

Abstract
Menkes disease (MD) is an X-linked recessive disorder caused by mutations in ATP7A. Patients with MD exhibit severe neurological and connective tissue disorders due to copper deficiency and typically die before 3 years of age. Early treatment with copper injections during the neonatal period, before the occurrence of neurological symptoms, can alleviate neurological disturbances to some degree. We investigated whether early symptoms can help in the early diagnosis of MD. Abnormal hair growth, prolonged jaundice, and feeding difficulties were observed during the neonatal period in 20 of 69, 16 of 67, and 3 of 18 patients, respectively. Only three patients visited a physician during the neonatal period; MD diagnosis was not made at that point. The mean age at diagnosis was 8.7 months. Seven patients, who were diagnosed in the prenatal stage or soon after birth, as they had a family history of MD, received early treatment. No diagnosis was made based on early symptoms, highlighting the difficulty in diagnosing MD based on symptoms observed during the neonatal period. Patients who received early treatment lived longer than their elderly relatives with MD. Three patients could walk and did not have seizures. Therefore, effective newborn screening for MD should be prioritized.
Keywords: Menkes disease, Early treatment, Early symptom, Prognosis
Abbreviations: MD, Menkes disease; NBS, newborn screening; PCR, polymerase chain reaction; WISC-IV, Wechsler Intelligence Scale for Children, fourth edition; DEDTC, diethyldithiocarbamate
1. Introduction
Menkes disease (MD, MNK; MIN# 309400) is an X-linked recessive disorder of copper metabolism characterized by progressive neurodegeneration, connective tissue disorders, and abnormal hair growth [[1], [2], [3]]. MD is extremely rare, with an incidence of 1 in 360,000 individuals in Japan [4]. MD is caused by a dysfunction in ATP7A due to mutations in ATP7A (MIM# 300011), located on chromosome Xq13.3 [5,6]. ATP7A transports copper from the cytosol to the Golgi apparatus, which is then incorporated into copper-dependent enzymes and secreted from the cells. An ATP7A mutation in intestinal cells leads to the accumulation of copper in the intestine and failure to absorb copper, resulting in copper deficiency in the body, including the brain. Copper deficiency leads to reduced activities of copper-dependent enzymes, such as cytochrome c oxidase, lysyl oxidase, and dopamine β-hydroxylase, which are associated with most of the clinical features of MD [3,7]. Without treatment, the neurological disturbances appear from 2 to 3 months of age. Most of these patients remain bedridden and suffer from intractable seizures, eating difficulties, and/or intracranial bleeding due to arterial abnormalities [8,9]. Most patients who do not receive a timely diagnosis of or treatment for MD die by the age of 3 years (Fig. 1).
Fig. 1.
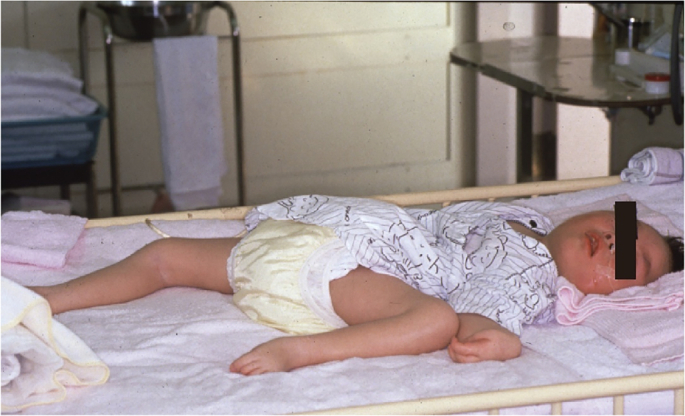
A 3-year-old patient with Menkes disease. He is bedridden and suffers from intractable seizures.
The current treatment for MD is parenteral administration of copper-histidine injections. Although the initiation of treatment after the neonatal period does not alleviate neurological disturbances, if therapy is initiated during the neonatal period, the neurological symptoms can be prevented to some degree with an improved clinical course [[10], [11], [12], [13], [14], [15], [16], [17], [18], [19]]. Thus, the early initiation of treatment is particularly important. A history of MD in family members, such as elder brothers, cousins, or uncles previously diagnosed with MD, makes it easier to make a diagnosis in infants during the prenatal or neonatal period. However, these cases are extremely rare. In most cases, a diagnosis of MD is made when the patient is 2 months old following the appearance of neurological disturbances, including seizures.
To diagnose MD in the neonatal period, it is crucial to pay attention to the mild symptoms and signs of MD and then examine the patient for MD. To help realize such early detection, in this study, we investigated whether symptoms and signs can serve as useful clues for the diagnosis of MD before the onset of neurological disturbances. Herein, we describe the clinical characteristics of seven patients with MD who received early treatment. Our results suggest that it is difficult to diagnose MD during the neonatal period, although some symptoms can be referred to during diagnosis in this period. Three of the seven patients who have been treated since the neonatal period are still alive at 10–14 years of age, and their neurological disturbances are mild, highlighting the importance of early diagnosis and treatment.
2. Material and methods
2.1. Subjects
Clinical data of 69 patients with MD were obtained from a questionnaire survey performed in major hospitals throughout Japan in 2013 [20]. The survey was performed with the assistance and written informed consent of guardians of patients between the ages of 0 months and approximately 15 years and 6 months.
