

Abstract
The development of immunotherapeutic monoclonal antibodies targeting checkpoint inhibitory receptors, such as programmed death 1 (PD-1), or their ligands, such as PD-L1, has transformed the oncology landscape. However, durable tumor regression is limited to a minority of patients. Therefore, combining immunotherapies with those targeting checkpoint inhibitory receptors is a promising strategy to bolster antitumor responses and improve response rates. Natural killer (NK) cells have the potential to augment checkpoint inhibition therapies, such as PD-L1/PD-1 blockade, because NK cells mediate both direct tumor lysis and T cell activation and recruitment. However, sourcing donor-derived NK cells for adoptive cell therapy has been limited by both cell number and quality. Thus, we developed a robust and efficient manufacturing system for the differentiation and expansion of high-quality NK cells derived from induced pluripotent stem cells (iPSCs). iPSC-derived NK (iNK) cells produced inflammatory cytokines and exerted strong cytotoxicity against an array of hematologic and solid tumors. Furthermore, we showed that iNK cells recruit T cells and cooperate with T cells and anti-PD-1 antibody, further enhancing inflammatory cytokine production and tumor lysis. Because the iNK cell derivation process uses a renewable starting material and enables the manufacturing of large numbers of doses from a single manufacture, iNK cells represent an “off-the-shelf” source of cells for immunotherapy with the capacity to target tumors and engage the adaptive arm of the immune system to make a “cold” tumor “hot” by promoting the influx of activated T cells to augment checkpoint inhibitor therapies.
One sentence summary:
iPSC NK cells prime T cells for anti-PD-1 therapy
Introduction
Over the past 15 years the development of cancer treatment approaches designed to manipulate the immune system, collectively termed “immunotherapies”, has resulted in a paradigm shift in cancer therapy. Although malignant cells can be immunogenic, cytotoxic immune cells often fail to properly engage with tumors and eliminate them (1). There are myriad mechanisms that interfere with cytotoxic lymphocyte function in the context of cancer, including extrinsic suppression mediated by myeloid-derived suppressor cells, regulatory T cells, and suppressive cytokines, as well as intrinsic dysfunction induced by excessive antigen stimulation resulting in anergy or exhaustion (2–4). A hallmark of T cell exhaustion is high abundance of multiple inhibitory receptors, including programmed cell death 1 (PD-1) (5). The discovery that antibody-mediated PD-1 blockade partially reverses T cell exhaustion and influences viral titers or tumor load was a key breakthrough (6, 7). The ligand for PD-1 is PD-L1, which is often abundant on tumor cells (8). Recent clinical trials show that blocking the PD-L1/PD-1 pathway enhances antitumor immunity across different types of malignancies, leading to objective responses, some of which are sustained (9–13). However, complete remissions are not experienced by most patients receiving anti-PD-1 or anti-PD-L1 treatment, with some exhibiting no clinical response.
The clinical activity of checkpoint blockade generally correlates with three major factors. The first factor is the number and type of somatic mutations acquired by tumor cells and the degree to which these mutations cause presentation of immunogenic neoantigens, which depends on neoantigen production, HLA class I proteins and beta-2-microglobulin (B2M) (14–16). Clinical trials have shown that B2M mutations can result in resistance to checkpoint blockade therapy through disruption of antigen presentation (17). The second factor is the amount of PD-L1 in the tumor microenvironment. High amounts of PD-L1 on tumor cells or tumor-infiltrating immune cells is associated with better clinical response to PD-L1/PD-1 blockade (18). The third factor is the frequency of actively proliferating CD8+ T cells, which can be detected with Ki67, relative to tumor burden. Patients with increased numbers of Ki67+CD8+ T cells after PD-1 blockade are more likely to experience a clinical benefit (19). Quantification and localization of immune cells within the tumor microenvironment have been proposed as measurements that reflect tumor immunogenicity, with three typical scenarios described: (i) Immune desert tumors have no immune infiltration (“cold”), (ii) immune-excluded tumors have T cells present at the tumor margins but absent in the tumor core, and (iii) inflamed tumors have extensive immune infiltration (“hot”) (20). Thus, strategies aimed at targeting cells with low or no HLA class I, increasing the immunogenicity of tumors, or promoting T cell trafficking and activation should be prioritized for combination with checkpoint blockade to maximize the likelihood of therapeutic benefit (21).
One potential approach to promote immune infiltration and augment checkpoint blockade is combination with adoptive transfer of natural killer (NK) cells. NK cells directly lyse malignantly transformed or virally infected cells and secrete inflammatory cytokines, such as tumor necrosis factor (TNF) and interferon (IFN)-γ (22). Such cytokines promote cytotoxic immunity. Unlike CD8+ T cells, NK cells do not undergo somatic DNA rearrangement events during development to generate receptor diversity, but instead detect missing or altered “self” through an array of germline-encoded inhibitory and activating receptors (23). This unique ability enables NK cells to directly target HLA class I-low or -null cancer cells that are no longer recognized by T cells and thus refractory to checkpoint blockade. Through the release of the cytokine Flt3-ligand, NK cells at tumor sites recruit stimulatory dendritic cells (SDCs) which, in turn, stimulate cytotoxic T cells to promote antitumor responses (24). Furthermore, NK cell frequency at tumor sites correlates with melanoma patient responsiveness to PD-1 blockade (24) and superior survival for advanced head and neck squamous cell carcinoma (HNSCC) patients (25). NK cells within tumors also produce the chemokines CCL5 and XCL1 to recruit the conventional type 1 dendritic cell subset (cDC1), which can efficiently take up tumor cell antigens and prime CD8+ T cells. The presence of high cDC1 and NK cell gene signatures positively correlates with cancer patient survival (26). Thus, NK cells are important for both direct cytotoxicity against tumors and for orchestrating the recruitment and priming of other cell subsets for enhanced immune attack.
The administration of NK cells in combination with checkpoint blockade may be particularly effective to target tumor cells that have alterations in antigen presentation or HLA downregulation. Such alterations are presumed represent a common mechanism of immune escape for tumors (27). Additionally, genes encoding HLA molecules can be downregulated through reversible epigenetic mechanisms (28). Exome sequencing analysis of paired samples from 15 patients at initial presentation with acute myeloid leukemia (AML) and at relapse after hematopoietic cell transplantation revealed downregulation of major histocompatibility complex (MHC) class II genes to amounts that were 3 to 12 times lower at relapse relative to the amounts at initial presentation. This downregulation was reversible, because treatment of cells with IFN-γ fully restored MHC class II protein abundance in almost all AML blasts analyzed. AML cells with reduced expression of MHC class II genes did not stimulate CD4+ T cells in vitro (29). Because NK cell activation and cytotoxicity are not MHC-restricted (30), they represent an important complement to T cell-mediated cytotoxicity.
Allogeneic NK cell adoptive transfer has shown clinical benefit in patients with advanced cancer (31–34). However, there are inherent limitations with respect to the number of NK cells that can be isolated during an apheresis and marked variability in the quantity and quality of NK cells between donors, making treatment with multiple doses of cells impossible. To overcome these barriers, we developed a manufacturing strategy for large scale expansion of NK cells derived from induced pluripotent stem cells (iPSCs) and showed that these cells effectively combined with a PD-1 antibody for the treatment of multiple tumor types. We refer to these cells as “iNK” (iPSC-derived NK) cells. We showed that iNK cells are highly functional and have broad cytotoxic activity against hematological and solid tumors. We found that iNK cells recruit both CD4+ and CD8+ T cells, enabling them to respond to PD-1 blockade to enhance inflammatory cytokine secretion and tumor lysis. Our data showed that iNK cells represent a promising new approach to adoptive NK cell immunotherapy and that iNK cells can be provided as an off-the-shelf cellular therapy and combined with antibodies against inhibitory checkpoint receptors to augment antitumor responses.
Results
iNK cells expand robustly and are transcriptionally similar to peripheral blood NK cells
Our goal was to develop a clinical culture system that would support robust and consistent NK cell differentiation and expansion from iPSCs for use in immunotherapy. To this end, we generated transgene-free iPSCs from human fibroblast cells using previously published methods (35, 36). We cultured the iPSCs in a cocktail of small molecules and cytokines to induce differentiation towards CD34+ hematopoietic progenitor cells (HPCs). Enriched CD34+ HPCs were then co-cultured with stroma cells previously transduced with Notch ligand and in the presence of cytokines that support HPC proliferation and differentiation toward the NK cell lineage. Notch signaling “instructs” HPCs towards the NK cell lineage (37). In the final stage of the culture system, cells were co-cultured with modified K562 cells for further expansion (Fig. 1A). At day 21 of the iNK cell differentiation culture, most cells had an NK cell phenotype defined as CD3−CD56+ (average = 68.9%; range = 40% to 92.9%). The iNK cells continued to mature during co-culture with modified K562 feeder cells, and the cells had a CD3−CD56+ NK cell phenotype at day 42 of the NK cell differentiation culture (average = 95.8%; range = 91.5% to 99.2%) (Fig. 1B). We observed consistently high rates of proliferation throughout the iNK cell culture period. When accounting for the entire process (iPSC to expanded iNK cells), total expansion reached 1 × 105 to 1 × 106-fold (Fig. 1C).
Fig. 1. iNK cells efficiently differentiate and expand in culture and are phenotypically and transcriptionally similar to primary peripheral blood NK cells.
