

Abstract
Expansions of a G4C2 repeat in the C9ORF72 gene are the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), two devastating adult-onset neurodegenerative disorders. Using C9-ALS/FTD patient derived cells and C9ORF72 BAC transgenic mice, we have generated and optimized antisense oligonucleotides (ASOs) that selectively blunt expression of G4C2 repeat containing transcripts and effectively suppress tissue levels of polyGP dipeptides. ASOs with reduced phosphorothioate content showed improved tolerability without sacrificing efficacy. In a single patient harboring mutant C9ORF72 with the G4C2 repeat expansion, repeated dosing by intrathecal delivery of the optimal ASO was well tolerated, leading to significant reductions in levels of CSF polyGP. This report provides insight into the impact of nucleic acid chemistry on toxicity and for the first time demonstrates the feasibility of clinical suppression of the C9ORF72 gene. Further clinical trials will be required to demonstrate safety and efficacy of this therapy in patients with C9ORF72 gene mutations.
Introduction
A GGGGCC (G4C2) hexanucleotide repeat expansion (HRE) in the first intron of the C9ORF72 (C9) gene is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), two devastating adult-onset neurodegenerative disorders1,2. Proposed disease mechanisms include a partial loss of the C9ORF72 protein function (C9ORF72 haploinsufficiency) and acquired toxicity of the repeat expansion3. Transcription of C9ORF72 gene generates three transcript variants, V1, V2 and V3 (Fig. 1a). V1 is translated to produce a short protein isoform (222 amino acids) while V2 and V3 generate the most predominant C9ORF72 protein (481 amino acids), which functions in vesicular trafficking4. Located adjacent to the promoter region of the most abundant V2 transcript variant, the G4C2 repeat expansion impairs its transcription, leading to C9ORF72 protein haploinsufficiency5,6, impaired function of myeloid cells7,8 and diminished neuronal viability9. Both sense and antisense transcripts encompassing the HRE in V1 and V3 generate RNA foci and undergo translation into atypical, aggregation-prone dipeptide repeat (DPR) proteins in all open reading frames10,11. These unusual DPRs are toxic in several experimental model systems12–15. Despite important advances in elucidating the molecular pathology of the expanded hexanucleotide repeats, there are no meaningful therapies for C9ORF72-related ALS or FTD.
Figure 1. G4C2-targeting LNA- and MOE-modified ASOs reduce the C9ORF72 repeat-containing transcripts in patient derived fibroblasts, C9BAC mouse derived neurons, and C9BAC mice.