Copper concentration determination in cultured fibroblasts and gene analysis were performed at the Department of Pediatrics, Teikyo University School of Medicine. All diagnoses of MD were made following clinical examinations, copper concentration measurement in cultured fibroblasts, clinical symptom assessments, and/or genetic analyses. Mutations in ATP7A were identified in 66 patients [14]. Six pairs of patients were siblings, and one patient had a maternal uncle with MD. The remaining patients were unrelated to each other.
Control skins were obtained at the time of operation from pediatric patients who had no metabolic or operative diseases such as inguinal hernia.
2.2. Diagnosis of MD
2.2.1. Serum concentrations of copper and ceruloplasmin
The serum concentration of copper was determined using a colorimetric method [21], and the ceruloplasmin concentration was determined by immunonephelometry using a Behring BNII nephelometer (Dade Behring GmbH, Marburg, Germany).
2.2.2. Copper concentration in cultured skin fibroblasts
The copper concentration in cultured skin fibroblasts was analyzed according to a previously described method [22]. In brief, fibroblasts were obtained via biopsy from patients with MD or control subjects and were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). The cells were cultured in humidified air with a CO2 level of 5% and a temperature of 37 °C. Skin fibroblasts derived from 28 patients with MD and 21 control subjects were used in this analysis. When the cultured skin fibroblasts reached confluence, the culture medium was replaced with fresh culture medium. After incubation for 17 h, the cells were harvested using 0.25% trypsin solution, washed, and disrupted by sonification. The disrupted cell solution was digested with 60% nitric acid, and the copper concentration was determined using atomic absorption spectrometry (Z-8100; Hitachi, Tokyo, Japan). The protein concentration in the cell solution was measured using the Lowry method [23].
2.2.3. Analysis of ATP7A mutations
Genomic DNA and/or total RNA were isolated from blood, cord blood, Epstein-Barr virus-transformed lymphoblasts, cultured skin fibroblasts, or amniotic fluid cells using standard protocols. Polymerase chain reaction (PCR)-single strand conformation polymorphism analysis was performed in the 5′-upstream region, each of the 23 exons, and the adjacent intronic sequences of ATP7A. The PCR products were then directly sequenced using a genetic analyzer (ABI PRISM 3130xl Genetic Analyzer; Applied Biosystems, Foster City, CA, USA) as described previously [14].
2.3. Clinical data
Clinical data of all patients were obtained from medical records or medical record summaries courtesy of their pediatricians.
2.4. Statistical analysis
Data of patients with MD were compared with those of control infants/children. Group means were compared using the t-test of BellCurve for Excel. A two-tailed P value of <0.05 was considered to indicate statistical significance.
3. Results
3.1. Diagnosis of MD
3.1.1. Serum concentrations of copper and ceruloplasmin
The serum concentrations of copper and ceruloplasmin in healthy infants under the age of 3 months were lower than those in older infants (Table 1) [13]. The serum concentrations of copper and ceruloplasmin were considerably lower in most patients with MD under 3 months of age than in healthy infants. However, their concentrations in some patients were indistinguishable from those in the controls, as shown in Table 1.
Table 1.
Serum concentrations of copper and ceruloplasmin at diagnosis.
| 0–3 months of age |
>3 months of age |
|||
|---|---|---|---|---|
| Menkes diseasea (n = 8) | Controlb | Menkes disease (n = 52) | Control | |
| Copper (μg/dL) | 16.4 ± 7.4 (8–30) | 20–70 | 6.9 ± 3.4 (3–12) | 75–150 |
| Ceruloplasmin (mg/dL) | 6.9 ± 3.4 (3–12) | 5–20 | 7.4 ± 3.3 (2.8–20) | 20–45 |
Data represent mean ± standard deviation; the range (minimum–maximum) is shown in parentheses.
Control values are based on the data from Kaler and DiStasio [13].
3.1.2. Copper concentration in cultured fibroblasts
The copper concentration in the cultured fibroblasts from patients with MD (149.31 ± 16.1 ng/mg protein, range 73.4–520.9 ng/mg protein) was significantly higher than that in the cultured fibroblasts from the control subjects (17.05 ± 1.52 ng/mg protein, range 6.45–29.4 ng/mg protein, p = 6.88 × 10−9). The copper concentration in the cultured fibroblasts from all patients was significantly higher than the maximum concentration in the cultured fibroblasts from the control subjects.
3.1.3. Gene analysis
Following gene analysis, mutations in ATP7A were identified in 65 of the 69 patients. However, a diagnosis could not be confirmed based on the genetic analysis in four patients (5%) because no mutations were detected [14]. In these patients, the diagnosis was based on clinical manifestations and biochemical examinations. A diagnosis of MD was made for seven patients during the prenatal or neonatal period, because their elder brother, cousin, or uncle had previously been diagnosed with MD.
3.2. Clinical manifestations
3.2.1. Status at birth
Table 2 shows the gestational age and birth weight of each patient with MD. Thirty-four percent of the patients with MD were born at less than 37 weeks of gestation, which is substantially higher than the national rate of 6.1% in Japan [15]. The prevalence of low birth weight in patients with MD was 33%, compared with 8.2% in the control individuals. In addition, 19% of the patients with MD were not only born preterm at less than 37 weeks gestation but also presented low birth weights (<2500 g). This phenomenon is likely to be more frequent in patients with MD than in healthy individuals.
Table 2.
