

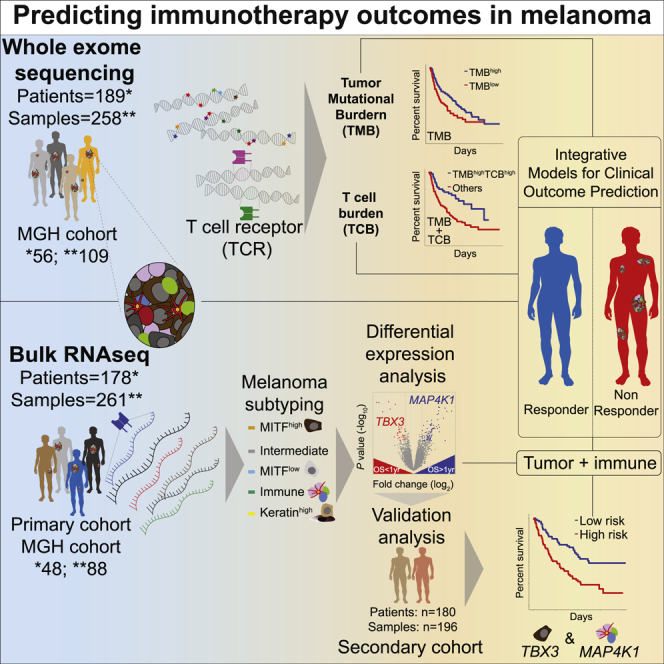
Summary
Immune checkpoint blockade (CPB) improves melanoma outcomes, but many patients still do not respond. Tumor mutational burden (TMB) and tumor-infiltrating T cells are associated with response, and integrative models improve survival prediction. However, integrating immune/tumor-intrinsic features using data from a single assay (DNA/RNA) remains underexplored. Here, we analyze whole-exome and bulk RNA sequencing of tumors from new and published cohorts of 189 and 178 patients with melanoma receiving CPB, respectively. Using DNA, we calculate T cell and B cell burdens (TCB/BCB) from rearranged TCR/Ig sequences and find that patients with TMBhigh and TCBhigh or BCBhigh have improved outcomes compared to other patients. By combining pairs of immune- and tumor-expressed genes, we identify three gene pairs associated with response and survival, which validate in independent cohorts. The top model includes lymphocyte-expressed MAP4K1 and tumor-expressed TBX3. Overall, RNA or DNA-based models combining immune and tumor measures improve predictions of melanoma CPB outcomes.
Keywords: cancer immunotherapy, immune checkpoint blockade, melanoma, cancer genomics, tumor mutational burden, TMB, T cell receptor, melanoma subtype, integrative model
Graphical abstract
Highlights
-
•
T and B cell burden (TCB/BCB) can be calculated from rearranged TCR/Ig DNA sequences
-
•
Combining DNA-based TCB or BCB with TMB measurements improves outcome prediction
-
•
RNA subtypes reflect tumor differentiation or immune abundance and predict survival
-
•
Gene-pair MAP4K1 and TBX3 expression predict outcome and validate in independent data
Freeman et al. perform a meta-analysis of DNA and RNA-sequencing data from melanoma patients treated with checkpoint inhibitors to identify combinations of factors associated with outcome. Using data from either a DNA or RNA assay, they conclude that models incorporating tumor and immune measurements can improve predictions of immunotherapy outcomes.
Introduction
Why only some patients respond to checkpoint blockade therapies is still unclear. For example, patients with microsatellite instability (MSI), which have high indel and mutation burden, have higher response rates than non-MSI cases of the same tumor type, but the predictive value of TMB is not always strong.7, 8, 9, 10 Also, while T cells are crucial for responses, their presence alone does not dictate whether patients will benefit from checkpoint blockade (CPB).4,7,11, 12, 13, 14, 15 Studies of acquired resistance have discovered rare mutations associated with resistance,3,16, 17, 18, 19 but these do not explain the majority of cases.
Many studies have identified mechanisms of response or resistance to CPB. Initially, TMB was identified as a predictor of melanoma CPB response.1,20 A later study demonstrated an association between TMB as a continuous variable and overall survival (OS) for multiple tumor types,9 but melanoma patients with high TMB (above the 20th or 30th percentile) did not have longer OS in this study.9,21 Additionally, others have identified sun exposure or melanoma subtype as factors confounding the association of TMB with CPB response,7,22 but these results were not validated in independent cohorts. Mutations in SERPINB3/4 were associated with CPB outcomes in two cohorts,19 but this finding was not reproduced in a meta-analysis.23 This meta-analysis found that many CPB predictors were not significant when analyzing multiple cohorts, and even fewer were significant in independent data, but their predictor validated in independent cohorts.23 Thus, meta-analysis of large cohorts and validation in independent cohorts are crucial for identifying robust features underlying CPB response.
While most studies have analyzed either malignant3,4,7,9,10,16,24, 25, 26, 27, 28 or microenvironmental11,12,29, 30, 31, 32, 33, 34 features, integrative models have improved predictions of CPB outcomes. For example, integrating TMB with immune expression signatures in multimodal datasets improved stratification of melanoma OS after CPB in multiple studies.35,36 Additionally, TMB from tumor or cfDNA combined with staining for PD-L1 improved prediction of lung cancer response to anti-PD-L137 or combination CTLA-4/PD-1.38 A limitation of these studies is the use of multiple assays that require large samples and multiple nucleic acid isolation techniques.
To address some of these limitations, we analyzed tumor exomes and transcriptomes from patients with melanoma receiving CPB and derived several DNA or RNA-based pre-treatment features predictive of OS and response. First, combining TMB with quantification of T or B cell abundance using only whole-exome sequencing (WES) data identified a subgroup of patients with high immune infiltration and high TMB that are more likely to benefit from CPB. Next, using transcriptomic data, we found the combination of transcription factor TBX3, expressed in poorly differentiated melanomas, with MAP4K1, expressed in lymphocytes and dendritic cells, to be predictive of OS and response in a meta-analysis and an independent secondary meta-analysis cohort. Overall, this study serves as a resource for investigating CPB outcome predictors and improves knowledge of potential mechanisms of response or resistance to immunotherapy.
Results
DNA and RNA meta-analysis in melanoma patients treated with CPB
To identify factors that predict CPB response and OS, we sequenced DNA and RNA from melanoma samples before and after CPB. We performed WES of 109 samples from 56 patients (of which 37 patients had matched pre/post-treatment biopsies) and bulk RNA sequencing (RNA-seq) of 88 samples from 48 patients. We aggregated these data with published WES1,2,3 and bulk RNA-seq1,4,5 (Table S1; Figure S1). In total, we analyzed 258 DNA WES samples from 189 patients (52 with matched pre/post-treatment samples) and 261 bulk RNA-seq samples from 178 patients (68 with matched pre/post-treatment samples; Table S1). Overall, 59 patients had both pre-treatment WES and RNA-seq data and 154 patients had pre-treatment RNA-seq. For MGH patients, we determined response based on a combination of radiographic measurements routinely performed on all patients with clinical evaluations (range 4–12 weeks after start of treatment), and we defined OS from initiation of therapy until death or last follow-up. For the published WES and RNA-seq cohorts, we used OS and their definitions for response (STAR Methods).
Combining TMB with DNA-based measures of immune infiltration improves predictions of CPB outcomes
Our analysis of WES data (n = 189 patients, Figure S1) identified significantly mutated genes, somatic copy number alterations, and mutation signatures (Table S2), similar to previous studies.39 As others observed,1,4,20 we found that patients with TMB above median (TMBhigh) or TMB above 10 mutations/Mb had longer OS after CPB (TMBhigh log-rank p = 0.015, HR 1.56, Figure 1A; Figure S2). However, TMB was not significantly higher in responders than non-responders (Wilcoxon p = 0.13, Figure 1A). Neoantigen burden and clonal TMB highly correlated with TMB (rho = 0.99 and 0.97 respectively) but did not provide predictive power over TMB (Figure S2). Though aneuploidy is associated with poor CPB outcomes in some studies,2,40 we found that tumor ploidy was not associated with OS (log-rank p = 0.35). Also, survival models using the mutation status of single genes did not identify associations passing multiple hypothesis correction (Figure S2), likely due to lack of power.10 We identified somatic mutations in B2M that were present in WES data of tumors biopsied after progression and absent from pretreatment biopsies or cell lines derived from pre-treatment samples3,16 but did not identify novel genes with mutations exclusive to post-treatment tumors (Figures S2 and S3). Consistent with prior work,7 we found that tumor purity below median was associated with OS (log-rank p = 0.00094) but not response (Figure 1B). Since purity and TMB were not correlated (rho = −0.03, Figure 1C), we combined these factors and found that the subgroup with TMBhigh and low tumor purity had longer OS (log-rank p = 0.0037, Figure 1D; Figure S2) but not higher response (Figure 1E). While single-gene analyses did not identify important features, the analysis of tumor purity and TMB suggests that combining tumor and immune features may improve outcome models.
Figure 1.

TMB, tumor purity, and their combination associate with CPB outcomes
(A and B) Kaplan-Meier curve (left) and responder/non-responder box-plots (right) for patients with high (above median) or low (below median) TMB (A) or tumor purity (B). P values in right panels from Wilcoxon tests.
(C) Correlation between TMB and tumor purity with P value for spearman correlation.
(D and E) Kaplan-Meier curve (D) or response (E) for the TMBhigh, low tumor purity subgroup with P value from Fisher's exact test.
Since T and B cell infiltration are associated with response to CPB and are inversely correlated with tumor purity,1,2,4,12,32,33,35,41 we next considered the predictive value of T and B cell infiltration quantified using rearranged T cell receptor (TCR) and immunoglobulin (Ig) sequences, respectively, from RNA. Deep TCR and Ig repertoire sequencing is required for analysis of clonotype diversity, but analysis of TCR and Ig levels in bulk RNA-seq or WES data can quantify T or B cell infiltration. High TCR or Ig read counts in pre-treatment RNA-seq samples (n = 154) (TCRRNA and IgRNA respectively) were associated with OS and response (Figures 2A and 2B; Table S3). Moreover, TCRRNA and IgRNA correlated highly with expression of T or B cell markers (Figures 2C ad 2D). Thus, we created RNA-based metrics of T or B cell burden (TCBRNA or BCBRNA) using the number of rearranged TCR or Ig reads, respectively (STAR Methods), which were consistent across cohorts and correlated with each other (rho = 0.67; Figure S4). Patients with high TCBRNA and BCBRNA had longer OS but not increased response (Figure 2E; Figure S4).
Figure 2.

TCR/Ig rearrangements in DNA and RNA quantify immune infiltration and predict CPB outcome when combined with TMB
(A and B) Kaplan-Meier curve for patients with high/low TCRRNA (A) or IgRNA (B) and TCRRNA (A) or IgRNA (B) for responders and non-responders. P values in right panels from Wilcoxon tests.
(C and D) Correlation between TCRRNA and T cell gene expression (C) or IgRNA and B cell gene expression (D), with P values for spearman correlations.
(E) Kaplan-Meier curve for RNA T cell burden (TCBRNA) high, B cell burden (BCBRNA) high subgroup.
(F and G) Correlation between TCBRNA and TCBDNA (F) or BCBRNA and TCBDNA (G) for patients with DNA and RNA extracted from the same location in the tumor, with P values for spearman correlations.
(H) Fraction of cases with TCR or Ig CDR3 clonotypes shared between RNA and DNA, for patients with DNA and RNA extracted from the same location in the tumor.
(I–M) Kaplan-Meier curve for patients with high/low TCBDNA (I), high/low BCBDNA (J), TCBDNAhigh, BCBDNAhigh subgroup (K), TMBhigh, TCBDNAhigh subgroup (L), and all TMB and TCBDNA subgroups (M).
(N) Response rate for the TMBhigh, TCBDNAhigh subgroup with P value from Fisher's exact test.
To extend the association between TCB/BCB and outcome to a larger cohort, we generated metrics from DNA, TCBDNA and BCBDNA (Table S3). Since we did not perform targeted TCR sequencing but rather used WES data, we first verified that the RNA and DNA-based metrics were correlated using 35 cases with DNA and RNA extracted from the same area (Figures 2F and 2G; Table S3). Also, we detected shared TCR and Ig CDR3 sequences across DNA and RNA (Figure 2H), with increased sharing in samples with higher TCBDNA or BCBDNA. As TCBDNA and BCBDNA levels differed between cohorts, we classified samples as above/below median within each cohort and found that TCBDNA and BCBDNA associated with OS, as did their combination (Figures 2I–2K; Figure S5). Interestingly, a subset of samples had higher BCBRNA than BCBDNA. When we compared BCB and TCB levels for DNA and RNA, we found that TCBDNA was higher than BCBDNA, consistent with CIBERSORTx42; however, BCBRNA was higher than TCBRNA (Figures S5J–S5L). We compared BCBRNA and single-cell RNA (scRNA)-derived B cell signatures43 and found that BCBRNA correlated more strongly with a plasma B cell signature than a naive B cell signature (Figures S5M and S5N). This suggests that BCBRNA is partially driven by the high expression of Igs in plasma B cells, and BCBDNA may reflect B cell frequency more accurately since only one copy of a rearranged Ig is present in DNA per cell. These results show that lymphocyte infiltration can be quantified with rearranged TCR/Ig reads from tumor exomes alone and is associated with OS.
