

Abstract
Protein switches perform essential roles in many biological processes and are exciting targets for de novo protein design, which aims to produce proteins of arbitrary shape and functionality. However, the biophysical requirements for switch function — multiple conformational states, fine-tuned energetics, and stimuli-responsiveness — pose a formidable challenge for design by computation (or intuition). A variety of methods have been developed towards tackling this challenge, usually taking inspiration from the wealth of sequence and structural information available for naturally occurring protein switches. More recently, modular switches have been designed computationally, and new methods have emerged for sampling unexplored structure space, providing promising new avenues towards the generation of purpose-built switches and de novo signaling systems for cellular engineering.
Introduction
Protein switches are ubiquitous and essential facilitators of biological function that link input signals to output responses. Most broadly, signal transduction can be described as a conformational response to some input perturbation that leads to a functional output (Figure 1A). Inputs for biomolecular switches span diverse stimuli such as ligand binding, environmental changes (e.g., temperature, pH), and post-translational modifications. Outputs can also be diverse, including changes in oligomeric state, affinity for binding partners, and the exposure or burial of functional sites. Structural responses range from subtle (e.g., shifts in populations of conformational states, coupled rearrangements of sidechains) to striking (e.g., involving large hinge-like motions of secondary structure elements or entire domains, and fold switching). The mechanisms underlying conformational coupling associated with switching are typically intricate, with fine-tuned energetics to regulate the kinetics, dynamic range, and specificity of the switch.
Figure 1 |. What makes a protein switch?.
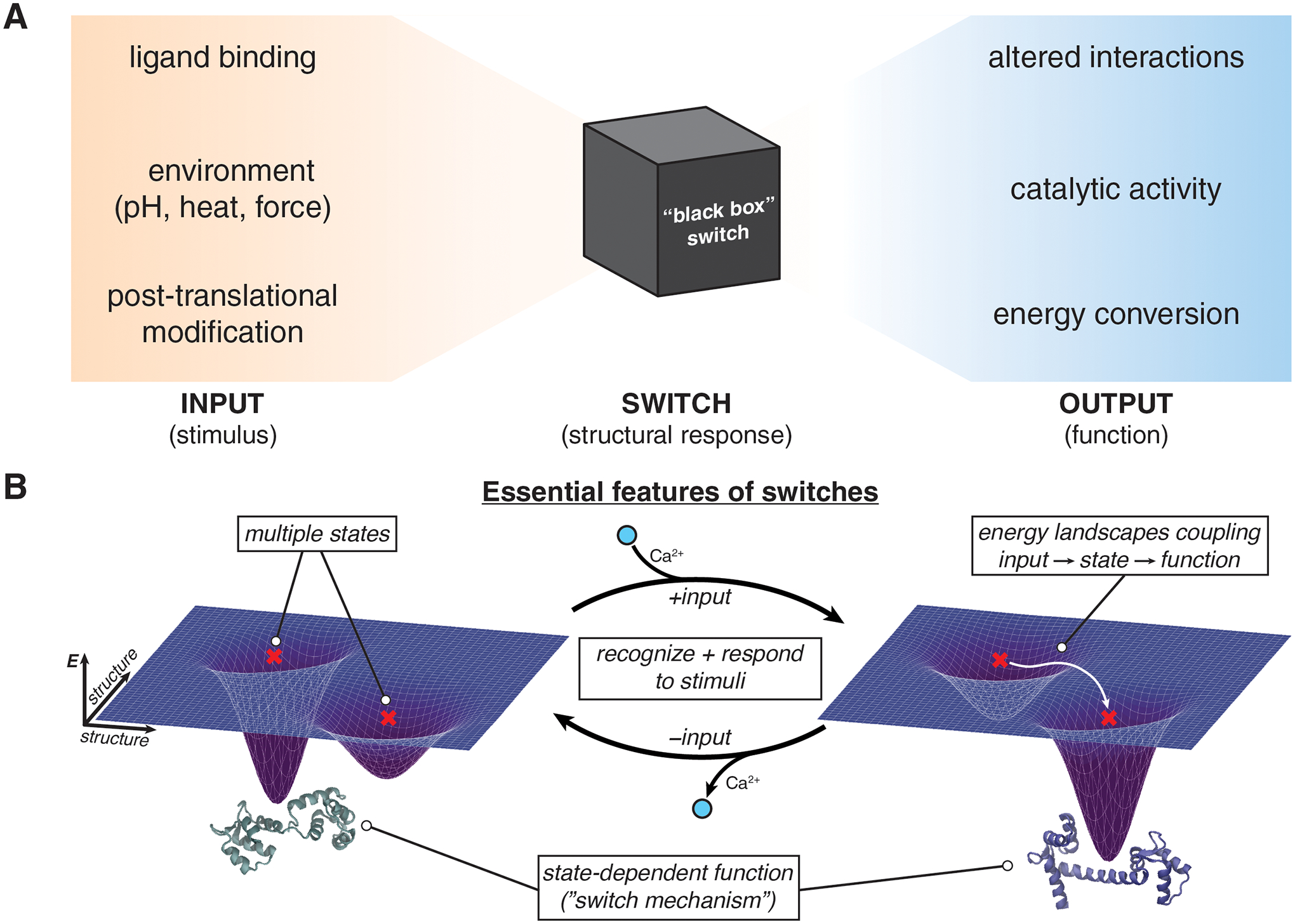
A, Protein switches couple inputs to functional outputs via structural responses. Due to the diversity of inputs and outputs, and the ability of variable structural responses to connect different stimuli and functions, protein switches are often envisioned as a “black box”. In contrast, as illustrated in B, engineering controllable switches de novo requires a specific and “designable” structural definition of the switch mechanism. Protein switch function arises from four essential components: the presence of multiple structural states, with distinct functions, that are differentially populated in response to an input, owing to their suitable energy landscapes. We illustrate these components with the natural example of calmodulin, a Ca2+-binding signaling protein.