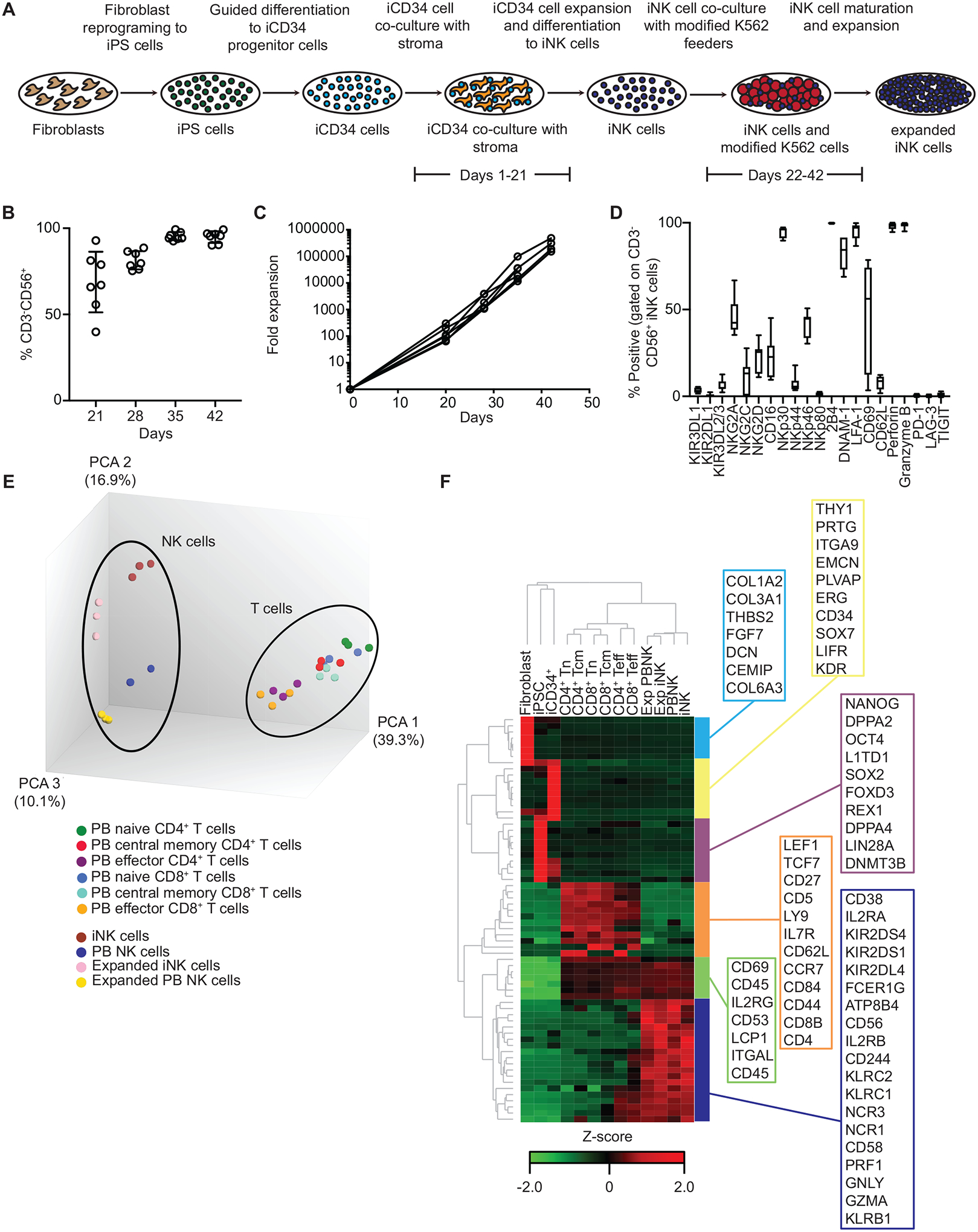
(A) Overview of the steps leading from human fibroblasts to expanded iNK cells. (B) The percentages of cells in culture with a CD3−CD56+ phenotype at the indicated days after the initiation of iNK cell differentiation and expansion from iCD34+ HPCs (N = 6 independent expansions from 1 iPSC clone). (C) iNK cells were differentiated and expanded from iCD34+ HPCs over the course of 42 days. Shown are calculated fold expansion values for total cells in culture relative to day 0 (N = 6). (D) The percentage of CD3−CD56+ iNK cells positive for each surface receptor or intracellular granule component at the end of culture (n = 7). (E) Principal component analysis of microarray gene expression analyses of the indicated primary lymphocyte and iNK cell populations. PB, peripheral blood. (F) Unsupervised hierarchical clustering of RNA transcripts associated with NK cell identity, as determined by microarray analysis, for the indicated cell populations. CD4+ Tn, naïve CD4+ T cells from peripheral blood; CD4+ Tcm, CD4+ central memory T cells from peripheral blood; CD8+ Tn, naïve CD8+ T cells from peripheral blood; CD8+ Tcm, central memory CD8+ T cells from peripheral blood; CD4+ Teff, effector CD4+ T cells from peripheral blood; CD8+ Teff, effector CD8+ T cells from peripheral blood; Exp PNBK, expanded NK cells from peripheral blood; Exp iNK, expanded iNK cells; PB NK, NK cells from peripheral blood (not expanded); iNK, iNK cells not expanded. Genes shown in the boxes represent signature genes for each cell type.
Phenotypically, iNK cells displayed various NK cell surface markers, including low frequency subsets positive for killer immunoglobulin-like receptors (KIRs) and moderate frequency subsets positive for killer cell lectin-like receptors (NKG2A, NKG2C, NKG2D) and CD16 (Fig. 1D). After infusion, these frequencies are dynamic and can change in vivo. Indeed, we observed a higher frequency of iNK cells positive for KIRs 14 days after adoptive transfer into NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice compared to the frequency of these cells prior to infusion (fig. S1), consistent with in vivo maturation of NK cells differentiated from cord blood CD34+ progenitors (38). The presence of most cytotoxicity receptors was variable on iNK cells, although the cells were consistently positive for NKp30 (Fig. 1D). Furthermore, all iNK cells were positive for intracellular granzyme B and perforin, as well as for the adhesion molecules DNAM-1 and LFA-1 and the activating receptor 2B4. The inhibitory checkpoint receptors PD-1, LAG-3, and TIGIT were not detected on iNK cells (Fig. 1D). In co-culture assays with OVCAR8 ovarian carcinoma cells as targets individually blocking NKG2D, NKp30, DNAM-1, or LFA-1 had little effect on iNK function, as measured by the percent of cells positive for IFN-γ; however, blocking NKG2D in combination with either NKp30 or DNAM-1 reduced the number of cells producing IFN-γ by ~50% (fig. S2). Thus, these data indicated that iNK cells signal through multiple known activating receptors in the presence of tumor cells.
We also performed microarrays to compare the transcriptome of iNK cells to that of primary T cell and NK cell populations, K562 feeder cell-expanded primary NK cells, fibroblasts, iPSCs, and iCD34+ cells. In a principal component analysis (PCA) of the transcriptomic data, peripheral blood T cell populations formed a distinct cluster, as did resting and expanded peripheral blood NK cells and iNK cells (Fig. 1E). Unsupervised hierarchal clustering of whole transcriptome data showed a similar pattern in which iNK cells and peripheral blood NK cells (expanded or not) had similar global transcriptional profiles that were distinct from the profiles of T cells, fibroblasts, iPSCs, and iCD34+ cells. Peripheral blood NK cells and iNK cells also shared a core NK cell gene signature that was distinct from that of all other cells included in the analysis (Fig. 1F). Collectively, our data showed that iNK cells robustly expand and mature in culture and are transcriptionally similar to primary peripheral blood NK cells.
iNK cells are functional against an array of tumor lines and infiltrate tumor spheroids
For an NK cell-based immunotherapy to have broad clinical application, strong effector responses to multiple tumor types is important. We assayed iNK cell function in 2-dimensional live-imaging IncuCyte assays. We transduced the following cancer cell lines with NucLight Red lentivirus: A549 lung carcinoma cells, HepG2 hepatocyte carcinoma cells, SKOV-3 ovarian adenocarcinoma cells, K562 myeloid leukemia cells, and SK-MEL2 melanoma cells. These transduced cells served as targets for iNK cells at various effector cell: tumor cell (E:T) ratios. We detected iNK cell-mediated killing over the course of 66 hours against all target cell lines tested and found that killing increased proportionally with E:T ratios (Fig. 2). We performed similar assays with iNK cells and K562 feeder-expanded peripheral blood NK cells, each set of effector cells thawed from cryopreservation and used as effectors against SKOV-3 targets in a 48-hour killing assay. Although thawed iNK cells and peripheral blood NK cells exhibited similar killing activity early in the assay, iNK cells maintained better antitumor function at later time points (fig. S3).
Fig. 2. iNK cells are functional against multiple hematopoietic and solid tumor cell lines.
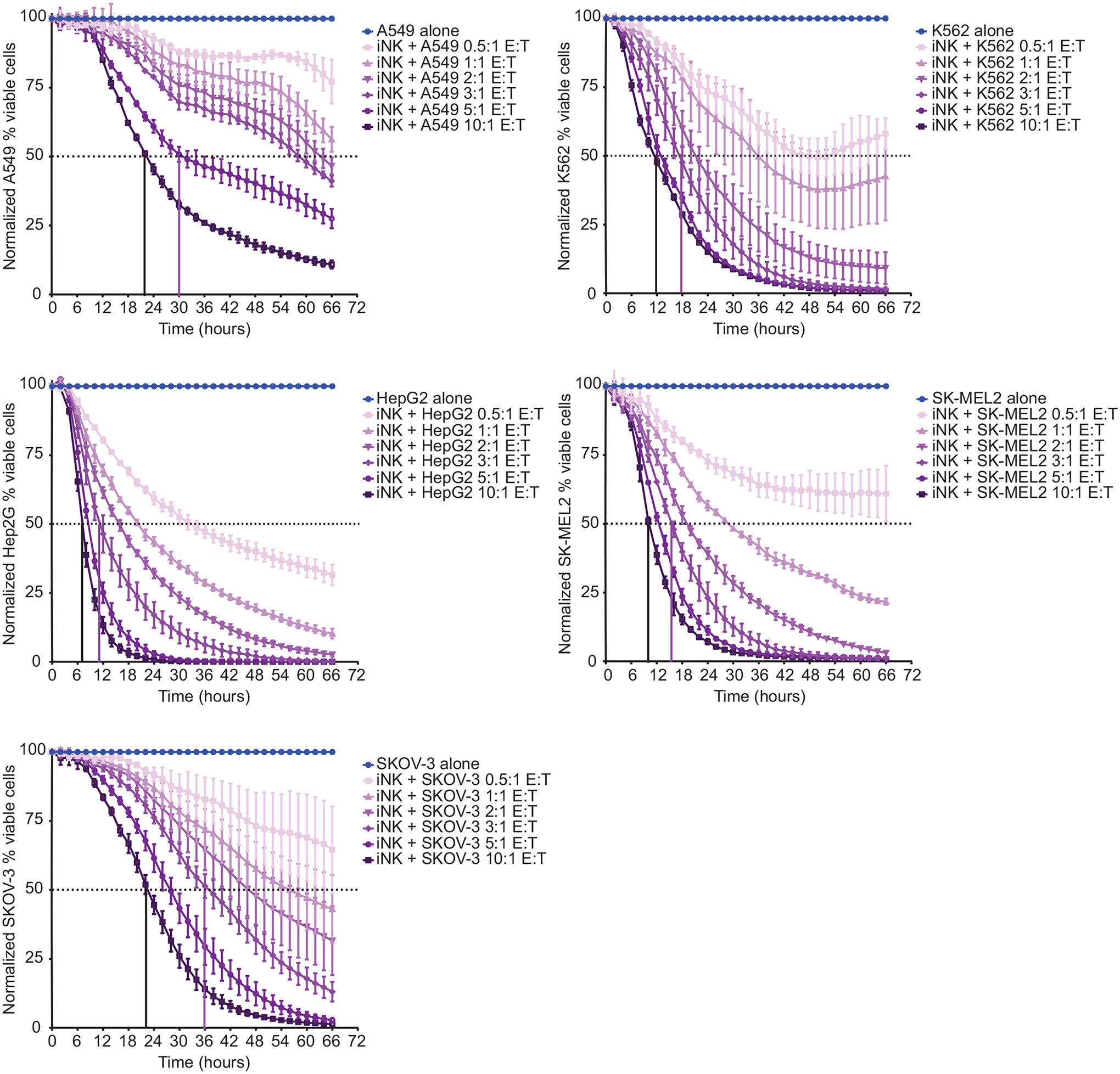
iNK cells were used as effectors at the indicated effector cell: tumor cell (E:T) ratios in 66-hour IncuCyte-based function assays with A549 lung carcinoma cells, HepG2 hepatocyte carcinoma cells, SKOV-3 ovarian adenocarcinoma cells, K562 myeloid leukemia cells, and SK-MEL2 melanoma cells. Data are representative of at least two independent experiments with each tumor cell line and are shown as mean ± SD (n = 3 technical replicates).
