


Abstract
CRISPR-Cas systems provide prokaryotic organisms with an adaptive defense mechanism that acquires immunological memories of infections. This is accomplished by integration of short fragments from the genome of invaders such as phages and plasmids, called ‘spacers’, into the CRISPR locus of the host. Depending on their genetic composition, CRISPR-Cas systems can be classified into six types, I-VI, however spacer acquisition has been extensively studied only in type I and II systems. Here, we used an inducible spacer acquisition assay to study this process in the type III-A CRISPR-Cas system of Staphylococcus epidermidis, in the absence of phage selection. Similarly to type I and II spacer acquisition, this type III system uses Cas1 and Cas2 to preferentially integrate spacers from the chromosomal terminus and free dsDNA ends produced after DNA breaks, in a manner that is enhanced by the AddAB DNA repair complex. Surprisingly, a different mode of spacer acquisition from rRNA and tRNA loci, which spans only the transcribed sequences of these genes and is not enhanced by AddAB, was also detected. Therefore, our findings reveal both common mechanistic principles that may be conserved in all CRISPR-Cas systems, as well as unique and intriguing features of type III spacer acquisition.
INTRODUCTION
CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats—CRISPR associated proteins) are prokaryotic adaptive immune systems that protect bacteria and archaea from infection by mobile genetic elements such as viruses and plasmids (1,2). The CRISPR locus is composed of short (∼40 bp) repetitive sequences separated by equally short ‘spacer’ sequences that match the genomes of these invaders (3–5). Spacers are transcribed and processed to generate CRISPR RNAs (crRNAs) (6,7). CrRNAs are incorporated into Cas nucleases, where they are used as guides to facilitate the recognition and destruction of complementary nucleic acids, also known as ‘protospacers’ (8–13). Depending on the cas gene content CRISPR-Cas systems can be classified into six types (I-VI), each of which present different variations in the molecular mechanism of target destruction (14).
Types I, II and III are not only the most abundant (14) but also the most studied CRISPR systems. In type I and type II systems, Cas nucleases use the crRNA guide to find and cleave a DNA target (9,11,12). DNA cleavage requires the presence of a short sequence motif that flanks the target, known as the protospacer-adjacent motif (PAM) (15,16). In contrast, during type III CRISPR-Cas immunity, the crRNA binds to complementary RNA molecules (10). As a consequence of this mechanism, transcription of the target is required for effective immunity (17,18). Upon recognition of a target transcript, two activities of the Cas10 subunit are triggered: single-stranded DNase that degrades accessible ssDNA in a sequence-independent manner (18,19), and a cyclase that produces cyclic oligoadenylates (cA) (20,21). These cA molecules function as second messengers to activate non-specific nucleases that degrade indiscriminately both host and invader nucleic acids (22–24). This activity is responsible for the growth arrest of the host cell and facilitates the clearance of the invader (22). Finally, the Csm3 RNase subunit cuts the transcript protospacer sequence (10), thus eliminating binding of the crRNA to its target and terminating both activities of Cas10 (19). It is not clear whether target recognition by type III effector complexes requires the presence of a PAM. One study investigated targeting by the type III-B system of Pyrococcus furiosus and found a motif requirement for the activation of the ssDNase domain of Cas10 but not for the cleavage of the protospacer RNA (25). On the other hand, experiments that analyzed the sequence space flanking a target of the type III-A system of Staphylococcus epidermidis (26) and Thermus thermophilus (27), failed to find a PAM.
The hallmark of the CRISPR-Cas immune response is the acquisition of new spacer sequences into the CRISPR array during infection (1). This process allows the host to survive infection and adapt to its environment, and therefore is called ‘adaptation’. Spacer acquisition has been extensively studies in type I and type II CRISPR-Cas systems (28), which were shown to have a preference to use accessible dsDNA ends as spacer substrates. In phages, it has been shown that the cos DNA end of the injected viral genome can be a substrate for spacer acquisition, presumably before it mediates genome circularization (29). In the bacterial host, sequences in the vicinity of double strand DNA breaks (DSBs) introduced at the chromosomal terminus during replication are preferentially incorporated as new spacers (29,30). Free DNA ends are processed by the RecBCD (in Gram negative bacteria) or AddAB (Gram positive) complexes to achieve the repair of DSBs through homologous recombination (31). Processing by these complexes stops at chi sequences distributed along the bacterial chromosome and generates 3′ overhangs for homologous recombination (32). As a consequence of this, hotspots of spacer acquisition are limited by free DNA ends and chi sites (29,30). At the molecular level, spacer acquisition is achieved by integration of short genomic segments of the invading genome into the CRISPR array via the activity of the conserved Cas1–Cas2 integrase complex (33). Whereas this complex is sufficient for type I CRISPR adaptation (34), the addition of new spacers requires also cas9 and csn2 during the type II CRISPR-Cas immune response (35,36).
In contrast to type I and II systems, much less is known about spacer acquisition in type III systems. A subset of these systems contain a reverse transcriptase (RT) associated with their CRISPR locus (37). The marine bacterium Marinomonas mediterranea encodes a Cas1 nuclease that is fused to an RT domain (RT-Cas1), and has been used to study spacer acquisition in type III systems (38,39). When self-acquisition events (in the absence of infection, also referred as ‘autoimmunity’) were investigated using overexpression of RT-Cas1 and Cas2 and next-generation sequencing analysis of CRISPR loci, a strong correlation between the transcription and the acquisition levels of the different protospacers sequences was found, which depended on the presence of the RT domain of Cas1. More surprisingly, additional genetics and biochemistry experiments showed that spacers were acquired not only from DNA, but also from RNA molecules (39). Since type III systems require annealing of the crRNA to their target RNA, their spacer (and thus crRNA) sequences must be complementary to the transcript sequence to initiate immunity. While the activity of RT-Cas1 ensures that new spacers arise from transcribed regions, the results did not show an insertion bias that would favor the generation of crRNAs with complementary sequences to the target transcript. Moreover, an enrichment for non-functional spacers (generating the crRNAs with the same sequence as M. mediterranea transcripts) was observed, presumably due to the autoimmunity triggered by functional spacers (40). A more recent study investigated CRISPR adaptation during infection of T. thermophilus with the phage phiFa (27). This thermophile bacterium carries a type III-A CRISPR-cas locus with a canonical cas1 gene. Next generation sequencing of the PCR products corresponding to the expanded CRISPR array showed a marked enrichment of spacers originating from the long terminal repeat (LTR) region, which harbors genes that are expressed early during phiFa lytic cycle. However, while all acquired spacers were shown to be fully functional, the reasons behind their strong selection are unknown.