In nature, switches are employed as regulatory elements, mediators of energy conversion in molecular machines, and drivers of mechanical mobility. This broad utility (but also challenging functionality) of natural switches has rendered this class of proteins particularly attractive as targets for de novo design. De novo design could generate “idealized” and modular switches that can be more easily controlled and coupled to diverse in- and outputs. Conceptually, the design of new switches includes (1) defining a switch mechanism, (2) generating the desired endstates/conformational ensembles, (3) engineering an input to the switch, and (4) tuning the energy landscapes underpinning switch function (Figure 1B). We begin with a classification of switch mechanisms occurring in nature, then summarize switches that have been designed (often inspired by natural switches, Table 1), and end with a perspective for the future of designed switches, highlighting recent methodological advances and unsolved challenges.
Table 1 |.
Summary of designed protein switches inspired from natural examples
| Scale of structural change | Natural examples | Design objective | Design methods |
|---|---|---|---|
| Allostery/local changes in geometry | Many receptors (e.g. GPCRs), enzymes (e.g. kinases), and signaling domains (e.g. GTPases) | Sequence compatible with multiple geometries (ranging from conformational changes of side chains to those of entire secondary structure elements) | Saturation mutagenesis of key structural residues and experimental screening [25] |
| Design of mutually incompatible, conformationally frustrated states [26,33] | |||
| Design on backbone ensemble approximating conformational landscape [34] | |||
| Introduction of phosphorylation sites [42] | |||
| Fold switching | XCL1, KaiB, Mad2, CLIC1 | Balance between multiple intrachain contact networks corresponding to distinct topologies | Computational modeling of the sequence fitness landscape [21,43] ⱡ |
| Sequence hybridization [4,5,19,20,24] | |||
| Rational mutation of key structural residues [27] | |||
| Explicit design of distinct intrachain contact networks [11] | |||
| Differential domain assembly | SHP-2, Src family kinases | Stimulus-triggered formation or dissociation of interdomain contacts | Modular allostery [15] |
| Tuning of intrachain contact vs. ligand binding strength [16,30] |
Purely in silico without experimental validation of switch designs
Natural switches
Allostery and local changes in geometry
Perhaps the most common mechanism of switching in nature are residue-level conformational changes, often resulting in altered loop conformations or distinct spatial orientations of secondary structure elements, that change specific geometries important for function (Figure 2A). Although these changes are often subtle and relatively local, they can have drastic functional consequences, resulting in the activation or inactivation of enzymes, signaling domains, and receptors such as kinases [1], GTPases [2], and G-protein coupled receptors (GPCRs) [3]. Perhaps counterintuitively, while dramatic global topological changes have been designed successfully [4,5], bistable and more subtle local conformational changes have yet to be realized in de novo design of functional switches. It is precisely the level of subtlety that makes de novo switch design at this scale difficult to achieve, as it is challenging to accurately capture the relatively small energy differences between conformational states during computational design and structure prediction.
Figure 2 |. Examples of switch mechanisms found in nature.