Gestation age and birth weight of patients with Menkes disease (prevalence).
| Number (%) | Mean ± SD | Range | National data in Japan (1998–2011)a | |
|---|---|---|---|---|
| Gestation age (n = 69) | 37 w 2 d ± 12 d | 28 w–40 w 1 d | ||
| <37 weeks | 23 (34) | 35 w 5 d ± 2 d | 28 w–36 w 6 d | 6.1% |
| ≥37 weeks | 46 (68) | 38 w 1 d ± 6 d | 37 w–40 w 1 d | 93.9% |
| Birth weight (n = 68) | 2634 ± 397 g | 1105–3682 g | 3076 g | |
| <2500 g | 22 (33) | 2212 ± 107 g | 1105–2496 g | 8.2% |
| ≥2500 g | 45 (67) | 2836 ± 240 g | 2505–3682 g | 91.8% |
| <37 weeks and <2500 g | 13 (19) | 3.8% |
mhlw.go.jp: https://www.mhlw.go.jp/toukei/list/81-1.html (1998–2011) mean values [15].
3.2.2. Comparison between age at first visit to a physician due to symptoms and age at diagnosis
Fig. 2 shows the age at first visit of the patients to a physician due to the onset of symptoms and signs and the age at which MD was diagnosed in 64 patients (the seven patients who had a family history of MD were excluded from this analysis). The mean ages of patients at their first visit to a physician (in relation to minor symptoms and signs) and diagnosis of MD were 3.4 (range 0–9) and 8.7 (range 0–84) months, respectively. The mean time from the first visit of the patients to a physician in relation to minor symptoms and signs to diagnosis was 5.8 (range 0–81) months.
Fig. 2.

Age of 64 patients at the first visit to a physician due to the onset of symptoms and signs and the age at which they were diagnosed with Menkes disease. <1 month represents 0–29 days old, 1 month represents 30–59 days old, and so on.
Only three individuals, excluding those who had a family history of MD, visited a physician in relation to minor symptoms and signs during the neonatal period. These patients were diagnosed with MD between the ages of 1 and 80 months. One patient who had convulsions and respiratory distress at the first visit was subsequently diagnosed with MD at under 1 month of age. Although two of the three patients displayed hair abnormalities (pili torti, kinky, and depigmented hair), hypotonia, and/or respiratory distress, the diagnosis of MD was only confirmed at 11 and 80 months of age, respectively. Only three patients visited a physician in relation to minor symptoms and signs between the ages of 30 and 59 days. Ten patients with symptoms and signs between the ages of 2 and 3 months visited physicians; their symptoms were hypothermia, poor sucking, and hair growth abnormalities. One of these patients who was diagnosed with MD soon after presentation to a physician also suffered from convulsions. The diagnosis for the other nine patients was made between the ages of 3 and 7 months. Only 26 of the 64 patients (38%) were diagnosed with MD within 1 month of their first visit to a physician. However, it took more than 6 months for 11 of the 68 patients (16%) to receive the diagnosis following their first visit to a physician, and three of these patients (27%) exhibited some symptoms at birth or during the neonatal period.
3.2.3. Clinical presentations
Table 3 shows the cumulative number of patients who experienced each symptom according to age. During the neonatal period (<1 month of age), 29% of the patients had abnormal hair growth, 24% had prolonged jaundice, 17% had feeding difficulties, 15% had hypothermia, and 3% experienced seizures. The prevalence of these symptoms increased with age. At diagnosis (0–84 months of age), most symptoms had manifested. In addition, at diagnosis, most patients exhibited artery abnormalities (54/61: 89%), cerebral atrophy (39/40: 98%), and bladder diverticulum (47/58: 81%). Magnetic resonance angiography also indicated tortuosity of the blood vessels, aneurysmal dilatations, stenoses, and ruptures of arteries in the brain and other organs.
Table 3.
Cumulative number of patients experiencing each symptom.
| Age at onset and cumulative numbers (%) of each symptom |
At diagnosis (0–84 months) | ||||
|---|---|---|---|---|---|
| 0–1 month | ~2 months | ~3 months | ≥4 months | ||
| Abnormal hair (n = 69) | 20 (29) | 26 (38) | 34 (49) | 42 (61) | 68 (99) |
| Hypothermia (n = 53) | 8 (15) | 12 (23) | 13 (25) | 14 (26) | 25 (47) |
| Muscle weakness (n = 37) | 1 (3) | 1 (3) | 3 (8) | 11 (29) | 38 (100) |
| Hypopigmented skin (n = 39) | 4 (10) | 5 (13) | 12 (31) | 15 (38) | 35 (90) |
| Seizure (n = 66) | 2 (3) | 11 (17) | 24 (36) | 38 (58) | 64 (97) |
| Developmental retardation (n = 62) | 4 (6) | 8 (13) | 14 (23) | 32 (52) | 57 (92) |
| Feeding difficulties (n = 17) | 3 (17) | 8 (44) | 12 (67) | 16 (89) | 16 (89) |
| Hypotonia (n = 15) | 2 (13) | 4 (25) | 7 (44) | 13 (81) | 13 (81) |
| Jaundice (n = 67) | 16 (24) | 16 (24) | 16 (24) | 16 (24) | 16 (24) |
| Loose skin (n = 9) | 1 (11) | 1 (11) | 1 (11) | 2 (22) | 9 (100) |
3.2.4. Clinical characteristics of seven patients who received early treatment
Table 4 shows the clinical characteristics of the seven patients who received treatment with copper-histidine injections before the age of 1 month. Following continuous treatment with copper-histidine injections, their serum concentrations of copper and ceruloplasmin were maintained at normal levels.
Table 4.
