

Abstract
Objectives:
Acute injuries or insults to the cortex, such as trauma, subarachnoid hemorrhage, lobar hemorrhage, can cause seizures or status epilepticus(SE). Neocortical SE is associated with coma, worse prognosis, delayed recovery, and the development of epilepsy. The anatomical structures progressively recruited during neocortical-onset status epilepticus (SE) is unknown. Therefore, we constructed large-scale maps of brain regions active during neocortical SE.
Methods:
We used a neocortical injury-induced SE mouse model. We implanted cobalt (Co) in the right supplementary motor cortex (M2). We 16 hours later administered a homocysteine injection (845 mg/kg, intraperitoneal) to C57Bl/6J mice to induce SE and monitored it by video and EEG. We harvested animals for 1 hr (early-stage) and 2 hrs (late-stage) following homocysteine injections. To construct activation maps, we immunolabeled whole-brain sections for cFos and NeuN, imaged them using a confocal microscope and quantified cFos immunoreactivity (IR).
Results:
SE in the early phase consisted of discrete, focal intermittent seizures, which became continuous and bilateral in the late stage. In this early stage, cFos IR was primarily observed in the right hemisphere, ipsilateral to the Co lesion, specifically in the motor cortex, retrosplenial cortex, somatosensory cortex, anterior cingulate cortex, lateral and medial septal nuclei, and amygdala. We observed bilateral cFos IR in brain regions during the late stage, indicating the bilateral spread of focal seizures. We found increased cFOS IR in the bilateral somatosensory cortex and the motor cortex and subcortical regions, including the amygdala, thalamus, and hypothalamus. There was noticeably different, intense cFos IR in the bilateral hippocampus compared to the early stage. In addition, there was higher activity in the cortex ipsilateral to the seizure focus during the late stage compared with the early one.
Conclusion:
We present a large-scale, high-resolution map of seizure spread during neocortical injury-induced SE. Cortico-cortical and cortico subcortical re-entrant circuits sustain neocortical SE. Neuronal loss following neocortical SE, distant from the neocortical focus, may result from seizures.
Keywords: cFos immunostaining, neocortical status epilepticus, neuronal activation maps, neuronal injury, supplementary motor cortex
Introduction
Status epilepticus (SE) is a neurological emergency characterized by prolonged self-sustaining seizures that can lead to brain injury if not terminated (ILAE definition). There is growing recognition that acute cortical insults, such as subarachnoid hemorrhage, lobar hemorrhage, and trauma, cause seizures (Herman et al. 2015); (Vespa et al. 2016; Vespa et al. 2003). Acute seizures are associated with worse prognosis, delayed recovery, and the development of epilepsy (Klein et al. 2018; Herman et al. 2015). In a study of patients with moderate to severe traumatic brain jury, those with SE suffered a higher mortality rate than those who did not have seizures (Vespa et al. 1999). Seizures after aneurysmal subarachnoid hemorrhage are positively associated with increased mortality (Rush et al. 2016). After prolonged episodes of seizures, patients become comatose, during which the seizures become nonconvulsive (Claassen et al., 2007). It is observed that nonconvulsive seizures following traumatic brain injury cause a delayed increase in intracranial pressure, cerebral perfusion pressure, and metabolic crisis and trigger inflammation (Claassen et al., 2013). Patients with seizures had a higher likelihood of hydrocephalous, paresis, and comatose compared with the no-seizure cohort. Seizures cause secondary neuronal injuries by increasing metabolic demand, resulting in elevated cerebral blood flow and neuronal death via neurochemical excitotoxicity (Vespa et al. 2016). These seizures are associated with physiological complications and poorer neurological outcomes. Presumably, preventing or terminating SE in these patients will improve their prognosis by limiting secondary neocortical injury. We need to understand underlying mechanisms at the circuit, cellular and molecular levels to develop novel therapies.
An initial step is to identify neuronal circuits active during neocortical SE. For example, in the past, the dentate gate hypothesis was proposed as a potential circuit breaker for limbic system-driven SE (Lothman et al., 1990; Lothman et al., 1991). This hypothesis was then extensively studied at the cellular and molecular levels to understand mechanisms underlying limbic SE (Kapur and Macdonald 1997a; Joshi and Kapur 2012b; Kittler and Moss 2003; Terunuma et al., 2008). This perspective encourages us to follow a similar approach in the case of neocortical SE.
We recently characterized a mouse model of neocortical onset SE (Singh et al., 2020). Previously, we performed brain-wide mapping of neuronal activation with cellular level details using immunohistochemistry and high-resolution imaging with automated image analysis (Dabrowska et al., 2019; Brodovskaya et al., 2021). Here we present the first large-scale functional anatomy of neuronal activation during neocortical injury-induced SE.
Materials and Methods
We performed all studies following protocols approved by the Animal Care and Use Committee of the University. Adult C57BL/6J mice (Charles River Laboratories, Wilmington, MA, USA) of either sex, 23 – 27 g, 8–10 weeks old, were used for these studies. All chemicals were purchased from Sigma‐Aldrich or otherwise stated. We pooled the data from male and female mice since the results were similar, as reported previously (Singh et al., 2020).
Induction of SE and EEG recordings
We implanted 2.1 mg, 0.5 mm diameter, 1.2 mm long Co wire (Alfa Aesar, Haverhill, MA, USA) orthogonally into the right supplementary motor cortex (M2) (AP, +2.6 mm; ML, +1.8 mm) under isoflurane anesthesia as reported previously. We also implanted stainless‐steel electrodes in the right and (Fi) and left (Fc) frontal cortices at coordinates AP, +1.6 mm; ML, ±1.8 mm; and DV, +1.0 mm and bilateral parietal electrodes (Pi and Pc) at coordinates AP, −2.6 mm; ML, ±1.8 mm; and DV +1.0 mm and a reference electrode over the cerebellum. Animals received ketoprofen (1 mg/kg i.p.) to minimize pain and discomfort. We initiated continuous video and EEG monitoring 30 min after the surgery (Grass ARUA LTM64 using Twin software, Grass, Warwick, RI, USA). We administered homocysteine (845 mg/kg, intraperitoneal), 16–18h, following Co implantation to induce SE. We exported EEG data to Lab Chart 7 (ADI Instruments, Colorado, USA). We generated a power spectrum using cortical EEG recorded from the ipsilateral frontal electrode and expressed power as mV2.
Cobalt baseline
Brain injury can increase cfos expression (Dragunow and Robertson 1988). Co causes acute injury at the premotor cortex. We implanted cobalt in three (two male and one female) 8–10 weeks old mice. We administered diazepam (10 mg/kg, intraperitoneal) at the 15th hour after cobalt implantation. The animals were perfused transcardially after one hour later. This group of mice, which we call Co-baseline, did not receive homocysteine.
cFos immunohistochemistry
Co-implanted animals received homocysteine and developed SE. The animals of Co-homocysteine and the Co-baseline groups were transcardially perfused with 4% PFA in 0.1 M PB either at 60 min (early stage) or 120 min (late stage) after homocysteine injection. We studied five animals in the early-stage group and five animals in the late-stage group. The brains were harvested and postfixed in 4% PFA solution overnight and incubated in 30% sucrose for 48 h for cryoprotection. We prepared Forty-micron-thick coronal sections on a Leica cryostat and processed every fourth section for immunohistochemistry. Anti-cFos (1:1000, ab190289, Abcam) and anti-NeuN antibodies (1:500, MAB377, Millipore, Burlington, MA, USA) were used.
Statistical analysis and Image acquisition
The EEG data were processed using Graph Pad Prism version 8 (San Diego, CA 92108, USA). The coronal sections for cFos and NeuN immunoreactivity were scanned at 10x magnification using a confocal microscope (Nikon Eclipse Ti-U at 10x magnification, 0.45 NA). Excitation lasers were 488 nm for green and 561 nm for red. Images were tiled as stacks with optical section separation (Z interval) of 10μm and stitched using NIS Elements software. We used Imaris 9.3.0 (Bitplane) for visualizing and Adobe Photoshop CC for cropping the original image and display.
We semi-automatically counted neurons that expressed c-Fos immunoreactivity in key brain regions. We first opened the z-stack of a two-channel (NeuN and cFos) image of a slice using Imaris. We computed the maximum intensity projection (MIP) image for each channel. We focused on the region of interest in a MIP image by magnifying the image by 10 times (100X). Next, we saved one magnified MIP image per channel and opened each of them via ImageJ, and converted it to grayscale for counting. Images in the figures are MIP generated by the Imaris software. The cFos intensity profile of a neuron varied based on the slice and region. cFos was also expressed in non-neuronal cells. Our objective was to count only cFos+ neurons. So, we did not measure the brightness of neuronal cFos.
For a slice with a large variation in intensity, we saved the MIP image, opened Matlab, masked the desired region, and loaded the masked image back to ImageJ for auto-thresholding. To eliminate the possibility of missing neurons because of varying intensities and a single global threshold, we separated very bright regions from comparatively low-intensity regions by applying masks (manually-drawn contours). Next, we applied an auto thresholding function on the individual masked image. We applied a watershed algorithm and a set of morphological operations to ensure at least one-pixel width separation between two neighboring cells. We segmented NeuN and cFos separately and then maximized region overlap to determine the neurons. We also manually confirmed the accuracy of the counts in a couple of slices.
We considered at least three slices per region for each of the five animals in each stage. We maintained coronal slices’ coordinates the same for both stages to avoid any bias of region volume incurred by inconsistent slice selection. We verified the locations from the Allen atlas. For each animal, we considered 4 slices for the motor cortex, 4 for the somatosensory cortex, 5 for the entorhinal cortex, 5 for the visual cortex, 4 for the retrosplenial cortex, 3 for the hippocampus and DG, 3 for the hypothalamus, 3 for the thalamus, 4 for the piriform and 4 for the amygdala.
We compared the c-Fos activity between sites, ipsilateral to Co implantation, and the contralateral sites at the early stage. We performed paired t-test with Welch’s correction for unequal variance and tested normality using the Shapiro-Wilks test as well as Q-Q plot. We considered coronal slices from their anterior and posterior (4 slices each) for motor and somatosensory cortices to investigate the anterior to posterior seizure spread. We compared the c-Fos IR between the early and the late stages for each region using one-way ANOVA with Bonferroni’s correction for multiple comparisons. We set the significance level at 5% and performed all statistical tests using GraphPad Prism 8 (San Diego, CA 92108, USA).
