

Abstract
Dysregulated type I interferon responses play crucial roles in the development of multiple forms of autoimmunity. Many patients with lupus, systemic sclerosis, Sjogren’s syndrome, and dermatomyositis demonstrate enhanced type I IFN signaling. Type I IFN excess is associated with disease severity, autoantibodies, and could potentially predict response to newer therapies targeting type I IFN pathways. Herein, we provide an overview of the signaling pathway and immune functions of type I IFNs in health and disease. We also review the systemic autoimmune diseases classically associated with type I IFN upregulation and current therapeutic strategies targeting the type I IFN system.
Keywords: Type I IFN, IFN signature, Autoimmunity, systemic lupus erythematosus, Sjogren’s syndrome, systemic sclerosis, dermatomyositis
1. Introduction
The type I interferon (IFN-I) system is crucial for antiviral and antitumoral responses. However, dysregulated IFN-I responses contribute to the development of autoimmunity. Increased IFN-I signaling or high circulating IFN levels is a feature of various systemic autoimmune diseases. However, the clinical manifestations and mechanisms of injury vary widely among these disorders. In this review, we provide an overview of the biology, signaling pathways, and immune functions of IFN-I, as well as their roles in triggering and perpetuating systemic autoimmunity. We also review the main systemic autoimmune diseases associated with dysregulated IFN-I responses and current therapeutic strategies targeting the IFN-I system.
2. The biology of IFN-I
IFN-I are a family of functionally and structurally related cytokines that bind the IFN-I receptor (IFNAR). In humans, the IFN-I family consists of IFN-α (comprised of 13 different subtypes), IFN-β, IFN-ɛ, IFN-κ, and IFN-ω (Gibbert et al., 2013, Li S. F. et al., 2018). Production of the specific subtypes of IFN-I depends on the cell type and the microenvironment (Ali et al., 2019). For example, plasmacytoid dendritic cells (pDC) are the most potent producers of IFN-α in response to viruses and other endosomal TLR triggers, whereas IFN-β is more ubiquitously expressed. Although most IFN-I are primarily produced following stimulation by pathogens or other damage signals, keratinocytes have been shown to constitutively produce IFN-κ (LaFleur et al., 2001, Siegal et al., 1999, Zhang et al., 2016). IFN-ω is structurally similar to IFN-α and is produced in response to viral infections. IFN-ɛ is primarily expressed in the glandular endometrial epithelium and is regulated by estrogens and progesterone during the menstrual cycle (Fung et al., 2013).
In normal immunity, IFNs are produced when exogenous or pathogen-derived particles are recognized by pattern recognition receptors (PRR), including the cytosolic nucleic acid sensors and Toll-like receptors (TLR) (Kawai and Akira, 2006). After PRR activation, interferon regulatory factors (IRF) translocate to the nucleus, enhancing the expression of genes encoding IFN-I. Activation of TLR-7 by single-stranded RNA or TLR-9 by unmethylated CpG-rich DNA in the endosomal compartments of pDCs leads to MyD88-dependent phosphorylation of IRF5 and/or IRF7. In the nucleus, IRF5 induces the transcription of pro-inflammatory cytokines such as IL-6 and TNF, whereas IRF7 stimulates the expression of IFN-I (Jensen and Niewold, 2015). In phagocytes and other immune cells, IRF3 is preferentially activated upon stimulation of TLR3 by double-stranded RNA and TLR4 by bacterial lipopolysaccharide. The cytosolic nucleic acid sensors cGAS-STING (DNA sensor) and RIG-I or MDA5 (RNA sensors) also activate IRF3 via the common signaling adaptor protein MAVS (Wu and Hur, 2015, Yu and Liu, 2021). PRRs can also recognize endogenous nucleic acids derived from defective nucleic acid metabolism, expression from endogenous virus-like genomic repeat elements, or autoantibody-containing immune complexes (Mavragani et al., 2016, Sisirak et al., 2016).
All IFN-I bind and activate IFNAR, a heterodimeric receptor constituted by IFNAR1 and IFNAR2. These subunits are constitutively associated with members of the Janus kinase (JAK) family by non-covalent interactions (Platanias, 2005). Through the canonical IFN-I signaling pathway, binding of IFN-I to IFNAR results in autophosphorylation and activation of TyK2 and JAK1, followed by recruitment, phosphorylation and dimerization of the signal transducers and activators of transcription (STAT) proteins. Once phosphorylated, STATs form dimers, translocate to the nucleus, and bind specific DNA domains to modulate the expression of IFN-stimulated genes (ISG).
IFN-I signaling is tightly regulated. The nature of the response is determined by a delicate balance between enhancing and suppressive mechanisms that allow an effective antimicrobial response while avoiding deleterious effects to host tissues (Ivashkiv and Donlin, 2014). Certain stimuli such as pro-inflammatory cytokines, viruses, and tumor cells can impair the IFN-I response via internalization from the cell membrane, downregulation of expression and enhanced degradation of IFNAR, and by increasing the expression of negative regulators, including the suppressor of cytokine signaling (SOC) and the ubiquitin carboxy-terminal hydrolase 18 (USP18) proteins (Liu et al., 2009, Mayer-Barber and Yan, 2017, Sarasin-Filipowicz et al., 2009). In contrast, induction of the ISGs STAT1 and IRF9 by IL-6, type I and type II IFNs, and positive regulation of JAK1 by spleen tyrosine kinase (SYK) and protein tyrosine kinase 2 (PYK2) amplify IFN-I responses (Gough et al., 2010, Hu et al., 2002, Tassiulas et al., 2004).
3. Factors influencing IFN-I interferon pathways in autoimmunity
3.1. Genetic associations
Polymorphisms in genes involved in IFN-I-mediated pathways are overrepresented in patients with several forms of autoimmunity, which generally follow a pattern of complex polygenic diseases. Family members of patients with SLE and other autoimmune diseases have an increased risk of developing these disorders (Alarcón-Segovia et al., 2005, Kuo et al., 2015, Sinicato et al., 2019). Previous studies have identified a relative risk of over 300 for the development of SLE in identical twins (Kuo et al., 2015), and an overall heritability estimate of 44–66% (Kuo et al., 2015, Lawrence et al., 1987). In this sense, IFN-I levels are a heritable trait in SLE and juvenile dermatomyositis (DM) families (Niewold et al., 2007, Niewold et al., 2011), supporting the idea that there is a genetic component influencing the activation of t IFN-I pathways.
Several functional gene variants known to impact IFN-I production and response associate with an increased risk of several autoimmune disorders (Ghodke-Puranik and Niewold, 2015) (Figure 1). For example, gain-of-function variants in IFIH1, the gene encoding the RNA cytosolic sensor MDA5, are associated with psoriasis and SLE (Harley et al., 2008, Robinson et al., 2011, Strange et al., 2010). Similarly, functional polymorphisms affecting the IRFs that modulate the induction of IFN-I transcripts downstream of TLRs and cytosolic nucleic acid receptors have been found to heighten susceptibility to systemic autoimmunity (Matta et al., 2017). Genetic variants in IRF5 are not only associated with overt SLE and progression to clinical disease in asymptomatic ANA-positive individuals (Alarcón-Riquelme et al., 2016, Cherian et al., 2012), but also with Sjogren’s syndrome, systemic sclerosis, dermatomyositis, and certain organ-specific autoimmune disorders such as cutaneous lupus (Chen et al., 2014, Graham et al., 2006, Ito et al., 2009, Järvinen et al., 2010, Miceli-Richard et al., 2007, Radstake et al., 2010). In addition, IRF5 and IRF7 or IRF7/PHRF1 risk haplotypes associate with the presence of autoantibodies and high serum IFN-alpha activity in patients with SLE (Fu et al., 2011, Harley et al., 2008, Niewold et al., 2008, Salloum et al., 2010). Interestingly, an even greater effect on IFN-I levels is seen in patients with SLE-associated autoantibodies and IRF5 or IRF7 polymorphisms, as an example of gene-environment interaction in autoimmunity (Niewold et al., 2012, Salloum et al., 2010). Genetic variants of IRF7 have also been described in systemic sclerosis (Carmona et al., 2012). Rare variants in BLK and its epistatic partner BANK1 are also associated with SLE via impaired repression and nuclear translocation of IRF5, and subsequent enhancement of IFN-β expression (Jiang et al., 2019). Furthermore, genetic variation near the IFNK locus has been shown to have sex-specific associations with SLE susceptibility and specific skin genotypes (Harley et al., 2010).
Figure 1. Risk alleles affecting type I interferon-related pathways in systemic autoimmune diseases.
