

Abstract
Background:
Left ventricular noncompaction cardiomyopathy (LVNC) was discovered half a century ago as a cardiomyopathy with excessive trabeculation and a thin ventricular wall. In the decades since, numerous studies have demonstrated that LVNC primarily impacts left ventricles (LVs), and is often associated with LV dilation and dysfunction. However, owing in part to the lack of suitable mouse models that faithfully mirror the selective LV vulnerability in patients, mechanisms underlying susceptibility of LV to dilation and dysfunction in LVNC remain unknown. Genetic studies have revealed that deletions and mutations in PRDM16 cause LVNC, but previous conditional Prdm16 knockout mouse models do not mirror the LVNC phenotype in patients, and importantly, the underlying molecular mechanisms by which PRDM16 deficiency causes LVNC are still unclear.
Methods:
Prdm16 cardiomyocyte (CM)-specific knockout (Prdm16cKO) mice were generated and analyzed for cardiac phenotypes. RNA sequencing and ChIP sequencing were performed to identify direct transcriptional targets of PRDM16 in CMs. Single cell RNA sequencing in combination with Spatial Transcriptomics were employed to determine CM identity at single cell level.
Results:
CM-specific ablation of Prdm16 in mice caused LV-specific dilation and dysfunction, as well as biventricular noncompaction, which fully recapitulated LVNC in patients. Mechanistically, PRDM16 functioned as a compact myocardium-enriched transcription factor, which activated compact myocardial genes while repressing trabecular myocardial genes in LV compact myocardium. Consequently, Prdm16cKO LV compact myocardial CMs shifted from their normal transcriptomic identity to a transcriptional signature resembling trabecular myocardial CMs and/or neurons. Chamber-specific transcriptional regulation by PRDM16 was in part due to its cooperation with LV-enriched transcription factors Tbx5 and Hand1.
Conclusions:
These results demonstrate that disruption of proper specification of compact CM may play a key role in the pathogenesis of LVNC. They also shed light on underlying mechanisms of LV-restricted transcriptional program governing LV chamber growth and maturation, providing a tangible explanation for the susceptibility of LV in a subset of LVNC cardiomyopathies.
Keywords: LVNC, PRDM16, HEY2, MYCN, NPPA, cardiomyopathy, cardiogenesis, spatiotemporal gene regulation, cell identity maintenance, scRNA-seq, spatial transcriptomics
Introduction
Left ventricular noncompaction cardiomyopathy (LVNC) is the third most prevalent cardiomyopathy characterized by excessive trabeculation (noncompaction) and thin ventricular wall1–3, which predominantly impacts left ventricle4–6. Trabeculation is a meshwork of myocardial protrusions (trabeculae) that extend from the compact ventricular wall into the ventricular lumen in developing heart7, 8. From late embryonic to perinatal stages, trabecular myocardium undergoes compaction, resulting in disappearance of trabeculae and thickening of a tightly-packed ventricular wall, creating a relatively smooth endocardial surface9. However, mounting evidence suggests thinning and dilation of compact myocardium can independently predict adverse clinical outcomes10, 11, whereas noncompaction alone without ventricular dilation is usually asymptomatic12, suggesting that left ventricle (LV) dilation is the most important feature of symptomatic LVNC13, 14.
Classified as a primary genetic cardiomyopathy by AHA15, LVNC has been linked to mutations in genes encoding sarcomeric and cytoskeletal proteins, nuclear membrane proteins, ion channels and transcription factors2, 16, but the mechanisms by which these mutant proteins lead to LVNC have been poorly defined. This is at least in part due to the lack of suitable mouse models to faithfully mirror disease conditions in patients (i.e., selective LV vulnerability) as mouse models for LVNC constructed to date, including Mib1cKO 17, Nb/NblcKO 18 and Nae1cKO 19, all manifest biventricular noncompaction and dilation.
LVNC has been associated with 1p36 syndrome, which has been further linked to deletions in PRDM1620, a gene encoding for a member of the PR domain-containing (PRDM) family of transcriptional regulators and histone methyltransferases21. Mutations in PRDM16 are also known to cause dilated cardiomyopathy20, 22, which is in line with the localization of PRDM16 in murine and human CM nuclei20. This suggests that PRDM16 may function as an important transcriptional regulator in CM. However, PRDM16 has also been elucidated to play versatile roles in brown adipose tissue determination and differentiation23, neural stem cell maintenance and migration24, 25 and hematopoiesis26. Consequently, Prdm16 global knockout mice develop multi-system defects, including ventricular hypoplasia, and die shortly after birth27. Thus, it remains unclear whether PRDM16 causes LVNC via a cell autonomous mechanism in CM or due to a systematic failure of multiple organs.
To address the potential CM autonomous contribution to LVNC, CM-specific Prdm16 knockout mice have been generated with Myh6-Cre, but unlike the germline null mutation, the mutant mice manifested dilated cardiomyopathy 5 months after birth28. Consistent with this postnatal phenotype, another conditional Prdm16 knockout mouse line was created with Mesp1-Cre, which drives Cre expression in anterior mesoderm, including cardiac progenitor cells, and these mice showed age-dependent onset of hypertrophic cardiomyopathy29, a less severe phenotype than that of Myh6-Cre CM-specific Prdm16 knockout mice. Together, these observations point to the possibility that PRDM16 deficiency-induced LVNC may not solely reflect a CM autonomous requirement.
As part of our long-term interest in understanding mechanisms underlying LVNC, we have embarked on our own efforts to create Prdm16 CM-specific knockout mice by using two independent Cre lines: Xmlc2-Cre and cTnT-Cre, which are activated in CM as early as E7.5 with high efficiency30, 31. Contrary to the aforementioned mouse models, we found that both of these lines of CM-specific ablation of Prdm16 (Prdm16cKO) developed LV-specific dilation and biventricular noncompaction, typical of LVNC phenotypes in patients. This is reminiscent of the observation that different CM-specific Cre lines show distinct cardiac phenotypes due to variable recombination efficiencies in different conditional knockout genes32. More importantly, we seized this opportunity to dissect the disease mechanism autonomous to CM, revealing that PRDM16 functions as a compact myocardium-enriched transcription factor required to maintain compact myocardial CM identity. This is accomplished by activating a subset of essential compact myocardial genes (herein referred to as ‘compact genes’) while repressing a subset of trabecular myocardial genes (herein referred to as ‘trabecular genes’). Consequently, the loss of PRDM16 leads to a switch in gene signature associated with compact CM identity to that characteristic of trabecular CM and/or neurons in LV compact myocardium. Our data thus shed critical lights on an LV-specific transcriptional program instituted by PRDM16 and underscore the role of defective compact myocardium in the pathogenesis of LVNC.
Methods
All high-throughput sequencing data have been made publicly available at the Gene Expression Omnibus (GEO, accession No. GSE179393). Other data and study materials are available from the corresponding author upon reasonable request. The sources of reagents and detailed methods are provided in the online-only Data Supplement. All animal procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the University of California San Diego with approved protocol # S01049.
Results
Cardiomyocyte-specific deletion of PRDM16 recapitulates left ventricular noncompaction
To study the function of PRDM16 in the heart, we generated cardiomyocyte (CM)-specific Prdm16 knockout (KO) mice (Prdm16fl/fl::Xmlc2-Cre+/−, hereafter Prdm16cKO) by crossing Prdm16fl/fl mice33 with Xmlc2-Cre mice, which takes advantage of the Xenopus myosin light chain 2 promoter with dramatically enhanced activity and specificity in mouse CM31, 34. Fluorescence in situ hybridization (FISH) revealed Prdm16 expression overlapped with that of Hey2, a well-established marker for compact myocardium35, 36 (Figure 1A). Immunofluorescence (IF) further demonstrated PRDM16 was predominantly localized to CM nuclei of compact myocardium and interventricular septum (IVS) (Figure 1B). In contrast, cardiac transcription factor Nkx2–5 was expressed evenly between compact and trabecular CMs (Figure 1B). The mRNA and protein levels of PRDM16 were greatly reduced in Prdm16cKO mice, confirming specificity of Prdm16 FISH probes and PRDM16 antibody (Figure S1A–S1B). Western blot further confirmed the absence of PRDM16 in whole heart (Figure S1C).
Figure 1. Cardiomyocyte-specific deletion of PRDM16 recapitulates left ventricular noncompaction.
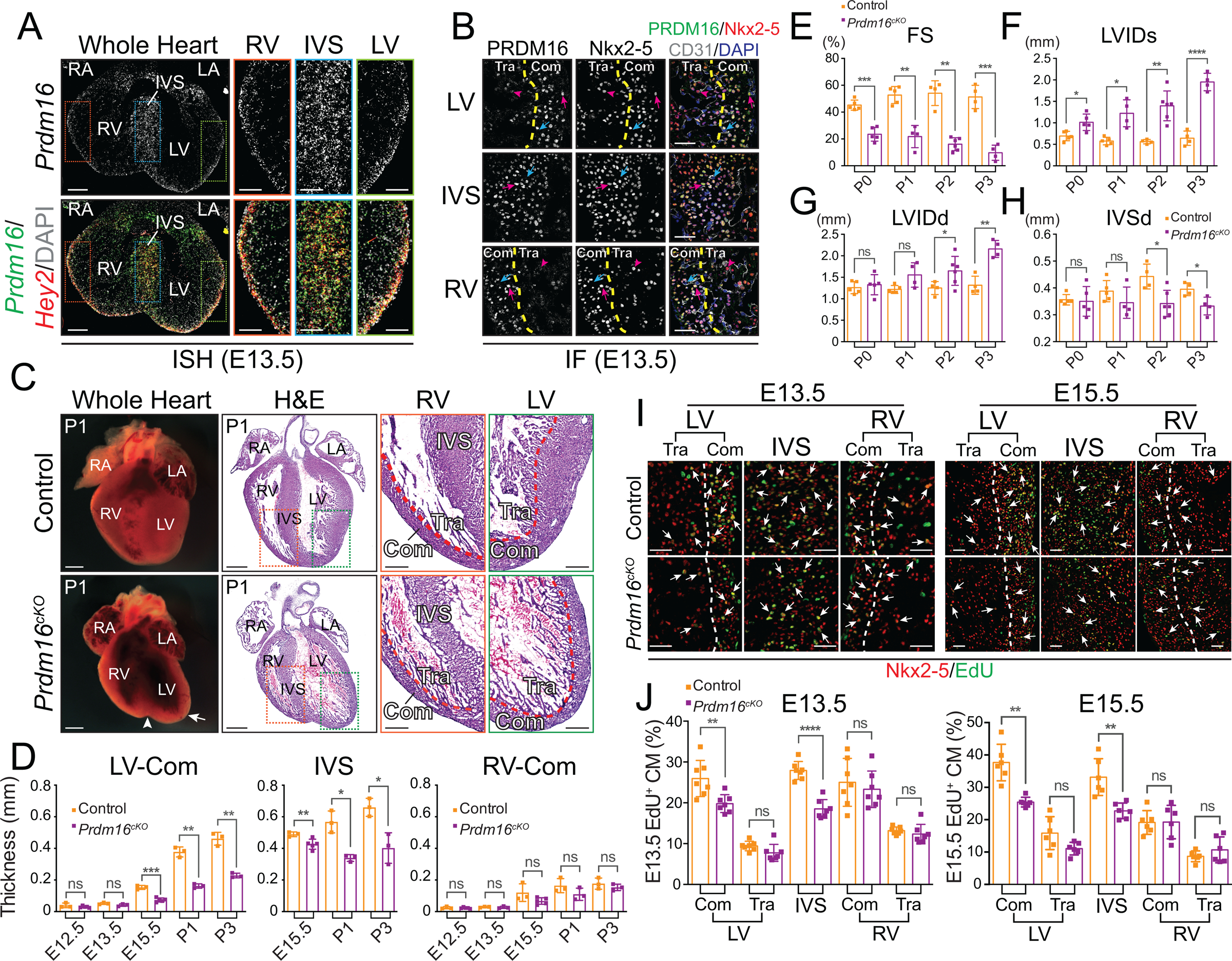
(A) Representative images of Prdm16 and Hey2 in situ hybridization (ISH) performed on E13.5 mouse embryonic heart cryosections. DNA was stained with DAPI. LV, left ventricle. RV, right ventricle. IVS, interventricular septum. LA, left atrium. RA, right atrium. Scale bars, 0.2 mm (whole heart view) and 0.1 mm (magnified view). See also Figure S1A.
