

Abstract
Methodological advances over the last three decades have led to a profound transformation in our understanding of the genetic origins of neuropsychiatric disorders. This is exemplified by the study of autism spectrum disorders (ASDs) for which microarrays, whole exome sequencing and whole genome sequencing have yielded over a hundred causal loci. Genome-wide association studies in ASD have also been fruitful, identifying 5 genome-wide significant loci thus far and demonstrating a substantial role for polygenic inherited risk. Approaches rooted in systems biology and functional genomics have increasingly placed genes implicated by risk variants into biological context. Genetic risk affects a finite group of cell-types and biological processes, converging primarily on early stages of brain development (though, the expression of many risk genes persists through childhood). Coupled with advances in stem cell-based human in vitro model systems, these findings provide a basis for developing mechanistic models of disease pathophysiology.
Introduction
A national survey of US children, conducted between 2009 and 2017, estimated the prevalence of neurodevelopmental disorders (NDDs) at about 1 in 6, or 16% (1). The onset of NDDs is early in life by definition and treatment options remain limited, so many affected individuals experience debilitating symptoms for the majority of their lives. Over the last 30 years, the steadily decreasing cost of genetic analysis, most prominently sequencing, has fueled tremendous gene discovery efforts in the study of NDDs. In this review, we reflect on recent progress and consider a significant challenge in the field for the next 30 years: how to move from genes to mechanisms to drive biological understanding and inform therapeutic development. We focus on autism spectrum disorder (ASD), an NDD that affects nearly 1 in 68 US children (1). We wrestle with how to move from genes to biological mechanisms by considering what genes have been implicated in ASD, where and when they are expressed and how they may relate to each other and, ultimately, to behavior.
ASD is typically diagnosed before the age of 3 based on symptomology in two core areas: social interaction and restrictive/repetitive behaviors (2). Social symptoms often manifest as lack of interest in social interactions and difficulty responding to social and nonverbal cues. Restrictive and repetitive behaviors may include stereotyped motor movements, difficulty with change to routine and intense fixation on restricted tasks or objects. These are often accompanied by hyper- and hypo-sensitivity to sensory stimuli. In addition to these core symptom domains, numerous other allied medical conditions are frequently co-morbid with ASD, including: seizure disorders (3–5), food sensitivities and gastrointestinal symptoms (6), sleep disorders (7), additional psychiatric diagnoses and intellectual disability (3). The significant heterogeneity in symptom presentation and severity is reflected by the term ‘autisms’ (8) and motivates the development of personalized treatment options. Early behavioral interventions are the most common treatments for disabling symptoms in ASD and show promise in a subset of patients (9,10). No pharmaceutical agents have been approved for the treatment of core social impairments (11,12). Significant efforts have been made to identify canonical biomarkers, but none are yet recognized (11,13), except perhaps macrocephaly in a subset of individuals (4,14). Thus, investigation into the genetics of autisms offers great promise in advancing our definition of autisms and developing much needed therapeutic options.
ASD as a Heritable Disorder
ASD has been recognized as a heritable condition since the mid-1970s (15), due to its co-occurrence in monozygotic twins, which is well summarized in a recent meta-analysis (16) and the persistence of sub-threshold traits in first degree family members (17–17). Further evidence of genetic etiology in ASD come from its association with dozens of genetic syndromes and chromosomal rearrangements (20–22,8,23,24). Twin studies have since estimated the heritability (h2) of broadly defined autisms anywhere from 0.45 to 0.9 (25–30), with a reasonable consensus around 0.7–0.8. The genetic architecture of psychiatric diseases, including ASD, has been extensively reviewed and this range of heritability observed in ASD is very similar to, but on the higher end of, the range of heritability for other common neuropsychiatric disorders (31,32).
Thus, it is no surprise that genetic discovery in ASD over the last three decades has been very fruitful, defining and refining a new neurobiological understanding of ASD (11,33). These discoveries have been especially fueled by technological advances (33). In retrospect, it is not surprising that most gene discovery occurred after the era of linkage analysis, based on higher resolution methods. These remarkable genetic findings, which have accelerated over the last decade, have shown that the genetic architecture of ASD differs from later onset disorders, such as schizophrenia and major depression. Mendelian and de novo variants contribute less liability in adult-onset conditions, whereas it is predicted that ~15% of ASD cases are caused by major effect mutations (11,32,34–36). Rare de novo CNVs (11,23,35,37,38) and single-nucleotide variants (34,39,40) that disrupt the protein coding components of genes, likely leading to haploinsufficiency (11), each account for ~5% of cases. Slightly more than 50% of liability is estimated to come from heritable, common, polygenic risk (26). The remainder of genetic risk is yet to be defined but is predicted to encompass additional major effect mutations (34,35), rare inherited variation (41), epistasis and additional inherited common genetic risk. Furthermore, as we discuss below, common and rare genetic risk likely act together in an additive fashion in many cases, and as suggested by the recent work of Robinson and colleagues (42).
Hundreds of Risk Loci Implicated by Rare De Novo Variation
Over the last 15 years, consortium efforts led by the Simons Simplex Collection (33,35,37–40,43,44), Autism Sequencing Consortium (45–47) and Autism Genetic Resource Exchange (23,41) have enabled large-scale sequencing studies that have implicated hundreds of genes and copy number variants (CNVs) in ASD based on the occurrence of de novo variants within a particular gene. Such methods can also incorporate predicted tolerance to the loss of function and the prevalence of protein truncating variants in the general population (48). These efforts have been the primary drivers of gene discovery in ASD to date. In the largest and most recent cohort, Satterstrom et al. (36) analyzed whole exome sequencing (WES) data from over 35 000 people (11.9 K of whom have ASD) and identified 102 ASD risk-genes, many of which overlap with previously published studies (34,45). These high-confidence risk genes overlap with small insertions and deletions that have been previously associated with ASD (23,35,38), but the relationship between specific genes and phenotypes implicated in most CNVs, particularly those spanning more than 100 kb and harboring many genes, remains unknown. Attributing the effect of large CNVs to one gene has been complicated by evidence that multiple risk genes likely exist within large CNV loci implicated in ASD (36).