To investigate iNK cell infiltration and cytotoxicity in a more physiological context that better reflects the 3-dimensional architecture of the tumor, we performed live-imaging IncuCyte assays using SKOV-3 spheroids. NucLight Red-transduced SKOV-3 cells were added to wells in tissue culture plates and left to form spheroids over the course of 72 hours, after which dye-labeled iNK cells were added to wells at various E:T ratios. Without the addition of iNK cells, SKOV-3 cells formed spheroids over 48 – 72 hours and the spheroids remained stable in size for the remainder of the experiment. Co-culture with iNK cells led to marked loss of SKOV-3 spheroid fluorescence intensity in a manner proportional to the E:T ratio (Fig. 3, A and B). To determine the degree to which iNK cells infiltrated SKOV-3 spheroids and the kinetics of infiltration, we measured the integrated intensity of the green signal (given off by dye-labeled iNK cells) within the spheroid structures during the first 24 hours after addition of iNK cells. We observed rapid infiltration of iNK cells into SKOV-3 spheroids that began immediately upon co-culture and persisted for 20 hours at the higher E:T ratios (Fig. 3C). Collectively, these data showed that iNK cells are cytotoxic toward both hematologic and solid tumor cell lines in 2D culture and rapidly infiltrate and kill SKOV-3 cells in 3D tumor spheroids.
Fig. 3. iNK cells infiltrate and destroy solid tumor spheroids.
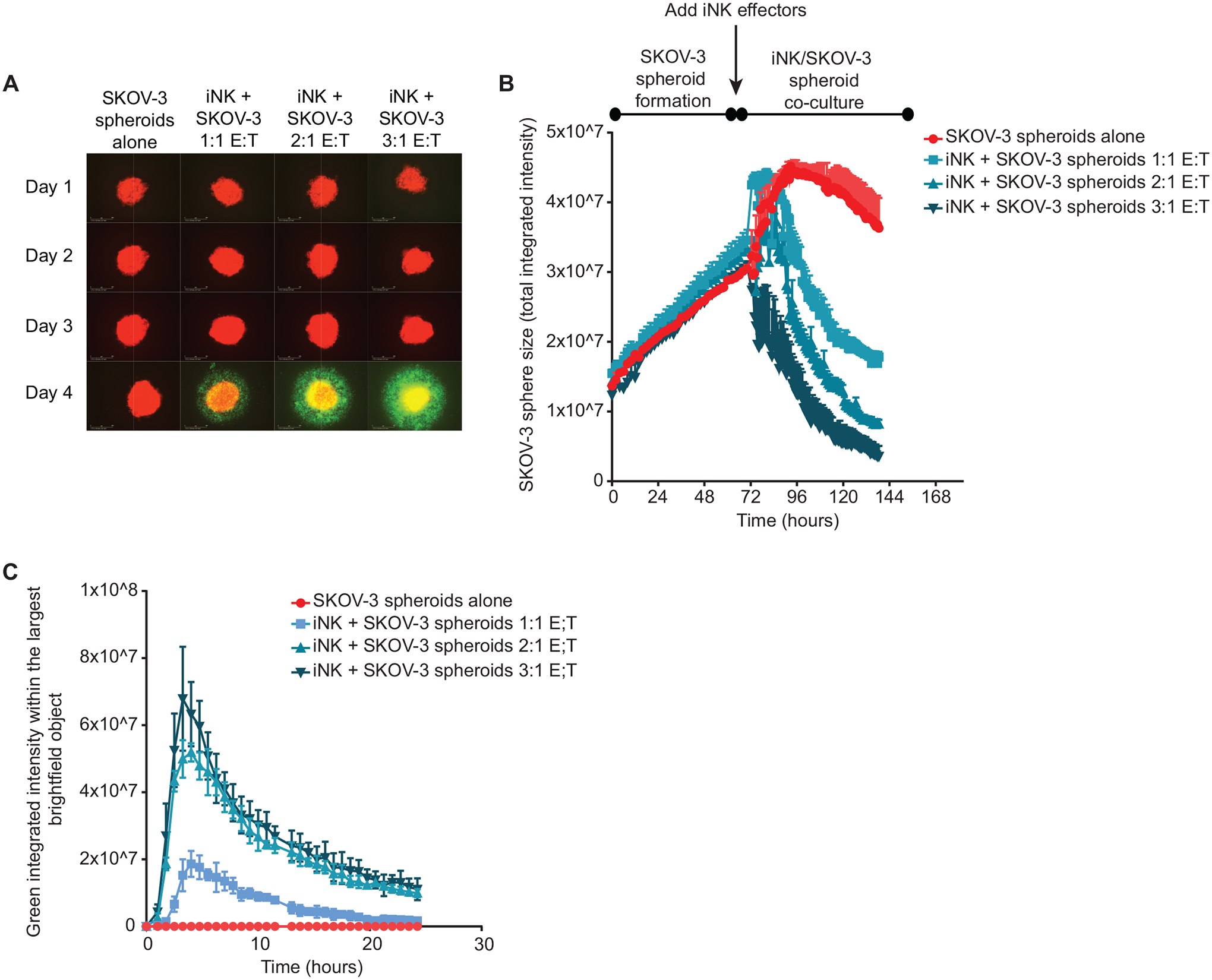
SKOV-3 cells were seeded into tissue culture plate wells where they formed spheroids over the course of 72 hours. iNK cells were then added as effectors at the indicated E:T ratios. (A) Representative images are shown from wells containing SKOV-3 spheroids alone or with the addition of iNK cells at each day of the assay. SKOV-3 cells are labeled red, and iNK cells are labeled green. (B) Cumulative data are shown of SKOV-3 spheroid size throughout the assay for each condition. (C) Quantification of iNK cell infiltration of SKOV-3 spheroids as indicated by integrated green signal within each spheroid. Data in B and C are representative of two independent experiments and are shown as mean ± SD (n = 3 technical replicates).
iNK cells delay tumor progression after single- or multi-dose administration
Having tested iNK cell function in vitro, we assessed the ability of iNK cells to mediate antitumor responses in vivo. To this end, we set up a xenogeneic model of ovarian cancer. Human OVCAR8 cells transduced with the gene encoding firefly luciferase were injected into the peritoneal cavities of NSG mice and allowed to establish tumors over the course of 3 days. Mice were then sublethally irradiated to facilitate iNK cell engraftment. The next day, mice in the treatment group received 1 dose of iNK cells harvested directly from the end of the differentiation and expansion culture along with interleukin (IL)-2. Both iNK cells and IL-2 was administered intraperitoneally (i.p.). IL-2 was i.p. injected twice per week for two weeks to support iNK cell survival. Tumor bioluminescence was measured weekly throughout the first 3 weeks, after which mice were monitored for survival (Fig. 4A). Relative to the control OVCAR8 alone group, tumor burden was significantly reduced in mice treated with a single dose of iNK cells and two weeks of IL-2 (p ≤ 0.001) (Fig. 4B, C). Furthermore, median survival was longer in the treated group relative to the control group (53 days versus 35 days; p = 0.01) (Fig. 4D).
Fig. 4. Adoptive transfer of iNK cells delays tumor progression in a xenogeneic model of ovarian cancer.
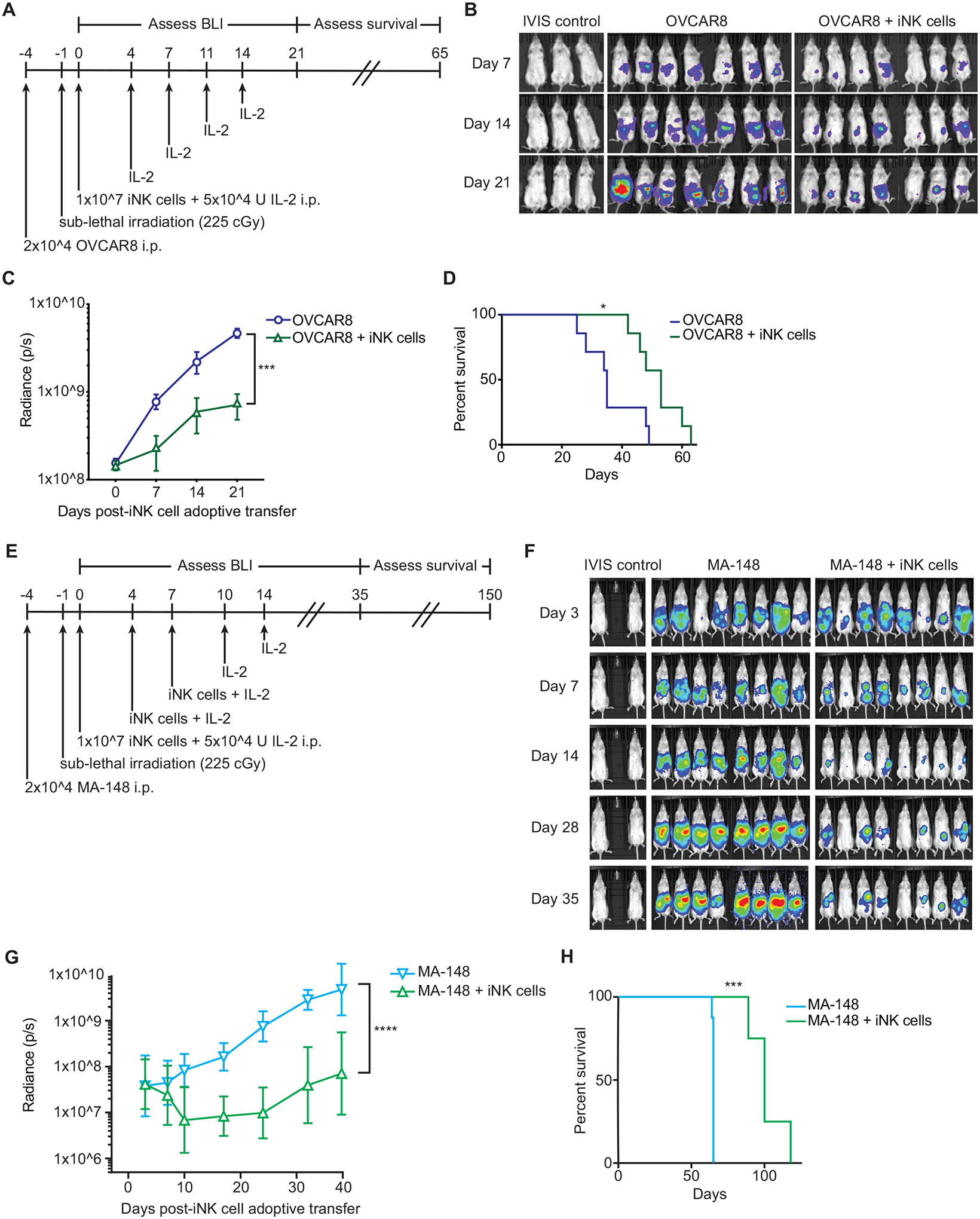
(A) Schematic of the experimental design to test in vivo antitumor function of iNK cells immediately post-culture. (B) Bioluminescence images of groups of mice that were not engrafted with tumor (IVIS control, n = 3), were engrafted with OVCAR8 cells without treatment (n = 7), or were engrafted with OVCAR8 cells and treated with iNK cells and IL-2 (n =7). Images were taken at the indicated days as defined in A. (C) Cumulative radiance for the OVCAR8 alone and OVCAR8 + iNK groups over the first 21 days of the experiment. Statistical significance was determined by two-way ANOVA. ***p ≤ 0.001. (D) Kaplan Meier analysis of overall survival. Statistical significance was determined by Log-rank (Mantel-Cox) tests. *p ≤ 0.05. (E) Schematic of the experimental design to test in vivo antitumor function with multiple doses of thawed iNK cells. (F) Bioluminescence images of groups of mice that were not engrafted with tumor (IVIS control, n = 2), were engrafted with MA-148 cells without treatment (n = 8), or were engrafted with MA-148 cells and treated with three doses of cryopreserved iNK cells and IL-2 (n = 8). Images were taken at the indicated days as defined in E. (G) Cumulative radiance for each group over the first 35 days of the experiment. Statistical significance was determined by two-way ANOVA. ****p ≤ 0.0001. (H) Kaplan Meier analysis of overall survival. Statistical significance was determined by Log-rank (Mantel-Cox) tests. ***p ≤ 0.001. BLI, bioluminescence imaging
Because more than 1 × 1011 iNK cells can be obtained from a single differentiation and expansion culture at current capacity, this system enables a multi-dosing approach to adoptive transfer. To set up a model that would approximate an iNK cell multi-dosing regimen to treat patients with cancer, human MA-148 ovarian carcinoma cells transduced with the gene encoding firefly luciferase were injected into the peritoneal cavities of NSG mice and allowed to establish tumors over the course of 3 days. Mice were then sublethally irradiated. The next day, mice in the treatment group received 1 dose of iNK cells that were thawed from cryopreserved stock and immediately injected i.p. along with IL-2. Mice then received 2 additional i.p. injections of thawed iNK cells and IL-2 on days 4 and 7. During the subsequent week, mice received 2 additional i.p. injections of IL-2. Tumor bioluminescence was measured weekly out to 35 days followed by monitoring for survival (Fig. 4E). Relative to the MA-148 alone group, tumor burden was markedly and sustainably reduced in mice given multiple thawed iNK cell injections along with IL-2 (Fig. 4F, G). Additionally, median survival was better in the treated group relative to the control group (100 days versus 65 days; p = 0.0001) (Fig. 4H). These data, along with previous work by our group (39), showed that iNK cells exhibit potent antitumor function in vivo and retain function after a freeze/thaw cycle, making multiple dosing strategies feasible.