(B) Representative images of PRDM16, Nkx2–5 and CD31 immunofluorescences (IF) performed on E13.5 mouse embryonic heart cryosections. Compact myocardium (Com) or IVS CMs (Nkx2–5-positive) with high expression of PRDM16 are indicated by magenta arrows. Trabecular myocardium (Tra) CMs with low expression of PRDM16 are indicated by magenta arrowheads. Endothelial cells (CD31-positive) without expression of PRDM16 are indicated by cyan arrows. The approximal boundaries between compact myocardium and trabecular myocardium are depicted by yellow dashed lines. Scale bars, 50 μm. See also Figure S1B.
(C) Representative wholemount and H&E stained cryosection images of P1 control and Prdm16cKO mouse hearts. White arrowhead indicates a cleft at the apex of Prdm16cKO heart. White arrow indicates the enlarged left ventricle of Prdm16cKO heart. Scale bars, 0.5 mm (whole heart view of wholemount and H&E) and 0.2 mm (magnified view on H&E stained cryosections). See also Figure S1D–G.
(D) Measurements of left ventricular compact myocardium (LV-Com), IVS and right ventricular compact myocardium (RV-Com) thicknesses on control and Prdm16cKO mice (n = 3–5 per group) heart sections (n = 4 per heart) from E12.5 to P3 (IVS thicknesses were measured from E15.5 to P3). Data are represented as mean ± SEM. Statistical significance was determined with two-tailed Student’s t test (ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001).
(E-H) Echocardiographic parameters fraction shortening (FS) (E), left ventricle internal dimension, systolic (LVIDs) (F), left ventricle internal dimension, diastolic (LVIDd) (G) and interventricular septum thickness, diastolic (IVSd) (H) of control and Prdm16cKO neonatal mice (n = 4–6 per group) from P0 to P3. Data are represented as mean ± SEM. Statistical significance was determined with two-tailed Student’s t test (ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
(I-J) Representative images (I) and quantitative analyses (J) of EdU positive (EdU+) CMs from Nkx2–5 and EdU IF performed on E13.5 (left panels) and E15.5 (right panels) control and Prdm16cKO hearts (E13.5: n = 7 hearts per group, n = 3 sections per heart; E15.5: n = 6 hearts per group, n = 3 sections per heart). Arrows indicate examples of proliferating CMs positive for both Nkx2–5 and EdU. White dashed lines indicate the approximal boundaries between compact myocardium and trabecular myocardium. Scale bars, 50 μm. Data are represented as mean ± SEM. Statistical significance was determined with two-tailed Student’s t test (ns, not significant; **p < 0.01, ****p < 0.0001).
In line with PRDM16’s prominent expression in heart, we found that Prdm16cKO mice exhibited cardiac abnormalities, including clefts at the apices of the hearts and enlarged left ventricles as early as embryonic day (E) 15.5 (Figures 1C and S1D), with progressive cardiac dysfunction leading to death of all Prdm16cKO mice before postnatal (P) day 7 (Figure S1I). This fully recapitulates the abnormal heart morphology observed in Prdm16 global KO mice27, indicating that the lost cell autonomous function of PRDM16 in CM is sufficient to account for the morphological defects. By H&E staining, we noticed that Prdm16cKO mice developed biventricular noncompaction (Figures 1C and S1E–S1H), as seen in previous LVNC mouse models17–19. However, unlike the previous models, we also observed dramatic dilation that occurred only in LV of Prdm16cKO mice (Figures 1C and S1E–S1G). Corroborating these observations, measurements of compact myocardium and interventricular septum (IVS) thickness on heart sections demonstrated thinner LV compact myocardium and IVS in Prdm16cKO mice, starting from E15.5, whereas the thickness of RV compact myocardium was not significantly different between Prdm16cKO mice and littermate controls (Figure 1D). As LVNC accompanied by LV dilation and dysfunction in human patients leads to far worse clinical outcomes compared with isolated LVNC12, we then sought to determine the functional consequences of observed abnormal heart morphology in Prdm16cKO mice. To this end, we performed echocardiography on postnatal day (P) 0 to P3 neonatal Prdm16cKO mice and littermate controls, and detected severe cardiac contractile dysfunction (Figure 1E), increased left ventricular chamber size (Figure 1F–1G), and thinned IVS (Figure 1H). These data demonstrated that our model largely mirrored the LVNC phenotype in patients.
Role of PRDM16 in embryonic versus adult cardiomyocyte
Thinned LV compact myocardium and IVS may result from decreased cell proliferation or increased apoptosis in Prdm16cKO mice. To differentiate between these possibilities, we used EdU labeling37 to measure the percentage of proliferating CMs in Prdm16cKO mice and littermate controls. Concordant with specific decreases in thicknesses of LV compact myocardium and IVS, we found significant reductions in EdU-positive CM percentages only in LV compact myocardium and IVS of Prdm16cKO mice at both E13.5 and E15.5 (Figure 1I–1J), developmental stages respectively preceding and following overt morphological abnormalities observed in Prdm16cKO hearts (Figure S1D–S1G). On the other hand, we did not observe any differences in CM apoptosis as measured by cleaved caspase 3 (cCSP3) immunofluorescence38 (Figure S1J–S1K), indicating that thinning of LV compact myocardium and IVS result largely from the reduction in CM proliferation in the critical regions coincident with PRDM16 expression. This is in direct contrast to the lack of impact on CM proliferation when conditional Prdm16fl/fl mice were bred with Mesp1-Cre mice29, suggesting that the observed late onset of the disease phenotype in this mouse model may result from inefficient depletion of the gene during development. To further establish the CM autonomous effect of Prdm16 KO, we used an independent cTnT-Cre line30 to generate Prdm16 CM-specific KO, which again displayed postnatal lethality before P7 and dramatically dilated left ventricle (Figure S1L), indicating that cardiac phenotypes we observed in Prdm16cKO mice were not Cre driver line dependent. Taken together, these findings suggested that PRDM16 was essential to ventricle growth and maturation during embryonic and neonatal heart developmental stages.
Because our data clearly show developmental defects in early embryos, we next sought to elaborate the function of PRDM16 in adult CMs. To this end, we generated tamoxifen-inducible CM-specific Prdm16 knockout mice (Prdm16fl/fl::αMHC-MerCreMer+/−, Prdm16icKO)39 to circumvent perinatal lethality of Prdm16cKO mice. To our surprise, Prdm16icKO mice did not show overt cardiac phenotypes, even 36 weeks following induction (Figure S2A), despite efficient deletion of PRDM16 (Figures S2B). We noticed that PRDM16 protein levels were much lower in adult hearts than postnatal hearts (Figure S2B). To investigate the developmental expression pattern of PRDM16 in the heart, we performed western blot analyses using antibody to PRDM16 with wild type heart samples from embryonic to adult stages, and found that PRDM16 protein levels dramatically decreased after birth (Figure S2C–S2D). This observation is consistent with recently published human and mouse heart RNA-seq data across multiple developmental and adult stages (Figure S2E–S2F)40. Collectively, our findings indicate that PRDM16 plays a negligible role in adult CMs, consistent with the developmental origin of LVNC41.
Loss of PRDM16 leads to widespread gene misregulation in developing heart
The requirement of PRDM16 for ventricle growth and maturation in developing heart suggests that its regulated gene expression program is pivotal to these processes. To determine gene expression differences between control and Prdm16cKO mice, we collected embryonic heart ventricles at E13.5, a developmental stage before Prdm16cKO hearts showed overt cardiac phenotypes, for RNA-seq analysis. In addition, to investigate the underlying mechanism for the selective impact of Prdm16 ablation on LV relative to RV, we performed RNA-seq on dissected LV and RV from E13.5 Prdm16cKO and littermate control mice. Our RNA-seq data verified chamber-specific expression of LV genes (e.g., Hand1 and Tbx5)42, 43 and RV genes (e.g., Pcsk6)44 (Table S1), indicating our data faithfully reflected gene expression differences between LV and RV. Principal component analysis (PCA) revealed a clear separation between Prdm16cKO and control on LV samples but less so on RV samples (Figure 2A), indicating the transcriptomic differences were most pronounced in LV upon loss of PRDM16. Using false discovery rate (FDR) < 0.05, we determined differentially expressed genes (DEGs) in LV with 1,448 downregulated and 1,508 upregulated, which greatly outnumbered those in RV or with whole ventricles (Figure 2B and Table S2). We further noted that commonly altered gene expression events in both chambers tended to be more pronounced in LV than those detected in RV (Figure 2C), suggesting that PRDM16 is involved in a chamber-specific gene expression program.
Figure 2. Loss of PRDM16 leads to widespread gene misregulation in developing heart.