Characteristics of the seven patients who were treated during the neonatal period.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Mutation in ATP7A | NE⁎ | IVS8 + 6, t > c, splice site, skip exon 8 | g.2429-2430del, p.Leu762Aspfs*9, exon 10, | g.2298 T > A, p.Leu718*, exon 8 | NE⁎ | g.4060C > G, p.Asp1305Glu, exon 20 | g.3802 del, p.Asp1219Glufs*7, exon 18 |
| Family history | Elder brother | Elder brother | Elder brother | Elder brother | Elder brother | Uncle | Elder brother |
| Gestational age (weeks) | 37 | 37 | 3 | 37 | 34 | 36 | 37 |
| Birth weight (g) | 2148 | 2855 | 1752 | 2804 | 2128 | 2536 | 2510 |
| Age of treatment start (days) | 13 | 2 | 4 | 30 | 14 | 21 | 7 |
| Symptom at the start | Hair abnormalities | Hair abnormalities | None | Hypothermia | Hypothermia, Hair abnormalities | Hair abnormalities | Hair abnormalities |
| Age of head control (months) | No | 5 | Never | − | 4 | 6 | 9 |
| Age of sitting (months) | − | − | Never | − | 16 | 15 | 18 |
| Age of walking alone (years) | − | − | − | − | 8 | 2 | 9 |
| Mental retardation | severe | Severe | severe | Severe | mild | mild | mild |
| Speech | − | − | − | − | + | + | + |
| Convulsion | + | No | + | + | no | no | + |
| Bladder diverticula | + | + | − | + | + | + | + |
| Arterial abnormalities | + | + | + | + | + | + | |
| Skin laxity | + | + | + | + | + | + | + |
| Osteoporosis | + | + | NE | NE | + | + | + |
| Hypotonia | + | + | +++ | + | + | + | + |
| Status | Sudden death at 5 years | Death at 6 years | Death at 1 year and 10 months; trachea spasm | Death at 14 years due to sepsis | Alive at 14 years of age | Alive at 11 years of age | Alive at 10 years of age |
⁎NE: not examined.
All these patients had a family history of MD, thereby facilitating the diagnosis of MD in the prenatal stage or soon after birth. A mutation in ATP7A was identified in five of the seven patients. The type of mutation was different in each patient. Although developmental delays and neurological disturbances were observed in all patients, the severity of these symptoms varied among the patients. Patients 1–4 showed severe neurological disturbances and died between the ages of 5 and 14 years. In Patient 2, the mutation was a splice-site mutation, but neurological disturbances were severe, and the patient died at 6 years of age. However, this patient lived longer than his elder brother (who died at 1 year and 7 months) who began to receive copper-histidine injections at 5 months of age. Patients 5–7 are alive, and their neurological disturbances are mild (Fig. 3). Seizures were observed in four patients but were not observed in the other three patients (Patients 2, 5, and 6).
Fig. 3.

Image on the left is of Patient 5 at 14 years of age. Image in the center is of Patient 6 at 8 years of age. Image on the right is of Patient 7 at 10 years of age.
Patient 6 could use language to communicate at the age of 1 year and 6 months. This patient walked without support at 2 years of age, and his neurodevelopmental dysfunctions were mild at the age of 2 years and 11 months. Patient 4 also had growth hormone deficiencies and has therefore been receiving growth hormone injections since the age of 3 years. Connective disorders, including bladder diverticula and arterial abnormalities, were observed in all patients. Patient 6 had stones in the right kidney, hydronephrosis, and renal dysfunction at the age of 5 years and required a pyelolithotomy at the age of 6 years. The IQ of Patient 5 was FSIQ 71 and 58 based on the Wechsler Intelligence Scale for Children, fourth edition (WISC-IV) at the age of 5 and 14 years, respectively. The IQ of Patient 6 was FSIQ 54 (WISC-IV) at the age of 11 years. The IQ of Patient 7 was FSIQ 64, Verbal Comprehension Index 80, Perceptual Reasoning Index 66, Working Memory Index 71, and Processing Speed Index 64 at the age of 10 years.
4. Discussion
MD is generally diagnosed following the identification of a mutation in ATP7A [12]. Most cases can be diagnosed using gene analysis. Indeed, in our study, direct sequencing of ATP7A facilitated the diagnosis of 95% of the patients with MD. However, gene mutations could not be identified in 5% of the patients [14]. Although the copper concentration in cultured fibroblasts provides a definitive diagnosis of MD, this method requires expertise and thus is only carried out in a few countries. Generally, diagnoses are based on clinical symptoms, biochemical data, and imaging findings.
The current standard-of-care treatment for MD is the parenteral administration of copper-histidine. When this therapy is initiated during the neonatal period, neurological symptoms can be prevented to some degree [10]. Thus, treatment at an early stage is crucial. In this study, we examined whether symptoms and signs can provide clues that will facilitate MD diagnosis before the onset of neurological disturbances.
Premature delivery and low birth weight are frequently observed in patients with MD [2,3,16]. The results of our current study and a previous study [14] showed that more than half of the patients with MD were delivered normally without any complications. The mean gestational age was 37 weeks and 2 days and the mean birth weight was 2634 g, indicating that these patients were not severely premature. Moreover, some symptoms, including hyperbilirubinemia, hypothermia, poor sucking of milk, and abnormal hair, are observed in premature babies unaffected by MD [2,3,16]. Therefore, a diagnosis of MD can be missed in premature babies. However, as patients with MD do not tend to be severely premature, neonates who have these symptoms should be considered for the differential diagnoses of MD.
