.
DISCUSSION
Prokaryotes must capture and integrate short fragments of foreign DNA into their genomic CRISPR arrays to attain heritable CRISPR-Cas immunity against mobile genetic elements. Despite the widespread occurrence of type III CRISPR-Cas systems in diverse bacterial and archaeal species, we are only now beginning to understand how type III systems acquire new CRISPR spacers. Here, we demonstrate that the native Sth type III-A system is active for adaptation against plasmid and phage invaders. By comparing the adaptation properties of a type III-A and a type II system that co-exist in the strain, we show that adaptation by type III-A systems fundamentally differs from type II (and other studied type I and II CRISPR systems) in that it does not rely on a protospacer adjacent motif (PAM) and functions with an apparent lack of integration orientation bias. However, by challenging Sth with a variety of plasmids and phage invaders, we found that type III-A spacer choice is not random and that DNA with predicted secondary structures (e.g. imperfect DNA hairpins formed on single-stranded rolling-circle replication intermediates or as a result of transient R-loop formation during transcription) is preferentially targeted. As detailed below (and see Figure 8), this type of spacer selection strategy could make key components of mobile genetic elements particularly vulnerable to adaptation.
Figure 8.

Model illustrating features of mobile genetic elements that could be targeted for spacer uptake by a type III-A system. Mobile genetic elements invade prokaryotic cells by conjugation, transduction and transformation. The stem-loop structures present in the ssDNA of mobile genetic elements during invasion, replication, and gene expression are highlighted with stars.
The type III-A spacer acquisition machinery
Cas1 and Cas2 interact to form an integrase complex (23,25,28) found to be essential for acquiring new spacers in all adaptation-competent CRISPR-Cas systems tested thus far (23,24,26–28,31,32,48,100). Many systems also require additional Cas proteins in order to adapt. For example, the type I-F Csy effector complex of Pseudomonas aeruginosa (101) and the type II-A effector nuclease Cas9 and Csn2 protein in Streptococcus thermophilus (26) and S. pyogenes (27) are essential for efficient adaptation. Our genetic analyses revealed that Cas1 and Cas2 are necessary and sufficient Cas components for type III-A adaptation (Figures 2–5 and Supplementary Figure S5). However, it remains to be seen whether type III-A Cas 1 and Cas2 have an intrinsic ability to recognize and capture pre-spacers from regions of DNA with secondary structures or whether they collaborate with non-Cas host proteins to achieve this observed property. Future experiments with engineered DNA hairpins will be necessary to explore these potential mechanisms.
Role of counterselection in shaping type III CRISPR spacer composition
Because we were able to characterize adaptation in an interference-null strain (e.g. the Δcsm1-6 strain) alongside the wild-type (WT) strain, we could look beyond survival biases and identify different features that promoted uptake of new spacers (Figures 3–5 and Supplementary Figure S5). Adaptation assays performed in the absence of the type III-A interference pathway genes (csm1-6), revealed that spacers are normally integrated in both possible orientations but a strand bias arises in WT strains, likely due to negative selection against cells that incorporate self-targeting spacers capable of triggering lethal autoimmunity or plasmid loss under selective growth conditions (Figure 3). These findings are in agreement with observations from the type III-A system in Thermus thermophilus (51); there they also found evidence for orientation-independent spacer acquisition followed by survivor bias in favor of cells which had acquired properly oriented spacers. Taken together, the evidence suggests that type III-A systems initially acquire a relatively wide range of spacers, which is then winnowed by survival selection through pressures such as phage predation and lethal autoimmunity. Other factors, such as dynamics of CRISPR spacer loss and gain through recombination (Figure 2), could also contribute to the observed property that endogenous type III CRISPR arrays almost always encode for functional crRNAs (Figure 3B and (55)).
DNA hairpins and other transiently formed DNA secondary structures as preferred substrates for type III spacer acquisition machinery
A prominent and intriguing feature of type III-A adaptation was the high level of spacer uptake from DNA regions predicted to form transient secondary structures through intra-strand base-pairing of inverted repeats. Transient hairpins in ssDNA are often involved in horizontal gene transfer or replication of mobile genetic elements like plasmids and phages/viruses (102) (Figure 8). For example, we observed that structurally diverse, single-stranded origins (sso) of replication were highly targeted by the type III adaptation machinery in both wild-type and interference-null strain (Figure 4). Both strains sampled spacers throughout the sso, but interestingly, in the absence of interference (Δcsm1-6 strain), spacers at the predicted RNA polymerase binding site were particularly abundant (Figure 4A,B). Given that we observed counter-selection bias elsewhere in our data (Figures 3–5), we speculate that the difference in sso spacer abundances is evidence of interference in the wildtype strain: the sso transcription start site is the most heavily sampled site in both strains, but in the wild-type strain some spacers are lost due to interference or disruption of plasmid replication and thus are less visible in our assay results. This would imply that type III spacers against these natural ssos are competent for plasmid interference, a prediction that awaits experimental confirmation. Most natural plasmids found in gram-positive bacteria, including Sth, and many of those found in gram-negative bacteria and archaea are RCR plasmids (80,103). In addition, many phages use RCR and have a sso functionally similar to that of the pWAR plasmid that we used in this work (102). Notably, while the type III-A adaptation system was selective for acquiring spacers from the sso vs. the dso (double-stranded origin of replication) of RCR plasmids, the inverse was observed when spacer acquisition was investigated for the type I adaptation system of Pyrococcus furiosus (45). In the case of P. furiosus, the newly acquired spacers were preferentially captured from the dso region centered on the site that is nicked by the Rep protein to generate the free 3′ DNA end used for priming plus strand replication.