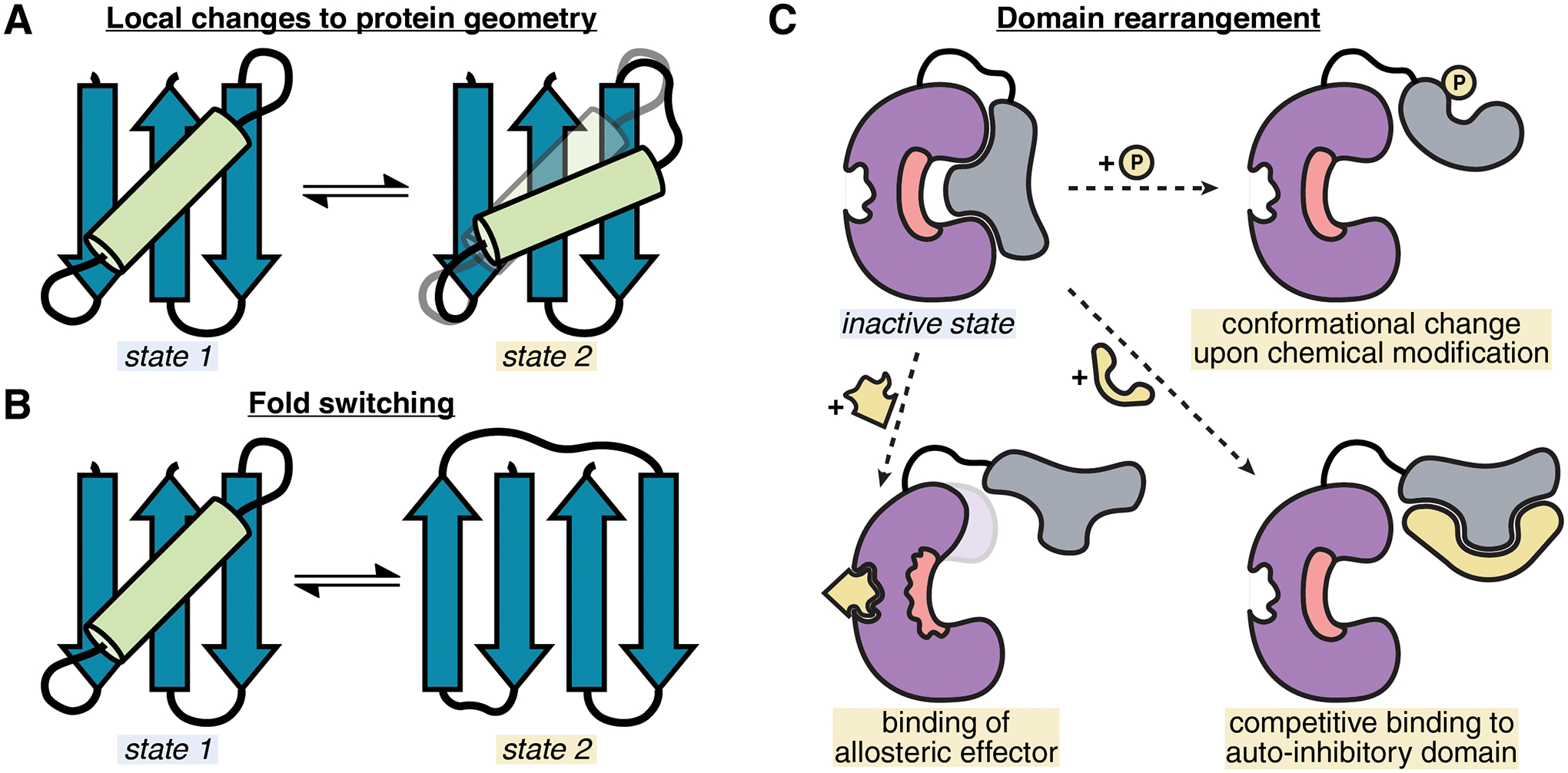
A, Local changes in geometry between two distinct conformations are commonly used as toggles for activity. B, Fold switching proteins involve exchange of entire secondary structure elements and can exhibit different functions specific for each state. C, Rearrangement of entire domains can occur via various mechanisms and regulate activity by increasing access to the domain and/or active site.
Fold switching
Metamorphic proteins undergo more dramatic structural changes, reversibly adopting two different native topologies (identity and connectivity of secondary structure elements) that have distinct biological functions (Figure 2B). Natural fold switch proteins such as lymphotactin [6], KaiB [7], or Mdm2 [8] often have binding interfaces specific to one fold, and are hence able to form favorable interactions in the presence of other proteins upon fold switching. It may be possible to use this principle to engineer controllable protein conformational switches by tuning the free energy difference between folds and their binding interactions. Structural studies combined with ancestral sequence reconstruction of lymphotactin have suggested how fold switching may have evolved through optimizing intra-chain interactions for desired interconversion rates without trapping the protein in either fold [9]. In contrast, there are also cases of irreversible fold switching driven by the strain associated with local sequence-structure incompatibility. One prominent example of this mechanism is the conversion of influenza hemagglutinin from inactive to fusogenic form [10]. Further investigation into the requisite energetic and mechanical transformations of fold switching proteins should enable the design of new switches which either emulate [11] or deviate significantly from switch mechanisms in natural examples.
Differential domain assembly
A third type of switching mechanism occurring in nature involves the rearrangement of individual domains of larger multi-domain proteins (Figure 2C). Conformational changes at this scale, where the moving parts largely behave as rigid bodies, are common in the regulation of signaling proteins and often involve changes in interactions between catalytic and auto-inhibitory domains [12]. For instance, if the catalytic site is physically obstructed by a bound auto-inhibitory domain, activating inputs may cause conformational changes that allow access to the active site. An example is the regulation of the SH2-containing phosphatase 2 in which phosphorylation of the SH2 domain causes dissociation from the phosphatase active site [13]. The broader principle of regulation of multi-domain assemblies through domain rearrangements has been termed “modular allostery” [14] and has provided ample inspiration for synthetic signaling systems [15] and protein-based biosensors [16]. Modular switch mechanisms can also be coupled to within-domain allosteric changes in the catalytic domain conformation, as seen in Src family kinases [17], but this type of regulation has not yet been engineered de novo.
