

Abstract
Vorolanib (CM082) is a multi-targeted tyrosine kinase receptor inhibitor with a short half-life and limited tissue accumulation that has been shown to reduce choroidal neovascularization in rats. In this preclinical study, vorolanib demonstrated competitive binding and inhibitory activities with KDR, PDGFRβ, FLT3, and C-Kit, and inhibited RET and AMPKα1 more weakly than sunitinib, indicating more stringent kinase selectivity. Vorolanib inhibited vascular endothelial growth factor (VEGF)-induced proliferation of human umbilical vein endothelial cells (HUVECs) and HUVEC tube formation in vitro. In mouse xenograft models, vorolanib inhibited tumor growth of MV-4-11, A549, 786-O, HT-29, BxPC-3, and A375 cells in a dose-dependent fashion. Complete tumor regression was achieved in the MV-4-11 xenograft model. No significant toxicities were observed in vorolanib groups, whereas a significant negative impact on body weights was observed in the sunitinib group at a dose of 40 mg/kg qd. Overall, vorolanib is a novel multi-kinase receptor inhibitor with potent preclinical anti-angiogenic and anti-tumor activity that is potentially less toxic than other similar kinase inhibitors.
Keywords: angiogenesis, anti-tumor, vorolanib, PDFGR, VEGFR
Graphical abstract
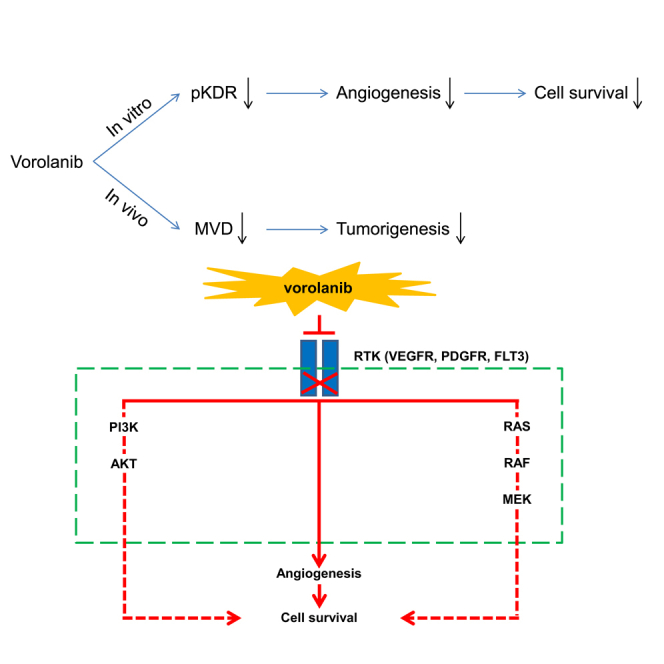
Vorolanib, a novel multi-targeted tyrosine kinase inhibitor (TKI) with anti-tumor and anti-neovascularization effects, binds to the TK receptor of two different kinase receptors to provide more effective tumor inhibition than drugs that bind to a single receptor. It is also potentially less toxic than other multi-targeted TKIs.
Introduction
Receptor tyrosine kinase (RTK) signaling is frequently dysregulated in human cancers. Vascular endothelial growth factor receptor (VEGFR) overexpression is frequently found in several cancer types, such as lung, breast, kidney, and ovarian, and thus has become a prominent therapeutic target for cancer.1,2 The VEGF RTK family are composed of three transmembrane proteins: VEGFR-1 (also called FLT-1), VEGFR-2 (also known as kinase insert domain receptor [KDR] or FLK-1), and VEGFR-3 (also called FLT-4).3
VEGFR tyrosine kinase inhibitors (TKIs) can be classified as selective or non-selective inhibitors based on their in vitro potency. Non-selective TKIs are inhibitors that have multiple targets and can have relatively low (sorafenib), intermediate (sunitinib), or high (cabozantinib and lenvatinib) in vitro potency against VEGFRs; selective TKIs are those have intermediate (pazopanib) or high (axitinib and tivozanib) in vitro inhibitory activity against VEGFRs.4 Lenvatinib can inhibit VEGFR-1/3 and FGFR-1/4 pathways and has been used in combination with everolimus as a second-line strategy in patients with advanced renal cell carcinoma (RCC) after prior anti-VEGF therapy.5 Tivozanib is a derivative structurally related to lenvatinib with improved potency and selectivity for the VEGFR-1, -2, and -3.6 However, these VEGFR TKIs can cause hypertension and can lead to myocardial ischemia, left ventricular systolic dysfunction, and heart failure in patients with other risk factors.7,8 Therefore, an unmet clinical need for an RTK with an improved safety profile that maintains good efficacy is present.
Vorolanib (CM082) is a TKI that targets all VEGFR and platelet-derived growth factor receptor (PDGFR) isoforms.9 It has a shorter half-life and limited tissue accumulation compared with other TKIs.10 Enhanced chemotherapeutic drug efficacy has also been shown by inhibition of the drug efflux function of ABCG2, a potential ATP-binding cassette transporter.11 Several pharmacodynamic and pharmacokinetic studies and/or phase I clinical trials of vorolanib, alone or combined with other targeted therapy or chemotherapeutic agents,12 have been performed in advanced RCC and lung cancer.10,12, 13, 14, 15 A randomized phase 2/3, double-blinded, multi-center trial of vorolanib and everolimus in patients with pretreated metastatic RCC is ongoing.