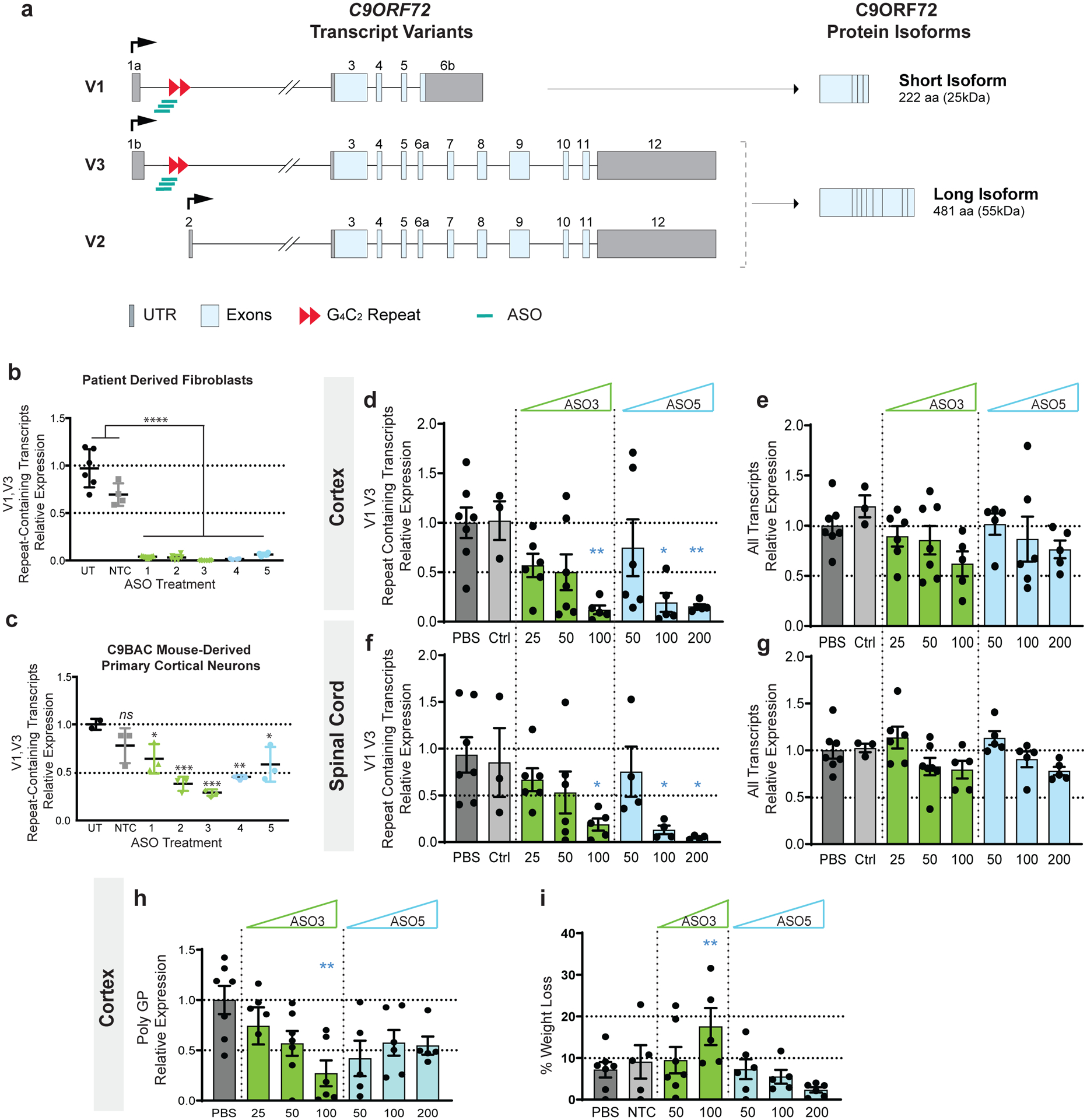
(a) Schematic of C9ORF72 transcript variants and of the two protein isoforms as named in PubMed. The repeat expansion (red triangles) located in the first intron is expressed in variants 1 and 3 (V1-V3). Variant 2 (V2), the most abundant, starts form a different transcription start site (black arrow) and does not include the repeat expansion. V2 and V3 encode the main C9ORF72 protein isoform. Grey boxes represent untranslated regions (UTR); light blue boxes, exons; black lines, introns. ASOs used in this study (green bars) target the intronic region flanking the repeat expansion. (b) LNA and 2’-O-MOE ASOs dramatically reduce the level of repeat-containing transcripts at a dose of 100nM 72 hrs after lipid-mediated delivery in patient derived fibroblasts as measured by qRT-PCR. n=6, each data point is derived from a separate well. ****p<0.0001, comparing any treatment group to either control group, based on one-way ANOVA with Dunnett’s multiple comparisons test. (c) LNA and 2’-O-MOE ASOs significantly reduce the level of repeat-containing transcripts after two weeks treatment with 1 μM ASO without lipid assistance (gymnotic delivery) in primary cortical neurons derived from C9BAC mice. n=3, each data point is derived from a separate well. ns, not significant (p=0.23), comparing NTC group to PBS group, based on one-way ANOVA with Dunnett’s multiple comparisons test. (d-g) Expression of V1-V3 repeat containing transcripts (d, f) and all transcripts (e, g) in cortex and spinal cord quantified by qRT-PCR in mice infused with PBS (dark grey), NTC ASO (light grey), ASO3 (green) or ASO5 (blue) at the indicated dose. For each dose level, n= 5–7, except NTC group (n=3). (h) Relative expression of polyGP in the cortex of mice treated with ASO3 (green) and ASO5 (blue) assayed by sandwich immunoassay. Data are represented as mean ± SEM. (i) % of body weight loss at end point relative to before treatment. In all panels, *p<0.05, **p<0.01, ***p<0.001, comparing treatment groups to PBS group, based on one-way ANOVA with Dunnett’s multiple comparisons test. All replicates shown as individual data points. In panels d-i, each data point is derived from a separate animal.
Antisense oligonucleotides (ASOs) can drive therapeutic effects by mechanisms that include splice-modulation or, if the ASO contains DNA, activation of endogenous RNase H16 to degrade the target RNA. The broad bioavailability of ASOs in the CNS, including both neurons and glial cells17 has prompted development of ASOs as therapy for dominantly transmitted genetic disorders of the central nervous system (e.g. ALS caused by mutations in the SOD1 gene).