Designed switches
Computational methodologies applied to switch design
Modern de novo protein design methods typically start with the construction of a backbone structure with the desired topology/geometry [18]. Sidechain design is then performed on this backbone (fixed or flexible) to optimize sequences predicted to fold (i.e., adopt its lowest energy conformation) into that structure in accordance with Anfinsen’s principle. This approach is only partially transferable to the de novo design of protein switches, which instead must attempt to simultaneously design (1) multiple interconvertible geometries, (2) at different local energy minima, (3) whose relative populations shift in response to a stimulus, (4) with all of the requisite structural mechanics incorporated. These requirements place additional demands on sampling, scoring, and our ability to represent multi-state ensembles with controlled functionality and energetics. Thus, switch design requires sampling over both sequence and structure space seeking single sequences that simultaneously achieve all of the above properties, posing not only a formidable combinatorial problem but also requiring high accuracy. Consequently, most designed switches to date have borrowed heavily from nature, using the backbones of known proteins and/or information derived from multiple sequence alignments to guide design. This wealth of sequence and structural data, as well as particularly well-characterized natural switches, also provide important benchmarks for computational methods. However, current approaches for switch design remain limited in scope and number; methodological developments are needed to realize arbitrary structures and functions.
Sequence landscape-based design
Recapitulating conformational responses in engineered proteins can range from performing relatively small searches in sequence space between existing structures to designing completely de novo folds unseen in nature. At one end of the spectrum, the most conservative design strategy involves transitioning between two well-defined folds that are known to be designable via stepwise mutational trajectories. Some of the first switches designed were based on merging the well-known motifs of zinc finger and all-helical folds and testing hybrid designs for switching behavior [19,20]. The computational RosettaDesign algorithm was used later to achieve a similar switch behavior [4]. While the straightforward approach of threading single sequences onto multiple backbones and scoring all states simultaneously to evaluate fitness was successful, the design strategy was facilitated by the facts that the endstate backbones were well defined and that zinc fingers commonly undergo folding/unfolding transitions in response to Zn2+ ions (Figure 3A). Generalizing this approach to arbitrary backbones will be an important step forward to designing functional switches de novo, though the use of metals or cofactors to mediate folding/conformational changes is itself likely to be an important mechanism underlying many target functions.
Figure 3 |. Designed protein switches.
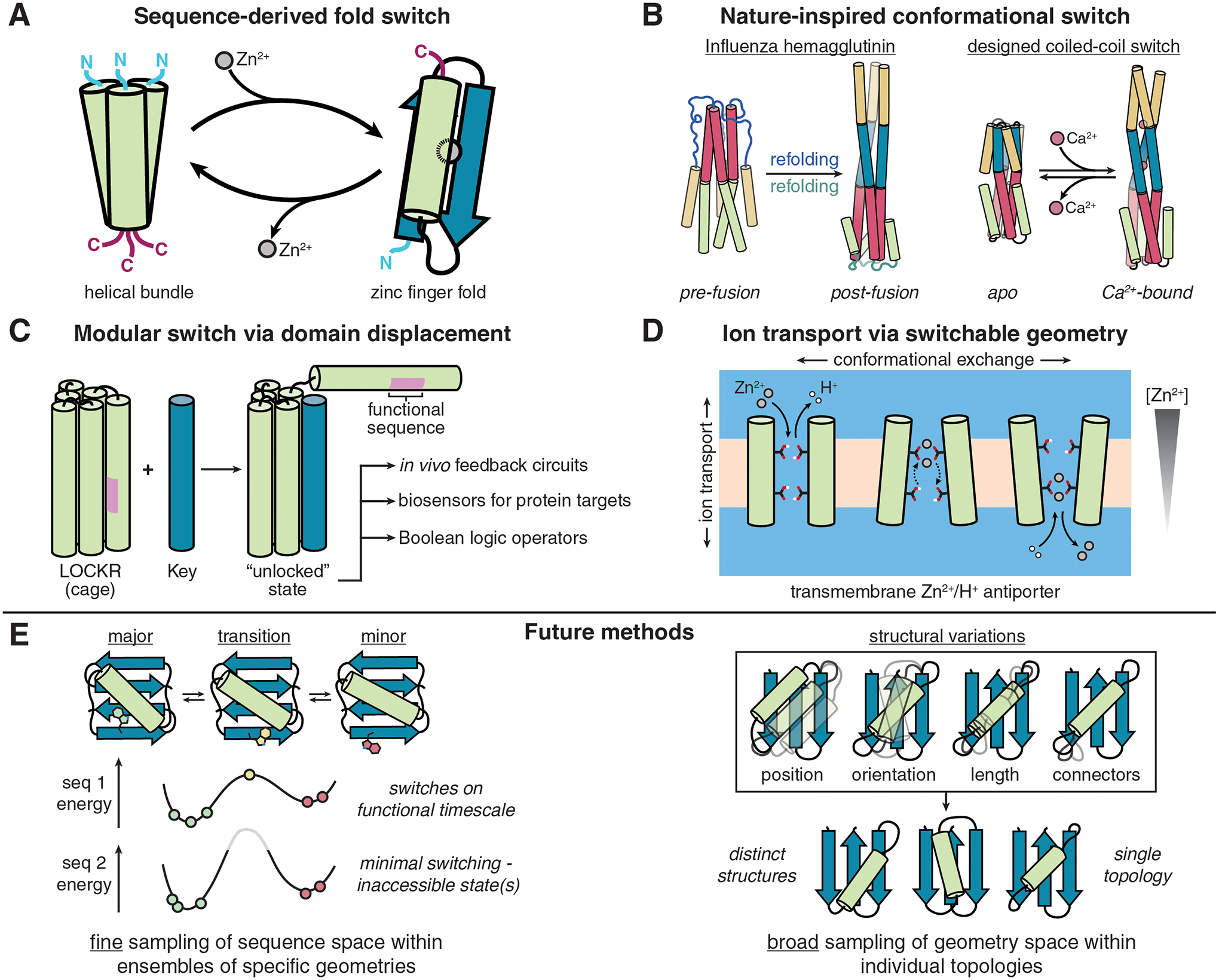
A, Sequence-based switch design. These designs are heavily informed by homologous protein sequences to infer basic requirements for adopting one fold over another. This example [4] used sequence motifs of zinc finger proteins and helical bundles to design a Zn2+-dependent fold switch. B, Nature-inspired conformational switches. Switches in this category typically aim to emulate specific conformational changes seen in nature. Here, we depict a coiled-coil switch designed to mimic hemagglutinin, using Ca2+ binding as a trigger [11]. C, De novo designed modular protein switches. The LOCKR class of designed proteins [30] utilizes domain displacement as a switch mechanism and has been applied towards a number of different uses. D, Functionally coupled changes in geometry. By taking advantage of the knowledge of helical packing and metal binding motifs, the ROCKER protein [33] undergoes conformational exchange between different states, passing Zn2+ ions in one direction while antiporting H+ ions, much in the same way as some natural channels do. E, The future of switch design will necessitate the ability to widely sample geometry space, define energy landscapes, and explicitly incorporate kinetic considerations. The recent advances highlighted here (left: DANCER [34], right: LUCS [36]) have brought us closer to these goals, but a greater number of methods with broader applicability are required to make advanced protein switch design routine.
Recently, the joint fitness landscape of the GA (3α) and GB (4β+α) binding domains of streptococcal protein G [5] was characterized by using the observed sequence variation of protein homologues to identify mutational pathways bridging the two distinct folds via Monte Carlo simulations [21]. By applying stability requirements on transitional sequences, it may be possible to identify putative fold switches more generally. The REstrained CONvergence (RECON) multistate design algorithm likewise uses a restrained search over the sequence fitness landscape between states to find sequences compatible with multiple distinct design goals (e.g., alternative conformations) by allowing each state to independently sample sequence space while driven to convergence through ramping restraints [22]. When applied to proteins known to be capable of large conformational changes, the sequence space sampled by RECON was found to indeed resemble the evolutionary sequence space of the starting structures [23]. An advantage to using RECON over explicitly incorporating evolutionary data is the possibility of targeting de novo structures as alternative conformational states. We note that these methods have been tested only in silico but provide exciting tools for experimental validation and future application. A recent experimental study using sequence alignment-based algorithms for fold switch design characterized proteins that adopted one fold in a truncated form (56 amino acids) and an alternative fold in a longer form (~90 amino acids). While the focus of the study was not on identifying a single bistable amino acid sequence for each fold pair, the authors do note possible spontaneous interconversion between folds for some expressed designs [24].
Integrative redesign
Currently, methods to engineer residue-level ‘microswitches’ capable of transducing signals rely on some combination of evolutionary information, experimental feedback, and molecular dynamics (MD) simulations to inform design. This integrative approach is needed to compensate for the difficulty of exerting fine control over protein geometry from first principles. For instance, to design new GPCR variants with altered conformational dynamics, Barth et al. first predicted residue-residue coupling by MD simulations before systematically assessing all possible amino acid combinations in silico [25]. Similar approaches relying on mutation of key structural residues based on evolutionary data or manual inspection of starting structures have also been successful in creating switchable peptides and proteins [26,27]. Such methods do not define the precise conformation of alternative states and are highly dependent on the topology of the starting structure. A benefit of such designs, however, is the identification of sequence-structure relationships that may improve our understanding of native proteins and aid in future efforts to reprogram switching behavior.