iNK cells recruit T cells in vitro and in vivo
We evaluated whether iNK cell adoptive transfer could be used as a combinatorial strategy to improve immunotherapeutic approaches that rely on T cells and to overcome resistance to checkpoint blockade therapy. As a first step, we tested whether iNK cells recruit allogeneic T cells in vitro. We set up transwell experiments in which CD3+ T cells isolated from peripheral blood and stimulated with beads conjugated with antibodies against CD3 and CD28 (anti-CD3/CD28) were seeded above the transwell, and the bottom of each transwell contained either control media alone, or conditioned media from K562 cell cultures, iNK cell cultures, or a 1:1 mixture of iNK cells and K562 cells. We confirmed that the stimulated T cells had a phenotype indicative of strong activation, positive for CD38, CD45RO, CD27, CCR7, and PD-1 (fig. S4). Although T cell migration through the transwell was low in the media alone and K562-conditioned media conditions, significant migration occurred in the iNK cell conditioned medium condition (p = 0.0213) and migration was enhanced further using conditioned media from the iNK cell and K562 cell co-cultures (Fig. 5A). In an attempt to identify chemokines secreted by iNK cells that could promote T cell recruitment, we performed additional transwell assays with a neutralizing antibody against CCL3 and CCL4. We did not observe any statistically significant effect for CCL3 and CCL4 neutralization in these assays, suggesting that other chemokines or combinations of chemokines mediate T cell recruitment (fig. S5).
Fig. 5. iNK cells recruit peripheral blood T cells both in vitro and in vivo.
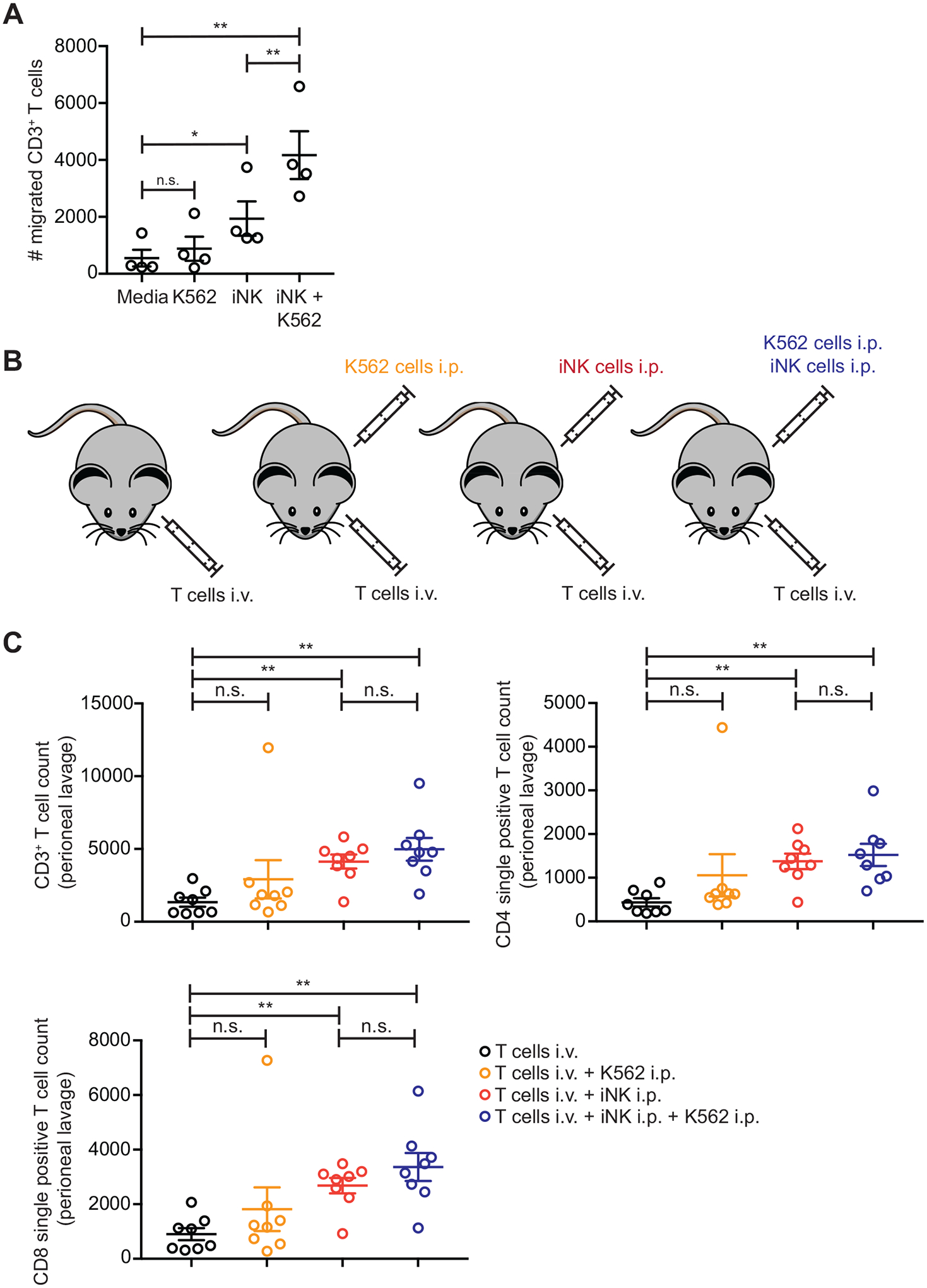
(A) CD3- and CD28-stimulated CD3+ T cells were seeded into transwells with either control media or conditioned media from K562 cell cultures, iNK cell cultures, or iNK and K562 cell co-cultures. Cumulative data of the numbers of CD3+ T cells that migrated across the transwell in each condition are shown. Statistical significance was determined by paired t tests (n = 4). *p ≤ 0.05, **p ≤ 0.01, n.s., not significant. (B) Schematic of cell injections to test T cell recruitment from the circulating blood to the peritoneum. (C) CD3- and CD28-stimulated CD3+ T cells were injected intravenously (i.v.) into mice with or without additional intraperitoneal (i.p.) injections of either K562 cells, iNK cells, or a 1:1 mixture of iNK cells and K562 cells. Mice were euthanized after 5 days, and the numbers of total CD3+ T cells, CD3+CD4−CD8+ T cells, and CD3+CD4+CD8− T cells in the peritoneal lavages were counted. Shown are cumulative data from 2 independent experiments. Statistical significance was determined by unpaired t tests corrected for multiple comparisons (n = 8). *p ≤ 0.05, **p ≤ 0.01
To model this in vivo, we set up groups of NSG mice in which peripheral blood CD3+ T cells stimulated with anti-CD3/CD28 beads were injected intravenously (i.v.). The control group received only T cell injections. The other 3 groups were injected i.p with either iNK cells, K562 cells, or a 1:1 mixture of iNK cells and K562 cells (Fig. 5B). Mice were euthanized after 4 days, and the numbers of T cells in the peritoneal cavity of all mice were determined by flow cytometry. Very few CD3+ T cells were found in the peritoneal cavity of mice receiving only i.v. T cells, and no enhancement of CD3+ T cell numbers was observed in mice that received i.p. injections of K562 cells (Fig. 5C). In contrast, there were significantly increased numbers of CD3+ T cells in the peritoneal cavity of mice that received i.p. injections of iNK cells (p < 0.01). In contrast to the in vitro transwell system, no additive effect of CD3+ T cell recruitment was observed in mice that received i.p. injections of iNK cells and K562 cells together. Similar trends were observed for CD4+ T cells and CD8+ T cells, indicating that both types of T cells migrated to the peritoneum (Fig. 5C).
iNK cells cooperate with T cells and PD-1 blockade
Because T cells migrated to injected iNK cells, we hypothesized that iNK cells cooperate with T cells to enhance T cell effector functions. To test this hypothesis, we performed experiments in which SKOV-3 spheroids were cultured alone or in combination with iNK cells and activated CD4+ or CD8+ peripheral blood T cells. We measured production of the chemokines CCL3 and CCL4 and the cytokines TNF and IFN-γ in supernatants from these cultures by Mesoscale Discovery electrochemiluminescence assay (Fig. 6A). As expected, none of these chemokines or cytokines were measured in cultures that contained SKOV-3 cells alone. Moderate amounts of CCL3, CCL4, TNF, and IFN-γ were detected in SKOV-3 co-cultures with iNK cells or activated T cells. Importantly, greater increases in the amounts of both chemokines and cytokines occurred in co-cultures that contained combinations of SKOV-3 cells, iNK cells, activated T cells, and anti-PD-1 antibody.