Results
The following sections present the results of brain-wide cFos immunoreactivity (IR). We used the convention of ipsilateral and contralateral as the right and left hemispheres of a mouse brain, respectively. The cobalt was implanted on the right hemisphere. All observations from the right hemisphere, i.e, ipsilateral to the cobalt lesion, referred to as “ipsi” in the figures.
cFos immunoreactivity due to cortical injury
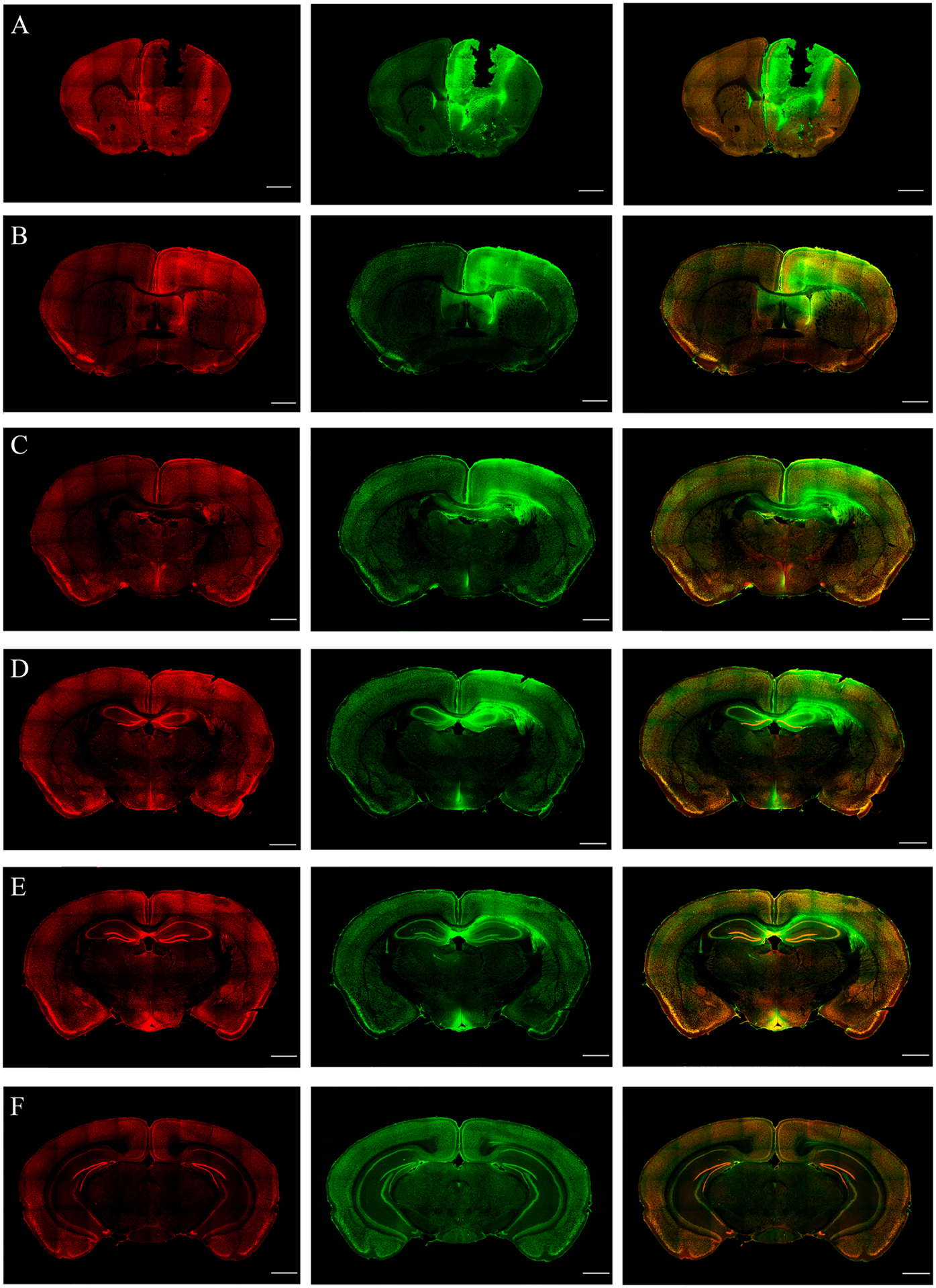
The cFos IR in the brain slices of the Co-baseline group was shown in the top panel of Fig.1. We found considerable cFos IR in the ipsilateral motor cortex (Fig. 1(A)), hypothalamus (Fig. 1(C)), prelimbic, and infralimbic (Fig. 1(B)) cortices. We included the baseline cFos IR of the motor cortex and hypothalamus while comparing against the early stage. We found very sparse cFos IR in other brain regions, including the visual cortex, posterior parietal association areas, somatosensory cortex, paraventricular thalamus, hippocampus and dentate gyrus. We omitted the baseline cFos IRs of these regions because they did not fit the same scale of cFos IR during the early and late stages. Three whole brain coronal slices containing NeuN and cFos expressions were shown in Fig. 1(E).
Figure 1.
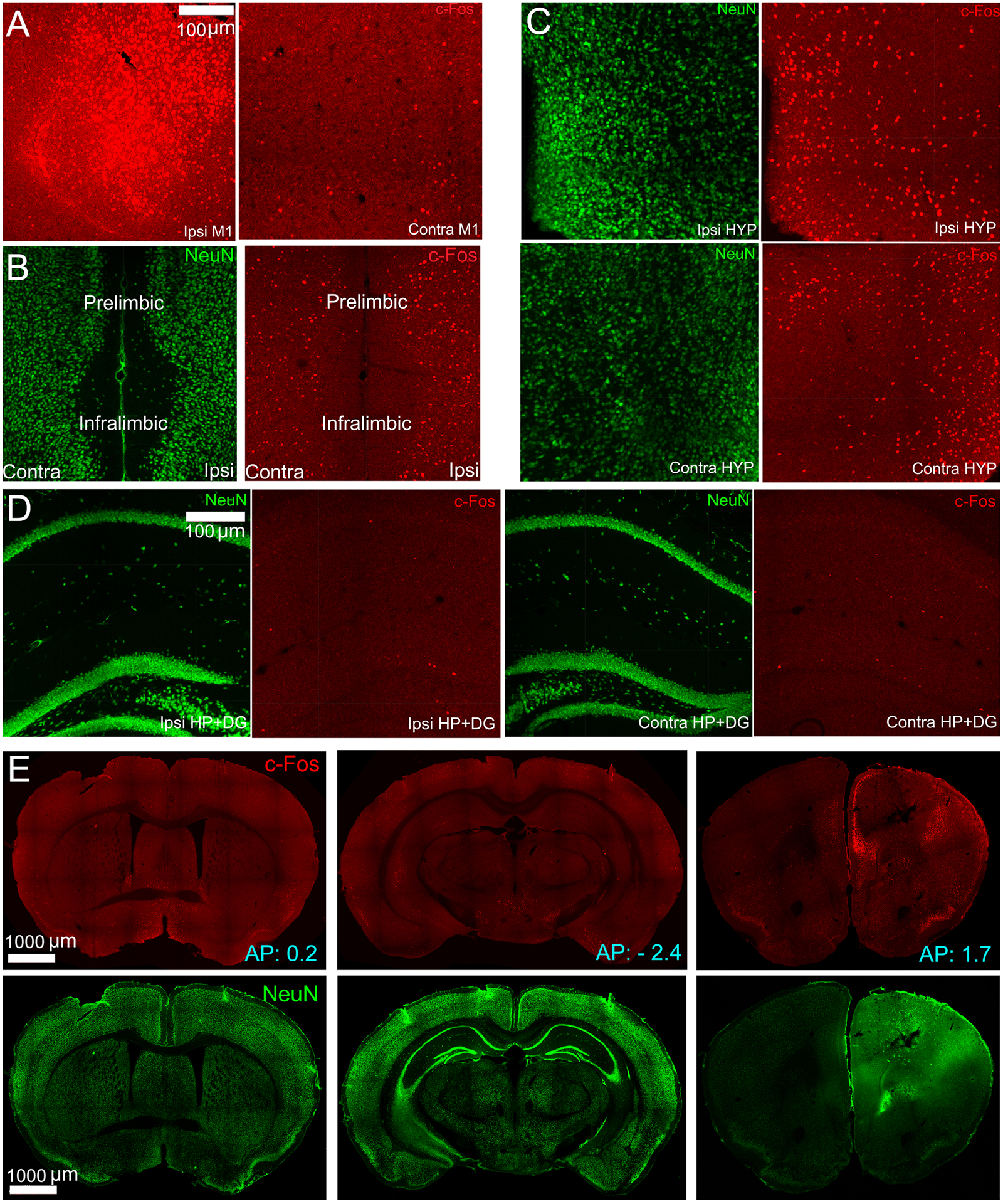
Panel A, B, C, D show key brain regions that expressed substantial cFos IR due to cobalt injury in mice in the Co-baseline group. In all the figures, the green and red channels correspond to NeuN and cFos expressions. The scale bar is set as 100 μm. Ipsi is the right hemisphere, Contra is the left hemisphere, M1 is the primary motor cortex, HYP is the hypothalamus, HP+DG is the hippocampus and dentate gyrus. Co is cobalt baseline group. (A) cFos IR at the ipsilateral and contralateral sides of the primary motor cortex due to cobalt injury. The ipsilateral side showed intense cFos IR. (B) Prelimbic and infralimbic cortical areas were found to have cFos+ cells due to cobalt injury. (C) cFos and NeuN in the hypothalamus on both hemispheres, showing considerable cFos IR due to cobalt injury. (D) The figure shows sparse cFos IR in the hippocampus and dentate gyrus due to cobalt injury. (E) Neun and cFos immunostainings of coronal brain slices, selected at three different anterior-posterior (AP) coordinates. The coordinates are obtained from an online mouse brain atlas (http://labs.gaidi.ca/mouse-brain-atlas/).
cFos immunoreactivity at cortical areas
In a recent study, we described Co /Homocysteine-induced SE’s evolution (Singh et al., 2020). Briefly, SE started with brief, discrete focal seizures that became longer. Electrographically, rhythmic spike-wave discharges were recorded from electrodes in the frontal and parietal lobes. After 4 to 8s, spike-wave discharges appeared on both parietal electrodes. EEG was suppressed in all four electrodes after the spikes disappeared, marking the end of the seizure. Power spectrum analysis of EEG recorded from the right frontal electrode (Fig. 2(A)), near the cobalt focus, demonstrated sudden bursts of high-power activity associated with early focal intermittent seizures. The EEG traces recorded during focal seizures were recorded (Fig. 2(B–C)), marking distinct behavior patterns (forepaw clonus, focal dystonia with clonus).
Figure 2.
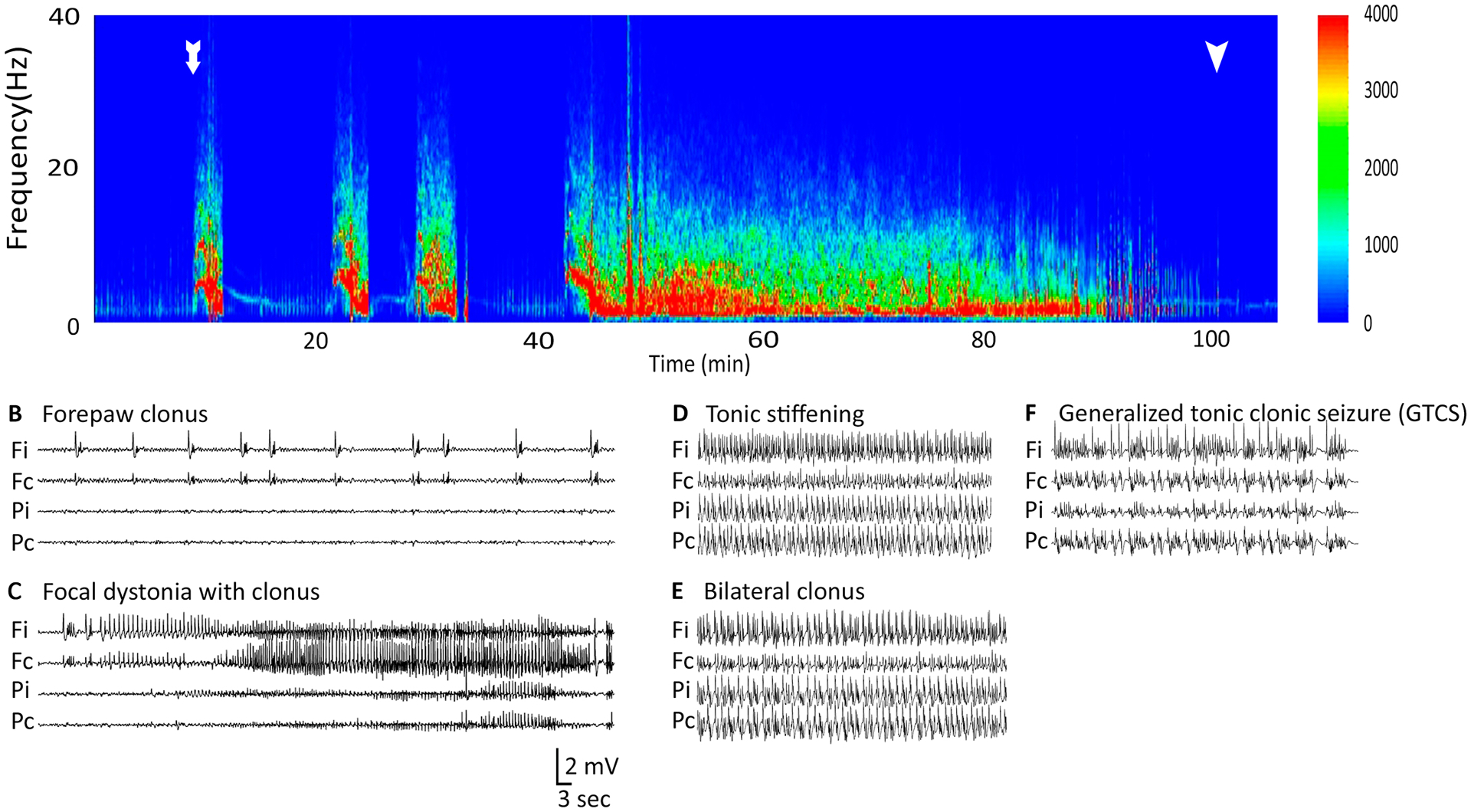
This figure shows that power spectrogram and corresponding EEG traces observed during early focal intermittent seizures, continuous generalized convulsive seizures and final coma phase during Co-homocysteine-induced SE. The EEG electrodes were placed in four cortical locations - frontal ipsilateral (Fi), frontal contralateral (Fc), parietal ipsilateral (Pi), and parietal contralateral (Pc). (A) The total power of EEG recorded at Fi was plotted against time. Time t=0 corresponds the instant of homocysteine injection. The onset of first focal seizure is shown with a white arrow. Each focal seizure was followed by baseline EEG trace. The generalized seizure started around 42 mins from the homocysteine injection and continued for a long time (~50 mins). The end of the late stage is shown with another white arrow. (B-C) EEG traces correspond to two behavioral patterns during the early phase. Please note the changes in EEG recorded at the frontal electrodes between B and C. (D-E) EEG traces corresponding to tonic stiffening and bilateral clonus during the late phase. (F) EEG traces associated with generalized tonic clonic seizures. (G) Pie chart showing the proportion of seizures with distinct behavioral patterns observed in our experiments.