Since TMB did not correlate with TCBDNA (rho = 0.03, Figure S6), we tested a model combining these tumor and immune features and found that patients with TMBhigh and TCBDNAhigh survived longer (p = 3.6 × 10−4, HR 2.28, Figures 2L and 2M) and had a higher response rate (p = 0.028, OR = 2.18, Figure 2N). This combined model was superior to models incorporating TMB or TCBDNA alone (likelihood ratio test [LRT] p = 0.0036 and 0.038, respectively). Similarly, patients with TMBhigh and BCBDNAhigh had longer OS and higher response rates (log-rank p = 1.6 × 10−3, Fisher p = 0.021, Figures S6C and S6D), but a model with TMB, TCBDNA, and BCBDNA did not provide additional predictive value (Figures S6F–S6I). To assess why the TMB and TCB model outperformed the TMB and purity model, we estimated cell-type composition in RNA with CIBERSORTx. The proportion of melanoma cells in RNA correlated with the DNA-based estimate of tumor purity. While non-tumor cells consisted of both immune and stromal cells, immune cells were more abundant than stromal cells (Wilcoxon p = 0.016), and we observed that both proportions negatively correlated with melanoma cell fraction (Figures S7A–S7F). Thus, tumor purity captures immune and stromal components, while TCB/BCB only reflect immune cell abundance. We also analyzed stage III/IV melanomas from The Cancer Genome Atlas (TCGA, Table S3, Figure S8) and found that the TMBhigh, TCBDNAhigh subgroup had increased survival (log-rank p = 0.025, Figures S8D–S8K). Thus, through combining tumor and immune features by quantifying TMB and TCBDNA from WES alone, we were able to identify patients with a higher chance of benefiting from CPB.
Previous studies have demonstrated expansion of T cell clones after CPB11,44 and that immune infiltration is associated with outcome in patients receiving anti-CTLA-4 prior to anti-PD-1 but not in CTLA-4-naive patients.7 Analysis of paired pre/post-treatment samples showed that TCB but not BCB increased after treatment (Table S3; Figure S9). Additionally, increases in TCBRNA but not TCBDNA were specific to CTLA-4 naive patients. Our results are consistent with the association of pre-treatment levels of both T and B cells with outcome but suggest that CPB may induce T cell but not B cell expansion.
RNA-seq analysis identifies melanoma subtypes associated with immunotherapy survival
Expression-based cancer subtypes have been linked with survival both with or without immunotherapy.15,39,45, 46, 47 Using bulk RNA-seq from 469 TCGA melanoma specimens (101 primary and 368 metastatic biopsies39), we identified 5 robust tumor subtypes with Bayesian non-negative matrix factorization (NMF) clustering48 (Table S4; Figure S10). As expected from previous studies,1,4 one subtype had high levels of immune infiltrate (Immune), and a second had high levels of keratin expression (Keratin-high), likely due to keratinocytes. The other three subtypes were associated with the degree of melanocyte differentiation. Two subtypes were classified by expression of MITF (MITF-low and MITF-high) and the third by intermediate melanocyte differentiation (Intermediate) (Figure S10). The MITF-low and Immune subtypes were concordant with TCGA subtypes,39 and the MITF-high, Intermediate and MITF-low subtypes were closely related to differentiation states identified in melanoma cell lines49 (Figure S10). The poorly differentiated MITF-low subtype resembles neural crest stem cells and is associated with resistance to targeted therapies50,51 and immunotherapies.5 TCBRNA and BCBRNA were higher in Immune subtype tumors (Figure S10). In TCGA data, the subtypes were strongly associated with survival for all (log-rank p = 2 × 10−10, Figure S10K) and for stage III/IV patients (p = 2.18 × 10−6, Figure 3A). In RNA-seq data from pre-immunotherapy patients (n = 154), after batch-effects correction between cohorts (Table S5; Figures S11A–S11G), we found that tumor subtypes were associated with post-immunotherapy OS (log-rank p = 0.019, Figures 3B and 3C) but not response (Figure S11H), with the Immune subtype associated with the longest OS (Fisher p = 0.035, HR = 1.73, Figure 3D).
Figure 3.

Melanoma gene-expression markers of survival
(A) Kaplan-Meier curve by expression subtype for TCGA melanoma stage III/IV patients
(B) Heatmap of marker gene expression for pre-immunotherapy (primary cohort n = 154) patients grouped by subtype
(C and D) Kaplan-Meier curve by subtype for primary cohort (C) and for immune subtype patients (D)
(E) Differential expression between patients with OS >1 year (long OS) and patients with OS <1 year (short OS) in the primary cohort using DESeq2.
(F) Expression of differentially expressed genes in melanoma CCLE cell lines and Human Protein Atlas blood cell types
Identification and validation of gene-pair models combining tumor and immune genes to predict CPB outcomes
To pinpoint gene-expression markers of outcome, we identified genes differentially expressed between patients with OS >1 year (long OS) and patients with OS <1 year (short OS), irrespective of subtype (Figure 3E, Table S5, q < 0.05). We identified 83 genes differentially expressed between long and short OS patients (55 overexpressed in long OS patients, 28 overexpressed in short OS patients). Genes associated with long OS included T and B lymphocyte expressed genes (CD3E, CD3G, LTB, SELL, SLAMF6, CD52, CD79A, CXCL13, MAP4K1), and genes associated with short OS included multiple tumor-expressed genes (TBX3, EFNB2, NREP, S100A2, AGER). We also identified 101 genes differentially expressed between responders and non-responders, which overlapped with the 83 genes associated with OS (29/101, p = 8.16 × 10−41, Figure S12). We next analyzed the 55 genes associated with long OS and found that most were expressed in immune cells, including lymphocytes and memory CD8 T cells, which are critical for anti-tumor immunity12 (Figure 3F; Figure S12). In contrast, the 28 genes associated with short OS were highly expressed in melanoma cell lines, with the highest expression in the MITF-low subtype (Figure 3F; Figures S12G and S12H). We found similar patterns for genes differentially expressed between responders and non-responders (Table S5; Figure S13).
Since previous studies combined tumor and immune features to improve the prediction of CPB outcomes35, 36, 37, 38 and the combination of TMB with TCBDNA was associated with outcome, we created models using gene pairs by combining immune and tumor-associated genes. We tested all pairwise combinations of the 83 OS differentially expressed genes as predictors of OS and response (Table S5). Additionally, we tested a metagene-pair model which averaged the normalized expression of the 55 long OS genes as one metagene and the 28 short OS genes as the other metagene. This metagene-pair model was highly predictive of response and OS (Figures S13F and S13G). Finally, we found that models based on pairs of short OS genes were significantly worse than other gene-pair models (Table S5; Figures S13H–S13J).
After testing all pairwise models, we identified 3 gene pairs significantly associated with OS and response (Bonferroni-corrected p < 0.05, Figure 4A; Figures S13 and S14). The three pairs were MAP4K1&TBX3, MAP4K1&AGER and the metagene-pair model. MAP4K1 is expressed in multiple immune cell types including T and B lymphocytes as well as dendritic cells.52,53 In contrast, TBX3, AGER, and the short OS metagene are most highly expressed in the dedifferentiated MITF-low melanoma subtype (Figure S14). Next, we compared the three gene-pair models and TCBRNA to six published models of CPB outcomes: CD274 (PD-L1) expression, GEP, CYT, IMPRES, TIDE, and MHC II.7,35,54, 55, 56, 57 We computed values for each predictor for each patient, and, when we clustered patients, one cluster had high immune infiltration and high values for immune-based models (Figure 4B; Table S5). Additionally, many patients with low immune infiltration and high values for tumor-associated predictors had Intermediate or MITF-low subtype tumors (Figure 4B). We found that the three gene-pair models outperformed the previous models in predictions of response and OS (Figures 4C and 4D; Figures S14I–S14K). We also tested the addition of TMB to the gene-pair models, though there were few cases (n = 59) with both WES and RNA-seq data. We found that TMB did not add to the response models (DeLong’s test p > 0.05) or the metagene-pair model for OS (LRT p = 0.13), but TMB significantly improved the MAP4K1&AGER and MAP4K1&TBX3 models for OS (LRT p = 0.03, 0.02, respectively, Figures S14A–S14D). Studies of larger cohorts with DNA and RNA profiling will be required to evaluate combining TMB with these models. Finally, to assess the robustness of the gene-pair models, we performed a cross-validation analysis and found that increasing the training set size (1) increased the number of gene-pair models discovered, (2) increased the robustness of the long OS metagene, but the short OS metagene was still variable, and (3) the top gene-pair models were repeatedly discovered in training sets but were rarely significant in the held out validation sets, supporting the need for larger datasets (Table S5; Figure S15).
Figure 4.

Development and validation of RNA-based gene-pair models to predict CPB outcomes
(A) Performance of gene-pair models in predictions of OS (Cox model log-rank P value) and response (logistic regression AUC P value) in the primary cohort, with three models with Bonferroni p < 0.05 labeled.
(B) Heatmap of values for published immunotherapy models and top gene pairs.
(C and D) Performance of gene-pair models in comparison to published models in significance (C) and effect size (D) of predictions of response and OS in the primary cohort.
(E) Schematic of independent secondary cohort.
(F) Kaplan-Meier curve of immune subtype patients in the secondary cohort.
(G and H) Performance of gene-pair models in comparison to published models in significance (G) and effect size (H) of predictions of response and OS in the secondary cohort.
(I and J) Forest plot of MAP4K1&TBX3 OS model performance in the primary (I) and secondary cohorts (J). Error bars represent 95% confidence intervals for hazard ratio estimates and starred P values are from Wald tests for each gene.
(K and L) OS of patients stratified to high/low risk using MAP4K1 and TBX3 expression in the primary (K) and secondary cohort (L).
(M) Analysis of melanoma cell lines shows that TBX3 forms a gradient of expression across melanoma differentiation states.
To confirm our findings in independent data, we merged two cohorts of melanoma patients receiving PD-1 or combination CTLA-4/PD-16,7 (n = 180), which we refer to as the “secondary cohort” (Figure 4E). We found that patients with Immune subtype tumors in the secondary cohort had longer OS than other patients (log-rank p = 0.022, Figure 4F; Table S6; Figure S16). When we tested the performance of the three gene-pair models, all three validated in the secondary cohort with Bonferroni p < 0.05 for predictions of OS and response. However, for predictions of response in the secondary cohort, their performance was statistically equivalent to those of previous models (DeLong’s test p > 0.05), with AUC and C-index values between 0.6 and 0.7 (Figures 4G and 4H; Table S6; Figures S17A–S17D).
The top performing gene-pair model in the secondary cohort was the MAP4K1&TBX3 model (Figures 4G and 4H). In Cox models incorporating MAP4K1 and TBX3, both genes were significant in both the primary and the secondary cohorts (Figures 4I and 4J). As expected, patient stratification into risk groups using MAP4K1 and TBX3 expression was also associated with OS in both cohorts (Figures 4K and 4L; Figure S17). Treatment (PD-1 versus combination CTLA-4/PD-1) was associated with OS in the secondary cohort, but in a Cox model incorporating MAP4K1, TBX3, and treatment, all three were significant (Figures S17J and S17K). When we analyzed the models in individual cohorts, their performance was variable (Table S6; Figure S18). Additionally, by analyzing patients grouped by therapy, we observed that the gene-pair models were predictive of OS for patients treated with either CTLA-4 or PD-1, demonstrating that these models are predictive in multiple treatment contexts (Figure S19). Similar to published models, the gene-pairs models were not predictive of OS for patients treated with combination PD-1/CTLA-4. However, in contrast to published models, the gene-pair models were predictive of OS for patients treated with PD-1 and no prior CTLA-4. This treatment context is relevant to current clinical practice as CTLA-4 monotherapy is not used in the first line for melanoma. In summary, the top gene-pair models were able to predict outcomes, and the simplicity of these models points to potential biological connections between gene expression and outcome.
TBX3 is a marker of poorly differentiated melanomas
To better understand the role of TBX3, we analyzed multiple melanoma datasets. First, TBX3 was expressed in most melanoma cell lines49 except for well-differentiated ones with high MITF expression (Figure 4M), in concordance with high TBX3 expression in MITF-low tumors (Figure S14). Second, genes negatively correlated with TBX3 expression in melanoma cell lines58 were enriched for pigmentation gene sets (Table S6; Figure S20A). Third, TBX3 was expressed in melanoma cells but rarely in non-tumor cells based on melanoma scRNA-seq data51,59 and was more highly expressed in melanoma cells expressing neural crest marker NGFR (Figures S20B–S20F), consistent with prior work showing that NGFR+ melanoma cells are resistant to CPB.60 Functional studies have shown that overexpressing TBX3 in melanoma cells enhances tumor formation and invasion in vivo.61 In summary, TBX3 is a tumor-specific gene expressed in poorly differentiated melanomas, and a model combining lymphocyte-expressed MAP4K1 with TBX3 is associated with patient outcomes after CPB. Overall, our results suggest that models combining a metric of immune infiltration with a tumor-derived metric associated with poor melanoma differentiation can predict melanoma immunotherapy outcomes.
Discussion
By extracting biological features from tumor DNA and RNA, we uncovered factors predicting melanoma CPB outcomes: (1) tumor purity and TMB, (2) T/B cell infiltration combined with TMB from DNA alone, (3) expression subtyping of tumors, and (4) expression of MAP4K1 and TBX3 (as well other gene pairs involving an immune-associated and a tumor-associated gene). While some features are correlated (e.g., tumor purity and T/B cell burden), specific combinations of uncorrelated features improved predictions of response and OS.
Our data show that integrative models measuring immune infiltration (MAP4K1) and expression of tumor-associated genes (TBX3) are predictive of patient outcomes. These simple gene-pair models were as or more accurate than complex models and provide insight into biological features. This is consistent with previous studies that found that multivariate models combining multiple data types can outperform single feature models of post-immunotherapy OS (such as TMB, PD-L1 staining, or T cells alone).7,25,35,37,38 We also show that high TMB (providing more neoantigens) combined with high TCB (indicating T cell response) can predict outcome. Our results suggest that including TCR and Ig sequences in targeted sequencing panels, along with genes that allow TMB estimation, may be useful for prediction of outcome using a single DNA assay. Patients with multiple positive prognostic factors may be better served by PD-1/PD-L1 monotherapy, whereas those with negative factors may benefit from more aggressive combinations of therapies, but this would need to be studied in clinical trials.