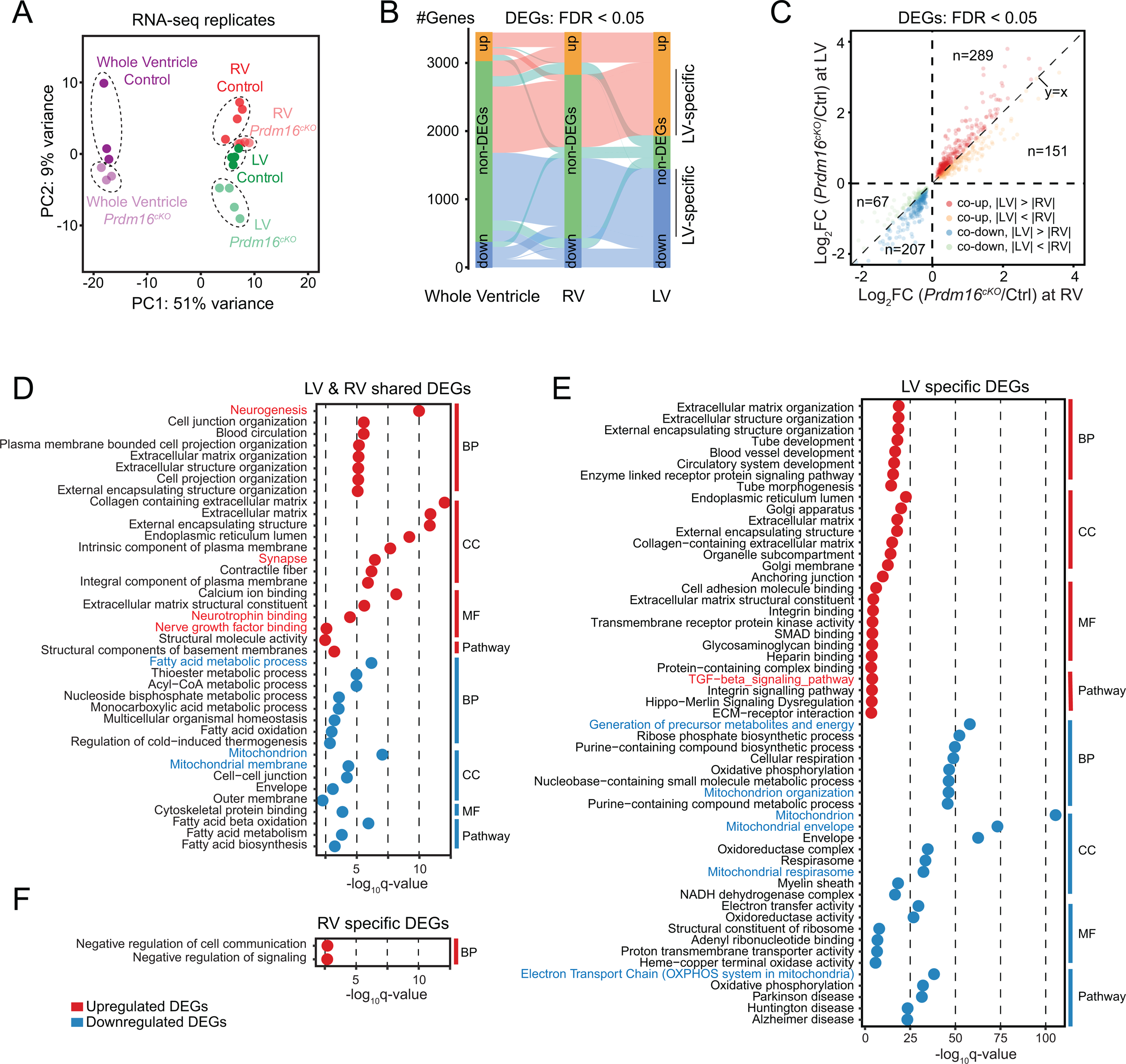
(A) Principal component analysis of RNA-seq data from whole ventricle, left ventricle (LV) and right ventricle (RV) samples of E13.5 Prdm16cKO and littermate control mice.
(B) Diagram showing the overlap of upregulated or downregulated differentially expressed genes (DEGs) from whole ventricle, left ventricle (LV) and right ventricle (RV) samples of E13.5 Prdm16cKO mice compared with littermate controls (FDR < 0.05). See also Table S2.
(C) Correlation analysis of shared DEGs between LV and RV of E13.5 Prdm16cKO mice. Upregulated or downregulated DEGs with bigger fold changes in either LV or RV are indicated by dots with different colors. Gene number of each category is indicated.
(D-F) Gene ontology analysis of LV-RV shared DEGs (D), LV-specific DEGs (E) and RV-specific DEGs (F), which was divided into four categories: biological process (BP), cellular component (CC), molecular function (MF) and KEGG pathway. Notable GO terms or pathways are highlighted in red (upregulated) or blue (downregulated).
See also Figure S3.
Gene ontology (GO) analysis revealed some of the most enriched GO terms in both whole- ventricle-downregulated DEGs and LV-RV-shared downregulated DEGs were associated with mitochondrial biogenesis and function (Figures S3A and 2D), as observed earlier in heart of Prdm16fl/fl::Mesp1-Cre mice29. We further noted that while specifically downregulated genes in LV were mostly enriched in mitochondrial pathways (Figure 2E), those specifically downregulated in RV did not show any significant GO enrichment (Figure 2F). Since mitochondria accounts for nearly all ATP production supporting heart contraction45, impaired mitochondrial function likely contributed to decreased heart contractile function in Prdm16cKO mice (see Figure 1E). We also found that genes involved in fatty acid metabolism were downregulated in both LV and RV (Figure 2D), suggesting that PRDM16 shares some common transcriptional regulatory functions between CM and adipocyte46.
Compared to downregulated genes, the most enriched GO terms in whole-ventricle-upregulated and LV-RV-shared upregulated genes were related to neuronal development pathways (Figures S3A and 2D), indicating that PRDM16 may repress alternative cell fates such as neurons in CMs, similar to the observations made with Nkx2–547 and Tbx2048. We confirmed that Sox3, a gene encoding a transcription factor expressed in neural progenitor cells and critical for neural stem cell maintenance49, was upregulated in E13.5 Prdm16cKO ventricles by immunofluorescence microscopy. Interestingly, similar to PRDM16, Sox3 was predominantly localized to CM nuclei in compact myocardium and its upregulation in Prdm16cKO was more prominent in LV than RV (Figure S3B). Last, but not least, the TGF-β signaling pathway was specifically upregulated in LV compared to RV (Figure 2E), and given the negative role of TGF-β signaling in CM proliferation50, this may partially account for the LV-restricted decrease in CM proliferation in Prdm16cKO mice.
PRDM16 functions as a bifunctional transcription regulator in the genome
DEGs in Prdm16cKO mice may be directly or indirectly regulated by PRDM16. To identify direct targets for PRDM16, we performed PRDM16 chromatin immunoprecipitation deep sequencing (ChIP-seq) on E13.5 mouse heart ventricles. We identified 17,587 peaks in control samples (Figure 3A), compared to only 330 peaks in Prdm16cKO samples, indicative of the high specificity with our ChIP-seq experiments (Figure S4A). Among PRDM16-specific ChIP-seq peaks, 35.7% reside in intergenic regions, 45% in introns, 2.9% in exons and 12.3% at promoters (Figure 3A). Intersection of the PRDM16 ChIP-seq peaks with publicly available histone modification ChIP-seq profiles51 revealed co-occupation of PRDM16 with H3K27ac, an active enhancer marker, H3K4me1, a poised and active enhancer marker, H3K4me3, a marker for promoters, as well as other histone modifications associated with enhancers and promoters (Figure 3B). This is further illustrated on the Nppb-Nppa locus (Figure 3C), which encodes for atrial natriuretic factor and brain natriuretic peptide critical for heart development52.
Figure 3. PRDM16 functions as a bifunctional transcription regulator in the genome.
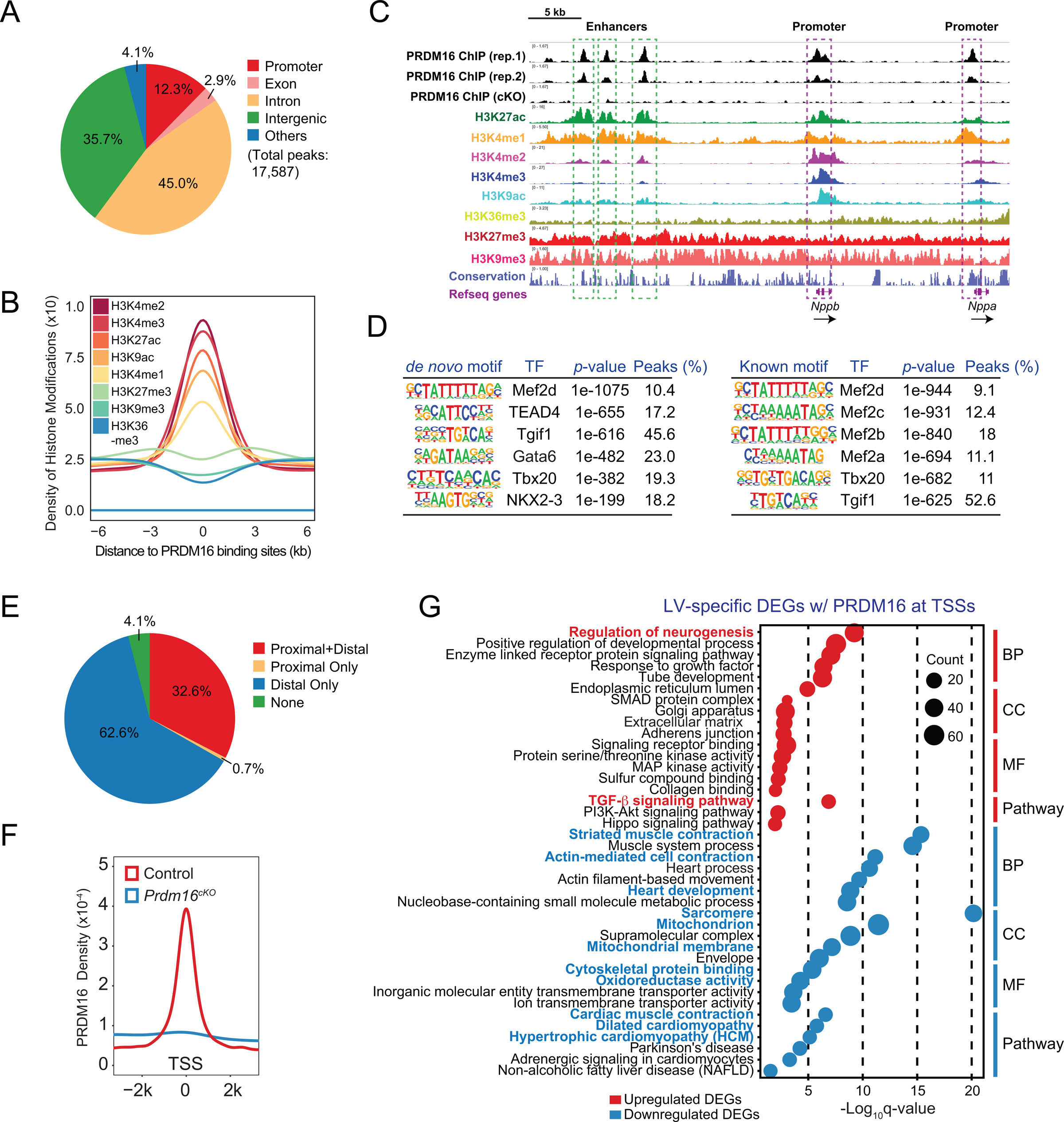
(A) Genomic distributions of PRDM16 ChIP-seq peaks.
(B) H3K4me2, H3K4me3, H3K27ac, H3K9ac, H3K4me1, H3K27me3, H3K9me3 and H3K36me3 profiles within a 12 kb window centered on PRDM16 binding sites.
(C) Integrative genomics viewer (IGV) view showing histone modifications and PRDM16 binding at Nppb-Nppa locus. Enhancers and promoters are indicated by green and purple boxes, respectively. Direction of transcription is indicated by black arrows.
(D) De novo (left panel) and known (right panel) motif analyses of PRDM16 ChIP-seq peaks. See also Figure S4B.
(E) Pie chart showing percentages of whole ventricle DEGs with PRDM16 binding sites within 2 kb of their transcription start sites (TSSs) (proximal), with PRDM16 binding sites within 2 kb-200 kb of their TSSs (distal), with proximal and distal PRDM16 binding sites (proximal+distal) and DEGs without any PRDM16 binding sites within 200 kb of their TSSs (None). See also Figure S4C–D.
(F) PRDM16 binding profiles of control and Prdm16cKO ventricles within a 4 kb window centered on TSSs of whole ventricle DEGs.
(G) Gene ontology analysis of LV-specific DEGs with promoter PRDM16 binding divided into four categories: biological process (BP), cellular component (CC), molecular function (MF) and KEGG pathway. Notable GO terms or pathways are highlighted in red (upregulated) or blue (downregulated).