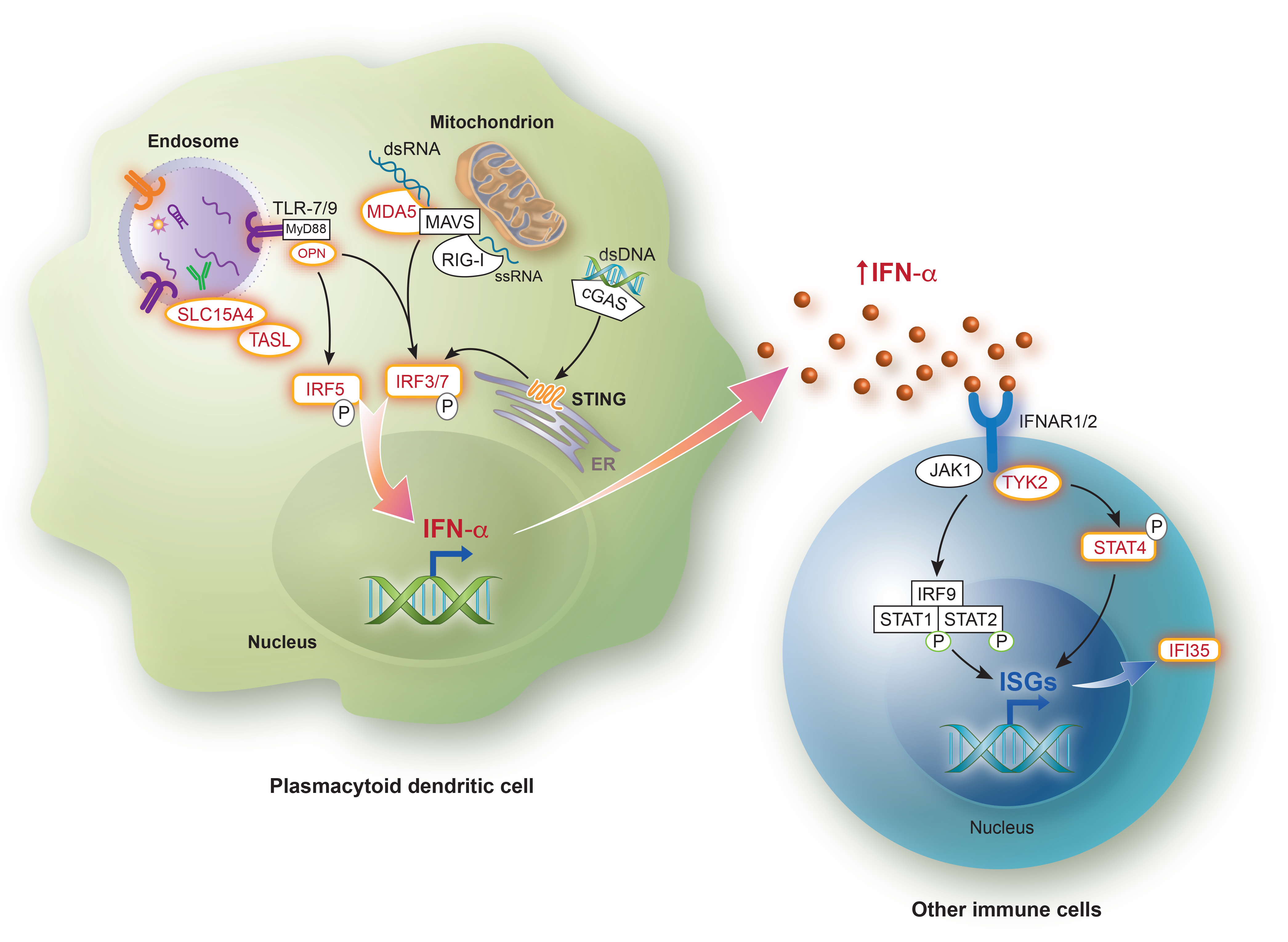
Risk variants directly impacting the production of type I IFN or response that have been identified in SLE, Sjogren’s syndrome, systemic sclerosis, or dermatomyositis are shown in red. Immune complexes containing autoantibodies and self nucleic acids (or antibodies and viral particles) are endocytosed by plasmacytoid dendritic cells, leading to ligand-triggered activation of endosomal Toll-like receptors. The adaptor interacting with SLC15A4 on the Lysosome (TASL, encoded by the CXorf21 gene) is necessary for the recruitment and activation of IRF5 induced by ligand binding to the endosomal TLRs. TASL escapes chromosome X inactivation and is overexpressed in females compared to males, whereas SLC15A4 has been identified as an SLE risk loci. Following TLR-7 and TLR-9 activation, MyD88 and OPN are recruited, which allows for subsequent phosphorylation and nuclear translocation of IRF5 and/or IRF7. Polymorphisms in SPP1 (OPN), IRF5, IRF7 have also been identified in various type I IFN-related autoimmune diseases. Cytosolic nucleic acids are recognized by cGAS (double-stranded DNA), MDA5 (double-stranded RNA) or RIG-I (single-stranded RNA), after which the adaptor proteins MAVS (for MDA5 and RIG-I) and STING (for cGAS) are recruited, allowing for IRF3 phosphorylation and translocation to the nucleus. Activation of IFNAR by type I interferons, including IFN-α, results in phosphorylation of JAK1 and TYK2 and subsequent recruitment and phosphorylation of STAT proteins, which translocate to the nucleus and enhance expression of multiple interferon-stimulated genes (ISGs). TYK2 represents a susceptibility locus in SLE. STAT4 risk alleles have been identified in SLE and systemic sclerosis. The ISG IFI35, a negative regulator of the cytosolic nucleic acid sensor RIG-I, has been shown to be associated with dermatomyositis.
cGAS, cyclic GMP–AMP synthase; dsDNA, double-stranded DNA; dsRNA, double-stranded RNA; IFI35, Interferon Induced Protein 35; IFN, Interferon; IFNAR, Interferon α/β receptor; IRF, Interferon regulatory factor; ISGs, Interferon-stimulated genes; JAK, Janus kinase; MAVS, mitochondrial antiviral-signaling protein; MDA5, Melanoma differentiation-associated protein 5; MyD88, myeloid differentiation factor 88; OPN, Osteopontin; RIG-I, Retinoic acid-inducible gene I; ssRNA; single-stranded RNA; STAT, Signal transducer and activator of transcription; STING, Stimulator of interferon genes; TASL, TLR adaptor interacting with SLC15A4 on the Lysosome; TLR, Toll-like receptor; TYK2, Tyrosine kinase 2.
Illustration assistance provided by Jan Ruvido Stebbins, Ruvido Medical Illustration, Dexter, MI.
Most commonly, autoimmune rheumatic diseases such as SLE, DM, and Sjogren’s syndrome, are considered complex polygenic diseases. However, several monogenic conditions involving IFN-I pathway hyperactivation have been described in the last decade (i.e., monogenic interferonopathies). For example, Aicardi–Goutieres syndrome (AGS) is caused by functional mutations in genes related to nucleic acid degradation or sensing mechanisms, including TREX1, the gene encoding the intracellular 3’−5’ repair exonuclease 1, and IFIH1 (Oda et al., 2014, Rice et al., 2007). AGS manifests as an early-onset encephalopathy and chilblain lesions in distal extremities and ears. Interestingly, a heterozygous missense mutation in TREX1 was identified in individuals with familial chilblain lupus, which also manifests with ulcerating acral skin lesions but without the neurologic affectation of AGS (Lee-Kirsch et al., 2007). Similarly, a heterozygous gain-of-function mutation in TMEM173 (the gene encoding the protein STING) was also recently found as a monogenic cause of familial chilblain lupus (König et al., 2017). Germline dominant gain-of-function mutations in TMEM173 are also responsible for the development of STING-associated vasculopathy with onset in infancy (SAVI), another form of monogenic interferonopathy which manifests as cutaneous vasculopathy and interstitial lung disease (Jeremiah et al., 2014, Liu et al., 2014).
By focusing on IFN-I activity as a molecular sub-phenotype, additional susceptibility loci were discovered on GWAS when comparing patients with SLE and high versus low IFN-α activity (Kariuki et al., 2015), which had not been previously identified in case-control studies (Alarcón-Riquelme et al., 2016, Harley et al., 2008). The top susceptibility genes in European ancestry patients include the PNP rs7897633 risk variant, a loss-of-function polymorphism that induces IFN-I pathway activation in lymphocytes (Ghodke-Puranik et al., 2017), and the PRKG1 rs7897633 variant, for which mechanistic studies are needed to elucidate the connection to IFN-I production and signaling. Polymorphisms in various genes associated with high IFN-I activity in SLE have also been found in association with increased circulating IFN-α levels in juvenile DM, including the osteopontin (OPN or SPP1) and tumor necrosis factor (TNF)-α−308 G/A variants (Kariuki et al., 2009b, Niewold et al., 2010).
Gene variants affecting IFN-I downstream signaling pathways have also been described. TYK2 represents an independent susceptibility locus for SLE (Gateva et al., 2009, Sigurdsson et al., 2005). An epistatic interaction between the TYK2 and IRF5 genes has also been postulated in SLE (Hellquist et al., 2009, Suarez-Gestal et al., 2010, Tang et al., 2015). The STAT4 risk haplotype correlates with greater STAT4 expression and is strongly associated with SLE, rheumatoid arthritis, and systemic sclerosis (Dieudé et al., 2009, Gourh et al., 2009, Harley et al., 2008, Remmers et al., 2007, Rueda et al., 2009, Sigurdsson et al., 2008, Zheng et al., 2013). In SLE, the STAT4 rs7574865 polymorphism is associated with low circulating IFN-I, increased sensitivity to IFN-α, and a more severe SLE phenotype, whereas the rs7582694 risk allele is associated with the presence of anti-dsDNA antibodies and shows an additive effect with two independent IRF5 risk alleles (Kariuki et al., 2009a, Sigurdsson et al., 2008, Zheng et al., 2013). Furthermore, the STAT4 rs7574865 and IRF5 rs2004640 alleles have an additive effect for susceptibility to systemic sclerosis and systemic sclerosis-associated interstitial lung disease (Dieudé et al., 2009). In DM, the ISG IFI35 has also been proposed as a potential risk locus (Bianchi et al., 2021).