Fig. 6. iNK cells cooperate with T cells and anti-PD-1 antibody to eliminate tumor spheroids and release inflammatory cytokines.

SKOV-3 cells were seeded into 96-well tissue culture wells where they formed spheroids over the course of 72 hours. The indicated combinations of iNK cells, CD3-and CD28-activated CD3+CD4+ or CD3+CD8+ T cells, and anti-PD-1 antibody were then added to the wells. Experiments were performed with T cells from 3 different peripheral blood donors. (A) Quantification is shown for the indicated chemokines and cytokines collected from individual wells at the end of the assay. (B) Representative images are shown of wells containing SKOV-3 spheroids and combinations of iNK cells, CD3+ T cells, and anti-PD-1 antibody. SKOV-3 cells are labeled red, and CD3+ T cells are labeled green. (C) Cumulative data are shown from a representative experiment showing SKOV-3 viability, quantified by red intensity, in each coculture condition over the course of 150 hours. Data are shown as mean ± SD of 3 images per timepoint. Flow cytometry was used to count the number of remaining SKOV-3 cells in each well at the end of each IncuCyte assay. Shown are cumulative data from 2 independent experiments. Statistical significance was determined by unpaired t tests corrected for multiple comparisons (n = 3–19). **p ≤ 0.01, ***p≤ 0.001****, p ≤ 0.0001
To determine whether iNK cells cooperate with T cells and PD-1 blockade for antitumor cytotoxicity, we used SKOV-3 cells in 7-day live-imaging IncuCyte assays, because SKOV-3 cells have surface PD-L1 that can inhibit CD8+ T cell function (40). SKOV-3 cells expressing NucLight Red were seeded in wells and allowed to form into spheroids over the course of 3 days. Once SKOV-3 spheroids were established, we added iNK cells, CD3+ peripheral blood T cells stimulated with anti-CD3/CD28 beads, and anti-PD-1 antibody in various combinations. In wells that contained iNK cells, but no T cells, the iNK cells were dye-labeled. In all wells that contained T cells, the T cells (but not iNK cells) were dye-labeled. At the lower iNK cell numbers used in this assay (compared to higher E:T ratios in Fig. 3), addition of iNK cells led to a ~ 50% reduction in SKOV-3 spheroid size relative to the size of SKOV-3 spheroids in culture alone (Fig. 6B, C). The addition of anti-PD-1 antibody to iNK cell and SKOV-3 spheroid co-cultures had minimal effect, which was expected because PD-1 is not typically present on iNK cells. Activated T cells did not reduce the size of SKOV-3 spheroids with or without the addition of anti-PD-1 antibody. However, the combination of iNK cells and activated T cells led to more effective cytotoxicity against SKOV-3 spheroids, and the combination of iNK cells, activated T cells and anti-PD-1 antibody led to more effective elimination of SKOV-3 spheroids. We observed similar responses with T cells isolated from 3 different donors (Fig. 6B). The cumulative reduction in SKOV-3 cells was quantified by monitoring the reduction in red color intensity (Fig. 6C, left). We also harvested wells at the end of the assay to quantify the number of remaining SKOV-3 cells by flow cytometry. In line with quantified imaging data, we counted the lowest number of remaining SKOV-3 cells in wells containing the combination of iNK cells, activated T cells, and anti-PD-1 antibody (Fig. 6C, right). We observed a 99% reduction in tumor cell counts in the presence of all three (iNK cells, activated T cells, and anti-PD-1 antibody) compared to an ~60% reduction in the presence of iNK cells and activated T cells without PD-1 blockade.
To validate these results in an in vivo model, we injected OVCAR8 tumor cells into the peritoneal cavities of NSG mice. Following sublethal irradiation, we treated groups of mice with either anti-PD-1 antibody alone, activated CD3+ T cells alone, iNK cells alone, or various combinations of these (Fig. 7A). IL-2 was injected i.p. twice per week for two weeks into all mice except those in the tumor alone group. Tumor bioluminescence was measured weekly for 5 weeks (Fig. 7B). Mice treated with either anti-PD-1 antibody or activated CD3+ T cells alone exhibited similar rates of tumor growth over time relative to the untreated group. Modest tumor control was observed in the group of mice that received the combination of activated CD3+ T cells and anti-PD-1 antibody, although the effect was only significant on day 28 (p < 0.05). In mice that received iNK cells alone, significant tumor control was observed over the first 28 days (p < 0.001) but was not sustained at later time points. Tumor control was similar between groups of mice that received iNK cells alone and mice that received the combination of iNK cells and anti-PD-1 antibody. Durable tumor control at time points past day 21 were observed in groups of mice that received either the combination of iNK cells and activated CD3+ T cells or all three iNK cells, activated CD3+ T cells, and anti-PD-1 antibody. However, at day 35, tumor bioluminescence was significantly lower in the group of mice receiving the triple therapy (iNK cells, activated CD3+ T cells, and anti-PD-1 antibody) relative to the group that received iNK cells and activated CD3+ T cells (p < 0.05). Of note, symptoms of graft versus host disease (GvHD) were observed at later time points in all groups of mice that received T cells. Some mice from each group receiving T cells were euthanized before the end of the experiment because of GvHD. Collectively, these data showed that iNK cells can recruit and activate T cells to tumors, making them responsive to checkpoint receptor blockade to enhance inflammatory cytokine production and tumor elimination.
Fig. 7. Combining iNK cells with T cells and anti-PD-1 antibody promotes durable tumor control in vivo.
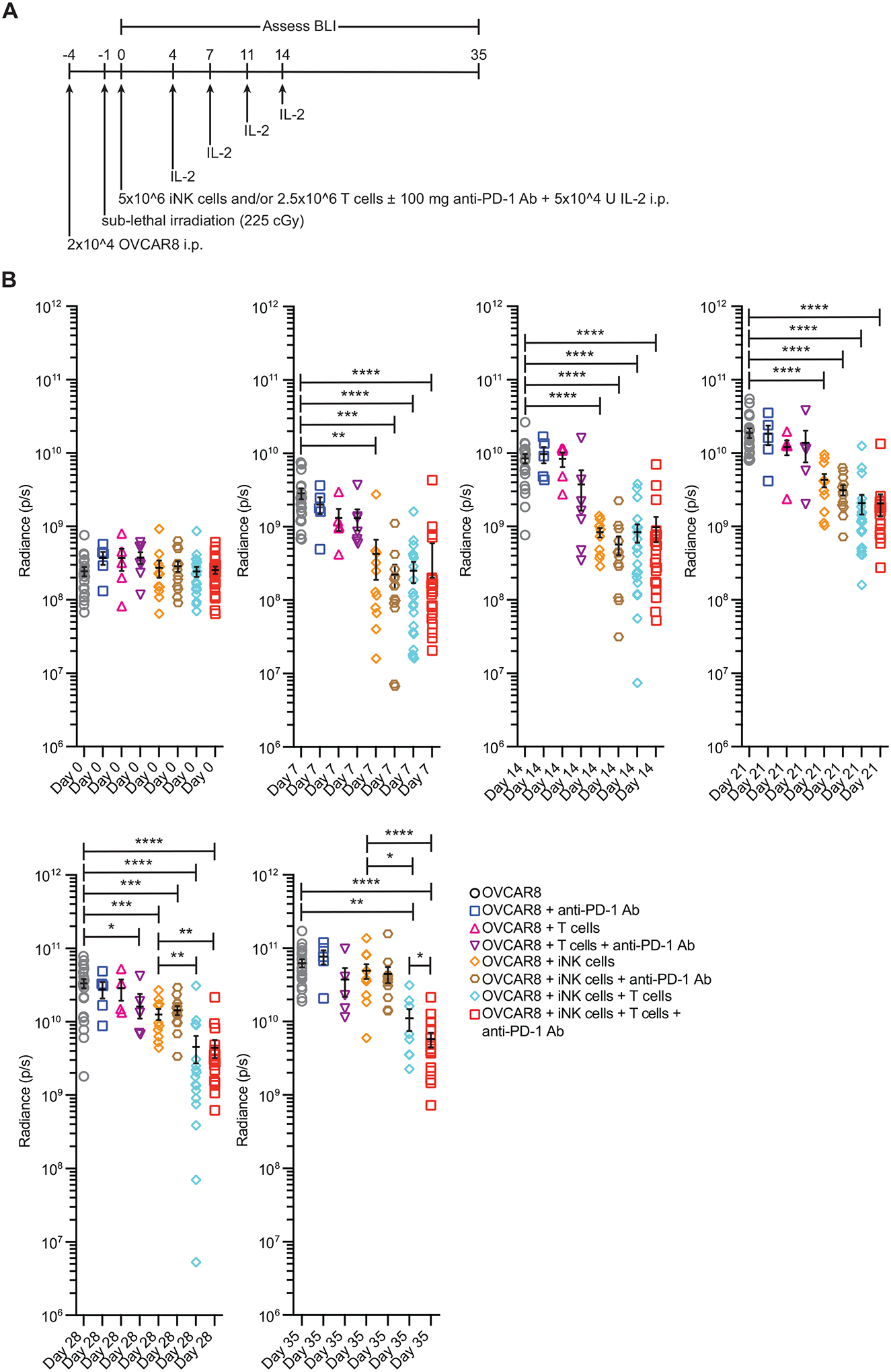
(A) Schematic of the experimental design to test the effectiveness of iNK cells, CD3- and CD28-activated CD3+ T cells, and anti-PD-1 antibody treatment in vivo. (B) Cumulative radiance for the OVCAR8 group and each treatment group at the indicated time points. Data are from 3 independent experiments. Statistical significance was determined using unpaired t tests corrected for multiple comparisons (n = 4–20). * p ≤ 0.5, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001
Discussion
Allogeneic NK cells represent an attractive and complementary alternative to T cell immunotherapies because of their ability to target malignant cells in the body without the need for extensive prior sensitization, their unique ability to prime the adaptive immune system, low risk for GvHD, and lower risk of cytokine release syndrome (CRS). Compared to T cells, NK cells are attractive candidates for allogeneic off-the-shelf immunotherapy because HLA matching is not a requisite for target cell killing. Accumulating data suggests that NK cell immunotherapy may provide a complementary approach to inhibitory receptor checkpoint blockade (41). For NK cell immunotherapy to advance beyond its current state, new approaches are needed to reliably obtain large numbers of highly functional cells that can be banked (cryopreserved), distributed to multiple centers, and thawed at the bedside for “off-the-shelf” use.
Data presented here showed that large numbers of iPSC-derived NK cells could be consistently produced, with up to 1 × 106-fold expansion from initial seeding of iPSCs. iNK cells were phenotypically and transcriptionally similar to peripheral blood and divergent from all other cell types analyzed. iNK cells secreted high amounts of cytokines and chemokines when stimulated and exhibited high cytotoxic function against diverse hematologic and solid tumor cell lines in cellular cytotoxicity assays. The cell lines tested included lung carcinoma cells, hepatocyte carcinoma cells, ovarian adenocarcinoma cells, melanoma cells, and myeloid leukemia cells. Currently, antibody-mediated checkpoint blockade has FDA approval for the treatment of metastatic non-small cell lung cancer (NCLC), advanced hepatocellular carcinoma (HCC), and melanoma. Furthermore, in a xenograft model of ovarian cancer, iNK cells controlled tumor growth and extended median survival rates. These data indicated that iNK cells are highly effective at targeting and eliminating cancer cells, which is reflective of their propensity to contain high amounts of both granzyme and perforin.