See also Figure S4.
Motif analysis of PRDM16 ChIP-seq peaks using Homer53 revealed binding motifs for several cardiac transcription factors (TFs), such as MEF2 family TFs, Tbx20, TEAD and GATA family TFs (Figure 3D), pivotal for heart development54, 55. Of note, we did not retrieve the putative PRDM16 binding motif56, which echoes other studies suggesting that PRDM16 functions as a cofactor with other DNA-binding TFs57, 58. Indeed, we observed extensive co-occupation of PRDM16 with Mef2c, Tbx5, Nkx2–559 and Gata460 in embryonic mouse heart (Figure S4B).
By examining the positions of PRDM16 ChIP-seq peaks relative to transcription start sites (TSSs) of E13.5 whole ventricle DEGs, we found that approximately one-third of DEGs had PRDM16 peaks at their promoters and for the 62.6% of DEGs without PRDM16 binding at their promoters, we were able to identify at least one PRDM16 peak within 200 kb of their TSSs (Figure 3E), indicating that PRDM16 may regulate a subset of DEGs by occupying their enhancers. Corroborating this idea, PRDM16 peaks were found on two in vivo validated enhancers of Hey260, a downregulated DEG encoding a transcription factor that has important roles in ventricular wall morphogenesis61 (Figure S4C), as well as on two in vivo validated enhancers of Tbx362, a upregulated DEG which encodes a transcription factor regulating a myriad of gene programs in the cardiac conduction system63 (Figure S4D). These observations implied that PRDM16 is a bifunctional transcriptional regulator, acting to activate or repress gene expression in a context-dependent manner, likely through cooperating with different sets of TFs in different action sites in the genome.
To further establish such context-dependent action of PRDM16, we chose to focus on DEGs that showed PRDM16 binding at their promoters (Figure 3F), thus likely to be its direct targets (Table S3). Ontology analysis on LV-specific, RV-specific or LV-RV common DEGs with PRDM16 binding at their promoters revealed LV-specific downregulated target genes were enriched in GO terms directly related to heart development and function (Figure 3G), whereas similar analysis performed on RV-specific or LV-RV-shared downregulated target genes did not recover any of these GO terms (Figure S4E). On the other hand, both LV-specific and LV-RV-shared upregulated target genes were enriched for neuronal development pathways (Figures 3G and S4E), suggesting that PRDM16 directly suppresses the expression of neural genes in both ventricles. Together, these findings suggest that PRMD16 may act as a transcription activator to drive cardiogenesis, but as a transcription repressor to restrict neurogenesis in developing heart.
Cooperation of PRDM16 with LV-enriched TFs
While PRDM16 depletion clearly was more impactful on LV than RV, our FISH, immunostaining, and RNA-seq studies found little significant difference in PRDM16 expression between LV and RV (see Figures 1A, 1B and Table S1). To further confirm this, we dissected LVs and RVs from E13.5 mouse embryos and performed western blots to compare their PRDM16 protein levels. Concordant with our previous data, western blot analyses of PRDM16 levels showed that LV had the same amount of PRDM16 as RV. In comparison, using Tbx5 as a known LV-enriched TF42, we confirmed that LV had more than twice the amount of Tbx5 than RV (Figure 4A–4B).
Figure 4. Cooperation of PRDM16 with LV-enriched TFs.
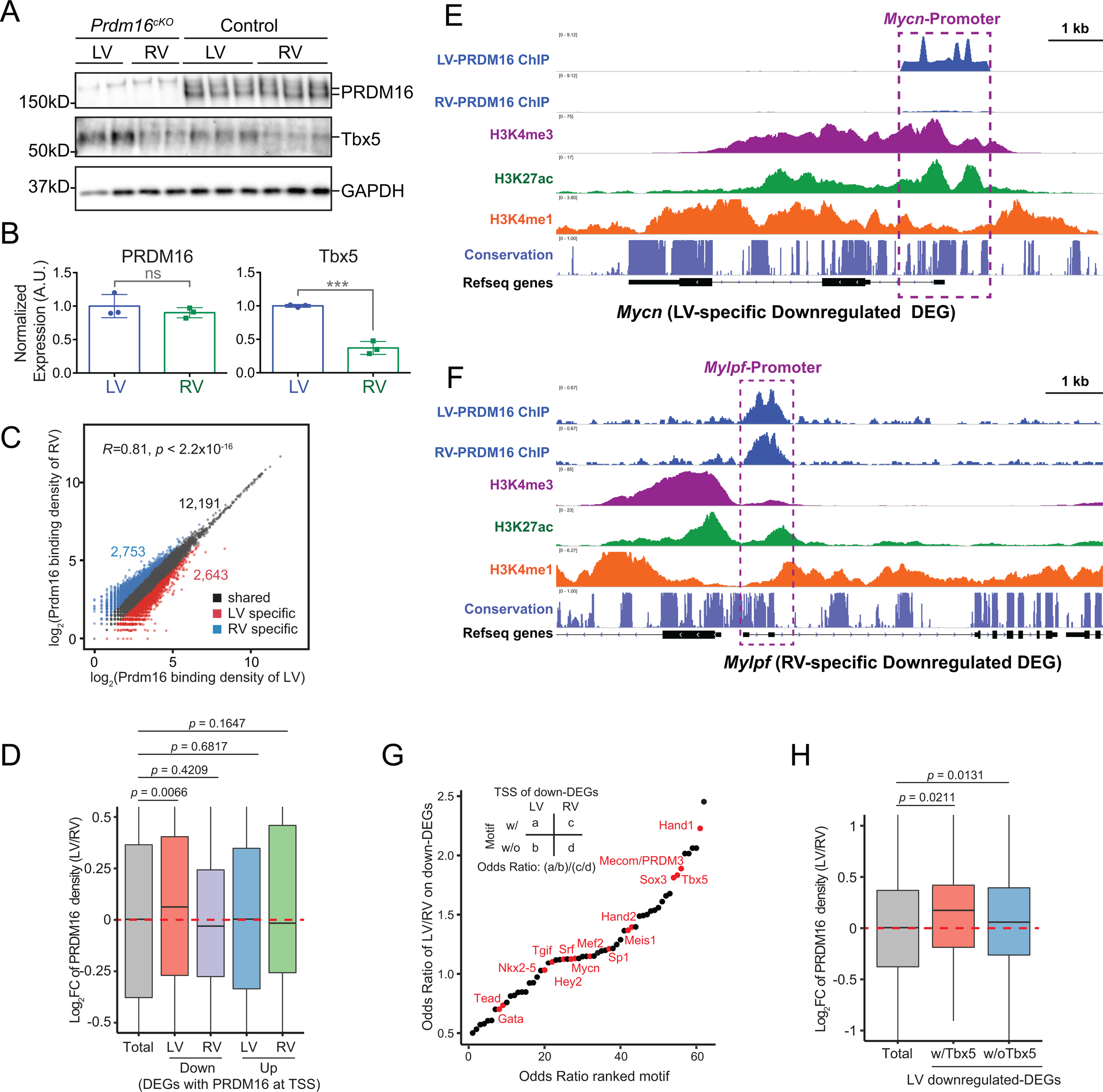
(A-B) PRDM16 and Tbx5 western blots (A) and their densitometry quantification results (B) normalized to GAPDH using protein samples extracted from LV or RV of E13.5 wild type embryos (n = 3 per group). Samples from LV or RV of Prdm16cKO embryos were used as negative controls. Data are represented as mean ± SEM. Statistical significance was determined with two-tailed Student’s t test (ns, not significant; ***p < 0.001).
(C) Correlation analysis of LV and RV PRDM16 binding intensity at PRDM16 ChIP-seq peaks calculated from whole ventricle PRDM16 ChIP-seq data. The threshold for defining LV- or RV-specific peaks was set at 1.5 folds.
(D) Ratio of PRDM16 binding density between LV and RV at genomic regions as indicated. Statistical significance was determined with permutation test with 1,000,000 iterations.
(E-F) IGV view showing histone modifications and LV-, RV-PRDM16 binding at Hey2 (E) and Mylpf (F) loci. Promoters are indicated by purple boxes.
(G) Relative transcription factor motif enrichment at TSS of LV-specific or RV-specific downregulated DEGs. Notable TF motifs were highlighted in red. See also Figure S5.
(H) Ratio of PRDM16 binding density between LV and RV at promoters of LV-specific downregulated DEGs, with or without the presence of Tbx5 motif. Statistical significance was determined with permutation test with 1,000,000 iterations.
Since the LV-restricted regulation of cardiac genes cannot be explained by chamber-specific expression of PRDM16 in LV, we then asked if PRDM16 had differential affinity for key target genes in LV versus RV. To this end, we performed PRDM16 ChIP-seq separately on LV or RV dissected from E13.5 mouse embryos. A majority of LV and RV PRDM16 peaks had similar binding intensities (Figure 4C), indicating that PRDM16 generally occupies similar genomic loci in both ventricles. However, we identified 2,643 LV-specific peaks and 2,753 RV-specific peaks (Figure 4C), underscoring the chamber-specific binding preference of PRDM16 at these loci. We then compared LV and RV PRDM16 binding intensities at the promoters of LV-specific or RV-specific DEGs, and found PRDM16 binding in LV was significantly stronger than that observed in RV at promoters of LV-specific downregulated DEGs (Figure 4D). In contrast, PRDM16 binding at promoters of RV-specific downregulated DEGs or LV-, RV-specific upregulated DEGs were not significantly different between two chambers (Figure 4D). For example, the promoter of Mycn, which was specifically downregulated in LV of Prdm16cKO mice, showed much greater PRDM16 binding in LV than RV (Figure 4E). On the contrary, Mylpf, which was specifically downregulated in RV of Prdm16cKO mice (Table S2), had nearly identical PRDM16 binding at its promoter between LV and RV (Figure 4F). These findings indicated that PRDM16 had higher affinity for LV-specific downregulated genes in LV than RV, which may explain why the loss of PRDM16 in Prdm16cKO mice has a much larger impact on the expression of these genes in LV.
We then analyzed TF binding motifs at promoters of LV- or RV-specific downregulated DEGs. Interestingly, we found motifs of LV-enriched TF Hand1 and Tbx5 were among the highest-ranked TF motifs for LV-specific downregulated DEGs (Figure 4G). In fact, PRDM16 peaks harboring the Tbx5 motif generally had higher density in LV than RV at the promoters of LV-specific downregulated DEGs (Figure 4H), suggesting that PRDM16 may cooperate with LV-enriched TFs, such as Hand1 and Tbx5, to activate the expression of its target genes in LV. On the other hand, Hand1 and Tbx5 were less enriched at promoters of LV-specific upregulated DEGs (Figure S5), indicating these LV-enriched TFs mainly function as activators when cooperating with PRDM16 in LV. Interestingly, another high-rank TF motif associated with LV-specific downregulated DEGs corresponded to the Mecom/PRDM3 motif (Figure 4G). As the PRDM16 DNA binding motif is almost identical to the PRDM3 motif56, this result suggested that PRDM16 may act as a direct DNA-binding TF at a subset of LV-specific downregulated DEGs.