3.2. Sex bias in IFN-I-related pathways
Accumulating evidence seems to suggest that the X chromosome is at least partially responsible for the female bias in certain autoimmune diseases (Weckerle and Niewold, 2011). An X chromosome gene-dose effect is supported by the increased risk of developing SLE and Sjogren’s syndrome in patients with numerically abnormal karyotypes, such as trisomy X (47, XXX) and Klinefelter syndrome (47, XXY) (Harris et al., 2016, Liu et al., 2016, Scofield et al., 2008). In addition, mammalian X chromosomes are enriched for immune-related genes, and several of these escape X chromosome inactivation (Mousavi et al., 2020), including the TLR7 gene. In this sense, the role of the X chromosome complement is illustrated by an enhanced TLR-7 induced IFN-I production by pDC in females, which is exacerbated by estrogens (Laffont et al., 2014, Seillet et al., 2012). Similarly, the CXorf21 gene, which commonly escapes chromosome X inactivation (Zhang et al., 2013), is overexpressed in females compared to males (Odhams et al., 2019). The CXorf21 gene encodes for the TLR adaptor interacting with SLC15A4 on the Lysosome (TASL), a necessary adaptor for the recruitment and activation of IRF5 induced by ligand binding to the endosomal TLRs (Heinz et al., 2020).
Additional mechanisms of the sex bias in autoimmunity have been proposed. In a recent study, the transcription cofactor vestigial like family member 3 (VGLL3), which is enriched in the epidermis of women compared to men’s (Liang et al., 2017), was postulated as key orchestrator of the female bias in autoimmunity. Overexpression of VGLL3 in mice was associated with enhanced IFN-I signaling and inflammatory changes that mimicked cutaneous lupus, as well as SLE-like features including autoantibody production and immune complex deposition in the kidneys (Billi et al., 2019). In agreement with these observations, Skopelja-Gardner, et al. demonstrated that UV light-irradiated mice skin showed an early IFN-I response that was approximately 10-fold higher in females compared to male mice (Skopelja-Gardner et al.). Altogether, these findings provide a plausible explanation for the female predominance of cutaneous, and possibly systemic, autoimmune diseases.
3.3. Non-genetic contributions to IFN-I dysregulation in autoimmunity
Although the genetic contribution to IFN-I-related autoimmune diseases is widely accepted, it is generally insufficient for the development of clinical autoimmunity. Environmental triggers such as infections, cigarette smoking, occupational exposures, as well as the microbiome are likely to play an important role in the development and heterogeneity of autoimmune diseases (Barbhaiya and Costenbader, 2016, Björk et al., 2020, De Martinis et al., 2016, Li B. et al., 2018). Epigenetic modifications have been proposed as a link between environmental exposures and modulation of expression in immune-related genes. Hypomethylation and overexpression of ISGs such as IFIT1, MX1, and USP18 have been identified in T cells from patients with primary Sjogren’s syndrome, SLE, and systemic sclerosis (Altorok et al., 2014, Chen et al., 2019, Coit et al., 2013, Imgenberg-Kreuz et al., 2016). Although some epigenetic modifications are cell-specific, genes related to IFN-I pathway activation can be hypomethylated in more than one cell type; for example, hypomethylation of IRF7 has been identified in both myeloid and lymphoid cells from SLE patients (Absher et al., 2013). Interestingly, the hypomethylation at ISGs has been shown to be more pronounced in patients with active SLE compared to those with inactive disease, and in patients with Sjogren’s syndrome (Imgenberg-Kreuz et al., 2018). Given the lack of prospective epigenetic data assessing the progression from preclinical to established clinical autoimmunity, it remains unclear whether the observed DNA methylation changes are a cause or a consequence of increased IFN-I signaling. Moreover, genetic ancestry, risk polymorphisms, and certain medications such as glucocorticoids may alter DNA methylation patterns and be important confounders in epigenetic studies in autoimmunity (Lanata et al., 2018).
Recent studies have provided a link between intestinal dysbiosis and the pathogenesis of several autoimmune diseases (Rosser and Mauri, 2016). It is thought that a restricted intestinal microbiota and impaired enteral barrier could lead to bacterial translocation and consequently to increased IFN-I levels and autoantibody production (Silverman, 2019). Compared to healthy individuals, patients with SLE or primary Sjogren’s syndrome have been found to have a less diverse gut microbiota (van der Meulen et al., 2019). Furthermore, a specific pattern of intestinal dysbiosis characterized by a greater representation of Ruminococcus gnavus and restrictions in taxonomic diversity was recently found to be associated with higher SLE disease activity scores, particularly in lupus nephritis (Azzouz et al., 2019). In this study, there was also a significant correlation between the levels of serum IgG against a specific non-protein bacterial antigen and IFN-α2 levels, providing a potential mechanism linking intestinal dysbiosis and enhanced IFN-I pathway activation in SLE.
4. IFN-I in systemic autoimmune disorders
4.1. SLE
Multiple lines of evidence have implicated IFN-I in the pathogenesis of SLE (Postal et al., 2020). Up to 80% of patients with SLE show overexpression of IFN-I-related genes in circulating immune cells (i.e., IFN signature), and about 50% have chronically elevated IFN-I levels that are detectable in plasma or serum (Baechler et al., 2003, Weckerle et al., 2011). Patients with SLE and evidence of high IFN-I activity tend to have higher disease activity scores and a greater risk of relapse while in remission (Kirou et al., 2005, Mathian et al., 2019). Increased circulating IFN-I levels also associates with specific organ involvement in SLE, including lupus nephritis, cutaneous manifestation, and arthritis, as well as autoantibody patterns (Hua et al., 2006, Oke et al., 2019).
The source of IFN-I in SLE has been debated. pDCs are best known for their ability to effectively produce IFN-α in response to endosomal TLR activation (Reizis, 2019). In SLE, immune complexes formed by autoantibodies and endogenous nucleic acids are thought to result in pDC stimulation and subsequent production of IFN-I, which could contribute to the early break in tolerance and disease initiation (Mavragani et al., 2016, Munroe et al., 2016, Niewold et al., 2007). Abnormalities in the processing and clearance of extracellular DNA, such as it occurs in genetic or acquired cases of DNAse1L3 deficiency, affect tolerance to self-DNA and associate with anti-dsDNA antibody production and clinical manifestations of SLE (Hartl et al., 2021, Soni and Reizis, 2019). Interestingly, the generation of antibody-forming cells in this setting is facilitated by IFN-I in a pDC-dependent manner, further confirming the essential role of these cells in triggering and perpetuating autoimmunity (Soni et al., 2020). Accordingly, in early phase studies, pDC depletion has been shown to correlate with decreased IFN-I activity and improved disease activity (Furie et al., 2019, Karnell et al., 2021).
However, the isolation and identification of IFN-α-producing pDCs in patients with SLE have remained difficult (Der et al., 2019). Interestingly, SLE patients with high IFN-I have reduced circulating pDC numbers (Thanarajasingam et al., 2019). Furthermore, it was recently shown that circulating pDCs from patients with established SLE, Sjogren’s syndrome, and patients in the preclinical stage are not only reduced in number but also are dysfunctional, with loss of all immunologic functions and features of senescence (Psarras et al., 2020). It is thus conceivable that pDC are not the only cell type involved in IFN-I dysregulation in SLE and other immune cells are also contributing. For example, monocytes are a source of IFN-I after UVB-triggered injury of mouse skin (Sontheimer et al., 2017). Follicular dendritic cells also produce IFN-α in response to self immune-complexes, mediated by TLR7 and IRF5 activation in murine models of lupus (Das et al., 2017). Neutrophils have been shown to mediate subclinical renal inflammation with transient proteinuria after UV light exposure to the skin, with evidence of reverse transmigration (i.e., after initially entering the irradiated skin, some neutrophils return to the bloodstream and infiltrate the kidney). Moreover, low-density granulocytes, a highly pro-inflammatory neutrophil subtype in SLE, have an enhanced ability to release neutrophil extracellular traps (NET), which can also trigger IFN-I production by pDCs (Garcia-Romo et al., 2011, Rahman et al., 2019).
Keratinocytes are also likely to play important roles in generating a robust IFN-I response in preclinical autoimmunity and established SLE via the production of large amounts of IFN-κ (and/or IFNβ) (Psarras et al., 2020, Sarkar et al., 2018). For example, increased levels of IFN-κ, which is seen in cutaneous and systemic lupus, may enhance the sensitivity of epithelia to IFN-α and UV irradiation (Sarkar et al., 2018). In addition, chronic ultraviolet B (UVB) light exposure-triggered IFN-I signaling has been shown to be independent of pDC recruitment in mice (Sontheimer et al., 2017). Interestingly, Skopelja-Garner et al. recently demonstrated that acute UVB exposure in the skin could trigger both local and systemic IFN-I responses via the cGAS/STING DNA sensing pathway, suggesting a potential rationale behind SLE flares induced by sunlight exposure in certain susceptible patients (Foering et al., 2013, Skopelja-Gardner et al.). These findings underscore the complex system linking IFN-I and various immune and non-immune cell types in the pathogenesis of SLE.