We also observed type III spacer enrichment around the origin of transfer (oriT) of the pNT1 plasmid, which contains conserved inverted repeats and is predicted to form a transient hairpin-like structure (Figure 4C,D) (84). In the context of its native conjugative plasmid, this oriT is recognized and nicked, bound by a relaxase protein, and then the plasmid is unwound and transferred to the recipient cell as ssDNA. Upon transfer, the oriT hairpin structure may then also act as a promoter for transcription of plasmid genes (102). We propose that hairpin formation is what makes the oriT of pNT1 suitable for spacer uptake and that this is another example wherein a key component for proliferation of a mobile genetic element is targeted by type III adaptation.
As described above, hairpins in an sso or oriT are transient, and so the same region of DNA would often be in dsDNA form. We also assayed adaptation in samples after exogenous addition of ssM13 plasmid, which is expected to remain single-stranded in the gram-positive host, S. thermophilus. Though rare, we saw uptake of ssM13-derived spacers, specifically at confirmed stem-loop structures in the regulatory sequences of the lacZ gene (89) (Supplementary Table S2), which lends additional support to the hypothesis that structured ssDNA is targeted. Recently, the type III-A adaptation machinery of Thermus thermophilus was found to acquire spacers from a discrete region of a phage genome noted as containing early-expressed genes but also encompassing long terminal repeats (LTRs) (51). Our evidence for a preference for structured DNA regions raises the possibility that DNA secondary structures associated with the LTRs contributed to the pattern of phage genome spacer sampling observed in this study (51).
Structured DNA associated with expressed genes is also a preferred substrate for type III adaptation
A second highly targeted feature was DNA coding for riboswitch transcriptional attenuators (Figure 5 and Supplementary Figure S6 and Table S3). These cis-acting RNAs are widely employed in bacteria and archaea to regulate expression of genes involved in the metabolism or uptake of amino acids, nucleotides, and metal ions (104). Typically, the riboswitch lies in the 5′UTR of the regulated mRNA and interactions between a signaling molecule and the riboswitch control formation of a transcriptional terminator hairpin (104) (Figure 5A–C). We identified18 riboswitches with statistically significant enrichment of type III spacer clusters upstream of the ORF (Figure 5F,G; Supplementary Table S3 and Figure S6). What about these regions makes them hotspots for type III CRISPR spacer uptake? With no reverse transcriptase domain fused to either Cas1 or Cas2 and no predicted Sth genomic reverse transcriptase, there is no obvious mechanism for direct capture of riboswitch RNA as a pre-spacer. Furthermore, we found that overall RNA abundance did not correlate well with spacer uptake (Figure 6E,F). However, the same inverted repeats that promote RNA folding for riboswitches may also promote formation of non-B conformation DNA structures in the non-template strand of transcribed DNA (Figure 8). In a typical transcription scenario, the non-template strand would be single-stranded only within the transcription bubble, which is generally <25 nucleotides long (102). Beyond this small stretch, B-form dsDNA is thought to predominate, and that would preclude formation of ssDNA hairpins or other structures. However, in certain cases, non-B form DNA is observed beyond the confines of the transcription bubble, and it appears to be aided by DNA torsion due to negative supercoiling. As transcribing RNA polymerase moves along a gene, unwinding leads to positively supercoiled DNA in front of the polymerase and negatively supercoiled DNA behind it (upstream) (105). Negative supercoiling promotes strand separation, and in turn the liberated ssDNA can assume localized structures (106,107). For example, in E. coli, upstream torsion due to transcription at a downstream promoter can induce DNA with inverted repeats to form a cruciform (108). Localized DNA structures upstream of the transcribing RNA polymerase can also occur as the result of co-transcriptional strand invasion of the nascent transcript with the DNA template strand leading to extended R-loop formation (three-stranded nucleic acid structures, consisting of a RNA:DNA hybrid and the extruded non-template single-stranded DNA). The displaced non-template (coding) DNA strand of R-loop structures would be available to engage in intra-strand pairing to form DNA secondary structures and thus could be preferred type III adaption substrates, based on our model (Figure 8).