The majority of sequencing efforts have focused on simplex families wherein only one child has been diagnosed with ASD (34,35,37–40,43–46). However, multiplex families, in which multiple individuals have been diagnosed with ASD, exhibit distinct genetic architecture (41,49–52). In 2016, Leppa et al. (50) demonstrated rare inherited variation in multiplex families that is obscured in simplex families. Furthermore, they show that de novo variation contributes less to ASD liability in multiplex families compared with simplex families (41), although it does occur. In addition, there is non-transmission of rare de novo or inherited large effect mutations in multiplex families, highlighting the complexity of the genetic architecture of ASD. Rare inherited risk variants, identified in multiplex families, significantly overlap with de novo hits within protein–protein interaction (PPI) networks; though, they highlight distinct biological elements, such as the microtubule cytoskeleton and cell-cycle regulation (41). Given the identification of similar pathways in recent analysis of simplex families (36), it is possible that current differences in mutational impact on biological pathways reflect sample sizes rather than true biological differences.
Initial pathways from RDNV analysis implicated high-confidence ASD risk genes in convergent biological pathways relating to transcription, chromatin structure and synaptic function (Fig. 1A and B; (35,45,52–55)). As sequenced cohorts have grown, this list has expanded to include genes implicated in axonal outgrowth and cytoskeletal and vesicle machinery, likely also related to synaptic signaling (36,41,55). Many of the earliest ASD risk genes to be identified were known to explicitly function at the synapse (such as NRXN1, GABRB3, SHANK3 and NLGN3) and/or cause structural and functional synaptic phenotypes in mouse models and human tissue. Synaptic deficits are also consistently observed in mouse and iPSC models of syndromic and RDNV models of ASD (11). These observations contributed to the now widespread idea that an imbalance in excitatory and inhibitory synaptic transmission in the brain is an important contribution to ASD etiology (56). A distinct subset of ASD risk genes are chromatin-modifiers (ex. CHD8, SETD5) and RNA-binding proteins (ex. FMR1, RBFOX), many of which may be upstream regulators of other known ASD risk genes, including those involved in synaptic function (45,57–60).
Figure 1 .
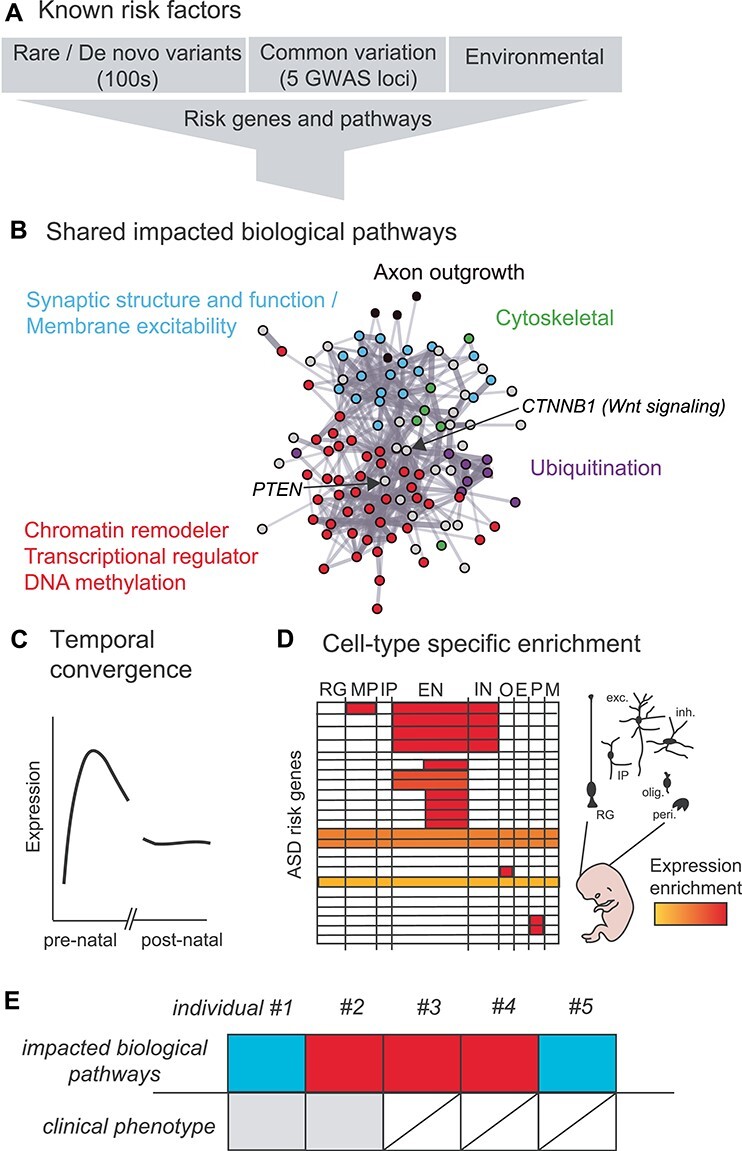
ASD risk factors converge onto shared biological pathways and cell-types during early brain development. (A) Rare inherited, de novo and common inherited variants contribute to ASD liability, in addition to environmental risk factors. (B) Genetic (and likely environmental) factors converge onto biological pathways as schematized in this PPI network made using the STRING database (62) derived from risk genes in (36,41). Genes implicated in synaptic transmission (blue), transcriptional and epigenetic regulation (red), protein ubiquitination (purple), axon outgrowth (black) and the cellular cytoskeleton (green) are highlighted. Risk genes that do not fall into those categories are shown in gray and include those related to Wnt signaling, such as CTNNB1, and the cell cycle, such as PTEN. (C) The expression of risk genes peaks during prenatal development but is not exclusive to this period. Adapted from (63). (D) Expression of risk genes is concentrated in developing glutamatergic neurons. RG—radial glia; MP—migrating progenitor; IP—intermediate progenitor; EN—excitatory neuron; IN—inhibitory neuron; O—oligodendrocyte precursor cell; E—endothelial cell; P—pericyte; M—microglia. Adapted from (64). (E) Impacted biological pathways and phenotypes will vary across individuals and in their convergence and divergence on clinical manifestation.
Down-regulation of gene co-expression programs implicated in neuronal activity and synaptic transmission has been reported in human post-mortem tissue and is enriched in ASD Genome-wide association study (GWAS) signals (61). Up-regulation of genes expressed in astrocytes and microglia has also been consistently observed in ASD post-mortem brain but is not associated with genetic risk and may be a compensatory or environmentally induced signature (61,65). The first single-cell study of gene expression in ASD tissue reinforced many of these observations, identifying differentially expressed synaptic genes in cortical neurons and interneurons, as well as altered gene expression profiles in astrocytes and microglia (66).