Our study evaluated the affinity of vorolanib in a variety of RTKs, assessing kinase activity inhibition in vitro. We also investigated the functional significance of inhibiting VEGF-induced proliferation on human umbilical vein endothelial cell (HUVEC) vessel formation, in vitro, and tumor growth in a mouse xenograft model using several established human tumor cell lines.
Results
Vorolanib showed competitive binding with the kinases
The half maximal inhibitory concentration (IC50) of vorolanib for PDGFRβ was the same as that of sunitinib. The IC50 values of vorolanib for KDR, FLT3, and C-Kit were 4.7- to 15.4-fold lower than those of sunitinib. On the contrary, the IC50 values of vorolanib for RET and AMPKα1 were 1.1- to 2.4-fold higher than those of sunitinib (Table 1). More comprehensive vorolanib results against a total of 38 human recombinant kinases are presented in a supplementary table, indicating that vorolanib showed similar inhibitory spectrum and higher selectivity for kinases than sunitinib (Table S1).
Table 1.
IC50 values of competitive binding between vorolanib (CM082) and sunitinib with different kinases
| IC50 (nM) | KDR | PDGFRβ | FLT3 | C-Kit | RET | AMPKα1 |
|---|---|---|---|---|---|---|
| Sunitinib | 17.25 | 0.13 | 2.93 | 1.22 | 177 | 398.9 |
| Vorolanib | 1.12 | 0.13 | 0.63 | 0.14 | 74.1 | 352.2 |
AMPKα1, AMP-activated protein kinase α1; FLT3, FMS-like tyrosine kinase 3; KDR, vascular endothelial growth factor receptor 2 (VEGFR2); PDGFRβ, platelet-derived growth factor receptor β; RET, rearranged during transfection; .
They showed higher affinity with vorolanib compared with other kinases tested in the KINOMEscan in vitro competition kinase binding assays.
Vorolanib inhibited VEGF-induced proliferation and tube formation of HUVECs
Vorolanib demonstrated a dose-dependent VEGF165-induced proliferation of HUVEC cell line and primary HUVECs in vitro with IC50 values of 64.13 and 92.37 nM, respectively (Table 2). Meanwhile, vorolanib had no significant effect on the proliferation of primary HUVECs without VEGF165 induction, and the IC50 value was more than 2,000 nM. At both 50 and 100 nM, vorolanib inhibited the VEGF165-induced tube formation of HUVECs, in vitro, with inhibitory strengths comparable with that of sunitinib (Figure 1). Similar to sunitinib, vorolanib also inhibited the VEGF-induced KDR phosphorylation of HUVECs (Figure S1). These results indicated that the main anti-tumor mechanism of vorolanib may be anti-angiogenesis.
Table 2.
IC50 values of vorolanib (CM082) on human umbilical vein endothelial cell proliferation with or without VEGF165
| IC50 (nM) | Sunitinib | CM082 | Staurosporine |
|---|---|---|---|
| Primary HUVECs | — | >2,000 | 8.12 |
| VEGF165-induced Primary HUVECs | 12.55 | 92.37 | — |
| VEGF165-induced HUVEC cell lines | 39.84 | 64.13 | — |
HUVEC, human umbilical vein endothelial cell; VEGF, vascular endothelial growth factor.
Staurosporine was used as positive control.
Figure 1.

Effects of vorolanib on human umbilical vein endothelial cells (HUVECs)
Effects of vorolanib on vascular endothelial growth factor (VEGF)-induced in vitro tube formation of primary HUVECs. ##p < 0.01 versus control group, ∗∗p < 0.01 versus VEGF group.
In order to investigate whether the inhibitory effects of vorolanib on tube formation were caused by its direct toxicity to tumor cells, the toxicity of vorolanib on tumor cells was tested in vitro. The results showed that vorolanib had almost no significant inhibitory activity on A375, 786-O, HT-29, HCT-116, BXPC-3, and A549 cell lines, which was inferior compared with sunitinib. The kinases, including KDR, PDGFRβ, KIT, and FLT3, were almost not expressed in these cell lines,16 whereas vorolanib showed an inhibitory effect on MV-4-11 cells, with an IC50 of 140 nM, but it is still weaker than sunitinib, with an IC50 of 14.81 nM (Table 3). The MV-4-11 cells were derived from biphenotypic B myelomonocytic leukemia with a FLT3 internal tandem duplication (ITD) mutation.17 As a growth factor receptor, FLT3 can regulate cell growth and proliferation via various signaling pathways, including the Ras/MAPK, PI3K/Akt, and JAK/STAT pathways.18 Therefore, vorolanib may repress the cell growth by selectively inhibiting tyrosine kinases mutated in cancer cells.
Table 3.
IC50 values of vorolanib for different cancer cell lines
| IC50 (nM) | BxPC-3 | HT-29 | HCT-116 | A375 | A549 | 786-O | MV-4-11 |
|---|---|---|---|---|---|---|---|
| Sunitinib | 4,963 | 4,420 | 2,522 | 2,879 | 4,430 | 6,041 | 14.81 |
| CM082 | >9,000 | >9,000 | >9,000 | >9,000 | >9,000 | >9,000 | 140 |
The effects of vorolanib on the proliferation of different cancer cell lines, including MV-4-11, HT-29, HCT-116, BxPC-3, A549, A375, and 786-O cells, were tested using a cell proliferation assay.