Previous work has demonstrated that clinical variables and single-cell profiling can significantly improve models of immunotherapy outcomes.7,12 While our models were predictive and validated in independent data, their performance is modest (AUC and C-index of 0.6–0.7). In addition to the features we explored, others have identified immune subtypes and germline factors associated with immune infiltration as associated with CPB outcomes, and additional studies are warranted to validate these findings and test whether these factors lead to distinct resistance mechanisms.62,63 Future studies will require deeper clinical, tumor, and immune characterization of larger cohorts to discover genetic and non-genetic predictors. To achieve this goal, the community will need to generate and share genomic, transcriptomic, and outcome data from patients receiving immunotherapy.
Limitations of the study
Although we developed RNA-based models that predicted outcomes in an independent cohort, these models were trained and tested on data from patients with varied clinical histories who were treated with different therapies. Thus, these models are not optimized for patients receiving a specific treatment. Additionally, since our meta-analysis was based on multiple published cohorts, clinical annotations were limited and heterogeneous, making it impossible to assess whether predictors were independent from clinical risk factors. Therefore, larger homogeneous cohorts are needed to develop more robust predictors that are applicable to patients receiving the current standard of care, and validation in prospective cohorts will be necessary.
A second limitation is the use of bulk RNA-seq which cannot assign gene expression to a given cell type. Bulk RNA-seq has limited resolution to identify rare cell populations. In recent years, scRNA-seq has become standard as it can detect rare cell types and states associated with clinical phenotypes.6,12
Third, while we identified several genes associated with clinical outcomes, whether these genes can affect tumor-intrinsic and extrinsic mechanisms of immune evasion or the tumor microenvironment remains to be determined in future functional studies. Leveraging human data to select candidate genes together with in vivo CRISPR screens will enable testing of thousands of hypotheses in a single experiment and link gene-expression associations with potential causal roles using model systems.
STAR★Methods
Key resources table
| Reagent or resource | Source | Identifier |
|---|---|---|
| Biological samples | ||
| MGH cohort (bulk DNA and RNA) used in this study are detailed in Table S1 | Massachusetts General Hospital and MD Anderson Cancer Center | N/A |
| Critical commercial assays | ||
| QIAGEN AllPrep DNA/RNA Mini Kit | QIAGEN | Cat# 80204 |
| TrueSeq exome kit | Illumina | Cat# 20020615 |
| Deposited data | ||
| Primary cohort- MGH samples (bulk DNA and RNA) data | This paper | dbGAP: phs002683.v1.p1. https://zenodo.org/record/5528497 |
| Primary cohort- Van Allen | Van Allen et al. 20151 | dbGAP: phs000452.v3.p1 |
| Primary cohort- Roh | Roh et al. 20172 | BioProject: PRJNA369259 |
| Primary cohort- Zaretsky | Zaretsky et al. 20163 | BioProject: PRJNA324705 or SRA: SRP076315 |
| Primary cohort- Riaz | Riaz et al. 20174 | BioProject: PRJNA356761 or SRA: SRP094781 |
| Primary cohort- Hugo | Hugo et al. 20165 | GEO: GSE78220 or SRA: SRP070710 |
| Secondary cohort- Gide | Gide et al. 20196 | ENA: PRJEB23709 |
| Secondary cohort- Liu | Liu et al. 20197 | dbGAP: phs000452.v3.p1 |
| Jerby-Arnon scRNA data | Jerby-Arnon et al. 201859 | GEO: GSE115978 |
| https://singlecell.broadinstitute.org/single_cell/study/SCP109/melanoma-immunotherapy-resistance | ||
| Human Protein Atlas Blood RNA-Seq | Uhlen et al., 201964 | https://www.proteinatlas.org/download/rna_blood_cell_sample_tpm_m.tsv.zip |
| CCLE | Barretina et al., 201258 | https://data.broadinstitute.org/ccle/CCLE_RNAseq_rsem_genes_tpm_20180929.txt.gz |
| TCGA Melanoma | Cancer Genome Atlas Network. 201539 | dbGAP: phs000178.v5.p5. https://app.terra.bio/#workspaces/broad-firecloud-tcga/TCGA_SKCM_ControlledAccess_V1-0_DATA |
| Software and algorithms | ||
| CGA WES Characterization pipeline | Birger et al. 201765 | https://app.terra.bio/#workspaces/broad-fc-getzlab-workflows/CGA_WES_Characterization_OpenAccess |
| ContEst | Cibulskis et al. 201166 | http://software.broadinstitute.org/cancer/cga/contest |
| CrossCheckFingerprints | Broad Institute Picard Tools | https://broadinstitute.github.io/picard/ |
| https://software.broadinstitute.org/gatk/documentation/tooldocs/4.0.1.0/picard_fingerprint_CrosscheckFingerprints.php | ||
| MuTect | Cibulskis et al. 201367 | https://github.com/broadinstitute/mutect |
| Strelka | Saunders et al. 201268 | https://github.com/genome-vendor/strelka |
| DeTiN | Taylor-Weiner et al. 201869 | https://github.com/getzlab/deTiN |
| Oncotator | Ramos et al. 201570 | https://github.com/broadinstitute/oncotator |
| OxoG and FFPE Orientation Bias filters | Costello et al. 201371 | http://software.broadinstitute.org/cancer/cga/dtoxog |
| BLAT Realignment filter | Kent et al. 200272 | https://github.com/djhshih/blat |
| MutSig2CV | Lawrence et al. 201473 | https://github.com/getzlab/MutSig2CV |
| SignatureAnalyzer | Kim et al. 201674 | https://github.com/broadinstitute/getzlab-SignatureAnalyzer |
| POLYSOLVER | Shukla et al. 201575 | http://software.broadinstitute.org/cancer/cga/polysolver |
| NetMHCPan 4.0 | Jurtz et al. 201776 | http://www.cbs.dtu.dk/services/NetMHCpan-4.0/ |
| GATK version 4.0.8.0 | Mckenna et al. 201077 | https://newreleases.io/project/github/broadinstitute/gatk/release/4.0.8.0 |
| GISTIC 2.0 | Mermel et al. 201178 | https://github.com/broadinstitute/gistic2 |
| ABSOLUTE | Carter et al. 201279 | http://software.broadinstitute.org/cancer/cga/absolute |
| PhylogicNDT | Leshchiner et al. 201980 | https://github.com/broadinstitute/PhylogicNDT |
| GTEx RNA-Seq pipeline | GTEx Consortium 202081 | https://github.com/broadinstitute/gtex-pipeline/ |
| RNA-SeqQC | Graubert et al. 202182 | https://github.com/getzlab/rnaseqc |
| ComBat | Johnson et al. 200783 | https://bioconductor.org/packages/release/bioc/html/sva.html |
| DESeq2 | Love et al. 201484 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| CIBERSORTx | Newman et al. 201942 | https://cibersortx.stanford.edu |
| MixCR v3.0.3 | Bolotin et al. 201585 | https://github.com/milaboratory/mixcr/releases |
| Melanoma dedifferentiation signature resource | Tsoi et al. 201849 | https://systems.crump.ucla.edu/dediff/index.php |
Resource availability
Lead contact
Requests for information and resources should be directed to and will be fulfilled by the lead contact, Nir Hacohen (nhacohen@mgh.harvard.edu).
Materials availability
This study did not generate new reagents.
Experimental model and subject details
Patient specimens and consent
All patients analyzed in this study (referred as the MGH cohort) provided written informed consent for the collection of tissue and matched normal blood samples for research and genomic profiling, as approved by the Dana-Farber/Harvard Cancer Center Institutional Review Board (DF/HCC Protocol 11-181) and UT MD Anderson Cancer Center (IRB LAB00-063 and 2012-0846). All samples in this study are from patients with metastatic melanoma treated with checkpoint blockade therapy (Table S1) at Massachusetts General Hospital (Boston, MA) and University of Texas MD Anderson Cancer Center (Houston, TX). Patient response status at initial restaging examination for the MGH cohort was defined based on a combination of radiographic measurements routinely performed on all patients with clinical evaluations (range 4-12 weeks after initiation of treatment), with patients achieving PR or CR for responders and SD or PD for nonresponders. Subsequent responses for biopsies taken during treatment were determined on the date of biopsy acquisition based on a combination of clinical and radiographic measurements. Overall survival (OS) was defined from the date of treatment initiation until the date of death or last follow-up.
Clinical outcomes from previously published cohorts
For the analysis of outcomes of patients from previously published cohorts, we used the published overall survival and clinical response data. Overall survival data was reported in each study. For clinical response, we used the binary classification of clinical outcome from each individual study. For Hugo et al., patients with PR/CR by irRECIST criteria were classified as responders, and patients with PD by irRECIST criteria were classified as non-responders5. For Zaretsky et al., the 4 patients were classified as PR/CR by irRECIST criteria and were classified as responders3. For Roh et al., we used the same response definitions as stated in their paper: responders had either complete resolution of tumors or partial reduction of at least 30% in tumor size (based on imaging) or stable disease for over 6 months, and nonresponders had an increase of at least 20% in tumor size (based on imaging) or stable disease for under 6 months2. We used these definitions for each sample (anti-CTLA-4 response and anti-PD-1 response) from Roh et al. Table S1B. For Riaz et al., the authors separated patients with RECIST PD, SD and PR/CR, so we classified patients in the same manner as the MGH cohort with RECIST PR/CR as responders and patients with RECIST PD or SD as non-responders4. Additionally, for the two patients in the Riaz cohort (Pt76_pre and Pt23_pre) that died prior to disease assessment (after 10 and 52 days respectively), we included these patients as non-responders. For Van Allen et al., patients with durable clinical benefit (DCB, PR/CR or SD with OS > 1 year) were classified as responders and patients with no durable clinical benefit (NDB, PD or SD with OS < 1 year) were classified as non-responders1. For four patients in this cohort with no annotated RECIST response but very short overall survival (MEL-IPI_Pat157, MEL-IPI_Pat166, MEL-IPI_Pat168 and MEL-IPI_Pat175 with deaths after 86, 77, 67 and 90 days respectively), we included these patients as non-responders. Additionally, one patient with no RECIST annotation and an overall survival of 1326 days (MEL-IPI_Pat29) was included as a responder. For Liu et al., patients with RECIST PR, CR or listed as MR (mixed response) were classified as responders, and patients with RECIST PD or SD were classified as non-responders7. For Gide et al., patients with RECIST PR/CR or SD with PFS over 6 months were classified as responders, and patients with RECIST PD or SD with PFS under 6 months were classified as non-responders6. Overall survival was available for all patients (except for one patient from the Hugo at el. study for which survival data was missing).
Method details
Whole exome sequencing
Whole exome sequencing for MGH cohort samples was performed at the Broad Institute genomic platform on 109 tumor and matched normal blood samples from 56 patients using a protocol previously described16. Briefly, 150-500ng of gDNA was extracted using QIAGEN AllPrep DNA/RNA Mini Kit (cat# 80204). DNA was fragmented using acoustic shearing followed by size selection to achieve library insert size distribution in the range of 300-650bp. Libraries for all samples were prepared using the Kapa HyperPrep kit, according to manufacturer’s specifications, followed by quantification and normalization using PicoGreen to ensure equal concentration. Adaptor ligation was performed using the TrueSeq DNA exome kit from Illumina according to the manufacturer’s instructions. Libraries were sequenced using the HiSeq2500 with paired end 76bp reads, followed by analysis with RTA v.1.12.4.2.
Whole transcriptome sequencing of bulk tumor samples
Whole transcriptome sequencing for MGH cohort samples was performed at the Broad Institute genomic platform on 88 bulk tumor samples from 48 metastatic melanoma patients treated with CPB therapy (53% anti-CTLA-4; 26% anti-PD-1; 11% anti-PD-L1 and 9% anti-CTLA-4+PD-1), using the Transcriptome Capture method (FFPE compatible) as previously described16. 250-500ng of purified total RNA with DV200 scores > 30% was considered acceptable for library preparation and sequencing. First a stranded cDNA library from isolated RNA was constructed followed by hybridization of the library to a set of DNA oligonucleotide probes to enrich the library for mRNA transcript fragments (capturing 21,415 genes). The normalized, pooled libraries were loaded onto HiSeq2500 for a target of 50 million 2x76bp paired reads per sample.
Quantification and statistical analysis
Whole-exome mutation calling
Using the Broad Picard pipeline, we aligned bams from all blood normal samples and tumor samples to hg19 using bwa 0.5.986. For somatic mutation calling, we used the CGA WES Characterization pipeline within the Firecloud framework (https://app.terra.bio/#workspaces/broad-fc-getzlab-workflows/CGA_WES_Characterization_OpenAccess,65). For mutation calling, we assessed the Agilent exome target regions for the Van Allen samples and the Illumina Capture Exome (ICE) target regions for samples from all other cohorts. We first assessed cross-sample contamination levels using ContEst66 and checked for sample swaps using Picard CrossCheckFingerprints (https://software.broadinstitute.org/gatk/documentation/tooldocs/4.0.1.0/picard_fingerprint_CrosscheckFingerprints.php). We then called somatic single nucleotide variants using MuTect67 with the cross-sample contamination estimates as lower bounds for allele fraction and called somatic indels using MuTect2 and Strelka (https://github.com/broadinstitute/gatk/tree/master/scripts/mutect2_wdl; https://github.com/genome-vendor/strelka68). Next, we applied DeTiN to detect cases with evidence of tumor in the exome normal sample and recover somatic mutations that had been incorrectly filtered out due to alternate reads present in the normal sample69 and we annotated somatic mutations consequences using Oncotator70. We merged adjacent SNV calls and annotated them as di-nucleotide variants as CC > TT mutations are frequently detected in sun-exposed melanomas39. To filter the somatic mutation calls, we applied OxoG and FFPE Orientation Bias filters71 and we filtered out mutations present in a panel of TCGA normal samples or a panel of Illumina Capture Exome (ICE) normal samples87. We then used a BLAT Realignment filter72 to filter out mutations that were only supported by reads that had mapped ambiguously and finally filtered out mutations using a final panel of normal samples87 that included FFPE normal samples.