Here, we report development of antisense oligonucleotides targeting C9ORF72 to treat ALS and FTD. Using different C9 related model systems, including patient-derived samples and two C9BAC transgenic mouse models18,19, we have generated ASOs that specifically reduce levels of the transcripts harboring the HRE as well as their DPR products, with minimal effects on the most abundant V2 isoform, which does not contain the HRE. We show that modification of a subset of the phosphodiester internucleoside linkages significantly improves ASO tolerability without impairing potency. We demonstrate that in a single patient harboring mutant C9ORF72 with the G4C2 repeat expressions, repeated intrathecal dosing of the optimal ASO was well tolerated and led to significant and durable reduction in levels of CSF polyGP.
Results
ASO suppresses C9ORF72 in fibroblasts and mouse neurons
Because haploinsufficiency of C9ORF72 is thought to be adverse, we developed ASOs that target only the 5’end of transcripts V1 and V3 that bear the G4C2 repeat expansion, sparing transcript V2. As it is not fully clear whether the repeat-containing intron is retained or spliced out, we focused our effort on ASO sequences targeting the intron-repeat junction (Fig. 1a). Others have previously tested ASOs against this target region in patient derived samples with success both in vitro20,21 and in vivo in mice22–25 and flies26. We designed locked nucleic acid (LNA) and 2’-O-methoxyethyl (MOE) gapmer ASOs (Table 1). All cytosine residues were 5-methylated to reduce immunogenicity.
Table 1.
Antisense oligonucleotide sequences and modification patterns.
| Name | Sequences and modification pattern1 |
|---|---|
| ASO1 | CCCTAGCGCGCGACT |
| ASO2 | CCCGGCCCCTAGCGCGCGAC |
| ASO3 | GCCCCTAGCGCGCGACTC |
| ASO4 | CCCGGCCCCTAGCGCGCGAC |
| ASO5 | GCCCCTAGCGCGCGACTC |
| ASO5-1 | GCoCoCoCoTAGCGCGCoGoAoCoTC |
| ASO5-2 | GCoCoCoCTAGCGCGCGoAoCoTC |
normal text, DNA; bold italic text, LNA; bold underlined text, MOE. All C residues are 5-methyl cytidine. All linkages are PS except where PO linkages are indicated by “o”.
We tested ASOs in multiple steps. We first developed a dual luciferase screen for ASOs that suppress expression of transcripts V1,V2 and V3 (Fig. S1a), and used this assay to narrow our focus to five ASOs. We then treated primary C9-ALS/FTD patient-derived fibroblasts (containing >1000 repeats, Fig. S1b, and showing visible nuclear C9ORF72 foci, Fig. S1c) with ASOs 1–5 at a dose of 100 nM by lipid transfection. After 72 hours, all five ASOs reduced V1-V3 expression to almost undetectable levels (Fig. 1b). Silencing was dose-dependent in patient-derived fibroblasts (Fig. S1d) and in HEK293 cells expressing the dual luciferase reporter (Fig. S1e).
A hallmark of C9-ALS/FTD is the presence of repeat-containing RNA foci1,2,20,21,27,28. Three days after ASO treatment, the number of cells with foci was reduced from 80% (untreated) to 20–40% (treated). Moreover, fewer foci per cell were detected, showing that all five ASOs were potent inhibitors of G4C2 RNA foci (Fig. S1f–g).
Each of the five ASOs was also active by lipid-free delivery to neurons. Primary cortical neurons were derived from E15.5 C9BAC embryos and treated with 1μM ASO at 5 days in vitro (DIV). 15 days after treatment, expression of human repeat containing transcript was significantly reduced in all treated conditions as compared to the non-targeting control or untreated condition (Fig. 1c).
ASOs suppress C9ORF72 in C9BAC neurons
We next evaluated the properties of these ASOs in vivo in wild-type (WT) and C9ORF72 transgenic mice. ASOs can be delivered to the brain tissue and spinal cord through the surrounding cerebral spinal fluid (CSF) via an intracerebroventricular (ICV) bolus injection or osmotic pump infusion29. ASO3 and ASO5 distributed broadly throughout the mouse CNS and showed neuronal uptake (Fig. S2).
We first assessed ASO tolerability in wild-type C57BL/6 mice. Each animal received a single ICV bolus dose of one of our five ASOs (Table 1). Mice injected with ASOs 1, 2 and 4 had severe seizure-like phenotypes upon recovery from anesthesia or did not survive 24 hours post-injection, while mice treated with ASOs 3 and 5 recovered well post-injection.