Fig. 2 shows that only 3 out of 64 patients without a family history of MD visited a physician when they were less than 1 month old. Although they had symptoms, including hair abnormalities, hypothermia, and prolonged jaundice, during the neonatal period and the first visit to a physician, a diagnosis of MD was not initially considered for these patients.
The symptoms described in Table 3 have been reported from several countries [4,12,13]. However, the frequency of each symptom at different months of age has rarely been reported. Table 3 shows the frequency of each symptom in neonates, indicating that it is difficult to suspect MD based on symptoms.
Here, correct diagnoses were only made following the appearance of convulsions, when it is too late for treatment with copper-histidine injections to be successful. One of the reasons for the late diagnoses may be the fact that most pediatricians lack MD-related knowledge and experience, as MD is an extremely rare disease.
Hair growth abnormalities, hypothermia, muscle weakness, feeding difficulties, and prolonged jaundice were also observed in some patients during the neonatal period. However, most of the patients did not visit a clinic during the neonatal period, even with the presence of these symptoms.
In this study, seven patients were diagnosed and treated in the neonatal period, and all of them were diagnosed based on the family history but not symptoms. It is also true that almost all patients who were treated from the neonatal period were diagnosed based on the family history but not symptoms, consistent with the results reported by Kaler et al. [10] and Parad et al. [30]. These findings suggest that neonatal mass screening is needed for this disease.
Screening tests for MD include analyzing the serum concentrations of copper and ceruloplasmin, in addition to the plasma dopamine/norepinephrine ratio [17,18] and/or the urinary homovanillic acid to vanillylmandelic acid ratio [18,19]. Vario et al. [12] conducted a meta-analysis of studies on neonatal diagnosis and found that the plasma catecholamine assays, such as determination of the dopamine/norepinephrine ratio and dihydroxyphenylacetic acid/dihydroxyphenylglycol ratio, showed a particularly high sensitivity. However, using plasma catecholamine analysis as a screening tool for all neonates is difficult; the analysis cannot be performed using newborns' dried blood spots, which have been used for detecting some inherited metabolic diseases. Recently, Parad et al. reported targeted next-generation sequencing for newborn screening of MD using dried blood spots, and pathogenic ATP7A variants were identified in 21/22 patients (95.5%). These findings suggest that this method could emerge as a strategy for mass screening of MD.
When copper-histidine therapies are initiated during the neonatal period, outcomes improve, but the degree of improvement varies among patients [[8], [9], [10],[24], [25], [26], [27]]. Kaler et al. [24,28] reported that seizures were observed in only 12.5% of patients who were treated early, whereas seizures were observed in 87.5%–100% of patients who received late or no treatment. Some patients exhibited near-normal neurodevelopment and neurological functions [9,10]. Moreover, the effect of early treatment may be dependent on the type of mutations in ATP7A [[28], [29], [30]]. Splice-site mutations and missense mutations display high sensitivity to early treatment, whereas the response of patients with missense mutations appears to be dependent on the effect of the mutation on the ATP7A structure [27]. Our results also showed that neurological disturbances were different among patients who had different mutations. For example, in Patient 2, the mutation in ATP7A was a splice-site mutation, which suggested skipping of exon 8. However, the patient's neurological development and outcome were poor. In contrast, the outcomes of Patient 4, who had a nonsense mutation in exon 8, and Patient 7, who had a deletion in exon 18, were good. Kaler et al. [10] suggested that patients with residual ATP7A activity had better outcomes following early copper-histidine treatments. However, currently, there is no method to examine the activity of ATP7A.
Therefore, for the early diagnosis of MD, it is important to find signs that are observed before clear clinical symptoms appear. In our study, the diagnosis of MD was confirmed for all patients who received early treatment based on a positive family history and not early symptoms, suggesting that it is exceedingly difficult to diagnose MD based on early symptoms alone. However, newborn screening (NBS) using dried blood spots has now been adopted to detect many diseases, and it can enable pre-symptomatic identification of a disease. Recently DNA-based NBS has been adopted for screening MD. Parad et al. [30] described a targeted next-generation sequencing method for the NBS of MD. However, it will take some time before a screening system using next-generation sequencers is established in Japan. Further research is required to aid the establishment of NBS for MD.
Copper-histidine injections initiated during the neonatal period are effective in preventing the onset of neurological disturbances. However, the administration of this treatment after the neonatal period is ineffective. Through experiments in macular mice, an animal model for MD, it has been found that copper accumulates in astrocytes and blood vessels in the brain, that is, components of the blood–brain barrier. Copper has also been shown to accumulate in the choroid plexus and epidermal cells in the mouse model [[31], [32], [33], [34]]. These findings suggest that copper cannot transfer from the blood vessels to neurons after the maturation of the blood–brain barrier (Fig. 4).
Fig. 4.

Copper metabolism in the intestine (upper figure) and brain (lower figure) in patients with Menkes disease. It is well known that in the intestine, copper accumulates in the epithelial cells and is not transferred inside the body. Additionally, in the brain, copper in the blood vessels presumably accumulates at the blood–brain barrier (the vascular endothelial cells and astrocytes) and is not transported to neurons.