To exclude exome samples that were low quality, we excluded samples with tumor or normal mean bait coverage below 30 or cross-sample contamination values above 5%. WES samples from the Hugo cohort had high cross-sample contamination levels, so we did not include the WES DNA data from this cohort10. We retained one sample (Case4-Relapse) from the Zaretsky cohort with a contamination fraction of 5.4% as this represented an important clinical scenario (acquired resistance to PD-1 therapy after a response). Moreover, we excluded samples with tumor in normal estimates above 15%, as these samples would have reduced sensitivity for somatic mutation calling. In order to maintain high sensitivity for somatic variant calling, we excluded samples with ABSOLUTE estimated tumor purity below 10% (described in the ABSOLUTE section below79. After applying these filters, we retained a total of 258 WES samples from 189 patients as passing QC. Additionally, for analysis of outcomes, in order to use a single WES sample per patient for patients with multiple samples, if the patient had multiple WES samples we used the earliest biopsy available unless the patient was a non-responder for the first line of immunotherapy but a responder for a subsequent line of immunotherapy, in which case we used the later sample which was directly preceding or in some cases after the response. Finally, for patient 33 in the Roh cohort, we used the post-treatment sample (33C) rather than the pre-treatment sample (33A) as the pre-treatment sample had tumor purity at our QC threshold level (10% tumor purity) which severely limited the analysis of clonality. This resulted in a total of 189 WES samples for analyses of outcomes using the combined WES data from the MGH, Van Allen, Roh and Zaretsky cohorts combined. For analyses of immune infiltration or purity and outcome in the WES data, we excluded the two cell line samples from the Zaretsky cohort, leaving 187 WES samples for these analyses.
We collected mean target coverage (MTC) and mean bait coverage (MBC) statistics (Table S2) for WES samples from the MGH cohort (mean tumor MTC 199.03, standard deviation [sd] 94.66, median tumor MTC 188.61, mean tumor MBC 341.32, sd 164.65, median tumor MBC 302.60, mean normal MTC 106.67, sd 33.57, median normal MTC 101.14, mean normal MBC 148.30, sd 49.44, median normal MBC 137.51). For the MGH samples sequenced with Illumina Capture Exome baits, the target territory was 28,665,628 bases. We also collected coverage statistics from the Roh cohort (mean tumor MTC 115.85, sd 16.26, median tumor MTC 115.40, mean tumor MBC 183.10, sd 26.98, median tumor MBC 185.87, mean normal MTC 69.80, sd 14.79, median normal MTC 67.97, mean normal MBC 95.00, sd 18.11, median normal MBC 91.93), Van Allen cohort (mean tumor MTC 137.07, sd 44.61, median tumor MTC 145.54, mean tumor MBC 369.26, sd 187.71, median tumor MBC 327.16, mean normal MTC 121.19, sd 51.57, median normal MTC 124.99, mean normal MBC 295.44, sd 79.68, median normal MBC 283.12) and Zaretsky cohort (mean tumor MTC 105.19, sd 30.88, median tumor MTC 101.42, mean tumor MBC 178.86, sd 53.09, median tumor MBC 178.25, mean normal MTC 95.88, sd 26.74, median normal MTC 100.92, mean normal MBC 148.77, sd 38.51, median normal MBC 156.99) (Table S2).
Mutation significance analysis
We used MutSig2CV73,88 to identify significantly mutated driver genes in the 189 melanoma WES samples. This meta-analysis cohort was smaller than previously published melanoma analyses which included patients not treated with immunotherapy and melanoma has a high mutation burden, so we were underpowered to discover novel melanoma drivers. We used a MutSig2CV q < 0.1 threshold to classify genes as significantly mutated. Additionally, we excluded significantly mutated genes with a median log2(TPM+1) below 1 in CCLE melanoma cell lines (analysis of CCLE melanoma RNA-Seq data described below).
Mutation signature analysis
We used the SignatureAnalyzer Bayesian NMF approach to identify mutation signatures present in the somatic coding SNVs in the 189 melanoma WES samples74,89,90. As melanoma has a high mutation burden, we used the SignatureAnalyzer with the hypermutation option, and we excluded one sample (from patient 16 in the Roh cohort) which had evidence of an MSI signature (COSMIC Signature 2691,92). We ran the Bayesian NMF with 50 random initializations, and converged to k = 3 signatures, 44 converged to k = 4 and 4 converged to k = 5, so we chose the solution with k = 4 with the maximum posterior probability. We compared the four signatures to COSMIC mutational signatures using cosine similarity and we assigned mutations to signatures based on the association probability and computed the counts and fractions of mutations assigned to signatures by sample. As expected, we identified a UV signature and a CpG signature in many samples as well as a temozolomide (TMZ) signature (as a subset of patients had prior TMZ treatment in the Van Allen cohort)1. Additionally, we identified a fourth signature which primarily consisted of T to C mutations and had some similarity to COSMIC signature 26, but this signature was only present as a small proportion of mutations in a subset of samples.
Neoantigen and TMB analysis
To identify neoantigens, we used normal WES samples to call germline MHC Class I alleles using POLYSOLVER75. We considered somatic single nucleotide and di-nucleotide variants as potential neoantigens and predicted the binding affinity of all possible 9mers and 10-mer peptide sequences that overlapped the mutated residue using NetMHCPan 4.076,93,94. We predicted binding affinities to all six germline MHC Class I alleles. We counted predicted binders as neoantigens if they had a NetMHCPan 4.0 percentile rank of 2 or lower (note that certain mutations may lead to multiple neoantigens). To compute tumor mutation burden (TMB), we counted the number of non-silent somatic SNVs, DNVs and Indels per sample, and we used log10(TMB) in continuous Cox models with overall survival. For the analysis of TMB above or below 10 mutations/Mb in the 189 WES samples, we obtained the number of mutations per megabase by dividing the number of non-silent somatic SNVs, DNVs and Indels by the ICE target territory and multiplying by 106, and we split samples using a threshold of 10 mutations/Mb.
Association of SNVs with outcome
To investigate the relationship between somatic mutations and outcomes in the 189 WES samples, we associated mutation status with survival using Cox models which included mutation status and log10(TMB) and we associated mutation status with response using logistic regression models which incorporated mutation status and log10(TMB). For the associations with survival and response, we tested loss of function mutation status (loss of function mutation or wild-type) for all genes with 3 or more loss of function mutations and non-synonymous mutation status for all genes with 3 or more non-synonymous mutations. For the survival models, we used the P value of the mutation status in the combined Cox model with log10(TMB) and for the logistic regression response models, we used the P value of mutation status in the combined logistic regression model with log10(TMB).
Somatic copy number alteration analysis
To assess somatic copy number alterations (SCNAs) in autosomes from WES data, we used GATK4 CNV (with GATK version 4.0.8.0, https://github.com/gatk-workflows/gatk4-somatic-cnvs 77). We created a panel of normals to normalize the copy number data using GATK4 CNV CNV Somatic Panel Workflow using a set of 928 normal WES samples including TCGA melanoma, the matched normals from the MGH, Roh, Zaretsky and Hugo cohorts (the Hugo samples were included to improve the normalization of the Zaretsky samples sequenced by the same group, even though the Hugo samples failed cross-sample contamination QC) and a set of FFPE normal samples. We set the GATK CNV bin_length to 0 to skip binning (as recommended for WES data). Additionally, in order to be able to compare copy number results across cohorts, we used the hg19 Illumina Capture Exome (ICE) target set with 250bp of padding as the target list for all samples for collecting read counts for copy number calling. As this created a more heterogeneous distribution of coverage per target, we set --maximum-zeros-in-interval-percentage=1 (the threshold of the minimum percentage of samples in the panel of normals with zero-coverage in a target, and intervals failing this filter are removed).
We then used the GATK4 CNV Somatic Pair Workflow incorporating GATK ACNV to estimate somatic copy number alterations using the described panel of normals for all WES samples. To allow for heterozygous SNPs to be used in estimating allelic coverage, we set minimum-total-allele-count=10 (the minimum coverage required in the tumor to collect allelic counts at heterozygous SNP sites). To estimate allelic copy number in GATK4 CNV, we used the GATK set of frequently polymorphic SNP sites (gs://gatk-test-data/cnv/somatic/common_snps.interval_list). Additionally, we filtered potential germline or artifactual segments that appeared in both the germline blood normal and the tumor sample and segments at recurrent copy number segment breakpoints. After removing these events, gaps were imputed if the two neighboring segments had the same copy ratio and were 2.5Mb apart. Additionally, we filtered out allelic copy number segments supported by zero heterozygous SNPs.
GISTIC analysis of significant SCNAs
We used GISTIC 2.0 to identify recurrently amplified and deleted regions in the 189 melanoma WES samples78. Similar to the mutation significance analysis, we were underpowered to discover novel melanoma drivers in comparison to previous studies. We ran GISTIC 2.0 on the GATK4 CNV segment mean log2 copy ratio (after the post-processing to remove potential germline and artifactual events as described).
Analysis of tumor purity and ploidy using ABSOLUTE
In order to estimate tumor purity and ploidy as well as integer copy number, we used ABSOLUTE79,95. Using the called somatic mutations and somatic copy number alterations as input, we then used ABSOLUTE to generate candidate purity/ploidy solutions. We then manually reviewed these potential ABSOLUTE solutions and selected the solution most concordant with the copy number and mutation data. Finally, after obtaining the integer somatic copy number segments, we imputed gaps by extending the neighboring segments to meet in the middle of the gap, and we annotated the integer copy number and LOH status of GENCODE v19 genes. For analyses of purity and outcomes in the primary cohort, we excluded two samples from the Zaretsky cohort (Case2-Baseline and Case3-Baseline) as they were derived from cell lines.
Analysis of longitudinal samples using PhylogicNDT
To investigate clonal dynamics in longitudinal WES samples, we used ABSOLUTE and PhylogicNDT (https://github.com/broadinstitute/PhylogicNDT)96. After calling somatic mutations in all 123 samples from the 54 patients with WES data at multiple time points, we then took the union of mutations called in each patient and then calculated the number of reference and alternate reads present at the union of the mutated sites in each sample from the patient. For this analysis of the union of mutated sites, we used non-duplicate reads with mapping quality of at least 5 and with a recalibrated base quality scores of at least 20 at the mutated site. We then called ABSOLUTE solutions for all samples and clustered the mutation clonality across time points using a multidimensional Dirichlet process model implemented in PhylogicNDT to estimate the mutation cancer cell fractions (CCFs) and somatic copy number alteration clonality across samples. We plotted clonality of mutations and gene level copy number in paired biopsies, with 108 biopsies in total from 54 patients (Table S1; Figure S3). For patients in the MGH cohort with three or more biopsies, we used the earliest two WES samples available, except for patients 208T and 272T as these patients both had responses but then developed resistance at the time of the third sample. For patient 208T, we used samples 208A and 208C and for patient 272T we used samples 272A and 272C16. In addition to the analysis of longitudinal samples, we also separately applied the PhylogicNDT mutation clustering to single samples in order to estimate the mutation CCFs, and we counted mutations as clonal if they had estimated CCF ≥ 0.85.
RNA-Seq gene-expression analysis
In order to quantify gene expression in RNA-Seq data, we used the GTEx RNA-Seq pipeline (https://github.com/broadinstitute/gtex-pipeline/)81, which uses STAR97 2 pass alignment followed by quantification of TPMs using RSEM98. We quantified gene expression in TPM for all transcripts using the GENCODE v19 reference transcriptome. We derived quality control metrics from the aligned bams using RNA-SeqQC (https://github.com/getzlab/rnaseqc)82, and we excluded samples with below 15000 genes detected (where genes with 5 or more unambiguous reads were considered detected) or Exon CV MAD > 1. The Exon CV MAD is the median absolute deviation of the coefficient of variation of the exonic coverage (which excludes the first and last 500bp of a gene). Additionally, we performed PCA on the log2(TPM + 1) values for all protein coding genes using the MGH pre-treatment samples (including only pre-treatment A RNA-Seq samples and not post-treatment B or C samples), and we excluded two additional samples that were PCA outliers (samples 346AR and 9AR). We also excluded the one post-treatment sample from the Hugo cohort from analysis (Pt16-OnTx). Additionally, we re-processed the TCGA melanoma (SKCM) RNA-Seq data using the GTEx pipeline to obtain log2(TPM+1) values.
When we performed PCA on the log2(TPM+1) values of protein coding genes (excluding genes with zero expression in all samples) for the 154 pre-treatment samples from the primary cohort, we noted significant batch effects between cohorts (Figure S11A). The cohorts clustered in batches based on the RNA-Seq method (polyA selection for Hugo and Riaz versus transcriptome capture for MGH and Van Allen), so we applied ComBat83 in order to remove this batch effect. We also set all negative values following ComBat batch correction to zero, as the input log2(TPM+1) values were non-negative. After this batch effects correction, the separate cohorts were overlapping in PCA space (Figure S11A).