4.2. Systemic Sclerosis
Multiple studies have identified an IFN-I signature in whole blood or peripheral blood mononuclear cells from as many as 50% of patients with systemic sclerosis, even in early (pre-fibrotic) phases of the disease (Brkic et al., 2016, Duan et al., 2008, Higgs et al., 2011, Rudnik et al., 2021, York et al., 2007). The presence of an IFN-I signature or increased circulating IFN-α levels have also been shown to correlate with serologic subtypes, specifically with anti-topoisomerase and anti-U1 RNP autoantibody positivity (Assassi et al., 2010), and more severe vascular manifestations and lung involvement (Eloranta et al., 2010, Liu et al., 2013). Target organs such as the skin and lung also demonstrate upregulation of ISGs in patients with systemic sclerosis (Christmann et al., 2014, Farina et al., 2010, Valenzi et al., 2021). Interestingly, as seen in SLE, there have been cases of systemic sclerosis developing as a complication of IFN-α therapy for myeloproliferative disorders or hepatitis C infection (Beretta et al., 2002, Solans et al., 2004).
Circulating pDC numbers are reduced in patients with systemic sclerosis (Ah Kioon et al., 2018), similar to what has been described in SLE (Thanarajasingam et al., 2019). Conversely, pDCs infiltrate the skin of systemic sclerosis patients and demonstrate abnormal TLR8 expression, which likely leads to dysregulated IFN-α production by these cells (Ah Kioon et al., 2018). Furthermore, circulating and skin infiltrating pDCs have been shown to produce large amounts of the chemokine CXCL4, which exacerbates the TLR-mediated IFN-α response in pDCs (Ah Kioon et al., 2018, van Roon et al., 2014). In scleroderma mouse models, depletion of pDCs has also been associated with decreased IFN response in the skin and a reduction in skin fibrosis (Ah Kioon et al., 2018, Ross et al., 2021). Various IFN-I-related gene variants have also been associated with an increased risk of systemic sclerosis, including IRF5, IRF7, and STAT4 (Skaug and Assassi, 2020). Limited data on anifrolumab use in systemic sclerosis suggest that IFN-I blockade can suppress T cell activation and collagen accumulation (Guo et al., 2015). These findings support the idea that IFN-I pathway dysregulation represents a pathogenic driver in systemic sclerosis. Clinical trials involving direct or indirect IFN-I antagonists may benefit at least a subset of patients with systemic sclerosis, most likely in the early pre-fibrotic phases.
4.3. Dermatomyositis
A prominent IFN signature has been identified in the skin, muscle, and blood of patients with adult and juvenile DM (Bilgic et al., 2009, Chen et al., 2008, Greenberg et al., 2005, Higgs et al., 2011, Moneta et al., 2019, Wong et al., 2012). The IFN-I gene signature also seems to correlate with disease activity in adult DM (Bilgic et al., 2009, Walsh et al., 2007). Certain subsets of patients with DM, such as those with anti-melanoma differentiation-associated gene 5 (MDA5)+ disease, have been shown to have a lower level of ISG expression in muscle tissue compared to MDA5− patients (Allenbach et al., 2016). In contrast, MDA5+ patients seem to have a stronger IFN-I signature in blood and skin than MDA5− patients (Cassius et al., 2020, Ono et al., 2019, Zhang et al., 2019). As MDA5+ patients have relatively mild muscle involvement and worse skin and lung disease compared to MDA5− patients, it is plausible that the IFN-I signature differences identified in these studies relate to the degree of organ affectation. In addition, MDA5+ patients exhibit overexpression of the IFN-ĸ gene in the skin, suggesting keratinocytes as an important local source of IFN-I (Cassius et al., 2020).
pDCs are part of the inflammatory infiltrate in muscle and skin biopsies from DM patients, suggesting a potential local source of IFN-α, presumably induced by autoantibody-containing immune complexes (Greenberg et al., 2005, McNiff and Kaplan, 2008, Shrestha et al., 2010). In this sense, serum IFN-α activity (measured with a sensitive bioassay) was found to be associated with elevation of muscle enzymes and shorter duration of untreated disease in patients with newly diagnosed juvenile DM (Niewold et al., 2009), as well as with anti-Ro, anti-La, and anti-Sm/RNP (Balboni et al., 2013). Conversely, elevated levels of circulating IFN-β (determined by ELISA), but not IFN-α, were found to correlate with blood IFN signature in DM patients (Liao et al., 2011). The contrasting findings are likely due to the limitations of measuring circulating IFN-I by ELISA, potential differences between local versus circulating levels of these cytokines, as well as differences in the timing of disease, serologic subtype, therapeutic agents used, and age at the time of diagnosis.
5. Available therapeutic agents targeting IFN-I-related pathways in autoimmunity
Most therapeutic agents targeting IFN-I that have entered clinical development were initially aimed at treating SLE (Paredes and Niewold, 2020). Anifrolumab, a monoclonal antibody against IFNAR1, has been shown to be superior to placebo in reducing disease activity, glucocorticoid dose, and severity of skin disease in SLE (Furie et al., 2017, Furie et al., 2021, Morand et al., 2020). Previously, the anti-IFN-α monoclonal antibody rontalizumab failed to achieve its primary and secondary endpoints in SLE (Kalunian et al., 2016). Other anti-IFN-α agents, sifalimumab and AGS-009, were assessed in early phase clinical trials but did not enter further development stages despite promising preliminary findings on safety and efficacy (Khamashta et al., 2016, Tcherepanova et al., 2013). The IFN-α kinoid, an immunotherapeutic vaccine aimed to elicit high titers of anti-IFN-α neutralizing antibodies, also failed to meet its primary endpoint in SLE, but met clinically relevant secondary outcomes, including prednisone daily dose reduction and a greater proportion of patients in lupus low disease activity state compared to placebo (Houssiau et al., 2020). However, anifrolumab is likely superior to anti-IFN-α-directed therapies given the broader IFN-I antagonism in the former, since all IFN-I molecules signal through IFNAR. Various other agents targeting IFN-I dysregulation currently being assessed in clinical trials for safety and efficacy in SLE, systemic sclerosis, and DM are shown in Table 1.
Table 1.
Molecules in early-phase clinical trials targeting the type I interferon pathway in systemic autoimmune diseases.
| Drug name | Mechanism of action | Clinical trial phase | Main outcomes | Disease focus | References |
|---|---|---|---|---|---|
| Tofacitinib | JAKi | Pilot study | All subjects met the IMACS group definition of improvement. Improvement in cutaneous disease activity by CDASI. Decreased IFN score in 5/8 patients (muscle only). |
DM, refractory | (Paik et al., 2021) |
| VIB7734 | pDC-depleting agent (anti-ILT7 mAb) | Phase 1 | Unpublished results. | DM, SjS, SSc | Completed, pending results (NCT03817424) |
| VIB7734 | pDC-depleting agent (anti-ILT7) | Phase 1 | Adequate safety and tolerability. Rapid and durable depletion of pDCs. Reduction in type I IFN activity in skin and CLASI-A scores in CLE. |
CLE, SLE, DM, SjS, SSc | (Karnell et al., 2021) |
| BIIB059 | pDC-depleting agent (anti-BDCA2 mAb) | Phase 1 | Tolerability and a favorable safety profile. Decreased expression of IFN response genes in blood. Reduction of immune infiltrates in skin and decreased CLASI-A scores in SLE. |
SLE | (Furie et al., 2019) |
| Tofacitinib | JAKi | Phase 1 | Adequate safety and tolerability. The effect of JAK inhibition was more robust in subjects with the STAT4 risk allele. Improvement in cardiometabolic and immunologic parameters associated with premature atherosclerosis in SLE. |
SLE | (Hasni et al., 2021) |
| Baricitinib | JAKi | Phase 2 | More SLE patients achieved resolution of arthritis or rash with baricitinib (4 mg dosing) than placebo. | SLE | (Wallace et al., 2018) |
| BMS-986165 | TYK2i | Phase 2 | Unpublished results. | SLE | Active, not recruiting (NCT03252587) Recruiting (NCT03920267) |
| Filgotinib | JAKi | Phase 2 | Unpublished results. | SjS | Completed, pending results (NCT03100942) |
| Anifrolumab (MEDI-546) | Anti-IFNAR mAb | Phase 1 | Adequate tolerability and safety. Sustained inhibition of the type I IFN gene signature. |
SSc | (Goldberg et al., 2014) |
BDCA2, Blood dendritic cell antigen 2; CLASI-A, Cutaneous Lupus Erythematosus Disease Area and Severity Index Activity score; CLE, Cutaneous lupus erythematosus; DM, Dermatomyositis; JAKi, Janus kinase inhibitor; IFN, Interferon; IFNAR, Interferon alpha/beta receptor; ILT7, Immunoglobulin-like transcript 7; mAb, Monoclonal antibody; pDC, Plasmacytoid dendritic cell; SjS, Sjogren's syndrome; SLE, Systemic lupus erythematosus; SSc, Systemic Sclerosis; TYK2i, Tyrosine kinase 2 inhibitor.