Here, we studied spacer acquisition by the type III-A CRISPR-Cas system of S. epidermidis. We used a heterologous set up in which this system is expressed from two plasmids in Staphylococcus aureus (41). This strategy allowed us to (i) use a single repeat instead of a full CRISPR array, facilitating the detection of spacer acquisition events with very low frequency, (ii) induce the expression of cas1 and cas2 to control the timing of acquisition and capture the full spectrum of new spacers before selection can enrich spacers according to their immunity properties, and (iii) investigate the importance of different genes for type III-A CRISPR adaptation. We found two modes of spacer acquisition. One is similar to the previously described mechanisms for type I and II systems, where spacers are preferentially acquired from the chromosomal terminus and free dsDNA ends, presumably generated at DSBs, a process that is enhanced by the activity of AddAB. A second mode, exclusive to the S. epidermidis type III-A system, showed high level of spacer acquisition spacers from rRNA and tRNA loci, and was limited to the transcribed region of these genes, without apparent AddAB-dependent expansion of the acquisition hotspot.
MATERIALS AND METHODS
Bacterial strains and growth conditions
Cultivation of S. aureus RN4220 (42) and derivatives JW263 and JW418 (29), and S. aureus Newman (43) was carried out in brain-heart infusion (BHI) medium at 37°C. Media was supplemented with chloramphenicol at 10 μg/ml, erythromycin at 10 μg/ml, kanamycin at 25 μg/ml or spectinomycin at 250 μg/ml for plasmid maintenance. Media was supplemented with 1 mM IPTG to induce expression of genes under the control of the IPTG-inducible Pspank-hy promoter. See Supplementary Table S1 for a full list of bacterial strains and plasmids used in this study.
Plasmid construction
The plasmids used in this study are listed in Supplementary Table S1. The sequences of oligonucleotides used in this study are listed in Supplementary Table S2. The plasmid cloning strategies are listed in Supplementary Table S3.
Inducible adaptation assay and amplification of newly acquired spacers by DR-PCR for high-throughput sequencing
Overnight culture of the indicated S. aureus strains harboring pCas1-2 and pCRISPR/pDR were diluted 1:100 in 50 ml BHI and grown 1 h shaking at 37°C. An uninduced sample was obtained by pelleting 10 ml of the culture and removing supernatant. Pellets were kept at −80°C until all time points were collected. Then, freshly made IPTG was added to the remaining culture before it was placed back shaking at 37°C. Every hour, for 3–5 h post-induction, 10 ml of the culture was pelleted and frozen, OD600 was measured and calibrated back to ∼0.3 by adding the appropriate volume of BHI supplemented with 1 mM IPTG. The remaining of the culture was left shaking at 37°C overnight. The next day, 1.5 ml of the overnight culture was pelleted and frozen. Plasmids were isolated from S. aureus pellets with a modified QIAprep Spin Miniprep Kit (Qiagen) protocol: bacterial cell pellets were resuspended in 250 μl P1 buffer supplemented with lysostaphin at a final concentration of 107 μg/ml (AMBI Products) and incubated at 37°C for 15 min followed by the standard QIAprep protocol. 250 ng of each sample were used as input for DR-PCR, using the Phusion DNA Polymerase (Thermo) with primers NA101/NA102 or NA169/170 for type III-A or type II-A, respectively (see Supplementary Table S2 for a full list of primers used in this study). Primer annealing temperature was set to 64 and 54°C for type III-A and II-A, respectively, and extension time was 10 s. Both plasmid isolation and DR-PCR preparation were performed in a PCR-free room to avoid contaminations. After amplification, 5 ul of DR-PCR products were analyzed by agarose gel electrophoresis to verify successful amplification. The remaining sample underwent cleanup with the MinElute PCR Purification Kit (Qiagen) and size selection using PippnHT 3% cassette with a timed protocol set at extraction between 26 and 35 min. Size selected products were then prepared for sequencing with the TrueSeq Nano DNA Library Prep protocol (Illumina). For maintaining the small sized product, 2.2× Sample Purification Beads (Ilumina) were used after end repair. Illumina libraries underwent high-throughput sequencing with the MiSeq platform.
RNA sequencing
Overnight culture of RN4220 was diluted 1:200 in 10 ml BHI and grown 1:10 h shaking at 37°C. Culture was then pelleted, and supernatant was removed. Pellets were resuspended in 100ul RNase free PBS supplemented with 100 μg/ml lysostaphin (AMBI Products), incubated for 5 min at 37°C, and sarkosyl was added at 1%. RNA was purified from lysed pellets using the Zymo Direct-Zol RNA miniprep plus kit, and genomic DNA was removed by Ambion Turbo DNA-free kit. For the rRNA-depleted sample, Illumina Ribo-Zero rRNA removal (Bacteria) kit was used to remove rRNA. Untreated and rRNA-depleted RNA samples were prepared for sequencing using the TruSeq Stranded mRNA Library prep kit, beginning at the RNA fragmentation step. Illumina libraries underwent high-throughput sequencing with the MiSeq platform.
High throughput sequencing data analysis
Spacers were extracted from MiSeq FASTQ files using a Python code that finds all sequences flanked by two DR sequences. The sequences, location and abundance of all spacers detected in this study are provided in the Supplementary Data File spreadsheet. For generating Weblogos, a python code searching for fully aligned spacers gave an output of spacer sequence, strand, start and end positions, spacer length, 20 bp upstream and downstream of spacer, and number of reads for that unique spacer sequence. Then, spacers were filtered to length of 35 or 30 bp in type III-A or type II-A, respectively, and motif search was performed by WebLogo (version 3.7.4.)
For generating genome alignment maps, all spacers were aligned to the indicated bacterial chromosome using bowtie2. Genome positions covered by aligned spacers were counted and aggregated using the Python pysam package (version 0.15.3). For spacers that aligned to more than one position (n > 1) counts were divided by number of aligned position (1/n). Genome was either divided to 10 or 1 kb bins, or analyzed at a single nucleotide resolution, as indicated in each figure legend. RPM values were calculated as chromosomal reads within a bin per million total aligned reads. RNA-seq sequences underwent the same pipeline of analysis.
RESULTS
Detection of spacer acquisition into the type III-A CRISPR locus of S. epidermidis using an inducible system
We investigated the mechanism of spacer acquisition in the type III-A system of S. epidermidis (Figure 1A). To minimize spacer selection bias, we designed an inducible adaptation assay that enabled the control of the timing of spacer acquisition. This system is composed of two plasmids: (i) pCas1-2, encoding the cas1 and cas2 spacer integrase genes under the IPTG-inducible Pspank-hy promoter (44); and (ii) pCRISPR, encoding all other cas genes of the system along with a single direct repeat (DR) instead of the full CRISPR locus (Figure 1B). This plasmid contains the 700 bp that are located upstream of the first repeat of the CRISPR array in the S. epidermidis genome to preserve potential regulatory regions such as promoter and leader sequences. These plasmids were introduced in S. aureus RN4220 and DNA was isolated at different times after addition of IPTG and used as template for PCR amplification with forward and reverse primers that anneal to the DR facing opposite directions (Figure 1C). This method, known as DR-PCR (45), amplifies a PCR product only after a spacer has integrated into the array and the DR has duplicated (Figure 1C). Next generation sequencing of the PCR products is then used to obtain the sequences and number of reads of the acquired spacer and align them to the bacterial chromosome to generate a pattern of spacer acquisition (Figure 1D). The sequences, location and abundance of all spacers detected in this study are provided in the Supplementary Data File spreadsheet. Using this method, we were able to detect a PCR product (Figure 1E) and obtain the sequences of the new spacers acquired 3 h after induction. On average, spacers were 35.48 ± 0.98 (mean ± SD) bp long, with 54% of the spacers matching the positive strand of the S. aureus chromosome (Figure 1F). Importantly, we were unable to detect any conserved motifs in either of the target flanking sequences (Figure 1G). This observation confirms previous studies on the S. epidermidis (26) and T. thermophilus (27) type III-A systems, which failed to detect a distinct PAM required for the targeting of plasmids and phages, respectively. As a control, we implemented the same experimental design to capture the spacers acquired by the Streptococcus pyogenes type II-A system (35) (Supplementary Figure S1A-D). In contrast to the results obtained for type III-A, we found the expected spacer length of ∼30 bp (Supplementary Figure S1E) and a very strong conservation of the NGG motif downstream of the target sequence (Supplementary Figure S1F), with 52% of the spacers matching to the positive strand (Supplementary Figure S1E).