The density of NK cells in tumor tissue correlates positively with prognosis. This was first described in colorectal cancer (42) and has subsequently been reported for many other types of cancers, including hepatocellular carcinoma, lung adenocarcinoma, gastric carcinoma, renal cancer, and squamous cell cancers of the vulva and lung (43–48). Although direct lysis of tumor cells by NK cells likely contributes to reduced tumor burden and better outcomes, NK cell densities within solid tumor tissue are generally modest (below 100 cells per square millimeter). Thus, a substantial amount of the beneficial effect of tumor-infiltrating NK cells could be mediated through modulation of the tumor microenvironment and promoting T cell recruitment and activation. Early studies using mixed lymphocyte cultures showed that NK cells were required for the differentiation and activation of CD8+ T cells with cytotoxic function. Direct contact between NK cells and T cells was required for this effect, because antibody blockade of NK cells reduced cytotoxic T lymphocyte induction in mixed lymphocyte cultures (49). This phenomenon was corroborated in mouse experiments in which in vivo depletion of NK cells prior to immunization with B16 melanoma cells prevented CD8+ T cells from differentiating into tumor-specific cytotoxic T lymphocytes after in vitro re-stimulation and abrogated protective immunity against re-challenge with melanoma cells (50). Similarly, NK cells play an important role in the polarization of CD4+ to cells to a pro-inflammatory Th1 (T helper cell type 1) state (51). IFN-γ production by tumor-infiltrating NK cells also increases MHC class I abundance at the surface of tumor cells and boosts tumor-specific cytotoxic T lymphocyte activity (52).
The results of our current study showing a critical role for NK cells in promoting T cell antitumor responses is supported by previous clinical observations and experimental models. In this study, we showed that iNK cells have an inherent ability to target HLA-low or -null tumor cells, as indicated by our results with SKOV-3 targets, and recruit T cells in vitro and in vivo to support T cell homing and trafficking. Perhaps most importantly, our in vitro co-culture and in vivo xenogeneic adoptive transfer experiments demonstrated that iNK cells recruit and activate T cells making them responsive to PD-1 blockade for the enhanced inflammatory cytokine production and tumor elimination. Collectively, our work extends previous findings and highlights the unique ability of iNK cells to potentially overcome checkpoint blockade resistance.
One of the major limitations of NK cell therapy is that, historically, only one-to-two cell doses could be harvested from a single related donor for use in adoptive transfer. NK cells from a donor apheresis are difficult to export, and cell processing is very costly. These challenges have limited the ability to definitively test NK cells as an effective anticancer therapy. Several groups are now developing alternative approaches to sourcing therapeutic doses of NK cells for off-the-shelf use. These approaches include expansion of peripheral blood NK cells with engineered feeder lines (53), differentiation and expansion of NK cells from cord blood progenitors (54, 55) and therapeutic use of the NK-92 cell line (56, 57). The manufacturing system that we have developed to generate iNK cells should enable efficient cell distribution to multiple clinical sites and multi-dosing to treat patients with advanced cancer. We have completed all translational steps for initiation of a phase I first-in-human, first-in-class clinical trial for solid tumor and lymphoma patients who have failed standard-of-care checkpoint blockade. Manufacturing scale-up has been successful, and prospectively established lot release criteria have been met. The IND has been approved by the FDA, and the trial is currently accruing patients (NCT03841110). Advanced cancer patients will be treated with three weekly doses of iNK cells following outpatient lymphodepletion therapy and will receive a dose of checkpoint antibody blockade with their iNK cell administration.
Although the interaction between iNK and allogeneic T cells is definitively shown, a limitation of this study is that we did not determine which chemokines produced by iNK cells were responsible for driving T cell recruitment. We performed transwell experiments with cell-conditioned media that included conditions with a blocking antibody against CCL3 and CCL4 but did not observe a statistically significant effect on T cell transmigration. This is not surprising given the complex composition of chemokines and other factors found in supernatants. A more comprehensive analysis of the iNK cell chemokine secretion profile and manipulation of chemokine production by iNK cells will be needed to further address this important question.
Our data supports the notion that iNK cells maintain robust antitumor function after cryopreservation have the ability to target cancer cells that have evaded T cell recognition through HLA downregulation, and can prime T cells that may otherwise remain inactive. Through these mechanisms, iNK cells can convert a “cold” tumor microenvironment into a “hot” microenvironment by increasing T cell infiltration and restoring responsiveness to checkpoint blockade therapy. Continued scale-up of the iNK cell manufacturing process will enable banking of “livable drugs” for on-demand immunotherapy with multiple cycles or as maintenance therapy, similar to strategies used for all standard chemotherapy regimens. The work presented here also represents a step towards future off-the-shelf strategies that include gene modification of iNK cells to enhance persistence and subvert allo-rejection, as well as methods to generate antigen specificity with introduction of rationally designed versions of CD16 or NK cell-tailored chimeric antigen receptors (58, 59). We are entering into a new and exciting era of cell therapy with the potential to change standards of care.
Materials and Methods
Study design
The objectives of this study were to (i) develop a manufacturing system for generating large numbers of iPSC-derived NK cells for immunotherapy and (ii) to characterize these cells with respect to in vitro and in vivo antitumor function and ability to compliment checkpoint receptor blockade. Toward the first objective, we used our established methods for generating high-quality iPSCs for differentiation into CD34+ hematopoietic progenitor cells (35, 36, 60) and differentiated these cells along the NK cell lineage using stroma cells and media preparations that have been previously published (61). This system was developed by our research group and has been scaled up to generate clinical-grade cells in the GMP facility at the University of Minnesota. Toward the second objective, we determined NK cell receptor expression levels on iNK cells by flow cytometry and analyzed the transcriptome of these cells compared to other immune and non-immune populations by microarray. The antitumor efficacy of iNK cells was studied extensively with flow cytometry-based function assays and live imaging of cytotoxicity using IncuCyte Live Cell Analysis Systems (Essen BioScience). Xenogeneic adoptive transfer experiments were performed to measure the ability of iNK cells to promote T cell homing and the ability of iNK cells to exert antitumor function alone and in combination with T cells and inhibitory checkpoint receptor blockade in vivo. Studies were planned with the minimum number of animals per treatment group to reproducibly observe statistically significant differences (n = 5 to 8 mice per arm per experiment). All murine experiments were replicated at least twice using iNK cells from different cultures. Tumor engraftment was defined by baseline BLI before iNK cell adoptive transfer, and mice were randomized into treatment groups. No data were excluded at any point after adoptive transfer. Researchers imaging and collecting data from xenogeneic experiments were unaware of what each treatment group represented, but data were not analyzed in a blinded manner.
iNK cell culture and phenotyping
Human iPSC culture and differentiation to iCD34+ cells were performed as previously described (35, 36, 60). At the beginning of the iNK cell differentiation culture, iCD34 cells were plated on stroma cells in B0 media supplemented with cytokines that support NK cell differentiation from hematopoietic progenitors (61). After 21 days of culture, iNK cells were harvested and co-cultured with irradiated K562 cells transduced with membrane-bound IL-21 and 4–1BBL constructs (62) in supplemented B0 media for 2 – 3 weeks. K562 cells were propagated in RPMI 1640 media (Corning) containing 10% fetal bovine serum (FBS) (Hyclone). The following fluorescently-conjugated antibodies were used for phenotypic analysis of iNK cells: anti-KIR3DL1 (DX9), anti-KIR2DL1 (HP-MA4), anti-KIR3DL2/3 (DX27), anti-NKG2D (D11), anti-CD16 (3G8), anti-NKp44 (p44–8), anti-NKp46 (9E2), anti-NKp80 (5D12), anti-perforin (B-D48), anti-granzyme b (QA16A02), anti-2B4 (C1.7), anti-LFA-1 (HI111), anti-CD69 (FN50), anti-CD62L (DREG-56), anti-PD-1 (EH12.2H7), anti-LAG-3 (11C3C65), anti-TIGIT (A15153G) (all from Biolegend), anti-NKG2A (Z199; Beckman Coulter), anti-NKp30 (p30–15; BD Biosciences), and anti-NKG2C (134591; R&D Systems).
Global gene expression analysis
Total RNA isolation was conducted using Arcturus PicoPure RNA Isolation Kit (Applied Biosystems) and then diluted to approximately 5.0 ng/μL. RNA integrity was assessed for all RNA samples on a Bioanalyzer 2100 using RNA Pico Chips (Agilent Technologies). Only RNA samples with a RIN score of 7 or higher were used. Genome-wide expression analysis on 10 ng input of total RNA was performed following standard Affymetrix protocols. In brief, total RNA was processed using the Affymetrix GeneChip WT Pico Reagent Kit for target preparation according to the manufacturer’s recommended protocol. Fragmented and labeled cRNA was hybridized for 17 hours at 45°C to Affymetrix GeneChip HTA 2.0 Arrays (Affymetrix). Arrays were washed and stained on a FS450 Fluidic station (Affymetrix) and scanned on a Gene Chip 7G Scanner (Affymetrix) using the standard protocol. Probe intensities were normalized using a signal space transformation robust multi-array analysis method (SST-RMA, Transcriptome Analysis Console 4.0.1, Thermo Fisher Scientific). Differential gene expression analysis was performed using PCA, hierarchical clustering of differentially expressed genes, and z-score gene expression heatmap with Spotfire 7.7 (Perkin-Elmer).
Activating receptor blockade assays
iNK cells were incubated for 30 minutes at 37°C with the following monoclonal antibodies: anti-NKG2D (1D11) at 20 μg/ml, anti-NTB-A (NT7) at 5 μg/ml, anti-NKp30 (P30–15) at 10 μg/ml, anti-LFA-1 (HI111) at 10 μg/ml (all from Biolegend), and anti-DNAM-1 at 10 μg/ml (102511) (R&D Systems) alone or in various combinations. iNK cells were then co-cultured with OVCAR8 cells for 4 hours in B0 media and analyzed by flow cytometry for intracellular IFN-γ production using a fluorescently conjugated anti-IFN-γ (B27) (Biolegend) antibody.
Live kinetic analysis of tumor cell killing
SKOV-3 cells were transduced with red fluorescent protein using the NucLight Red lentiviral reagent (NLR; Essen BioScience). For kinetic analysis of tumor cell killing, each NLR-transduced cell line was individually plated at a concentration of 4 × 103 cells per well in 96-well flat bottom plates. The following day, NK cells were added at E:T ratios ranging from 0.5:1 to 10:1. All co-cultures were performed in B0 media. The number of viable target cells was monitored by hourly fluorescence imaging over 66 hours using an IncuCyte Live Cell Analysis System (Essen BioScience). Live cell numbers were quantified by IncuCyte S3 software (Essen BioScience) and normalized to the number of live cells remaining in the target cell-only control group.