Vorolanib inhibited microvessel density and choroidal neovascularization development
To further evaluate the effect of vorolanib on angiogenesis, immunofluorescent staining of CD31 was performed (Figure 2). Many blood vessels were intensely stained by the CD31 antibody in the xenograft models. However, the microvessel density (MVD) of the mouse was significantly inhibited by sunitinib. Similarly, vorolanib suppressed the MVD of the mouse in a dose-dependent manner, and the quantitative analysis showed that the inhibitory activity of high-dose vorolanib (160 mg/kg twice daily [bid]) was comparable with sunitinib.
Figure 2.

Representative images of immunofluorescent labeling of CD31 in the xenograft models
Scale bar, 1 μm.
We next determined whether pharmacologic inhibition of VEGF could prevent choroidal neovascularization (CNV) development in this model by administering vorolanib (10 and 30 mg/kg) 10 to 20 days after Matrigel injection. In the control group, the CNV area on day 20 was 3.7 times larger than that on day 10 (CNV area: 1326.95 ± 583.71 × 103 μm2, n = 6, versus 282.40 ± 99.20 × 103 μm2, n = 7; Figures 3A and 3B), indicating that the development of CNV in every eye was notably observed. However, CNV development was repressed in the eyes of animals dosed with vorolanib 10 mg/kg (CNV area: 493.58 ± 317.17 × 103 μm2, n = 6) and 30 mg/kg (CNV area: 357.70 ± 276.97 × 103 μm2, n = 6; Figures 3C and 3D).
Figure 3.

Vorolanib inhibited choroidal neovascularization (CNV) development
Vorolanib (10 and 30 mg/kg) was given days 10–20 after Matrigel injection. Control animals were untreated. Eyes were harvested 10 days after Matrigel injection. CNV (arrowheads) was detected in every eye of the control group on days 10 (A) and 20 (B), while CNV was significantly inhibited in the eyes of the vorolanib-treated groups (C). (D) Quantitative analysis shows that the CNV area of the treated group was significantly less than that of the control eyes (p < 0.001). R, retina; M, Matrigel; C, choroid. Scale bar, 100 μm. ##p < 0.01 versus control group on day 10, ∗p < 0.05, ∗∗p < 0.01 versus control group on day 20.
Vorolanib inhibited tumor growth in athymic mice in a dose-dependent manner
We further evaluated the in vivo anti-tumor activity of vorolanib in multiple tumor-cell-line-derived xenograft models. Sunitinib was used as a positive control in these experiments and demonstrated an inhibitory profile on these tumors. In the xenograft models, especially in the MV-4-11 model, vorolanib showed similar inhibitory power at a dose of 160 mg/kg (80 mg/kg bid) to that of sunitinib at a dose of 40 mg/kg every day (qd) (Figure 4A). Furthermore, vorolanib exerted stronger inhibition of tumor growth in the MV-4-11 xenograft tumors than in the other xenograft tumors. One possible reason may be that MV-4-11 cells harbor an FLT3-ITD mutation. At 10 mg/kg bid, vorolanib began to show significant inhibition of MV-4-11 xenograft tumor growth. The efficacious dose in the HT-29-cell-derived xenograft models was 40–160 mg/kg bid (Figure 4C). The effect of vorolanib on other xenografts, including HCT-116, BxPC-3, A375, and 786-O, is similar to that on HT-29 tumor xenografts (Figure S2). Orally administered vorolanib was well tolerated by the animals in the study, with no obvious body weight loss or other significant toxicity observed. However, severe body weight loss was observed in the sunitinib group at the dose of 40 mg/kg qd (Figures 4B and 4D).
Figure 4.

Effects of vorolanib on mouse xenograft tumors
(A and B) Oral administration of vorolanib inhibited the growth of established MV-4-11 mouse xenograft tumors. (C and D) Oral administration of vorolanib inhibited the growth of established HT-29 mouse xenograft tumors. ∗p < 0.05, ∗∗p < 0.01 versus vehicle control group.