Figure 1.

An inducible system for studying spacer acquisition by the S. epidermidis type III-A CRISPR-Cas system. (A) Schematic representation of the S. epidermidis type III-A CRISPR-Cas system. (B) Schematic representation of the two-plasmid system used in the inducible adaptation assay for S. epidermidis type III-A CRISPR-Cas. (C) Sensitive DR-PCR design with repeat-annealing primers (arrows) that allows for detection of rare acquisition evens. (D) After next-generation sequencing of DR-PCR products and extraction of spacer sequences, spacers are mapped to the bacterial genome. (E) Agarose gel electrophoresis of DR-PCR products sampled at different time points. (F) Spacers acquired 3 h post induction of the S. epidermidis type III-A acquisition machinery are 35.48 ± 0.98 (mean ± SD) bp long. 54% and 46% match the plus and minus strand of the bacterial genome, respectively. (G) Weblogo analysis of genome-aligned spacers acquired by the S. epidermidis type III-A CRISPR-Cas system, along with 20 bp upstream and downstream. The y-axis was modified (from the default of 2 bits to 0.5 bit) to allow for better resolution, hence the height of the letters does not represent a significantly enriched motif. Only 35 bp long spacers were included in this alignment, n = 1460.
Spacer acquisition by the S. epidermidis type III-A CRISPR-Cas system is concentrated at specific genomic hotspots
We then mapped the spacer reads to the S. aureus RN4220 genome (46) and found that the majority of them match the chromosome terminus region (the dif site, labeled as ‘c’ in Figure 2A), similar to the case for the reads coming from spacers acquired by the type II-A system, as previously reported (29) (Figure 2B). However, type III-A spacer sequences mapped to several distinct hotspots within the bacterial genome, many of which corresponded to the five loci encoding ribosomal RNAs (rRNAs) (labeled as ‘a’, ‘b’, ‘d’, ‘e’, ‘f’ in Figure 2A). It is important to note that the sequences of the 5S, 16S and 23S rRNA genes are highly similar between the five rRNA loci of S. aureus. Therefore, we cannot determine the exact origin of spacer reads sequences that match to all of them. Instead, we distributed spacers into multiple identical positions (n), assigning 1/n of the total number of reads to each position. RNA sequencing (RNA-seq) of S. aureus RN4220, with or without rRNA depletion, confirmed both the identity of these loci as well as their high level of transcription (Figure 2C). Examination of both the RNA-seq and the spacer acquisition pattern at single nucleotide resolution revealed that the majority of spacers match the 5S and 16S rRNA genes as well as associated tRNA genes, however less spacers were derived from the 23S rRNA gene (Figure 2D, Supplementary Figures S2A-D). Intriguingly, an additional region that was highly enriched for type III-A spacers was a tRNA cluster that is not associated with any of the rRNA loci (Supplementary Figure S2E). However, an adjacent locus positioned ∼20 kb upstream on the same strand and transcribed at comparable levels to the tRNA cluster (Supplementary Figure S2F), did not show high acquisition levels (Supplementary Figure S2E, F). Close inspection of the spacers matching tRNA genes revealed highly variable levels of acquisition between and within loci (Supplementary Figure S3).
Figure 2.

Spacer acquisition by the S. epidermidis type III-A CRISPR-Cas system preferentially occurs at the chromosome terminus, rRNA loci and tRNA clusters. (A) Abundance (in reads per million, RPM) of spacers acquired 3 h after induction of the S. epidermidis type III-A acquisition machinery, mapped to the bacterial chromosome (10 kb bins). Peak labeled ‘c’ corresponds to the dif site, peaks labeled ‘a’, ‘b’, ’d’, ‘e’, ‘f’ correspond to the five rRNA loci in the S. aureus genome. (B) Abundance (RPM) of spacer acquired 3 h post induction of the S. pyogenes type II-A acquisition machinery, mapped to the bacterial chromosome (10 kb bins). Peak labeled ‘c’ corresponds to the dif site. (C) Abundance (RPM) of S. aureus RN4220 RNA-seq reads, of either an rRNA depleted sample (purple) or an untreated sample (pink), mapped to the bacterial chromosome (10 kb bins). Peaks labeled ‘a’, ‘b’, ’d’, ‘e’, ‘f’ correspond to the five rRNA loci in the S. aureus genome. (D) Details of the RNA-seq (rRNA depleted sample, purple; untreated sample, pink), type III-A (green) and type II-A (blue) spacer acquisition data (RPM), obtained 3 h post-induction, for the ribosomal RNA locus rrnE (peak ‘f’ in (B)) of S. aureus RN4220.
To evaluate the possibility of an association between transcription and spacer acquisition, we aggregated the number of RNA-seq or spacer reads into 1 kb bins across the S. aureus genome, without accounting for strand directionality. We found that the genomic coverage by type III-A spacers is significantly correlated to level of RNA expression (P < 10–10; Figure 3A). Such correlation was not observed when examining the type II-A spacer coverage (not significant; Figure 3B). Notably, type II-A and type III-A spacers also correlate, which can be explained by their common acquisition hotspot at the dif site (Supplementary Figure S4A). Next, we tested whether the correlation between type III-A spacers and RNA expression leads to the generation of crRNA complementary to target transcripts; i.e., are able to trigger CRISPR immunity. We compared all combinations of forward and reverse reads (those that match the plus and minus strand of the genome, respectively) from RNA-seq and and type III-A spacer data, but we failed to detect a directionality preference (Figure 3C, Supplementary Figure S4B). Altogether, these data showed a general correlation between the transcription and acquisition of a DNA sequence, with some, but not all, highly transcribed regions forming hotspots of type III-A spacer acquisition.
Figure 3.