Common Inherited Variation
Although WES studies have been the primary drivers of gene discovery in ASD, known highly penetrant RDNVs account for <10% of disease liability (11,35), whereas common variation accounts for >50% (11,26,34,45,67,68). The importance of inherited variation in ASD is supported by the high recurrence of ASD (and autistic traits) in families (17), and observations that autistic behaviors exist on a continuum in the general population (17,69,69). Early ASD GWAS provided suggestive evidence linking a handful of SNPs to ASD, but no SNPs were reproducibly identified (71–76). This paucity of GWAS hits led to the suggestion that thousands of common variants, each with modest effect size, may modulate risk (71,73,77). In the most recent ASD GWAS, 5 genomic loci did reach genome-wide significance (63). As most GWAS signals occur in the non-coding genome (78), ‘hits’ must be linked to genes by functional annotation. Data from 3D chromatin conformation experiments in the developing brain (79) reveal that ASD GWAS hits are enriched in putative regulatory elements active in the prenatal cortex, consistent with prenatal cortical development being a window for particular ASD vulnerability (63). In addition, Walker et al. (80) showed that common inherited and de novo variation coalesce into a single early gene co-expression module, consistent with convergent biological effects (Fig. 1B and C). This module, which was functionally implicated in RNA splicing and chromatin organization, was also specifically enriched in upper layer developing neurons, as had been suggested by previous work (59). Analysis of both rare de novo and common variation has implicated the gene KMT2E in ASD; though, common risk loci identified by GWAS do not yet show significant overlap with genes implicated in rare and de novo variation (36). Similarly, ASD-associated common variants show limited overlap with RDNVs and transcriptomic signatures observed in ASD brain tissue (63). Polygenic risk has been proposed to module expressivity in rare genetic disorders (81). Common risk variants are also over-transmitted in individuals with RDNV, strongly suggesting additive interactions between rare and common risk variants (42). Altogether, these data suggest that there is very likely overlap in risk genes and biological pathways impacted by rare de novo, rare inherited and common variation, but limitations in sample size have impaired detection (Fig. 1B and C). Indeed, such functional convergence has been demonstrated for rare inherited and rare de novo variation in PPI networks (41) and in a specific fetal co-expression module that is enriched for superficial layer glutamatergic neurons (80).
Polygenic risk scores (PRSs), calculated using GWAS summary statistics, summarize the contributions of (typically) thousands of SNPs to the likelihood of developing a trait. LD score regression (82) can further stratify common variants into specific cell-types and tissues to incorporate biological context (83). Tremendous effort has been made to relate ASD PRS to trait variation within ASD and in the general population. ASD PRS has been consistently and positively associated with cognitive traits in the general population (63,72,84,85). Similarly, the ASD PRS has also been associated with anatomical features such as cortical thickness (86). Attempts to correlate the ASD PRS with specific cognitive impairments have been less successful (80,87–90). Broadly, though, there is significant shared common variation that increases risk for ASD, ADHD, depression (63) and schizophrenia (67,72). What is clear is that risk for ASD is related to variants that modulate social cognition in the general population (91).
Non-genetic Risk Factors
A significant and understudied amount of ASD liability is attributed to non-genetic, elements (11,26,27). Many environmental risk factors have been proposed including paternal age (39), maternal immune conditions (92), fever during pregnancy (93), prenatal exposure to air pollution (meta-analysis in (94)) and pesticides (95). Ingestion of some pharmaceutical agents during pregnancy, such as the anti-convulsant valproate, has also been associated with increased risk (96). The strength of evidence for such risk factors varies due to (relatively) small cohorts, difficulty parameterizing environmental exposures and magnitude of effects. Longitudinal cohorts such as Childhood Autism Risk from Genetics and Environment and Markers of Autism Risk in Babies—Learning Early Signs have and will continue to identify environmental risk factors. Beginning to articulate mechanisms by which pre- and peri-natal environmental exposures influence ASD risk will be a challenge for the future. Both genetic and environmental risk factors converge on the prenatal period and likely the cellular epigenome (97). As genetic analyses implicate specific cell-types, circuits and pathways in ASD, one can study the environmental impacts on these processes and their potential interactions with genetic risk factors (98,99). Understanding G × E interactions is currently limited by power, but provides a potentially important future avenue for risk mitigation.
Functional Annotation of Known Genetic Risk Factors Implicates Specific Epochs and Pathways Harboring Biological Risk
Databases that catalog functional genomic data, including gene expression across tissues and time, have become increasingly available over the last few years. By mapping known ASD risk genes onto these data, we have learned a lot about when and where neuropsychiatric risk genes are expressed (100). For example, in Parikshak et al. (59), the authors identified groups of co-expressed genes (modules), which generally capture cell-types and cell-states, present in bulk human neural tissue from mid-fetal development to late adulthood using the BrainSpan database. They identify two types of gene networks enriched for genetic ASD risk; one, relating to transcriptional regulation and expressed in mid-fetal development and the other expressed later in development (around birth) and functionally related to synaptic activity. These ASD-associated gene modules were highly expressed in developing excitatory neurons in a laminar specific-manner (59). Willsey et al. (101) took a more directed approach to identify modules in the BrainSpan dataset that correlated with high-confidence ASD risk genes and again spotlighted mid-fetal cortical excitatory neurons. Alternate bioinformatic approaches have continued to implicate developing cortical glutamatergic neurons (41,102), as well as cortical interneurons and striatal medium spiny neurons (103,104). Single-cell RNAseq experiments have since demonstrated enriched expression of RDNV ASD genes maturing excitatory neurons in the mid-fetal brain (Fig. 1D; (64)), as well as in early striatal and cortical interneurons (36,105). Expression of RDNV risk genes is rarely restricted to developing neurons, however, and recent work has raised the possibility that non-neuronal cells, such as pericytes and oligodendrocytes (as was suggested by some bulk analysis (104)), may also mediate risk (64). As WES cohorts continue to increase in size, additional rare inherited and de novo variants will inevitably be identified (36). And, while there is consensus in the field that developing neurons in the mid-fetal cortex are the major area of convergence for genetic risk for ASD, their association is by no means unique. In addition, there will almost certainly be rare variation that contributes to ASD risk in the non-coding genome. Whole genome sequencing (WGS) efforts to identify rare and de novo non-coding variation are ongoing, but must contend with the difficulty of functionally annotating the non-coding genome and statistical power (106,107).