Numerous regions of ssDNA with associated secondary structures have been detected in vivo. These non-B form structures require two features to form: sequence elements, for example, a G-skew or an AT rich stretch, and downstream transcription (and presumably the torsion it causes) (109). Higher expression of downstream genes is associated with more detectable non-B form DNA (109). Non-B form structures on the non-template (coding) strand of transcribed genes can co-occur with R-loops, and can control R-loop size and architecture (110). Furthermore, non-template DNA secondary structures are enriched just upstream from transcription pause sites, and experiments demonstrated that more stable structures induced stronger pausing (111). This suggested to the authors that the DNA secondary structures themselves may serve regulatory purposes through their induction of transcription pausing (111). Applying these different findings to our data, we speculate that when conditions are favorable, non-template DNA in riboswitch-coding regions can form hairpins that are heavily targeted by type III adaptation machinery. We extend this idea to the type III spacer hotspots observed in rRNA and tRNA genes: the non-template DNA of these highly expressed and highly folded RNA genes could also form structures targeted for adaptation. Since alternate (non-B) DNA structures such as hairpins are promoted by transcription, the net effect of type III adaptation targeting structure DNA would be a bias toward transcriptionally active DNA sequences.
We also observed, to a lesser extent, a general enrichment of type III spacers roughly 10–50 bp downstream from start codons (Figure 6A–D). Even in the absence of outright transcription attenuator modules, this region, along with the start codon itself, is often the site of temporary RNA polymerase pausing (112). In prokaryotes, transcription and translation are coupled and the ribosome appears to help push RNA polymerase forward from a promoter-proximal stalled position (113,114). In that interval when the polymerase is paused but still awaiting ribosome engagement, R-loops may form, leaving non-template DNA single stranded (114). Furthermore, the ribosome may assist in moving polymerase along the full length of the mRNA at a pace determined by conditions like codon availability (113). Given this overlap between spacer enrichment and pause site enrichment in the region ∼10–50 bp downstream from ORF start codons, along with the observed spacer hotspots over transcription attenuators, we propose that stalled transcription in general creates favorable conditions for type III adaptation, potentially by exposing non-template DNA in a single stranded form capable of forming secondary structures (Figure 8).
By targeting transient hairpins in ssDNA, the type III system could be biased towards sampling key components of mobile genetic elements. How do the putative hairpins of non-template, transcribed DNA fit in to this model? One interpretation of our data is that the type III system is tuned to target hairpins found in mobile genetic elements, and the self-targeting at transcribed genes that we observe is a deleterious side effect of this hairpin-directed adaptation. Alternatively, a preference for ssDNA associated with a stalled RNA polymerase could also bias adaptation towards transcribed foreign or invasive genes (Figure 8). Nutrient depletion, which can occur when a mobile genetic element starts to consume too many resources through its replication and/or gene expression, can make RNA polymerase more prone to pausing (113). In addition, foreign genetic elements may have codon usage patterns and/or transcriptional landscapes that are out of sync with those of their host cell, leading to inefficient and potentially paused or stalled transcription or translation (115). With this second interpretation, auto-immunity (deleterious adaptation of transcribed self-DNA) can occur, but transcribed foreign or parasitic DNA is relatively more vulnerable.
Type III adaptation vs. the relatively well-studied type I and II adaptation
Previous work with type I and II DNA targeting CRISPR systems has suggested that adaptation recognizes and samples widely from available PAM-containing DNA, but a few key characteristics guide the process and provide an imperfect means of differentiating foreign from self-DNA. For example, the relative abundance of sequence motifs in self-DNA of Escherichia coli, as compared to foreign DNA, can bias adaptation (42). The double-strand DNA break repair complex in E. coli, RecBCD (AddAB in gram positive bacteria), is thought to generate spacer substrates through DNA strand unwinding and breakdown of DNA, but it slows DNA degradation upon encountering an octameric sequence called a chi site (116,117). Since chi sites are common in the host chromosome, uptake of self-targeting spacers is disfavored compared to phage and plasmid targeting spacers (42,116). More generally, type I and II protospacer uptake mechanisms appears to target free DNA ends (e.g. linearized dsDNA) (42,45,118) and this may likewise help limit adaptation against the host chromosome, which is circular, and favor adaptation against mobile genetic elements like plasmids, phages and transposons that become linearized upon host cell uptake (via transduction, conjugation and transformation) or during replication or recombination with the host genome (45). Here we propose that adaptation in type III systems is also biased toward signature features associated with parasitic genetic elements, but through a different mechanism (Figure 8). Adaptation against transient ssDNA structures sets up the system to preferentially sample from replicating plasmids, phages and inefficiently transcribed genes. Moreover, the ways that mobile genetic elements enter prokaryotic cells (via conjugation, natural transformation and viral transduction) often involves entry of single-stranded DNA intermediates capable of forming secondary DNA structures (102), and this could increase the susceptibility of exogenous DNA to recognition by type III adaptation machinery (Figure 8). Given that recombination of foreign DNA with host chromosomes and mobilization of transposons also requires formation of DNA secondary structures, there are yet other potential steps for which mobile genetic element DNA may be vulnerable to the action of type III spacer acquisition. By this model, type III protospacer choice preferences are general enough that DNA is widely sampled, but still guide Cas1 and Cas2 towards potentially deleterious mobile genetic elements.
DATA AVAILABILITY
Sequence data were deposited in the NCBI Sequence Read Archive under the BioProject ID PRJNA762861.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Terns lab, Walter Woodside and Nicholas Jones, for construction of some bacterial strains used in this study, Cécile Phillipe and Katie Johnson for their work in characterizing the type III-A BIMs described in Figure 7 and Ryan Catchpole and Chris Noble-Molnar for comments on the manuscript. We thank the members of the Terns and Graveley labs for the helpful discussions. We are grateful to those researchers (mentioned in the Materials and Methods section) that generously provided plasmids, bacterial strains, and phage used in this study with special thanks to Sylvain Moineau (Laval University, Canada) for kindly sharing Sth reagents and expertise.
