

Abstract
Background:
Death-associated protein kinase 1 (DAPK1) is a widely distributed serine/threonine (Ser/Thr) kinase that is critical for cell death in multiple neurological disorders, including Alzheimer’s disease (AD) and stroke. However, little is known about the role of DAPK1 in the pathogenesis of Parkinson’s disease (PD), the second most common neurodegenerative disorder.
Methods:
We used western blot, immunohistochemistry to evaluate the alteration of DAPK1. Q-PCR and FISH were used to analyze the expression of miRNAs in PD mice and patient. Rotarod, open field and pole tests were used to evaluate the locomotor ability. Immunofluorescence, western blot and filter trap were used to evaluate synucleinopathy in PD mice.
Results:
we find that DAPK1 is post-transcriptionally upregulated by a reduction in miR-26a caused by a loss of the C/EBPα transcription factor. The overexpression of DAPK1 in PD mice is positively correlated with neuronal synucleinopathy. Suppressing miR-26a or up-regulating DAPK1 results in synucleinopathy, DA neuron cell death and motor disabilities in wild-type (wt) mice. In contrast, genetic deletion of DAPK1 in DA neurons by crossing the DAT-Cre mice with DAPK1 floxed mice effectively rescues the abnormalities in mice with chronic MPTP treatment. We further show that DAPK1 overexpression promotes PD-like phenotypes by direct phosphorylation of α-synuclein at Ser129 site. Correspondingly, a cell-permeable competing peptide that blocks the phosphorylation of α-synuclein prevents motor disorders, synucleiopathy and dopaminergic neuron loss in the MPTP mice.
Conclusions:
We conclude that miR-26a/DAPK1 signaling cascades are essential in the formation of the molecular and cellular pathologies in PD.
Keywords: Parkinson’s disease, DAPK1, miRNA, alpha-synuclein, MPTP, peptide
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease in people over 60 years old. In 2015, PD affected 6.2 million people and resulted in approximately 117,400 deaths globally(1). The most prominent clinical manifestations in PD are the motor symptoms, such as tremor, slowed movement (bradykinesia), rigidity, and postural instability(2), which result from the chronic loss of dopaminergic (DA) neurons in the basal ganglia, mainly in the substantia nigra (SN)(3). Pathologically, PD is also characterized by the presence of Lewy bodies (LBs) and Lewy neurites (LNs), which consist of cytoplasmic inclusions of aggregated α-synuclein in a hyperphosphorylated state(4).
Death-associated protein kinase 1 (DAPK1) belongs to a family of five serine/threonine (Ser/Thr) kinases and is regulated by calcium/calmodulin(5). DAPK1 was originally shown to be essential for interferon gamma (IFN-γ)-induced cell death in HeLa cells(6), and it is considered a positive mediator of apoptosis. Both internal and external apoptotic stimulants lead to activation of DAPK1, which in turn participates in both type I apoptotic (caspase-dependent) and type II autophagic (caspase-independent) cell death(7). Over expression of exogenous DAPK1 in cell cultures results in pronounced death-associated cellular changes(8). DAPK1 is also abundant in the brain and has been reported to play important roles in neural development and multiple neurological diseases. Importantly, activated DAPK1 directly binds to and phosphorylates NMDA receptor GluN2B subunit at Ser1303, which is implicated in the excitotoxicity in ischemic stroke via up-regulating NR1/NR2B receptor channel conductance at the extrasynaptic sites(9). Moreover, activated DAPK1 directly phosphorylates p53 at Ser23 and induces necrotic and apoptotic neuronal death in stroke(10). In Alzheimer’s disease (AD), hippocampal DAPK1 expression is markedly increased, which enhances tau protein stability via phosphorylation at multiple AD-related sites to mediate the pathological toxicity of tau(11). However, whether the expression of DAPK1 in PD is dysregulated and how it is involved in the death of DA neurons remain unclear.
In the current study, we found that in the brain of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mice, the protein amounts but not the mRNA levels of DAPK1 were increased. We showed that inhibition of miR-26a caused by suppression of the transcription factor C/EBPα was responsible for DAPK1 upregulation in PD mice and patients. Downregulation of miR-26a and upregulation of DAPK1 induced synucleinopathy and cell death of DA neurons in vivo. DAPK1 induces synucleiopathy by direct phosphorylation of α-synuclein at Ser129 site. Genetic deletion of DAPK1 in DA neurons rescued synucleinopathy in two PD mice models. Furthermore, the loss of DA neurons and the locomotor deficits can also be reversed by DAPK1 deletion in mice of chronic MPTP treatment. Finally, generation of a membrane-permeable peptide to directly disrupt the association of DAPK1 and α-synuclein ameliorated both the pathological and behavioral abnormalities in the MPTP mice. Thus, our findings provide a promising novel strategy for the therapeutic intervention of PD.
Methods and Materials
Animals
Adult male C57 BL/6 mice were purchased from the National Resource Center of Model Mice (Nanjing, China). The mice with dopaminergic neuron specific knockdown of DAPK1 were generated by crossing DAPK1-KDloxp/loxp transgenic mice and DAT-cre mice (Jax lab. No. 006660). The M83 transgenic mice expresses the mutant human A53T alpha-synuclein were purchased from the Jackson Laboratory(Jax lab.No.004479).All the mice were housed under a 12-h light/12-h dark cycle in a temperature-controlled room (22–24 °C) with free access to food and water. All of the experimental procedures were approved by the Institutional Animal Care and Use Committee of the Huazhong University of Science and Technology followed AALAC, DLAR, ARRIVE, and NIH Animal Care Guidelines on IACUC approved protocols.
Generation of a highly infectious virus and the mimics/antagomir
Mmu-miR-26a-5p mimics and the scrambled control were purchased from RiboBio (Guangzhou, China) Supplementary Table 2. The mimics and antagomirs to upregulate/downregulate the level of miR-26a were used as previously reported(12). The AAV2/9-DAPK1 (1*1013vg/mL) was purchased from Allabio technology (Shanghai, China). We injected the particles (1.5 µl at 0.2 µl/min) into each side of the SN (3.0 mm posterior to bregma; 1.0 mm lateral to midline; 4.3 mm below the dura). In this study, 28 days after the virus injection, mice were used for the phenotyping assays, including miRNA, mRNA and protein expression assays, and for the behavioral tests.
Fluorescence in situ hybridization (FISH)
Multiplexed miRNA fluorescence in situ hybridization (miRNA FISH) is an advanced method for visualizing differentially expressed miRNAs, together with other reference RNAs in fresh-frozen and archival tissues. Probes were purchased from TSINGKE Supplementary Table 2. For in situ hybridization in frozen brain slices, mice were perfused with 1xPBS and 4%(v/v) paraformaldehyde solution in PBS. The brain tissues were fixed in 4%(v/v) paraformaldehyde solution in PBS overnight at 4°C, subsequently cryoprotected in 30%(w/v) sucrose in PBS(DEPC-treated) overnight at 4°C and cryosectioned at 16µm thickness. The manufacturer’s protocol was applied in situ hybridization, skipping the dehydration/rehydration and proteinase QS treatment steps. Probes were diluted to 1:50. After completion of the in situ hybridization, brain slices were blocked for 1h in blocking buffer for immunostaining. The slices were stained overnight at 4°C using primary antibodies, washed three times with 1xPBS and incubated with secondary antibody for 1h at room temperature. DAPI was added to one of the following three washing steps with 1xPBS to visualize nuclei. Slices were then mounted for microscopy (14)
Patient population and CSF collection
The PD group consisted of 28 patients with Hoehn and Yahr (H&Y) stage 1 PD(15) who attended the Department of Neurology, Union Hospital Affiliated to Tongji Medical College (Wuhan, China). Patients with PD were diagnosed according to the United Kingdom Brain Bank criteria(16) and staged according to H&Y. The clinical diagnosis of idiopathic PD was established based on individual patient’s medical history, physical examination and laboratory results. The severity of motor symptoms was assessed in PD patients using the Unified Parkinson’s Disease Rating Scale (UPDRS) part 3(17). CSF from the control group or the PD group was collected via routine lumbar puncture following confirmation of informed consent from each patient and/or a family member. Following centrifugation at 755×g for 10 min, 3–4 ml CSF samples were aliquoted and stored at −80˚C until analysis. Every CSF sample was tested by routine biochemical examination, and the results were all within the normal range. The information of PD and control brain for western blot and immunohistochemistry studies are list in Supplementary Table 4.The present study was approved by the Ethics Committee of Tongji Medical College (Wuhan, China).