PRDM16 activates compact genes while repressing trabecular genes in LV compact myocardium and IVS
To gain further mechanistic insight into LV- and IVS-specific dilation in Prdm16cKO mice, we focused on LV-specific downregulated target genes Hey2 and Mycn as they encode compact myocardium-restricted transcription factors35, 64 essential for ventricle wall morphogenesis61, 65. We confirmed LV-specific downregulation of Hey2 and Mycn in Prdm16cKO mice using qRT-PCR (Figure S6A). Strikingly, by FISH, we found that the expression of both Hey2 and Mycn was drastically decreased in LV compact myocardium and IVS, but remained unchanged in RV myocardium of Prdm16cKO mice (Figure 5A–5B). Notably, the differential FISH signals appeared more pronounced than qRT-PCR signals, particularly with Hey2 (Figures 5A, 5B and S6A). Consequently, the expression of Mlc2a/Myl7 and Tbx5, which are normally repressed by Hey2 in compact myocardium35, 61, were increased in IVS and LV compact myocardium (Figure S6B). Furthermore, we observed the ectopic expression of the trabecular myocardium marker gene Nppa35 in the IVS and some regions of the LV compact myocardium of Prdm16cKO mice (Figure 5A–5B). Consistent with this, the trabecular-compact expression gradient, where the expression of trabecular genes (e.g., Nppa) gradually decreases from trabecular myocardium to compact myocardium44, disappeared in LVs of Prdm16cKO mice, as the expression of Nppa expanded to the base of the trabeculae (Figure 5A–5B). This suggests that the gradient of gene expression from trabecular myocardium to compact myocardium was disrupted. Moreover, the chemokine ligand gene Cxcl12 was also dramatically decreased in LV compact myocardium but not in LV trabeculae of Prdm16cKO mice (Figure 5C). Collectively, these findings indicated that PRDM16 helps maintain compact myocardial CM identity by activating expression of compact myocardial genes (e.g., Hey2 and Mycn), while repressing trabecular myocardial genes (e.g., Nppa) in LV compact myocardium and IVS (Figure 5D).
Figure 5. PRDM16 activates compact genes while repressing trabecular genes in LV compact myocardium and IVS.
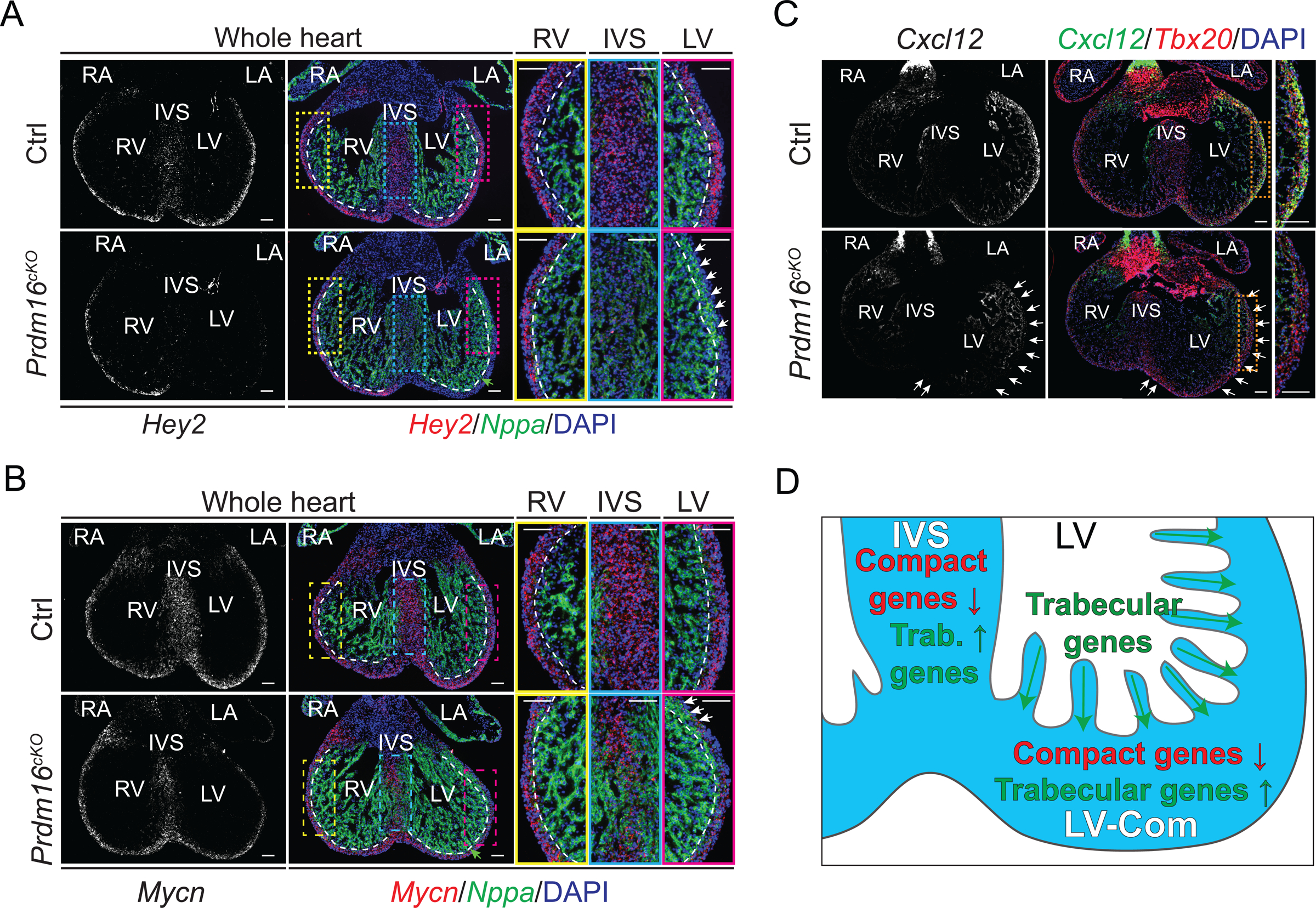
(A-B) Representative images of Hey2 (A) or Mycn (B) and Nppa in situ hybridization (ISH) performed on E13.5 mouse embryonic heart cryosections. DNA was stained with DAPI. LV, left ventricle. RV, right ventricle. IVS, interventricular septum. LA, left atrium. RA, right atrium. White arrows indicate ectopic expression of Nppa in LV compact myocardium. Green arrow indicates the expression of Nppa expands to base of trabeculae in LV. White dashed lines indicate the approximal boundaries between compact myocardium and trabecular myocardium. Scale bars, 0.1 mm.
(C) Representative images of Cxcl12 and Tbx20 (compact myocardium marker) in situ hybridization (ISH) performed on E13.5 mouse embryonic heart cryosections. LV compact myocardium where Cxcl12 was specifically reduced in Prdm16cKO is indicated by white arrows. Scale bars, 0.1 mm.
(D) Illustration depicting the expression of compact myocardium marker genes (compact genes) were reduced in compact myocardium and IVS of Prdm16cKO mice, whereas the expression of trabecular myocardium marker genes (trabecular genes) expanded from trabeculation to compact myocardium and IVS.
See also Figure S6.
Single-cell RNA-seq uncovers a PRDM16-anchored transcription network in LV
Reduced expression of a subset of compact CM marker genes and ectopic expression of a subset of trabecular CM marker genes suggest that PRDM16 deficiency may cause improper specification of compact CM identity in LV compact myocardium. To test this hypothesis, we performed single-cell RNA-seq (scRNA-seq), which provides much greater resolution than bulk RNA-seq66, to compare between Prdm16cKO and control mice. To focus on CMs in scRNA-seq, we crossed a Rosa26-tdTomato reporter transgene67 into the Prdm16cKO background to label CMs with tdTomato, which enabled fluorescence-activated cell sorting (FACS) to obtain pure single CM preparations (Figures 6A and S7A). We obtained 7,783 high quality scRNA-seq profiles from two control and two Prdm16cKO samples. Unsupervised clustering separated CMs (Nkx2–5+), epicardial cells (Tcf21+)68 and atrioventricular cushion mesenchyme cells (Postn+)69 (Figure 6B). Owing to the FACS enrichment of CMs prior to scRNA-seq, our dataset predominantly consists of CMs (Figure 6B). We also identified clusters of outflow tract (OFT) CMs that expressed the OFT marker gene Rspo344 (Figure S7B).
Figure 6. Single-cell RNA-seq uncovers a PRDM16-anchored transcription network in LV.
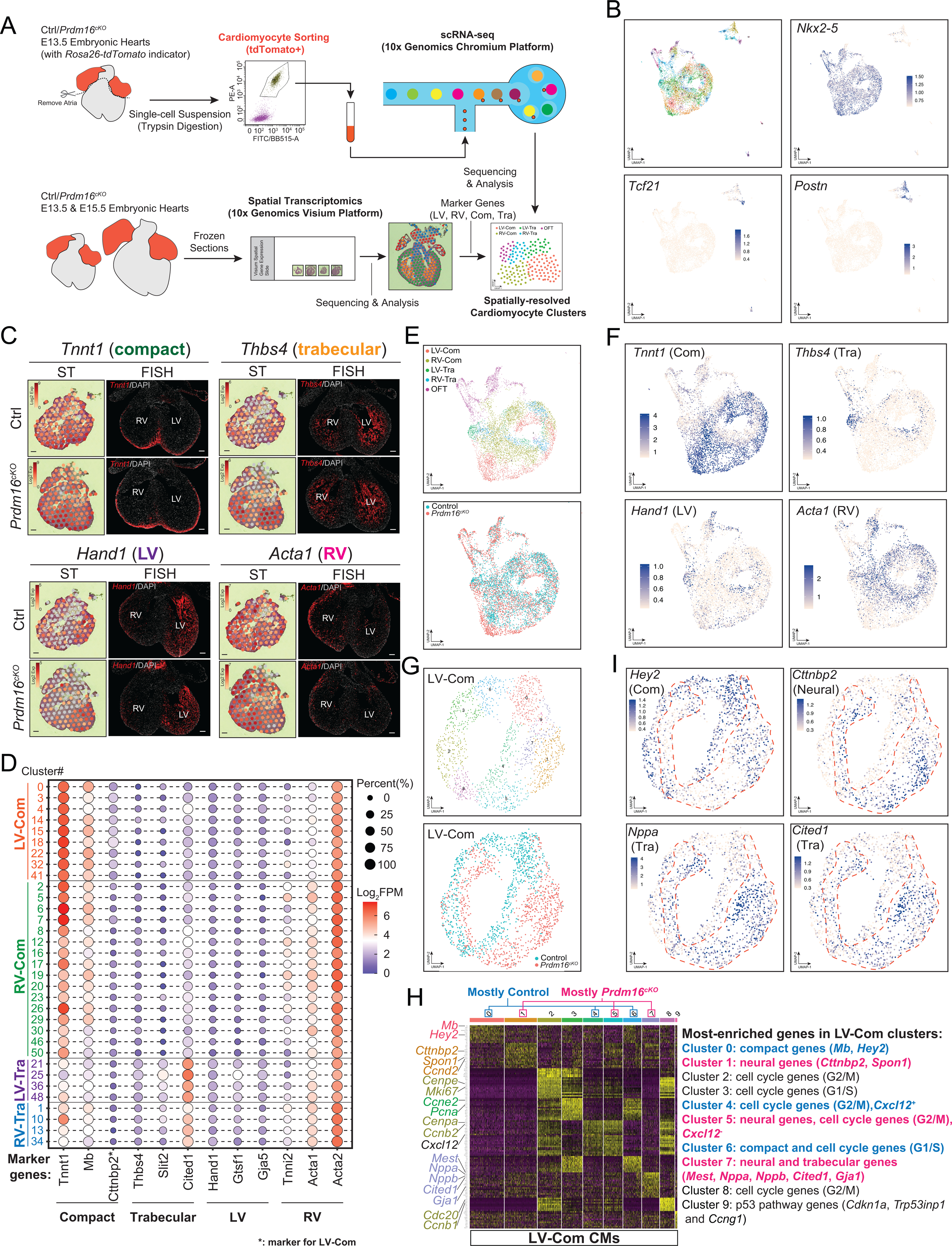
(A) Overview of single-cell RNA-seq (scRNA-seq) and spatial transcriptomics (ST) experimental design.