Discussion
Over the past few decades, scientific research has established that neoangiogenesis is needed to sustain tumor growth.19 Thus, targeting the angiogenic pathway is an effective strategy for anti-cancer therapy. Sunitinib is a multi-targeted kinase inhibitor that selectively targets important molecules in the angiogenic pathway, including VEGFR and PDGFR. It also inhibits two other members of the PDGFR RTK family, KIT and FLT3.20 However, serious side effects resulting from the off-target effects limit the clinical application and patient benefits. The treatment-related toxicities and the more rapid progression and poorer prognoses of some cancers caused by an interruption/discontinuation of sunitinib treatments due to toxicities pointed to the clinical need for a safer and more effective TKI. In clinical studies, dose-limiting toxicities (DLTs), including grade 3 fatigue, grade 3 hypertension, and grade 2 bullous skin toxicity, have been observed. Cardiotoxicities and hypothyroidism have also occurred.20, 21, 22 Pharmacokinetic and pharmacodynamic analyses revealed the accumulation of sunitinib in tissue.23, 24, 25 As a result of these toxicities, sunitinib is given as a 4-week-on and 2-week-off cycle; yet the impact of treatment-related toxicities can still be significant, and living longer does not always equate to living better for some patients. Moreover, sunitinib treatment interruptions or discontinuations can lead to endothelial cell proliferation proportionate to the time off of therapy and a more rapid progression and poorer prognosis of metastatic RCC.26,27
In our study, vorolanib potently inhibited KDR, PDGFRβ, FLT3, and KIT kinase activities with high specificity, similar to what has been seen with sunitinib. Of the three VEGFR family members, KDR appears to exert the major mitogenic, angiogenic, and permeability-enhancing effects of VEGF, which can regulate tumor angiogenesis, cancer stem cell function, and tumor initiation28 Platelet-derived growth factors (PDGFs) and PDGFRs are involved in growth-factor-mediated integrin activation, which is critical for tumor angiogenesis,29,30 and implicated in the pathogenesis of several different human tumors.31 For example, PDGFRα gene mutations were found in 5% of gastrointestinal stromal tumors (GISTs).32 Mutant PDGFRα proteins were shown to be mis-localized to intracellular compartments and to constitutively activate STAT1, -2, and -5.33 KIT gene mutations were more common than PDGFRα gene mutations in GISTs,34 and acute myeloid leukemia (AML) with ITD mutations of the FLT3 gene has been associated with poor outcomes.35 All of these results demonstrated that vorolanib is a powerful multi-kinase inhibitor. These kinases play important roles in solid tumors, such as RCC and lung cancer. For example, excess KDR and PDGFRβ activities, induced by VEGF and PDGF upregulation, have been shown in patients with clear cell RCC. These excess activities were shown to contribute to the highly vascular nature of tumors and were also associated with RCC tumor progression.36 PDGFR is intimately linked and related to KIT and FLT3, which explains why TKIs with activities against all RTK targets are effective in RCC therapies.26,27 Upregulation of VEGF and KDR has been observed in non-small cell lung cancer (NSCLC), and their expression is correlated with tumor angiogenesis and shorter survival times.37 Clinical trials using vorolanib, alone or combined with other targeted therapies or chemotherapeutic agents, are being conducted against these cancers. In contrast, the inhibitory effects of vorolanib on RET and AMPKα1 kinase activities were inferior to sunitinib. Because inhibiting RET and AMPKα1 has been implicated as the potential mechanism of sunitinib-associated side effects, such as fatigue, cardiotoxicity, and thyroid gland toxicity,38 our results, in addition to a shorter half-life and limited tissue accumulation observed in other studies,10 suggest that vorolanib might be less toxic because of its higher kinase binding and selective inhibitions.
Angiogenesis has a vital role in tumor development and metastasis. Preclinical studies in rats revealed that an oral administration of vorolanib could reduce CNV lesions in a laser-induced CNV rat model.9 In this study, vorolanib dose-dependently inhibited the KDR phosphorylation of HUVECs, thus notably repressed VEGF-induced proliferation of HUVECs. Moreover, vorolanib was found to inhibit the tube formation of HUVECs, further suggesting its role in inhibiting angiogenesis. This cellular anti-angiogenic effect of vorolanib could be due to its highly selective binding of KDR and potent inhibition of VEGF-stimulated phosphorylation. KDR appears to mediate almost all known cellular responses to VEGF and the biologic or tumorigenic cellular events associated with angiogenesis.39,40
In vitro cell proliferation assays revealed the lack of direct inhibition of cell proliferation by vorolanib in most tested human tumor cell line cells, including HT-29, HCT-116, BxPC-3, A549, A375, and 786-O cells. These results indicated that tumor growth inhibition could occur primarily by an indirect mechanism, such as the inhibition of tumor angiogenesis, in mouse xenograft tumors established from these cells. The only exception was seen with vorolanib in the significant direct inhibition of MV-4-11 cell proliferation in vitro and indirect inhibition of tumor growth in the mouse xenograft model with the MV-4-11 cell line. This dual inhibition mechanism might be due to high FLT3 expression on MV-4-11 cells, resulting in the increased inhibition of tumor angiogenesis in MV-4-11 tumors.41
In this study, vorolanib exhibited tumor growth inhibition in mouse xenograft models established from all tumor cell lines tested, including renal, colorectal, and pancreatic carcinomas, melanomas, and leukemias. Sunitinib demonstrated inhibitory profiles against these tumors that were consistent with previous reports.33,42,43 The tumor growth inhibition of vorolanib was dose-dependent in all tested xenografts, and the inhibition at the 160 mg/kg (80 mg/kg bid) dose was comparable with the sunitinib dose at 40 mg/kg qd. Vorolanib showed strong inhibition of the MV-4-11 xenograft tumors, and tumors completely disappeared at the 80 mg/kg bid dose. These results demonstrated the strong anti-tumor activity of vorolanib. In vivo studies further confirmed that vorolanib can exert both anti-angiogenic and anti-proliferative effects. In addition, vorolanib was well-tolerated by the animals in the study, whereas sunitinib had negative impacts on body weights at a dose of 40 mg/kg qd, again indicating that vorolanib is safer than sunitinib.