Statistical Analysis
All data were shown as mean ± SD and analyzed using SPSS version 16.0 (SPSS Inc., Chicago, IL). Normality was tested with Shapiro-Wilk test, and equal variance was evaluated before analysis of variance analysis. The difference between two groups was assessed using unpaired Student’s t test (two-tailed), and the variance among multiple groups was assessed by one- or two-way analysis of variance with/without repeated measures followed by Tukey, Dunnett, Newman-Keuls, or Bonferroni post hoc test, as indicated in the figure legends. The Mann-Whitney U test (two groups) and Kruskal-Wallis H test (three or more groups) were used for nonparametric testing. All experiments were repeated three times except those specified, and p <0.05 was considered as statistically significant.
Results
DAPK1 is increased in PD mice and is positively correlated with synucleinopathy
To determine the possible role of DAPK1 in PD, we first examined the expression level of DAPK1 in the acute and chronic MPTP-injected mice. We found that in the SN of the acute MPTP-injected mice, the expression of DAPK1 was significantly increased compared with vehicle-treated control mice (Fig. 1A). By using immunohistochemistry, we found that the enhanced staining of DAPK1 was mainly located in the cytoplasm of neurons (Fig. 1B). In the chronic MPTP mouse model, DAPK1 protein expression was increased to approximately 2.3-fold of the control (Fig. 1C, D), which was more apparent than the increase in the acute group. We also examined DAPK1 expression at different time points (1 month, 2 months, 3 months) after MPTP injection in a chronic model and found that the elevation of DAPK1 protein was peaked at 1 month (Supplementary Figure 1). These results show that DAPK1 is upregulated in the MPTP mouse model. As the chronic MPTP mice displayed more obvious PD-like behavioral features and pathological changes(20) and apparent DAPK1 upregulation, we utilized the model of chronic MPTP administration for 1 month in the following experiments. We then examined the potential role of DAPK1 upregulation in the pathological changes, especially in neuronal synucleinopathy, in PD mice. By using double-immunofluorescence labeling with anti-DAPK1 and anti-phospho-Ser129-α-synuclein (anti-p-syn) or anti-α-synuclein (anti-t-syn) antibodies, we found that in the chronic MPTP mice, the immunoreactivity of DAPK1, p-syn, and t-syn was much higher than that in the vehicle-treated mice. Particularly, the neurons with stronger staining of DAPK1 also displayed enhanced immunosignals for p-syn and t-syn, and quantitative analysis suggests positive correlations between DAPK1 and p-syn and between DAPK1 and t-syn in the PD mouse model (Fig. 1E-H). In addition, we found that the α-synuclein inclusions were more prominent in neurons with higher DAPK1 expression (Fig. 1I). In addition, upregulation of DAPK1 was detected mainly in the dopaminergic neurons (Supplementary Figure 2). These findings suggest that DAPK1 upregulation is positively correlated with the progression DA neuron synucleinopathy in PD mice. We also examined the changes of DAKP1 in the substantia nigra of PD patients. While western blot and immunohistochemical analysis did not reveal any significant changes in the expression of DAPK1 between PD and age-matched healthy control patients, Lewy bodies, the hallmark pathology of PD, were occasionally stained positive for DAPK1 (Supplementary Figure 3). Given that the extent of DAPK1 upregulation declined over time in the chronic MPTP mice (Supplementary Figure 1), and the PD samples were obtained from patients at their end stage of disease, these results likely suggest that upregulation of DAPK1 in the dopaminergic neurons occurred at the early stage of PD.
Figure 1. DAPK1 is increased in the MPTP mice and is positively correlated with synucleinopathy.
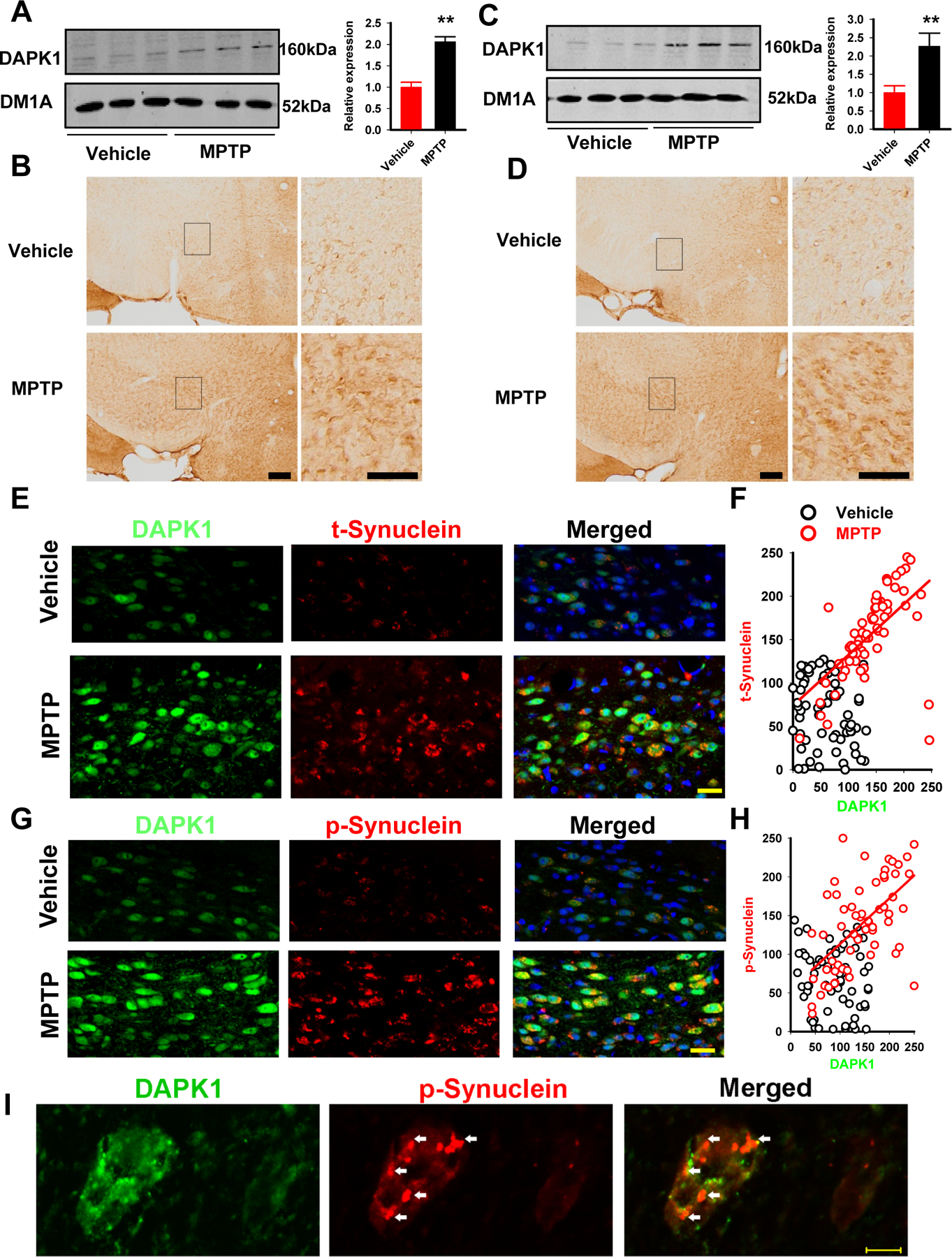
(A-B) Lysates from the SN of acute (A) and chronic (B) MPTP mice were examined by western blot with anti-DAPK1 and anti-DM1A antibodies. Representative images were shown. The quantitative analysis was performed with Student’s t-test. **P<0.01 vs vehicle-injected mice; N=3 independent experiments by using 6 mice per group.
(C-D) Immunohistochemical staining with anti-DAPK1 was performed in coronal slices from the acute (C) and chronic (D) MPTP-injected mice. The black rectangle regions in the left panels were shown in higher magnification at the right panels. Bar=50 µm; Student’s t-test; N=3 independent experiments by using 6 mice per group.