Insights from ASD and Future Directions
Investigation of the genetic etiology of ASD has spurred the rejection of early psychodynamic hypotheses including domineering mothers, in favor of true, causal factors. ASD genetic risk factors are complex and comprised of both rare and common variation, which likely interact with each other and environmental risk factors. The strong genetic component underlying ASD (and other neuropsychiatric conditions) raises the possibility of genetics informing nosology. There is already preliminary evidence that the ASD PRS overlaps with distinct subtypes of ASD (63) and that different classes of RDNV genes may be associated with ASD with and without intellectual disability (36). Understanding distinct genetic architectures present in different forms of autisms may facilitate the development of personalized therapies. Use of quantitative traits, rather than categorical diagnoses, for example, different metrics of social responsiveness/social cognition, has shown promise (17,91,108) and suggests that ASD in part represents a continuum with the normal distribution of social competency in the general population. Use of quantitative language metrics shows a similar relationship between language in ASD and the general population as well (109,110). Dissecting the components of polygenic risk into its differential effects with respect to specific components of human cognition and behavior, or biological pathways, represents an important frontier in this respect.
At this stage in the field, genetic (and, likely, non-genetic) ASD risk factors largely converge onto neurogenesis, transcriptional/epigenetic regulation and synaptogenesis in the developing cortex. Broad genetic psychiatric disease risk (67,111), and ASD genetic risk specifically, are strongly rooted during the period of neurogenesis and migration during early cortical development (59,63,85,101), primarily, in glutamatergic neurons (59,101,64); though, developing interneurons (36,103,104) and non-neuronal cells (64) have also been implicated by some risk genes. In addition to this convergence at the genetic level, there is strong evidence for convergence in genomic data from post-mortem ASD brain (61,65,112), motivating systems biology approaches. These data also emphasize the value of proper functional annotation of cell-type and stage-specific gene expression.
Moving forward, genetics will also help resolve the mysteries of ASD risk in the individual. How do the results supporting pathway level convergence which are derived from the population, map onto individual risk (Fig. 1E): Are there specific combinations of pathway/gene involvement that typically occur in the individual with ASD? Do different combinations of risk pathways manifest as different forms of ASD, which likely require different treatment? This is almost certainly the case with syndromic or rare variation. Furthermore, we must move from association to understanding causal mechanisms, which in addition to GWAS’ and WGS (DNA variation) requires bench experimentation and integration with multi-layered genomic data, transcriptomes, epigenomes and 3D chromatin structure to connect regulatory variation with genes. Recent work in ASD mouse and iPSC-derived models has shown great promise in contextualizing the functional impact of ASD-associated genetic syndromes and RDNVs in specific cell-types and circuits (11, 113–115). Tremendous effort has gone into developing 3D spheroid models that recapitulate key aspects of brain development and express known ASD risk genes (115–118), offering unprecedented experimental access to a model of pre- and peri-natal neurobiology. Highly parallel, high-throughput efforts using these systems will be important to move our understanding of disease mechanisms forward.
Contributor Information
Katherine W Eyring, Neurogenetics Program, Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Daniel H Geschwind, Neurogenetics Program, Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center For Autism Research and Treatment, Semel Institute, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Human Genetics and Institute for Precision Health, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA.
References
- 1. Zablotsky, B., Black, L.I., Maenner, M.J., Schieve, L.A., Danielson, M.L., Bitsko, R.H., Blumberg, S.J., Kogan, M.D. and Boyle, C.A. (2019) Prevalence and trends of developmental disabilities among children in the United States: 2009-2017. Pediatrics, 2019;144(4):e20190811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. American Psychiatric Association (2013) American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 5th edn. American Psychiatric Publishing, Arlington, VA. [Google Scholar]
- 3. Geschwind, D.H. (2009) Advances in autism. Annu. Rev. Med., 60, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pan, P.Y., Bolte, S., Kaur, P., Jamil, S. and Jonsson, U. (2021) Neurological disorders in autism: a systematic review and meta-analysis. Autism, 25, 812–830. [DOI] [PubMed] [Google Scholar]
- 5. Spence, S.J. and Schneider, M.T. (2009) The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatr. Res., 65, 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chaidez, V., Hansen, R.L. and Hertz-Picciotto, I. (2014) Gastrointestinal problems in children with autism, developmental delays or typical development. J. Autism Dev. Disord., 44, 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richdale, A.L. (1999) Sleep problems in autism: prevalence, cause, and intervention. Dev. Med. Child Neurol., 41, 60–66. [DOI] [PubMed] [Google Scholar]
- 8. Coleman, M. and Gillberg, C. (2012) The Autisms. Oxford University Press, New York. [Google Scholar]
- 9. Dawson, G. and Burner, K. (2011) Behavioral interventions in children and adolescents with autism spectrum disorder: a review of recent findings. Curr. Opin. Pediatr., 23, 616–620. [DOI] [PubMed] [Google Scholar]
- 10. Sigafoos, J. and Waddington, H. (2016) 6 year follow-up supports early autism intervention. Lancet, 388, 2454–2455. [DOI] [PubMed] [Google Scholar]
- 11. de la Torre-Ubieta, L., Won, H., Stein, J.L. and Geschwind, D.H. (2016) Advancing the understanding of autism disease mechanisms through genetics. Nat. Med., 22, 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Centers for Disease Control and Prevention . (2019) 'Treatment and Intervention Services for Autism Spectrum Disorder'. https://www.cdc.gov/ncbddd/autism/treatment.html#ref.