We used cFos immunohistochemistry to study the spatiotemporal spread of SE for several reasons. Following seizures, mRNA levels for different IEGs increases in cfos > jun-B > c-jun > jun-D. The jun family’s expression was less marked than c-fos (Williams and Jope 1994; Morgan et al. 1987). Anticonvulsants blocked c-fos immunoreactivity and the development of seizures. Analysis of tissue specimens from epileptic foci in humans also confirmed a correlation of cFos immunoreactivity with the frequency of interictal activity (Rakhade et al., 2007). Thus, based on these studies, The upregulation of cFos mRNA and protein expression generally takes approximately 30–45 mins (Peng and Houser, 2005). We, therefore, evaluated cFos expression in animals 60 minutes (early stage) and 120 minutes (late stage) after homocysteine injection to map the seizure activity over time. The seizure focus was in the supplementary motor cortex (M2), which has a variety of cortico-cortical connections with primary motor area (M1), somatosensory, auditory, and visual cortex and limbic/paralimbic areas including orbital, insular, perirhinal, entorhinal (EC), retrosplenial and presubiculum cortex (Reep et al., 1987; Zingg et al., 2014).
Early stage
In the early stage, there was intense unilateral cFos IR in the cortical region as shown in Fig. 3(A–F). The cFos was not uniformly expressed across all the cortical layers. For example, in the somatosensory cortex, the superficial layers 1 and 2/3 were densely cFos labeled compared to the deep layers. In contrast, the entorhinal cortex showed almost uniform expression across all layers (Fig. 3(F) and Fig. 6(A)). We found elevated cFos IR anteriorly around the lesion in the right supplementary motor cortex compared to its contralateral side (p = 0.0017). There was extensive cell death near the cobalt lesion. Even though we discarded the slices at the focus, we found slightly reduced cFos IR in the motor cortex, as shown in Fig. 5(A). The total number of cFos+ neurons in 4 slices was approximately 1423 ± 173.1 per animal on the ipsilateral motor cortex. The Co-baseline control group contained around 913.4 ± 164.4 cFos+ neurons per animal on the same side. cFos IR at the ipsilateral motor cortex (p = 0.0221), but not the contralateral side (p = 0.40), was different between the early stage and Co-baseline group. There was intense cFos IR ie right (ipsilateral) somatosensory cortex compared with the contralateral side (p = 0.0092, Fig. 5(C)). In the visual cortex also, cFos IR was different between hemispheres (p = 0.0336). The cFos IR for the visual and entorhinal (p = 0.035) cortical areas were shown in Fig. 6(A, C). Compared to the contralateral side, substantial cFos IR was also observed in other cortical areas, including the anterior cingulate (p = 0.0007), retrosplenial (p = 0.011), and other cortical areas, such as perirhinal, ectorhinal, piriform (p = 0.027) on the ipsilateral side. cFos IR appeared on the contralateral side was possibly due to the bilateral projections of the M2 cortex to perirhinal and entorhinal cortices (Reep et al. 1987). A similar explanation holds for the piriform cortex.
Figure 3.
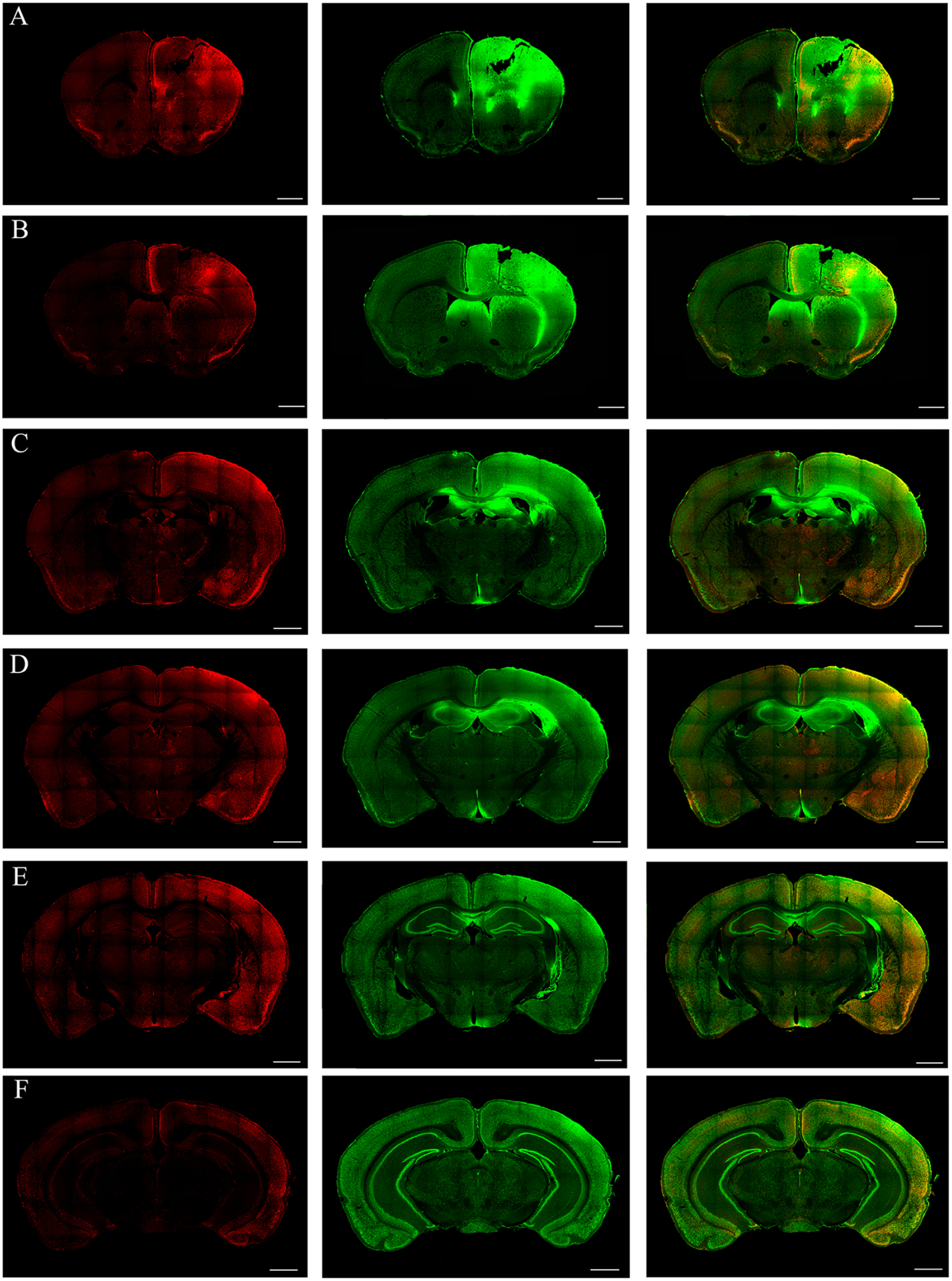
During the early stage, cFos immunoreactivity (IR) in the cortex, thalamus and amygdalar region during early intermittent focal seizures is shown in the coronal sections at the level of bregma 1.5, 1.0, −0.5, −1.0, −2.0, −3.0 mm (A to F, respectively; scale bar =1000 μm). The red and the green channels correspond to cFos and NeuN stained sections. (A-B) The cFos IR in the coronal sections at the level of Co lesion are shown. Intense unilateral cFos IR anteriorly around the lesion in the motor and somatosensory cortices are observed. (C-D) Coronal sections at the level of bregma −0.5 and −1.0 mm respectively, shows the activation of cortical (such as the retrospenial, somatosensory, entorhinal, perirhinal and piriform cortices) and amygdalar regions at the ipsilateral side. Sparse cFos activity was found in the thalamic regions, especially the paraventricular and paratenial and mediodorsal thalamic nuclei. (E-F) Coronal sections at the level of bregma −2.0 and −3.0 mm (E to F, respectively), presents cFos IR in posterior cortical regions, such as the posterior-parietal association area, primary somatosensory area, auditory area, temporal association area. Sparse cFos IR was observed in any of the hippocampus during early focal intermittent seizures.
Figure 6.
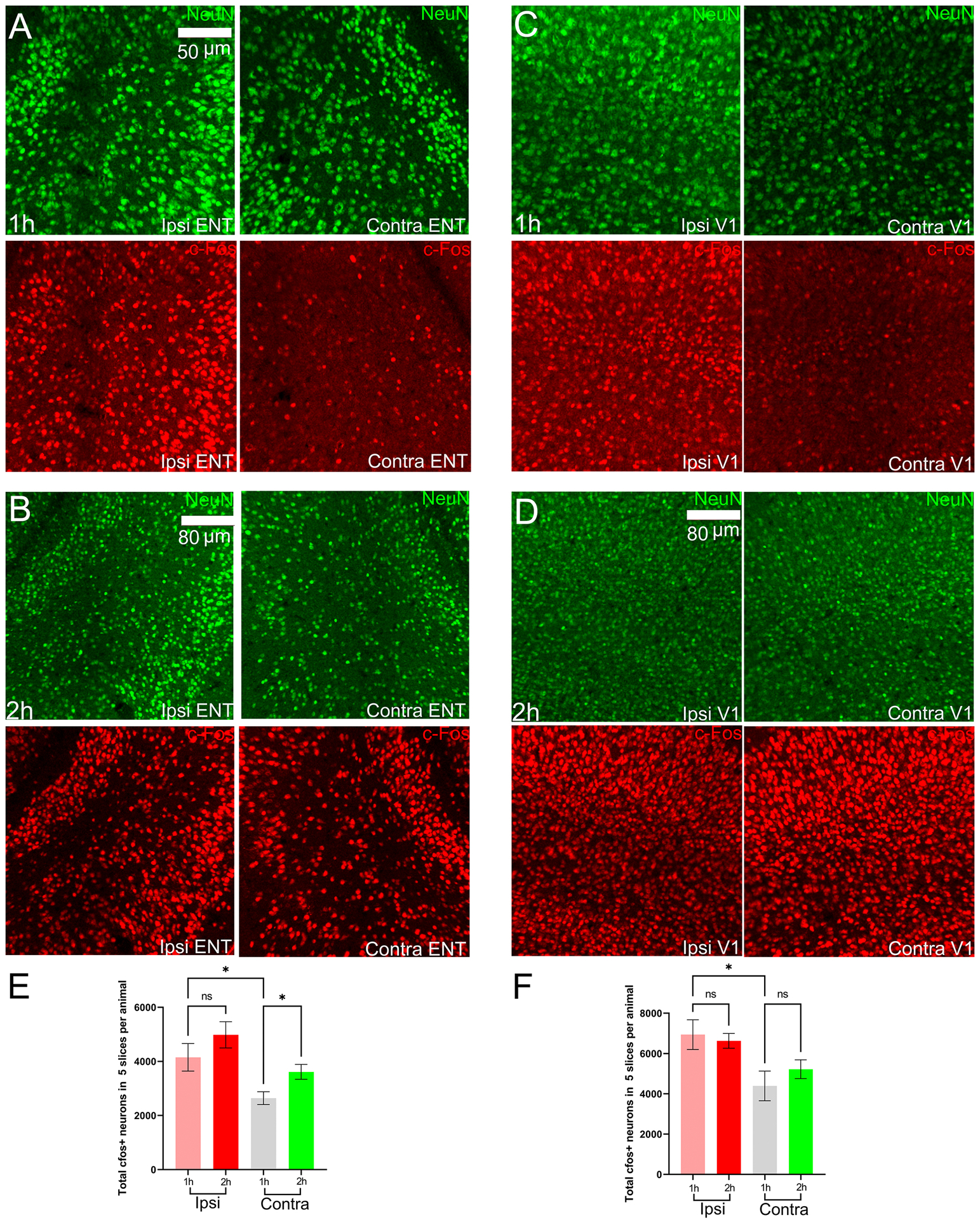
NeuN and cFos expression in the entorhinal and primary visual cortices. Ipsi is right hemisphere, Contra is left hemisphere, ENT is entorhinal cortex, S1 is primary visual cortex. (A)-(B) NeuN and cFos during the early stage (1h) and late-stage (2h) in the entorhinal cortex. (C)-(D) NeuN and cFos during the early stage (1h) and late-stage (2h) in the primary visual cortex. (E)-(F) cFos+ neuronal counts for the entorhinal cortex and primary visual cortex in mice of Co-homocysteine group.