Designing de novo switches
Computational protein design increasingly aims to design proteins entirely de novo, i.e. from structural principles without relying on a natural protein as a starting point. Helical bundles are particularly popular targets, as their highly predictable backbone geometries facilitate the design of alternate conformational states (Figure 3B–3D). Moreover, residue-level interactions are well characterized; tuning their strengths in different conformational states has proven to be a successful design strategy for design of helical bundle-based protein switches. The development of algorithms to design extensive hydrogen bond networks along helical interfaces—first employed for the design of mutually orthogonal oligomeric structures [28]—have been exploited to create new switchable architectures based on polar interactions, including pH-responsive oligomers with histidine-rich interfaces [29] and a Ca2+-dependent coiled-coil switch that mimics the conformational states of influenza hemagglutinin [11] (Figure 3B). One principal advantage of this approach is the inherent modularity of helical bundles and the differential strengths of hydrophobic interactions and polar networks, which are a central feature of ‘latching orthogonal cage-key proteins’ (LOCKR) [30]. Constructed from a trimer of helical hairpins, LOCKR proteins consist of a five-helix ‘cage’ with a sixth ‘latch’ helix bound via a polar interface. LOCKR switching follows the principle of domain displacements in modular allostery described above. Functional peptides can be threaded onto the latch such that they are sequestered within the helix until a helical ‘key’ peptide with cage-optimal interactions displaces the latch, causing the latch to unfold and expose the functional sequence(s) (Figure 3C). Originally shown to be capable of regulating protein degradation in vivo, variants of the LOCKR system designed by simply interchanging modular components have since been employed to create synthetic feedback circuits in cells [31], perform Boolean logic operations on the surfaces of live cells [32], and sense target proteins [16]. In an earlier example of functionally coupled geometry changes, a transmembrane four-helix bundle with engineered metal binding sites was designed to alternate between two degenerate states to yield a Zn2+/H+ antiporter [33] (Figure 3D). These applications underscore the utility of purpose-built switches. Though modular displacement of helical elements in designed helical bundle assemblies has now been realized to yield functional proteins, the complete end-to-end design of conformational switches in which arbitrary end states can be specified (perhaps most tantalizing in the form of de novo folds) remains unachieved and would begin to demonstrate a mastery of de novo switch design.
Future directions
Designs of modular switchable systems have proven useful as sensors, regulators, and mediators of cell signaling; however, de novo designed switch mechanisms beyond modular displacements with hinge-like rigid body movements have yet to be achieved. To engineer the kinds of ordered, subtle conformational changes integral to natural enzymes, receptors, and other protein-based machines, we must be able to exercise greater control both spatially and temporally. For instance, switches often operate on characteristic time scales. However, kinetic considerations are often entirely neglected in current design approaches, as transient intermediates and the exact pathway for conformational exchange are difficult to model, and design algorithms typically seek to find low-energy structures representing thermodynamic sinks. One (challenging) approach to designing exchange pathways between conformational endstates is to generate an ensemble of microstates representing the intermediate structures and use these states in design simulations. This approach is the basis underlying the engineered DANCER proteins (Figure 3E, left), which spontaneously interconvert between a major and minor state via a predefined transition trajectory [34]. However, these endstates were defined by changes in a single rotamer, and explicit control over switching kinetics remains challenging. One possible approach to this problem is to employ MD simulations to evaluate mutations likely to accelerate switching kinetics by destabilizing the ground states relative to transition states, as utilized to enhance the response time of a calbindin-based switch by up to 32-fold [35].
A more comprehensive list of design strategies for engineering conformational switching can be found in Table 1. However, it should be noted that many of these methods rely on mutation of the native fold and subsequent characterization of structure-function relationships, leaving the alternative conformation(s) undefined prior to design. Moving forward, we envision that the design of protein switches will build upon recent advances in sampling methodologies to generate endstate structures (or ensembles) from which individual sequences can be evaluated for switchability in silico, analogous to earlier successes using known natural backbones. Recently, a method termed loop-helix-loop unit combinatorial sampling (LUCS) was shown to exhaustively sample the length, orientation, and position of alpha helices on predefined protein scaffolds (Figure 3E, right), creating geometric variability within a given topology exceeding that of known natural proteins [36]. Similarly, a method for systematic sampling of NTF2-like fold geometries was developed to generate diverse pocket structures [37]. These algorithms generate ensembles of alternative secondary structure element geometries that may be used to target specific geometries for switching.
Ultimately, a biophysically intuitive approach to protein design will likely entail the concept of “designing on a landscape” [38], where sequence design considers multiple stable conformations, or even multiple landscapes representing pre-/post-stimulus states. Deep learning methods for protein structure prediction [39,40] are thought to operate by “smoothing out” folding landscapes, suggesting that it may become possible to evaluate the conformational landscapes of structures (and ensembles) and couple deep learning-based structure prediction with design [41]. This level of detailed control could enable the design of coupled networks of residue interactions and more complex conformational changes characteristic of evolved switches and machines. While many outstanding challenges remain, the ability to design de novo all of the components of switchable elements (Figure 1) would allow the design of new families of biological signaling systems with modular and tunable behavior. These systems could be interfaced with naturally occurring regulatory mechanisms to control existing biological processes or build up increasingly complex de novo machines with entirely new functions.
Acknowledgements
This work was supported by a grant from the National Institutes of Health (NIH) (R01-GM110089) to TK. ABG acknowledges support from a National Science Foundation (NSF) Graduate Research Program Fellowship. TK is a Chan Zuckerberg Biohub Investigator.
Footnotes
Declaration of interest
The authors declare no competing interests.