For the secondary cohort, we obtained log2(TPM+1) values in the same way using the GTEx pipeline. Upon inspecting the RNASeqQC metrics for the Liu cohort, we found that 14 of the initial 121 samples failed the 15000 genes detected threshold or the Exon CV MAD > 1 threshold, so we excluded these samples, leaving 107 samples. For the Gide cohort, no samples failed the QC thresholds. For the secondary cohort analysis of RNA-Seq samples, we combined the 107 Liu samples with the 73 Gide pre-treatment samples (excluding the 16 Gide early during treatment samples), leaving 180 samples in total. We observed similar batch effects in the PCA of log2(TPM+1) values for the Gide and Liu cohorts (Figure S16A), so we applied ComBat to the full secondary cohort which reduced the batch-specific clustering in PCA.
Quantification of TCR and Ig levels in WES and RNA-seq
To quantify T and B cell infiltration, we used MixCR v3.0.385,99. We used the MixCR analyze shotgun pipeline with DNA or RNA as the starting material for all WES and RNA-Seq samples. We excluded TCR or Ig clonotypes with rearrangements that had a stop codon or resulted in an out-of-frame sequence. We quantified the number of TCR or Ig reads by summing the clone counts of reads assigned to clonotypes. For TCRs, we included TCR alpha, beta, delta and gamma sequences. By counting these reads, we potentially included rearrangements from CD8 T cells, CD4 T cells (some of which may be regulatory T cells) and γδ T cells. For the analysis of RNA-Seq data, in rare cases clonotypes were aligned to both a TCR and an Ig (with MixCR V region alignment allVHitsWithScore containing both a TCR and an Ig), and we included these reads in both the TCR and the Ig counts.
As different samples had different depths of sequencing, we normalized the TCR and Ig read counts by sample coverage. For RNA-Seq, we used the number of mapped reads from RNASeqQC, and for WES we used the number of reads aligned from samtools idxstats. To create the T cell burden (TCB) and B cell burden (BCB) metrics, we computed TCB = (1+TCR read count)/(aligned reads/106) and BCB = (1+Ig read count)/(aligned reads/106), with appropriate TCR/Ig read counts and aligned read counts separately for DNA and RNA. Additionally, we plotted TCB and BCB on log10 scales and we used log10(TCB) and log10(BCB) for outcome analysis using Cox or logistic regression models.
TCBRNA values were slightly higher in samples from the Hugo cohort (Figure S4A) than in samples from other cohorts, but we saw significant batch effects for both TCBDNA and BCBDNA (Figure S5A). As a result, we decided to dichotomize TCBDNA and BCBDNA within cohorts, so we calculated median(TCBDNA) separately for each cohort and we labeled samples as TCBDNAhigh if they had TCBDNA > median(TCBDNA) using the cohort-specific median (with similar analysis for BCBDNA). For analyses of TCBDNA/BCBDNA and outcomes in the primary cohort, we again excluded the two cell line samples from the Zaretsky cohort (Case2-Baseline and Case3-Baseline).
When we compared TCBDNA and TCBRNA, there were a subset of samples in the MGH cohort for which DNA and RNA was extracted from different locations of a tumor biopsy. These samples could have different levels of TCB in RNA and DNA due to sampling rather than technical factors, so when we looked at the correlation between TCBDNA and TCBRNA (as well as the correlation between BCBDNA and BCBDNA), we only included Van Allen samples and MGH samples with DNA and RNA extracted from the same location (n = 35 total). For analysis of TCBDNA and BCBDNA from longitudinal WES samples, we analyzed samples from the same 54 patients with longitudinal WES samples as in the PhylogicNDT longitudinal analysis, and for patients in the MGH cohort with three or more biopsies, we used the earliest two WES samples available from each patient. Additionally, for analysis of TCBRNA and BCBRNA from longitudinal RNA-Seq, we analyzed 172 paired biopsies from 86 patients across the Riaz, Gide and MGH cohorts, and again for patients in the MGH cohort with three or more biopsies, we used the earliest two RNA-Seq samples available from each patient.
TCR/Ig overlap analysis
In order to further establish that TCRs and Igs were shared between WES and RNA-Seq data, we assessed the degree of overlap between TCR and Ig sequences in primary cohort samples with DNA and RNA extracted from the same location (n = 35 total). Using each DNA CDR3 region from the MixCR output, we performed pairwise global alignments using the Needleman-Wunsch algorithm (implemented in the R biostrings package in function pairwiseAlignment with gapOpening=10 and gapExtension=4) between the DNA CDR3 region and all RNA CDR3 regions in the MixCR output from the paired RNA sample. If the top alignment had a Needleman-Wunsch alignment score greater than 25 and the top alignment had Si = number of mismatches+total insertion length+total deletion length ≤ 2, then we counted the TCR or Ig pair as an overlap. We then calculated the number of patients with DNA/RNA overlaps for TCRs and for Igs.
B cell scRNA signature analysis
To assess the correlation between BCBRNA and B cell types in the primary cohort RNA-Seq data, we assessed scRNA-derived naive B cell and plasma B cell signatures. We used published cell-type marker genes for naive B cells and plasma B cells43. To calculate signature scores, we z-scored the log2(TPM+1) values for marker genes that overlapped with the genes in the batch-effects corrected TPM data from the primary cohort, and we then took the mean. We then assessed the correlation between the naive B cell signature or the plasma B cell signature and BCBRNA for the primary cohort samples.
CIBERSORTx deconvolution analysis
To assess the cell type composition of the bulk RNA-Seq samples in the primary cohort, we performed a deconvolution analysis using CIBERSORTx42. In order to deconvolute melanoma cell types accurately, we used published melanoma scRNA-Seq data from the Tirosh cohort, available on the CIBERSORTx web portal (https://cibersortx.stanford.edu)51. This scRNA reference data included both tumor and normal cell types. We ran CIBERSORTx using B-mode batch correction in absolute mode with 100 iterations. In order to obtain cell type fractions, we divided each sample’s cell type score by the sum of scores for the sample. For analysis of T cell fractions, we summed CD4 and CD8 T cell fractions. To assess the cell type fractions of different cell types we aggregated stromal cells (Endothelial cells and Cancer Associated Fibroblasts [CAFs]) and immune cells (CD8 T cells, CD4 T cells, NK cells, Macrophages and B cells) separately. Finally, we tested the correlation between the fraction of malignant cells in RNA from CIBERSORTx and the tumor purity in DNA from ABSOLUTE for the 35 primary cohort samples with DNA and RNA extracted from the same location.
RNA-seq tumor subtyping
In order to identify melanoma subtypes using bulk RNA-Seq data, we applied a Bayesian NMF based clustering method to 469 TGCA melanoma RNA-Seq samples39,47,48. We preprocessed the data by removing non-protein-coding genes and by retaining only the 25% of genes with the highest standard deviation of expression across samples, and we removed all genes that were expressed at 0 TPM in at least 10% of samples. After these steps, 2684 genes remained. Next, we transformed the matrix of TPMs into fold changes by subtracting the median of each gene from the log2(TPM+1) values for that gene. To cluster samples, we created a distance matrix by calculating the Spearman correlation between each pair of samples and then performed repeated hierarchical clustering with K (the number of clusters) between 2 and 10. We performed hierarchical clustering using 80% sampling and average linkage, and we repeated this clustering 500∗K times for each K. We created a consensus matrix MK by calculating the number of times that each pair of samples clustered together in the repeated hierarchical clusterings for a given K. Then, we summed all MK and normalized the final matrix M∗ by the total number of iterations. We then used Bayesian non-negative matrix factorization with a half-normal prior on the M∗ matrix to determine the optimal number of clusters K∗. In this setup where we are approximating M∗ ∼ HTH, the H matrix represents the association of samples to clusters. The most frequent solution was K∗ = 5, so we selected this clustering solution. We also obtained the normalized matrix H∗ by normalizing each column so that the values of each sample’s hij sample-to-cluster association sum to 1.
To identify marker genes for clusters, we took the full TCGA SKCM log2(TPM+1) matrix X with 19820 protein-coding genes and performed non-negative matrix factorization by approximating X ∼ WH∗. In this case, the values in W represent the association of a gene to each cluster. We obtained the normalized matrix W∗ by again normalizing each column to sum to 1.
We used a previously developed approach in order to select marker genes for each subtype and project the subtypes identified in TCGA samples to new data using the expression of the reduced set of marker genes47. In order to optimize the subtype classifier, we varied parameters for selecting cluster marker genes for the subtype classification. We did not consider genes with 0 TPM in 10% or more samples as candidate marker genes. When selecting marker genes, we used only marker genes which were overexpressed in a cluster relative to the other clusters. We selected marker genes which had normalized association to a cluster greater than a threshold parameter Wcut and we limited the number of marker genes for each cluster based on the threshold parameter genecut. For the marker genes in each cluster, we considered only genes with a mean difference in log2(fold change) across clusters of 0.5 or better. In order to identify the optimal classifier, we performed a parameter sweep with Wcut from 0.1 to 0.96 in increments of 0.01 and genecut from 30 to 250 in increments of 10. Using these parameters, we selected the reduced set of marker genes g, and then we performed the matrix factorization Xg∼W∗gH, and we assigned samples to clusters using this H matrix. We measured the performance of the classifier using the adjusted rand index when comparing the projected cluster labels to the original cluster labels. We identified Wcut = 0.71 and genecut = 65 as the optimal parameter setting, which resulted in an adjusted rand index of 0.653 for the TCGA SKCM samples.
We then used the marker genes and the subtype membership for TCGA samples to compare our melanoma subtypes to previous melanoma subtype classification schemes. The TCGA melanoma RNA-Seq subtyping identified 3 subtypes: an immune subtype, an MITF high subtype and a keratin subtype. The TCGA MITF low and Immune subtypes had strong overlap with two of our subtypes (Figure S10B), and the marker genes were consistent with many T cell genes identified as markers for one (CD2 and CD8A) and neural crest marker genes (such as AXL and NGFR) highly expressed in the other (Figures S10E and S10F). Based on these results, we denoted these subtypes Immune and MITF low. Another subtype had many keratin marker genes such as (KRT10, KRT5 and KRT19), suggesting a high degree of keratinocyte infiltration, so we denoted this subtype Keratin high. Next, when we compared our subtype assignments to a melanoma differentiation-based subtype classification scheme, we saw a high degree of overlap between some subtypes (Figure S10D).49 The analysis in the Tsoi paper attempted to remove non-tumor intrinsic signals including immune infiltration and keratin expression, whereas our goal was to incorporate both tumor-intrinsic markers and potential immune markers. The Tsoi melanocytic subtype strongly overlapped with one of our subtypes, and this subtype had the highest expression of melanocyte markers such as PMEL, MITF and MLANA, so we denoted this subtype MITF high. The MITF high and MITF low subtypes match the previously recognized melanoma differentiation axis with well differentiated melanocyte-like tumors expressing MITF and poorly differentiated neural-crest-like tumors expressing AXL49, 50, 51. The final subtype had the highest degree of overlap with the Tsoi transitory subtype (Figure S10D) which had an intermediate differentiation state, so we denoted this the Intermediate subtype (though the MITF expression level in this subtype is similar to that of the MITF high subtype). Additionally, we looked at the relationships between TMB (by reprocessing TCGA melanoma WES data using the same somatic mutation calling pipeline), TCBRNA, BCBRNA and tumor purity (from the previous TCGA melanoma analysis)39.
Finally, we sought to classify the tumors in the melanoma immunotherapy meta-analysis cohort by their subtype. We used the ComBat batch corrected log2(TPM+1) values for the 154 primary cohort RNA-Seq samples and preprocessed the data by removing genes with zero TPM values in 10% or more samples and transforming the TPM values to log2(fold change) values using median centering. This preprocessing removed a subset of the marker genes which were selected in the subtype classifier. Then, we used the weights inferred for the matrix W∗g and the log2(fold change) values for the selected genes in the immunotherapy expression data to perform the approximation Xg CPB∼W∗gHCPB from which we could identify the subtypes of the immunotherapy samples using the normalized matrix H∗CPB. We separately applied this same procedure to the ComBat batch corrected log2(TPM+1) values for the 180 secondary cohort samples in order to determine their subtype memberships.
Differential expression analysis
We used DESeq284 in order to identify differentially expressed genes from the primary cohort RNA-Seq data. Based on the previous batch effects that we identified between cohorts, we used the cohort as an additional covariate in all DESeq2-based differential expression analyses. We compared patients with long OS (overall survival > 1 year) versus short OS (overall survival < 1 year) and responders versus non-responders in 154 samples in the primary cohort (153 for overall survival as one patient did not have OS data), while including batch as a covariate. We analyzed genes with median log2(TPM+1) > 1 and we performed Benjamini-Hochberg multiple hypothesis correction using the DESeq2 P values. We considered genes with q < 0.05 as differentially expressed.
Analysis of CCLE and human protein atlas (RNA-seq data
To assess whether the differentially expressed genes were expressed in melanoma cells, immune cells or both cell types, we assessed gene expression in CCLE cell lines and bulk RNA-Seq data of HPA blood cell types58,64. We downloaded CCLE RNA-Seq TPM data from https://data.broadinstitute.org/ccle/CCLE_RNAseq_rsem_genes_tpm_20180929.txt.gz and used data from 49 melanoma cell lines. When we performed PCA on the log2(TPM+1) values, we noted 2 outliers in PCA (CJM_SKIN and LOXIMVI_SKIN), and we removed these two cell lines from the dataset, leaving 47 melanoma cell lines. Then, we computed the median log2(TPM+1) values for the differentially expressed genes of interest. To quantify expression of genes in immune cells, we downloaded the HPA blood expression data from https://www.proteinatlas.org/download/rna_blood_cell_sample_tpm_m.tsv.zip and took the median log2(TPM+1). We used thresholds of log2(TPM+1) of 1 for both cohorts to determine whether genes were expressed in immune cells, expressed in tumor cells, expressed in both or had low expression in both.