Figure 5.
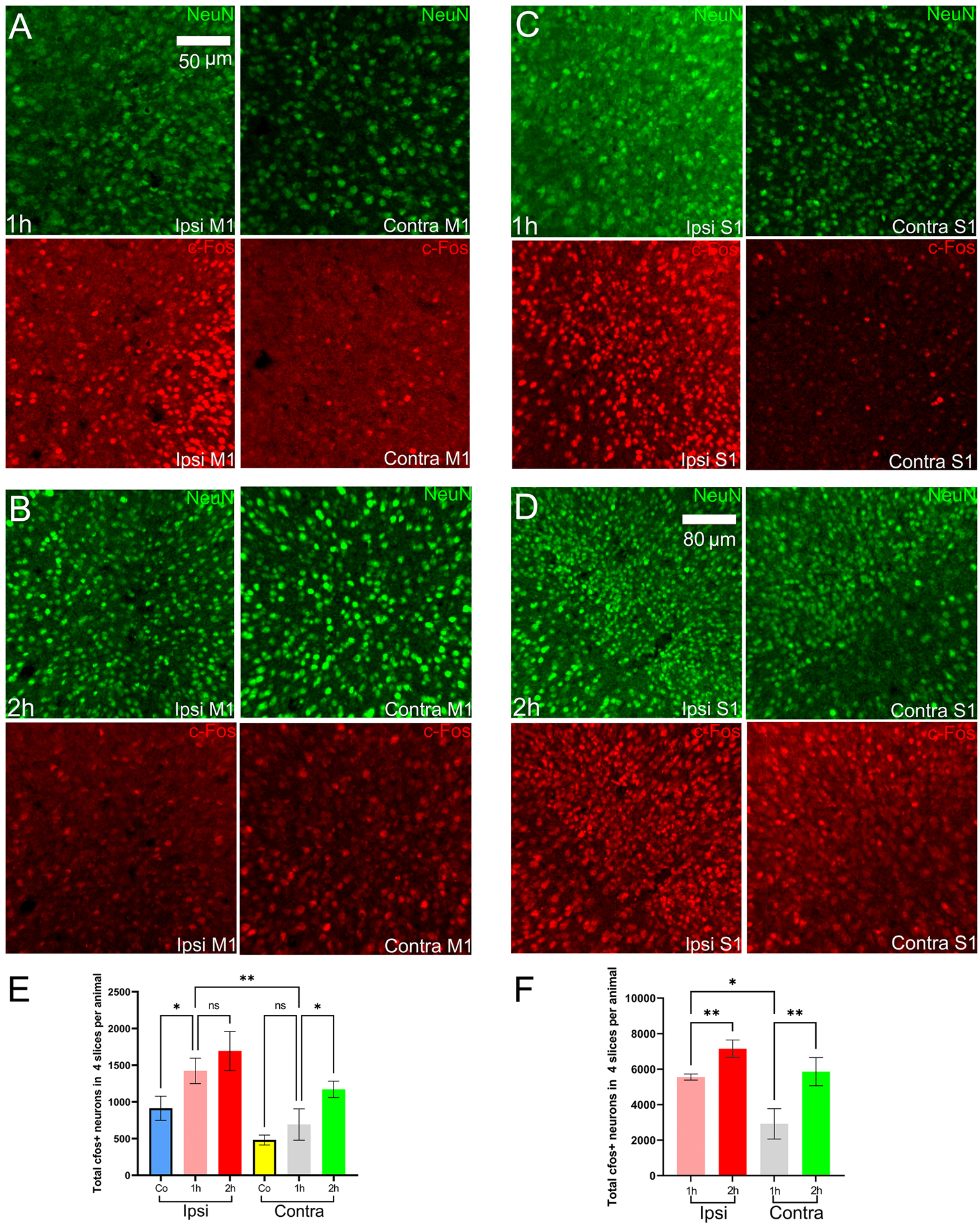
NeuN and cFos expression in the primary motor and primary somatosensory cortices. Ipsi is the right hemisphere, and Contra is the left hemisphere, M1 is the primary motor cortex, S1 is the primary somatosensory cortex. Co is cobalt baseline group. (A)-(B) NeuN and cFos during the early stage (1h) and late-stage (2h) in the primary motor cortex. (C)-(D) NeuN and cFos during the early stage (1h) and late-stage (2h) in the primary somatosensory cortex. (E)-(F) cFos+ neuronal counts for the primary motor cortex and primary somatosensory cortex in mice of the Co-homocysteine group.
Late stage
Continuous seizure activity marked the late phase. Ongoing generalized seizure activity transitioned to generalized periodic discharges interspersed with bursts of polyspike discharges and periods of relatively flat EEG background (Fig. 2(A)). The end of SE was determined by visual inspection of EEG traces. The end pf SE was marked when the frequency of spikes fell below 1 Hz, became arrhythmic, and had no sign of restoration of seizures. The EEG traces corresponding to behavior (tonic stiffening, bilateral clonus) during continuous seizures were given in Fig. 2(D–E). Coma followed this late phase with the corresponding EEG traces shown in Fig. 2(F).
We observed bilateral cortical cFos IR during the late stage. We observed that cortical areas, such as the motor (Fig. 5(B)), somatosensory (Fig. 5(D)), visual (Fig. 6(D)), and retrosplenial cortices (Fig. 9(C)), have a nearly uniform distribution of cFos across all layers. However, the entorhinal cortex (Fig. 6(B)) on both hemispheres showed dense cFos IR at layers 1, 2a, 2b, and 6a, whereas the rest of the layers showed sparse cFos IR (Fig. 4). We found differences in IR between early and late-stage SE in cortical regions, such as the somatosensory cortex (p < 0.0001), motor cortex (p=0.0008), anterior cingulate cortex (p = 0.002), and retrosplenial cortex (p = 0.007). In the case of the motor cortex, the contralateral side cFos IR was substantially increased. The cFos IR at the ipsilateral motor cortex (Fig. 5(E)) did not change (from 1423 ± 173.1 to 1693 ± 266.6, p = 0.469 Fig. 5(E)). The constant cFos IR was a likely effect of the cobalt lesion. The somatosensory cortex (Fig. 5(F)) showed a marked increase in cFos IR in both hemispheres (right : from 5558 ± 171.3 to 7152 ± 491.6, p = 0.0065; left: from 2919 ± 856.6 to 5861 ± 791.2, p = 0.0092). The cFos IR at the entorhinal cortex had the same pattern as that of the motor cortex (right: from 4153 ± 508.4 to 4893 ± 484, p = 0.2541; left: from 2641 ± 236.2 to 3614 ± 274.4, p = 0.0243). In the case of the visual cortex, there was no marked increase in cFos IR between stages. In the case of the retrosplenial cortex (Fig. 9(D)), there was an increase in cFos IR in the right (ipsilateral to cobalt) hemisphere (right: from 9906 ± 118.7 to 11277 ± 353.5, p = 0.0496; left: from 7324 ± 166.5 to 9056 ± 1702 , p = 0.4248 Fig. 9(D)). In addition, there were intense cFos IR in the perirhinal, ectorhinal, and piriform (p = 0.008) cortices. These results confirmed that the activation patterns were different between the stages.
Figure 9.
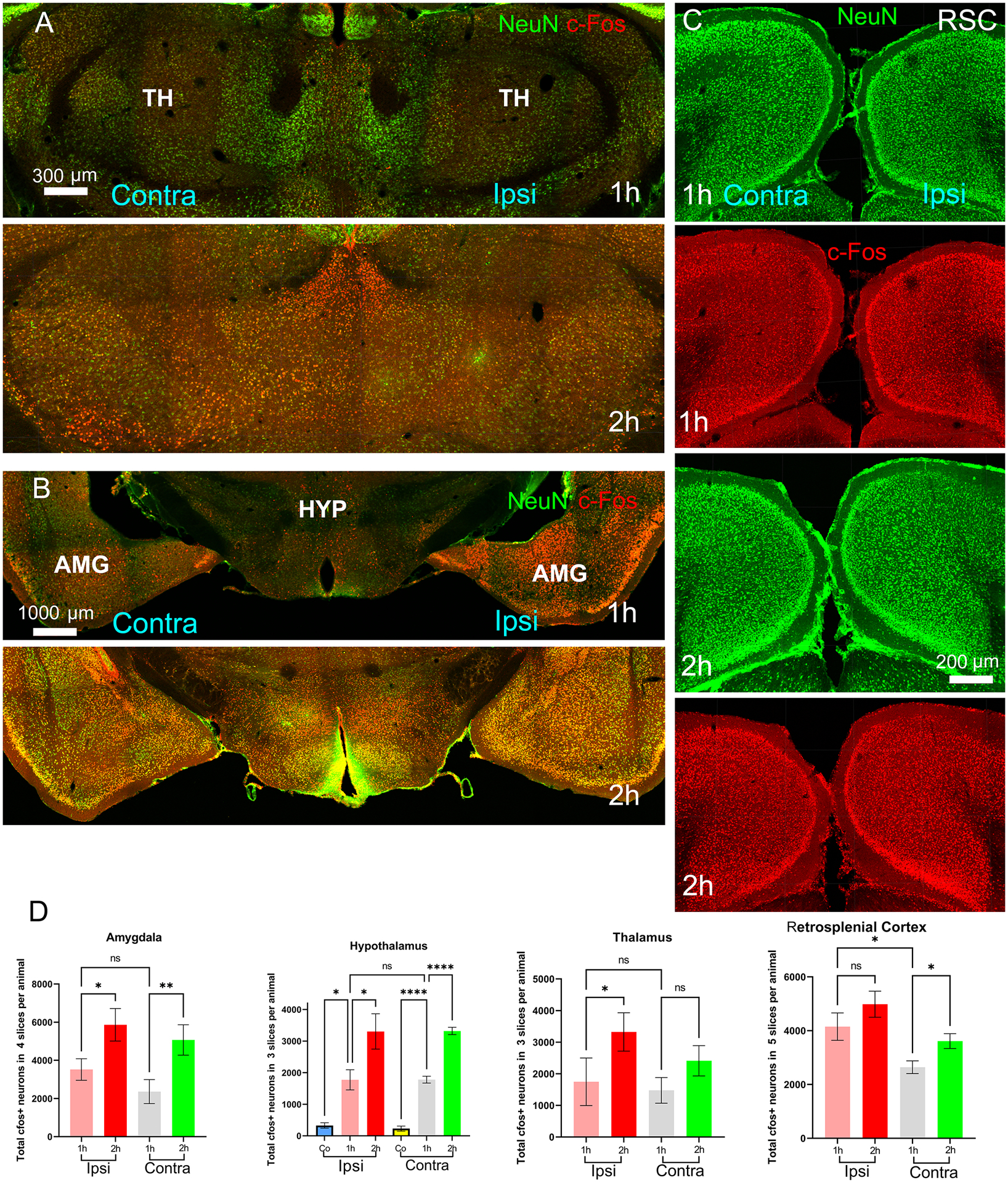
Co-expression of NeuN and cFos in brain regions – (A) thalamus and (B) amygdala and hypothalamus – are shown. (C) NeuN and cFos expression on the ipsilateral and contralateral sides are shown at the retrosplenial cortical areas. (D) cFos+ neuronal counts for the brain regions – amygdala, hypothalamus, thalamus and retrosplenial cortex – are shown in the bar graphs. TH is the thalamus; HYP is the hypothalamus; AMG is the amygdala; and RSC is the retrosplenial cortex.