If copper could be delivered to the Golgi apparatus within cells comprising the blood–brain barrier, it could be excreted from the components of the blood–brain barrier. The copper could then reach the neurons and be incorporated into cuproenzymes, including cytochrome c oxidase. This treatment would also be beneficial for connective tissue disorders. In MD, the connective disorders result from low lysyl oxidase activity, a secretory cuproenzyme that incorporates copper in the Golgi apparatus. If copper could be delivered to the Golgi apparatus, it would be incorporated into lysyl oxidase, and connective tissue disorders would be ameliorated.
A combination therapy using copper and lipophilic chelators, such as diethyldithiocarbamate (DEDTC), disulfiram (a dimer of DEDTC), and dimethyldithiocarbamate, has been studied using a macular mouse model. The results suggest that this combination therapy improves copper concentration and cuproenzyme activities in the brain of the macular mouse [[35], [36], [37], [38], [39]]. A combination therapy using copper injections and oral disulfiram was administered to two patients with MD at the age of 8 and 10 years, respectively. They were diagnosed with MD based on the identification of a mutation in ATP7A at the age of 8 and 6 months, respectively. Since then, they were treated with injections of copper-histidine. Neurological disturbances progressed and at the time of combination therapy initiation, they were already bedridden and suffered from convulsions. No major clinical or biochemical improvements were observed with disulfiram treatment. Therefore, initiating the combination therapy at an age of 9–13 years might be too late to alleviate neurological disturbances, such as emotional expression and behavior disorders [40]. Our results indicate that the development of an NBS test and effective treatment for MD is crucial.
5. Conclusions
Here, we report the clinical characteristics of 69 Japanese patients with MD, especially their symptoms in the neonatal period. Our results suggest that it is difficult to diagnose MD based on symptoms during the neonatal period, even in the presence of several characteristic symptoms, including hypothermia, muscle weakness, and feeding difficulties associated with MD. In our study, seven of the 69 patients with MD started treatment in the neonatal period. All of them were diagnosed based on their family histories. The neurological disturbances in the seven patients treated early were alleviated to some degree. The results suggest that effective NBS and treatment for MD should be prioritized.
Funding
This work was supported by the MHLW Research on Rare and Intractable Diseases program [grant number JPMH20FC1025], funded by the Ministry of Health, Labour, and Welfare in Japan.
Data availability statement
Data for comparison are available at Health and Labor Sciences Research Grants-in-Aid for Scientific Research on Intractable Diseases: Research on the Actual Condition of Menkes Disease and Occipital Horn Syndrome, Establishment of Early Diagnosis Criteria, and Development of Treatment Methods. 2012, Ministry of Health, Labour and Welfare: http://www.pediatric-world.com/menkes/book24nendo/soukatsu/HTML/list1.html
Author statement
All authors have read and agreed to the published version of the manuscript.
Declaration of Competing Interest
The study protocol was approved by the Institutional Review Board of Teikyo University School of Medicine (TEIRIN No. 12-014, No. 08-114, No. 10-053).The authors declare no conflict of interest. The funding body had no role in the study design; in the collection, analysis, and interpretation of the data; in the writing of the report; or in the decision to submit the paper for publication.
Acknowledgments
We thank Dr. Takashi Hamazaki (Department of Pediatrics, Osaka City University Graduate School of Medicine), Dr. Hideaki Shiraishi (Department of Pediatrics, Hokkaido University School of Medicine), and Dr. Kiyotaka Kosugiyama (Department of Pediatrics, Hokkaido University School of Medicine) for research collaboration.
Contributor Information
Chie Fujisawa, Email: chie.fujisawa@med.toho-u.ac.jp.
Hiroko Kodama, Email: h.kodama@thu.ac.jp.
References
- 1.Danks D.M., Campbell P.E., Walker-Smith J., Stevens B.J., Gillespie J.M., Blomfield J., Turner B. Menkes' kinky-hair syndrome. Lancet. 1972;299:1100–1103. doi: 10.1016/S0140-6736(72)91433-X. [DOI] [PubMed] [Google Scholar]
- 2.Chelly J., Tümer Z., Tønnesen T., Petterson A., Ishikawa-Brush Y., Tommerup N., Horn N., Monaco A.P. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet. 1993;3:14–19. doi: 10.1038/ng0193-14. [DOI] [PubMed] [Google Scholar]
- 3.Tümer Z., Møller L.B. Menkes disease. Eur. J. Hum. Genet. 2010;18:511–518. doi: 10.1038/ejhg.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu Y.H., Kodama H., Shiga K., Nakata S., Yanagawa Y., Ozawa H. A survey of Japanese patients with Menkes disease from 1990 to 2003: incidence and early signs before typical symptomatic onset, pointing the way to earlier diagnosis. J. Inherit. Metab. Dis. 2005;28:473–478. doi: 10.1007/s10545-005-0473-3. [DOI] [PubMed] [Google Scholar]