(E-F) Double immunofluorescence was performed in coronal slices from the chronic MPTP-injected mice by using the DAPK1 (green) and total α-synuclein (red) antibodies (E), and the correlation analysis (F) was performed via SigmaPlot after the fluorescent intensity measurements were acquired via ImageJ. Bar=50 µm; red and black circles are the fluorescent intensities from the MPTP- and vehicle-treated mice; R2=0.3401; P<0.001.
(G-H) Double immunofluorescence staining was performed in coronal slices from the chronic MPTP-injected mice using DAPK1 (green) and p-syn (red) antibodies (G), and the correlation analysis (H) was performed via SigmaPlot after the fluorescent intensity measurements were acquired via ImageJ. Bar=50 µm, red and black circles are the fluorescent intensities from MPTP- and vehicle-treated mice; R2=0.3728; P<0.001.
(I) Neurons with high magnification showing α-synuclein (red) inclusions (white arrow) and strong DAPK1 staining (green). The cell on the left side displays more intense DAPK1 staining. Bar=10 µm.
Reduction in miR-26a in the PD model induces DAPK1 overexpression by post-transcriptional regulation
We next explored the mechanisms underlying DAPK1 upregulation in PD mice. We found that there was no difference in dapk1 mRNA levels in both acute and chronic MPTP mouse models (Fig. 2A), suggesting that the upregulation of DAPK1 in PD mice is not caused by enhanced gene transcription. Recently, the post-transcriptional regulation of genes by microRNA (miRNA) has been well studied. We hypothesized that DAPK1 upregulation is mediated by the loss of specific miRNAs. We first analyzed the 3’ untranslated region (3’UTR) of the dapk1 gene via TargetScan7.0 and miRNA.org and found that miR-124, let-7/miR-98, miR-26a/b and miR-141 were scored the highest in both predicted outputs Supplementary Table 3. Using qPCR, we examined alterations of those miRNAs in the SN of the chronic MPTP mice. We found that the levels of miR-141 and miR-26a/b were dramatically decreased, the level of let-7e was increased, while the other members of the let-7 family, miR-98 and miR-124 showed no change (Fig. 2B). We further examined the levels of miR-141 and miR-26a/b in the CSF of PD patients, and found that only miR-26a was significantly decreased when compared to that of age-matched healthy subjects (Fig. 2C). To verify the post-transcriptional regulation of DAPK1 by miR-26a, we constructed the wild-type (wt) 3’UTR of dapk1, which contains the binding site to miR-26a, and a mutated version to the luciferase reporter vector, which were co-transfected into HEK293 cells with miR-26a mimics or a scrambled control (Fig. 2D). We found that miR-26a mimics suppressed the luciferase activity in the wt constructs but not in the mut constructs (Fig. 2E). Moreover, overexpression of miR-26a reduced, whereas the miR-26a inhibitor elevated DAPK1 protein expression in the N2a cells without changes in the dapk1 mRNA levels (Fig. 2F-H). Importantly, although the 3’UTR of DAPK1 has two predicted binding sites for miR-141, none of them displayed significant responses in the luciferase experiment. Neither the mRNA nor the protein of DAPK1 was altered upon the treatment of miR-141 mimics, which further ruled out the potential regulation of DAPK1 by miR-141 (Supplementary Figure 4). miR-26a expression and DAPK1 protein expression displayed a negative correlation in the MPTP mice (Fig. 2I). Furthermore, both the miR-26a transcript and the DAPK1 protein could be detected in the dopaminergic neurons of SN (Fig. 2J). These results suggest that miR-26a regulates DAPK1 expression at a post-transcriptional level in the DA neurons, and the loss of miR-26a mediates DAPK1 upregulation in PD mice.
Figure 2. Abnormal downregulation of miR-26 is responsible for DAPK1 elevation in PD.

(A) DAPK1 mRNA expression showed no change in the MPTP models.
(B) Alterations in predicted miRNAs that target DAPK1 in the SN of chronic MPTP-injected mice models. **P<0.01 vs vehicle; Student’s t-test; N=8 from 3 independent experiments.
(C) Alterations in miR-26a, miR-26b and miR-141 in the CSF of PD patients and aged-matched healthy subjects. **P<0.01 vs vehicle; Student’s t-test.
(D) The binding sites of miR-26a with DAPK1 3’UTR are conserved in mammalians. Mmu, mouse; Rno, rat; Rmk, Rhesus monkey; Hsa, human; Pig, pig.
(E) The wt and mut 3’UTR of DAPK1 were sub-cloned into the psi-CHECK vector and transfected into HEK293 cells, together with miR-26a mimics or its scrambled control. The luciferase intensity was measured. Unpaired Student’s t-test was used; **P<0.01 compared to the scrambled control-treated group; N=6 for each group.
(F-H) The N2a cells were treated with miR-26a mimics and its scrambled control or with the A-miR-26a and its scrambled control for 24 h. The cell lysates were collected for the detection of DAPK1 protein and mRNA expression. Representative blots (F), quantitative analysis (G), and the relative expression level of DAPK1 mRNA (H) are shown. **P<0.01 compared to the respective Con; N=6; Student’s t-test.
(I) Analysis of the correlation between the miR-26 level and DAPK1 protein levels in the brain tissue of chronic MPTP-injected mice.
(J) A representative image of localization of miR-26 and DAPK1 in the dopaminergic neurons. Bar=20µm.
A previous study identified that miR-26a transcription was positively regulated by the transcription factor CCAAT enhancer-binding protein α (C/EBPα)(21); therefore, we examined the protein levels of C/EBPα in the SN of MPTP mice and found that it decreased to ~40% of the level in the vehicle-treated mice (Supplementary Figure 5), indicating potential involvements of C/EBPα reduction in the loss of miR-26a in PD mice.
Loss of miR-26a or DAPK1 upregulation induces PD-like pathological and behavioral changes
We then asked whether the dysfunction of the miR-26a/DAPK1 signaling pathway plays an important role in the pathogenesis of PD. We injected the miR-26a antagomir (A-miR-26a) or adeno-associated virus-packaged full-length mouse DAPK1 cDNA (AAV-DAPK1) into the SN of wt mice (Fig. 3A). We found that both the A-miR-26a treatment and the AAV-DAPK1 treatment led to not only activation of DAPK1 (indicated by the intensity of pMLC according to previous studies (9)) but also hyperphosphorylation of α-synuclein at Ser129 and enhanced α-synuclein expression (Fig. 3B, C). Because abnormally hyperphosphorylated α-synuclein usually aggregates to form insoluble fibrils in LBs, a dominant pathological change in PD, we examined the solubility of α-synuclein. We found that the level of phosphorylated α-synuclein was dramatically increased in the radioimmunoprecipitation assay (RIPA) buffer fraction (soluble) and in the 70% fatty acid (FA) fraction (insoluble; Fig. 3D, E). Likewise, by using a filter trap assay, we found that α-synuclein oligomers were significantly increased in either miR-26a-inhibited or DAPK1-overexpressed mice (Fig. 3F, G). These findings suggest that the loss of miR-26a or the upregulation of DAPK1 plays an important role in DA neuron synucleinopathy in PD mice.
Figure 3. Overexpression of DAPK1 or inhibition of miR-26a induces synucleinopathy.

(A) Diagram of the virus injection site (upper panel) and a representative fluorescence image (lower panel).
(B-C) Effects of DAPK1 overexpression (B) and miR-26a inhibition (C) on the phosphorylation level and expression of α-synuclein in wt mice. The upper panels are representative blots, and the lower panels are the quantified data. Vector: AAV-EGFP virus; DAPK1: AAV-DAPK1-IRES-EGFP virus; Scramble: the scrambled control for miR-26a antagomer; A-miR-26a: miR-26a antagomer. **P<0.01 vs vector (B) or scramble (C); Student’s t-test; N=8 from 3 independent experiments.
(D-E) Effects of DAPK1 overexpression (D) and miR-26a inhibition (E) on the solubility of α-synuclein. The left panels are representative blots, and the right panels are the quantified data (left side of the blue dash for the 70% FA, right side for RIPA). **P<0.01 vs vector (D) or scramble (E); Student’s t-test; N=8 from 3 independent experiments.