Figure 4.
cFos immunoreactivity (IR) in the different brain regions during continuous generalized convulsive seizures are shown using the coronal sections at the level of bregma 1.5, 1.0, −0.5, −1.0, −2.0, −3.0 mm (A to F, respectively; scale bar =1000 μm). (A-B) Coronal sections at the level of bregma 1.5 and 1.0 mm (A and B, respectively) show the Co lesion and cFos immunoreactivity in the cortical and subcortical areas, such as the motor cortex, somatosensory cortex, septal nucleus and anterior cingulate gyrus. The activation is found to be bilateral. (C-D) Coronal sections at the level of bregma −0.5 and −1.0 mm (C and D, respectively) indicating the bilateral activation of cortical, amygdalar, and thalamic nuclei. (E-F) Coronal sections at the level of bregma −2.0 and −3.0 mm (E and F, respectively), indicating bilateral cFos IR in various cortical regions, such as the posterior-parietal association area, primary somatosensory area and auditory area, Please note that intense cFos IR was observed at the hippocampus.
cFos IR on the contralateral cortex increased substantially in the late stage (left column of Fig. 4) compared to the early stage (left column of Fig. 3). cFos IR in the ipsilateral cortex appeared to increase during continuous generalized convulsive seizures (late-stage) than during intermittent focal seizures (early stage). Therefore, we tested if the ipsilateral cFos IR increased in cortical areas proximal such as the somatosensory cortex and remote from the Co lesion like the rhinal and visual cortex.
In late-stage, cFos increased in somatosensory (p = 0.012) (Fig. 5(D)), retrosplenial (p = 0.043) (Fig. 9(C)) but not in the motor cortex(p = 0.63) (Fig. 5(B)). We discarded slices at the cobalt focus from counting. The primary motor cortex at the ipsilateral side was still affected by the extent of cobalt lesion, as observed in Fig. 3(A) and Fig. 4(A). To reduce the effect of the lesion, we normalized the cFos+ neuronal count by dividing it with the total neuronal count (using NeuN) at the motor cortex. We performed this procedure for the early stage and the late stage separately. We found a difference (p = 0.041) in the normalized cFos+ neuronal count at the ipsilateral motor cortex between both stages. Next, let us consider the somatosensory cortex in detail. At the contralateral side, SE elicited a substantial increase in per animal mean cFos IR from 2919 (early stage) to 5861 (late stage), marking a 2.14 folds increment (Fig. 5(F)). At the ipsilateral side, SE also induced an elevated mean cFos IR per animal from 5558 (early stage) to 7152 (late stage), indicating a 1.5 folds increment. This pattern of a greater increase in the contralateral cortex held true for other cortical areas, such as the retrosplenial and anterior cingulate cortices, close to the motor cortex. This observation suggested that prolonged seizures progressively activated more neurons over time in both hemispheres. In addition, There are cortico-cortical connections from M2 to these cortical regions, which are densely innervated by the perirhinal cortex and sparsely by the piriform cortex (Vismer et al. 2015).
cFos expression in the hippocampus
The hippocampus and DG were very sparsely activated during the early stage (Fig. 7(CII)) but intensely during the late stage (Fig. 7(DII)). Elaborately, we observed sparse activation of CA1, CA2, and CA3 and dentate gyrus (DG) in 4 out of 5 animals on both hemispheres during the early stage. In early stage, there was bilaterally equal cFos IR in CA1 (p = 0.84), CA2 (p = 0.91), CA3 (p = 0.46) and DG (p = 0.74). The magnified images of CA1, CA2, CA3, and DG are in Fig. 8(A–H). Each image contains the co-expression of NeuN and cFos. We found significant increase in the late stage, (multiple comparison Fig. 8(I)) at CA1 (p = 0.0013), CA2 (p = 0.014), CA3 (p = 0.024) and DG (p = 0.001). cFos IR of DG increased from 86.40 ± 9.3 (early stage) to 651.4 ± 28.16 (late stage), indicating a nearly eight-fold increase in the number of cFos expressing cells per image at the ipsilateral side. CA1 cFos IR increased from 81.20 ± 7.39 to 648.6 ± 52.96, CA2 increased five-fold from 52.7 ± 9.52 to 243.4± 30.85 on the ipsilateral side. Lastly, we observed eightfold cFos IR increment from 113.6 ± 14.84to 851.4± 99.65 in ipsilateral CA3. The fold of increment on the contralateral stayed the same as that of the ipsilateral side for CA1, CA2, CA3, and DG.
Figure 7.
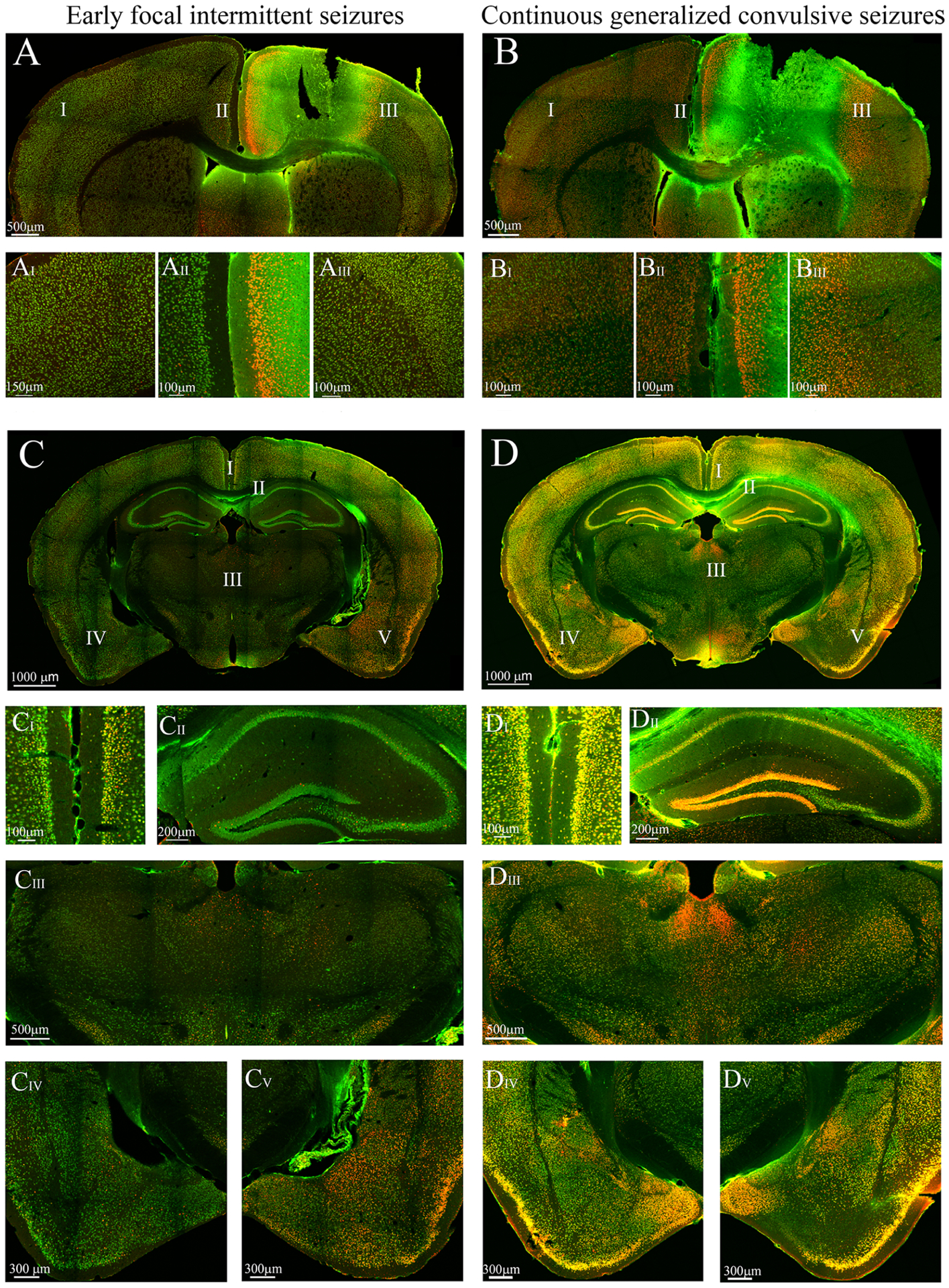
This figure presents cFos immunoreactivity (IR) of slices to compare neuronal activation between the ipsi and contralateral sides in each stage and activation between stages. Areas with roman numerals in the image are magnified for better visualization of the cFos IR. (A) Ipsilateral cFos IR observed in the anterior cingulate gyrus and primary somatosensory cortex during early focal intermittent seizures (B) Bilateral cFos IR in the anterior cingulate gyrus and somatosensory cortex during continuous generalized convulsive seizures. (C) Ipsilateral cFos IR observed primarily in cortical and amygdalar areas. Sparse cFos immunoreactivity was observed in the thalamus and hippocampus. (D) Bilateral cFos IR observed in the cortical and subcortical areas. There is a striking difference of cFos IR at the hippocampus, especially CA1, CA2 and the dentate gyrus (DG), between both stages. It can also be observed that cortical cFos IR (at the retrosplenial, somatosensory, auditory and piriform cortices) in the late stage was increased from the early stage. In addition, the cFos IR at the amygdala (CIV-V vs DIV-V) was elevated in the late stage compared the early stage and became bilateral.
Figure 8.