In summary, vorolanib is a novel multi-kinase TKI with potent preclinical anti-angiogenic and anti-tumor activities with potentially less toxic effects compared with sunitinib. Several clinical trials using vorolanib, alone or in combination with other therapeutic agents, are being conducted to investigate its anti-cancer effects in patients with advanced solid tumors.
Materials and methods
Compounds
Vorolanib was provided by Betta Pharmaceuticals (Hangzhou, China). Sunitinib was purchased from Pfizer (New York, NY, US). The purity for all compounds was ≥98%. Vorolanib was formulated as a suspension at a concentration of 10 mM in DMSO and stored at −20°C. VEGF165 was provided by R&D Systems (Minneapolis, MN, US).
Ethics statement
The animal protocol was reviewed by the board of the city government of Hangzhou, China, and approved for all experimental procedures. All experiments were conducted in a manner where discomfort, pain, distress, and suffering were avoided or minimized and in accordance with the Guide for the Care and Use of Laboratory Animals, as defined by the National Institutes of Health (NIH).
Cell cultures
HUVECs (ATCC, Manassas, VA, US) were cultured in F-12 media (Invitrogen, Carlsbad, CA, US) containing 10% fetal bovine serum (FBS), 18 U/mL heparin, and 30 μg/mL endothelial cell growth supplement (ECGS). Primary HUVECs (from Shanghai University of Traditional Chinese Medicine) were cultured in Medium 199 (Invitrogen) with 20% FBS, 18 U/mL heparin, and 30 μg/mL ECGS. HCT-116, BxPC-3, A549, and 786-O were purchased from ATCC, while HT-29, A375, and MV-4-11 cells were procured from the Cell Bank (Cell Institute, Sinica Academia Shanghai, Shanghai, China). HT-29, HCT-116, BxPC-3, A549, A375, and 786-O cells were cultured in DMEM (Invitrogen) with 10% FBS. MV-4-11 cells were cultured in Iscove's Modified Dulbecco's Medium (IMDM) (Invitrogen) with 10% FBS.
The KINOMEscan assay
The binding of the compounds including, vorolanib (CM082) and sunitinib (as the control), with 287 kinases covering about 55% of the predicted human protein kinome, was tested using the KINOMEscan assay (Ambit Biosciences, San Diego, CA). Binding affinities were quantified by measuring the amount of kinase captured in the test versus control samples using quantitative RT-PCR. The IC50 values for KDR, PDGFRβ, FLT3, C-Kit, RET, and AMPKα1 were calculated for vorolanib and sunitinib, respectively.
The EMD Millipore KinaseProfilerTM Service
The EMD Millipore KinaseProfiler Service (MERCK Millipore) was used to profile the inhibitory activity of vorolanib (at 0.1 and 1 μM) against a total of 38 recombinant human kinases, with sunitinib (at 0.1 and 1 μM) as the control.
Cell proliferation assays
HUVECs were seeded in 96-cell plates in triplicate at a density of 10,000 cells/well in 100 μL of F-12 media with 10% FBS and were allowed to adhere overnight. Supernatants were removed and replaced with 100 μL of culture media with 0.1% FBS and cultured for an additional 24 h. The supernatants were then replaced with 50 μL fresh culture media with 0.1% FBS, with or without vorolanib, CM082R, and sunitinib, at graded concentrations of 0.8, 3, 11.8, 46.8, and 20,000 nM. Another 50 μL of culture media with 0.1% FBS, with or without VEGF165, at a concentration of 80 ng/mL, was added to each well 45 min later. For the tumor cell lines, cells were seeded in 96-cell plates in triplicate at a density of 10,000 cells/well in 50 μL of media with 10% FBS and were allowed to adhere for 3–4 h. Another 50 μL of culture media, with or without vorolanib and sunitinib, at increasing concentrations of 74, 222.2, 666.6, 2,000, 6,000, 18,000, and 54,000 nM, were added to each well. Proliferation was determined at 72 h of culture under the conditions mentioned above using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS, Promega, WI, US), according to the manufacturer's protocol. The spectrophotometric absorbance of each sample was measured at 490 nm using a microplate reader. Percent proliferation relative to the controls was calculated based on the MTS readout; the IC50 value was defined as the drug concentration that produced a 50% reduction in absorbance relative to the control.
In vitro HUVEC tube formation assays
Primary HUVEC endothelial cells were seeded at the concentration of 10,000 cells per well in 100 μL of Medium 199 with 1% FBS and with or without vorolanib or sunitinib at concentrations of 50 and 100 nM, respectively. The cultures were incubated at 37°C with 5% CO2 for 15 min. VEGF165 or control medium was then added to a final concentration of 30 ng/mL and incubated at 37°C with 5% CO2 for 3 h. Tube formation was visualized using an inverted phase-contrast microscope at 4× magnification.