We then compared the tolerability and efficacy of ASOs 3 and 5 in C9BAC transgenic mice. C9BAC transgenic mice generated in our laboratory express approximately 600 G4C2 repeat motifs within a truncated human C9ORF72 gene (from exons 1–6). Although these mice do not develop a motor phenotype, they recapitulate disease hallmarks including repeat containing RNA foci and DPR18. In our C9BAC mice, we were not able to safely perform ICV bolus injections with more than 10 nmol of LNA-modified ASO3 due to induction of severe motor phenotypes. To overcome this limitation, we used osmotic pumps to compare the potency of ASO3 and ASO5. Doses ranging from 2.5 to 20 nmol per day of each ASO were continuously infused over 10 days into the right lateral ventricle of age-matched heterozygous C9BAC mice through a cannula using an implanted Alzet osmotic pump. The cortex and spinal regions of animals treated with ASO3 and ASO5 demonstrated potent, dose-dependent reduction in V1 and V3 repeat-containing transcripts in both the cortex and spinal cord regions (Fig. 1d, f). Importantly, despite their impact on V1 and V3, neither ASO3 nor ASO5 produced any substantial reduction of the level of the V2 transcript (and hence the total C9ORF72 transcript variants) (Fig. 1e, g). Poly-GP DPR was also reduced in the cortex of mice treated with both ASO3 and 5 (Fig. 1h).
Neither ASO produced adverse behavioral side effects; all animals remained healthy until they were sacrificed at 21 days. Routine clinical blood chemistry and liver and kidney morphology after H&E staining revealed no gross abnormalities. At the 100 nmol dose, mice receiving ASO3 showed more weight loss than the other groups (18%) (Fig. 1i). We therefore focused on ASO5 for further optimization.
PS reduction optimizes ASO5 safety and activity in mice
Bolus injections of 50 nmol or above induced severe motor phenotypes in the first hours following injection. This low MTD notably limits the dosing scheme and narrows the therapeutic window. We therefore sought to improve the efficacy and tolerability of ASO5.
ASO5 has a fully modified phosphorothioate (PS) backbone. The PS modification confers nuclease stability and increased protein binding, which leads to increased cellular uptake but can also lead to toxicity30. We sought to reduce the number of PS linkages to generate a less toxic ASO without compromising activity31–33. We reduced the PS content within the 5’ and 3’ MOE-modified “wings” of the ASO, maintaining PS modification throughout the DNA gap. ASO5-1 and ASO5-2 differed only in the presence or absence of a PS linkage between nucleotides 5–6 and 13–14 (Table 1). Both of these analogues showed reduced motor phenotypes and a higher MTD than ASO5. Elsewhere, we explore the mechanism of the motor phenotypes observed here and follow up on the improvement observed by mixed backbone modification patterns34.
We administered 30 nmol of each ASO via a bolus ICV injection in C9BAC mice and analyzed efficacy at 8 weeks (Fig. 2a–f). Mice treated with 30nmol of ASO5 and ASO5-2 had a significantly reduced level of V1 and V3 transcripts in cortex and spinal cord as compared to the PBS treated group (Fig. 2a,d) with minimal effect on total transcripts (Fig. 2b,e) showing that absence of PS inter-nucleotide linkages between two MOE modified nucleotides did not impair biological activity in vivo. By contrast, ASO5-1, which lacked the PS linkage at the junction between the MOE wings and the DNA gap, achieved only ~ 25% knock down of V1-V3 at a 30nmol dose (Fig. 2a,d). Dose-response studies confirmed the superior efficacy and potency of ASO5 and ASO5-2 relative to ASO5-1 (Fig. S3). ASO5-2 treatment also reduced polyGP levels in cortex and spinal cord (Fig 2c,f). Based on its efficacy and tolerability profile, we selected ASO5-2 as our lead compound.
Figure 2. Analogues of ASO5 with reduced phosphorothioate content maintain robust and durable biological activity after CNS administration in heterozygous C9BAC mice.