6. Conclusion
In summary, IFN-I dysregulation is a cornerstone of several systemic autoimmune diseases. Although multiple studies have proposed mechanisms by which IFN-I contribute to the pathogenesis of autoimmunity, many unanswered questions remain. The exact source and triggers of IFN-I, the specific role of genetic variation and interactions between genetic and environmental factors that lead to dysfunctional IFN-I responses are, in many cases, unknown. The heterogeneity of disease manifestations, medication use, exposures, and genetic background in autoimmunity further complicates the interpretation of study findings. As we better understand disease pathogenesis and its role on immune dysregulation and clinical heterogeneity, the role of precision medicine in IFN-I-related autoimmune diseases seems promising. Reassuringly, various therapeutic agents that target IFN-I pathways are in different phases of development or testing for various autoimmune diseases.
Acknowledgments:
TBN has grants from the Colton Center for Autoimmunity, NIH (AI167271, AR078416, AR065964), the Lupus Research Foundation, and the Lupus Research Alliance.
Conflicts of Interest:
TBN has received research grants from EMD Serono, Inc., and has consulted for Thermo Fisher, Progenetec, Roivant Sciences, Ventus, Toran, and Inova, all unrelated to the current manuscript. RFR has nothing to disclose.
Abbreviations:
- AGS
Aicardi–Goutieres syndrome
- BAFF
B-cell activating factor
- BILAG
British Isles Lupus Assessment Group
- BICLA
BILAG-based Composite Lupus Assessment
- DM
Dermatomyositis
- JAK
Janus kinase
- IFN-I
Type I interferon
- IFNAR
Interferon alpha/beta receptor
- ILT7
Immunoglobulin-like transcript 7
- ISG
Interferon-stimulated gene
- IRF
Interferon regulatory factor
- MDA5
Melanoma differentiation-associated protein 5
- pDC
Plasmacytoid dendritic cell
- PRR
Pattern recognition receptor
- RIG-I
Retinoic acid-inducible gene I
- SAVI
STING-associated vasculopathy with onset in infancy
- SLE
Systemic lupus erythematosus
- STAT
Signal transducer and activator of transcription
- STING
Stimulator of interferon genes
- TASL
TLR adaptor interacting with SLC15A4 on the Lysosome
- TLR
Toll-like receptor
- TYK2
Tyrosine kinase 2
- VGLL3
Transcription cofactor vestigial like family member 3
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Absher DM, Li X, Waite LL, Gibson A, Roberts K, Edberg J, et al. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genet 2013;9(8):e1003678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, et al. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med 2018;10(423). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón-Riquelme ME, Ziegler JT, Molineros J, Howard TD, Moreno-Estrada A, Sánchez-Rodríguez E, et al. Genome-Wide Association Study in an Amerindian Ancestry Population Reveals Novel Systemic Lupus Erythematosus Risk Loci and the Role of European Admixture. Arthritis Rheumatol 2016;68(4):932–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón-Segovia D, Alarcón-Riquelme ME, Cardiel MH, Caeiro F, Massardo L, Villa AR, et al. Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum 2005;52(4):1138–47. [DOI] [PubMed] [Google Scholar]
- Ali S, Mann-Nüttel R, Schulze A, Richter L, Alferink J, Scheu S. Sources of Type I Interferons in Infectious Immunity: Plasmacytoid Dendritic Cells Not Always in the Driver’s Seat. Front Immunol 2019;10:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allenbach Y, Leroux G, Suárez-Calvet X, Preusse C, Gallardo E, Hervier B, et al. Dermatomyositis With or Without Anti-Melanoma Differentiation-Associated Gene 5 Antibodies: Common Interferon Signature but Distinct NOS2 Expression. Am J Pathol 2016;186(3):691–700. [DOI] [PubMed] [Google Scholar]
- Altorok N, Coit P, Hughes T, Koelsch KA, Stone DU, Rasmussen A, et al. Genome-wide DNA methylation patterns in naive CD4+ T cells from patients with primary Sjögren’s syndrome. Arthritis Rheumatol 2014;66(3):731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assassi S, Mayes MD, Arnett FC, Gourh P, Agarwal SK, McNearney TA, et al. Systemic sclerosis and lupus: points in an interferon-mediated continuum. Arthritis Rheum 2010;62(2):589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, et al. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheum Dis 2019;78(7):947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 2003;100(5):2610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balboni I, Niewold TB, Morgan G, Limb C, Eloranta ML, Rönnblom L, et al. Interferon-α induction and detection of anti-ro, anti-la, anti-sm, and anti-rnp autoantibodies by autoantigen microarray analysis in juvenile dermatomyositis. Arthritis Rheum 2013;65(9):2424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbhaiya M, Costenbader KH. Environmental exposures and the development of systemic lupus erythematosus. Curr Opin Rheumatol 2016;28(5):497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beretta L, Caronni M, Vanoli M, Scorza R. Systemic sclerosis after interferon-alfa therapy for myeloproliferative disorders. Br J Dermatol. 147. England; 2002. p. 385–6. [DOI] [PubMed] [Google Scholar]
- Bianchi M, Kozyrev SV, Notarnicola A, Hultin Rosenberg L, Karlsson Å, Pucholt P, et al. Contribution of rare genetic variation to disease susceptibility in a large Scandinavian myositis cohort. Arthritis Rheumatol 2021. [DOI] [PubMed] [Google Scholar]
- Bilgic H, Ytterberg SR, Amin S, McNallan KT, Wilson JC, Koeuth T, et al. Interleukin-6 and type I interferon-regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum 2009;60(11):3436–46. [DOI] [PubMed] [Google Scholar]
- Billi AC, Gharaee-Kermani M, Fullmer J, Tsoi LC, Hill BD, Gruszka D, et al. The female-biased factor VGLL3 drives cutaneous and systemic autoimmunity. JCI Insight 2019;4(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk A, Mofors J, Wahren-Herlenius M. Environmental factors in the pathogenesis of primary Sjögren’s syndrome. J Intern Med 2020;287(5):475–92. [DOI] [PubMed] [Google Scholar]
- Brkic Z, van Bon L, Cossu M, van Helden-Meeuwsen CG, Vonk MC, Knaapen H, et al. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann Rheum Dis 2016;75(8):1567–73. [DOI] [PubMed] [Google Scholar]
- Carmona FD, Gutala R, Simeón CP, Carreira P, Ortego-Centeno N, Vicente-Rabaneda E, et al. Novel identification of the IRF7 region as an anticentromere autoantibody propensity locus in systemic sclerosis. Ann Rheum Dis 2012;71(1):114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassius C, Amode R, Delord M, Battistella M, Poirot J, How-Kit A, et al. MDA5(+) Dermatomyositis Is Associated with Stronger Skin Type I Interferon Transcriptomic Signature with Upregulation of IFN-κ Transcript. J Invest Dermatol 2020;140(6):1276–9.e7. [DOI] [PubMed] [Google Scholar]
- Chen S, Pu W, Guo S, Jin L, He D, Wang J. Genome-Wide DNA Methylation Profiles Reveal Common Epigenetic Patterns of Interferon-Related Genes in Multiple Autoimmune Diseases. Front Genet 2019;10:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang Q, Wu Z, Li Y, Li P, Sun F, et al. Genetic association study of TNFAIP3, IFIH1, IRF5 polymorphisms with polymyositis/dermatomyositis in Chinese Han population. PLoS One 2014;9(10):e110044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YW, Shi R, Geraci N, Shrestha S, Gordish-Dressman H, Pachman LM. Duration of chronic inflammation alters gene expression in muscle from untreated girls with juvenile dermatomyositis. BMC Immunol 2008;9:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian TS, Kariuki SN, Franek BS, Buyon JP, Clancy RM, Niewold TB. Brief Report: IRF5 systemic lupus erythematosus risk haplotype is associated with asymptomatic serologic autoimmunity and progression to clinical autoimmunity in mothers of children with neonatal lupus. Arthritis Rheum 2012;64(10):3383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christmann RB, Sampaio-Barros P, Stifano G, Borges CL, de Carvalho CR, Kairalla R, et al. Association of Interferon- and transforming growth factor β-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol 2014;66(3):714–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coit P, Jeffries M, Altorok N, Dozmorov MG, Koelsch KA, Wren JD, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naïve CD4+ T cells from lupus patients. J Autoimmun 2013;43:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Heesters BA, Bialas A, O’Flynn J, Rifkin IR, Ochando J, et al. Follicular Dendritic Cell Activation by TLR Ligands Promotes Autoreactive B Cell Responses. Immunity 2017;46(1):106–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Martinis M, Ciccarelli F, Sirufo MM, Ginaldi L. An overview of environmental risk factors in systemic sclerosis. Expert Rev Clin Immunol 2016;12(4):465–78. [DOI] [PubMed] [Google Scholar]
- Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol 2019;20(7):915–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieudé P, Guedj M, Wipff J, Ruiz B, Hachulla E, Diot E, et al. STAT4 is a genetic risk factor for systemic sclerosis having additive effects with IRF5 on disease susceptibility and related pulmonary fibrosis. Arthritis Rheum 2009;60(8):2472–9. [DOI] [PubMed] [Google Scholar]