Contributor Information
Xinfu Zhang, Department of Biochemistry and Molecular Biology, University of Georgia, Athens, GA 30602, USA.
Sandra Garrett, Department of Genetics and Genome Sciences, Institute for Systems Genomics, University of Connecticut Health Center, Farmington, CT 06030, USA.
Brenton R Graveley, Department of Genetics and Genome Sciences, Institute for Systems Genomics, University of Connecticut Health Center, Farmington, CT 06030, USA.
Michael P Terns, Department of Biochemistry and Molecular Biology, University of Georgia, Athens, GA 30602, USA; Department of Microbiology, University of Georgia, Athens, GA 30602, USA; Department of Genetics, University of Georgia, Athens, GA 30602, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35GM118160 to M.P.T., R35GM118140 to B.R.G.]. Funding for open access charge: NIH [R35GM118140].
Conflict of interest statement. None declared.
REFERENCES
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P.. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007; 315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 2. Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J.. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hale C.R., Zhao P., Olson S., Duff M.O., Graveley B.R., Wells L., Terns R.M., Terns M.P.. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009; 139:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marraffini L.A., Sontheimer E.J.. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008; 322:1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Makarova K.S., Wolf Y.I., Iranzo J., Shmakov S.A., Alkhnbashi O.S., Brouns S.J.J., Charpentier E., Cheng D., Haft D.H., Horvath P.et al.. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020; 18:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Amitai G., Sorek R.. CRISPR-Cas adaptation: insights into the mechanism of action. Nat. Rev. Microbiol. 2016; 14:67–76. [DOI] [PubMed] [Google Scholar]
- 7. McGinn J., Marraffini L.A.. Molecular mechanisms of CRISPR-Cas spacer acquisition. Nat. Rev. Microbiol. 2019; 17:7–12. [DOI] [PubMed] [Google Scholar]
- 8. Sternberg S.H., Richter H., Charpentier E., Qimron U.. Adaptation in CRISPR-Cas Systems. Mol. Cell. 2016; 61:797–808. [DOI] [PubMed] [Google Scholar]
- 9. Carte J., Wang R.Y., Li H., Terns R.M., Terns M.P.. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008; 22:3489–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nussenzweig P.M., Marraffini L.A.. Molecular mechanisms of CRISPR-Cas immunity in bacteria. Annu. Rev. Genet. 2020; 54:93–120. [DOI] [PubMed] [Google Scholar]
- 11. Hille F., Richter H., Wong S.P., Bratovic M., Ressel S., Charpentier E.. The biology of CRISPR-Cas: backward and forward. Cell. 2018; 172:1239–1259. [DOI] [PubMed] [Google Scholar]
- 12. Jackson R.N., van Erp P.B., Sternberg S.H., Wiedenheft B.. Conformational regulation of CRISPR-associated nucleases. Curr. Opin. Microbiol. 2017; 37:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shah S.A., Erdmann S., Mojica F.J., Garrett R.A.. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol. 2013; 10:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Estrella M.A., Kuo F.T., Bailey S.. RNA-activated DNA cleavage by the Type III-B CRISPR-Cas effector complex. Genes Dev. 2016; 30:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Samai P., Pyenson N., Jiang W., Goldberg G.W., Hatoum-Aslan A., Marraffini L.A.. Co-transcriptional DNA and RNA cleavage during Type III CRISPR-Cas immunity. Cell. 2015; 161:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Staals R.H., Zhu Y., Taylor D.W., Kornfeld J.E., Sharma K., Barendregt A., Koehorst J.J., Vlot M., Neupane N., Varossieau K.et al.. RNA targeting by the type III-A CRISPR-Cas Csm complex of Thermus thermophilus. Mol. Cell. 2014; 56:518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tamulaitis G., Kazlauskiene M., Manakova E., Venclovas C., Nwokeoji A.O., Dickman M.J., Horvath P., Siksnys V.. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol. Cell. 2014; 56:506–517. [DOI] [PubMed] [Google Scholar]
- 18. Elmore J.R., Sheppard N.F., Ramia N., Deighan T., Li H., Terns R.M., Terns M.P.. Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR-Cas system. Genes Dev. 2016; 30:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peng W., Feng M., Feng X., Liang Y.X., She Q.. An archaeal CRISPR type III-B system exhibiting distinctive RNA targeting features and mediating dual RNA and DNA interference. Nucleic Acids Res. 2015; 43:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Foster K., Grüschow S., Bailey S., White M.F., Terns M.P.. Regulation of the RNA and DNA nuclease activities required for Pyrococcus furiosus Type III-B CRISPR-Cas Immunity. Nucleic Acids Res. 2020; 48:4418–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kazlauskiene M., Kostiuk G., Venclovas C., Tamulaitis G., Siksnys V.. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science. 2017; 357:605–609. [DOI] [PubMed] [Google Scholar]
- 22. Niewoehner O., Garcia-Doval C., Rostol J.T., Berk C., Schwede F., Bigler L., Hall J., Marraffini L.A., Jinek M.. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature. 2017; 548:543–548. [DOI] [PubMed] [Google Scholar]