(B) UMAP plots of FACS-sorted E13.5 CMs, showing the expression of CM marker gene Nkx2–5, epicardial cell maker Tcf21 and atrioventricular cushion mesenchyme cell marker Postn. See also Figure S7B.
(C) ST expression profiles and representative FISH images of selected anatomical marker genes in E13.5 control and Prdm16cKO hearts. Scale bars, 0.1 mm. See also Figure S7I and Figure S7J.
(D) Dot plot of CM clusters that were grouped into LV compact myocardial CMs (LV-Com), RV compact myocardial CMs (RV-Com), LV trabecular myocardial CMs (LV-Tra) and RV trabecular myocardial CMs (RV-Tra) based on marker gene expression. FPM, fragments per million reads.
(E) UMAP plots of CM populations assigned to their original anatomical locations (upper panel) or genotypes (lower panel).
(F) UMAP plots of selected anatomical marker gene expression in CM populations. See also Figure S8A.
(G) UMAP plots of LV compact myocardial CMs assigned to subclusters (upper panel) or genotypes (lower panel). See also Figure S8B.
(H) Heatmap showing most-enriched genes in subclusters of LV compact myocardial CMs. See also Figure S8C.
(I) UMAP plots of LV compact myocardial CMs showing selected subcluster marker genes. Red dashed lines indicate the approximate positions of Prdm16cKO LV compact myocardial CMs in UMAP plot. See also Figure S8D.
To facilitate mapping CM clusters to their original locations in the heart, we also performed Spatial Transcriptomics (ST)70 on E13.5 and E15.5 control and Prdm16cKO hearts (Figure 6A). Unsupervised clustering of each sample generated four clusters that mapped back to their original locations (Figure S7C–S7D). To identify marker genes for trabecular myocardium and compact myocardium, we selected their corresponding ST spots and calculated their top enriched genes that were consistent across all samples (Figure S7E–S7F) and validated some of them by FISH (Figure 6C). We also discovered Cttnbp2, a gene encoding a neural-specific F-actin regulator71, as a specific marker for LV compact myocardium of Prdm16cKO hearts and validated it with FISH (Figure S7I). We then used similar approach, combining with LV and RV bulk RNA-seq data (Table S1), to identify LV and RV marker genes (Figure S7G–S7H), some of which were validated by FISH (Figures 6C and S7J).
The assignment of anatomical marker genes enabled us to assign scRNA-seq CM clusters to their original locations (Figures 6D–6F and S8A). As PRDM16 is predominantly expressed in compact myocardium (see Figure 1A–1B), we extracted and reclustered LV-Com CMs and RV-Com CMs and found that control and Prdm16cKO RV-Com CMs were transcriptionally indistinguishable (Figure S8B–S8C). By contrast, control and Prdm16cKO CMs of LV-Com were segregated into different clusters, underscoring their significant transcriptomic differences (Figure 6G). Corroborating our findings by bulk RNA-seq that LV compact CMs in Prdm16cKO ectopically expressed trabecular genes and neural genes, we found that the Prdm16cKO LV-Com CMs formed a cluster enriched with trabecular genes, such as Nppa, Cited1, Nppb and Mest (Cluster 7) as well as clusters enriched with neural genes, such as Cttnbp2 and Spon1 (Clusters 1, 5, 7 ) (Figures 6H- 6I and S8D), thus further establishing the role of PRDM16 in maintaining the compact CM identity by repressing alternative CM (trabeculation) and other cell fate (neuron) genes. Conversely, control LV-Com CMs showed two clusters (Clusters 0, 6) that were enriched with compact genes, such as Hey2 and Mb (Figure 6H), which were markedly decreased in Prdm16cKO LV-Com CMs (Figures 6I and S8D). These data confirmed that PRDM16 activates compact genes in LV compact myocardium. Interestingly, Cluster 4 enriched for Cxcl12 was exclusively included in control CMs (Figure 6G–6H), consistent with its specific downregulation in LV compact myocardium of Prdm16cKO mice (see Figure 5C).
To investigate the underlying gene regulatory network (GRN) in compact myocardial CMs, we applied SCENIC72 to our control and Prdm16cKO compact CMs single-cell data and found twenty TF regulatory modules in all compact CMs (Figure S8E). Interestingly, the activity of five TFs was specifically altered in LV-Com CMs but not in RV-Com CMs upon loss of PRDM16 (Figure S8F). Among the TFs whose activity decreased were Pgc-1α (Ppargc1a), Hey2 and Gata6. On the other hand, the activity of neural TF Sox3 and cardiac conduction system TF Tbx3 were increased. These findings thus provide evidence that PRDM16 plays a key role in orchestrating a transcriptional regulatory network in LV compact myocardium to promote compact CM gene expression while inhibiting inappropriate gene expression of alternative cell fate.
Discussion
The most intriguing question of LVNC is the susceptibility of the LV to disease, in particular dilation and dysfunction. This phenomenon has been poorly recapitulated in previous LVNC animal models in which noncompaction and dilation seem to occur at similar severity in both ventricles17–19. Our current work demonstrated that the Prdm16cKO LVNC model is inherently advantageous in studying LV susceptibility in LVNC as the dilation was restricted to LV. This enabled us to use RV as internal control for identifying critical molecular changes responsible for LV-specific phenotypes. Through RNA-seq and PRDM16 ChIP-seq, we identified compact myocardial marker genes Hey2 and Mycn as PRDM16 direct target genes, both of which are essential for CM proliferation in compact myocardium61, 65. In line with the specific reduction of CM proliferation in LV and IVS of Prdm16cKO mice, these genes were only reduced in these specific areas of the heart, suggesting that their regulation by PRDM16 is spatially restricted.
Although the biventricular noncompaction phenotype might be attributed to misregulation of mitochondrial genes and neural genes that was observed in both ventricles (Figure 2D), the LV-specific dilation indicated that PRDM16 might specifically regulate genes in LV, not in RV, that are essential to ventricular wall morphogenesis. As we did not find any significant differences in PRDM16 protein levels between the two ventricles (Figure 4A), how would PRDM16 selectively regulate these genes in LV? To investigate this, we performed RNA-seq and PRDM16 ChIP-seq independently on each ventricle and found that PRDM16 appeared to be ‘left-handed’, as loss of PRDM16 resulted in significantly decreased expression of a subset of critical target genes in LV, that were barely affected in RV. Motif analyses suggested that this preferential effect of PRDM16 on this subset of LV targets might occur through cooperation with LV-enriched TFs, like Tbx5 and Hand1. Our findings thus provide a tangible explanation for LV susceptibility in PRDM16-related LVNC and open a new avenue for future mechanistic studies on the etiology of LVNC.
To further tackle the spatially restricted function of PRDM16, we performed scRNA-seq in combination with spatial transcriptomic analysis. Using these advanced functional genomics technologies, we now systematically identified anatomical marker genes for E13.5 and E15.5 mouse hearts. We then applied them to our scRNA-seq data and successfully mapped CM clusters back to their anatomical locations, together illustrating how the identity of compact myocardial CMs was compromised in Prdm16cKO mice. To our knowledge, this is the first integration of scRNA-seq with Spatial Transcriptomics on embryonic mouse heart, which provides a rich resource for studying spatial gene expression at single-cell level during mammalian heart development.
Spatially resolved scRNA-seq data led to the discovery that some LV compact myocardial CMs of Prdm16cKO transcriptionally resembled trabecular myocardial CMs or even neurons, suggesting PRDM16 is instrumental in maintaining the identity of compact CM. Since compact myocardial CMs are generally more proliferative than trabecular CMs36, 73, the loss of compact CM identity in LV compact myocardium and IVS of Prdm16cKO animals thereby resulted in the reduction of CM proliferation that ultimately led to LV dilation and dysfunction according to our working model (Figure 7). This observation is reminiscent of PRDM16’s role in brown adipose tissue (BAT) where it activates BAT genes while repressing white adipose tissue genes46, and similarly in common progenitor cells of BAT and skeletal muscle, PRDM16 activates BAT genes while repressing skeletal muscle genes74. Together these findings all point to a general role of PRDM16 in determining cell fate by promoting a set of lineage-specific genes while inhibiting other sets of genes critical for inappropriate cell fates.
Figure 7. Working model of PRDM16’s transcriptional regulation in the heart.