- Duan H, Fleming J, Pritchard DK, Amon LM, Xue J, Arnett HA, et al. Combined analysis of monocyte and lymphocyte messenger RNA expression with serum protein profiles in patients with scleroderma. Arthritis Rheum 2008;58(5):1465–74. [DOI] [PubMed] [Google Scholar]
- Eloranta ML, Franck-Larsson K, Lövgren T, Kalamajski S, Rönnblom A, Rubin K, et al. Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis 2010;69(7):1396–402. [DOI] [PubMed] [Google Scholar]
- Farina G, Lafyatis D, Lemaire R, Lafyatis R. A four-gene biomarker predicts skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum 2010;62(2):580–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foering K, Chang AY, Piette EW, Cucchiara A, Okawa J, Werth VP. Characterization of clinical photosensitivity in cutaneous lupus erythematosus. J Am Acad Dermatol 2013;69(2):205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Zhao J, Qian X, Wong JL, Kaufman KM, Yu CY, et al. Association of a functional IRF7 variant with systemic lupus erythematosus. Arthritis Rheum 2011;63(3):749–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung KY, Mangan NE, Cumming H, Horvat JC, Mayall JR, Stifter SA, et al. Interferon-ε protects the female reproductive tract from viral and bacterial infection. Science 2013;339(6123):1088–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol 2017;69(2):376–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furie R, Morand EF, Askanase AD, Vital EM, Merrill JT, Kalyani RN, et al. Anifrolumab reduces flare rates in patients with moderate to severe systemic lupus erythematosus. Lupus 2021;30(8):1254–63. [DOI] [PubMed] [Google Scholar]
- Furie R, Werth VP, Merola JF, Stevenson L, Reynolds TL, Naik H, et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Invest 2019;129(3):1359–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 2011;3(73):73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet 2009;41(11):1228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodke-Puranik Y, Dorschner JM, Vsetecka DM, Amin S, Makol A, Ernste F, et al. Lupus-Associated Functional Polymorphism in PNP Causes Cell Cycle Abnormalities and Interferon Pathway Activation in Human Immune Cells. Arthritis Rheumatol 2017;69(12):2328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodke-Puranik Y, Niewold TB. Immunogenetics of systemic lupus erythematosus: A comprehensive review. J Autoimmun 2015;64:125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbert K, Schlaak JF, Yang D, Dittmer U. IFN-α subtypes: distinct biological activities in anti-viral therapy. Br J Pharmacol 2013;168(5):1048–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg A, Geppert T, Schiopu E, Frech T, Hsu V, Simms RW, et al. Dose-escalation of human anti-interferon-α receptor monoclonal antibody MEDI-546 in subjects with systemic sclerosis: a phase 1, multicenter, open label study. Arthritis Res Ther 2014;16(1):R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough DJ, Messina NL, Hii L, Gould JA, Sabapathy K, Robertson AP, et al. Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol 2010;8(4):e1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourh P, Agarwal SK, Divecha D, Assassi S, Paz G, Arora-Singh RK, et al. Polymorphisms in TBX21 and STAT4 increase the risk of systemic sclerosis: evidence of possible gene-gene interaction and alterations in Th1/Th2 cytokines. Arthritis Rheum 2009;60(12):3794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet 2006;38(5):550–5. [DOI] [PubMed] [Google Scholar]
- Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol 2005;57(5):664–78. [DOI] [PubMed] [Google Scholar]
- Guo X, Higgs BW, Bay-Jensen AC, Karsdal MA, Yao Y, Roskos LK, et al. Suppression of T Cell Activation and Collagen Accumulation by an Anti-IFNAR1 mAb, Anifrolumab, in Adult Patients with Systemic Sclerosis. J Invest Dermatol 2015;135(10):2402–9. [DOI] [PubMed] [Google Scholar]
- Harley IT, Niewold TB, Stormont RM, Kaufman KM, Glenn SB, Franek BS, et al. The role of genetic variation near interferon-kappa in systemic lupus erythematosus. J Biomed Biotechnol 2010;2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008;40(2):204–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris VM, Sharma R, Cavett J, Kurien BT, Liu K, Koelsch KA, et al. Klinefelter’s syndrome (47,XXY) is in excess among men with Sjögren’s syndrome. Clin Immunol 2016;168:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl J, Serpas L, Wang Y, Rashidfarrokhi A, Perez OA, Sally B, et al. Autoantibody-mediated impairment of DNASE1L3 activity in sporadic systemic lupus erythematosus. J Exp Med 2021;218(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasni SA, Gupta S, Davis M, Poncio E, Temesgen-Oyelakin Y, Carlucci PM, et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat Commun 2021;12(1):3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz LX, Lee J, Kapoor U, Kartnig F, Sedlyarov V, Papakostas K, et al. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7–9. Nature 2020;581(7808):316–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellquist A, Järvinen TM, Koskenmies S, Zucchelli M, Orsmark-Pietras C, Berglind L, et al. Evidence for genetic association and interaction between the TYK2 and IRF5 genes in systemic lupus erythematosus. J Rheumatol 2009;36(8):1631–8. [DOI] [PubMed] [Google Scholar]
- Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis 2011;70(11):2029–36. [DOI] [PubMed] [Google Scholar]
- Houssiau FA, Thanou A, Mazur M, Ramiterre E, Gomez Mora DA, Misterska-Skora M, et al. IFN-α kinoid in systemic lupus erythematosus: results from a phase IIb, randomised, placebo-controlled study. Ann Rheum Dis 2020;79(3):347–55. [DOI] [PubMed] [Google Scholar]
- Hu X, Herrero C, Li WP, Antoniv TT, Falck-Pedersen E, Koch AE, et al. Sensitization of IFN-gamma Jak-STAT signaling during macrophage activation. Nat Immunol 2002;3(9):859–66. [DOI] [PubMed] [Google Scholar]
- Hua J, Kirou K, Lee C, Crow MK. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum 2006;54(6):1906–16. [DOI] [PubMed] [Google Scholar]
- Imgenberg-Kreuz J, Carlsson Almlöf J, Leonard D, Alexsson A, Nordmark G, Eloranta ML, et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann Rheum Dis 2018;77(5):736–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imgenberg-Kreuz J, Sandling JK, Almlöf JC, Nordlund J, Signér L, Norheim KB, et al. Genome-wide DNA methylation analysis in multiple tissues in primary Sjögren’s syndrome reveals regulatory effects at interferon-induced genes. Ann Rheum Dis 2016;75(11):2029–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito I, Kawaguchi Y, Kawasaki A, Hasegawa M, Ohashi J, Hikami K, et al. Association of a functional polymorphism in the IRF5 region with systemic sclerosis in a Japanese population. Arthritis Rheum 2009;60(6):1845–50. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014;14(1):36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MA, Niewold TB. Interferon regulatory factors: critical mediators of human lupus. Transl Res 2015;165(2):283–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest 2014;124(12):5516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang SH, Athanasopoulos V, Ellyard JI, Chuah A, Cappello J, Cook A, et al. Functional rare and low frequency variants in BLK and BANK1 contribute to human lupus. Nat Commun 2019;10(1):2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järvinen TM, Hellquist A, Koskenmies S, Einarsdottir E, Koskinen LL, Jeskanen L, et al. Tyrosine kinase 2 and interferon regulatory factor 5 polymorphisms are associated with discoid and subacute cutaneous lupus erythematosus. Exp Dermatol 2010;19(2):123–31. [DOI] [PubMed] [Google Scholar]
- Kalunian KC, Merrill JT, Maciuca R, McBride JM, Townsend MJ, Wei X, et al. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis 2016;75(1):196–202. [DOI] [PubMed] [Google Scholar]
- Kariuki SN, Ghodke-Puranik Y, Dorschner JM, Chrabot BS, Kelly JA, Tsao BP, et al. Genetic analysis of the pathogenic molecular sub-phenotype interferon-alpha identifies multiple novel loci involved in systemic lupus erythematosus. Genes Immun 2015;16(1):15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariuki SN, Kirou KA, MacDermott EJ, Barillas-Arias L, Crow MK, Niewold TB. Cutting edge: autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-alpha in lupus patients in vivo. J Immunol 2009a;182(1):34–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariuki SN, Moore JG, Kirou KA, Crow MK, Utset TO, Niewold TB. Age- and gender-specific modulation of serum osteopontin and interferon-alpha by osteopontin genotype in systemic lupus erythematosus. Genes Immun 2009b;10(5):487–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnell JL, Wu Y, Mittereder N, Smith MA, Gunsior M, Yan L, et al. Depleting plasmacytoid dendritic cells reduces local type I interferon responses and disease activity in patients with cutaneous lupus. Sci Transl Med 2021;13(595). [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol 2006;7(2):131–7. [DOI] [PubMed] [Google Scholar]
- Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, et al. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis 2016;75(11):1909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum 2005;52(5):1491–503. [DOI] [PubMed] [Google Scholar]