Correlation between spacer acquisition and transcription of genomic DNA during type III-A CRISPR adaptation. (A) Correlation plot of genomic coverage (1 kb bins), comparing RNA-seq reads and spacer reads after 3 h of induction of the S. epidermidis type III-A acquisition machinery. (B) Correlation plot of genomic coverage (1 kb bins), comparing between RNA-seq reads and spacer reads after 3 h of induction of the S. pyogenes type II-A acquisition machinery. (C) R-values of correlation plots (Supplementary Figure S4B), comparing between type III-A spacers and RNA-seq next-generation sequencing reads, each either matching the plus strand (+) or the minus strand (–) of the bacterial genome.
The type III-A Cas1–Cas2 integrase is responsible for the spacer acquisition pattern
The S. epidermidis type III-A locus harbors genes that encode four proteins or protein complexes with distinct functions: cas1 and cas2 encode the spacer integrase complex (34), cas10 and csm2-5 encode the crRNA-guided Csm effector complex (18,47), cas6 processes crRNAs from longer precursor transcripts (41), and csm6 produces the auxiliary RNase of this CRISPR system (22,48) (Figure 1A). To dissect the contribution of each of these to the type II-A spacer acquisition pattern, we eliminated their function by mutating the pCRISPR plasmid and repeated the spacer acquisition assay. First, we eliminated RNase activity of Csm6, by introducing two mutations in its active site (R364A, H369A) generating ‘dead’ Csm6, or dCsm6 (pCRISPR-dcsm6) (48) (Figure 4A). The elimination of Csm6′s RNase activity, however, did not change the pattern of early-acquired spacers (Figure 4B, C).
Figure 4.
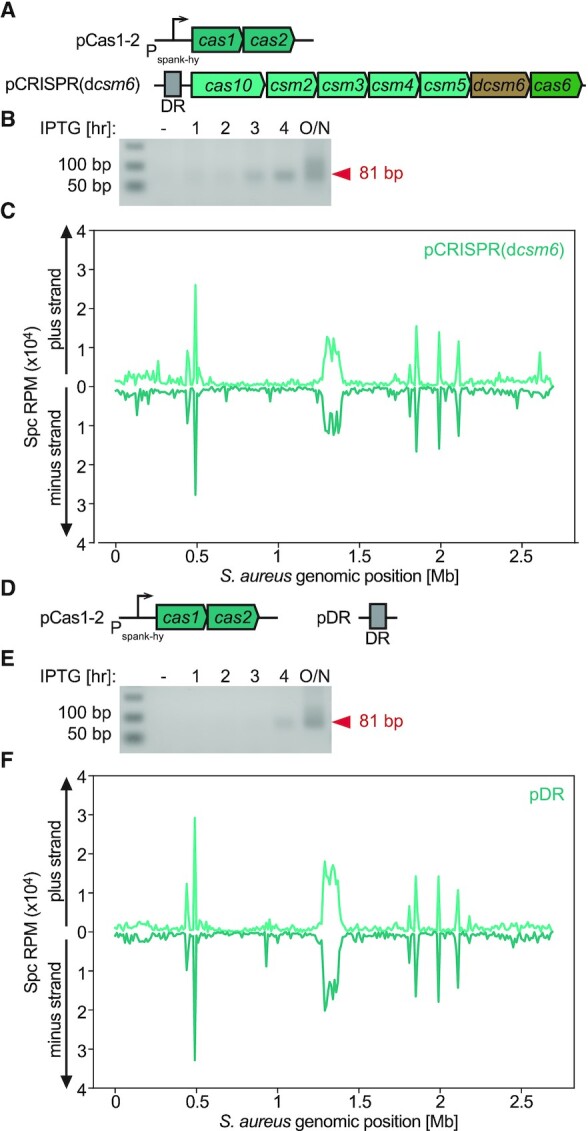
Cas1 and Cas2 are sufficient to determine the observed S. epidermidis type III-A acquisition pattern. (A) Schematic representation of the two-plasmid system used in the inducible adaptation assay for S. epidermidis type III-A dcsm6 CRISPR-Cas system. (B) Agarose gel electrophoresis DR-PCR products at different time points post induction of CRISPR adaptation in the presence of pCRISPR(dcsm6). (C) Abundance (RPM) of spacers acquired 3 h post induction of the S. epidermidis type III-A acquisition machinery in host cells harboring pCRISPR(dcsm6), mapped to the bacterial chromosome (10 kb bins). (D) Schematic representation of the two-plasmid system used in the inducible adaptation assay for S. epidermidis type III-A pDR (i.e. Δcas10-6 CRISPR-Cas system). (E) Agarose gel electrophoresis DR-PCR products at different time points post induction of CRISPR adaptation in the presence of pDR. (F) Abundance (RPM) of spacer acquired 3 h post induction of the S. epidermidis type III-A acquisition machinery in host cells harboring pDR, mapped to the bacterial chromosome (10 kb bins).
Next, we evaluated the impact of the Csm effector complex and cas6 on the pattern of spacer acquisition. We therefore deleted all cas genes from pCRISPR leaving just a mini array (pDR, Figure 4D). After performing the spacer acquisition assay in this strain we detected the same pattern as the one for the strain harboring the full CRISPR-Cas system (Figure 4E-F). We corroborated these findings in the clinical isolate S. aureus Newman (43), which was transformed with pCas1-2 and pDR (Supplementary Figure S5), indicating that the observed pattern is not unique to the RN4220, laboratory-generated, strain. These results demonstrate that Cas1 and Cas2 are sufficient to establish the observed S. epidermidis type III-A spacer acquisition pattern around the dif site and rRNA/tRNA loci. In addition, since we could not detect any spacer acquisition events with our sensitive DR-PCR approach when cas1-cas2 were not induced (Figures 1B, 4B, E), we can also conclude that the integrase is not only sufficient but also necessary for the expansion of the type III-A CRISPR array.
Autoimmunity influences the type III-A spacer acquisition pattern
The acquisition of spacers from the bacterial DNA would trigger both the attack of the chromosome by Csm complex (17) as well as the activation of cell dormancy by Csm6 (22). We limited these toxic events by sampling shortly (3 h) after induction of Cas1–Cas2. We wondered about how prolonged targeting would affect the spacer acquisition pattern and therefore collected data at ∼24 h after the addition of IPTG. We found a substantial reduction of the rRNA/tRNA derived peaks (Figure 5A). In contrast, the enrichment around the dif site was not diminished, suggesting that these spacers do not provide robust autoimmunity. This could be explained by the lack of uniform transcription across the dif site (Figure 5B), which would limit type III-A targeting in this region, allowing these spacers to be fixed in the bacterial population. Surprisingly, the enrichment for type II-A spacers around the terminus did not change (Supplementary Figure S6A), raising the possibility of the existence of an unknown mechanism that prevents targeting by different CRISPR types at these sites.
Figure 5.