- 13. Amaral, D.G., Schumann, C.M. and Nordahl, C.W. (2008) Neuroanatomy of autism. Trends Neurosci., 31, 137–145. [DOI] [PubMed] [Google Scholar]
- 14. Fombonne, E., Roge, B., Claverie, J., Courty, S. and Fremolle, J. (1999) Microcephaly and macrocephaly in autism. J. Autism Dev. Disord., 29, 113–119. [DOI] [PubMed] [Google Scholar]
- 15. Folstein, S. and Rutter, M. (1977) Infantile autism: a genetic study of 21 twin pairs. J. Child Psychol. Psychiatry, 18, 297–321. [DOI] [PubMed] [Google Scholar]
- 16. Tick, B., Bolton, P., Happe, F., Rutter, M. and Rijsdijk, F. (2016) Heritability of autism spectrum disorders: a meta-analysis of twin studies. J. Child Psychol. Psychiatry, 57, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Constantino, J.N., Zhang, Y., Frazier, T., Abbacchi, A.M. and Law, P. (2010) Sibling recurrence and the genetic epidemiology of autism. Am. J. Psychiatry, 167, 1349–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ruzich, E., Allison, C., Smith, P., Watson, P., Auyeung, B., Ring, H. and Baron-Cohen, S. (2016) Subgrouping siblings of people with autism: identifying the broader autism phenotype. Autism Res., 9, 658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wheelwright, S., Auyeung, B., Allison, C. and Baron-Cohen, S. (2010) Defining the broader, medium and narrow autism phenotype among parents using the autism Spectrum quotient (AQ). Mol. Autism., 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amir, R.E., Van den Veyver, I.B., Wan, M., Tran, C.Q., Francke, U. and Zoghbi, H.Y. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet., 23, 185–188. [DOI] [PubMed] [Google Scholar]
- 21. Blomquist, H.K., Bohman, M., Edvinsson, S.O., Gillberg, C., Gustavson, K.H., Holmgren, G. and Wahlstrom, J. (1985) Frequency of the fragile X syndrome in infantile autism. A Swedish multicenter study. Clin. Genet., 27, 113–117. [DOI] [PubMed] [Google Scholar]
- 22. European Chromosome 16 Tuberous Sclerosis Consortium (1993) Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell, 75, 1305–1315. [DOI] [PubMed] [Google Scholar]
- 23. Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T., Yamrom, B., Yoon, S., Krasnitz, A., Kendall, J. et al. (2007) Strong association of de novo copy number mutations with autism. Science, 316, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vorstman, J.A., Staal, W.G., van Daalen, E., van Engeland, H., Hochstenbach, P.F. and Franke, L. (2006) Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol. Psychiatry, 11, 18–28. [DOI] [PubMed] [Google Scholar]
- 25. Bailey, A., Le Couteur, A., Gottesman, I., Bolton, P., Simonoff, E., Yuzda, E. and Rutter, M. (1995) Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med., 25, 63–77. [DOI] [PubMed] [Google Scholar]
- 26. Gaugler, T., Klei, L., Sanders, S.J., Bodea, C.A., Goldberg, A.P., Lee, A.B., Mahajan, M., Manaa, D., Pawitan, Y., Reichert, J. et al. (2014) Most genetic risk for autism resides with common variation. Nat. Genet., 46, 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hallmayer, J., Cleveland, S., Torres, A., Phillips, J., Cohen, B., Torigoe, T., Miller, J., Fedele, A., Collins, J., Smith, K. et al. (2011) Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry, 68, 1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lichtenstein, P., Carlstrom, E., Rastam, M., Gillberg, C. and Anckarsater, H. (2010) The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am. J. Psychiatry, 167, 1357–1363. [DOI] [PubMed] [Google Scholar]
- 29. Lundstrom, S., Chang, Z., Rastam, M., Gillberg, C., Larsson, H., Anckarsater, H. and Lichtenstein, P. (2012) Autism spectrum disorders and autistic like traits: similar etiology in the extreme end and the normal variation. Arch. Gen. Psychiatry, 69, 46–52. [DOI] [PubMed] [Google Scholar]
- 30. Sandin, S., Lichtenstein, P., Kuja-Halkola, R., Larsson, H., Hultman, C.M. and Reichenberg, A. (2014) The familial risk of autism. JAMA, 311, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geschwind, D.H. and Flint, J. (2015) Genetics and genomics of psychiatric disease. Science, 349, 1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sullivan, P.F. and Geschwind, D.H. (2019) Defining the Genetic, genomic, cellular, and diagnostic architectures of psychiatric disorders. Cell, 177, 162–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abrahams, B.S. and Geschwind, D.H. (2008) Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet., 9, 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iossifov, I., O'Roak, B.J., Sanders, S.J., Ronemus, M., Krumm, N., Levy, D., Stessman, H.A., Witherspoon, K.T., Vives, L., Patterson, K.E. et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanders, S.J., Xin, H., Willsey, A.J., Ercan-Sencicek, A.G., Samocha, K.E., Cicek, A.E., Murtha, M.T., Bal, V.H., Bishop, S.L., Shan, D. et al. (2015) Insights into autism Spectrum disorder genomic architecture and biology from 71 risk loci. Neuron, 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Satterstrom, F.K., Kosmicki, J.A., Wang, J., Breen, M.S., De Rubeis, S., An, J.Y., Peng, M., Collins, R., Grove, J., Klei, L. et al. (2020) Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell, 180, 568. e23–568. e584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levy, D., Ronemus, M., Yamrom, B., Lee, Y.H., Leotta, A., Kendall, J., Marks, S., Lakshmi, B., Pai, D., Ye, K. et al. (2011) Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron, 70, 886–897. [DOI] [PubMed] [Google Scholar]
- 38. Sanders, S.J., Ercan-Sencicek, A.G., Hus, V., Luo, R., Murtha, M.T., Moreno-De-Luca, D., Chu, S.H., Moreau, M.P., Gupta, A.R., Thomson, S.A. et al. (2011) Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron, 70, 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. O'Roak, B.J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B.P., Levy, R., Ko, A., Lee, C., Smith, J.D. et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sanders, S.J., Murtha, M.T., Gupta, A.R., Murdoch, J.D., Raubeson, M.J., Willsey, A.J., Ercan-Sencicek, A.G., DiLullo, N.M., Parikshak, N.N., Stein, J.L. et al. (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature, 485, 237–U124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ruzzo, E.K., Perez-Cano, L., Jung, J.Y., Wang, L.K., Kashef-Haghighi, D., Hartl, C., Singh, C., Xu, J., Hoekstra, J.N., Leventhal, O. et al. (2019) Inherited and de novo genetic risk for autism impacts shared networks. Cell, 178, 850, e26–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weiner, D.J., Wigdor, E.M., Ripke, S., Walters, R.K., Kosmicki, J.A., Grove, J., Samocha, K.E., Goldstein, J.I., Okbay, A., Bybjerg-Grauholm, J. et al. (2017) Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet., 49, 