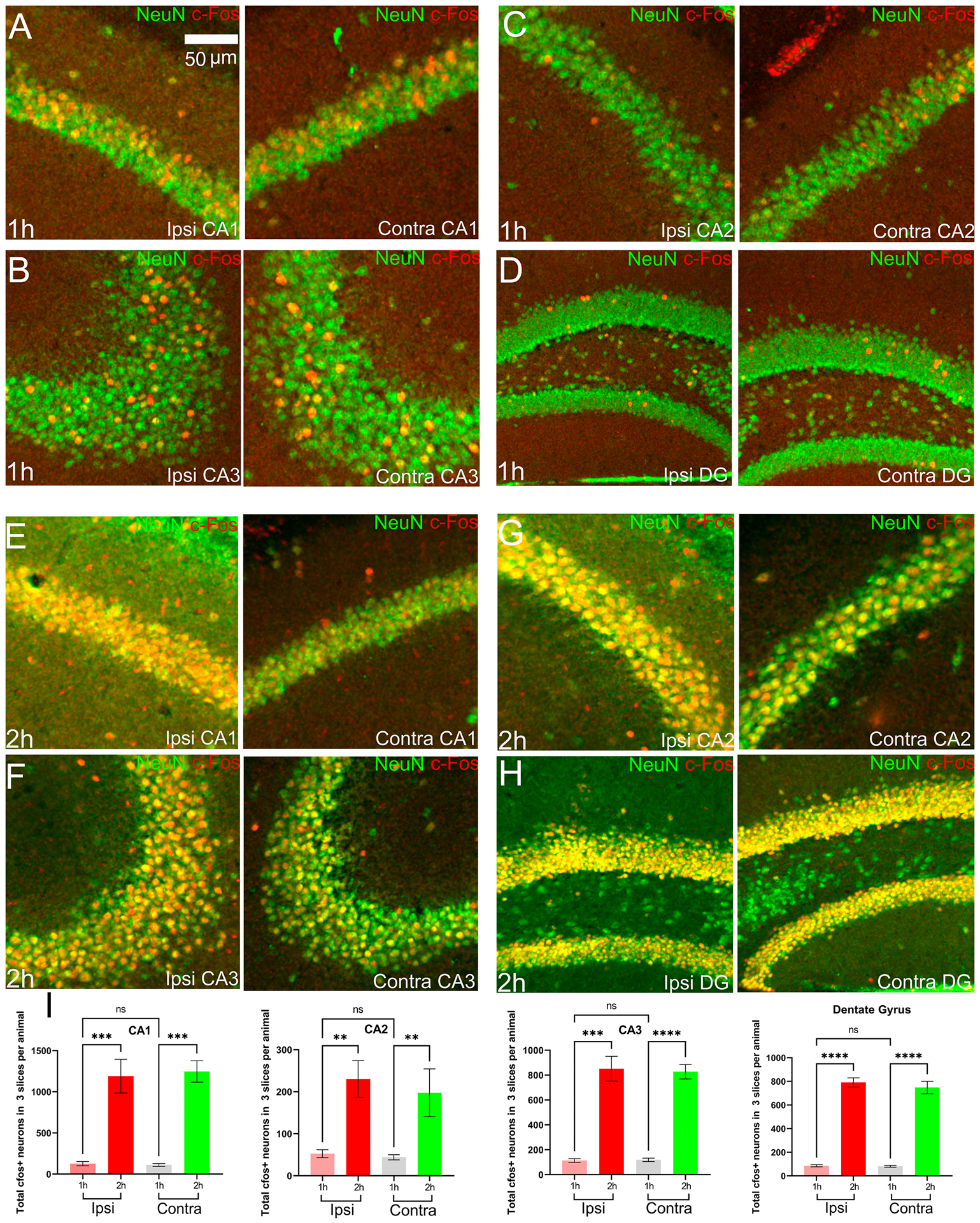
The figure shows cFos IR at the hippocampus (CA1, CA2 and CA3) and dentate gyrus. The images in panel A-H have both stainings, NeuN and cFos overlapped. (A)-(D) cFos IR as well as NeuN at CA1, CA3, CA2 and DG respectively in the early stage. There was very spase cFos IR in these regions. (E)-(H) cFos IR as well as NeuN at CA1, CA3, CA2 and DG respectively in the late stage. There was intense cFos IR in these regions. (I) cFos+ neuronal counts for the hippocampus and DG in mice of Co-homocysteine group.
cFos expression in other subcortical structures
Previous showed multiple pathways connecting the motor cortex to subcortical structures, such as the striatum, globus pallidus externa (GPe), globus pallidus interna (GPi), subthalamic nuclei (STN), substantia nigra reticulata (SNr) and thalamus studies (Brodovskaya et al., 2021; Dabrowska et al., 2019). There exists a direct reciprocal connection from the motor cortex to the motor thalamus, which consists of ventrolateral (VL), ventral anterior and ventromedial thalamic nuclei. The appearance of cFos IR at the motor thalamus is consistent with such projections (Fig. 9(A)).
We investigated the cFos expression in those regions and found sparse activation of the thalamus during the early stage, and the difference between the ipsilateral and contralateral sides did not reach significance (p = 0.32) (Fig. 9(D)). STN and GPe were also cFos labeled. However, we did not find region-wise differences of cFos IR between the ipsi- and contralateral sides in the early stage except STN (p = 0.03). Previous studies reported that electrical stimulation of the motor cortex activates the STN through direct excitatory, glutamatergic inputs, also known as a cortico-subthalamic hyper-direct pathway which projects primarily from the motor cortex (Inoue et al. 2012). These connections explained the intense STN activation in both early focal intermittent and continuous generalized convulsive seizure phase. When comparing the ipsilateral side between both the stages, we found substantial differences in cFos IR (STN: p = 0.005, GPe: p = 0.018).
Among other thalamic nuclei, the reticular thalamus (RT), midline thalamic nuclei, such as the paraventricular (PVT) and paratenial (PT) thalamic nuclei and the mediodorsal (MD) thalamic nucleus were cFos labeled during both stages. The prefrontal cortex (PFC) areas, including the infralimbic, prelimbic, and insular cortices, are a major source of input to the PT, and PVT nuclei (Kirouac 2015; Mitchell and Chakraborty 2013). Anatomical tracing studies confirmed that each subgroup of MD nuclei is interconnected to the PFC. The MD medial, central and lateral were found to have intense cFos activation (p = 0.043 for MD on the ipsilateral side compared to the contralateral side) in the early stage. The late-stage was marked with more cFos activity in MD (p = 0.01) compared to the early stage. The thalamus on both hemispheres was shown in Fig. 7 (CIII) (early stage) and Fig. 7(DIII) (late-stage) and magnified images are in Fig. 9(A). The cFos+ neuronal count is shown in Fig. 9(D).
Several amygdalar nuclei, such as the lateral amygdala, medial amygdalar nucleus, central amygdalar nucleus, cortical amygdalar area, and piriform-amygdalar nucleus, had intense cFos IR ipsilateral to the Co lesion during early stage (p = 0.029). There is a direct connection from the piriform cortex to the amygdala (Veening 1978; Wakefield 1980) and a diffuse polysynaptic connection from the amygdala to the piriform cortex via the endopiriform nucleus (Krettek and Price 1977). The difference between the left and right amygdala was found to be significant (p = 0.029) during the early stage. Amygdala is robustly activated during the late stage when compared to the early one (p = 0.004). The amygdala on both hemispheres is shown in Fig. 7, early-stage CIV-V, and late-stage DIV-V. The magnified images are shown in Fig. 9 (B) with counts in Fig. 9(D).
Hypothalamic regions on both sides contained sparse cFos IR. Cobalt injury-induced elevated cFos expression (Fig. 1(C)). Therefore, we included the count of cFos+ neurons as a control, as shown in Fig. 9(D). Both hemispheres during the early stage had significantly more cFos IR compared to the cobalt control. Statistical tests suggested no significant difference (p = 0.97) between the hemispheres during the initial stage. However, there was a remarkable increment in cFos IR during the late stage, specifically at the contralateral side. This effect was visible by comparing Fig. 9 (early-stage (1h) and late-stage (2h)). The cFos IR per animal was elevated (right: from 1773 ± 319.9 in the early stage to 3305 ± 559.1 in the late stage, p = 0.0205; left: from 1780 ± 107.6 in the early stage to 3320 ± 116.7 in the late stage, p < 0.0001).
Discussion
The present study provided the first detailed functional anatomy of seizure spread during neocortical injury-induced SE. There was intense cFos IR in the cerebral cortex ipsilateral to the seizure focus during early SE compared to the contralateral side. Major cortical regions included the motor, somatosensory, anterior cingulate, retrosplenial, visual, rhinal and piriform cortices and the posterior parietal association areas. Over time, seizures spread bilaterally and became multi-lobar, as demonstrated by bilateral cFos IR. We noticed a substantial cFos IR increase in the left hemisphere (contralateral side) during the late stage compared to the early stage. In addition, we found that the activity at the ipsilateral side was also increased during the late stage. Subcortical structures, such as several thalamic nuclei, hypothalamus, hippocampus, and the dentate gyrus (DG) that exhibited sparse cFos IR during the early stage showed strong cFos IR in both hemispheres during the late stage.
The unilateral activation during the early stage during which major cortical areas were active and subcortical structures were found to contain sparse cFos IR imply that the primary anatomical pathways for the propagation of focal seizures were cortio-cortical connections. The cortex has a laminar organization (Barbas 1986; Douglas and Martin 2004) containing a major population (~70%) of excitatory neurons (mostly the pyramidal and stellate neurons (Powell and Mountcastle 1959; Shepherd 2004) and a rich variety of inhibitory neurons (Gabbott and Somogyi 1986; Isaacson and Scanziani 2011), together forming ~80% by volume of human brain (Passingham 1982). Based on their morphology, pyramidal neurons form synapses with neurons within cortical layers, between cortical layers, project efferents to and receive afferents from several subcortical and midbrain structures. Apart from receiving excitatory inputs from cortical layers, inhibitory interneurons also receive long-range projections from subcortical areas. However, cortico-cortical projections are remarkably extensive in number than the efferents from the subcortex. Braintenberg and Schuz found that only 1 in 100 or even 1000 white matter fibers connect the cortex with subcortical areas; inter-and intra-hemispheric cortical projections constitute the rest of the white matter fibers (Schüz, and Braitenberg 1998). In general, a single excitatory cortical neuron is densely innervated by a large group of cortical neurons.
Based on the target output and the degree of inhibition by neighboring interneurons, the laminar topography of cortical excitatory neurons can be subdivided in two groups (Felleman and Essen, 1991) – layers L2 and L3 occasionally L4, and layers 5 and 6 (L5 and L6). L2–3 and L5–6 are strongly interconnected with reciprocal projections. There is also substantial diversity among inter-cortical long-range projections of excitatory neurons (Gerfen et al., 2018; Aronoff et al., 2010; Mao et al., 2011; Chakrabarti and Alloway 2006). As an example, using Channelrhodopsin, retrograde and anterograde labeling, authors found that mouse barrel cortex preferentially targets L2/3 and L5A of the motor cortex, whereas the projections from the motor cortex to the primary somatosensory (S1) area are diffused over all the layers, thereby forming multiple feedback loops between the superficial layers of vibrissal primary motor area and the vibrissal primary somatosensory area (Mao et al., 2011). Similar to the motor and somatosensory cortices, the visual cortex has extensive long-range connections with other cortical areas, such the motor, somatosensory and anterior cingulate cortices (Wang, Gao, and Burkhalter 2011; Froudarakis et al., 2019; Muir et al., 1996). An example of long-range reciprocal projections between the motor cortex (A24b and the secondary motor area, M2) and the primary visual cortex (V1) was studied in (Wang, Gao, and Burkhalter 2011). These long-range connections play crucial role in sensorimotor integration (Hoffer et al., 2003; Ferezou et al., 2007; Wang, Gao, and Burkhalter 2011). Routing seizures through such long- and short-range intra-cortical and inter-cortical projections may explain the activation of distant cortical areas (example, the somatosensory and visual cortices) during the early stage.
During the late stage, subcortical structures were intensely active compared to the early stage. Previous works suggested that subcortical were parts of multiple cortico-subcortical re-entrant circuits (Lothman et al., 1991; Brodovskaya et al., 2021). One such circuit involves the motor cortex that projects to the striatum (Haber 2016). Striatum, in turn, projects to its downstream structures based on the spiny neurons that express dopamine receptors D1 or D2. D1-expressing neurons directly project to GPi and SNr. D2-expressing neurons project to GPe, STN and then finally to SNr, forming an indirect pathway. The anterior part of the motor thalamus is densely innervated by GPe and SNr with GABAergic projections, thus forming a re-entrant circuit that starts from and ends at the motor cortex (DeLong and Wichmann 2007). In addition, direct cortico-pallidal projections exist (Brodovskaya et al., 2021; Karube et al., 2019). Likewise, STN also receives direct input from the motor cortex (Nambu et al., 2002; Inoue et al., 2012). Striatum acts as a large reservoir of GABAergic medium spiny neurons. In addition, SNr and GP also contain GABAergic neurons. During cortical seizure spread, such strong inhibition can prevent or delay the activation of subcortical structures in this circuit. Breakdown of inhibition due to prolonged seizures may be a potential reason for intense activation during the late stage.