(F-G) The filter trap experiments were used to detect α-synuclein oligomer formation with DAPK1 overexpression (F) or miR-26a inhibition (G).
As α-synuclein aggregates are known to be cytotoxic, causing cell death of DA neurons in the SN and the striatum of PD patients, we then examined the loss of DA neurons by immunostaining with anti-tyrosine hydroxylase (TH). We found that the total number of TH-positive neurons in the SN was dramatically reduced in the A-miR-26a- and AAV-DAPK1-treated mice (Fig. 4A-D). Because the loss of neurons in the SN is known to result in the motor symptoms in PD, we employed an open field arena test to observe spontaneous movements and a rotarod test to analyze motor coordination. In the rotarod tasks, the mice treated with A-miR-26a or AAV-DAPK1 demonstrated a significant decrease in the time spent on an accelerating rotarod and the overall rotarod performance (ORP) compared with those outcomes in the controls (P<0.05)(22, 23) (Fig. 4E-H). This locomotor abnormality was also manifested in the open field experiments. The total distance traveled, mean velocity and rearing time were all significantly decreased upon A-miR-26a or AAV-DAPK1 treatment (Fig. 4I-N). These findings suggest that the loss of miR-26a or DAPK1 overexpression not only leads to DA neuron death but also causes impairments in locomotor functions.
Figure 4. Overexpression of DAPK1 or inhibition of miR-26a induces DA neuron death and locomotor disabilities.

(A-B) Immunohistochemistry of TH staining in the SN of mice treated with A-miR-26a or the Con (scrambled control). (A) Representative image and (B) the quantification of TH-positive neurons. Bar=100 µm; **P<0.01 vs Con.
(C-D) Immunohistochemistry of TH staining in the SN of mice treated with AAV-DAPK1 or the vector control. Vector: AAV-EGFP virus; DAPK1: AAV-DAPK1-IRES-EGFP virus. (C) A representative image and (D) the quantification of TH-positive neurons. Bar=100 µm; ** P<0.01 vs vector; Student’s t-test.
(E-F) Mice were injected with A-miR-26a or the Con, and the rotarod test was performed. (E) Time spent on the rod by mice in each group at different rotation speeds. (F) ORP scores in two groups. **P<0.01 vs Con; N=10; one-way ANOVA with Bonferroni post hoc test was used.
(G-H) Mice were injected with AAV-DAPK1 or the control virus (vector), and the rotarod test was performed. (G) Time spent on the rod by mice in each group at different rotation speeds. (H) ORP scores in two groups. ** P<0.01 vs Con; N=10–11; one-way ANOVA with Bonferroni post hoc test was used.
(I-K) The total distance traveled (I), mean velocity (J) and percentage of time spent mobile (K) in the open field test of mice treated with A-miR-26a or a control. ** P<0.01 vs Con; N=10; Student’s t-test.
(L-N) The total distance traveled (L), mean velocity (M) and percentage of time spent mobile (N) in the open field test of mice treated with AAV-DAPK1 or the control virus (vector). **P<0.01 vs vector; N=10–11; Student’s t-test.
Activation of DAPK1 induces synucleinopathy by directly phosphorylating α-synuclein
We then wanted to determine whether DAPK1 activation promotes DA neuron synucleinopathy. Given the important role of DAPK1 in mediating autophagy(24), we first examined the degradation of α-synuclein. Cycloheximide (CHX) chase analysis demonstrated that overexpression of DAPK1 did not extend the α-synuclein half-life (2 h) compared to that of the pcDNA control (Fig. 5A, B), excluding the potential involvement of protein degradation by DAPK1 activation. DAPK1 is a Ser/Thr kinase and phosphorylates multiple substrates under different pathological conditions and the phosphorylation of α-synuclein is increased in mice exhibiting DAPK1 overexpression via miR-26a inhibition, AAV-DAPK1 infection or MPTP treatment. Therefore, we hypothesized that DAPK1 could phosphorylate α-synuclein directly. We then transfected the wt DAPK1 plasmid (wtDAPK1) or the wt α-synuclein vector into HEK293 cells, and found that DAPK1 overexpression dramatically increased the levels of p-syn (Fig. 5C). This effect is specific because mutating this serine residue to alanine diminished the phosphorylation and the elevation in total α-synuclein (Fig. 5D). Further, we examined the temporal sequence of changes in phospho-synuclein and total-synuclein. We found that upon the DAPK1 overexpression, the increment in the phospho-Ser129 of α-synuclein began at 12 h, but the increment of total α-synuclein began at 18 h (Fig. 5E-F), suggesting that DAPK1 inducedα-synuclein phosphorylation occurred prior to the aggregation. In cell free system, co-incubation of purified DAPK1 and α-synuclein protein for 30 minutes resulted in the hyperphosphorylation of α-synuclein at Ser129 site. Application of TC-DAPK 6, a specific DAPK1 inhibitor, blocked the hyperphosphorylation of α-synuclein (Fig. 5G). By using double-immunofluorescence staining, we verified the hyperphosphorylation of pS129-synuclein in wtDAPK1-overexpressed cells (Fig. 5H). The colocalization and correlation analysis indicated that the expression of DAPK1 was positively correlated with the phosphorylation levels of pS129-synuclein, suggesting a direct interaction of DAPK1 with α-synuclein (Fig. 5I, J). These findings demonstrated that DAPK1 activation promotes DA neuron synucleinopathy via enhancing the phosphorylation of α-synuclein.
Figure 5. DAPK1 directly promotes the phosphorylation of α-synuclein at Ser129.
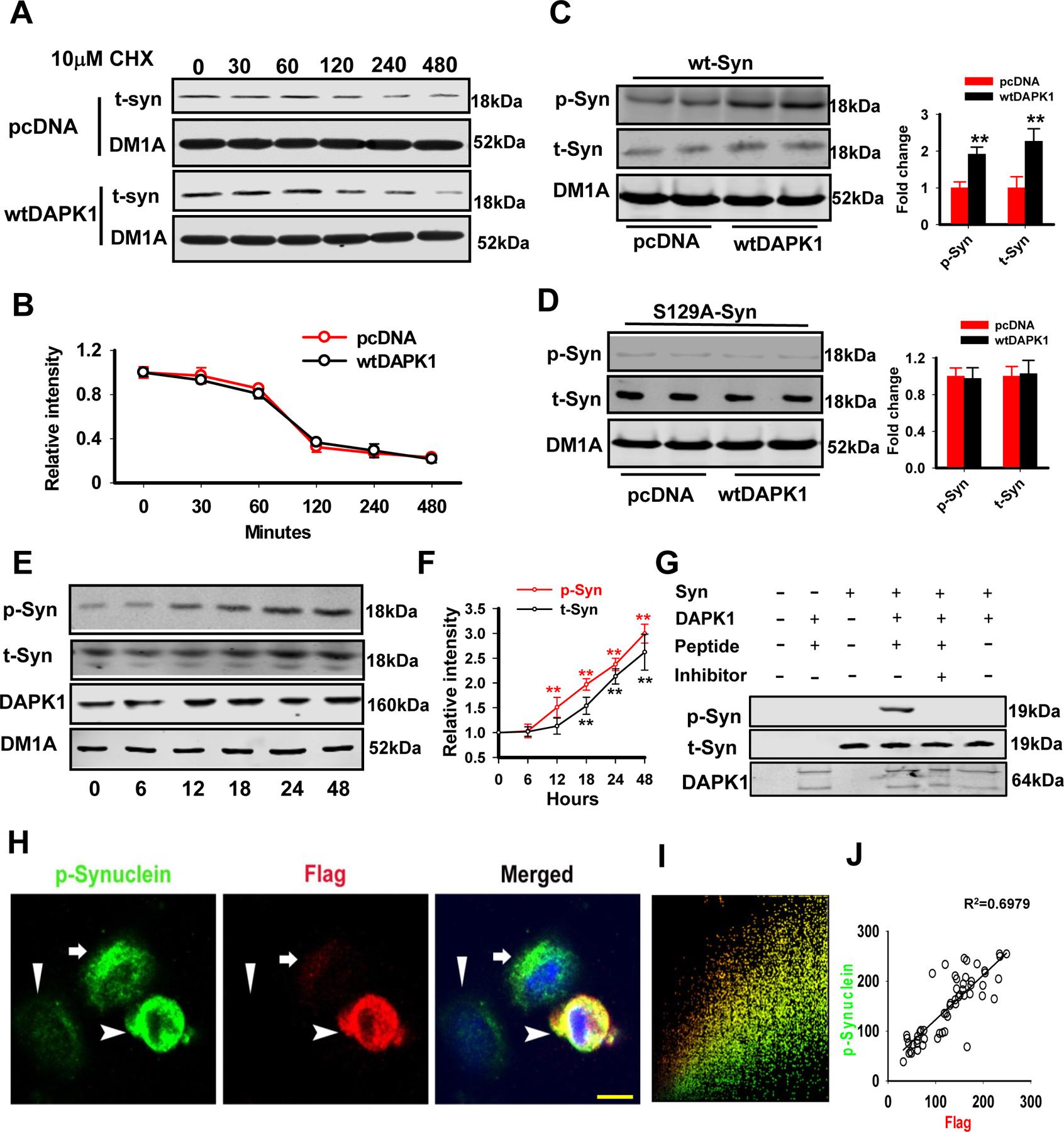
(A-B) A representative western blot image (A) of total α-synuclein protein levels during the CHX chase experiment. HEK293 cells were transfected with the wt α-synuclein plasmids with either wtDAPK1 or pcDNA. Twenty-four hours after transfection, cells were treated with CHX for the indicated time periods. Quantification of the data is shown (B). N=3 independent experiments; Student’s t-test.