- 23. Xiao Y., Ng S., Nam K.H., Ke A.. How type II CRISPR-Cas establish immunity through Cas1-Cas2-mediated spacer integration. Nature. 2017; 550:137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yosef I., Goren M.G., Qimron U.. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012; 40:5569–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nunez J.K., Kranzusch P.J., Noeske J., Wright A.V., Davies C.W., Doudna J.A.. Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014; 21:528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wei Y., Terns R.M., Terns M.P.. Cas9 function and host genome sampling in Type II-A CRISPR-Cas adaptation. Genes Dev. 2015; 29:356–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heler R., Samai P., Modell J.W., Weiner C., Goldberg G.W., Bikard D., Marraffini L.A.. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature. 2015; 519:199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu C., Tang D., Cheng J., Hu D., Yang Z., Ma X., He H., Yao S., Fu T.M., Yu Y.et al.. Mechanisms of spacer acquisition by sequential assembly of the adaptation module in Synechocystis. Nucleic Acids Res. 2021; 49:2973–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hale C.R., Majumdar S., Elmore J., Pfister N., Compton M., Olson S., Resch A.M., Glover C.V.C. III, Graveley B.R., Terns R.M.et al.. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol. Cell. 2012; 45:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lillestol R.K., Shah S.A., Brugger K., Redder P., Phan H., Christiansen J., Garrett R.A.. CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol. Microbiol. 2009; 72:259–272. [DOI] [PubMed] [Google Scholar]
- 31. Kim J.G., Garrett S., Wei Y., Graveley B.R., Terns M.P.. CRISPR DNA elements controlling site-specific spacer integration and proper repeat length by a Type II CRISPR-Cas system. Nucleic Acids Res. 2019; 47:8632–8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grainy J., Garrett S., Graveley B.R., Michael P.Terns. CRISPR repeat sequences and relative spacing specify DNA integration by Pyrococcus furiosus Cas1 and Cas2. Nucleic Acids. Res. 2019; 47:7518–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wei Y., Chesne M.T., Terns R.M., Terns M.P.. Sequences spanning the leader-repeat junction mediate CRISPR adaptation to phage in Streptococcus thermophilus. Nucleic Acids Res. 2015; 43:1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van Orden M.J., Newsom S., Rajan R.. CRISPR type II-A subgroups exhibit phylogenetically distinct mechanisms for prespacer insertion. J. Biol. Chem. 2020; 295:10956–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nunez J.K., Bai L., Harrington L.B., Hinder T.L., Doudna J.A.. CRISPR immunological memory requires a host factor for specificity. Mol. Cell. 2016; 62:824–833. [DOI] [PubMed] [Google Scholar]
- 36. Santiago-Frangos A., Buyukyoruk M., Wiegand T., Krishna P., Wiedenheft B.. Distribution and phasing of sequence motifs that facilitate CRISPR adaptation. Curr. Biol. 2021; 31:3515–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barrangou R., Dudley E.G.. CRISPR-based typing and next-generation tracking technologies. Annu. Rev. Food Sci. Technol. 2016; 7:395–411. [DOI] [PubMed] [Google Scholar]
- 38. Nunez J.K., Lee A.S., Engelman A., Doudna J.A.. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015; 519:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rollie C., Schneider S., Brinkmann A.S., Bolt E.L., White M.F.. Intrinsic sequence specificity of the Cas1 integrase directs new spacer acquisition. Elife. 2015; 4:e08716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shiimori M., Garrett S.C., Graveley B.R., Terns M.P.. Cas4 nucleases define the PAM, length, and orientation of DNA fragments integrated at CRISPR loci. Mol. Cell. 2018; 70:814–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ivancic-Bace I., Cass S.D., Wearne S.J., Bolt E.L.. Different genome stability proteins underpin primed and naive adaptation in E. coli CRISPR-Cas immunity. Nucleic Acids Res. 2015; 43:10821–10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Levy A., Goren M.G., Yosef I., Auster O., Manor M., Amitai G., Edgar R., Qimron U., Sorek R.. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015; 520:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wiegand T., Semenova E., Shiriaeva A., Fedorov I., Datsenko K., Severinov K., Wiedenheft B.. Reproducible antigen recognition by the Type I-F CRISPR-Cas system. CRISPR J. 2020; 3:378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garrett S., Shiimori M., Watts E.A., Clark L., Graveley B., Terns M.P.. Primed CRISPR DNA uptake in Pyrococcus furiosus. Nucleic Acids Res. 2020; 48:6120–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shiimori M., Garrett S.C., Chambers D.P., Glover C.V.C. 3rd, Graveley B.R., Terns M.P. Role of free DNA ends and protospacer adjacent motifs for CRISPR DNA uptake in Pyrococcus furiosus. Nucleic Acids Res. 2017; 45:11281–11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Silas S., Lucas-Elio P., Jackson S.A., Aroca-Crevillen A., Hansen L.L., Fineran P.C., Fire A.Z., Sanchez-Amat A.. Type III CRISPR-Cas systems can provide redundancy to counteract viral escape from type I systems. Elife. 