- 5.Vulpe C., Levinson B., Whitney S., Packman S., Gitschier J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 1993;3 doi: 10.1038/ng0193-7. 7–13-1310. [DOI] [PubMed] [Google Scholar]
- 6.Mercer J.F., Livingston J., Hall B., Paynter J.A., Begy C., Chandrasekharappa S., Lockhart P., Grimes A., Bhave M., Siemieniak D. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat. Genet. 1993;3:20–25. doi: 10.1038/ng0193-20. [DOI] [PubMed] [Google Scholar]
- 7.Kodama H., Murata Y., Kobayashi M. Clinical manifestations and treatment of Menkes disease and its variants. Pediatr. Int. 1999;41:423–429. doi: 10.1046/j.1442-200x.1999.01095.x. [DOI] [PubMed] [Google Scholar]
- 8.Kodama H., Fujisawa C., Bhadhprasit W. Inherited copper transport disorders: biochemical mechanisms, diagnosis, and treatment. Curr. Drug Metab. 2012;13:237–250. doi: 10.2174/138920012799320455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarkar B., Lingertat-Walsh K., Clarke J.T. Copper-histidine therapy for Menkes disease. J. Pediatr. 1993;123:828–830. doi: 10.1016/s0022-3476(05)80870-4. [DOI] [PubMed] [Google Scholar]
- 10.Kaler S.G., Holmes C.S., Goldstein D.S., Tang J., Godwin S.C., Donsante A., Liew C.J., Sato S., Patronas N. Neonatal diagnosis and treatment of Menkes disease. N. Engl. J. Med. 2008;358:605–614. doi: 10.1056/NEJMoa070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christodoulou J., Danks D.M., Sarkar B., Baerlocher K.E., Casey R., Horn N., Tümer Z., Clarke J.T. Early treatment of Menkes disease with parenteral copper-histidine: long-term follow-up of four treated patients. Am. J. Med. Genet. 1998;76:154–164. doi: 10.1002/(SICI)1096-8628(19980305)76:2<154::AID-AJMG9>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 12.Vairo F.P.E., Chwal B.C., Perini S., Ferreira M.A.P., de Freitas Lopes A.C., Saute J.A.M. A systematic review and evidence-based guideline for diagnosis and treatment of Menkes disease. Mol. Genet. Metab. 2019;126:6–13. doi: 10.1016/j.ymgme.2018.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Kaler S.G., DiStasio A.T. In: GeneReviews((R)) Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Mirzaa G., Amemiya A., editors. University of Washington; Seattle: 1993. ATP7A-related copper transport disorders; pp. 11–21. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved, Seattle. [PubMed] [Google Scholar]
- 14.Fujisawa C., Kodama H., Hiroki T., Akasaka Y., Hamanoue M. ATP7A mutations in 66 Japanese patients with Menkes disease and carrier detection: a gene analysis. Pediatr. Int. 2019;61:345–350. doi: 10.1111/ped.13817. [DOI] [PubMed] [Google Scholar]
- 15.Ministry of Health, Labour and Welfare in Japan . Ministry of Health, Labour and Welfare. Available, Ministry of Health, Labour and Welfare in Japan; 2019. Vital Statistics. [Google Scholar]
- 16.Culotta V.C., Gitlin J.D., Scriver C.R., Beaudet A.L., Sly W.S., Valle D. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. II. 2001. pp. 3105–3126. [Google Scholar]
- 17.Kaler S.G., Holmes C.S. Catecholamine metabolites affected by the copper-dependent enzyme dopamine-beta-hydroxylase provide sensitive biomarkers for early diagnosis of Menkes disease and viral-mediated ATP7A gene therapy. Adv. Pharmacol. 2013;68:223–233. doi: 10.1016/B978-0-12-411512-5.00011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuo M., Tasaki R., Kodama H., Hamasaki Y. Screening for Menkes disease using the urine HVA/VMA ratio. J. Inherit. Metab. Dis. 2005;28:89–93. doi: 10.1007/s10545-005-5083-6. [DOI] [PubMed] [Google Scholar]
- 19.Lee T., Yagi M., Kusunoki N., Nagasaka M., Koda T., Matsuo K., Yokota T., Miwa A., Shibata A., Morioka I., Kodama H., Takeshima Y., Iijima K. Standard values for the urine HVA/VMA ratio in neonates as a screen for Menkes disease. Brain Dev. 2015;37:114–119. doi: 10.1016/j.braindev.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 20.Kodama H. Health and Labor Sciences Research Grants- in Aid for Scientific Research on Intractable Diseases. Vol. 2013. Ministry of Health, Labour and Welfare; 2012. Research on the actual condition of Menkes disease and occipital horn syndrome, establishment of early diagnosis criteria, and development of treatment methods; pp. 1–72. 201231070A. [Google Scholar]
- 21.Abe A., Yamashita S., Noma A. Sensitive, direct colorimetric assay for copper in serum. Clin. Chem. (Baltimore, MD) 1989;35:552–554. doi: 10.1093/clinchem/35.4.552. [DOI] [PubMed] [Google Scholar]
- 22.Gu Y.H., Kodama H., Sato E., Mochizuki D., Yanagawa Y., Takayanagi M., Sato K., Ogawa A., Ushijima H., Lee C.C. Prenatal diagnosis of Menkes disease by genetic analysis and copper measurement. Brain Dev. 2002;24:715–718. doi: 10.1016/S0387-7604(02)00093-1. [DOI] [PubMed] [Google Scholar]
- 23.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. doi: 10.1016/S0021-9258(19)52451-6. [DOI] [PubMed] [Google Scholar]