Subretinal injections and CNV measurements
Preclinical studies in rats revealed that oral vorolanib, administered at the dose of 10 and 30 mg/kg/day, reduced the size of CNV in a rat model. Briefly, rats were anesthetized with ketamine (40 mg/kg, intraperitoneal [IP]) and xylazine (6 mg/kg, IP). A 33-gauge needle was inserted between the limbus and equator to reach the subretinal space. A blunt 33-gauge needle attached to a 10 μL microsyringe (Hamilton, Reno, NV) was then introduced into the subretinal space to inject 1.2 μL of Matrigel (BD Biosciences), diluted 3:1 with phosphate-buffered saline (PBS; Gibco, Invitrogen). The animals were euthanatized using CO2 inhalation and perfused with PBS, followed by a DiI solution (Sigma-Aldrich, St. Louis, MO) and 4% paraformaldehyde. The eyecups were embedded in 5% agarose. Thick (100 μm) serial sections were cut on a soft tissue microtome (VT1000S; Leica Microsystems, Bannockburn, IL) and examined with confocal microscopy. CNV areas were calculated throughout the entire Matrigel area. The CNV area of a section (Ci) was calculated by multiplying the width (Wi), the maximum measurement of CNV along the sclera, by the thickness of the section, Ti (Ci = TiWi). The height of CNV, the maximum distance between Bruch's membrane and the front edge of CNV, was not included because its variation was negligible. The thickness of each section (100 μm), Ti, was the same for all sections.
Xenograft models and tumor measurements
A mouse MV-4-11 xenograft model was established with male athymic BALB/c nude mice (6 weeks) by administering subcutaneous injections of 5 × 106 of MV-4-11 cancer cells into the flank next to the right forelimb. Mouse xenograft models of other cancer cell lines, including HT-29, HCT-116, BxPC-3, A375, and 786-O, were established in two steps. First, 5 × 106 of the cancer cells were injected subcutaneously into the flank of nude mice, and tumors were allowed to grow to a size of 100–300 mm3. Next, the tumors were extracted from the mice, minced into 1 mm3 pieces, and injected into the flank of another group of BALB/c nude mice. The resulting tumors were allowed to grow to a size of 100–300 mm3. A total of 32 mice were used for each xenograft model, including 16 in the blank control group, 8 in the vorolanib group, and 8 in the positive control (sunitinib) group. Vorolanib and sunitinib were administered at various doses orally for 3–4 weeks (6 weeks for the 786-O model). Percent tumor growth inhibition after vorolanib and sunitinib treatments was measured and calculated. The xenograft models were established, and inhibitory effect studies were repeated once each for the HT-29, HCT-116, BxPC-3, A375, and 786-O tumor cell lines and twice for the MV-4-11 cell line. All mice were allowed free access to disinfected water and food. The animal study was carried out according to the NIH guidelines for animal care and use.
Immunofluorescent staining
Tumor paraffin sections (8 μm) were incubated with Tris/EDTA buffer for 30 min after hydrated. The sections were treated and blocked with PBS containing 0.3% Triton X- and then primarily incubated with rabbit anti-mouse CD31 antibody (1:100, Abcam, Cambridge, UK) at 4°C overnight. After washing, the sections were then incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG secondary antibody (1:50, Abcam) for 2 h. For quantitative measurements of MVD, five slides, with each slide containing four fields (×200), were captured under a fluorescence microscope (Olympus Corporation, Tokyo, Japan) and analyzed using ImageJ software (NIH, Bethesda, MD, US).
Statistical analysis
The data were expressed as the mean ± SD. A one-way ANOVA analysis and Student's t test were performed to compute significant differences. All data were analyzed with SPSS software, version 22 (SPSS, Chicago, IL, US). p < 0.05 was considered statistically significant.
Acknowledgments
We express sincere gratitude to Dr. W. Sprague from iCoreMed Technology and Service LLC for medical writing support. The datasets generated and analyzed during the present study are available from the corresponding author on reasonable request.
Author contributions
C.L., L.M., and L.D. conceived the study and designed the experiments. C.L. performed the experiments and analyzed the data. X.Y. and Z.S. wrote the manuscript. Y.W. supervised the manuscript. Each author reviewed and revised the manuscript and provided final approval for submission of the final version.
Declaration of interests
All authors are employees of Betta Pharmaceuticals.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.01.001.