(C-D) HEK293 cells were co-transfected with the wt α-synuclein (C) or mut (D), together with either wtDAPK1 or pcDNA. The cell lysates were collected 48 h later for western blotting. ** P<0.01 vs pcDNA; N=4; Student’s t-test.
(E-F) The N2a cells were transfected with wild type DAPK1 and the cell lysates were collected at 0, 6, 12, 18, 24, 48 h for examination of pSer129-α-Synuclein (p-Syn) and total α-Synuclein (t-Syn). The representative blots were shown in (E) and the quantitative analysis were shown in (F). ** P<0.01 vs 0h. N=6; Bonferroni post hoc test after Repeated two-way ANOVA.
(G) The recombinant α-synulcein (human) and DAPK1 (human) were incubated in 30°C for 30 minutes, and the inhibitor at 69nM was added, which were then subjected to western blot. The pSer129-α-Synuclein (p-Syn), total α-Synuclein (t-Syn) and DAPK1 were detected.
(H-J) The N2a cells were transfected with Flag-DAPK1 plasmid and then double-immunostained with anti-p-syn (green) and anti-DAPK1 (red) antibodies. The nuclei were visualized by DAPI staining (H). A colocalization analysis (I) and correlation analysis (J) were performed. Arrowhead, cell with higher DAPK1 level; arrow, cell with moderate DAPK1 expression; triangle, cell without DAPK1 expression; bar=10 µm.
Genetic deletion of DAPK1 in DA neurons rescues the PD-like pathological and behavioral changes
We then wanted to know whether deletion of DAPK1 in DA neurons could rescue the PD-like pathological and behavioral changes in the chronic MPTP mice. We found that mice with DAPK1 knockout in the DA neurons (DD-KO/MPTP) displayed restored locomotor abilities in both the rotarod tests (increased time on the rod, the overall rod performance), the open field tasks (restored total distance, velocity and mobility time) and the pole test (shorter time to orient down (t-turn) and total time to descend the pole (t-total)) (Fig. 6A-H). Meanwhile, the enhanced phosphorylation and aggregation of α-synuclein were also attenuated in the SN of DD-KO/MPTP mice (Fig. 6I-J). In the aged (18 month) A53T α-synuclein mutant mice, injected of lentivirus that contains the effective siRNA for DAPK1 also repressed the hyperphosphorylation and aggregation of α-synuclein (Supplementary Fig. 6). Furthermore, deletion of DAPK1 reduced the loss of DA neurons in the SN (Fig. 6K-L) induced by MPTP.
Figure 6. Genetic deletion of DAPK1 rescues the PD-like behaviors and pathologies of MPTP mice.
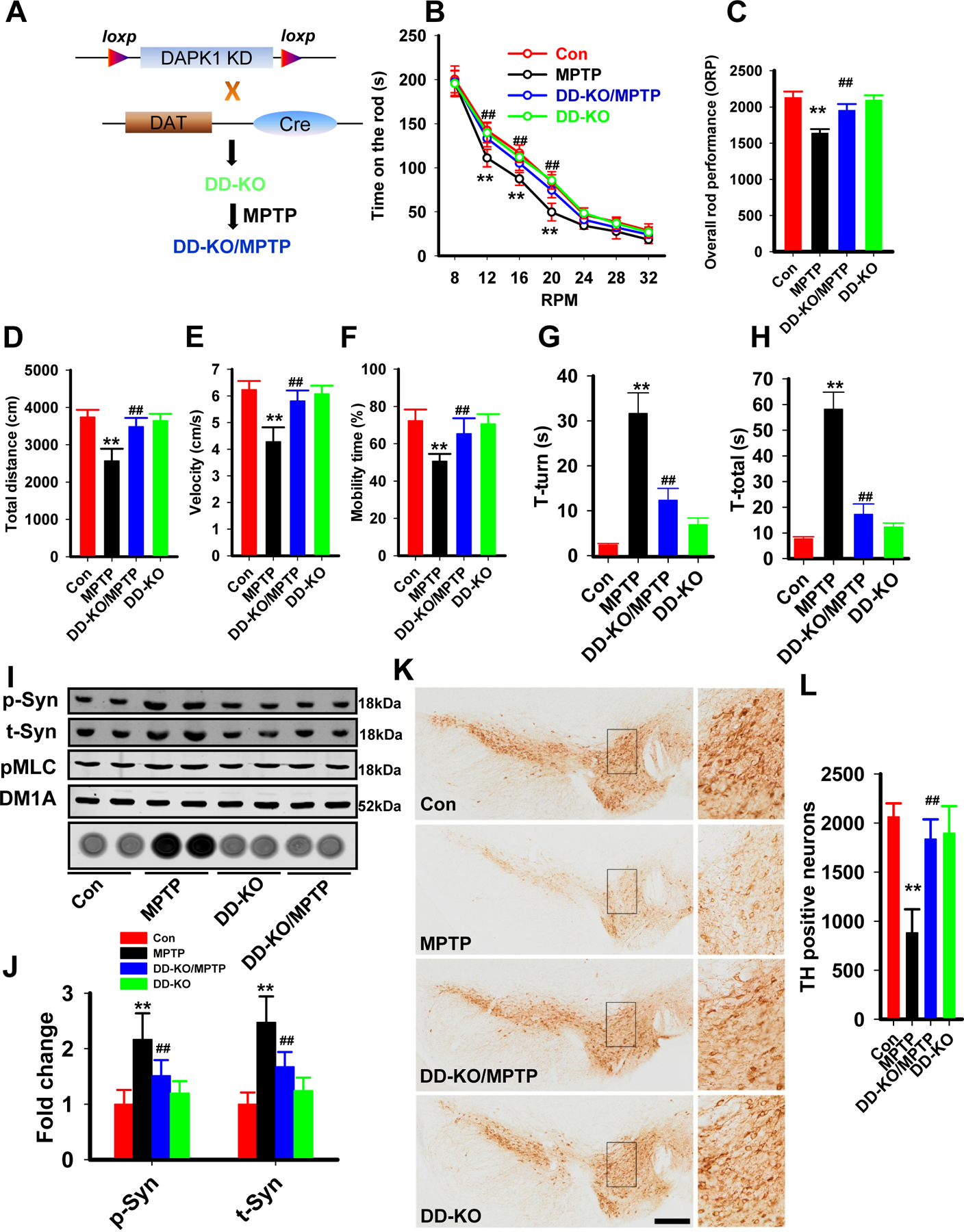
(A) A diagram for the generation of DD-KO mice and DD-KO/MPTP mice.