- 24.Kaler S.G., Liew C.J., Donsante A., Hicks J.D., Sato S., Greenfield J.C. Molecular correlates of epilepsy in early diagnosed and treated Menkes disease. J. Inherit. Metab. Dis. 2010;33:583–589. doi: 10.1007/s10545-010-9118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tümer Z., Petris M., Zhu S., Mercer J., Bukrinski J., Bilz S., Baerlocher K., Horn N., Møller L.B. A 37‐year‐old Menkes disease patient—residual ATP7A activity and early copper administration as key factors in beneficial treatment. Clin. Genet. 2017;92:548–553. doi: 10.1111/cge.13083. [DOI] [PubMed] [Google Scholar]
- 26.Verrotti A., Cusmai R., Darra F., Martelli P., Accorsi P., Bergamo S., Bevivino E., Coppola G., Freri E., Grosso S., Matricardi S., Parisi P., Sartori S., Spalice A., Specchio N., Carelli A., Zini D., Dalla Bernardina B., Giordano L. Epilepsy in Menkes disease: an electroclinical long-term study of 28 patients. Epilepsy Res. 2014;108:1597–1603. doi: 10.1016/j.eplepsyres.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Tümer Z., Horn N., Tønnesen T., Christodoulou J., Clarke J.T., Sarkar B. Early copper-histidine treatment for Menkes disease. Nat. Genet. 1996;12:11–13. doi: 10.1038/ng0196-11. [DOI] [PubMed] [Google Scholar]
- 28.Kaler S.G., Tang J., Donsante A., Kaneski C.R. Translational read-through of a nonsense mutation in ATP7A impacts treatment outcome in Menkes disease. Ann. Neurol. 2009;65:108–113. doi: 10.1002/ana.21576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Møller L.B., Bukrinsky J.T., Mølgaard A., Paulsen M., Lund C., Tümer Z., Larsen S., Horn N. Identification and analysis of 21 novel disease-causing amino acid substitutions in the conserved part of ATP7A. Hum. Mutat. 2005;26:84–93. doi: 10.1002/humu.20190. [DOI] [PubMed] [Google Scholar]
- 30.Parad R.B., Kaler S.G., Mauceli E., Sokolsky T., Yi L., Bhattacharjee A. Targeted next generation sequencing for newborn screening of Menkes disease. Mol. Genet. Metab. Rep. 2020;24:100625. doi: 10.1016/j.ymgmr.2020.100625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kodama H., Murata Y. Molecular genetics and pathophysiology of Menkes disease. Pediatr. Int. 1999;41:430–435. doi: 10.1046/j.1442-200x.1999.01091.x. [DOI] [PubMed] [Google Scholar]
- 32.Kodama H., Meguro Y., Abe T., Rayner M.H., Suzuki K.T., Kobayashi S., Nishimura M. Genetic expression of Menkes disease in cultured astrocytes of the macular mouse. J. Inherit. Metab. Dis. 1991;14:896–901. doi: 10.1007/BF01800470. [DOI] [PubMed] [Google Scholar]
- 33.Kodama H. Recent developments in Menkes disease. J. Inherit. Metab. Dis. 1993;16:791–799. doi: 10.1007/BF00711911. [DOI] [PubMed] [Google Scholar]
- 34.Lenartowicz M., Krzeptowski W., Lipiński P., Grzmil P., Starzyński R., Pierzchała O., Møller L.B. Mottled mice and non-mammalian models of Menkes disease. Front. Mol. Neurosci. 2015;8:72. doi: 10.3389/fnmol.2015.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhadhprasit W., Kodama H., Fujisawa C., Hiroki T., Ogawa E. Effect of copper and disulfiram combination therapy on the macular mouse, a model of Menkes disease. J. Trace Elem. Med. Biol. 2012;26:105–108. doi: 10.1016/j.jtemb.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Nomura S., Nozaki S., Hamazaki T., Takeda T., Ninomiya E., Kudo S., Hayashinaka E., Wada Y., Hiroki T., Fujisawa C., Kodama H., Shintaku H., Watanabe Y. PET imaging analysis with 64Cu in disulfiram treatment for aberrant copper biodistribution in Menkes disease mouse model. J. Nucl. Med. 2014;55:845–851. doi: 10.2967/jnumed.113.131797. [DOI] [PubMed] [Google Scholar]
- 37.Kodama H., Sato E., Gu Y.H., Shiga K., Fujisawa C., Kozuma T. Effect of copper and diethyldithiocarbamate combination therapy on the macular mouse, an animal model of Menkes disease. J. Inherit. Metab. Dis. 2005;28:971–978. doi: 10.1007/s10545-005-0150-6. [DOI] [PubMed] [Google Scholar]
- 38.Horn N., Møller L.B., Nurchi V.M., Aaseth J. Chelating principles in Menkes and Wilson diseases: choosing the right compounds in the right combinations at the right time. J. Inorg. Biochem. 2019;190:98–112. doi: 10.1016/j.jinorgbio.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 39.Yamagishi Y., Kudo T., Oyumi M., Sakamoto Y., Takahashi K., Akashi T., Kobayashi S., Kawakami T., Goda H., Sato Y., Mimaki M., Kodama H., Munakata M., Makino K., Takahashi H., Fukami T., Ito K. Pharmacokinetics of CuGTSM, a novel drug candidate, in a mouse model of Menkes disease. Pharm. Res. 2021;38:1335–1344. doi: 10.1007/s11095-021-03090-0. [DOI] [PubMed] [Google Scholar]
- 40.Ogawa E., Kodama H. Effects of disulfiram treatment in patients with Menkes disease and occipital horn syndrome. J. Trace Elem. Med. Biol. 2012;26:102–104. doi: 10.1016/j.jtemb.2012.04.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data for comparison are available at Health and Labor Sciences Research Grants-in-Aid for Scientific Research on Intractable Diseases: Research on the Actual Condition of Menkes Disease and Occipital Horn Syndrome, Establishment of Early Diagnosis Criteria, and Development of Treatment Methods. 2012, Ministry of Health, Labour and Welfare: http://www.pediatric-world.com/menkes/book24nendo/soukatsu/HTML/list1.html