References
- 1.Huse M, Kuriyan J: The Conformational Plasticity of Protein Kinases. Cell 2002, 109:275–282. [DOI] [PubMed] [Google Scholar]
- 2.Bourne HR, Sanders DA, McCormick F: The GTPase superfamily: conserved structure and molecular mechanism. Nature 1991, 349:117–127. [DOI] [PubMed] [Google Scholar]
- 3.Weis WI, Kobilka BK: The Molecular Basis of G Protein-Coupled Receptor Activation. Annu Rev Biochem 2018, 87:897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ambroggio XI, Kuhlman B: Computational Design of a Single Amino Acid Sequence that Can Switch between Two Distinct Protein Folds. 2006, doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- 5.Alexander PA, He Y, Chen Y, Orban J, Bryan PN: A minimal sequence code for switching protein structure and function. Proc Natl Acad Sci 2009, 106:21149–21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuloğlu ES, McCaslin DR, Markley JL, Volkman BF: Structural Rearrangement of Human Lymphotactin, a C Chemokine, under Physiological Solution Conditions *. J Biol Chem 2002, 277:17863–17870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang Y-G, Cohen SE, Phong C, Myers WK, Kim Y-I, Tseng R, Lin J, Zhang L, Boyd JS, Lee Y, et al. : A protein fold switch joins the circadian oscillator to clock output in cyanobacteria. Science (80-) 2015, 349:324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mapelli M, Massimiliano L, Santaguida S, Musacchio A: The Mad2 Conformational Dimer: Structure and Implications for the Spindle Assembly Checkpoint. Cell 2007, 131:730–743. [DOI] [PubMed] [Google Scholar]
- 9.Dishman AF, Tyler RC, Fox JC, Kleist AB, Prehoda KE, Babu MM, Peterson FC, Volkman BF: Evolution of fold switching in a metamorphic protein. Science (80-) 2021, 371:86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carr CM, Kim PS: A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 1993, 73:823–832. [DOI] [PubMed] [Google Scholar]
- 11.Wei KY, Moschidi D, Bick MJ, Nerli S, McShan AC, Carter LP, Huang P-S, Fletcher DA, Sgourakis NG, Boyken SE, et al. : Computational design of closely related proteins that adopt two well-defined but structurally divergent folds. Proc Natl Acad Sci 2020, 117:7208–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Demonstrates the design of conformational switches with different helical bundle structures inspired by the influenza hemagglutinin mechanism using in silico contact network modeling.
- 12.Kuriyan J, Cowburn D: MODULAR PEPTIDE RECOGNITION DOMAINS IN EUKARYOTIC SIGNALING. Annu Rev Biophys Biomol Struct 1997, 26:259–288. [DOI] [PubMed] [Google Scholar]
- 13.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE: Crystal structure of the tyrosine phosphatase SHP-2. Cell 1998, 92:441–450. [DOI] [PubMed] [Google Scholar]
- 14.Lim WA: The modular logic of signaling proteins: building allosteric switches from simple binding domains. Curr Opin Struct Biol 2002, 12:61–68. [DOI] [PubMed] [Google Scholar]
- 15.Dueber JE, Yeh BJ, Chak K, Lim WA: Reprogramming Control of an Allosteric Signaling Switch Through Modular Recombination. Science (80-) 2003, 301:1904–1908. [DOI] [PubMed] [Google Scholar]
- 16.Quijano-Rubio A, Yeh H-W, Park J, Lee H, Langan RA, Boyken SE, Lajoie MJ, Cao L, Chow CM, Miranda MC, et al. : De novo design of modular and tunable protein biosensors. Nature 2021, 591:482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boggon TJ, Eck MJ: Structure and regulation of Src family kinases. Oncogene 2004, 23:7918–7927. [DOI] [PubMed] [Google Scholar]
- 18.Pan X, Kortemme T: Recent advances in de novo protein design: Principles, methods, and applications. J Biol Chem 2021, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hori Y, Sugiura Y: Conversion of antennapedia homeodomain to zinc finger-like domain: Zn(II)-induced change in protein conformation and DNA binding. J Am Chem Soc 2002, 124:9362–9363. [DOI] [PubMed] [Google Scholar]
- 20.Cerasoli E, Sharpe BK, Woolfson DN: ZiCo: A Peptide Designed to Switch Folded State upon Binding Zinc. J Am Chem Soc 2005, 127:15008–15009. [DOI] [PubMed] [Google Scholar]
- 21.Tian P, Best RB: Exploring the sequence fitness landscape of a bridge between protein folds. PLOS Comput Biol 2020, 16:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Presents a method to sample evolutionary mutational paths between folds by using a multiple sequence alignment-derived joint fitness landscape.
- 22.Sevy AM, Jacobs TM, Crowe JE, Meiler J: Design of Protein Multi-specificity Using an Independent Sequence Search Reduces the Barrier to Low Energy Sequences. PLOS Comput Biol 2015, 11:e1004300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sauer MF, Sevy AM, Crowe JE Jr., Meiler J: Multi-state design of flexible proteins predicts sequences optimal for conformational change. PLOS Comput Biol 2020, 16:1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, He Y, Ruan B, Choi EJ, Chen Y, Motabar D, Solomon T, Simmerman R, Kauffman T, Gallagher DT, et al. : Rules for designing protein fold switches and their implications for the folding code. bioRxiv 2021, doi: 10.1101/2021.05.18.444643. [DOI] [Google Scholar]; ** Experimentally tests an approach to engineer fold switches between common small protein topolgies using a sequence-alignment based strategy.