Additionally, we wanted to determine which immune cell types and which melanoma subtypes had high expression of these differentially expressed genes of interest. First, we assessed which of the differentially expressed genes were coexpressed by calculating the pairwise spearman correlations of all genes in the 154 primary cohort samples (Figures S12D and S20A). Then, for the differentially expressed genes that were higher expressed in responders or patients with long OS (which were usually higher expressed in immune cells than in melanoma cell lines), we transformed the HPA log2(TPM+1) values to z-scores and took the mean of each gene’s z-scores for each cell type (Figures S12E and S13B). Additionally, for each of these genes, we ranked the 19 HPA cell types by log2(TPM+1) of the gene, with 1 being the highest expression and 19 being the lowest expression, allowing for ties. We then ordered each cell type by their median rank across the genes of interest (Figures S12F and S13C). Finally, using the primary cohort batch corrected log2(TPM+1) data and melanoma subtypes, we repeated the same analysis of mean z-scored expression by subtype and subtype ranking (Figures S12G, S12H, S13D, and S12E). These analyses suggested that lymphocytes had the highest expression of the genes overexpressed in responders and patients with long OS, and that the genes overexpressed in non-responder and patients with short OS were highest expressed in the MITF low melanoma subtype.
Construction of metagene models
To create gene signatures using the differentially expressed genes from the primary cohort, we constructed metagenes from the differentially expressed genes in the long OS versus short OS and the responder versus non-responder analyses. For each analysis, we split genes based on their effect size, consisting of 75 genes overexpressed in responders, 55 genes overexpressed in patients with long OS, 26 genes overexpressed in non-responders and 28 genes overexpressed in patients with short OS. We created a metagene using each of these four gene lists. In order to score individual patients, we transformed log2(TPM+1) values to z-scores for each gene across patients (so that genes with low expression would not be penalized) and calculated the mean of the gene z-scores for each individual patient.
Predicting survival and response using gene pair models
To create simple models to predict outcome using gene expression that could potentially incorporate both tumor and immune components, we considered the genes we identified as differentially expressed and tested all gene pairs as predictors of response and overall survival. For this analysis, we used the batch-effects corrected log2(TPM+1) values for primary cohort samples, whereas in the DESeq2 differential expression analysis we used the raw count data and included batch as a DESeq2 model covariate. For analysis of response, we used all 154 patients, but for analysis of survival, we analyzed 153 samples because one patient from the Hugo cohort did not have overall survival data. We performed the gene pair model analysis using 1) all 3403 unique pairs of the 83 genes differentially expressed between patients with long OS and short OS 2) all 5050 unique pairs of the 101 genes differentially expressed between responders and non-responders. For each analysis, we also considered the corresponding pair of metagenes as an additional model (for 1) the long OS and short OS metagenes and for 2) the R and NR metagenes). For each gene (or metagene) pair model, we tested the association with survival using Cox proportional hazards models incorporating two genes, and we calculated the significance with the log-rank P value and the performance with the C-index. Similarly, we tested the association between gene pairs and response using a logistic regression model incorporating two genes, and we calculated the significance with a P value testing whether the null hypothesis of an AUC = 0.5 can be rejected (implemented in the R verification package100) and the model performance with the model AUC (using the logistic regression ŷ values to rank samples).
Next, we performed multiple hypothesis corrections on the model significance values separately for logistic regression model P values and for Cox model P values. Since the tested gene pairs were genes associated with outcomes in the DESeq2 analysis, many of the gene pair models had low P values, and the distribution of gene pair model P values strongly deviated from a uniform distribution. Additionally, we wanted to very stringently select significant models, so we used a Bonferroni multiple hypothesis correction rather than a Benjamini-Hochberg correction. We performed Bonferroni correction on the P values for the response logistic regression models and the survival Cox models.
To identify gene pair models which predicted both response and survival, we selected gene models with Bonferroni corrected p < 0.05 for both response and survival predictions in the primary cohort. For the analysis using the 5050 gene pairs derived from the responder versus non-responder differential expression analysis and the response metagene pair model, 39 gene pair models had Bonferroni corrected p < 0.05 for prediction of response (one of which was the response metagene pair model), but 0 gene pair models had Bonferroni corrected p < 0.05 for predictions of survival. Thus, the gene pairs derived from the responder versus non-responder differential expression analysis were predictors of response but not survival. Additionally, when we looked at the performance of the gene pair models by the original differential expression effect size (R genes had higher expression in responders, and NR genes had higher expression in non-responders), gene pairs models with a R gene and an NR gene had higher C-index values than other model types but gene pair models with two NR genes had higher AUC values than other model types. For the analysis using the 3403 gene pairs derived from the long OS versus short OS differential expression analysis and the long/short OS metagene pair model, 67 gene pair models had Bonferroni corrected p < 0.05 for prediction of survival and 10 models had Bonferroni corrected p < 0.05 for predictions of response. There were 3 gene pair models that had Bonferroni-corrected p < 0.05 for both predictions of response and survival in the primary cohort: MAP4K1&TBX3, MAP4K1&AGER and the long OS/short OS metagene pair model (Figure 4A). These results suggest that some gene pairs derived from the long OS versus short OS differential expression analysis were able to predict both survival and response. Furthermore, gene pairs models with a long OS gene (higher expressed in patients with long OS) and a short OS gene (higher expressed in patients with short OS) had higher C-index values than other model types, but the gene pairs models with two short OS genes had very slightly higher AUC values than gene pair models with a long OS gene and a short OS gene, though the mean AUC values were very similar (mean AUC for models with a long OS gene and a short OS gene was 0.612 and mean AUC for models with two short OS genes was 0.619, Wilcoxon p = 0.026, Figures S13I and S13J). Additionally, the 3 gene pairs which passed Bonferroni-corrected P value thresholds for survival and response predictions were all pairs which incorporated a long OS gene and a short OS gene. These results suggest that gene model pairs which incorporate a long OS gene or an R gene (often immune-expressed genes) as well as a short OS gene or an NR gene (often tumor-expressed genes) have improved predictions of survival than other gene pair model types, but gene pair models with two poor outcome-associated genes can sometimes perform best in response prediction.
Finally, to determine whether tumor mutational burden improved the three gene pair models, we performed a combined DNA and RNA analysis. We considered the patients from the primary cohort with both WES data and RNA-Seq data, and we included patients for which the DNA and RNA were not taken from the same location (n = 59 total). We considered three component models with a gene pair plus log10(TMB), and we predicted overall survival using Cox models and response using logistic regression models. To determine whether TMB improved upon the gene pair models, we used likelihood ratio tests comparing Cox models using only a gene pair to Cox models with a gene pair and log10(TMB) for overall survival, and we used DeLong’s test to compare AUCs of response logistic regression models using only a gene pair to logistic regression models with a gene pair and log10(TMB) for response (Figures S14A–S14D). Based on the performance of the gene pair models, we decided to compare the performance of these three gene pair models to that of other expression-based immunotherapy models and assess their performance in an independent dataset.
Gene pair model cross-validation analysis
To assess the robustness of the gene pair model discovery, we performed a cross-validation analysis using the primary cohort data. We used different splits for training and validation sets of 80%/20%, 75%/25%, 70%/30%, 66%/33% and 50%/50%. Using training sets composed of subsets of the cohort, we repeated the differential expression analysis of responders versus non-responders and long OS versus short OS (DESeq2 q < 0.05). We tested the association between all differentially expressed gene pairs (within gene pair type, response or OS) and response and survival using logistic regression and Cox models as described. In addition to the gene pair models, we also constructed responder/non-responder and long OS/short OS metagene pair models using the differentially expressed genes discovered in each training set cross-validation differential expression analysis. Due to the reduced power in the training sets, there were sometimes few differentially expressed genes detected in the training sets, so we also constructed metagene pair models by ranking differentially expressed gene by P values and creating a metagene using the top 25 genes overexpressed in responders or patients with long OS and a separate metagene using the top 25 genes overexpressed in non-responders or patients with short OS (even if these genes had DESeq2 q > 0.05). Then, for each training set we performed the discovery analysis of gene pair models using each set of differentially expressed genes by identifying gene pairs with Bonferroni-corrected log-rank p < 0.05 and/or Bonferroni-corrected response AUC p < 0.05. Finally, we attempted to validate the discovered models in a validation set composed of all of the remaining samples in the cohort, checking for validation set Bonferroni-corrected log-rank p < 0.05 and Bonferroni-corrected response AUC p < 0.05.
Based on the results from this cross validation, we found that increasing the training set size increased the number of models discovered with Bonferroni-corrected p < 0.05 (Figures S15A and S15B). Even with 80% of the dataset as a training set, discovery was still increasing, which suggests that the full cohort analysis is likely underpowered. For the metagene models, increasing the training set size increased the robustness of the long OS metagene signatures, whereas the short OS metagene signatures were more variable across patients (Figures S15C–S15F). Finally, while the top gene pair models were frequently discovered training data subsets (Figures S15G and S15H), they were very rarely statistically significant in validation sets (Figures S15I and S15J), supporting the need for larger clinically annotated immunotherapy datasets.
Gene pair model validation analysis
We attempted to validate the three gene pair models (MAP4K1&TBX3, MAP4K1&AGER and the long OS/short OS metagene pair model) developed using the primary cohort samples in the independent secondary cohort. To be validated, we required these models to achieve significance below the Bonferroni corrected threshold of p = 0.05/3 = 0.0167 with both the OS log-rank P value < 0.0167 and the response AUC P value < 0.0167 in the secondary cohort. Each of the three gene pair models passed both of these thresholds in the secondary cohort (Figure 4G), so we considered them to be validated in an independent cohort.
In order to visualize the survival of high and low risk groups within the primary and secondary cohorts for the MAP4K1&TBX3 model, we divided patients into groups depending on whether their ŷ values in overall survival Cox models with MAP4K1&TBX3 were above or below median (Figure 4K).
Performance of external gene-expression models
In order to compare the performance of the top gene pair models to that of other predictors of immunotherapy outcomes, we evaluated a set of external models in both the primary and secondary cohorts. We also included TCBRNA and BCBRNA as additional predictors. Additionally, we included CD274 expression as a simple model. We tested the CYT model57 by taking the geometric mean of PRF1 and GZMB expression. We tested the GEP model35 by taking the mean expression of the GEP signature genes. We tested the IMPRES model55 by counting the number of IMPRES gene pairs that had the expected relationship (each IMPRES high expression gene having greater expression than the paired IMPRES low expression gene). We tested the MHC II model7 by performing ssGSEA using the MHC II gene list, and we z-scored the ssGESEA score so that hazard ratios would be more interpretable. Finally, we tested the TIDE model56 by calculating TIDE scores using the TIDE web portal (http://tide.dfci.harvard.edu/). The TIDE portal includes separate models for melanoma samples from patients that are immunotherapy naive or had prior immunotherapy. Additionally, mean centering is recommended as a preprocessing step. Thus, we separately mean-centered the batch-effects corrected log2(TPM+1) data for patients with prior CTLA-4 treatment and patients with no prior CTLA-4 treatment in the primary cohort, uploaded these matrices to the TIDE web portal and obtained TIDE scores. Separately, for the secondary cohort, we repeated the same process, and for TIDE we separately processed the patients with prior CTLA-4 in the Liu cohort (using the melanoma model for patients with prior immunotherapy) and then the combination of the CTLA4 naive patients in the Liu cohort with the Gide cohort (as there was no information regard prior immunotherapy treatment for the Gide cohort). For testing the associations between these models and outcome, we used the predictor values as described, but for visualizing the predictor values across patients, we z-scored the predictor values (Figure 4B; Figure S17A). After validating the three gene pair models, we compared the response classification performance of all pairs of models in the secondary cohort using DeLong’s test.
Finally, in order to further assess the robustness of these gene pair models, we considered a number of patient and treatment subsets. First, we tested models in the secondary cohort including the top gene pairs and treatment (PD-1 versus combined CTLA-4+PD-1) (Figures S17J and S17K). Additionally, to assess the robustness of models across cohorts, we evaluated the performance of all gene pair models and the external models within each cohort separately while including treatment (PD-1 versus CTLA-4+PD-1) as a covariate in the Gide cohort model (Figures S18A–S18K). Next, to determine the robustness of models across treatment groups, we subset all patients (primary and second cohorts combined) by treatment (CTLA-4, PD-1 or combined CTLA-4+PD-1). We included patients in the MGH cohort that were treated with PD-1+KIR combination therapy in the PD-1 patient subset. We combined the batch-effects corrected RNA-Seq data from the 154 patients in the primary cohort with batch-effects corrected RNA-Seq data from the 180 patients in the secondary cohort, and we tested the association between each model and overall survival for each treatment group. We also split PD-1 treated patients into PD-1 treated patients with prior CTLA-4 and PD-1 treated patients with no prior CTLA-47, and we repeated the Cox model analysis of overall survival for each predictive model. As we did not have prior treatment information for the Gide cohort, we did not include the Gide cohort in this analysis of PD-1 treated patients with or without prior CTLA-4 (Figures S19A–S19K).
TBX3 melanoma cell line GSEA
To identify pathways associated with TBX3 expression in melanoma, we again analyzed RNA-Seq from the 47 CCLE melanoma cell lines and ranked genes by their correlation with TBX3. Then, we performed ranked gene set enrichment analysis using the fgsea R package101 with all GO terms (Figure S20A). Negatively correlated genes were enriched for pigmentation pathways, which is consistent with TBX3 being expressed in poorly differentiated neural crest-like melanomas but not well differentiated MITF high melanomas.
Analysis of Jerby-Arnon melanoma scRNA data
To assess whether genes of interest were expressed in melanoma cells or melanoma tumor-infiltrating immune cells, we downloaded scRNA data from the Jerby-Arnon cohort59 from https://singlecell.broadinstitute.org/single_cell/study/SCP109/melanoma-immunotherapy-resistance. We plotted the expression of genes of interest using the TPM values and the pre-computed normal and malignant cell tSNE coordinates. To test whether TBX3 was higher expressed in NGFR-expressing melanoma cells (which would be consistent with TBX3 being higher expressed in poorly differentiated neural-crest-like melanoma cells), we performed Wilcoxon tests of TBX3 expression in melanoma cells versus immune cells and TBX3 expression in melanoma cells with an NGFR TPM of 0 versus TBX3 expression in melanoma cells with an NGFR TPM > 0 (Figures S20B–S20E).