Supplemental information
References
- 1.Moser C., Lang S.A., Stoeltzing O. The direct effects of anti-vascular endothelial growth factor therapy on tumor cells. Clin. Colorectal Cancer. 2007;6:564–571. doi: 10.3816/CCC.2007.n.023. [DOI] [PubMed] [Google Scholar]
- 2.Sigismund S., Avanzato D., Lanzetti L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018;12:3–20. doi: 10.1002/1878-0261.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaufman N., Dhingra S., Jois S.D., Vicente M. Molecular targeting of epidermal growth factor receptor (EGFR) and vascular endothelial growth factor receptor (VEGFR) Molecules. 2021;26:1076. doi: 10.3390/molecules26041076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fogli S., Porta C., Del Re M., Crucitta S., Gianfilippo G., Danesi R., Rini B.I., Schmidinger M. Optimizing treatment of renal cell carcinoma with VEGFR-TKIs: a comparison of clinical pharmacology and drug-drug interactions of anti-angiogenic drugs. Cancer Treat Rev. 2020;84:101966. doi: 10.1016/j.ctrv.2020.101966. [DOI] [PubMed] [Google Scholar]
- 5.Motzer R.J., Hutson T.E., Glen H., Michaelson M.D., Molina A., Eisen T., Jassem J., Zolnierek J., Maroto J.P., Mellado B., et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16:1473–1482. doi: 10.1016/S1470-2045(15)00290-9. [DOI] [PubMed] [Google Scholar]
- 6.Jamil M.O., Hathaway A., Mehta A. Tivozanib: status of development. Curr. Oncol. Rep. 2015;17:24. doi: 10.1007/s11912-015-0451-3. [DOI] [PubMed] [Google Scholar]
- 7.Abdel-Qadir H., Ethier J.L., Lee D.S., Thavendiranathan P., Amir E. Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: a systematic review and meta-analysis. Cancer Treat Rev. 2017;53:120–127. doi: 10.1016/j.ctrv.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Dobbin S., Cameron A.C., Petrie M.C., Jones R.J., Touyz R.M., Lang N.N. Toxicity of cancer therapy: what the cardiologist needs to know about angiogenesis inhibitors. Heart. 2018;104:1995–2002. doi: 10.1136/heartjnl-2018-313726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren C., Shi H., Jiang J., Liu Q., Du Y., He M., Cai W., Wei Q., Yu J. The effect of CM082, an oral tyrosine kinase inhibitor, on experimental choroidal neovascularization in rats. J. Ophthalmol. 2017;2017:6145651. doi: 10.1155/2017/6145651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bendell J.C., Patel M.R., Moore K.N., Chua C.C., Arkenau H.T., Dukart G., Harrow K., Liang C. Phase I, first-in-human, dose-escalation study to evaluate the safety, tolerability, and pharmacokinetics of vorolanib in patients with advanced solid tumors. Oncologist. 2019;24:455–e121. doi: 10.1634/theoncologist.2018-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu L., Huang J., Liu J., Xi Y., Zheng Z., To K., Chen Z., Wang F., Zhang Y., Fu L. CM082 enhances the efficacy of chemotherapeutic drugs by inhibiting the drug efflux function of ABCG2. Mol. Ther. Oncolytics. 2019;16:100–110. doi: 10.1016/j.omto.2019.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scarpelli M., Rampurwala M., Eickhoff J., Carmichael L., Heideman J., Binger K., Kolesar J., Perlman S., Harrow K., Dukart G., et al. Pharmacodynamic study using FLT PET/CT in advanced solid malignancies treated with a sequential combination of X-82 and docetaxel. Cancer Chemother. Pharmacol. 2018;82:211–219. doi: 10.1007/s00280-018-3599-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheng X., Yan X., Tang B., Chi Z., Guo J. A phase I clinical trial of CM082 (X-82) in combination with everolimus for treatment of metastatic renal cell carcinoma. J. Clin. Oncol. 2017;35:4575. [Google Scholar]
- 14.Yan X., Sheng X., Tang B., Chi Z., Guo J. Anti-VEGFR, PDGFR, and CSF1R tyrosine kinase inhibitor CM082 (X-82) in combination with everolimus for treatment of metastatic renal cell carcinoma: a phase I clinical trial. Lancet Oncol. 2017;18:S8. [Google Scholar]
- 15.Whisenant J., Beckermann K., Borghaei H., Owonikoko T., Horn L. Phase I/II study of nivolumab and vorolanib in patients with refractory thoracic tumors. J. Thorac. Oncol. 2019;14:S445–S446. [Google Scholar]
- 16.Nusinow D.P., Szpyt J., Ghandi M., Rose C.M., McDonald E.R., 3rd, Kalocsay M., Jané-Valbuena J., Gelfand E., Schweppe D.K., Jedrychowski M., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 2020;180:387–402.e16. doi: 10.1016/j.cell.2019.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Y., Chen Y., Xu X., Fu X., Zhao Z.J. SU11652 Inhibits tyrosine kinase activity of FLT3 and growth of MV-4-11 cells. J. Hematol. Oncol. 2012;5:72. doi: 10.1186/1756-8722-5-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J. Hematol. Oncol. 2011;4:13. doi: 10.1186/1756-8722-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Folkman J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 20.Faivre S., Demetri G., Sargent W., Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat. Rev. Drug Discov. 2007;6:734–745. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- 21.Cella D., Michaelson M.D., Bushmakin A.G., Cappelleri J.C., Charbonneau C., Kim S.T., Li J.Z., Motzer R.J. Health-related quality of life in patients with metastatic renal cell carcinoma treated with sunitinib vs interferon-alpha in a phase III trial: final results and geographical analysis. Br. J. Cancer. 2010;102:658–664. doi: 10.1038/sj.bjc.6605552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demetri G.D., van Oosterom A.T., Garrett C.R., Blackstein M.E., Shah M.H., Verweij J., McArthur G., Judson I.R., Heinrich M.C., Morgan J.A., et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 23.Rini B.I. Sunitinib. Expert Opin. Pharmacother. 2007;8:2359–2369. doi: 10.1517/14656566.8.14.2359. [DOI] [PubMed] [Google Scholar]