2017; 6:e27601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Majumdar S., Zhao P., Pfister N.T., Compton M., Olson S., Glover C.V. 3rd, Wells L., Graveley B.R., Terns R.M., Terns M.P. Three CRISPR-Cas immune effector complexes coexist in Pyrococcus furiosus. RNA. 2015; 21:1147–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Silas S., Mohr G., Sidote D.J., Markham L.M., Sanchez-Amat A., Bhaya D., Lambowitz A.M., Fire A.Z.. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science. 2016; 351:aad4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schmidt F., Cherepkova M.Y., Platt R.J.. Transcriptional recording by CRISPR spacer acquisition from RNA. Nature. 2018; 562:380–385. [DOI] [PubMed] [Google Scholar]
- 50. Gonzalez-Delgado A., Mestre M.R., Martinez-Abarca F., Toro N.. Spacer acquisition from RNA mediated by a natural reverse transcriptase-Cas1 fusion protein associated with a type III-D CRISPR-Cas system in Vibrio vulnificus. Nucleic Acids Res. 2019; 47:10202–10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Artamonova D., Karneyeva K., Medvedeva S., Klimuk E., Kolesnik M., Yasinskaya A., Samolygo A., Severinov K.. Spacer acquisition by Type III CRISPR-Cas system during bacteriophage infection of Thermus thermophilus. Nucleic Acids Res. 2020; 48:9787–9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Deveau H., Barrangou R., Garneau J.E., Labonte J., Fremaux C., Boyaval P., Romero D.A., Horvath P., Moineau S.. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008; 190:1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang J., Li J., Zhao H., Sheng G., Wang M., Yin M., Wang Y.. Structural and mechanistic basis of PAM-Dependent spacer acquisition in CRISPR-Cas systems. Cell. 2015; 163:840–853. [DOI] [PubMed] [Google Scholar]
- 54. Pyenson N.C., Gayvert K., Varble A., Elemento O., Marraffini L.A.. Broad targeting specificity during bacterial Type III CRISPR-Cas immunity constrains viral escape. Cell Host Microbe. 2017; 22:343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Millen A.M., Samson J.E., Tremblay D.M., Magadan A.H., Rousseau G.M., Moineau S., Romero D.A.. Lactococcus lactis type III-A CRISPR-Cas system cleaves bacteriophage RNA. RNA Biol. 2019; 16:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fontaine L., Dandoy D., Boutry C., Delplace B., de Frahan M.H., Fremaux C., Horvath P., Boyaval P., Hols P.. Development of a versatile procedure based on natural transformation for marker-free targeted genetic modification in Streptococcus thermophilus. Appl. Environ. Microbiol. 2010; 76:7870–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McKenzie R.E., Almendros C., Vink J.N.A., Brouns S.J.J.. Using CAPTURE to detect spacer acquisition in native CRISPR arrays. Nat. Protoc. 2019; 14:976–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S. . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Delorme C., Bartholini C., Luraschi M., Pons N., Loux V., Almeida M., Guedon E., Gibrat J.F., Renault P.. Complete genome sequence of the pigmented Streptococcus thermophilus strain JIM8232. J. Bacteriol. 2011; 193:5581–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Renye J.A. Jr, Somkuti G.A. Insertion of a heterologous gene construct into a non-functional ORF of the Streptococcus thermophilus chromosome. Biotechnol. Lett. 2009; 31:759–764. [DOI] [PubMed] [Google Scholar]
- 66. Garrett S.C. Pruning and tending immune memories: Spacer dynamics in the CRISPR array. Front. Microbiol. 2021; 12:664299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Deng L., Garrett R.A., Shah S.A., Peng X., She Q.. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol. Microbiol. 2013; 87:1088–1099. [DOI] [PubMed] [Google Scholar]
- 68. Goldberg G.W., Jiang W., Bikard D., Marraffini L.A.. Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature. 2014; 514:633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Liu T.Y., Iavarone A.T., Doudna J.A.. RNA and DNA Targeting by a Reconstituted Thermus thermophilus Type III-A CRISPR-Cas System. PLoS One. 2017; 12:e0170552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Samai P., Pyenson N., Jiang W., Goldberg G.W., Hatoum-Aslan A., Marraffini L.A.. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell. 2015; 161:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jiang W., Samai P., Marraffini L.A.. Degradation of phage transcripts by CRISPR-Associated RNases enables Type III CRISPR-Cas immunity. Cell. 2016; 164:710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sheppard N.F., Glover C.V. 3rd, Terns R.M., Terns M.P. The CRISPR-associated Csx1 protein of Pyrococcus furiosus is an adenosine-specific endoribonuclease. RNA. 2016; 22:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Foster K., Kalter J., Woodside W., Terns R.M., Terns M.P.. The ribonuclease activity of Csm6 is required for anti-plasmid immunity by Type III-A CRISPR-Cas systems. RNA Biol. 2019; 16:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Leenhouts K.J., Tolner B., Bron S., Kok J., Venema G., Seegers J.F.. Nucleotide sequence and characterization of the broad-host-range lactococcal plasmid pWVO1. Plasmid. 1991; 26:55–66. [DOI] [PubMed] [Google Scholar]