Analysis of Tsoi cell line data
To assess whether TBX3 was expressed in poorly differentiated melanoma cell lines49, we used the Tsoi web portal (https://systems.crump.ucla.edu/dediff/index.php) and checked the expression of MITF and AXL, which are known to be expressed in well and poorly differentiated melanoma cells respectively, as well as TBX3. TBX3 was expressed in poorly and intermediately differentiated melanomas but not in well differentiated melanomas (Figure 4M).
Acknowledgments
First, we would like to express our deep gratitude to the patients and their families for making this study possible. We thank the Broad Genomics Platform for the WES and bulk RNA-seq performed in this study. This study was supported by grants from Broad Next-10 (N.H.), Cancer Research Institute (Clinic and Laboratory Integration Program, N.H.), Adelson Medical Research Foundation (N.H.), U54 CA224068 (N.H.), the David P. Ryan, MD, Chair funded by a gift from Arthur, Sandra and Sarah Irving (N.H.), the Paul C. Zamecnick, MD, Chair in Oncology at MGH (G.G.), NIH/NHGRI 5U54HG003067 (S.G.), and NIH/NCI R01CA208756 (N.H). We also acknowledge the Tosteson & Fund for Medical Discovery fellowship (M.S.-F.) and NIH 1T32CA207021-01 (J.H.C.).
Author contributions
Conceptualization, S.S.F., M.S.-F., M.M., G.G., and N.H.; methodology, S.S.F., M.S.-F., J.K., M.M., G.G., and N.H.; software, S.S.F., J.K., C.S., A.R., M.B.A., K.Y., I.L., L.E., O.G.S., D.L., D.R., F.A., J.C.-Z., G.H., and Z.L.; validation, S.S.F., M.S.-F., A.L.K.G., I.G., T.J.L., and E.M.B.; formal analysis, S.S.F., M.S.-F., J.K., C.S., A.R., M.B.A., K.Y., I.L., L.E., O.G.S., D.L., D.R., F.A., J.C.-Z., G.H., and Z.L.; investigation, S.S.F., M.S.-F., A.L.K.G., I.G., T.J.L., and E.M.B.; resources, S.S.F., M.S.-F., A.L.K.G., I.G., T.J.L., E.M.B., D.T.F., T.S., J.H.C., M.B.-R., M.R.H., H.C.V.v.E., S.M.B., Y.J.J., S.G., D.P.L., L.M.D., A.O.S.-R., J.A.W., K.T.F., R.J.S., and G.M.B.; data curation, S.S.F., M.S.-F., and J.K.; writing – original draft, S.S.F., M.S.-F., M.M., G.G., and N.H.; writing – review/editing, S.S.F., M.S.-F., G.M.B., M.M., G.G., and N.H.; visualization, S.S.F. and M.S.-F.; supervision, M.M., G.G., and N.H.; funding acquisition, S.G., G.G., and N.H.
Declaration of interests
S.S.F., M.S.-F., and N.H. are inventors on provisional patent application No.62/866,261 related to methods for predicting outcomes of checkpoint inhibition described in this manuscript. I.L. is a consultant for PACT Pharma Inc. O.G.S. is an employee at Gritstone Oncology. J.A.W. is an inventor on a US patent application (PCT/US17/53.717) “Methods for enhancing immune checkpoint blockade therapy by modulating the microbiome” and a patent “Targeting B Cells To Enhance Response To Immune Checkpoint Blockade” UTSC.P1412US.P1 - MDA19-023. J.A.W. reports compensation for speaker’s bureau and honoraria from Imedex, Dava Oncology, Omniprex, Illumina, Gilead, PeerView, Physician Education Resource, MedImmune, and Bristol-Myers Squibb (BMS) and serves as a consultant/advisory board member for Roche/Genentech, Novartis, AstraZeneca, GlaxoSmithKline, BMS, Merck, and Ella Therapeutics. K.T.F. serves on the Board of Directors of Clovis Oncology, Strata Oncology, Kinnate, Checkmate Pharmaceuticals, and Scorpion; Scientific Advisory Boards of PIC Therapeutics, Apricity, Tvardi, xCures, Monopteros, and Vibliome; consultant to Lilly, Takeda, and Boston Biomedical; and research funding from Novartis and Sanofi. R.J.S. serves as a consultant/advisory board member for Asana Biosciences, AstraZeneca, BMS, Eisai, Lovane, Merck, Novartis, OncoSec, Pfizer, and Replimune and receives research support from Merck. G.M.B. has served on SAB and on the steering committee for Nektar Therapeutics. She has SRAs with Olink proteomics and Palleon Pharmaceuticals and served on SAB and as a speaker for Novartis. M.M. is the scientific advisory board chair of OrigiMed, is an inventor of a patent licensed to LabCorp for EGFR mutation diagnosis, and receives research funding from Bayer, Janssen, Novo, and Ono. G.G. receives research funds from IBM and Pharmacyclics, is an inventor on patent applications related to MuTect, ABSOLUTE, MutSig, MSMuTect, MSMutSig, MSIdetect, POLYSOLVER, and TensorQTL, and is a founder and consultant and has privately held equity in Scorpion Therapeutics. N.H. holds equity in BioNTech and is an equity holder and advisor for Danger Bio/Related Sciences.
Published: January 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100500.
Contributor Information
Gad Getz, Email: gadgetz@broadinstitute.org.
Nir Hacohen, Email: nhacohen@mgh.harvard.edu.
Supplemental information
Data and code availability
Raw sequencing data (WES and RNaseq) from this study (MGH cohort) is deposited in dbGAP with accession number phs002683.v1.p1, and processed data is available at https://zenodo.org/record/5528497. For the primary cohort, publicly available data was used. The Van Allen1 WES and bulk RNA dataset used in this study is available in dbGAP database under accession number phs000452.v3.p1. The Roh2 WES dataset is available under BioProject accession number PRJNA369259. The Zaretsky3 WES dataset is available in the National Center for Biotechnology Information Sequence Read Archive under accession number SRP076315. The Riaz4 bulk RNA dataset used in this study is available under BioProject accession number PRJNA356761 or SRA SRP094781. The Hugo5 bulk RNA dataset used in this study is available in GEO (Gene Expression Omnibus) under accession number GSE78220. For the secondary cohort, publicly available data from two recent publications was used. The Gide6 bulk RNA dataset is available in the European Nucleotide Archive (ENA) under accession number PRJEB23709. The Liu7 bulk RNA dataset is available in dbGAP under accession number phs000452.v3.p1. Finally, code for creating figures is available at https://zenodo.org/record/5528497.
References
- 1.Van Allen E.M., Miao D., Schilling B., Shukla S.A., Blank C., Zimmer L., Sucker A., Hillen U., Foppen M.H.G., Goldinger S.M., et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roh W., Chen P.L., Reuben A., Spencer C.N., Prieto P.A., Miller J.P., Gopalakrishnan V., Wang F., Cooper Z.A., Reddy S.M., et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017;9:eaah3560. doi: 10.1126/scitranslmed.aah3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaretsky J.M., Garcia-Diaz A., Shin D.S., Escuin-Ordinas H., Hugo W., Hu-Lieskovan S., Torrejon D.Y., Abril-Rodriguez G., Sandoval S., Barthly L., et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riaz N., Havel J.J., Makarov V., Desrichard A., Urba W.J., Sims J.S., Hodi F.S., Martín-Algarra S., Mandal R., Sharfman W.H., et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017;171:934–949. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gide T.N., Quek C., Menzies A.M., Tasker A.T., Shang P., Holst J., Madore J., Lim S.Y., Velickovic R., Wongchenko M., et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell. 2019;35:238–255. doi: 10.1016/j.ccell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Liu D., Schilling B., Liu D., Sucker A., Livingstone E., Jerby-Arnon L., Zimmer L., Gutzmer R., Satzger I., Loquai C., et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019;25:1916–1927. doi: 10.1038/s41591-019-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le D.T., Uram J.N., Wang H., Bartlett B.R., Kemberling H., Eyring A.D., Skora A.D., Luber B.S., Azad N.S., Laheru D., et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samstein R.M., Lee C.H., Shoushtari A.N., Hellmann M.D., Shen R., Janjigian Y.Y., Barron D.A., Zehir A., Jordan E.J., Omuro A., et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019;51:202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miao D., Margolis C.A., Vokes N.I., Liu D., Taylor-Weiner A., Wankowicz S.M., Adeegbe D., Keliher D., Schilling B., Tracy A., et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 2018;50:1271–1281. doi: 10.1038/s41588-018-0200-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V., et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sade-Feldman M., Yizhak K., Bjorgaard S.L., Ray J.P., de Boer C.G., Jenkins R.W., Lieb D.J., Chen J.H., Frederick D.T., Barzily-Rokni M., et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell. 2018;175:998–1013. doi: 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keenan T.E., Burke K.P., Van Allen E.M. Genomic correlates of response to immune checkpoint blockade. Nat. Med. 2019;25:389–402. doi: 10.1038/s41591-019-0382-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Havel J.J., Chowell D., Chan T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer. 2019;19:133–150. doi: 10.1038/s41568-019-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., Sosman J.A., McDermott D.F., Powderly J.D., Gettinger S.N., et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sade-Feldman M., Jiao Y.J., Chen J.H., Rooney M.S., Barzily-Rokni M., Eliane J.P., Bjorgaard S.L., Hammond M.R., Vitzthum H., Blackmon S.M., et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017;8:1136. doi: 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miao D., Margolis C.A., Gao W., Voss M.H., Li W., Martini D.J., Norton C., Bossé D., Wankowicz S.M., Cullen D., et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359:801–806. doi: 10.1126/science.aan5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.George S., Miao D., Demetri G.D., Adeegbe D., Rodig S.J., Shukla S., Lipschitz M., Amin-Mansour A., Raut C.P., Carter S.L., et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity. 2017;46:197–204. doi: 10.1016/j.immuni.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riaz N., Havel J.J., Kendall S.M., Makarov V., Walsh L.A., Desrichard A., Weinhold N., Chan T.A. Recurrent SERPINB3 and SERPINB4 mutations in patients who respond to anti-CTLA4 immunotherapy. Nat. Genet. 2016;48:1327–1329. doi: 10.1038/ng.3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snyder A., Makarov V., Merghoub T., Yuan J., Zaretsky J.M., Desrichard A., Walsh L.A., Postow M.A., Wong P., Ho T.S., et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valero C., Lee M., Hoen D., Wang J., Nadeem Z., Patel N., Postow M.A., Shoushtari A.N., Plitas G., Balachandran V.P., et al. The association between tumor mutational burden and prognosis is dependent on treatment context. Nat. Genet. 2021;53:11–15. doi: 10.1038/s41588-020-00752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gurjao C., Tsukrov D., Imakaev M., Luquette L.J., Mirny L.A. Limited evidence of tumour mutational burden as a biomarker of response to immunotherapy. BioRxiv. 2020 doi: 10.1101/2020.09.03.260265. [DOI] [Google Scholar]
- 23.Litchfield K., Reading J.L., Puttick C., Thakkar K., Abbosh C., Bentham R., Watkins T.B.K., Rosenthal R., Biswas D., Rowan A., et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell. 2021;184:596–614. doi: 10.1016/j.cell.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chowell D., Morris L.G.T., Grigg C.M., Weber J.K., Samstein R.M., Makarov V., Kuo F., Kendall S.M., Requena D., Riaz N., et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359:582–587. doi: 10.1126/science.aao4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Łuksza M., Riaz N., Makarov V., Balachandran V.P., Hellmann M.D., Solovyov A., Rizvi N.A., Merghoub T., Levine A.J., Chan T.A., et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017;551:517–520. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin D.S., Zaretsky J.M., Escuin-Ordinas H., Garcia-Diaz A., Hu-Lieskovan S., Kalbasi A., Grasso C.S., Hugo W., Sandoval S., Torrejon D.Y., et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017;7:188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehta A., Kim Y.J., Robert L., Tsoi J., Comin-Anduix B., Berent-Maoz B., Cochran A.J., Economou J.S., Tumeh P.C., Puig-Saus C., Ribas A. Immunotherapy Resistance by Inflammation-Induced Dedifferentiation. Cancer Discov. 2018;8:935–943. doi: 10.1158/2159-8290.CD-17-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodig S.J., Gusenleitner D., Jackson D.G., Gjini E., Giobbie-Hurder A., Jin C., Chang H., Lovitch S.B., Horak C., Weber J.S., et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018;10:eaar3342. doi: 10.1126/scitranslmed.aar3342. [DOI] [PubMed] [Google Scholar]
- 29.Gao J., Shi L.Z., Zhao H., Chen J., Xiong L., He Q., Chen T., Roszik J., Bernatchez C., Woodman S.E., et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167:397–404. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thommen D.S., Koelzer V.H., Herzig P., Roller A., Trefny M., Dimeloe S., Kiialainen A., Hanhart J., Schill C., Hess C., et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018;24:994–1004. doi: 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang A.C., Postow M.A., Orlowski R.J., Mick R., Bengsch B., Manne S., Xu W., Harmon S., Giles J.R., Wenz B., et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petitprez F., de Reyniès A., Keung E.Z., Chen T.W., Sun C.M., Calderaro J., Jeng Y.M., Hsiao L.P., Lacroix L., Bougoüin A., et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577:556–560. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 33.Cabrita R., Lauss M., Sanna A., Donia M., Skaarup Larsen M., Mitra S., Johansson I., Phung B., Harbst K., Vallon-Christersson J., et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 34.Wu T.D., Madireddi S., de Almeida P.E., Banchereau R., Chen Y.J., Chitre A.S., Chiang E.Y., Iftikhar H., O’Gorman W.E., Au-Yeung A., et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature. 2020;579:274–278. doi: 10.1038/s41586-020-2056-8. [DOI] [PubMed] [Google Scholar]