Autoimmunity shapes the type III-A spacer acquisition pattern. (A) Abundance (RPM) of spacers acquired overnight post induction of the S. epidermidis type III-A acquisition machinery, mapped to the bacterial chromosome (10 kb bins). (B) Abundance (RPM) of S. aureus RN4220 RNA-seq reads of either an rRNA depleted sample (purple) or an untreated sample (pink), in the vicinity of the dif site, single nucleotide resolution. (C) Abundance (RPM) of spacers acquired overnight post induction of the S. epidermidis type III-A acquisition machinery in hosts carrying pCRISPR(dcsm6), mapped to the bacterial chromosome (10 kb bins). (D) Abundance (RPM) of spacers acquired overnight post induction of the S. epidermidis type III-A acquisition machinery in the presence of pDR, mapped to the bacterial chromosome (10 kb bins).
The negative selection of spacers matching the rRNA/tRNA genes could be in principle a consequence of their ability to trigger either Cas10-mediated DNA degradation or Csm6-mediated growth arrest. To evaluate each of these possibilities, we obtained the spacer acquisition patterns after continued targeting in the presence of pCRISPR(dcsm6). We observed a similar pattern as the one obtained with the full CRISPR system (Figure 5C), suggesting that the Cas10 ssDNase activity is responsible for the toxicity of the spacers derived from these highly transcribed regions. In agreement with these findings, the elimination of the targeting mediated by the Csm complex, using pDR, restored the rRNA/tRNA-derived hotspots of spacer acquisition (Figure 5D). These results suggest that many of the early acquired spacers are not fixed in the bacterial population due to their ability to activate the Csm effector complex. To investigate this further, we compared combinations of forward and reverse RNA-seq reads and type III-A spacer reads sampled ∼24 h after IPTG induction. We detected stronger correlations (higher R-values) for reads matching the same strand of the genomic DNA; i.e., when the crRNA is not complementary to the transcript and cannot trigger immunity, in the pCRISPR and pCRISPR(dcsm6), but not pDR, samples (Supplementary Figure S6B-D). These findings highlight the value of inducible CRISPR adaptation assays to obtain patterns of spacer acquisition that exclusively reflect the activity and preferences of the adaptation machinery, without the skew introduced by the targeting properties of the integrated spacers.
AddAB facilitates type III-A spacer acquisition from free dsDNA ends
In type I and II systems, spacers are preferentially acquired from free DNA ends (29,30). For both systems, the acquisition hotspots at the chromosomal terminus (the dif and ter sites for the S. aureus and Escherichia coli hosts, respectively) have been attributed to the presence of DSBs (which generate two free dsDNA ends) that occur during replisome stalling at these sites. For both systems, the acquisition hotspot is limited by the chi sites flanking the DSBs, a result of the involvement of the host DNA repair systems in the generation of substrates for spacer acquisition (AddAB and RecBCD for S. aureus and E. coli, respectively) (29,30).
The presence of a similar hotspot at the dif site in the type III-A spacer acquisition pattern suggested a similar mechanism for this system. To investigate if AddAB is involved, we performed the inducible adaptation assay using a host strain harboring an inactivating mutation in the nuclease domain of AddA (addan) (29). In this genetic background, spacer acquisition was far less efficient and we could not detect a PCR product containing the newly acquired spacers at the 3 h time point with either type III-A (only expressing Cas1–Cas2) or type II-A CRISPR-Cas systems. For type III-A, a PCR band was detected after allowing acquisition to happen overnight, however for type II-A we had to extract and sequence an ‘invisible band’ (45) (Supplementary Figure S7A). For both systems, in the absence of a functional AddAB complex there was a marked reduction in the acquisition of spacers from the genome (Figure 6A, Supplementary Figure S7B). We also looked for the presence of chi sites limiting acquisition from the dif site. We found that in wild-type hosts this hotspot abruptly drops in the proximity of its flanking chi sites (Figure 6B, Supplementary Figure S7C). In the mutant host, however, chi sites no longer confined the pattern of spacer acquisition. Finally, to demonstrate that free DNA ends enhance type III-A CRISPR adaptation, we used a strain engineered to have an I-sceI cleavage site (29), and performed an inducible-adaptation assay with either a plasmid encoding an IPTG-inducible I-SceI endonuclease or an empty plasmid as a control. After addition of the inducer to the staphylococcal cultures, both type III-A and type II-A spacer acquisition patterns showed an additional hotspot at the I-sceI site (Figure 6C, Supplementary Figure S7D), which was limited by chi sites (Figure 6D, Supplementary Figure S7E). Taken together our data shows that, similarly to type I and type II CRISPR-Cas systems, free DNA ends promotes spacer acquisition by the type III-A CRISPR-Cas system, a process that is stimulated by the AddAB nuclease activity.
Figure 6.
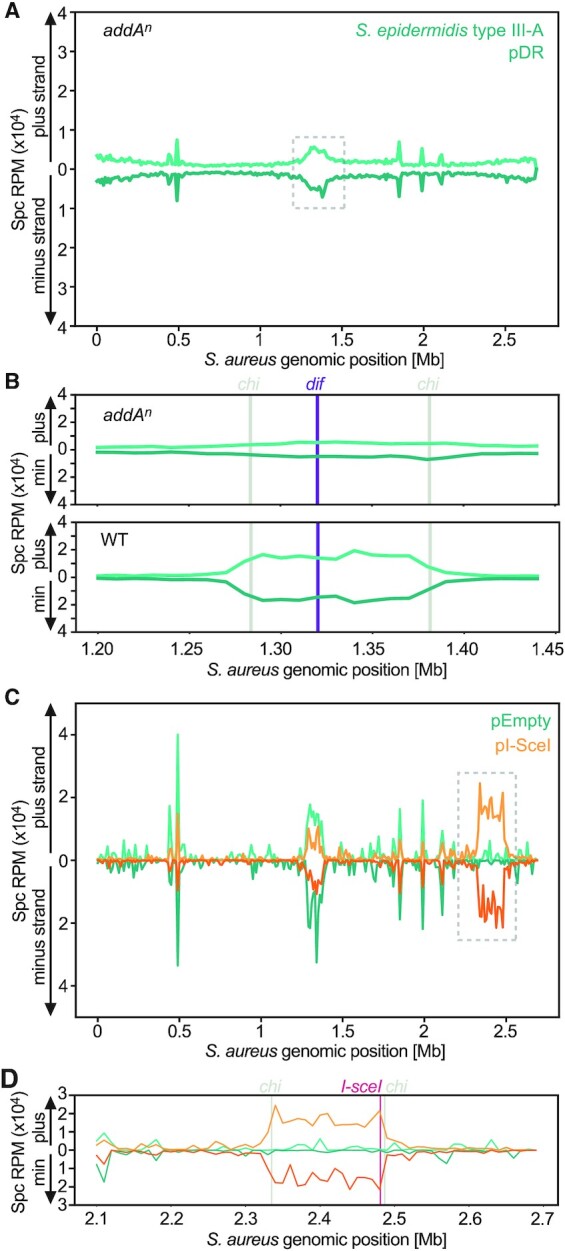
Adaptation in the S. epidermidis type III-A CRISPR-Cas system is facilitated by the AddAB helicase-nuclease and stimulated by free DNA ends. (A) Abundance (RPM) of spacers acquired in an addan mutant host, 24 h post induction of the S. epidermidis type III-A acquisition machinery in the presence of pDR, mapped to the bacterial chromosome (10 kb bins). (B) Details of the data within the dashed square shown in (A) (top panel) and for the same region of the pattern shown in Figure 5D (bottom panel), focusing on the chromosomal terminus (dif, purple line) and flanking chi sites (grey lines); (10 kb bins). (C) Abundance (RPM) of spacer acquired 3 h post induction of the S. epidermidis type III-A acquisition machinery in the presence of pDR, along with either an empty plasmid (pEmpty, green) or an IPTG-inducible I-SceI endonuclease (pI-SceI, orange), mapped to the bacterial chromosome (10 kb bins). (D) Details of the data within the dashed square shown in (C), focusing on the I-SceI cleavage site (red line) and flanking chi sites (grey lines); (10 kb bins).