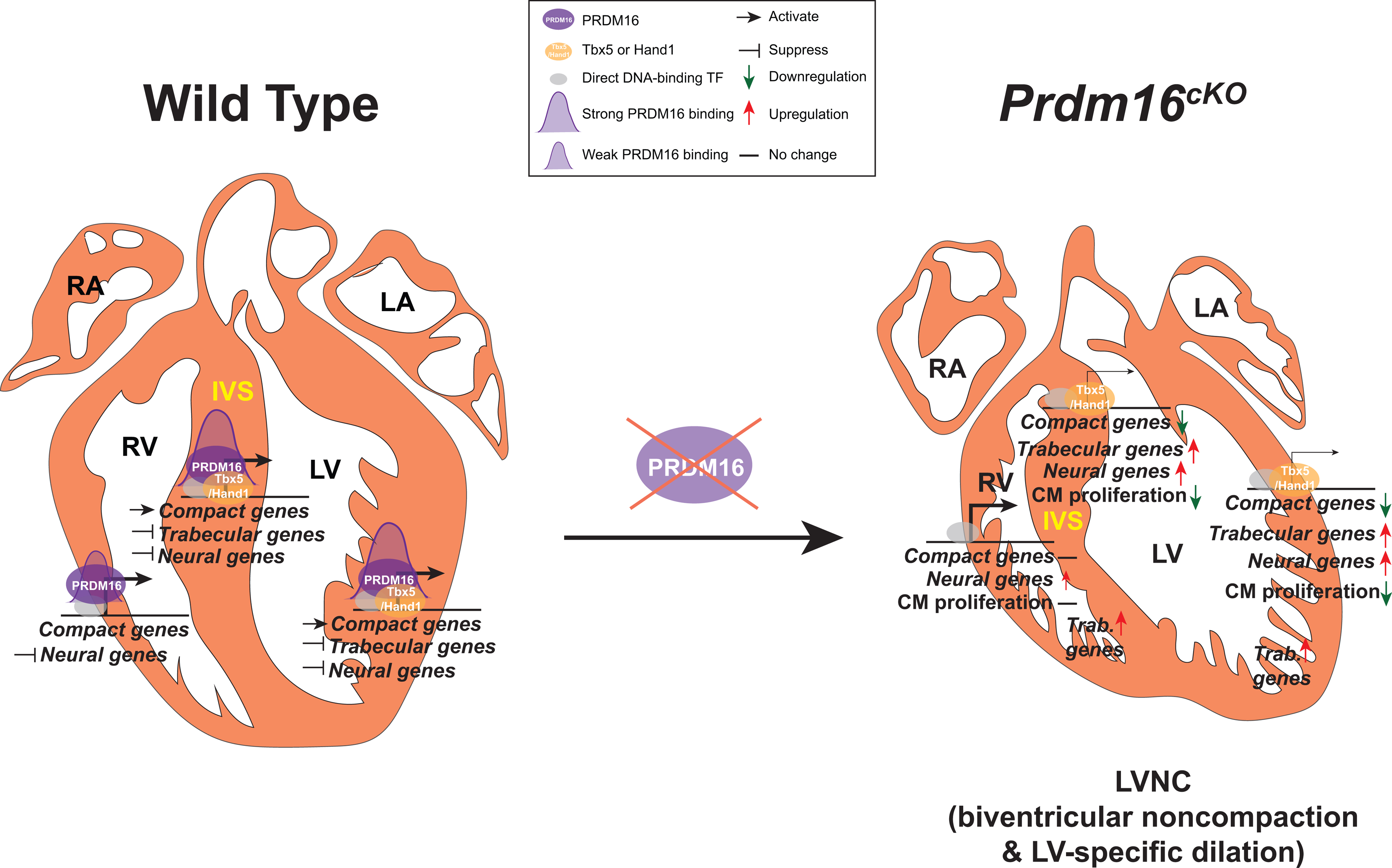
PRDM16 (purple ovals) expression is restricted to left ventricle (LV)-, right ventricle (RV)-compact myocardium and interventricular septum (IVS). PRDM16 is recruited by direct DNA-binding cardiac transcription factors (grey ovals) to the promoters of its target genes. In LV compact myocardium and IVS, PRDM16 cooperates with LV-enriched Tbx5 or Hand1 to activate the transcription of compact genes and has stronger binding at their promoters (PRDM16 binding intensity is depicted by the size of purple peaks). PRDM16 also suppresses the transcription of trabecular and neural genes in LV compact myocardium and IVS, as well as the transcription of neural genes in RV compact myocardium. The loss of PRDM16 in CMs led to dramatic downregulation of compact genes and upregulation of trabecular genes specifically in LV compact myocardium and IVS, whereas the expression of compact genes and trabecular genes in RV compact myocardium did not change. Despite neural genes were upregulated in both LV and RV compact myocardium, the upregulation tended to be smaller in RV. The compromised compact gene program in LV compact myocardium and IVS results in decreased CM proliferation and ultimately causes LVNC with LV-specific dilation in Prdm16cKO mice. The upregulation of trabecular genes in both LV and RV trabecular myocardium, along with other commonly misregulated genes in LV and RV, may account for the biventricular noncompaction observed in Prdm16cKO mice.
A major unsolved puzzle in pathogenesis of LVNC is the relationship between noncompaction and ventricular dilation. A recent study found that decreased CM proliferation in compact myocardium alone was sufficient to cause noncompaction36. Our findings seemed to validate this observation as we saw decreased CM proliferation in LV compact myocardium of Prdm16cKO mice, and noncompaction was indeed evident in LV. However, we also found noncompaction in RV without any changes in CM proliferation. More importantly, we only saw ventricular dilation in LV. These observations strongly suggest that noncompaction and ventricular dilation are independent phenomena, and noncompaction is not necessarily caused by defects in CM proliferation.
Our findings that LV compact CM of Prdm16cKO mice expressed decreased levels of compact CM marker genes but ectopically expressed trabecular CM marker genes (Figure 5D), are opposite to observations in previously characterized LVNC-mimicking Mib1cKO mice17, which barely expressed trabecular CM marker genes but ectopically expressed compact CM marker genes in their trabecular CMs. Although the trabecular/compact marker gene misregulation of Prdm16cKO and Mib1cKO largely took place in compact or trabecular myocardium, both models developed noncompaction and ventricular dilation in LV. Considered together, we propose that proper specification of either compact CM or trabecular CM is important for normal morphogenesis of the ventricular wall, and that disruption of specification of either leads to LVNC.
Supplementary Material
Clinical Perspective.
What is new?
Cardiomyocyte specific knockout of Prdm16 leads to left ventricular noncompaction cardiomyopathy with LV-specific dilation and dysfunction.
PRDM16 maintains compact myocardial cardiomyocyte transcriptomic identity by activating compact myocardial genes while repressing trabecular myocardial genes in LV.
PRDM16 cooperates with LV-enriched transcription factors to specifically regulate transcription in LV.
What are the clinical implications?
This study provides a unique LVNC mouse model for developing therapeutic interventions for LV dilation and dysfunction, which emerge as the most important disease features in LVNC.
Improper specification of compact or trabecular cardiomyocytes is likely a common mechanism in pathogenesis of LVNC.
Spatial single-cell gene expression profiles in normal or diseased conditions facilitate future studies for identifying new drug targets for LVNC.
Acknowledgements
We thank Patrick Seale (University of Pennsylvania) for providing PRDM16 antibody. We thank Yoshitake Cho (University of California San Diego) for providing wild type embryonic and adult mouse heart protein samples. We are grateful to Kristen Jepsen (UCSD IGM Genomics Center), Jennifer Santini (UCSD Microscopic Core Facility, supported by NIH grant P30NS047101) and Jesus Olvera (UCSD Stem Cell Genomics Core) for their technical assistance.
Sources of Funding
J.C. and S.M.E. are funded by grants from the National Heart, Lung, and Blood Institute and J.C. holds an American Heart Association Endowed Chair in Cardiovascular Research.
Non-standard Abbreviations and Acronyms
- LVNC
left ventricular noncompaction cardiomyopathy
- LV
left ventricle
- RV
right ventricle
- IVS
interventricular septum
- PRDM16
PR domain-containing 16
- HEY2
Hes related family bHLH transcription factor with YRPW motif 2
- MYCN
N-Myc proto-oncogene, bHLH transcription factor
- NPPA
natriuretic peptide A
- CM
cardiomyocyte
- DEG
differentially expressed genes
- ChIP-seq
chromatin immunoprecipitation deep sequencing
- TF
transcription factor
- TSS
transcription start site
- FACS
fluorescence-activated cell sorting
- scRNA-seq
single-cell RNA sequencing
- ST
spatial transcriptomics
- GRN
gene regulatory network
Footnotes
Disclosures
None.
Supplemental Materials
References:
- 1.Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, Davis AM, Kahler SG, Chow CW, Wilkinson JL, et al. The epidemiology of childhood cardiomyopathy in Australia. The New England journal of medicine. 2003;348:1639–1646. [DOI] [PubMed] [Google Scholar]
- 2.Finsterer J, Stollberger C, Towbin JA. Left ventricular noncompaction cardiomyopathy: cardiac, neuromuscular, and genetic factors. Nat Rev Cardiol. 2017;14:224–237. [DOI] [PubMed] [Google Scholar]
- 3.Towbin JA, Lorts A, Jefferies JL. Left ventricular non-compaction cardiomyopathy. Lancet. 2015;386:813–825. [DOI] [PubMed] [Google Scholar]
- 4.Nucifora G, Aquaro GD, Masci PG, Pingitore A, Lombardi M. Magnetic resonance assessment of prevalence and correlates of right ventricular abnormalities in isolated left ventricular noncompaction. Am J Cardiol. 2014;113:142–146. [DOI] [PubMed] [Google Scholar]
- 5.Pignatelli RH, McMahon CJ, Dreyer WJ, Denfield SW, Price J, Belmont JW, Craigen WJ, Wu J, El Said H, Bezold LI, et al. Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation. 2003;108:2672–2678. [DOI] [PubMed] [Google Scholar]
- 6.Tigen K, Karaahmet T, Gurel E, Cevik C, Basaran Y. Biventricular noncompaction: a case report. Echocardiography. 2008;25:993–996. [DOI] [PubMed] [Google Scholar]
- 7.Ben-Shachar G, Arcilla RA, Lucas RV, Manasek FJ. Ventricular trabeculations in the chick embryo heart and their contribution to ventricular and muscular septal development. Circ Res. 1985;57:759–766. [DOI] [PubMed] [Google Scholar]
- 8.Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol Rev. 2003;83:1223–1267. [DOI] [PubMed] [Google Scholar]
- 9.Sedmera D, Pexieder T, Vuillemin M, Thompson RP, Anderson RH. Developmental patterning of the myocardium. Anat Rec. 2000;258:319–337. [DOI] [PubMed] [Google Scholar]
- 10.Andreini D, Pontone G, Bogaert J, Roghi A, Barison A, Schwitter J, Mushtaq S, Vovas G, Sormani P, Aquaro GD, et al. Long-Term Prognostic Value of Cardiac Magnetic Resonance in Left Ventricle Noncompaction: A Prospective Multicenter Study. J Am Coll Cardiol. 2016;68:2166–2181. [DOI] [PubMed] [Google Scholar]
- 11.Amzulescu MS, Rousseau MF, Ahn SA, Boileau L, de Meester de Ravenstein C, Vancraeynest D, Pasquet A, Vanoverschelde JL, Pouleur AC, Gerber BL. Prognostic Impact of Hypertrabeculation and Noncompaction Phenotype in Dilated Cardiomyopathy: A CMR Study. JACC Cardiovasc Imaging. 2015;8:934–946. [DOI] [PubMed] [Google Scholar]
- 12.van Waning JI, Caliskan K, Michels M, Schinkel AFL, Hirsch A, Dalinghaus M, Hoedemaekers YM, Wessels MW, AS IJ, Hofstra RMW, et al. Cardiac Phenotypes, Genetics, and Risks in Familial Noncompaction Cardiomyopathy. J Am Coll Cardiol. 2019;73:1601–1611. [DOI] [PubMed] [Google Scholar]
- 13.Anderson RH, Jensen B, Mohun TJ, Petersen SE, Aung N, Zemrak F, Planken RN, MacIver DH. Key Questions Relating to Left Ventricular Noncompaction Cardiomyopathy: Is the Emperor Still Wearing Any Clothes? Can J Cardiol. 2017;33:747–757. [DOI] [PubMed] [Google Scholar]
- 14.Sen-Chowdhry S, McKenna WJ. Left ventricular noncompaction and cardiomyopathy: cause, contributor, or epiphenomenon? Curr Opin Cardiol. 2008;23:171–175. [DOI] [PubMed] [Google Scholar]
- 15.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB, American Heart A, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Chen H, Shou W. Potential Common Pathogenic Pathways for the Left Ventricular Noncompaction Cardiomyopathy (LVNC). Pediatr Cardiol. 2018;39:1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luxan G, Casanova JC, Martinez-Poveda B, Prados B, D’Amato G, MacGrogan D, Gonzalez-Rajal A, Dobarro D, Torroja C, Martinez F, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med. 2013;19:193–201. [DOI] [PubMed] [Google Scholar]
- 18.Hirai M, Arita Y, McGlade CJ, Lee KF, Chen J, Evans SM. Adaptor proteins NUMB and NUMBL promote cell cycle withdrawal by targeting ERBB2 for degradation. J Clin Invest. 2017;127:569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou J, Ma W, Li J, Littlejohn R, Zhou H, Kim IM, Fulton DJR, Chen W, Weintraub NL, Zhou J, et al. Neddylation mediates ventricular chamber maturation through repression of Hippo signaling. Proc Natl Acad Sci U S A. 2018;115:E4101–E4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arndt AK, Schafer S, Drenckhahn JD, Sabeh MK, Plovie ER, Caliebe A, Klopocki E, Musso G, Werdich AA, Kalwa H, et al. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet. 2013;93:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hohenauer T, Moore AW. The Prdm family: expanding roles in stem cells and development. Development. 2012;139:2267–2282. [DOI] [PubMed] [Google Scholar]