- 75. Mashburn-Warren L., Morrison D.A., Federle M.J.. The cryptic competence pathway in Streptococcus pyogenes is controlled by a peptide pheromone. J. Bacteriol. 2012; 194:4589–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Khan S.A. Plasmid rolling-circle replication: recent developments. Mol. Microbiol. 2000; 37:477–484. [DOI] [PubMed] [Google Scholar]
- 77. del Solar G., Giraldo R., Ruiz-Echevarria M.J., Espinosa M., Diaz-Orejas R.. Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev. 1998; 62:434–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. del Solar G.H., Puyet A., Espinosa M.. Initiation signals for the conversion of single stranded to double stranded DNA forms in the streptococcal plasmid pLS1. Nucleic Acids Res. 1987; 15:5561–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kramer M.G., Khan S.A., Espinosa M.. Plasmid rolling circle replication: identification of the RNA polymerase-directed primer RNA and requirement for DNA polymerase I for lagging strand synthesis. EMBO J. 1997; 16:5784–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ruiz-Maso J.A., Macho N.C., Bordanaba-Ruiseco L., Espinosa M., Coll M., Del Solar G.. Plasmid rolling-circle replication. Microbiol Spectr. 2015; 3:PLAS–0035–2014. [DOI] [PubMed] [Google Scholar]
- 81. Kramer M.G., Espinosa M., Misra T.K., Khan S.A.. Characterization of a single-strand origin, ssoU, required for broad host range replication of rolling-circle plasmids. Mol. Microbiol. 1999; 33:466–475. [DOI] [PubMed] [Google Scholar]
- 82. Kramer M.G., Espinosa M., Misra T.K., Khan S.A.. Lagging strand replication of rolling-circle plasmids: specific recognition of the ssoA-type origins in different gram-positive bacteria. Proc. Natl. Acad. Sci. USA. 1998; 95:10505–10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Duong T., Miller M.J., Barrangou R., Azcarate-Peril M.A., Klaenhammer T.R.. Construction of vectors for inducible and constitutive gene expression in Lactobacillus. Microb. Biotechnol. 2011; 4:357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vaillancourt K., Bedard N., Bart C., Tessier M., Robitaille G., Turgeon N., Frenette M., Moineau S., Vadeboncoeur C.. Role of galK and galM in galactose metabolism by Streptococcus thermophilus. Appl. Environ. Microbiol. 2008; 74:1264–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Biswas I., Jha J.K., Fromm N.. Shuttle expression plasmids for genetic studies in Streptococcus mutans. Microbiology. 2008; 154:2275–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Renye J.A. Jr, Somkuti G.A. Nisin-induced expression of pediocin in dairy lactic acid bacteria. J. Appl. Microbiol. 2010; 108:2142–2151. [DOI] [PubMed] [Google Scholar]
- 87. Lartigue M.F., Bouloc P.. A tetracycline-inducible expression vector for Streptococcus agalactiae allowing controllable gene expression. J. Microbiol. Methods. 2014; 96:16–18. [DOI] [PubMed] [Google Scholar]
- 88. Turgeon N., Moineau S.. Isolation and characterization of a Streptococcus thermophilus plasmid closely related to the pMV158 family. Plasmid. 2001; 45:171–183. [DOI] [PubMed] [Google Scholar]
- 89. Reckmann B., Grosse F., Urbanke C., Frank R., Blocker H., Krauss G.. Analysis of secondary structures in M13mp8 (+) single-stranded DNA by the pausing of DNA polymerase alpha. Eur. J. Biochem. 1985; 152:633–643. [DOI] [PubMed] [Google Scholar]
- 90. Le Chatelier E., Ehrlich S.D., Janniere L.. Countertranscript-driven attenuation system of the pAM beta 1 repE gene. Mol. Microbiol. 1996; 20:1099–1112. [DOI] [PubMed] [Google Scholar]
- 91. Brantl S. Antisense-RNA mediated control of plasmid replication - pIP501 revisited. Plasmid. 2015; 78:4–16. [DOI] [PubMed] [Google Scholar]
- 92. Rosinski-Chupin I., Sauvage E., Sismeiro O., Villain A., Da Cunha V., Caliot M.E., Dillies M.A., Trieu-Cuot P., Bouloc P., Lartigue M.F.et al.. Single nucleotide resolution RNA-seq uncovers new regulatory mechanisms in the opportunistic pathogen Streptococcus agalactiae. BMC Genomics. 2015; 16:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yakhnin H., Yakhnin A.V., Babitzke P.. Ribosomal protein L10(L12)4 autoregulates expression of the Bacillus subtilis rplJL operon by a transcription attenuation mechanism. Nucleic Acids Res. 2015; 43:7032–7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gutierrez-Preciado A., Henkin T.M., Grundy F.J., Yanofsky C., Merino E.. Biochemical features and functional implications of the RNA-based T-box regulatory mechanism. Microbiol. Mol. Biol. Rev. 2009; 73:36–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gagnon Y., Breton R., Putzer H., Pelchat M., Grunberg-Manago M., Lapointe J.. Clustering and co-transcription of the Bacillus subtilis genes encoding the aminoacyl-tRNA synthetases specific for glutamate and for cysteine and the first enzyme for cysteine biosynthesis. J. Biol. Chem. 1994; 269:7473–7482. [PubMed] [Google Scholar]