978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dong, S., Walker, M.F., Carriero, N.J., DiCola, M., Willsey, A.J., Ye, A.Y., Waqar, Z., Gonzalez, L.E., Overton, J.D., Frahm, S. et al. (2014) De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep., 9, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., Yamrom, B., Lee, Y.H., Narzisi, G., Leotta, A. et al. (2012) De novo gene disruptions in children on the autistic spectrum. Neuron, 74, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Rubeis, S., He, X., Goldberg, A.P., Poultney, C.S., Samocha, K., Cicek, A.E., Kou, Y., Liu, L., Fromer, M., Walker, S. et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu, L., Sabo, A., Neale, B.M., Nagaswamy, U., Stevens, C., Lim, E., Bodea, C.A., Muzny, D., Reid, J.G., Banks, E. et al. (2013) Analysis of rare, exonic variation amongst subjects with autism spectrum disorders and population controls. PLoS Genet., 9, e1003443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Neale, B.M., Kou, Y., Liu, L., Ma'ayan, A., Samocha, K.E., Sabo, A., Lin, C.F., Stevens, C., Wang, L.S., Makarov, V. et al. (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature, 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kosmicki, J.A., Samocha, K.E., Howrigan, D.P., Sanders, S.J., Slowikowski, K., Lek, M., Karczewski, K.J., Cutler, D.J., Devlin, B., Roeder, K. et al. (2017) Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet., 49, 504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bernier, R., Gerdts, J., Munson, J., Dawson, G. and Estes, A. (2012) Evidence for broader autism phenotype characteristics in parents from multiple-incidence autism families. Autism Res., 5, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leppa, V.M., Kravitz, S.N., Martin, C.L., Andrieux, J., Le Caignec, C., Martin-Coignard, D., DyBuncio, C., Sanders, S.J., Lowe, J.K., Cantor, R.M. and Geschwind, D.H. (2016) Rare inherited and de novo CNVs reveal complex contributions to ASD risk in multiplex families. Am. J. Hum. Genet., 99, 540–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Szatmari, P., MacLean, J.E., Jones, M.B., Bryson, S.E., Zwaigenbaum, L., Bartolucci, G., Mahoney, W.J. and Tuff, L. (2000) The familial aggregation of the lesser variant in biological and nonbiological relatives of PDD probands: a family history study. J. Child Psychol. Psychiatry, 41, 579–586. [DOI] [PubMed] [Google Scholar]
- 52. Virkud, Y.V., Todd, R.D., Abbacchi, A.M., Zhang, Y. and Constantino, J.N. (2009) Familial aggregation of quantitative autistic traits in multiplex versus simplex autism. Am. J. Med. Genet. B Neuropsychiatr. Genet., 150B, 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gilman, S.R., Iossifov, I., Levy, D., Ronemus, M., Wigler, M. and Vitkup, D. (2011) Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron, 70, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Glessner, J.T., Wang, K., Cai, G., Korvatska, O., Kim, C.E., Wood, S., Zhang, H., Estes, A., Brune, C.W., Bradfield, J.P. et al. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature, 459, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pinto, D., Delaby, E., Merico, D., Barbosa, M., Merikangas, A., Klei, L., Thiruvahindrapuram, B., Xu, X., Ziman, R., Wang, Z. et al. (2014) Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet., 94, 677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rubenstein, J.L. and Merzenich, M.M. (2003) Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav., 2, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cotney, J., Muhle, R.A., Sanders, S.J., Liu, L., Willsey, A.J., Niu, W., Liu, W., Klei, L., Lei, J., Yin, J. et al. (2015) The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun., 6, 6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Darnell, J.C., Van Driesche, S.J., Zhang, C., Hung, K.Y., Mele, A., Fraser, C.E., Stone, E.F., Chen, C., Fak, J.J., Chi, S.W. et al. (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell, 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Parikshak, N.N., Luo, R., Zhang, A., Won, H., Lowe, J.K., Chandran, V., Horvath, S. and Geschwind, D.H. (2013) Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell, 155, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sugathan, A., Biagioli, M., Golzio, C., Erdin, S., Blumenthal, I., Manavalan, P., Ragavendran, A., Brand, H., Lucente, D., Miles, J. et al. (2014) CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. U. S. A., 111, E4468–E4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Voineagu, I., Wang, X., Johnston, P., Lowe, J.K., Tian, Y., Horvath, S., Mill, J., Cantor, R.M., Blencowe, B.J. and Geschwind, D.H. (2011) Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature, 474, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., Jensen L.J., von Mering C. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res., 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Grove, J., Ripke, S., Als, T.D., Mattheisen, M., Walters, R.K., Won, H., Pallesen, J., Agerbo, E., Andreassen, O.A., Anney, R. et al. (2019) Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet., 51, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Polioudakis, D., de la Torre-Ubieta, L., Langerman, J., Elkins, A.G., Shi, X., Stein, J.L., Vuong, C.K., Nichterwitz, S., Gevorgian, M., Opland, C.K. et al. (2019) A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron, 103, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Parikshak, N.N., Swarup, V., Belgard, T.G., Irimia, M., Ramaswami, G., Gandal, M.J., Hartl, C., Leppa, V., Ubieta, L.T., Huang, J. et al. (2016) Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature, 540, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Velmeshev, D., Schirmer, L., Jung, D., Haeussler, M., Perez, Y., Mayer, S., Bhaduri, A., Goyal, N., Rowitch, D.H. and Kriegstein, A.R. (2019) Single-cell genomics identifies cell type-specific molecular changes in autism. Science, 364, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brainstorm, C., Anttila, V., Bulik-Sullivan, B., Finucane, H.K., Walters, R.K., Bras, J., Duncan, L., Escott-Price, V., Falcone, G.J., Gormley, P. et al. (2018) Analysis of shared heritability in common disorders of the brain. Science, 360, eaap8757. doi: 10.1126/science.aap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Krumm, N., Turner, T.N., Baker, C., Vives, L., Mohajeri, K., Witherspoon, K., Raja, A., Coe, B.P., Stessman, H.A., He, Z.X. et al. (2015) Excess of rare, inherited truncating mutations in autism. Nat. Genet., 47, 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Constantino, J.N. and Todd, R.D. (2003) Autistic traits in the general population: a twin study. Arch. Gen. Psychiatry, 60, 524–530. [DOI] [PubMed] [Google Scholar]