- 25.Chen K-YM, Keri D, Barth P: Computational design of G Protein-Coupled Receptor allosteric signal transductions. Nat Chem Biol 2020, 16:77–86. [DOI] [PubMed] [Google Scholar]; * Uses in silico saturation mutagenesis of key residues mediating allostery to reprogram signaling and alter stability of GPCRs.
- 26.Dawson WM, Lang EJM, Rhys GG, Shelley KL, Williams C, Brady RL, Crump MP, Mulholland AJ, Woolfson DN: Structural resolution of switchable states of a de novo peptide assembly. Nat Commun 2021, 12:1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campos LA, Sharma R, Alvira S, Ruiz FM, Ibarra-Molero B, Sadqi M, Alfonso C, Rivas G, Sanchez-Ruiz JM, Romero Garrido A, et al. : Engineering protein assemblies with allosteric control via monomer fold-switching. Nat Commun 2019, 10:5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyken SE, Chen Z, Groves B, Langan RA, Oberdorfer G, Ford A, Gilmore JM, Xu C, DiMaio F, Pereira JH, et al. : De novo design of protein homo-oligomers with modular hydrogen-bond network{\textendash}mediated specificity. Science (80-) 2016, 352:680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyken SE, Benhaim MA, Busch F, Jia M, Bick MJ, Choi H, Klima JC, Chen Z, Walkey C, Mileant A, et al. : De novo design of tunable, pH-driven conformational changes. Science (80-) 2019, 364:658–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langan RA, Boyken SE, Ng AH, Samson JA, Dods G, Westbrook AM, Nguyen TH, Lajoie MJ, Chen Z, Berger S, et al. : De novo design of bioactive protein switches. Nature 2019, 572:205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Describes the computational design of helical-bundle cage-key functional switches (LOCKR) relying on a helical displacement mechanism.
- 31.Ng AH, Nguyen TH, Gómez-Schiavon M, Dods G, Langan RA, Boyken SE, Samson JA, Waldburger LM, Dueber JE, Baker D, et al. : Modular and tunable biological feedback control using a de novo protein switch. Nature 2019, 572:265–269. [DOI] [PubMed] [Google Scholar]
- 32.Lajoie MJ, Boyken SE, Salter AI, Bruffey J, Rajan A, Langan RA, Olshefsky A, Muhunthan V, Bick MJ, Gewe M, et al. : Designed protein logic to target cells with precise combinations of surface antigens. Science (80-) 2020, 369:1637–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joh NH, Wang T, Bhate MP, Acharya R, Wu Y, Grabe M, Hong M, Grigoryan G, DeGrado WF: De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science (80-) 2014, 346:1520–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davey JA, Damry AM, Goto NK, Chica RA: Rational design of proteins that exchange on functional timescales. Nat Chem Biol 2017, 13:1280–1285. [DOI] [PubMed] [Google Scholar]
- 35.DeGrave AJ, Ha J-H, Loh SN, Chong LT: Large enhancement of response times of a protein conformational switch by computational design. Nat Commun 2018, 9:1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan X, Thompson MC, Zhang Y, Liu L, Fraser JS, Kelly MJS, Kortemme T: Expanding the space of protein geometries by computational design of de novo fold families. Science (80-) 2020, 369:1132–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Basanta B, Bick MJ, Bera AK, Norn C, Chow CM, Carter LP, Goreshnik I, Dimaio F, Baker D: An enumerative algorithm for de novo design of proteins with diverse pocket structures. Proc Natl Acad Sci 2020, 117:22135–22145. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Develops and demonstrates a computational design method for near-exhaustive sampling of accessible conformational space for loop-helix-loop geometries in de novo designed proteins.
- 38.Norn C, Wicky BIM, Juergens D, Liu S, Kim D, Koepnick B, Anishchenko I, Players F, Baker D, Ovchinnikov S: Protein sequence design by explicit energy landscape optimization. bioRxiv 2020, doi: 10.1101/2020.07.23.218917. [DOI] [Google Scholar]
- 39.Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D: Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci 2020, 117:1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, et al. : Accurate prediction of protein structures and interactions using a 3-track network. bioRxiv 2021, doi: 10.1101/2021.06.14.448402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anishchenko I, Chidyausiku TM, Ovchinnikov S, Pellock SJ, Baker D: De novo protein design by deep network hallucination. bioRxiv 2020, doi: 10.1101/2020.07.22.211482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith CA, Shi CA, Chroust MK, Bliska TE, Kelly MJS, Jacobson MP, Kortemme T: Design of a Phosphorylatable PDZ Domain with Peptide-Specific Affinity Changes. Structure 2013, 21:54–64. [DOI] [PubMed] [Google Scholar]
- 43.Au L, Green DF: Direct Calculation of Protein Fitness Landscapes through Computational Protein Design. Biophys J 2016, 110:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]