DISCUSSION
Here, we studied spacer acquisition into the type III-A CRISPR-cas locus of S. epidermidis. We used an inducible system that enabled us to control the timing of acquisition and sample the spacer content of the bacterial population at different times after induction. Using a similar setup, we compared the results with the previously characterized type II-A system of S. pyogenes, and found some common characteristics as well as distinct features. Both systems can use free dsDNA ends as substrates for spacer acquisition, and this process is enhanced by the activity of the DNA-repair machinery of the cell (AddAB in our experimental system). This is also the case for type I CRISPR-Cas systems (30), and therefore seems to be a general mechanism across diverse CRISPR-Cas systems, possibly due to the high conservation of the Cas1–Cas2 integrase (14), the complex thought to interact with pre-spacer DNA substrates (49,50). A consequence of this general mechanism is the acquisition of new spacer sequences from the chromosomal terminus by types I, II and III systems, as this region is subject to DSBs during replication termination (29,30). However, using our inducible spacer acquisition assay, we were able to demonstrate that the spacers acquired from this region by both the S. epidermidis type III-A and the S. pyogenes type II-A CRISPR systems, mediate inefficient self-targeting, as cells harboring these spacers are not negatively selected after prolonged induction of spacer acquisition. A possible explanation for the lack of targeting could be the abundance of DNA repair machinery as well as ssDNA templates for homologous recombination at the dif site (51), which could efficiently fix the DNA damage inflicted by the crRNA-guided Cas nuclease. If so, this would represent an evolved mechanism of the host to lower the cost of CRISPR autoimmunity at the chromosomal terminus. In addition to this possibility, the absence of uniform transcription across the dif site limits the activation of the Csm complexes and thus prevents type III-A targeting at the chromosomal terminus.
We also found several features of the spacer acquisition mediated by the S. epidermidis type III-A system that are not shared with all CRISPR types. One regards to the minimal module required for this process. Similar to type I systems (34,52), we found that cas1, cas2, and a leader-repeat sequence are necessary and sufficient for the expansion of the type III-A CRISPR array. This is different for type II systems, which require additional cas genes, namely csn2 and cas9 (35,36). Another unique characteristic revealed by this study is the lack of a sequence motif flanking the target of the spacers acquired by the type III-A system. This result corroborates previous studies that failed to establish a PAM requirement for efficient type III CRISPR immunity (26,27) and contrast with the strong presence of PAMs for the targets of type I and II spacers (35,53,54). A third difference is that, as opposed to type II-A spacer acquisition, there is a general correlation between the transcription of a given DNA region and the ability of the type III-A machinery to acquire spacers from it. While this does not mean that the new spacers are inserted in the appropriate direction to generate a functional crRNA (i.e. complementary to the transcript), nor that highly transcribed genes are always the best substrates for spacer acquisition (see below), this observation raises the possibility of an unknown mechanism that links these two cellular processes. A similar observation was reported for the type III-B CRISPR system of M. mediterranea (39,40), which harbors RT-Cas1. It was shown that RT-mediated spacer acquisition was correlated to expression, but the direction of integration appeared to be random (39).
The most striking unique attribute of type III-A spacer acquisition is the presence of hotspots at the rRNA and tRNA loci. Although much less pronounced than the hotspots detected in our system, high frequency of spacer acquisition from rRNA genes has been shown by the type I-A and I-G CRISPR systems of Pyrococcus furiosus (55). It was proposed that the high transcription of rRNA genes could lead to the formation of R-loops and DNA-nicking, which can result in DSBs upon replication and thus create substrates for spacer acquisition. However, if DSBs are the only driver of spacer acquisition for the type III-A system, we would expect the type II-A system to display enriched acquisition at these sites as well, as we observed at the I-SceI cleavage sites. In contrast, the rRNA and tRNA hotspots were exclusively observed in the type III-A system, implying that these spacers are acquired by a different mechanism, which does not stimulate acquisition by the type II-A system. Importantly, in contrast to the spacers acquired at the dif site, the rRNA/tRNA derived spacers were negatively selected in the presence of the Csm effector complex, a result that provides an explanation for the previous lack of detection of type III spacers that can be mapped to these loci (56). In addition, we found that strong DNA transcription is not sufficient to promote the high frequency of spacer acquisition by the type III-A system. One example of this is the variation in acquisition observed across tRNA genes, which are similarly transcribed, sometimes as a single transcriptional unit, yet there are different levels of spacer acquisition from these loci. We wonder whether tRNA processing and maturation (57,58), which could occur co-transcriptionally, can affect the access of the acquisition complex to the DNA of these genes. Another case is the 23S rRNA gene, which is immediately adjacent to the 16S gene and has similarly high transcription levels, but it is sampled by the CRISPR acquisition machinery much less often. Again, we wonder if variations in the co-transcriptional assembly of ribosomes at the different rRNA loci (59), could dissimilarly hinder the accessibility of this region to the acquisition machinery. These instances suggest that there is an interplay between the preference and the accessibility of the substrate DNA that dictates which sequences are incorporated as new spacers into the CRISPR array. While this manuscript was in revision, other work showed that type III-A spacer acquisition in Streptococcus thermophilus is facilitated by ssDNA secondary structures (60). It is possible that the DNA in rRNA and tRNA genes of staphylococci could form such secondary structures to enhance acquisition from these loci. More generally, other mechanisms that could link transcription of a given locus with spacer acquisition are the presence of regulatory cis sequences important for acquisition and/or host trans factors that link the acquisition machinery with specific promoters or RNAP. Such mechanisms could also influence the acquisition from each rRNA gene copy, which differences, if any, could not be detected in our study due to the impossibility to map spacer reads to identical genomic sequences. Interestingly, as opposed from the ∼150 kb-long spacer acquisition hotspots that originate from free dsDNA ends at the dif and I-sceI cleavage sites, the hotspots mapping to rRNA and tRNA genes were only ∼10 kb in length, only spanned the transcribed sequences of the DNA, and did not seem to be expanded by AddAB activity; i.e., are not limited by chi sites. The caveat of our analysis, which arbitrarily mapped spacer reads to all nearly-identical rDNA loci, also needs to be mentioned. There is the possibility that some of these loci are a more important source of spacers than others. If that is the case, this could constitute a useful tool to investigate the mechanistic details of this unique mode of spacer acquisition.