- 70. Constantino, J.N. and Todd, R.D. (2005) Intergenerational transmission of subthreshold autistic traits in the general population. Biol. Psychiatry, 57, 655–660. [DOI] [PubMed] [Google Scholar]
- 71. Anney, R., Klei, L., Pinto, D., Almeida, J., Bacchelli, E., Baird, G., Bolshakova, N., Bolte, S., Bolton, P.F., Bourgeron, T. et al. (2012) Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum. Mol. Genet., 21, 4781–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium (2017) Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism., 8, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Devlin, B., Melhem, N. and Roeder, K. (2011) Do common variants play a role in risk for autism? Evidence and theoretical musings. Brain Res., 1380, 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ma, D., Salyakina, D., Jaworski, J.M., Konidari, I., Whitehead, P.L., Andersen, A.N., Hoffman, J.D., Slifer, S.H., Hedges, D.J., Cukier, H.N. et al. (2009) A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann. Hum. Genet., 73, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J.T., Abrahams, B.S., Salyakina, D., Imielinski, M., Bradfield, J.P., Sleiman, P.M. et al. (2009) Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature, 459, 528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weiss, L.A., Arking, D.E., Hopkins Gene Discovery Project of Johns and the Autism Consortium, Daly, M.J. and Chakravarti, A. (2009) A genome-wide linkage and association scan reveals novel loci for autism. Nature, 461, 802–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Klei, L., Sanders, S.J., Murtha, M.T., Hus, V., Lowe, J.K., Willsey, A.J., Moreno-De-Luca, D., Yu, T.W., Fombonne, E., Geschwind, D. et al. (2012) Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism., 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Maurano, M.T., Humbert, R., Rynes, E., Thurman, R.E., Haugen, E., Wang, H., Reynolds, A.P., Sandstrom, R., Qu, H., Brody, J. et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science, 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Won, H., de la Torre-Ubieta, L., Stein, J.L., Parikshak, N.N., Huang, J., Opland, C.K., Gandal, M.J., Sutton, G.J., Hormozdiari, F., Lu, D. et al. (2016) Chromosome conformation elucidates regulatory relationships in developing human brain. Nature, 538, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Walker, R.L., Ramaswami, G., Hartl, C., Mancuso, N., Gandal, M.J., de la Torre-Ubieta, L., Pasaniuc, B., Stein, J.L. and Geschwind, D.H. (2019) Genetic control of expression and splicing in developing human brain informs disease mechanisms. Cell, 179, 750.e22–750.e771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Oetjens, M.T., Kelly, M.A., Sturm, A.C., Martin, C.L. and Ledbetter, D.H. (2019) Quantifying the polygenic contribution to variable expressivity in eleven rare genetic disorders. Nat. Commun., 10, 4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bulik-Sullivan, B.K., Loh, P.R., Finucane, H.K., Ripke, S., Yang, J., Consortium Schizophrenia Working Group of the Psychiatric Genomics, Patterson, N., Daly, M.J., Price, A.L. and Neale, B.M. (2015) LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet., 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Finucane, H.K., Reshef, Y.A., Anttila, V., Slowikowski, K., Gusev, A., Byrnes, A., Gazal, S., Loh, P.R., Lareau, C., Shoresh, N. et al. (2018) Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet., 50, 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Clarke, T.K., Lupton, M.K., Fernandez-Pujals, A.M., Starr, J., Davies, G., Cox, S., Pattie, A., Liewald, D.C., Hall, L.S., MacIntyre, D.J. et al. (2016) Common polygenic risk for autism spectrum disorder (ASD) is associated with cognitive ability in the general population. Mol. Psychiatry, 21, 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schork, A.J., Won, H., Appadurai, V., Nudel, R., Gandal, M., Delaneau, O., Revsbech Christiansen, M., Hougaard, D.M., Baekved-Hansen, M., Bybjerg-Grauholm, J. et al. (2019) A genome-wide association study of shared risk across psychiatric disorders implicates gene regulation during fetal neurodevelopment. Nat. Neurosci., 22, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Khundrakpam, B., Vainik, U., Gong, J., Al-Sharif, N., Bhutani, N., Kiar, G., Zeighami, Y., Kirschner, M., Luo, C., Dagher, A. and Evans, A. (2020) Neural correlates of polygenic risk score for autism spectrum disorders in general population. Brain Commun, 2(2), fcaa092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Aguilar-Lacasaña, S., Vilor-Tejedor, N., Jansen, P.R., López-Vicente, M., Bustamante, M., Burgaleta, M., Sunyer, J. and Alemany, S. (2020) Polygenic risk for ADHD and ASD and their relation with cognitive measures in school children. Psychol. Med., 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chang, S., Yang, L., Wang, Y. and Faraone, S.V. (2020) Shared polygenic risk for ADHD, executive dysfunction and other psychiatric disorders. Transl. Psychiatry, 10, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nudel, R., Christiani, C.A.J., Ohland, J., Uddin, M.J., Hemager, N., Ellersgaard, D.V., Spang, K.S., Burton, B.K., Greve, A.N., Gantriis, D.L. et al. (2020) Language deficits in specific language impairment, attention deficit/hyperactivity disorder, and autism spectrum disorder: an analysis of polygenic risk. Autism Res., 13, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Reed, Z.E., Mahedy, L., Jackson, A., Smith, G.D., Penton-Voak, I., Attwood, A.S. and Munafo, M.R. (2021) Examining the bidirectional association between emotion recognition and social autistic traits using observational and genetic analyses. J. Child Psychol. Psychiatry. doi: 10.1111/jcpp.13395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Robinson, E.B., Pourcain, B.S., Anttila, V., Kosmicki, J.A., Bulik-Sullivan, B., Grove, J., Maller, J., Samocha, K.E., Sanders, S.J., Ripke, S. et al. (2016) Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat. Genet., 48, 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Estes, M.L. and McAllister, A.K. (2015) Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci., 16, 469–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zerbo, O., Iosif, A.M., Walker, C., Ozonoff, S., Hansen, R.L. and Hertz-Picciotto, I. (2013) Is maternal influenza or fever during pregnancy associated with autism or developmental delays? Results from the CHARGE (CHildhood autism risks from genetics and environment) study. J. Autism Dev. Disord., 43, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lam, J., Sutton, P., Kalkbrenner, A., Windham, G., Halladay, A., Koustas, E., Lawler, C., Davidson, L., Daniels, N., Newschaffer, C. and Woodruff, T. (2016) A systematic review and meta-analysis of multiple airborne pollutants and autism