(B-H) The wild type mice (con) and the DD-KO mice (DD-KO) received a total of 10 doses of MPTP hydrochloride (25 mg/kg in saline, s.c.) on a 5-week schedule. The mice were then subjected to rotarod test (B-C), open field test (D-F) and pole test. Time spent on the rod (B) and ORP (C) were measured. The total distance traveled (D), mean velocity (E) and mobility time (F) were evaluated in the open field test. The time to orient down (G, T-turn) and total time to descend the pole (H, T-total) were evaluated in the pole test. **P<0.01 vs Con; ##P<0.01 vs MPTP; one-way ANOVA with Bonferroni post hoc test was used.
(I-J) The mice were treated as described in (b-h), and the homogenates from the SN were extracted for western blotting (upper four blots) and the filter trap analysis (bottom dot blot). The representative blots (I) and the quantitative analysis for p-syn and t-syn (J) are shown. **P<0.01 vs Con; ##P<0.01 vs MPTP; N=6; one-way ANOVA with Bonferroni post hoc test was used.
(K-L) Mice were treated as described in (B-H), and the coronal slices from those mice were prepared for TH staining. Representative images of the SN of different groups (K) and the quantitative analysis (L). **P<0.01 vs Con; ##P<0.01 vs MPTP; N=6; one-way ANOVA with Bonferroni post hoc test was used.
Beneficial effects of the siP-Syn peptide in PD mice
Finally, we disrupted the DAPK1 activation-induced hyperphosphorylation of α-synuclein by using a cell-permeable peptide and evaluated its effect on PD-like pathological and locomotor abnormalities. The peptide was generated by fusion of an HIV Tat (trans-activator of transcription) signal (YGRKKRRQRRR)(25) to the competing peptide(26, 27) that spans amino acids 125–135 (YEMPSEEGYQD) and contains the phosphorylation site (Ser129) of α-synuclein (siP-Syn; Fig. 7A). We first examined the phosphorylation of α-synuclein with different concentrations of siP-Syn peptide delivery to N2a cells. We found that application of 10 µM siP-Syn reduced the phosphorylation of α-synuclein, as well as the aggregation of α-synuclein caused by DAPK1 overexpression in N2a cells (Fig. 7B, C). We then injected siP-Syn once every three days at a dose of 10 mg/kg for 6 weeks. As expected, the siP-Syn peptide not only rescued the locomotor abnormalities in the MPTP mice (Fig. 7D-H) but also reduced the hyperphosphorylation and aggregation of α-synuclein (Fig. 7I, J). In addition, siP-Syn injection significantly promoted the survival of DA neurons in the brain (Fig. 7K). Thus, blocking DAPK1-dependent phosphorylation of α-synuclein effectively rescues the PD-like pathological and behavioral changes.
Figure 7. siP-Syn attenuates the behavioral and pathological abnormalities in PD mice.
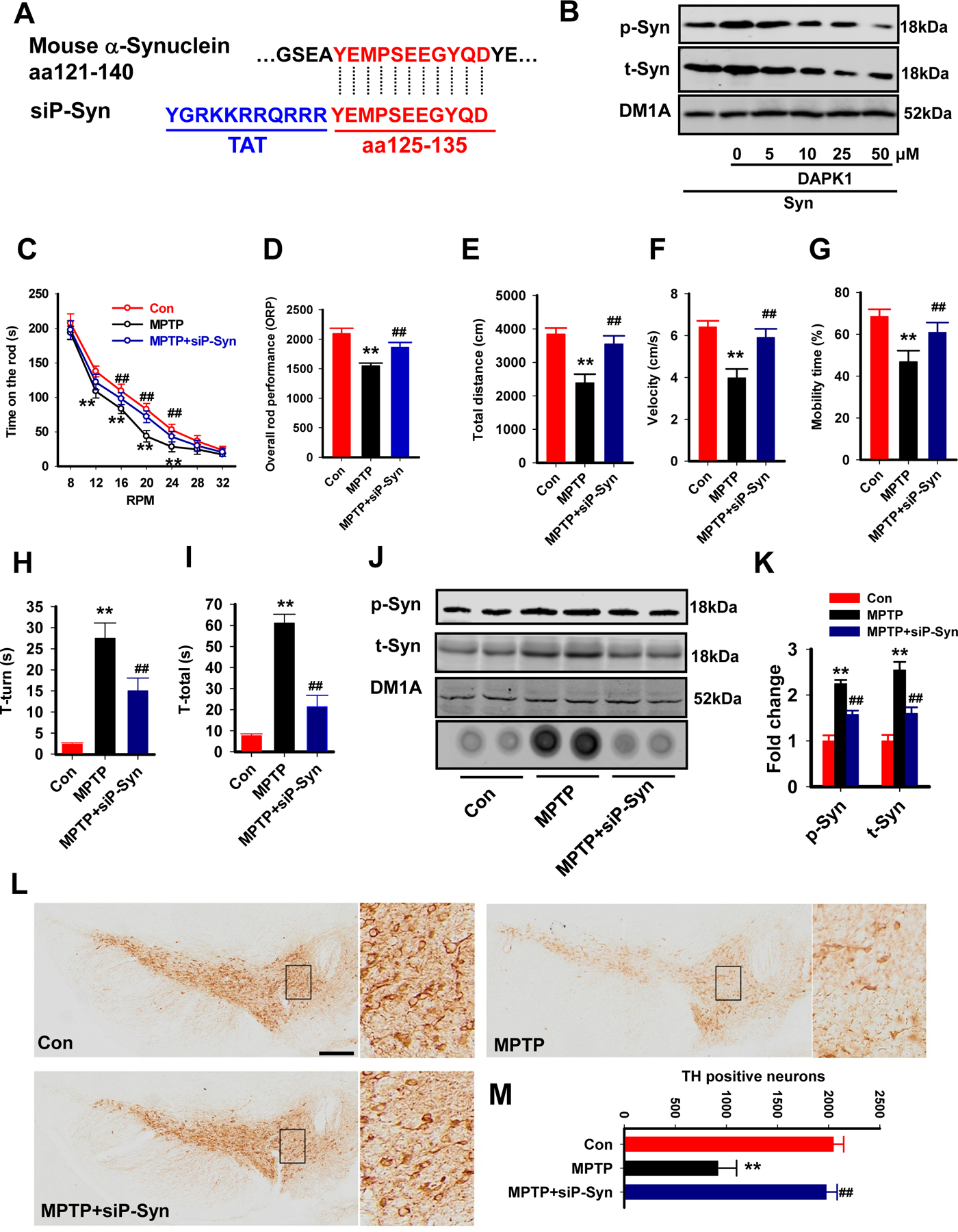
(A) A diagram of the siP-Syn design. Blue words, Tat sequence.
(B) The siP-Syn peptide blocked the phosphorylation of α-synuclein in vitro. The N2a cells were transfected with α-synuclein and DAPK1, and the siP-Syn peptide was applied at different doses as indicated. At 24 h later, the cell lysates were collected for western blotting.
(C-I) The siP-syn peptide and its Con peptide were injected into MPTP mice, and the mice were subjected to the rotarod test, the open field test and the pole test. The time on the rod (C) and ORP (D) were evaluated in the rotarod test. The total distance traveled (E), mean velocity (F) and mobility time (G) were evaluated in the open field test. The time to orient down (H, T-turn) and total time to descend the pole (I, T-total) were evaluated in the pole test. **P<0.01 vs Con; ##P<0.01 vs MPTP; N=9, one-way ANOVA with Bonferroni post hoc test was used.
(J-K) The mice were treated as described in (C-I), and the homogenates from the SN were extracted for western blotting (upper three blots) and the filter trap analysis (lower dot blot). The representative blots (J) and the quantitative analysis for p-syn and t-syn (K) are shown. **P<0.01 vs Con; ##P<0.01 vs MPTP; N=6; one-way ANOVA with Bonferroni post hoc test was used.
(L-M) The mice were treated as described in (C-I), and the coronal slices from those mice were prepared for TH staining. Representative images of the SN of different groups (L) and the quantitative analysis (M). **P<0.01 vs Con; ##P<0.01; vs MPTP; N=6; one-way ANOVA with Bonferroni post hoc test was used.