In the cobalt homocysteine model, cobalt caused local hypoxic injury to neurons. This caused an increase in neuronal excitability. Later, the administration of homocysteine caused blood-brain barrier damage, which caused seizures and induced SE (Singh et al., 2020). The exact molecular mechanism of how blood-brain barrier damage leads to seizures is still unknown.
We observed that the superficial cortical layers had intense cFos IR early. This may imply that the superficial layers of the cortex were a potential region for seizure initiation. In our model, the cobalt wire was 1.2 mm in length. Therefore, after implantation, the cobalt came in contact with layers 1, 2/3, 5 and 6a of the supplementary motor cortex (verified from the Allen atlas). However, we observed intense cFos IR in the superficial layers (primary motor cortex and somatosensory cortex) compared to the deep layers. This result is in agreement with (Wenzel et al. 2017), where investigators used fast, two-photon imaging and multi-array LFP measurements to trace the spread of locally-induced seizures by 4-AP in adult C57/BL6 mice.
On the other hand, this result contrasts with the findings by a group of investigators (Connors 1984; Pinto 2005) who used coronal slices of the somatosensory cortex in animals (adult guniea pigs Sprague Dowley rats). Seizures were elicited by using GABA-A antagonists (bicuculline or picrotoxin). Their findings suggested that deep layers are the potential regions for seizure initiation. In our model of neocortical injury-induced SE, whether layer 2/3 was the site for seizure initiation or not needs further in vivo experiments.
The intense cFos IR of the superficial cortical layers also indicates that the seizures spread unilaterally through cortico-cortical connections in the superficial layers. During the late stage, the superficial layers progressively activated the deep cortical layers. Studies using human EEG measured by multi-electrode arrays (Schevon et al. 2012) found that foci of cortical seizures in chronic epileptic patients have inhibitory restraint surrounding the foci. The inhibitory surrounding of a seizure focus limits the pace at which neurons are recruited for seizure spread. This characteristic of seizure foci in the cortex had also been reported in animals (Schwartz and Bonhoeffer 2001) with acute models of seizures. Using only cFos IR, it is difficult to assert how seizures spread from the focus. However, activation of cortical regions, which are remote from the cobalt implantation site, suggests that seizures promptly followed long-range excitatory cortico-cortical projections for propagation. The mechanism remains to be determined whether runway excitatory neurotransmission or breakdown of inhibitory neurotransmission or both in the cortex overcome the inhibitory surrounding of a seizure focus.
In animal models of SE, where SE can be induced by administering chemo-convulsants or performing electrical stimulation of limbic structures (Lothman et al., 1991; Lothman et al., 1990), the sensitivity towards benzodiazepine (such as lorazepam and diazepam) was reduced during SE (Kapur and Macdonald 1997b). Benzodiazepine facilitates the GABAergic inhibition of principal neurons by targeting GABAA type receptors. GABA is the major source of inhibition in the cortex, and the reduction of sensitivity indicates the decrease in the inhibitory neurotransmission. It has been proposed that internalization (removal of receptors from the plasma membrane) of GABAA receptors during SE via dephosphorylation may lead to such reduction (Joshi and Kapur 2012a; Kittler and Moss 2003; Terunuma et al. 2008). There is also evidence of resistance to benzodiazepines due to accumulation of chloride ions (Burman et al., 2019; Moore et al., 2018). Activation of AMPA (Rajasekaran et al., 2012; Joshi et al., 2017) and NMDA receptors (Naylor et al., 2013; Wasterlain et al., 2013) may also contribute to the accelerated internalization of GABARs. The breakdown of inhibition may trigger an increase in excitatory, glutamatergic transmission. Further studies are needed in order to confirm these findings in the context of neocortical SE.
Conclusion
In conclusion, this study provides an unbiased large-scale evaluation of neuronal circuits active during neocortical injury-induced SE for the first time. Neocortex injury can sustain prolonged seizures through several cortico-cortical and cortico-subcortical re-entrant circuits. However, what molecular and cellular mechanisms help SE sustain through neocortical circuits remains studied.
A large-scale, high-resolution map of neuronal activity during neocortical-onset SE.
Cortico-cortical re-entrant circuits sustain early SE
Bilateral subcortical and cortical structures were active during the late stage.
Funding and acknowledgments
National Institutes of Health grants R37 NS119012, R01NS120945, R01 NS 044370, and UVA Brain Institute grants JK supported this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
The authors report no competing interests.
References
- Aronoff Rachel, Matyas Ferenc, Mateo Celine, Ciron Carine, Schneider Bernard, and Petersen Carl C.H.. 2010. “Long-Range Connectivity of Mouse Primary Somatosensory Barrel Cortex: Long-Range Connectivity of Barrel Cortex.” European Journal of Neuroscience 31 (12): 2221–33. 10.1111/j.1460-9568.2010.07264.x. [DOI] [PubMed] [Google Scholar]
- Barbas H 1986. “Pattern in the Laminar Origin of Corticocortical Connections.” The Journal of Comparative Neurology 252 (3): 415–22. 10.1002/cne.902520310. [DOI] [PubMed] [Google Scholar]
- Braitenberg Valentino, Schüz A, and Braitenberg Valentino. 1998. Cortex: Statistics and Geometry of Neuronal Connectivity. 2nd thoroughly rev. ed. Berlin ; New York: Springer. [Google Scholar]
- Brodovskaya Anastasia, Shiono Shinnosuke, and Kapur Jaideep. 2021. “Activation of the Basal Ganglia and Indirect Pathway Neurons during Frontal Lobe Seizures.” Brain, March, awab119. 10.1093/brain/awab119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman Richard J, Selfe Joshua S, Lee John Hamin, van den Berg Maurits, Calin Alexandru, Codadu Neela K, Wright Rebecca, et al. 2019. “Excitatory GABAergic Signalling Is Associated with Benzodiazepine Resistance in Status Epilepticus.” Brain 142 (11): 3482–3501. 10.1093/brain/awz283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti Shubhodeep, and Alloway Kevin D.. 2006. “Differential Origin of Projections from SI Barrel Cortex to the Whisker Representations in SII and MI.” The Journal of Comparative Neurology 498 (5): 624–36. 10.1002/cne.21052. [DOI] [PubMed] [Google Scholar]
- Claassen J, Jette N, Chum F, Green R, Schmidt M, Choi H, Jirsch J, et al. 2007. “Electrographic Seizures and Periodic Discharges after Intracerebral Hemorrhage.” Neurology 69 (13): 1356–65. 10.1212/01.wnl.0000281664.02615.6c. [DOI] [PubMed] [Google Scholar]
- Claassen Jan, Perotte Adler, Albers David, Kleinberg Samantha, Schmidt J. Michael, Tu Bin, Badjatia Neeraj, et al. 2013. “Nonconvulsive Seizures after Subarachnoid Hemorrhage: Multimodal Detection and Outcomes: Claassen et al: Effects of Seizures after SAH.” Annals of Neurology 74 (1): 53–64. 10.1002/ana.23859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors Barry W. 1984. “Initiation of Synchronized Neuronal Bursting in Neocortex.” Nature 310 (5979): 685–87. 10.1038/310685a0. [DOI] [PubMed] [Google Scholar]
- Dabrowska Natalia, Joshi Suchitra, Williamson John, Lewczuk Ewa, Lu Yanhong, Oberoi Samrath, Brodovskaya Anastasia, and Kapur Jaideep. 2019. “Parallel Pathways of Seizure Generalization.” Brain, June, awz170. 10.1093/brain/awz170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felleman Daniel J., and Van Essen David C.. n.d. “Distributed Hierarchical Processing in the Primate Cerebral Cortex.” 1991. 1.1: 1–47. [DOI] [PubMed] [Google Scholar]
- DeLong Mahlon R., and Wichmann Thomas. 2007. “Circuits and Circuit Disorders of the Basal Ganglia.” Archives of Neurology 64 (1): 20. 10.1001/archneur.64.1.20. [DOI] [PubMed] [Google Scholar]
- Douglas Rodney J., and Martin Kevan A.C.. 2004. “NEURONAL CIRCUITS OF THE NEOCORTEX.” Annual Review of Neuroscience 27 (1): 419–51. 10.1146/annurev.neuro.27.070203.144152. [DOI] [PubMed] [Google Scholar]
- Dragunow M, and Robertson HA. 1988. “Brain Injury Induces C-Fos Protein(s) in Nerve and Glial-like Cells in Adult Mammalian Brain.” Brain Research 455 (2): 295–99. 10.1016/0006-8993(88)90088-1. [DOI] [PubMed] [Google Scholar]
- Ferezou Isabelle, Haiss Florent, Gentet Luc J., Aronoff Rachel, Weber Bruno, and Petersen Carl C.H.. 2007. “Spatiotemporal Dynamics of Cortical Sensorimotor Integration in Behaving Mice.” Neuron 56 (5): 907–23. 10.1016/j.neuron.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Froudarakis Emmanouil, Fahey Paul G., Reimer Jacob, Smirnakis Stelios M., Tehovnik Edward J., and Tolias Andreas S.. 2019. “The Visual Cortex in Context.” Annual Review of Vision Science 5 (1): 317–39. 10.1146/annurev-vision-091517-034407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbott PLA, and Somogyi P. 1986. “Quantitative Distribution of GABA-Immunoreactive Neurons in the Visual Cortex (Area 17) of the Cat.” Experimental Brain Research 61 (2). 10.1007/BF00239522. [DOI] [PubMed] [Google Scholar]
- Gerfen Charles R., Economo Michael N., and Chandrashekar Jayaram. 2018. “Long Distance Projections of Cortical Pyramidal Neurons.” Journal of Neuroscience Research 96 (9): 1467–75. 10.1002/jnr.23978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber Suzanne N. 2016. “Corticostriatal Circuitry.” Dialogues in Clinical Neuroscience 18 (1): 7–21. 10.31887/DCNS.2016.18.1/shaber. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman Susan T., Abend Nicholas S., Bleck Thomas P., Chapman Kevin E., Drislane Frank W., Emerson Ronald G., Gerard Elizabeth E., et al. 2015. “Consensus Statement on Continuous EEG in Critically Ill Adults and Children, Part I: Indications.” Journal of Clinical Neurophysiology 32 (2): 87–95. 10.1097/WNP.0000000000000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffer Zachary S., Hoover John E., and Alloway Kevin D.. 2003. “Sensorimotor Corticocortical Projections from Rat Barrel Cortex Have an Anisotropic Organization That Facilitates Integration of Inputs from Whiskers in the Same Row.” The Journal of Comparative Neurology 466 (4): 525–44. 10.1002/cne.10895. [DOI] [PubMed] [Google Scholar]
- Inoue Ken ichi, Koketsu Daisuke, Kato Shigeki, Kobayashi Kazuto, Nambu Atsushi, and Takada Masahiko. 2012. “Immunotoxin-Mediated Tract Targeting in the Primate Brain: Selective Elimination of the Cortico-Subthalamic ‘Hyperdirect’ Pathway.” PLoS ONE 7 (6): 1–9. 10.1371/journal.pone.0039149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson Jeffry S., and Scanziani Massimo. 2011. “How Inhibition Shapes Cortical Activity.” Neuron 72 (2): 231–43. 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi Suchitra, and Kapur Jaideep. 2012a. “GABAA Receptor Plasticity during Status Epilepticus.” [PubMed]