3D tumor spheroid modeling
To generate tumor microspheroids, 1 × 104 SKOV-3 cells transduced to stably express NucLight Red fluorescent protein (Essen Bioscience), were cultured in clear, round-bottom 96-well ultra-low attachment plates (Corning) in DMEM media containing 2.5% FBS and 2.5% Matrigel. After centrifugation for 10 min at 1,600 × g, plates were placed in the IncuCyte S3 for monitoring. After 3 days of culture, spheroids were used as targets for cytotoxicity assays. Effector cells were pre-labeled with IncuCyte CytoLight Rapid Green Reagent (Essen BioScience), added to the plates with spheroids in B0 media in the absence or presence of 10 μg/ml anti-PD-1 antibody (pembrolizimab, Invivogen), and monitored at 30 – 60-minute intervals. NK and T cells were added at defined E:T ratios based on the number of target cells present at the time of effector cell addition. In the mixed iNK and T cell micro-spheroid assays, only T cells were pre-labeled, while iNK cells remained unstained. Effector cell infiltration into tumor spheroids was monitored for 24 hours and analyzed with the EssenBio spheroid software module Green MFI algorithm. Following imaging of microspheroids and effector cell co-cultures, spheroid plates were centrifuged, and the remaining cells were collected. Media was collected from the plates, spheroids were disseminated by TrypLE Express solution (Gibco), and the number of viable Nuclear Red target cells was quantified by Flow Cytometry. Spheroid culture supernatants were assessed for pro-inflammatory cytokines and chemokines using a Meso Scale Discovery multiplex electrochemiluminescent assay (Meso-Scale Discovery, MSD) according to the manufacturer’s instructions.
Transwell chemotaxis assays
Magnetically purified CD3+ T cells were activated using anti-CD3/CD28 antibody-coated Human T-Expander Beads (Gibco) at a 2:1 bead:T cell ratio for 48 hours. Stimulated T cells (5 × 104 in 70 μl serum-free medium) were placed in the upper chambers of 96-well transwell plates (5 μM pore size) (Corning). Cell-free conditioned media (235 μl) collected from iNK cells alone, K562 cells alone, or iNK cells and K562 cells cultured overnight at a 1:1 ratio was placed in the lower chamber of transwell plate. Media alone were used as a background control Plates were incubated overnight at 37°C and 5% CO2, and migrated T cells from the lower chamber were harvested and stained with antibodies against CD45-BV395 (H130; BD Biosciences), CD3 (SK7; BD Biosciences), CD4 (SK3; BD Biosciences), or CD8 (RPA-T8; BD Biosciences). The numbers of migrated T cells were quantified by flow cytometry using counting beads (ACFP-70–5, Spherotech Inc.). For blocking experiments, 10 μg/ml anti-CCL3/CCL4 (MAB270; R&D Systems) was added.
In vivo T cell migration
All in vivo studies were conducted in compliance with the Institutional Animal Care and Use Committee (IACUC)-approved protocols at the University of Minnesota and Fate Therapeutics. K562 cells (5 × 106) were injected into the peritoneal cavities of NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. Four days after tumor transplant, 5 × 106 CD3+ T cells were administered i.v. by retro-orbital injection. Prior to injection, total CD3+ T cells were activated for 48 hours with anti-CD3/CD28 antibody beads (Human T-Expander Beads, Gibco) and then cultured in 100U/mL IL-2 for 4 additional days after bead removal. The following fluorescently-conjugated antibodies were used for phenotypic analysis of T cells: anti-CD4 (OKT4), anti-CD8 (SK1), anti-CCR7 (G043H7), anti-CD45RA (HI100), anti-CD127 (A019D5), anti-CD38 (HIT2), anti-CD27 (QA17A18), anti-CD45RO (UCHL1) (all from Biolegend), and anti-CD3 (UCHT1) (Thermo Fisher Scientific). iNK cells (1 × 107) and IL-2 (5 × 104 U) were injected directly into the peritoneal cavity on the same day. Five days after iNK and T cell injections, mice were euthanized, and peritoneal lavage was harvested. Cells were washed and stained with anti-human CD3 (S7, BD Biosciences), anti-human CD45 (H130, BD Biosciences), and anti-mouse CD45 (30-F11, Biolegend) antibodies and analyzed by flow cytometry. Human T cells in the peripheral blood and peritoneal cavity were identified as hCD3+hCD45+mCD45−.
In vivo tumor xenograft models
For in vivo experiments testing iNK cell function against OVCAR8 tumor cells in a single infusion model, 6-to-8-week-old (NSG) mice purchased from Jackson Laboratories were injected i.p. with 2 × 104 luciferase-expressing OVCAR8 tumor cells. Three days later mice were conditioned with 225 cGray radiation, bioluminescent imaging (BLI) was performed and mice were distributed into two groups with similar starting BLI values. The following day, 1 × 107 iNK cells harvested fresh after K562 expansion were injected i.p. into tumor-bearing mice along with 5 × 104 U IL-2, or mice were left intreated. IL-2 was then administered i.p. twice weekly for the next two weeks, and BLI was performed weekly to monitor tumor progression. For experiments testing iNK cells in combination with T cells and checkpoint inhibitory receptor blockade, groups of mice received i.p. injections of 5 × 106 iNK cells harvested fresh after K562 expansion, 2.5 × 106 CD3+ T cells pre-activated by the same method used for T cell recruitment experiments, with or without 100 mg pembrolizumab (Merck) per mouse along with the first injection of IL-2.
For in vivo experiments testing iNK cell function against MA-148 tumor cells in a multi-infusion model, 6-to-8-week-old NSG mice were injected i.p. with 2 × 104 luciferase-expressing MA-148 cells. Three days later mice were conditioned with 225 cGray radiation, BLI was performed, and mice were distributed into two groups with similar starting BLI values. The following day, 1 × 107 iNK cells were recovered from cryopreserved stock and immediately injected i.p into tumor-bearing mice along with 5 × 104 U IL-2, or mice were left untreated. iNK injections were repeated 4 and 7 days later, and BLI was performed weekly to monitor tumor progression.
BLI was conducted using an IVIS Spectrum, and images were analyzed using Live Imaging Software (Perkin Elmer). Survival was graphed as Kaplan-Meier curves, and the log-rank (Mantel-Cox) test was used to determine statistical significance.
Statistical analysis
Statistical analyses were performed using GraphPad Prism. All statistical tests are two-tailed, and log-rank Mantel-Cox tests were used to determine statistical significance for survival in xenogeneic experiments. Multiple unpaired t tests were used for analysis of BLI readouts from experimental groups compared to controls. The two-stage step-up method of Benjamini, Krieger and Yekutieli was used for determination of False Discovery Rates in the analysis of microarray data. Original data are provided in data file S1.
Supplementary Material
Figure S1. iNK cells acquire KIR expression after adoptive transfer into NSG mice.
Figure S2. Partial inhibition of NK cell activation by blockade of NKG2D in combination with NKp30 or DNAM-1.
Figure S3. iNK cells and expanded primary NK cells exhibit comparable tumor cell killing post-thaw.
Figure S4. CD3/CD28-stimulated T cells have a phenotype consistent with activation and have high PD-1 expression.
Figure S5. Neutralization of CCL3 and CCL4 does not impact T cell migration to iNK cell conditioned medium.
Data file S1. Original data.
Acknowledgments:
We would like to acknowledge the flow cytometry core at the University of Minnesota for excellent technical assistance. Editorial services were provided by Nancy R. Gough (BioSerendipity, LLC, Elkridge, MD).
Funding:
This work was supported by NIH R00 HL123638 (F.C.), NIH P01 CA111412 (J.S.M.), NIH P01 CA65493 (J.S.M.), and research funds provided by Fate Therapeutics.
Competing interests:
F.C., D.S.K. and J.S.M. are paid consultants for Fate Therapeutics and they receive research funds from this relationship. J.S.M. serves on the Scientific Advisory Board of OnkImmune, Nektar, Magenta and is a paid consultant consult for GT BioPharma and Vycellix (all unrelated to the content of this manuscript). He receives research funds from these relationships. B.R.B is a paid consultant/advisor for Regeneron Pharmaceuticals, Inc, Incyte Corp., Obsidian Therapeutics, Bristol-Myers Squibb and Bluerock. He receives research support from Tmunity, Bluerock, Kadmon, and Rheos and is co-founder of Tmunity. None of these relationships conflict with the content of the published research. He also receives research support from Fate Therapeutics for research unrelated to the content of this report. R.B., S.G., S.M., R.A., R.C., L.S., P.R., M.G., M.R., B.R., D.L.R., T.T.L., and B.V. are employees of Fate Therapeutics. Fate Therapeutics owns patents (METHODS AND COMPOSITIONS FOR INDUCING HEMATOPOIETIC CELL DIFFERENTIATION; Patent No. 10,626,372) covering the iPSC derived NK cells reported here. All other authors declare that they have no competing interests.
Data and materials availability:
Microarray data are available in the Gene Expression Omnibus curated by the National Center for Biotechnology Information (ncbi.nlm.nih.gov/geo) under the accession number GSE157327. All other data associated with this study are present in the paper or the Supplementary Materials.