Discussion
In the present study, we report that the DAPK1 protein is abnormally upregulated, a change which is positively correlated with DA neuron synucleinopathy in PD mice. We demonstrate that the loss of miR-26a caused by a reduction in the transcription factor C/EBPα in PD mice and patients leads to an elevation in DAPK1 expression post-transcriptionally. We have also revealed that both the loss of miR-26a and overexpression of DAPK1 induces α-synuclein hyperphosphorylation, aggregation and inclusion formation, which further result in the death of DA neurons and locomotor abnormalities. Finally, by deletion of DAPK1 in DA neurons or administering siP-Syn, a specific competing peptide, to MPTP mice, we found that neuronal synucleinopathy, DA neuron death, and the locomotor disabilities were dramatically attenuated or recovered. Thus, our study has not only uncovered a novel role for miR-26a and DAPK1 in the pathological and behavioral abnormalities related to PD, but also identified their potential application in the treatment for PD.
MiRNAs are endogenous and short, non-coding RNA molecules, 21–24 nucleotides in length, which play an important function in post-transcriptional regulation of gene expression through sequence-specific binding of the 3’UTR of target messenger RNA during neuronal development, differentiation and maturation(28). Dysregulation of miRNAs has been implicated in the pathogenesis of multiple neurodegenerative diseases, including PD(29). As such, miR-7 and miR-153 have been shown to regulate α-synuclein levels synergistically, and depletion of these two miRNAs results in a concomitant increase in α-synuclein levels in a PD brain. Moreover, miR-7 has a protective role by preventing oxidative stress, and miR-7 inhibition causes cell death(30, 31). In addition to miR-7 and miR-153, miR-26a has also been reported to be abnormally downregulated in PD patients(32) and cell lines(33). Here, we revealed that the expression of miR-26a was decreased both in the MPTP-induced PD mice and in the CSF of PD patients. We also showed that the transcriptional factor C/EBPα, which was reported to control the transcription of miR-26a, was significantly reduced in the MPTP-treated mice. We therefore propose that the loss of C/EBPα causes a reduction in the transcription of miR-26a, leading to a reduced expression of miR-26a in PD. Furthermore, we find that miR-26a specifically binds to the 3’UTR of DAPK1 and represses DAPK1 translation. Inhibition of miR-26a expression in vivo resulted in synucleinopathy and DA neuron loss, as well as locomotor disabilities, whereas an increase in miR-26a effectively ameliorates those abnormalities in PD mice. These findings strongly suggest a critical role for miR-26a in the pathological and behavioral changes in PD.
As an important mediator of cell death in response to ceramide, ischemia and glutamate toxicity, DAPK1 is implicated in the pathogenesis of multiple neurological diseases, such as epilepsy, AD and ischemic brain injury. In AD, overexpression of DAPK1 promotes tau phosphorylation at multiple AD-related sites and increases its stability(11). Additionally, DAPK1 activation enhances tau phosphorylation at Ser262 and results in tau insolubility and accumulation in dendritic spines, leading to synaptic dysfunction in ischemic stroke(34). Here, we report that activated DAPK1 in PD promotes the phosphorylation of α-synuclein at the Ser129, a site related to the neurotoxicity of α-synuclein. In LBs, a key pathological hallmark in PD brains, approximately 90% of α-synuclein is hyperphosphorylated at Ser129(35). Both in Drosophila and rat PD models(36, 37), p-syn has been well studied for its participation in neuronal toxicity especially in DA neuronal death. In this study, the phosphorylation level of p-syn was enhanced by DAPK1, resulting in the loss of DA neurons in the SN region. Thus, reduction in the phosphorylation level of p-syn could be a potential therapeutic approach for alleviating the pathological changes in PD. To this end, we generated the DD-KO mice to inhibit DAPK1-induced α-synuclein hyperphosphorylation, and demonstrated that both strategies mitigate the previously observed synucleinopathy and DA neuron death, as well as locomotor disabilities. Moreover, intravenous delivery of a membrane-permeable competing peptide also attenuated the synucleinopathy pathology and locomotor abnormalities. As one of the cell-penetrating peptides (CPP), Tat has been used to introduce multiple neuroprotective proteins to reduce cerebral ischemic damage and protect against ischemia in brain injury(38). In addition to the application in animal models, CPP-mediated drug delivery has also been clinically investigated. For example, KAI-9803, a specific inhibitor of δPKC, and KAI-1678, a specific inhibitor of εPKC, were fused with Tat. These reagents exhibited acceptable safety and tolerability profiles(39) and were currently under phase II clinical trials for myocardial infarction and pain, respectively. Therefore, the competing peptide used in this study may provide a possible treatment strategy for PD.
Overall, our study demonstrates an important role of the miR-26a/DAPK1 signaling pathway in neuronal synucleinopathy, DA neuron loss and locomotor disability in PD (Fig. 8). Findings from this work indicate novel therapeutic targets and treatment strategies for PD.
Figure 8. Schematic illustration of the effects of miR-26a.
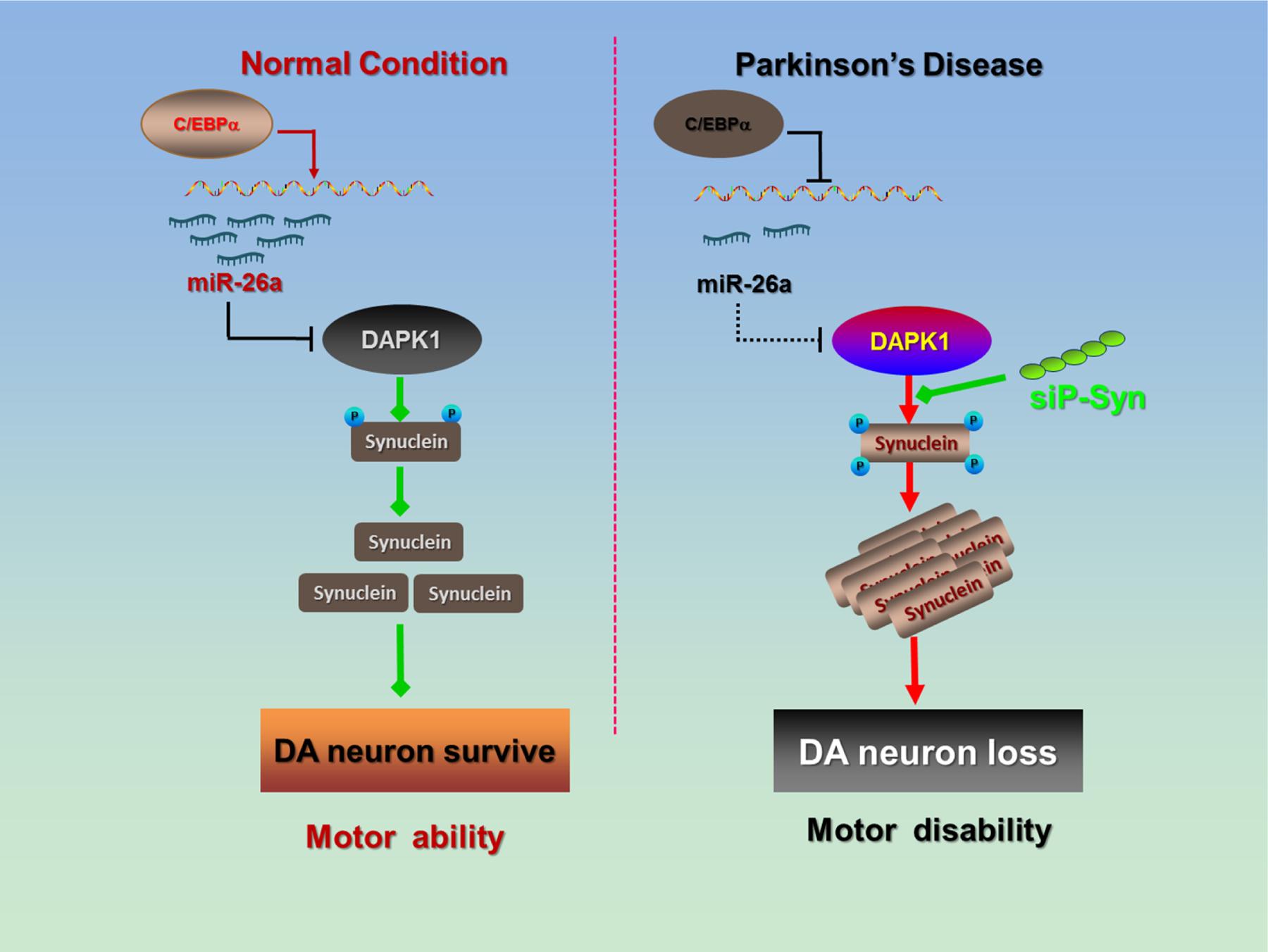
In the PD brain, the C/EBPα transcription factor is suppressed, and the transcription and expression of miR-26a is decreased. The loss of miR-26a induces post-transcriptional DAPK1 overexpression, resulting in the hyperphosphorylation of α-synuclein and α-synuclein aggregation. The toxic synucleinopathy finally causes DA neuron death and locomotor disabilities in PD.