- 22.Long PA, Evans JM, Olson TM. Diagnostic Yield of Whole Exome Sequencing in Pediatric Dilated Cardiomyopathy. J Cardiovasc Dev Dis. 2017;4(3):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, Tavernier G, Langin D, Spiegelman BM. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007;6:38–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimada IS, Acar M, Burgess RJ, Zhao Z, Morrison SJ. Prdm16 is required for the maintenance of neural stem cells in the postnatal forebrain and their differentiation into ependymal cells. Genes Dev. 2017;31:1134–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baizabal JM, Mistry M, Garcia MT, Gomez N, Olukoya O, Tran D, Johnson MB, Walsh CA, Harwell CC. The Epigenetic State of PRDM16-Regulated Enhancers in Radial Glia Controls Cortical Neuron Position. Neuron. 2018;98:945–962 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aguilo F, Avagyan S, Labar A, Sevilla A, Lee DF, Kumar P, Lemischka IR, Zhou BY, Snoeck HW. Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood. 2011;117:5057–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bjork BC, Turbe-Doan A, Prysak M, Herron BJ, Beier DR. Prdm16 is required for normal palatogenesis in mice. Hum Mol Genet. 2010;19:774–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nam JM, Lim JE, Ha TW, Oh B, Kang JO. Cardiac-specific inactivation of Prdm16 effects cardiac conduction abnormalities and cardiomyopathy-associated phenotypes. Am J Physiol Heart Circ Physiol. 2020;318:H764–H777. [DOI] [PubMed] [Google Scholar]
- 29.Cibi DM, Bi-Lin KW, Shekeran SG, Sandireddy R, Tee N, Singh A, Wu Y, Srinivasan DK, Kovalik JP, Ghosh S, et al. Prdm16 Deficiency Leads to Age-Dependent Cardiac Hypertrophy, Adverse Remodeling, Mitochondrial Dysfunction, and Heart Failure. Cell reports. 2020;33:108288. [DOI] [PubMed] [Google Scholar]
- 30.Jiao K, Kulessa H, Tompkins K, Zhou Y, Batts L, Baldwin HS, Hogan BL. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003;17:2362–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breckenridge R, Kotecha S, Towers N, Bennett M, Mohun T. Pan-myocardial expression of Cre recombinase throughout mouse development. Genesis. 2007;45:135–144. [DOI] [PubMed] [Google Scholar]
- 32.Chen JW, Zhou B, Yu QC, Shin SJ, Jiao K, Schneider MD, Baldwin HS, Bergelson JM. Cardiomyocyte-specific deletion of the coxsackievirus and adenovirus receptor results in hyperplasia of the embryonic left ventricle and abnormalities of sinuatrial valves. Circ Res. 2006;98:923–930. [DOI] [PubMed] [Google Scholar]
- 33.Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell. 2014;156:304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu C, Spinozzi S, Chen JY, Fang X, Feng W, Perkins G, Cattaneo P, Guimaraes-Camboa N, Dalton ND, Peterson KL, et al. Nexilin Is a New Component of Junctional Membrane Complexes Required for Cardiac T-Tubule Formation. Circulation. 2019;140:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koibuchi N, Chin MT. CHF1/Hey2 plays a pivotal role in left ventricular maturation through suppression of ectopic atrial gene expression. Circ Res. 2007;100:850–855. [DOI] [PubMed] [Google Scholar]
- 36.Tian X, Li Y, He L, Zhang H, Huang X, Liu Q, Pu W, Zhang L, Li Y, Zhao H, et al. Identification of a hybrid myocardial zone in the mammalian heart after birth. Nat Commun. 2017;8(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guimaraes-Camboa N, Stowe J, Aneas I, Sakabe N, Cattaneo P, Henderson L, Kilberg MS, Johnson RS, Chen J, McCulloch AD, et al. HIF1alpha Represses Cell Stress Pathways to Allow Proliferation of Hypoxic Fetal Cardiomyocytes. Dev Cell. 2015;33:507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu T, Mu Y, Bogomolovas J, Fang X, Veevers J, Nowak RB, Pappas CT, Gregorio CC, Evans SM, Fowler VM, et al. HSPB7 is indispensable for heart development by modulating actin filament assembly. Proc Natl Acad Sci U S A. 2017;114:11956–11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. [DOI] [PubMed] [Google Scholar]
- 40.Cardoso-Moreira M, Halbert J, Valloton D, Velten B, Chen C, Shao Y, Liechti A, Ascencao K, Rummel C, Ovchinnikova S, et al. Gene expression across mammalian organ development. Nature. 2019;571:505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choquet C, Nguyen THM, Sicard P, Buttigieg E, Tran TT, Kober F, Varlet I, Sturny R, Costa MW, Harvey RP, et al. Deletion of Nkx2–5 in trabecular myocardium reveals the developmental origins of pathological heterogeneity associated with ventricular non-compaction cardiomyopathy. PLoS Genet. 2018;14:e1007502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruneau BG, Logan M, Davis N, Levi T, Tabin CJ, Seidman JG, Seidman CE. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev Biol. 1999;211:100–108. [DOI] [PubMed] [Google Scholar]
- 43.Barnes RM, Firulli BA, Conway SJ, Vincentz JW, Firulli AB. Analysis of the Hand1 cell lineage reveals novel contributions to cardiovascular, neural crest, extra-embryonic, and lateral mesoderm derivatives. Dev Dyn. 2010;239:3086–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li G, Xu A, Sim S, Priest JR, Tian X, Khan T, Quertermous T, Zhou B, Tsao PS, Quake SR, et al. Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Dev Cell. 2016;39:491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dorn GW, 2nd, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015;29:1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chi J, Cohen P. The Multifaceted Roles of PRDM16: Adipose Biology and Beyond. Trends Endocrinol Metab. 2016;27:11–23. [DOI] [PubMed] [Google Scholar]
- 47.Luna-Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MA, He D, Small EM, Mileikovsky M, et al. Complex Interdependence Regulates Heterotypic Transcription Factor Distribution and Coordinates Cardiogenesis. Cell. 2016;164:999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakabe NJ, Aneas I, Shen T, Shokri L, Park SY, Bulyk ML, Evans SM, Nobrega MA. Dual transcriptional activator and repressor roles of TBX20 regulate adult cardiac structure and function. Hum Mol Genet. 2012;21:2194–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bergsland M, Ramskold D, Zaouter C, Klum S, Sandberg R, Muhr J. Sequentially acting Sox transcription factors in neural lineage development. Genes Dev. 2011;25:2453–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kodo K, Ong SG, Jahanbani F, Termglinchan V, Hirono K, InanlooRahatloo K, Ebert AD, Shukla P, Abilez OJ, Churko JM, et al. iPSC-derived cardiomyocytes reveal abnormal TGF-beta signalling in left ventricular non-compaction cardiomyopathy. Nat Cell Biol. 2016;18:1031–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Man J, Barnett P, Christoffels VM. Structure and function of the Nppa-Nppb cluster locus during heart development and disease. Cell Mol Life Sci. 2018;75:1435–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bruneau BG. Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb Perspect Biol. 2013;5:a008292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishikata I, Sasaki H, Iga M, Tateno Y, Imayoshi S, Asou N, Nakamura T, Morishita K. A novel EVI1 gene family, MEL1, lacking a PR domain (MEL1S) is expressed mainly in t(1;3)(p36;q21)-positive AML and blocks G-CSF-induced myeloid differentiation. Blood. 2003;102:3323–3332. [DOI] [PubMed] [Google Scholar]
- 57.Harms MJ, Lim HW, Ho Y, Shapira SN, Ishibashi J, Rajakumari S, Steger DJ, Lazar MA, Won KJ, Seale P. PRDM16 binds MED1 and controls chromatin architecture to determine a brown fat transcriptional program. Genes Dev. 2015;29:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baizabal JM, Mistry M, Garcia MT, Gomez N, Olukoya O, Tran D, Johnson MB, Walsh CA, Harwell CC. The Epigenetic State of PRDM16-Regulated Enhancers in Radial Glia Controls Cortical Neuron Position. Neuron. 2018; 99(1):239–24. [DOI] [PubMed] [Google Scholar]
- 59.Akerberg BN, Gu F, VanDusen NJ, Zhang X, Dong R, Li K, Zhang B, Zhou B, Sethi I, Ma Q, et al. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat Commun. 2019;10(1):4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He A, Gu F, Hu Y, Ma Q, Ye LY, Akiyama JA, Visel A, Pennacchio LA, Pu WT. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat Commun. 2014;5:4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xin M, Small EM, van Rooij E, Qi X, Richardson JA, Srivastava D, Nakagawa O, Olson EN. Essential roles of the bHLH transcription factor Hrt2 in repression of atrial gene expression and maintenance of postnatal cardiac function. Proc Natl Acad Sci U S A. 2007;104:7975–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Weerd JH, Badi I, van den Boogaard M, Stefanovic S, van de Werken HJ, Gomez-Velazquez M, Badia-Careaga C, Manzanares M, de Laat W, Barnett P, et al. A large permissive regulatory domain exclusively controls Tbx3 expression in the cardiac conduction system. Circ Res. 2014;115:432–441. [DOI] [PubMed] [Google Scholar]
- 63.Bakker ML, Boukens BJ, Mommersteeg MT, Brons JF, Wakker V, Moorman AF, Christoffels VM. Transcription factor Tbx3 is required for the specification of the atrioventricular conduction system. Circ Res. 2008;102:1340–1349. [DOI] [PubMed] [Google Scholar]
- 64.Moens CB, Stanton BR, Parada LF, Rossant J. Defects in heart and lung development in compound heterozygotes for two different targeted mutations at the N-myc locus. Development. 1993;119:485–499. [DOI] [PubMed] [Google Scholar]
- 65.Harmelink C, Peng Y, DeBenedittis P, Chen H, Shou W, Jiao K. Myocardial Mycn is essential for mouse ventricular wall morphogenesis. Dev Biol. 2013;373:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paik DT, Cho S, Tian L, Chang HY, Wu JC. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat Rev Cardiol. 2020;17:457–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tandon P, Miteva YV, Kuchenbrod LM, Cristea IM, Conlon FL. Tcf21 regulates the specification and maturation of proepicardial cells. Development. 2013;140:2409–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Snider P, Hinton RB, Moreno-Rodriguez RA, Wang J, Rogers R, Lindsley A, Li F, Ingram DA, Menick D, Field L, et al. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ Res. 2008;102:752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Asp M, Giacomello S, Larsson L, Wu C, Furth D, Qian X, Wardell E, Custodio J, Reimegard J, Salmen F, et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell. 2019;179:1647–1660 e19. [DOI] [PubMed] [Google Scholar]
- 71.Shih PY, Lee SP, Chen YK, Hsueh YP. Cortactin-binding protein 2 increases microtubule stability and regulates dendritic arborization. J Cell Sci. 2014;127:3521–3534. [DOI] [PubMed] [Google Scholar]
- 72.Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li G, Tian L, Goodyer W, Kort EJ, Buikema JW, Xu A, Wu JC, Jovinge S, Wu SM. Single cell expression analysis reveals anatomical and cell cycle-dependent transcriptional shifts during heart development. Development. 2019;146(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454(7207):961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.