In conclusion, although admittedly engineered, our inducible CRISPR adaptation system allowed us to directly compare between two different CRISPR-Cas systems, type III-A and type II-A, using different genetic backgrounds for the same host with the same controlled induction conditions. Our results revealed two distinct forms of type III-A spacer acquisition. One originating at DSBs within poorly transcribed DNA, enhanced by AddAB and limited by chi sites, and another targeting highly transcribed rRNA and tRNA genes and independent of AddAB activity. Future work on hosts that naturally harbor type III-A CRISPR-Cas systems will corroborate and possibly further develop our findings.
DATA AVAILABILITY
Datasets generated from Illumina sequencing has been made publicly available at NCBI, BioProject accession number PRJNA769698. The authors declare that all other data supporting the findings of this study are available within the article and its Supplementary Information files, or are available from the authors upon request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the members of the Marraffini laboratory for scientific discussions and feedback. We thank Pascal Maguin for plasmid pPM145 and for his cloning advice.
Contributor Information
Naama Aviram, Laboratory of Bacteriology, the Rockefeller University, 1230 York Ave, New York, NY 10065, USA.
Ashley N Thornal, Laboratory of Bacteriology, the Rockefeller University, 1230 York Ave, New York, NY 10065, USA.
David Zeevi, Department of Plant and Environmental Sciences, Weizmann Institute of Science, Rehovot 7610001, Israel.
Luciano A Marraffini, Laboratory of Bacteriology, the Rockefeller University, 1230 York Ave, New York, NY 10065, USA; Howard Hughes Medical Institute, The Rockefeller University, 1230 York Ave, New York, NY 10065, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health Director's Pioneer Award [1DP1GM128184-01]; Burroughs Wellcome Fund PATH Award (to L.A.M.); L.A.M. is an Investigator of the Howard Hughes Medical Institute; N.A. is a Junior Fellow of the Simons Society of Fellows; Simons Foundation [578759]; D.Z. is supported by a research grant from the Center for New Scientists at the Weizmann Institute of Science. Funding for open access charge: NIH.
Conflict of interest statement. L.A.M. is a founder and advisor of Intellia Therapeutics, CRISPR Biotechnologies and Eligo Biosciences.
REFERENCES
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P.. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007; 315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 2. Marraffini L.A., Sontheimer E.J.. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008; 322:1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bolotin A., Quinquis B., Sorokin A., Ehrlich S.D.. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005; 151:2551–2561. [DOI] [PubMed] [Google Scholar]
- 4. Mojica F.J., Diez-Villasenor C., Garcia-Martinez J., Soria E.. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005; 60:174–182. [DOI] [PubMed] [Google Scholar]
- 5. Pourcel C., Salvignol G., Vergnaud G.. CRISPR elements in yersiniapestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005; 151:653–663. [DOI] [PubMed] [Google Scholar]
- 6. Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J.. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hale C., Kleppe K., Terns R.M., Terns M.P.. Prokaryotic silencing (psi)RNAs in Pyrococcusfuriosus. RNA. 2008; 14:2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B., Shmakov S., Makarova K.S., Semenova E., Minakhin L.et al.. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016; 353:aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gasiunas G., Barrangou R., Horvath P., Siksnys V.. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E2579–E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hale C.R., Zhao P., Olson S., Duff M.O., Graveley B.R., Wells L., Terns R.M., Terns M.P.. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009; 139:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Westra E.R., van Erp P.B., Kunne T., Wong S.P., Staals R.H., Seegers C.L., Bollen S., Jore M.M., Semenova E., Severinov K.et al.. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by cascade and cas3. Mol. Cell. 2012; 46:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A.et al.. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015; 163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Makarova K.S., Wolf Y.I., Iranzo J., Shmakov S.A., Alkhnbashi O.S., Brouns S.J.J., Charpentier E., Cheng D., Haft D.H., Horvath P.et al.. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020; 18:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deveau H., Barrangou R., Garneau J.E., Labonte J., Fremaux C., Boyaval P., Romero D.A., Horvath P., Moineau S.. Phage response to CRISPR-encoded resistance in streptococcusthermophilus. J. Bacteriol. 2008; 190:1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Semenova E., Jore M.M., Datsenko K.A., Semenova A., Westra E.R., Wanner B., van der Oost J., Brouns S.J., Severinov K.. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:10098–10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goldberg G.W., Jiang W., Bikard D., Marraffini L.A.. Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature. 2014; 514:633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Samai P., Pyenson N., Jiang W., Goldberg G.W., Hatoum-Aslan A., Marraffini L.A.. Co-transcriptional DNA and RNA cleavage during type III CRISPR-Cas immunity. Cell. 2015; 161:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kazlauskiene M., Tamulaitis G., Kostiuk G., Venclovas C., Siksnys V.. Spatiotemporal control of type III-A CRISPR-Cas immunity: coupling DNA degradation with the target RNA recognition. Mol. Cell. 2016; 62:295–306. [DOI] [PubMed] [Google Scholar]
- 20. Kazlauskiene M., Kostiuk G., Venclovas C., Tamulaitis G., Siksnys V.. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science. 2017; 357:605–609. [DOI] [PubMed] [Google Scholar]
- 21. Niewoehner O., Garcia-Doval C., Rostol J.T., Berk C., Schwede F., Bigler L., Hall J., Marraffini L.A., Jinek M.. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature. 2017; 548:543–548. [DOI] [PubMed] [Google Scholar]
- 22. Rostol J.T., Marraffini L.A.. Non-specific degradation of transcripts promotes plasmid clearance during type III-A CRISPR-Cas immunity. Nat Microbiol. 2019; 4:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rostol J.T., Xie W., Kuryavyi V., Maguin P., Kao K., Froom R., Patel D.J., Marraffini L.A.. The card1 nuclease provides defence during type III CRISPR immunity. Nature. 2021; 590:624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sheppard N.F., Glover C.V. 3rd, Terns R.M., Terns M.P.. The CRISPR-associated csx1 protein of pyrococcus furiosus is an adenosine-specific endoribonuclease. RNA. 2016; 22:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elmore J.R., Sheppard N.F., Ramia N., Deighan T., Li H., Terns R.M., Terns M.P.. Bipartite recognition of target RNAs activates DNA cleavage by the type III-B CRISPR-Cas system. Genes Dev. 2016; 30:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pyenson N.C., Gayvert K., Varble A., Elemento O., Marraffini L.A.. Broad targeting specificity during bacterial type III CRISPR-Cas immunity constrains viral escape. Cell Host Microbe. 2017; 22:343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Artamonova D., Karneyeva K., Medvedeva S., Klimuk E., Kolesnik M., Yasinskaya A., Samolygo A., Severinov K.. Spacer acquisition by type III CRISPR-Cas system during bacteriophage infection of thermus thermophilus. Nucleic Acids Res. 2020; 48:9787–9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McGinn J., Marraffini L.A.. Molecular mechanisms of CRISPR-Cas spacer acquisition. Nat. Rev. Microbiol. 2018; 17:7–12. [DOI] [PubMed] [Google Scholar]