- ———. 2012b. “GABAA Receptor Plasticity during Status Epilepticus.” [PubMed]
- Joshi Suchitra, Rajasekaran Karthik, Sun Huayu, Williamson John, and Kapur Jaideep. 2017. “Enhanced AMPA Receptor-Mediated Neurotransmission on CA1 Pyramidal Neurons during Status Epilepticus.” Neurobiology of Disease 103 (July): 45–53. 10.1016/j.nbd.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur Jaideep, and Macdonald Robert L.. 1997a. “Rapid Seizure-Induced Reduction of Benzodiazepine and Zn 2+ Sensitivity of Hippocampal Dentate Granule Cell GABA A Receptors.” The Journal of Neuroscience 17 (19): 7532–40. 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1997b. “Rapid Seizure-Induced Reduction of Benzodiazepine and Zn 2+ Sensitivity of Hippocampal Dentate Granule Cell GABA A Receptors.” The Journal of Neuroscience 17 (19): 7532–40. 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karube Fuyuki, Takahashi Susumu, Kobayashi Kenta, and Fujiyama Fumino. 2019. “Motor Cortex Can Directly Drive the Globus Pallidus Neurons in a Projection Neuron Type-Dependent Manner in the Rat.” ELife 8 (November): e49511. 10.7554/eLife.49511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirouac Gilbert J. 2015. “Placing the Paraventricular Nucleus of the Thalamus within the Brain Circuits That Control Behavior.” Neuroscience and Biobehavioral Reviews 56: 315–29. 10.1016/j.neubiorev.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Kittler Josef T, and Moss Stephen J. 2003. “Modulation of GABAA Receptor Activity by Phosphorylation and Receptor Trafficking: Implications for the Efficacy of Synaptic Inhibition.” Current Opinion in Neurobiology 13 (3): 341–47. 10.1016/S0959-4388(03)00064-3. [DOI] [PubMed] [Google Scholar]
- Krettek JE, and Price JL. 1977. “Projections from the Amygdaloid Complex to the Cerebral Cortex and Thalamus in the Rat and Cat.” Journal of Comparative Neurology 172 (4): 687–722. 10.1002/cne.901720408. [DOI] [PubMed] [Google Scholar]
- Lewczuk Ewa, Joshi Suchitra, Williamson John, Penmetsa Mouna, Shan Sarah, and Kapur Jaideep. 2018. “Electroencephalography and Behavior Patterns during Experimental Status Epilepticus.” Epilepsia 59 (2): 369–80. 10.1111/epi.13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothman Eric W., Bertram Edward H., Kapur Jaideep, and Stringer Janet L.. 1990. “Recurrent Spontaneous Hippocampal Seizures in the Rat as a Chronic Sequela to Limbic Status Epilepticus.” Epilepsy Research 6 (2): 110–18. 10.1016/0920-1211(90)90085-A. [DOI] [PubMed] [Google Scholar]
- Lothman Eric W., Bertram Edward H., and Stringer Janet L.. 1991. “Functional Anatomy of Hippocampal Seizures.” Progress in Neurobiology 37 (1): 1–82. 10.1016/0301-0082(91)90011-O. [DOI] [PubMed] [Google Scholar]
- Mao Tianyi, Kusefoglu Deniz, Hooks Bryan M., Huber Daniel, Petreanu Leopoldo, and Svoboda Karel. 2011. “Long-Range Neuronal Circuits Underlying the Interaction between Sensory and Motor Cortex.” Neuron 72 (1): 111–23. 10.1016/j.neuron.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyas Ferenc, Sreenivasan Varun, Marbach Fred, Wacongne Catherine, Barsy Boglarka, Mateo Celine, Aronoff Rachel, and Petersen Carl C. H.. 2010. “Motor Control by Sensory Cortex.” Science 330 (6008): 1240–43. 10.1126/science.1195797. [DOI] [PubMed] [Google Scholar]
- Mitchell Anna S., and Chakraborty Subhojit. 2013. “What Does the Mediodorsal Thalamus Do?” Frontiers in Systems Neuroscience 7 (JUL): 1–19. 10.3389/fnsys.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore Yvonne E., Deeb Tarek Z., Chadchankar Heramb, Brandon Nicholas J., and Moss Stephen J.. 2018. “Potentiating KCC2 Activity Is Sufficient to Limit the Onset and Severity of Seizures.” Proceedings of the National Academy of Sciences 115 (40): 10166–71. 10.1073/pnas.1810134115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan J, Cohen D, Hempstead J, and Curran T. 1987. “Mapping Patterns of C-Fos Expression in the Central Nervous System after Seizure.” Science 237 (4811): 192–97. 10.1126/science.3037702. [DOI] [PubMed] [Google Scholar]
- Muir Janice L., Everitt Barry J., and Robbins Trevor W.. 1996. “The Cerebral Cortex of the Rat and Visual Attentional Function: Dissociable Effects of Mediofrontal, Cingulate, Anterior Dorsolateral, and Parietal Cortex Lesions on a Five-Choice Serial Reaction Time Task.” Cerebral Cortex 6 (3): 470–81. 10.1093/cercor/6.3.470. [DOI] [PubMed] [Google Scholar]
- Nambu Atsushi, Tokuno Hironobu, and Takada Masahiko. 2002. “Functional Significance of the Cortico–Subthalamo–Pallidal ‘Hyperdirect’ Pathway.” Neuroscience Research 43 (2): 111–17. 10.1016/S0168-0102(02)00027-5. [DOI] [PubMed] [Google Scholar]
- Naylor David E., Liu Hantao, Niquet Jerome, and Wasterlain Claude G.. 2013. “Rapid Surface Accumulation of NMDA Receptors Increases Glutamatergic Excitation during Status Epilepticus.” Neurobiology of Disease 54 (June): 225–38. 10.1016/j.nbd.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passingham RE 1982. The Human Primate. Oxford ; San Francisco: W.H. Freeman. [Google Scholar]
- Pinto DJ 2005. “Initiation, Propagation, and Termination of Epileptiform Activity in Rodent Neocortex In Vitro Involve Distinct Mechanisms.” Journal of Neuroscience 25 (36): 8131–40. 10.1523/JNEUROSCI.2278-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell TP, and Mountcastle VB. 1959. “Some Aspects of the Functional Organization of the Cortex of the Postcentral Gyrus of the Monkey: A Correlation of Findings Obtained in a Single Unit Analysis with Cytoarchitecture.” Bulletin of the Johns Hopkins Hospital 105 (September): 133–62. [PubMed] [Google Scholar]
- Rajasekaran Karthik, Todorovic Marko, and Kapur Jaideep. 2012. “Calcium-Permeable AMPA Receptors Are Expressed in a Rodent Model of Status Epilepticus.” Annals of Neurology 72 (1): 91–102. 10.1002/ana.23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade Sanjay N., Shah Aashit K., Agarwal Rajeev, Yao Bin, Asano Eishi, and Loeb Jeffrey A.. 2007. “Activity-Dependent Gene Expression Correlates with Interictal Spiking in Human Neocortical Epilepsy.” Epilepsia 48 (s5): 86–95. 10.1111/j.1528-1167.2007.01294.x. [DOI] [PubMed] [Google Scholar]
- Reep RL, Corwin JV, Hashimoto A, and Watson RT. 1987. “Efferent Connections of the Rostral Portion of Medial Agranular Cortex in Rats.” Brain Research Bulletin 19 (2): 203–21. 10.1016/0361-9230(87)90086-4. [DOI] [PubMed] [Google Scholar]
- Rush Barret, Wiskar Katie, Fruhstorfer Clark, and Hertz Paul. 2016. “Association between Seizures and Mortality in Patients with Aneurysmal Subarachnoid Hemorrhage: A Nationwide Retrospective Cohort Analysis.” Seizure 41 (October): 66–69. 10.1016/j.seizure.2016.07.008. [DOI] [PubMed] [Google Scholar]
- Schevon Catherine A., Weiss Shennan A., McKhann Guy, Goodman Robert R., Yuste Rafael, Emerson Ronald G., and Trevelyan Andrew J.. 2012. “Evidence of an Inhibitory Restraint of Seizure Activity in Humans.” Nature Communications 3 (1): 1060. 10.1038/ncomms2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz Theodore H., and Bonhoeffer Tobias. 2001. “In Vivo Optical Mapping of Epileptic Foci and Surround Inhibition in Ferret Cerebral Cortex.” Nature Medicine 7 (9): 1063–67. 10.1038/nm0901-1063. [DOI] [PubMed] [Google Scholar]
- Shepherd Gordon M., ed. 2004. The Synaptic Organization of the Brain. Oxford University Press. 10.1093/acprof:oso/9780195159561.001.1. [DOI] [Google Scholar]
- Singh Tanveer, Joshi Suchitra, Williamson John M., and Kapur Jaideep. 2020. “Neocortical Injury–Induced Status Epilepticus.” Epilepsia 61 (12): 2811–24. 10.1111/epi.16715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terunuma M, Xu J, Vithlani M, Sieghart W, Kittler J, Pangalos M, Haydon PG, Coulter DA, and Moss SJ. 2008. “Deficits in Phosphorylation of GABAA Receptors by Intimately Associated Protein Kinase C Activity Underlie Compromised Synaptic Inhibition during Status Epilepticus.” Journal of Neuroscience 28 (2): 376–84. 10.1523/JNEUROSCI.4346-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening JG 1978. “Cortical Afferents of the Amygdaloid Complex in the Rat: An HRP Study.” Neuroscience Letters 8 (3): 191–95. 10.1016/0304-3940(78)90120-9. [DOI] [PubMed] [Google Scholar]
- Vespa PM, O’Phelan K, Shah M, Mirabelli J, Starkman S, Kidwell C, Saver J, et al. 2003. “Acute Seizures after Intracerebral Hemorrhage: A Factor in Progressive Midline Shift and Outcome.” Neurology 60 (9): 1441–46. 10.1212/01.WNL.0000063316.47591.B4. [DOI] [PubMed] [Google Scholar]
- Vespa Paul M., Nuwer Marc R., Nenov Valeriy, Ronne-Engstrom Elisabeth, Hovda David A., Bergsneider Marvin, Kelly Daniel F., Martin Neil A., and Becker Donald P.. 1999. “Increased Incidence and Impact of Nonconvulsive and Convulsive Seizures after Traumatic Brain Injury as Detected by Continuous Electroencephalographic Monitoring.” Journal of Neurosurgery 91 (5): 750–60. 10.3171/jns.1999.91.5.0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vespa Paul, Tubi Meral, Claassen Jan, Buitrago-Blanco Manuel, McArthur David, Velazquez Angela G., Tu Bin, Prins Mayumi, and Nuwer Marc. 2016. “Metabolic Crisis Occurs with Seizures and Periodic Discharges after Brain Trauma: Metabolic Crisis after TBI.” Annals of Neurology 79 (4): 579–90. 10.1002/ana.24606. [DOI] [PubMed] [Google Scholar]
- Vismer Marta S., Forcelli Patrick A., Skopin Mark D., Gale Karen, and Koubeissi Mohamad Z.. 2015. “The Piriform, Perirhinal, and Entorhinal Cortex in Seizure Generation.” Frontiers in Neural Circuits 9 (May). 10.3389/fncir.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield Caroline. 1980. “The Topographical Organization and Laminar Origin of Some Cortico-Amygdaloid Connections.” Neuroscience Letters 20 (1): 21–24. 10.1016/0304-3940(80)90227-X. [DOI] [PubMed] [Google Scholar]
- Wang Q, Gao E, and Burkhalter A. 2011. “Gateways of Ventral and Dorsal Streams in Mouse Visual Cortex.” Journal of Neuroscience 31 (5): 1905–18. 10.1523/JNEUROSCI.3488-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasterlain Claude G., Naylor David E., Liu Hantao, Niquet Jerome, and Baldwin Roger. 2013. “Trafficking of NMDA Receptors during Status Epilepticus: Therapeutic Implications.” Epilepsia 54 (September): 78–80. 10.1111/epi.12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel Michael, Hamm Jordan P., Peterka Darcy S., and Yuste Rafael. 2017. “Reliable and Elastic Propagation of Cortical Seizures In Vivo.” Cell Reports 19 (13): 2681–93. 10.1016/j.celrep.2017.05.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams Mary B., and Jope Richard S.. 1994. “Distinctive Rat Brain Immediate Early Gene Responses to Seizures Induced by Lithium plus Pilocarpine.” Molecular Brain Research 25 (1–2): 80–89. 10.1016/0169-328X(94)90281-X. [DOI] [PubMed] [Google Scholar]
- Zingg Brian, Hintiryan Houri, Gou Lin, Song Monica Y., Bay Maxwell, Bienkowski Michael S., Foster Nicholas N., et al. 2014. “Neural Networks of the Mouse Neocortex.” Cell 156 (5): 1096–1111. 10.1016/j.cell.2014.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]