- 35.Cristescu R., Mogg R., Ayers M., Albright A., Murphy E., Yearley J., Sher X., Liu X.Q., Lu H., Nebozhyn M., et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362:eaar3593. doi: 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anagnostou V., Bruhm D.C., Niknafs N., White J.R., Shao X.M., Sidhom J.W., Stein J., Tsai H.L., Wang H., Belcaid Z., et al. Integrative Tumor and Immune Cell Multi-omic Analyses Predict Response to Immune Checkpoint Blockade in Melanoma. Cell Rep Med. 2020;1:100139. doi: 10.1016/j.xcrm.2020.100139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gandara D.R., Paul S.M., Kowanetz M., Schleifman E., Zou W., Li Y., Rittmeyer A., Fehrenbacher L., Otto G., Malboeuf C., et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018;24:1441–1448. doi: 10.1038/s41591-018-0134-3. [DOI] [PubMed] [Google Scholar]
- 38.Hellmann M.D., Nathanson T., Rizvi H., Creelan B.C., Sanchez-Vega F., Ahuja A., Ni A., Novik J.B., Mangarin L.M.B., Abu-Akeel M., et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell. 2018;33:843–852. doi: 10.1016/j.ccell.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cancer Genome Atlas Network Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davoli T., Uno H., Wooten E.C., Elledge S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017;355:eaaf8399. doi: 10.1126/science.aaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Helmink B.A., Reddy S.M., Gao J., Zhang S., Basar R., Thakur R., Yizhak K., Sade-Feldman M., Blando J., Han G., et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577:549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen J., Tan Y., Sun F., Hou L., Zhang C., Ge T., Yu H., Wu C., Zhu Y., Duan L., et al. Single-cell transcriptome and antigen-immunoglobin analysis reveals the diversity of B cells in non-small cell lung cancer. Genome Biol. 2020;21:152. doi: 10.1186/s13059-020-02064-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen P.-L., Roh W., Reuben A., Cooper Z.A., Spencer C.N., Prieto P.A., Miller J.P., Bassett R.L., Gopalakrishnan V., Wani K., et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016;6:827–837. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mariathasan S., Turley S.J., Nickles D., Castiglioni A., Yuen K., Wang Y., Kadel E.E., III, Koeppen H., Astarita J.L., Cubas R., et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S.T., Cristescu R., Bass A.J., Kim K.M., Odegaard J.I., Kim K., Liu X.Q., Sher X., Jung H., Lee M., et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018;24:1449–1458. doi: 10.1038/s41591-018-0101-z. [DOI] [PubMed] [Google Scholar]
- 47.Kim J., Kwiatkowski D., McConkey D.J., Meeks J.J., Freeman S.S., Bellmunt J., Getz G., Lerner S.P. The Cancer Genome Atlas Expression Subtypes Stratify Response to Checkpoint Inhibition in Advanced Urothelial Cancer and Identify a Subset of Patients with High Survival Probability. Eur. Urol. 2019;75:961–964. doi: 10.1016/j.eururo.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 48.Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R., et al. TCGA Research Network Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. 2018;174:1033. doi: 10.1016/j.cell.2018.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsoi J., Robert L., Paraiso K., Galvan C., Sheu K.M., Lay J., Wong D.J.L., Atefi M., Shirazi R., Wang X., et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell. 2018;33:890–904. doi: 10.1016/j.ccell.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konieczkowski D.J., Johannessen C.M., Abudayyeh O., Kim J.W., Cooper Z.A., Piris A., Frederick D.T., Barzily-Rokni M., Straussman R., Haq R., et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4:816–827. doi: 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hernandez S., Qing J., Thibodeau R.H., Du X., Park S., Lee H.M., Xu M., Oh S., Navarro A., Roose-Girma M., et al. The Kinase Activity of Hematopoietic Progenitor Kinase 1 Is Essential for the Regulation of T Cell Function. Cell Rep. 2018;25:80–94. doi: 10.1016/j.celrep.2018.09.012. [DOI] [PubMed] [Google Scholar]
- 53.Liu J., Curtin J., You D., Hillerman S., Li-Wang B., Eraslan R., Xie J., Swanson J., Ho C.P., Oppenheimer S., et al. Critical role of kinase activity of hematopoietic progenitor kinase 1 in anti-tumor immune surveillance. PLoS ONE. 2019;14:e0212670. doi: 10.1371/journal.pone.0212670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ayers M., Lunceford J., Nebozhyn M., Murphy E., Loboda A., Kaufman D.R., Albright A., Cheng J.D., Kang S.P., Shankaran V., et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Auslander N., Zhang G., Lee J.S., Frederick D.T., Miao B., Moll T., Tian T., Wei Z., Madan S., Sullivan R.J., et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med. 2018;24:1545–1549. doi: 10.1038/s41591-018-0157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang P., Gu S., Pan D., Fu J., Sahu A., Hu X., Li Z., Traugh N., Bu X., Li B., et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rooney M.S., Shukla S.A., Wu C.J., Getz G., Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jerby-Arnon L., Shah P., Cuoco M.S., Rodman C., Su M.J., Melms J.C., Leeson R., Kanodia A., Mei S., Lin J.R., et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell. 2018;175:984–997. doi: 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boshuizen J., Vredevoogd D.W., Krijgsman O., Ligtenberg M.A., Blankenstein S., de Bruijn B., Frederick D.T., Kenski J.C.N., Parren M., Brüggemann M., et al. Reversal of pre-existing NGFR-driven tumor and immune therapy resistance. Nat. Commun. 2020;11:3946. doi: 10.1038/s41467-020-17739-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peres J., Prince S. The T-box transcription factor, TBX3, is sufficient to promote melanoma formation and invasion. Mol. Cancer. 2013;12:117. doi: 10.1186/1476-4598-12-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bagaev A., Kotlov N., Nomie K., Svekolkin V., Gafurov A., Isaeva O., Osokin N., Kozlov I., Frenkel F., Gancharova O., et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell. 2021;39:845–865. doi: 10.1016/j.ccell.2021.04.014. [DOI] [PubMed] [Google Scholar]
- 63.Sayaman R.W., Saad M., Thorsson V., Hu D., Hendrickx W., Roelands J., Porta-Pardo E., Mokrab Y., Farshidfar F., Kirchhoff T., et al. Germline genetic contribution to the immune landscape of cancer. Immunity. 2021;54:367–386. doi: 10.1016/j.immuni.2021.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uhlen M., Karlsson M.J., Zhong W., Tebani A., Pou C., Mikes J., Lakshmikanth T., Forsström B., Edfors F., Odeberg J., et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science. 2019;366:eaax9198. doi: 10.1126/science.aax9198. [DOI] [PubMed] [Google Scholar]
- 65.Birger C., Hanna M., Salinas E., Neff J., Saksena G., Livitz D., Rosebrock D., Stewart C., Leshchiner I., Baumann A., et al. FireCloud, a scalable cloud-based platform for collaborative genome analysis: Strategies for reducing and controlling costs. bioRxiv. 2017 doi: 10.1101/209494. [DOI] [Google Scholar]
- 66.Cibulskis K., McKenna A., Fennell T., Banks E., DePristo M., Getz G. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics. 2011;27:2601–2602. doi: 10.1093/bioinformatics/btr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saunders C.T., Wong W.S., Swamy S., Becq J., Murray L.J., Cheetham R.K. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 69.Taylor-Weiner A., Stewart C., Giordano T., Miller M., Rosenberg M., Macbeth A., Lennon N., Rheinbay E., Landau D.A., Wu C.J., Getz G. DeTiN: overcoming tumor-in-normal contamination. Nat. Methods. 2018;15:531–534. doi: 10.1038/s41592-018-0036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramos A.H., Lichtenstein L., Gupta M., Lawrence M.S., Pugh T.J., Saksena G., Meyerson M., Getz G. Oncotator: cancer variant annotation tool. Hum. Mutat. 2015;36:E2423–E2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Costello M., Pugh T.J., Fennell T.J., Stewart C., Lichtenstein L., Meldrim J.C., Fostel J.L., Friedrich D.C., Perrin D., Dionne D., et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013;41:e67. doi: 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kent W.J. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lawrence M.S., Stojanov P., Mermel C.H., Robinson J.T., Garraway L.A., Golub T.R., Meyerson M., Gabriel S.B., Lander E.S., Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim J., Mouw K.W., Polak P., Braunstein L.Z., Kamburov A., Kwiatkowski D.J., Rosenberg J.E., Van Allen E.M., D’Andrea A., Getz G. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016;48:600–606. doi: 10.1038/ng.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shukla S.A., Rooney M.S., Rajasagi M., Tiao G., Dixon P.M., Lawrence M.S., Stevens J., Lane W.J., Dellagatta J.L., Steelman S., et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 2015;33:1152–1158. doi: 10.1038/nbt.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jurtz V., Paul S., Andreatta M., Marcatili P., Peters B., Nielsen M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017;199:3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carter S.L., Cibulskis K., Helman E., McKenna A., Shen H., Zack T., Laird P.W., Onofrio R.C., Winckler W., Weir B.A., et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leshchiner I., Livitz D., Gainor J.F., Rosebrock D., Spiro O., Martinez A., Mroz E., Lin J.J., Stewart C., Kim J., et al. Comprehensive analysis of tumour initiation, spatial and temporal progression under multiple lines of treatment. bioRxiv. 2019 doi: 10.1101/508127. [DOI] [Google Scholar]
- 81.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Graubert A., Aguet F., Ravi A., Ardlie K.G., Getz G. RNA-SeQC 2: Efficient RNA-seq quality control and quantification for large cohorts. Bioinformatics. 2021;37:3048–3050. doi: 10.1093/bioinformatics/btab135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 84.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bolotin D.A., Poslavsky S., Mitrophanov I., Shugay M., Mamedov I.Z., Putintseva E.V., Chudakov D.M. MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods. 2015;12:380–381. doi: 10.1038/nmeth.3364. [DOI] [PubMed] [Google Scholar]
- 86.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ellrott K., Bailey M.H., Saksena G., Covington K.R., Kandoth C., Stewart C., Hess J., Ma S., Chiotti K.E., McLellan M., et al. MC3 Working Group. Cancer Genome Atlas Research Network Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018;6:271–281. doi: 10.1016/j.cels.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A., et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kasar S., Kim J., Improgo R., Tiao G., Polak P., Haradhvala N., Lawrence M.S., Kiezun A., Fernandes S.M., Bahl S., et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat. Commun. 2015;6:8866. doi: 10.1038/ncomms9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tan V.Y.F., Févotte C. Automatic relevance determination in nonnegative matrix factorization with the β-divergence. IEEE Trans. Pattern Anal. Mach. Intell. 2013;35:1592–1605. doi: 10.1109/TPAMI.2012.240. [DOI] [PubMed] [Google Scholar]
- 91.Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., et al. Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alexandrov L.B., Nik-Zainal S., Wedge D.C., Campbell P.J., Stratton M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246–259. doi: 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hoof I., Peters B., Sidney J., Pedersen L.E., Sette A., Lund O., Buus S., Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nielsen M., Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8:33. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Landau D.A., Carter S.L., Stojanov P., McKenna A., Stevenson K., Lawrence M.S., Sougnez C., Stewart C., Sivachenko A., Wang L., et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Leshchiner I., Livitz D., Gainor J.F., Rosebrock D., Spiro O., Martinez A., Mroz E., Lin J.J., Stewart C., Kim J., et al. Comprehensive analysis of tumour initiation, spatial and temporal progression under multiple lines of treatment. bioRxiv. 2019 doi: 10.1101/508127. [DOI] [Google Scholar]
- 97.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bolotin D.A., Poslavsky S., Davydov A.N., Frenkel F.E., Fanchi L., Zolotareva O.I., Hemmers S., Putintseva E.V., Obraztsova A.S., Shugay M., et al. Antigen receptor repertoire profiling from RNA-seq data. Nat. Biotechnol. 2017;35:908–911. doi: 10.1038/nbt.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mason S.J., Graham N.E. Areas beneath the relative operating characteristics (ROC) and relative operating levels (ROL) curves: Statistical significance and interpretation. Q. J. R. Meteorolog. Soc. A. 2002 doi: 10.1256/003590002320603584. [DOI] [Google Scholar]
- 101.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.M., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021 doi: 10.1101/060012. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data (WES and RNaseq) from this study (MGH cohort) is deposited in dbGAP with accession number phs002683.v1.p1, and processed data is available at https://zenodo.org/record/5528497. For the primary cohort, publicly available data was used. The Van Allen1 WES and bulk RNA dataset used in this study is available in dbGAP database under accession number phs000452.v3.p1. The Roh2 WES dataset is available under BioProject accession number PRJNA369259. The Zaretsky3 WES dataset is available in the National Center for Biotechnology Information Sequence Read Archive under accession number SRP076315. The Riaz4 bulk RNA dataset used in this study is available under BioProject accession number PRJNA356761 or SRA SRP094781. The Hugo5 bulk RNA dataset used in this study is available in GEO (Gene Expression Omnibus) under accession number GSE78220. For the secondary cohort, publicly available data from two recent publications was used. The Gide6 bulk RNA dataset is available in the European Nucleotide Archive (ENA) under accession number PRJEB23709. The Liu7 bulk RNA dataset is available in dbGAP under accession number phs000452.v3.p1. Finally, code for creating figures is available at https://zenodo.org/record/5528497.