References and Notes
- 1.Pauken KE, Wherry EJ, Overcoming T cell exhaustion in infection and cancer. Trends. Immunol 36, 265–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mellman I, Coukos G, Dranoff G, Cancer immunotherapy comes of age. Nature 480, 480–490 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheitinger A, Greenberg PD, Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends. Immunol 35, 51–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim PS, Ahmed R, Features of responding T cells in cancer and chronic infection. Curr. Opin. Immunol 22, 223–230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wherry EJ, T cell exhaustion. Nat. Immunol 12, 492–499 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmad R, Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Hirano F, Kaneko K, Tamura H, Dong H, Want S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, Tamada K, Chen L, Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 65, 1089–1096 (2005). [PubMed] [Google Scholar]
- 8.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N, Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy with PD-L1 blockade. Proc. Natl. Acad. Sci. U. S. A 99, 12293–12297 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Ki XN, Kang SP, Ribas A, Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med 369, 134–144 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM, Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med 366, 2455–2465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, Smylie M, Dummer R, Hill A, Hogg D, Haanen J, Carlino MS, Bechter O, Maio M, Marquez-Rodas I, Guidoboni M, McArthur G, Lebbé C, Ascierto PA, Long GV, Cebon J, Sosman J, Postow MA, Callahan MD, Walker D, Rollin L, Bhor R, Hodi FS, Larkin J, Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med 377, 1345–1356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, Shen X, Boyd Z, Hegde PS, Chen DS, Vogelzang NJ, MPD3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515, 558–562 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, Zhu L, Grosso JF, Kim SY, Timmerman JM, Shipp MA, Armand P, PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl J. Med 372, 311–319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gubin MM, Zhang X, Shuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, Lam SS, Korman AJ, Allison JP, Freeman GJ, Sharpe AH, Pearce EL, Schumacher TN, Aebersold R, Rammensee HG, Melief CJ, Mardis ER, Gillanders WE, Artyomov MN, Schreiber RD, Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wont P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA, Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Bikbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, Mitter R, Akarca AU, Linares J, Marafioti T, Henry JY, Van Allen EM, Miao D, Schilling B, Schadendorf D, Garraway LA, Makarov V, Rizvi NA, Snyder A, Hellmann MD, Merghoub T, Wolchok JD, Shukia SA, Wu CJ, Peggs KS, Chan TA, Hadrup SR, Quezada SA, Swanton C, Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, Frederick DT, Hazar-Rethinam M, Nadres BA, Van Seventer EE, Shukla SA, Yizhak K, Ray JP, Rosenbrock D, Livitz D, Adalsteinsson V, Getz G, Duncan LM, Li B, Corcoran RB, Lawrence DP, Stemmer-Rachamimov A, Boland GM, Landau DA, Flaherty KT, Sullivan RJ, Hacohen N, Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun 8, 1136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, Rost S, Leabman M, Xiao Y, Mokatrin A, Koeppen H, Hegde PS, Mellman I, Chen DS, Hodi FS, Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 561–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, Adamow M, Kuk D, Panageas KS, Carrera C, Wong P, Quagliarello F, Wubbenhorst B, D’Andrea K, Pauken KE, Herati RS, Staupe RP, Schenkel JM, McGettigan S, Kothari S, George SM, Vonderheide RJ, Amaravadi RK, Karakousis GC, Schuchter LM, Xu X, Nathanson KL, Wolchok JD, Gangadhar TC, Wherry EJ, T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lanitis E, Dangaj D, Irving M, Coukos G, Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol 28, xxi18–xii32 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Zappasodi R, Merghoub T, Wolchok JD, Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 33, 581–598 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smyth MJ, Hayakawa Y, Takeda K, Yagita H, New aspects of natural-killer-cell surveillance and therapy of cancer. Nat. Rev. Cancer 2, 850–861 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Lanier LL, NK cell receptors. Annu. Rev. Immunol 16, 359–393 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, Nelson AE, Loo K, Kumar R, Rosenblum MD, Alvarado MD, Wolf DM, Bogunovic D, Bhardwaj N, Daud AI, Ha PK, Ryan WR, Pollack JL, Samad B, Asthana S, Chan V, Krummel MF, A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med 24, 1178–1191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandal R, Senbabaoglu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N, Lee KW, Ganly I, Hakimi AA, Chan TA, Morris LG, The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 1, e89829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Böttcher JP, Bonavita E, Chakravarty P, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, Reis e Sousa C, NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 172, 1022–1037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunn GP, Old LJ, Schreiber RD, The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21, 137–148 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Masuda K, Hiraki A, Fujii N, Watanabe T, Tanaka M, Matsue K, Ogama Y, Ouchida M, Shimizu K, Ikeda K, Tanimoto M, Loss or down-regulation of HLA class I expression at the allelic level in freshly isolated leukemic blasts. Cancer Sci 98, 102–108 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christopher MJ, Petti AA, Rettig MP, Miller CA, Chendamarai E, Duncavage EJ, Klco JM, Helton NM, O’Laughlin M, Fronick CC, Fulton RS, Wilson RK, Wartman LD, Welch JS, Heath SE, Baty JD, Payton JE, Graubert TA, Link DC, Walter MJ, Westervelt P, Ley TJ, DiPersio JF, Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N. Engl. J. Med 379, 2330–2341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kärre K, Ljunggren HG, Piontek G, Kiessling R, Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319, 675–678 (1986). [DOI] [PubMed] [Google Scholar]
- 31.Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, Orchard PJ, Blazar BR, Wagner JE, Slungaard A, Weisdorf DJ, Okazaki IJ, McGlave PB, Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 105, 3051–3057 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lewis D, Hippen K, McGlave P, Weisdorf DJ, Blazar BR, Miller JS, Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood 123, 3855–3863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, Rondon G, Chen J, Soebbing D, Kaur I, Gulbis A, Ahmed S, Rezvani K, Shpall EJ, Lee DA, Champlin RE, Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood 130, 1857–1868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, Leong JW, Abdel-Latif S, Schneider SE, Willey S, Neal CC, Yu L, Oh ST, Lee YS, Mulder A, Cooper MA, Fehniger TA, Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med 8, 357ra123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valamehr B, Abujarour R, Robinson M, Le T, Robbins D, Shoemaker D, Flynn P, A novel platform to enable the high-throughput derivation and characterization of feeder-free human iPSCs. Sci. Rep 2, 213 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valamehr B, Robinson M, Abujarour R, Rezner B, Vranceanu F, Le T, Medcalf A, Lee TT, Fitch M, Robbins D, Flynn P, Platform for induction and maintenance of transgene-free hiPSCs resembling ground state pluripotent stem cells. Stem Cell Reports 2, 366–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaleco AC, Neves H, Hooijberg E, Gameiro P, Clode N, Haury M, Henrique D, Parreira L, Differential effects of Notch ligands Delta-1 and Jagged-1 in human lymphoid differentiation. J. Exp. Med 194, 991–1002 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cany J, van der Waart AB, Spanholtz J, Tordoir M, Jansen JH, van der Voot R, Schaap NM, Dolstra H, Combined IL-15 and IL-12 drives the generation of CD34+-derived natural killer cells with superior maturation and alloreactivity potential following adoptive transfer. Oncoimmunology 4, e1017701 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hermanson DL, Bendzick L, Pribyl L, McCullar V, Vogel RI, Miller JS, Geller MA, Kaufman DS, Induced Pluripotent Stem Cell-Derived Natural Killer Cells for Treatment of Ovarian Cancer. Stem Cells 34, 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qu QX, Xie F, Huang Q, Zhang XG, Membranous and Cytoplasmic Expression of PD-L1 in Ovarian Cancer Cells. Cell. Physiol. Biochem 43, 1893–1906 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Souza-Fonseca-Guimaraes F, Cursons J, Huntington ND, The Emergence of Natural Killer Cells as a Major Target in Cancer Immunotherapy. Trends. Immunol 40, 142–158 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, Martos JA, Moreno M, The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 79, 2320–2328 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Taketomi A, Shimada M, Shirabe K, Kajiyama K, Gion T, Sugimachi K, Natural killer cell activity in patients with hepatocellular carcinoma: a new prognostic indicator after hepatectomy. Cancer 83, 58–63 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Takanami I, Takeuchi K, Giga M, The prognostic value of natural killer cell infiltration in resected pulmonary adenocarcinoma. J. Thorac. Cardiovasc. Surg 121, 1028–1063 (2001). [DOI] [PubMed] [Google Scholar]
- 45.Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, Aridome K, Hokita S, Aikou T, Prognostic value of intratumoral natural killer cells in gastric cancer. Cancer 88, 577–583 (2000). [PubMed] [Google Scholar]
- 46.Cózar JM, Canton J, Tallada M, Concha A, Cabrera T, Garrido F, Ruiz-Cabello Osuna F, Analysis of NK cells and chemokine receptors in tumor infiltrating CD4 lymphocytes in human renal carcinomas. Cancer Immunol. Immunother 54, 858–866 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sznurkowski JJ, Zawrocki A, Biernat W, Subtypes of cytotoxic lymphocytes and natural killer cells infiltrating cancer nests correlate with prognosis in patients with vulvar squamous cell carcinoma. Cancer Immunol. Immunother 63, 297–303 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villegas FR, Coca S, Villarrubia VG, Jiménez R, Chillón MJ, Jareño J, Zuil M, Callol L, Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer 35, 23–28 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Kos FJ, Engleman EG, Requirement for natural killer cells in the induction of cytotoxic T cells. J. Immunol 155, 578–584 (1995). [PubMed] [Google Scholar]
- 50.Kurosawa S, Harada M, Matsuzaki G, Shinomiya Y, Terao H, Kobayashi N, Nomoto K, Early-appearing tumor-infiltrating natural killer cells play a crucial role in the generation of anti-tumor T lymphocytes. Immunology 85, 338–346 (1995). [PMC free article] [PubMed] [Google Scholar]
- 51.Manetti R, Parronichi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri G, Romagnani S, Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J. Exp. Med 177, 1199–1204 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goding SR, Yu S, Lotze MT, Basse PH, Adoptive transfer of natural killer cells promotes the anti-tumor efficacy of T cells. Clin Immunol 177, 76–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, Rondon G, Chen J, Soebbing D, Kaur I, Gulbis A, Ahmed S, Rezvani K, Shpall EJ, Lee DA, Champlin RE, Phase 1 clinical trial using mbIL-21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood 130, 1857–1868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, Maas F, Leenders M, Bohme F, Kok N, Trilsbeek C, Paardekooper J, van der Waart AB, Westerweel PE, Snijders TJF, Cornelissen J, Bos G, Pruijt HFM, de Graaf AO, van der Reijden BA, Jansen JH, van der Meer A, Huls G, Cany J, Preijers F, Blijlevens NMA, Schaap NM, Successful Transfer of Umbilical Cord Blood CD34(+) Hematopoietic Stem and Progenitor-derived NK Cells in Older Acute Myeloid Leukemia Patients. Clin. Cancer Res 23, 4107–4118 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, Nandivada V, Kaur I, Nunez Cortes A, Cao K, Daher M, Hosing C, Cohen EN, Kebriaei P, Mehta R, Neelapu S, Nieto Y, Wang M, Wierda W, Keating M, Champlin R, Shpall EJ, Rezvani K, Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med 382, 545–553 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gong JH, Maki G, Klingemann HG, Characteristics of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 8, 652–658 (1994). [PubMed] [Google Scholar]
- 57.Uherek C, Tonn T, Uherek B, Becker S, Schnierle B, Klingemann HG, Wells W, Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 100, 1265–1273 (2002). [PubMed] [Google Scholar]
- 58.Jing Y, Ni Z, Wu J, Higgins L, Markowski TW, Kaufman DS, Walcheck B, Identification of an ADAM17 cleavage region in human CD16 (FcγRIII) and the engineering of a non-cleavable version of the receptor in NK cells. PLoS One 10, e0121788 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y, Hermanson DL, Moriarity BS, Kaufman DS, Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 23, 181–192 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsutsui H, Valamehr B, Hindoyan A, Qiao R, Ding X, Guo S, Witte ON, Liu X, Ho CM, Wu H, An optimized small molecule inhibitor cocktail supports long-term maintenance of human embryonic stem cells. Nat. Commun 2, 167 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cichocki F, Miller JS, In vitro development of human Killer-Immunoglobulin Receptor-positive NK cells. Methods Mol. Biol 612, 15–26 (2010). [DOI] [PubMed] [Google Scholar]
- 62.Wang X, Lee DA, Wang Y, Wang L, Yao Y, Lin Z, Cheng J, Zhu S, Membrane-bound interleukin-21 and CD137 ligand induce functional human natural killer cells from peripheral blood mononuclear cells through STAT-3 activation. Clin. Exp. Immunol 172, 104–112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. iNK cells acquire KIR expression after adoptive transfer into NSG mice.
Figure S2. Partial inhibition of NK cell activation by blockade of NKG2D in combination with NKp30 or DNAM-1.
Figure S3. iNK cells and expanded primary NK cells exhibit comparable tumor cell killing post-thaw.
Figure S4. CD3/CD28-stimulated T cells have a phenotype consistent with activation and have high PD-1 expression.
Figure S5. Neutralization of CCL3 and CCL4 does not impact T cell migration to iNK cell conditioned medium.
Data file S1. Original data.
Data Availability Statement
Microarray data are available in the Gene Expression Omnibus curated by the National Center for Biotechnology Information (ncbi.nlm.nih.gov/geo) under the accession number GSE157327. All other data associated with this study are present in the paper or the Supplementary Materials.