Supplementary Material
Acknowledgement
This study is supported partially by the National Natural Science Foundation of China (81761138043, 81829002, 91632114, 81771150 and 31571039 to L.-Q.Z., 31721002 to Y.L., the NSFC-NIH joint program (NO.81361120404 to B.T, 81871108 to D.L.), Key Project of National Science Foundation of China(NO.81430023 to B.T),National Program for Support of Top-Notch Young Professionals and Academic Frontier Youth Team of Huazhong University of Science and Technology to Dr. Ling-Qiang Zhu, Program for Changjiang Scholars and Innovative Research Team in University (No. IRT13016).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The peptide used to block synuclein phosphorylation submitted to the Patent Office of the People’s Republic of China by Dr. Ling-Qiang Zhu, Dr. Man-Fei Deng, Dr. Dan Liu, Dr. Ao-Ji Xie and Dr.Ya-Fan Zhou (Application No. 2018100396990). All other authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Mortality GBD, Causes of Death C (2016): Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388:1459–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xia R, Mao ZH (2012): Progression of motor symptoms in Parkinson’s disease. Neurosci Bull 28:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michel PP, Hirsch EC, Hunot S (2016): Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 90:675–691. [DOI] [PubMed] [Google Scholar]
- 4.Dexter DT, Jenner P (2013): Parkinson disease: from pathology to molecular disease mechanisms. Free Radic Biol Med 62:132–144. [DOI] [PubMed] [Google Scholar]
- 5.Bialik S, Kimchi A (2006): The death-associated protein kinases: structure, function, and beyond. Annu Rev Biochem 75:189–210. [DOI] [PubMed] [Google Scholar]
- 6.Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A (1995): Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev 9:15–30. [DOI] [PubMed] [Google Scholar]
- 7.Shohat G, Spivak-Kroizman T, Cohen O, Bialik S, Shani G, Berrisi H, et al. (2001): The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J Biol Chem 276:47460–47467. [DOI] [PubMed] [Google Scholar]
- 8.Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A (2002): DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol 157:455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, et al. (2010): DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 140:222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pei L, Shang Y, Jin H, Wang S, Wei N, Yan H, et al. (2014): DAPK1-p53 interaction converges necrotic and apoptotic pathways of ischemic neuronal death. J Neurosci 34:6546–6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim BM, You MH, Chen CH, Lee S, Hong Y, Hong Y, et al. (2014): Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function. Cell Death Dis 5:e1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Liu D, Huang HZ, Wang ZH, Hou TY, Yang X, et al. (2017): A Novel MicroRNA-124/PTPN1 Signal Pathway Mediates Synaptic and Memory Deficits in Alzheimer’s Disease. Biol Psychiatry [DOI] [PubMed] [Google Scholar]
- 13.Zhu LQ, Liu D, Hu J, Cheng J, Wang SH, Wang Q, et al. (2010): GSK-3 beta inhibits presynaptic vesicle exocytosis by phosphorylating P/Q-type calcium channel and interrupting SNARE complex formation. J Neurosci 30:3624–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sambandan S, Akbalik G, Kochen L, Rinne J, Kahlstatt J, Glock C, et al. (2017): Activity-dependent spatially localized miRNA maturation in neuronal dendrites. Science 355:634–637. [DOI] [PubMed] [Google Scholar]
- 15.Hoehn MM, Yahr MD (2001): Parkinsonism: onset, progression, and mortality. 1967. Neurology 57:S11–26. [PubMed] [Google Scholar]
- 16.Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992): Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P, et al. (2008): Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord 23:2129–2170. [DOI] [PubMed] [Google Scholar]
- 18.Kao SH, Wang WL, Chen CY, Chang YL, Wu YY, Wang YT, et al. (2015): Analysis of Protein Stability by the Cycloheximide Chase Assay. Bio Protoc 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang E, Kuret J (2008): Detection and quantification of tau aggregation using a membrane filter assay. Anal Biochem 373:330–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petroske E, Meredith GE, Callen S, Totterdell S, Lau YS (2001): Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Neuroscience 106:589–601. [DOI] [PubMed] [Google Scholar]
- 21.Mohamed JS, Lopez MA, Boriek AM (2010): Mechanical Stretch Up-regulates MicroRNA-26a and Induces Human Airway Smooth Muscle Hypertrophy by Suppressing Glycogen Synthase Kinase-3 beta. Journal of Biological Chemistry 285:29336–29347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rozas G, Guerra MJ, Labandeira-Garcia JL (1997): An automated rotarod method for quantitative drug-free evaluation of overall motor deficits in rat models of parkinsonism. Brain Res Brain Res Protoc 2:75–84. [DOI] [PubMed] [Google Scholar]
- 23.Rozas G, Lopez-Martin E, Guerra MJ, Labandeira-Garcia JL (1998): The overall rod performance test in the MPTP-treated-mouse model of Parkinsonism. J Neurosci Meth 83:165–175. [DOI] [PubMed] [Google Scholar]
- 24.Zalckvar E, Berissi H, Eisenstein M, Kimchi A (2009): Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 5:720–722. [DOI] [PubMed] [Google Scholar]
- 25.Bechara C, Sagan S (2013): Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett 587:1693–1702. [DOI] [PubMed] [Google Scholar]
- 26.Moles A, Sanchez AM, Banks PS, Murphy LB, Luli S, Borthwick L, et al. (2013): Inhibition of RelA-Ser536 phosphorylation by a competing peptide reduces mouse liver fibrosis without blocking the innate immune response. Hepatology 57:817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oakley F, Teoh V, Ching ASG, Bataller R, Colmenero J, Jonsson JR, et al. (2009): Angiotensin II activates I kappaB kinase phosphorylation of RelA at Ser 536 to promote myofibroblast survival and liver fibrosis. Gastroenterology 136:2334–2344 e2331. [DOI] [PubMed] [Google Scholar]
- 28.O’Carroll D, Schaefer A (2013): General principals of miRNA biogenesis and regulation in the brain. Neuropsychopharmacology 38:39–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoss AG, Labadorf A, Beach TG, Latourelle JC, Myers RH (2016): microRNA Profiles in Parkinson’s Disease Prefrontal Cortex. Front Aging Neurosci 8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM (2009): Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A 106:13052–13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doxakis E (2010): Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem 285:12726–12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martins M, Rosa A, Guedes LC, Fonseca BV, Gotovac K, Violante S, et al. (2011): Convergence of miRNA expression profiling, alpha-synuclein interacton and GWAS in Parkinson’s disease. PLoS One 6:e25443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Chen H, Chen F, Li F, Wang M, Wang L, et al. (2013): Effects of glial cell line-derived neurotrophic factor on microRNA expression in a 6-hydroxydopamine-injured dopaminergic cell line. J Neural Transm (Vienna) 120:1511–1523. [DOI] [PubMed] [Google Scholar]
- 34.Pei L, Wang S, Jin H, Bi L, Wei N, Yan H, et al. (2015): A Novel Mechanism of Spine Damages in Stroke via DAPK1 and Tau. Cereb Cortex 25:4559–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, et al. (2002): Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am J Pathol 160:1655–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen L, Feany MB (2005): Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci 8:657–663. [DOI] [PubMed] [Google Scholar]
- 37.Yamada M, Iwatsubo T, Mizuno Y, Mochizuki H (2004): Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson’s disease. J Neurochem 91:451–461. [DOI] [PubMed] [Google Scholar]
- 38.Zou LL, Ma JL, Wang T, Yang TB, Liu CB (2013): Cell-penetrating Peptide-mediated therapeutic molecule delivery into the central nervous system. Curr Neuropharmacol 11:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guidotti G, Brambilla L, Rossi D (2017): Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol Sci 38:406–424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.