spectrum disorder. PLoS One, 11, e0161851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Roberts, E.M., English, P.B., Grether, J.K., Windham, G.C., Somberg, L. and Wolff, C. (2007) Maternal residence near agricultural pesticide applications and autism spectrum disorders among children in the California Central Valley. Environ. Health Perspect., 115, 1482–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Christensen, J., Gronborg, T.K., Sorensen, M.J., Schendel, D., Parner, E.T., Pedersen, L.H. and Vestergaard, M. (2013) Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA, 309, 1696–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. LaSalle, J.M. (2013) Epigenomic strategies at the interface of genetic and environmental risk factors for autism. J. Hum. Genet., 58, 396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dunaway, K.W., Islam, M.S., Coulson, R.L., Lopez, S.J., Vogel Ciernia, A., Chu, R.G., Yasui, D.H., Pessah, I.N., Lott, P., Mordaunt, C. et al. (2016) Cumulative impact of polychlorinated biphenyl and large chromosomal duplications on DNA methylation, chromatin, and expression of autism candidate genes. Cell Rep., 17, 3035–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Volk, H.E., Kerin, T., Lurmann, F., Hertz-Picciotto, I., McConnell, R. and Campbell, D.B. (2014) Autism spectrum disorder: interaction of air pollution with the MET receptor tyrosine kinase gene. Epidemiology, 25, 44–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hernandez, L.M., Kim, M., Hoftman, G.D., Haney, J.R., de la Torre-Ubieta, L., Pasaniuc, B. and Gandal, M.J. (2021) Transcriptomic insight into the polygenic mechanisms underlying psychiatric disorders. Biol. Psychiatry, 89, 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Willsey, A.J., Sanders, S.J., Li, M., Dong, S., Tebbenkamp, A.T., Muhle, R.A., Reilly, S.K., Lin, L., Fertuzinhos, S., Miller, J.A. et al. (2013) Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell, 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hartl, C.L., Ramaswami, G., Pembroke, W.G., Muller, S., Pintacuda, G., Saha, A., Parsana, P., Battle, A., Lage, K. and Geschwind, D.H. (2020) The architecture of brain co-expression reveals the brain-wide basis of disease susceptibility. bioRxiv. 10.1101/2020.03.05.965749 biorxiv;2020.03.05.965749v1, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chang, J., Gilman, S.R., Chiang, A.H., Sanders, S.J. and Vitkup, D. (2015) Genotype to phenotype relationships in autism spectrum disorders. Nat. Neurosci., 18, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Xu, X., Wells, A.B., O'Brien, D.R., Nehorai, A. and Dougherty, J.D. (2014) Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J. Neurosci., 34, 1420–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nowakowski, T.J., Bhaduri, A., Pollen, A.A., Alvarado, B., Mostajo-Radji, M.A., Di Lullo, E., Haeussler, M., Sandoval-Espinosa, C., Liu, S.J., Velmeshev, D. et al. (2017) Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science, 358, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. An, J.Y., Lin, K., Zhu, L., Werling, D.M., Dong, S., Brand, H., Wang, H.Z., Zhao, X., Schwartz, G.B., Collins, R.L. et al. (2018) Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science, 362, eaat6576. doi: 10.1126/science.aat6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Werling, D.M., Brand, H., An, J.Y., Stone, M.R., Zhu, L., Glessner, J.T., Collins, R.L., Dong, S., Layer, R.M., Markenscoff-Papadimitriou, E. et al. (2018) An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet., 50, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lowe, J.K., Werling, D.M., Constantino, J.N., Cantor, R.M. and Geschwind, D.H. (2015) Social responsiveness, an autism endophenotype: genomewide significant linkage to two regions on chromosome 8. Am. J. Psychiatry, 172, 266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Alarcon, M., Abrahams, B.S., Stone, J.L., Duvall, J.A., Perederiy, J.V., Bomar, J.M., Sebat, J., Wigler, M., Martin, C.L., Ledbetter, D.H. et al. (2008) Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet., 82, 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Vernes, S.C., Newbury, D.F., Abrahams, B.S., Winchester, L., Nicod, J., Groszer, M., Alarcon, M., Oliver, P.L., Davies, K.E., Geschwind, D.H., Monaco, A.P. and Fisher, S.E. (2008) A functional genetic link between distinct developmental language disorders. N. Engl. J. Med., 359, 2337–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lee, S.H., Ripke, S., Neale, B.M., Faraone, S.V., Purcell, S.M., Perlis, R.H., Mowry, B.J., Thapar, A., Goddard, M.E., Witte, J.S. et al. (2013) Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet., 45, 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gandal, M.J., Haney, J.R., Parikshak, N.N., Leppa, V., Ramaswami, G., Hartl, C., Schork, A.J., Appadurai, V., Buil, A., Werge, T.M. et al. (2018) Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science, 359, 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Cederquist, G.Y., Tchieu, J., Callahan, S.J., Ramnarine, K., Ryan, S., Zhang, C., Rittenhouse, C., Zeltner, N., Chung, S.Y., Zhou, T. et al. (2020) A multiplex human pluripotent stem cell platform defines molecular and functional subclasses of autism-related genes. Cell Stem Cell, 27, 35, e6–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Jin, X., Simmons, S.K., Guo, A., Shetty, A.S., Ko, M., Nguyen, L., Jokhi, V., Robinson, E., Oyler, P., Curry, N. et al. (2020) In vivo perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science, 370, eaaz6063. doi: 10.1126/science.aaz60603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Khan, T.A., Revah, O., Gordon, A., Yoon, S.J., Krawisz, A.K., Goold, C., Sun, Y., Kim, C.H., Tian, Y., Li, M.Y. et al. (2020) Neuronal defects in a human cellular model of 22q11.2 deletion syndrome. Nat. Med., 26, 1888–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Gordon, A., Yoon, S.J., Tran, S.S., Makinson, C.D., Park, J.Y., Andersen, J., Valencia, A.M., Horvath, S., Xiao, X., Huguenard, J.R., Pasca, S.P. and Geschwind, D.H. (2021) Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci., 24, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lancaster, M.A., Renner, M., Martin, C.A., Wenzel, D., Bicknell, L.S., Hurles, M.E., Homfray, T., Penninger, J.M., Jackson, A.P. and Knoblich, J.A. (2013) Cerebral organoids model human brain development and microcephaly. Nature, 501, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Pasca, A.M., Sloan, S.A., Clarke, L.E., Tian, Y., Makinson, C.D., Huber, N., Kim, C.H., Park, J.Y., O'Rourke, N.A., Nguyen, K.D. et al. (2015) Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods, 12, 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]