- Kuo CF, Grainge MJ, Valdes AM, See LC, Luo SF, Yu KH, et al. Familial Aggregation of Systemic Lupus Erythematosus and Coaggregation of Autoimmune Diseases in Affected Families. JAMA Intern Med 2015;175(9):1518–26. [DOI] [PubMed] [Google Scholar]
- König N, Fiehn C, Wolf C, Schuster M, Cura Costa E, Tüngler V, et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann Rheum Dis 2017;76(2):468–72. [DOI] [PubMed] [Google Scholar]
- Laffont S, Rouquié N, Azar P, Seillet C, Plumas J, Aspord C, et al. X-Chromosome complement and estrogen receptor signaling independently contribute to the enhanced TLR7-mediated IFN-α production of plasmacytoid dendritic cells from women. J Immunol 2014;193(11):5444–52. [DOI] [PubMed] [Google Scholar]
- LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, et al. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J Biol Chem 2001;276(43):39765–71. [DOI] [PubMed] [Google Scholar]
- Lanata CM, Chung SA, Criswell LA. DNA methylation 101: what is important to know about DNA methylation and its role in SLE risk and disease heterogeneity. Lupus Sci Med 2018;5(1):e000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JS, Martins CL, Drake GL. A family survey of lupus erythematosus. 1. Heritability. J Rheumatol 1987;14(5):913–21. [PubMed] [Google Scholar]
- Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, et al. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med (Berl) 2007;85(5):531–7. [DOI] [PubMed] [Google Scholar]
- Li B, Selmi C, Tang R, Gershwin ME, Ma X. The microbiome and autoimmunity: a paradigm from the gut-liver axis. Cell Mol Immunol 2018;15(6):595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SF, Gong MJ, Zhao FR, Shao JJ, Xie YL, Zhang YG, et al. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell Physiol Biochem 2018;51(5):2377–96. [DOI] [PubMed] [Google Scholar]
- Liang Y, Tsoi LC, Xing X, Beamer MA, Swindell WR, Sarkar MK, et al. A gene network regulated by the transcription factor VGLL3 as a promoter of sex-biased autoimmune diseases. Nat Immunol 2017;18(2):152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao AP, Salajegheh M, Nazareno R, Kagan JC, Jubin RG, Greenberg SA. Interferon β is associated with type 1 interferon-inducible gene expression in dermatomyositis. Ann Rheum Dis 2011;70(5):831–6. [DOI] [PubMed] [Google Scholar]
- Liu J, HuangFu WC, Kumar KG, Qian J, Casey JP, Hamanaka RB, et al. Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 2009;5(1):72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Kurien BT, Zimmerman SL, Kaufman KM, Taft DH, Kottyan LC, et al. X Chromosome Dose and Sex Bias in Autoimmune Diseases: Increased Prevalence of 47,XXX in Systemic Lupus Erythematosus and Sjögren’s Syndrome. Arthritis Rheumatol 2016;68(5):1290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Mayes MD, Tan FK, Wu M, Reveille JD, Harper BE, et al. Correlation of interferon-inducible chemokine plasma levels with disease severity in systemic sclerosis. Arthritis Rheum 2013;65(1):226–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 2014;371(6):507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathian A, Mouries-Martin S, Dorgham K, Devilliers H, Yssel H, Garrido Castillo L, et al. Ultrasensitive serum interferon-α quantification during SLE remission identifies patients at risk for relapse. Ann Rheum Dis 2019;78(12):1669–76. [DOI] [PubMed] [Google Scholar]
- Matta B, Song S, Li D, Barnes BJ. Interferon regulatory factor signaling in autoimmune disease. Cytokine 2017;98:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavragani CP, Sagalovskiy I, Guo Q, Nezos A, Kapsogeorgou EK, Lu P, et al. Expression of Long Interspersed Nuclear Element 1 Retroelements and Induction of Type I Interferon in Patients With Systemic Autoimmune Disease. Arthritis Rheumatol 2016;68(11):2686–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Barber KD, Yan B. Clash of the Cytokine Titans: counter-regulation of interleukin-1 and type I interferon-mediated inflammatory responses. Cell Mol Immunol 2017;14(1):22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNiff JM, Kaplan DH. Plasmacytoid dendritic cells are present in cutaneous dermatomyositis lesions in a pattern distinct from lupus erythematosus. J Cutan Pathol 2008;35(5):452–6. [DOI] [PubMed] [Google Scholar]
- Miceli-Richard C, Comets E, Loiseau P, Puechal X, Hachulla E, Mariette X. Association of an IRF5 gene functional polymorphism with Sjögren’s syndrome. Arthritis Rheum 2007;56(12):3989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moneta GM, Pires Marafon D, Marasco E, Rosina S, Verardo M, Fiorillo C, et al. Muscle Expression of Type I and Type II Interferons Is Increased in Juvenile Dermatomyositis and Related to Clinical and Histologic Features. Arthritis Rheumatol 2019;71(6):1011–21. [DOI] [PubMed] [Google Scholar]
- Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med 2020;382(3):211–21. [DOI] [PubMed] [Google Scholar]
- Mousavi MJ, Mahmoudi M, Ghotloo S. Escape from X chromosome inactivation and female bias of autoimmune diseases. Mol Med 2020;26(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munroe ME, Lu R, Zhao YD, Fife DA, Robertson JM, Guthridge JM, et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann Rheum Dis 2016;75(11):2014–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun 2007;8(6):492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewold TB, Kariuki SN, Morgan GA, Shrestha S, Pachman LM. Elevated serum interferon-alpha activity in juvenile dermatomyositis: associations with disease activity at diagnosis and after thirty-six months of therapy. Arthritis Rheum 2009;60(6):1815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewold TB, Kariuki SN, Morgan GA, Shrestha S, Pachman LM. Gene-gene-sex interaction in cytokine gene polymorphisms revealed by serum interferon alpha phenotype in juvenile dermatomyositis. J Pediatr 2010;157(4):653–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum 2008;58(8):2481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewold TB, Kelly JA, Kariuki SN, Franek BS, Kumar AA, Kaufman KM, et al. IRF5 haplotypes demonstrate diverse serological associations which predict serum interferon alpha activity and explain the majority of the genetic association with systemic lupus erythematosus. Ann Rheum Dis 2012;71(3):463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewold TB, Wu SC, Smith M, Morgan GA, Pachman LM. Familial aggregation of autoimmune disease in juvenile dermatomyositis. Pediatrics 2011;127(5):e1239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, et al. Aicardi-Goutières syndrome is caused by IFIH1 mutations. Am J Hum Genet 2014;95(1):121–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odhams CA, Roberts AL, Vester SK, Duarte CST, Beales CT, Clarke AJ, et al. Interferon inducible X-linked gene CXorf21 may contribute to sexual dimorphism in Systemic Lupus Erythematosus. Nat Commun 2019;10(1):2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oke V, Gunnarsson I, Dorschner J, Eketjäll S, Zickert A, Niewold TB, et al. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res Ther 2019;21(1):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono N, Kai K, Maruyama A, Sakai M, Sadanaga Y, Koarada S, et al. The relationship between type 1 IFN and vasculopathy in anti-MDA5 antibody-positive dermatomyositis patients. Rheumatology (Oxford) 2019;58(5):786–91. [DOI] [PubMed] [Google Scholar]
- Paik JJ, Casciola-Rosen L, Shin JY, Albayda J, Tiniakou E, Leung DG, et al. Study of Tofacitinib in Refractory Dermatomyositis: An Open-Label Pilot Study of Ten Patients. Arthritis Rheumatol 2021;73(5):858–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes JL, Niewold TB. Type I interferon antagonists in clinical development for lupus. Expert Opin Investig Drugs 2020;29(9):1025–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 2005;5(5):375–86. [DOI] [PubMed] [Google Scholar]
- Postal M, Vivaldo JF, Fernandez-Ruiz R, Paredes JL, Appenzeller S, Niewold TB. Type I interferon in the pathogenesis of systemic lupus erythematosus. Curr Opin Immunol 2020;67:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psarras A, Alase A, Antanaviciute A, Carr IM, Md Yusof MY, Wittmann M, et al. Functionally impaired plasmacytoid dendritic cells and non-haematopoietic sources of type I interferon characterize human autoimmunity. Nat Commun 2020;11(1):6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radstake TR, Gorlova O, Rueda B, Martin JE, Alizadeh BZ, Palomino-Morales R, et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet 2010;42(5):426–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, Sagar D, Hanna RN, Lightfoot YL, Mistry P, Smith CK, et al. Low-density granulocytes activate T cells and demonstrate a non-suppressive role in systemic lupus erythematosus. Ann Rheum Dis 2019;78(7):957–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizis B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019;50(1):37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 2007;357(10):977–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet 2007;81(4):713–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson T, Kariuki SN, Franek BS, Kumabe M, Kumar AA, Badaracco M, et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-α and serologic autoimmunity in lupus patients. J Immunol 2011;187(3):1298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RL, Corinaldesi C, Migneco G, Carr IM, Antanaviciute A, Wasson CW, et al. Targeting human plasmacytoid dendritic cells through BDCA2 prevents skin inflammation and fibrosis in a novel xenotransplant mouse model of scleroderma. Ann Rheum Dis 2021;80(7):920–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosser EC, Mauri C. A clinical update on the significance of the gut microbiota in systemic autoimmunity. J Autoimmun 2016;74:85–93. [DOI] [PubMed] [Google Scholar]