- 96. Rosinski-Chupin I., Sauvage E., Fouet A., Poyart C., Glaser P.. Conserved and specific features of Streptococcus pyogenes and Streptococcus agalactiae transcriptional landscapes. BMC Genomics. 2019; 20:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wu Z., Wu C., Shao J., Zhu Z., Wang W., Zhang W., Tang M., Pei N., Fan H., Li J.et al.. The Streptococcus suis transcriptional landscape reveals adaptation mechanisms in pig blood and cerebrospinal fluid. RNA. 2014; 20:882–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wang Z., Gerstein M., Snyder M.. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009; 10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Duplessis M., Russell W.M., Romero D.A., Moineau S.. Global gene expression analysis of two Streptococcus thermophilus bacteriophages using DNA microarray. Virology. 2005; 340:192–208. [DOI] [PubMed] [Google Scholar]
- 100. Fagerlund R.D., Wilkinson M.E., Klykov O., Barendregt A., Pearce F.G., Kieper S.N., Maxwell H.W.R., Capolupo A., Heck A.J.R., Krause K.L.et al.. Spacer capture and integration by a type I-F Cas1-Cas2-3 CRISPR adaptation complex. Proc. Natl. Acad. Sci. USA. 2017; 114:E5122–E5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Vorontsova D., Datsenko K.A., Medvedeva S., Bondy-Denomy J., Savitskaya E.E., Pougach K., Logacheva M., Wiedenheft B., Davidson A.R., Severinov K.et al.. Foreign DNA acquisition by the I-F CRISPR-Cas system requires all components of the interference machinery. Nucleic Acids Res. 2015; 43:10848–10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bikard D., Loot C., Baharoglu Z., Mazel D.. Folded DNA in action: hairpin formation and biological functions in prokaryotes. Microbiol. Mol. Biol. Rev. 2010; 74:570–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Khan S.A. Rolling-circle replication of bacterial plasmids. Microbiol. Mol. Biol. Rev. 1997; 61:442–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Henkin T.M. Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev. 2008; 22:3383–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Liu L.F., Wang J.C.. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA. 1987; 84:7024–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ma J., Wang M.D.. DNA supercoiling during transcription. Biophys. Rev. 2016; 8:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Brown R.V., Hurley L.H.. DNA acting like RNA. Biochem. Soc. Trans. 2011; 39:635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Krasilnikov A.S., Podtelezhnikov A., Vologodskii A., Mirkin S.M.. Large-scale effects of transcriptional DNA supercoiling in vivo. J. Mol. Biol. 1999; 292:1149–1160. [DOI] [PubMed] [Google Scholar]
- 109. Kouzine F., Wojtowicz D., Baranello L., Yamane A., Nelson S., Resch W., Kieffer-Kwon K.R., Benham C.J., Casellas R., Przytycka T.M.et al.. Permanganate/S1 nuclease footprinting reveals Non-B DNA structures with regulatory potential across a mammalian genome. Cell Syst. 2017; 4:344–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Carrasco-Salas Y., Malapert A., Sulthana S., Molcrette B., Chazot-Franguiadakis L., Bernard P., Chedin F., Faivre-Moskalenko C., Vanoosthuyse V.. The extruded non-template strand determines the architecture of R-loops. Nucleic Acids Res. 2019; 47:6783–6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Szlachta K., Thys R.G., Atkin N.D., Pierce L.C.T., Bekiranov S., Wang Y.H.. Alternative DNA secondary structure formation affects RNA polymerase II promoter-proximal pausing in human. Genome Biol. 2018; 19:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Larson M.H., Mooney R.A., Peters J.M., Windgassen T., Nayak D., Gross C.A., Block S.M., Greenleaf W.J., Landick R., Weissman J.S.. A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science. 2014; 344:1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Proshkin S., Rahmouni A.R., Mironov A., Nudler E.. Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science. 2010; 328:504–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Irastortza-Olaziregi M., Amster-Choder O.. Coupled transcription-translation in prokaryotes: an old couple with new surprises. Front. Microbiol. 2020; 11:624830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Goz E., Mioduser O., Diament A., Tuller T.. Evidence of translation efficiency adaptation of the coding regions of the bacteriophage lambda. DNA Res. 2017; 24:333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Dillingham M.S., Kowalczykowski S.C.. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 2008; 72:642–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Radovcic M., Killelea T., Savitskaya E., Wettstein L., Bolt E.L., Ivancic-Bace I.. CRISPR-Cas adaptation in Escherichia coli requires RecBCD helicase but not nuclease activity, is independent of homologous recombination, and is antagonized by 5′ ssDNA exonucleases. Nucleic Acids Res. 2018; 46:10173–10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Modell J.W., Jiang W., Marraffini L.A.. CRISPR-Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature. 2017; 544:101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data were deposited in the NCBI Sequence Read Archive under the BioProject ID PRJNA762861.