- 29. Modell J.W., Jiang W., Marraffini L.A.. CRISPR-Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature. 2017; 544:101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Levy A., Goren M.G., Yosef I., Auster O., Manor M., Amitai G., Edgar R., Qimron U., Sorek R.. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015; 520:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dillingham M.S., Kowalczykowski S.C.. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 2008; 72:642–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chedin F., Handa N., Dillingham M.S., Kowalczykowski S.C.. The AddAB helicase/nuclease forms a stable complex with its cognate chi sequence during translocation. J. Biol. Chem. 2006; 281:18610–18617. [DOI] [PubMed] [Google Scholar]
- 33. Nunez J.K., Lee A.S., Engelman A., Doudna J.A.. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015; 519:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yosef I., Goren M.G., Qimron U.. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichiacoli. Nucleic Acids Res. 2012; 40:5569–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heler R., Samai P., Modell J.W., Weiner C., Goldberg G.W., Bikard D., Marraffini L.A.. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature. 2015; 519:199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wei Y., Terns R.M., Terns M.P.. Cas9 function and host genome sampling in type II-A CRISPR-Cas adaptation. Genes Dev. 2015; 29:356–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mohr G., Silas S., Stamos J.L., Makarova K.S., Markham L.M., Yao J., Lucas-Elio P., Sanchez-Amat A., Fire A.Z., Koonin E.V.et al.. A reverse transcriptase-cas1 fusion protein contains a cas6 domain required for both CRISPR RNA biogenesis and RNA spacer acquisition. Mol. Cell. 2018; 72:700–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Silas S., Makarova K.S., Shmakov S., Paez-Espino D., Mohr G., Liu Y., Davison M., Roux S., Krishnamurthy S.R., Fu B.X.H.et al.. On the origin of reverse transcriptase-using CRISPR-Cas systems and their hyperdiverse, enigmatic spacer repertoires. MBio. 2017; 8:e00897-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Silas S., Mohr G., Sidote D.J., Markham L.M., Sanchez-Amat A., Bhaya D., Lambowitz A.M., Fire A.Z.. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science. 2016; 351:aad4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Silas S., Lucas-Elio P., Jackson S.A., Aroca-Crevillen A., Hansen L.L., Fineran P.C., Fire A.Z., Sanchez-Amat A.. Type III CRISPR-Cas systems can provide redundancy to counteract viral escape from type i systems. Elife. 2017; 6:e27601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hatoum-Aslan A., Maniv I., Samai P., Marraffini L.A.. Genetic characterization of antiplasmid immunity through a type III-A CRISPR-Cas system. J. Bacteriol. 2014; 196:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kreiswirth B.N., Lofdahl S., Betley M.J., O’Reilly M., Schlievert P.M., Bergdoll M.S., Novick R.P.. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983; 305:709–712. [DOI] [PubMed] [Google Scholar]
- 43. Baba T., Bae T., Schneewind O., Takeuchi F., Hiramatsu K.. Genome sequence of staphylococcusaureus strain newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 2008; 190:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Britton R.A., Eichenberger P., Gonzalez-Pastor J.E., Fawcett P., Monson R., Losick R., Grossman A.D.. Genome-wide analysis of the stationary-phase sigma factor (sigma-H) regulon of bacillus subtilis. J. Bacteriol. 2002; 184:4881–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McKenzie R.E., Almendros C., Vink J.N.A., Brouns S.J.J.. Using CAPTURE to detect spacer acquisition in native CRISPR arrays. Nat. Protoc. 2019; 14:976–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nair D., Memmi G., Hernandez D., Bard J., Beaume M., Gill S., Francois P., Cheung A.L.. Whole-genome sequencing of staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J. Bacteriol. 2011; 193:2332–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hatoum-Aslan A., Samai P., Maniv I., Jiang W., Marraffini L.A.. A ruler protein in a complex for antiviral defense determines the length of small interfering CRISPR RNAs. J. Biol. Chem. 2013; 288:27888–27897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiang W., Samai P., Marraffini L.A.. Degradation of phage transcripts by CRISPR-associated RNases enables type III CRISPR-Cas immunity. Cell. 2016; 164:710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim S., Loeff L., Colombo S., Jergic S., Brouns S.J.J., Joo C.. Selective loading and processing of prespacers for precise CRISPR adaptation. Nature. 2020; 579:141–145. [DOI] [PubMed] [Google Scholar]
- 50. Xiao Y., Ng S., Nam K.H., Ke A.. How type II CRISPR-Cas establish immunity through Cas1–Cas2-mediated spacer integration. Nature. 2017; 550:137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wiktor J., Gynna A.H., Leroy P., Larsson J., Coceano G., Testa I., Elf J.. RecA finds homologous DNA by reduced dimensionality search. Nature. 2021; 597:426–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Arslan Z., Hermanns V., Wurm R., Wagner R., Pul U.. Detection and characterization of spacer integration intermediates in type I-E CRISPR-Cas system. Nucleic Acids Res. 2014; 42:7884–7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee H., Zhou Y., Taylor D.W., Sashital D.G.. Cas4-dependent prespacer processing ensures high-fidelity programming of CRISPR arrays. Mol. Cell. 2018; 70:48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shiimori M., Garrett S.C., Graveley B.R., Terns M.P.. Cas4 nucleases define the PAM, length, and orientation of DNA fragments integrated at CRISPR loci. Mol. Cell. 2018; 70:814–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shiimori M., Garrett S.C., Chambers D.P., Glover C.V.C. 3rd, Graveley B.R., Terns M.P.. Role of free DNA ends and protospacer adjacent motifs for CRISPR DNA uptake in Pyrococcus furiosus. Nucleic Acids Res. 2017; 45:11281–11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shmakov S.A., Sitnik V., Makarova K.S., Wolf Y.I., Severinov K.V., Koonin E.V.. The CRISPR spacer space is dominated by sequences from species-specific mobilomes. MBio. 2017; 8:e01397-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nicholson A.W. Function, mechanism and regulation of bacterial ribonucleases. FEMS Microbiol. Rev. 1999; 23:371–390. [DOI] [PubMed] [Google Scholar]
- 58. de Crecy-Lagard V., Jaroch M.. Functions of bacterial tRNA modifications: from ubiquity to diversity. Trends Microbiol. 2021; 29:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Davis J.H., Williamson J.R.. Structure and dynamics of bacterial ribosome biogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017; 372:20160181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang X., Garrett S., Graveley B.R., Terns M.P.. Unique properties of spacer acquisition by the type III-A CRISPR-Cas system. Nucleic Acids Res. 2021; 10.1093/nar/gkab1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets generated from Illumina sequencing has been made publicly available at NCBI, BioProject accession number PRJNA769698. The authors declare that all other data supporting the findings of this study are available within the article and its Supplementary Information files, or are available from the authors upon request.