- Rudnik M, Rolski F, Jordan S, Mertelj T, Stellato M, Distler O, et al. Regulation of Monocyte Adhesion and Type I Interferon Signaling by CD52 in Patients With Systemic Sclerosis. Arthritis Rheumatol 2021;73(9):1720–30. [DOI] [PubMed] [Google Scholar]
- Rueda B, Broen J, Simeon C, Hesselstrand R, Diaz B, Suárez H, et al. The STAT4 gene influences the genetic predisposition to systemic sclerosis phenotype. Hum Mol Genet 2009;18(11):2071–7. [DOI] [PubMed] [Google Scholar]
- Salloum R, Franek BS, Kariuki SN, Rhee L, Mikolaitis RA, Jolly M, et al. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-alpha activity in lupus patients. Arthritis Rheum 2010;62(2):553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarasin-Filipowicz M, Wang X, Yan M, Duong FH, Poli V, Hilton DJ, et al. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol Cell Biol 2009;29(17):4841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis 2018;77(11):1653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield RH, Bruner GR, Namjou B, Kimberly RP, Ramsey-Goldman R, Petri M, et al. Klinefelter’s syndrome (47,XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum 2008;58(8):2511–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seillet C, Laffont S, Trémollières F, Rouquié N, Ribot C, Arnal JF, et al. The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor α signaling. Blood 2012;119(2):454–64. [DOI] [PubMed] [Google Scholar]
- Shrestha S, Wershil B, Sarwark JF, Niewold TB, Philipp T, Pachman LM. Lesional and nonlesional skin from patients with untreated juvenile dermatomyositis displays increased numbers of mast cells and mature plasmacytoid dendritic cells. Arthritis Rheum 2010;62(9):2813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999;284(5421):1835–7. [DOI] [PubMed] [Google Scholar]
- Sigurdsson S, Nordmark G, Garnier S, Grundberg E, Kwan T, Nilsson O, et al. A risk haplotype of STAT4 for systemic lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Hum Mol Genet 2008;17(18):2868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson S, Nordmark G, Göring HH, Lindroos K, Wiman AC, Sturfelt G, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet 2005;76(3):528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman GJ. The microbiome in SLE pathogenesis. Nat Rev Rheumatol 2019;15(2):72–4. [DOI] [PubMed] [Google Scholar]
- Sinicato NA, de Oliveira L, Lapa AT, Postal M, de Oliveira Pelicari K, Costallat LTL, et al. Familial aggregation of childhood and adulthood-onset Systemic Lupus Erythematosus. Arthritis Care Res (Hoboken) 2019. [DOI] [PubMed] [Google Scholar]
- Sisirak V, Sally B, D’Agati V, Martinez-Ortiz W, Özçakar ZB, David J, et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 2016;166(1):88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaug B, Assassi S. Type I interferon dysregulation in Systemic Sclerosis. Cytokine 2020;132:154635. [DOI] [PubMed] [Google Scholar]
- Skopelja-Gardner S, An J, Tai J, Tanaka L, Sun X, Hermanson P, et al. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. [DOI] [PMC free article] [PubMed]
- Solans R, Bosch JA, Esteban I, Vilardell M. Systemic sclerosis developing in association with the use of interferon alpha therapy for chronic viral hepatitis. Clin Exp Rheumatol 2004;22(5):625–8. [PubMed] [Google Scholar]
- Soni C, Perez OA, Voss WN, Pucella JN, Serpas L, Mehl J, et al. Plasmacytoid Dendritic Cells and Type I Interferon Promote Extrafollicular B Cell Responses to Extracellular Self-DNA. Immunity 2020;52(6):1022–38.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni C, Reizis B. Self-DNA at the Epicenter of SLE: Immunogenic Forms, Regulation, and Effects. Front Immunol 2019;10:1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer C, Liggitt D, Elkon KB. Ultraviolet B Irradiation Causes Stimulator of Interferon Genes-Dependent Production of Protective Type I Interferon in Mouse Skin by Recruited Inflammatory Monocytes. Arthritis Rheumatol 2017;69(4):826–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet 2010;42(11):985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Gestal M, Calaza M, Gonzalez A. Lack of interaction between systemic lupus erythematosus-associated polymorphisms in TYK2 and IRF5. J Rheumatol. 37. Canada; 2010. p. 676–7; author reply 8. [DOI] [PubMed] [Google Scholar]
- Tang L, Wan P, Wang Y, Pan J, Chen B. Genetic association and interaction between the IRF5 and TYK2 genes and systemic lupus erythematosus in the Han Chinese population. Inflamm Res 2015;64(10):817–24. [DOI] [PubMed] [Google Scholar]
- Tassiulas I, Hu X, Ho H, Kashyap Y, Paik P, Hu Y, et al. Amplification of IFN-alpha-induced STAT1 activation and inflammatory function by Syk and ITAM-containing adaptors. Nat Immunol 2004;5(11):1181–9. [DOI] [PubMed] [Google Scholar]
- Tcherepanova I, Curtis M, Sale M, Miesowicz F, Nicolette C. SAT0193 Results of a randomized placebo controlled phase ia study of AGS-009, a humanized anti-interferon-α monoclonal antibody in subjects with systemic lupus erythematosus. Annals of the Rheumatic Diseases 2013;71(Suppl 3):536–7. [Google Scholar]
- Thanarajasingam U, Muppirala AN, Jensen MA, Ghodke-Puranik Y, Dorschner JM, Vsetecka DM, et al. Type I Interferon Predicts an Alternate Immune System Phenotype in Systemic Lupus Erythematosus. ACR Open Rheumatol 2019;1(8):499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzi E, Tabib T, Papazoglou A, Sembrat J, Trejo Bittar HE, Rojas M, et al. Disparate Interferon Signaling and Shared Aberrant Basaloid Cells in Single-Cell Profiling of Idiopathic Pulmonary Fibrosis and Systemic Sclerosis-Associated Interstitial Lung Disease. Front Immunol 2021;12:595811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen TA, Harmsen HJM, Vila AV, Kurilshikov A, Liefers SC, Zhernakova A, et al. Shared gut, but distinct oral microbiota composition in primary Sjögren’s syndrome and systemic lupus erythematosus. J Autoimmun 2019;97:77–87. [DOI] [PubMed] [Google Scholar]
- van Roon JA, Tesselaar K, Radstake TR. Proteome-wide analysis and CXCL4 in systemic sclerosis. N Engl J Med. 370. United States; 2014. p. 1563–4. [DOI] [PubMed] [Google Scholar]
- Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2018;392(10143):222–31. [DOI] [PubMed] [Google Scholar]
- Walsh RJ, Kong SW, Yao Y, Jallal B, Kiener PA, Pinkus JL, et al. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum 2007;56(11):3784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckerle CE, Franek BS, Kelly JA, Kumabe M, Mikolaitis RA, Green SL, et al. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum 2011;63(4):1044–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckerle CE, Niewold TB. The unexplained female predominance of systemic lupus erythematosus: clues from genetic and cytokine studies. Clin Rev Allergy Immunol 2011;40(1):42–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D, Kea B, Pesich R, Higgs BW, Zhu W, Brown P, et al. Interferon and biologic signatures in dermatomyositis skin: specificity and heterogeneity across diseases. PLoS One 2012;7(1):e29161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Hur S. How RIG-I like receptors activate MAVS. Curr Opin Virol 2015;12:91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York MR, Nagai T, Mangini AJ, Lemaire R, van Seventer JM, Lafyatis R. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and toll-like receptor agonists. Arthritis Rheum 2007;56(3):1010–20. [DOI] [PubMed] [Google Scholar]
- Yu L, Liu P. Cytosolic DNA sensing by cGAS: regulation, function, and human diseases. Signal Transduct Target Ther 2021;6(1):170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LJ, Sen GL, Ward NL, Johnston A, Chun K, Chen Y, et al. Antimicrobial Peptide LL37 and MAVS Signaling Drive Interferon-β Production by Epidermal Keratinocytes during Skin Injury. Immunity 2016;45(1):119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SH, Zhao Y, Xie QB, Jiang Y, Wu YK, Yan B. Aberrant activation of the type I interferon system may contribute to the pathogenesis of anti-melanoma differentiation-associated gene 5 dermatomyositis. Br J Dermatol 2019;180(5):1090–8. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Castillo-Morales A, Jiang M, Zhu Y, Hu L, Urrutia AO, et al. Genes that escape X-inactivation in humans have high intraspecific variability in expression, are associated with mental impairment but are not slow evolving. Mol Biol Evol 2013;30(12):2588–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Yin J, Huang R, Petersen F, Yu X. Meta-analysis reveals an association of STAT4 polymorphisms with systemic autoimmune disorders and anti-dsDNA antibody. Hum Immunol 2013;74(8):986–92. [DOI] [PubMed] [Google Scholar]