- 24.Faivre S., Delbaldo C., Vera K., Robert C., Lozahic S., Lassau N., Bello C., Deprimo S., Brega N., Massimini G., et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J. Clin. Oncol. 2006;24:25–35. doi: 10.1200/JCO.2005.02.2194. [DOI] [PubMed] [Google Scholar]
- 25.Di Lorenzo G., Porta C., Bellmunt J., Sternberg C., Kirkali Z., Staehler M., Joniau S., Montorsi F., Buonerba C. Toxicities of targeted therapy and their management in kidney cancer. Eur. Urol. 2011;59:526–540. doi: 10.1016/j.eururo.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Powles T., Kayani I., Sharpe K., Lim L., Peters J., Stewart G.D., Berney D., Sahdev A., Chowdhury S., Boleti E., et al. A prospective evaluation of VEGF-targeted treatment cessation in metastatic clear cell renal cancer. Ann. Oncol. 2013;24:2098–2103. doi: 10.1093/annonc/mdt130. [DOI] [PubMed] [Google Scholar]
- 27.Griffioen A.W., Mans L.A., de Graaf A., Nowak-Sliwinska P., de Hoog C., de Jong T., Vyth-Dreese F.A., van Beijnum J.R., Bex A., Jonasch E. Rapid angiogenesis onset after discontinuation of sunitinib treatment of renal cell carcinoma patients. Clin. Cancer Res. 2012;18:3961–3971. doi: 10.1158/1078-0432.CCR-12-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goel H.L., Mercurio A.M. VEGF targets the tumour cell. Nat. Rev. Cancer. 2013;13:871–882. doi: 10.1038/nrc3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jitariu A.A., Cimpean A.M., Kundnani N.R., Raica M. Platelet-derived growth factors induced lymphangiogenesis: evidence, unanswered questions and upcoming challenges. Arch. Med. Sci. 2015;11:57–66. doi: 10.5114/aoms.2015.49217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hill R.A., Patel K.D., Medved J., Reiss A.M., Nishiyama A. NG2 cells in white matter but not gray matter proliferate in response to PDGF. J. Neurosci. 2013;33:14558–14566. doi: 10.1523/JNEUROSCI.2001-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heldin C.H., Lennartsson J., Westermark B. Involvement of platelet-derived growth factor ligands and receptors in tumorigenesis. J. Intern. Med. 2018;283:16–44. doi: 10.1111/joim.12690. [DOI] [PubMed] [Google Scholar]
- 32.Heinrich M.C., Corless C.L., Duensing A., McGreevey L., Chen C.J., Joseph N., Singer S., Griffith D.J., Haley A., Town A., et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 33.Bahlawane C., Eulenfeld R., Wiesinger M.Y., Wang J., Muller A., Girod A., Nazarov P.V., Felsch K., Vallar L., Sauter T., et al. Constitutive activation of oncogenic PDGFRα-mutant proteins occurring in GIST patients induces receptor mislocalisation and alters PDGFRα signalling characteristics. Cell Commun. Sign. 2015;13:21. doi: 10.1186/s12964-015-0096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang W., Zhu C., Yu J., Ye X., Ma Z. KIT gene mutations in gastrointestinal stromal tumor. Front. Biosci. (Landmark Ed) 2015;20:919–926. doi: 10.2741/4346. [DOI] [PubMed] [Google Scholar]
- 35.Muppidi M.R., Portwood S., Griffiths E.A., Thompson J.E., Ford L.A., Freyer C.W., Wetzler M., Wang E.S. Decitabine and sorafenib therapy in FLT-3 ITD-mutant acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2015:S73–S79. doi: 10.1016/j.clml.2015.02.033. [DOI] [PubMed] [Google Scholar]
- 36.Christensen J.G. A preclinical review of sunitinib, a multitargeted receptor tyrosine kinase inhibitor with anti-angiogenic and antitumour activities. Ann. Oncol. 2007;18:x3–x10. doi: 10.1093/annonc/mdm408. [DOI] [PubMed] [Google Scholar]
- 37.Yuan A., Yu C.J., Kuo S.H., Chen W.J., Lin F.Y., Luh K.T., Yang P.C., Lee Y.C. Vascular endothelial growth factor 189 mRNA isoform expression specifically correlates with tumor angiogenesis, patient survival, and postoperative relapse in non-small-cell lung cancer. J. Clin. Oncol. 2001;19:432–441. doi: 10.1200/JCO.2001.19.2.432. [DOI] [PubMed] [Google Scholar]
- 38.Aparicio-Gallego G., Blanco M., Figueroa A., García-Campelo R., Valladares-Ayerbes M., Grande-Pulido E., Antón-Aparicio L. New insights into molecular mechanisms of sunitinib-associated side effects. Mol. Cancer Ther. 2011;10:2215–2223. doi: 10.1158/1535-7163.MCT-10-1124. [DOI] [PubMed] [Google Scholar]
- 39.Holmes D.I., Zachary I. The vascular endothelial growth factor (VEGF) family: angiogenic factors in health and disease. Genome Biol. 2005;6:209. doi: 10.1186/gb-2005-6-2-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmes K., Roberts O.L., Thomas A.M., Cross M.J. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19:2003–2012. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 41.Naoe T., Kiyoi H. Normal and oncogenic FLT3. Cell Mol. Life Sci. 2004;61:2932–2938. doi: 10.1007/s00018-004-4274-x. [DOI] [PubMed] [Google Scholar]
- 42.Mendel D.B., Laird A.D., Xin X., Louie S.G., Christensen J.G., Li G., Schreck R.E., Abrams T.J., Ngai T.J., Lee L.B., et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- 43.O'Farrell A.M., Abrams T.J., Yuen H.A., Ngai T.J., Louie S.G., Yee K.W., Wong L.M., Hong W., Lee L.B., Town A., et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101:3597–3605. doi: 10.1182/blood-2002-07-2307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.