(a-f) A single-dose infusion shows robust silencing of V1-V3 repeat containing transcripts (a, d) with minimal change in overall C9ORF72 RNA levels (b, e) in cortex and spinal cord quantified by qRT-PCR, along with a robust drop in levels of polyGP (c,f) in mice treated with PBS (dark grey), ASO5 (light blue), ASO5-1 (medium blue) and ASO5-2 (dark blue) eight weeks after administration of 30nmol of each ASO. For each ASO group, n=5–7. (g-h) Dose-dependent silencing of C9ORF72 transcripts (g) and polyGP (h) after a single injection of 1, 5, 15, 30, or 60 nmol of ASO5-2 relative to PBS control. ASO 5–2 was administered into the lateral ventricle of 5–6-month-old heterozygous C9BAC mice and expression of RNA and dipeptide levels was evaluated after three weeks. For each ASO group, n=5–7. (i-j) A time course experiment was performed in heterozygous C9BAC mice treated with 30nmol of ASO5-2: tissues were collected and analyzed at 24 h or 3, 8, 12 and 20 weeks after treatment; an 80 nmol dose was also harvested at 20 weeks only. Expression of V1,V3 repeat-containing transcripts (i) and polyGP (j) was analyzed in cortex 24 h or 3, 8, 12 or 20 weeks after a single dose injection of ASO5-2. In all panels, *p<0.05, **p<0.01, ***p<0.001, comparing treatment groups to PBS group, based on one-way ANOVA with Dunnett’s multiple comparisons test. In panels a-f, h-j, each data point is derived from a separate animal.
Sustained potency of mixed-backbone ASO5-2 two mouse models.
We next tested the in vivo dose dependence of ASO5-2, which revealed an ED50 of 4.75nmol (Fig. 2g) for V1-V3 RNA silencing in the cortex of C9BAC mice three weeks after treatment. At the same timepoint, ASO5-2 also significantly and dose-dependently reduced levels of polyGP in a dose-dependent manner (Fig. 2h).
We also evaluated the duration of effect of ASO5-2. Our initial studies showed that absence of PS linkages flanked by MOE nucleotides (as in ASO5-2) did not impair biological activity in vitro and in vivo three weeks after treatment. However, since PS linkages also protect ASOs from nuclease degradation, we wondered if the duration of effect of ASO5-2 would be comparable to its fully PS modified parent ASO5. To address this question, we injected C9BAC transgenic mice with 30 nmol of ASO5-2 and analyzed the level of V1-V3 transcripts 24 h or 3, 8 or 20 weeks after injection (Fig. 2i). No effect was observed on the V1-V3 target RNA 24 hours after injection. However, a significant, specific dose-dependent reduction of ~80% of V1-V3 transcripts but not total C9 transcripts was observed 3 weeks after injection and, importantly, this was sustained up to 20 weeks in the cortex (Fig. 2i). Analogously, we also observed a sustained reduction in the levels of the polyGP proteins, approaching 90% reduction even at 20 weeks after the single injection of 30 nmol ASO5-2 (Fig. 2j).
No significant body weight loss or behavioral adverse events (as defined in the methods section) were detected in animals treated with ASO5-2 or the PBS control (Fig. S4a). Likewise, no change in liver or spleen weight or morphology was observed (Fig. S4b–d). Finally, to further assay the tolerability of ASO5-2 treatment, we analyzed the coordinated motor functions of mice treated with ASO5-2 or injected with vehicle PBS. Seven mice per group were tested in a blinded manner on their rotarod performance weekly after treatment for 19 weeks. No motor deficit was observed in the treated group, underlining the tolerability of ASO treatment (Fig. S4e).
From these observations we concluded that ASO5-2 can be safely administered to transgenic C9BAC mice via intracerebroventricular delivery and that this durably suppresses the offending V1 and V3 transcripts but not the V2 transcript of C9ORF72, as well as the toxic polyGP dipeptides.
ASO5-2 is non-toxic in large animals
Encouraged by safety and efficacy data in the C9BAC mice, we next obtained additional preclinical toxicity profiles in large animals focusing on ASO5-2 as our lead ASO. As a pilot behavioral study, we performed an intrathecal injection of ASO5-2 at 2 mg/kg in four sheep. For intrathecal injection in sheep, it was necessary to thread a microcatheter up though the intrathecal space and deliver the ASO directly into the cisterna magna (see Methods)34. Intracisternal contrast injection and cone beam computed tomography confirmed the correct catheter position prior to ASO injection. At one month, the sheep showed no neurological abnormalities.
We then purchased a batch of GMP-grade ASO5-2 (ChemGenes). The sequence and modification pattern of the GMP ASO was confirmed by mass spectrometry fragment analysis (Fig. S5).
We engaged an outside laboratory (Charles River Laboratories) to conduct a GLP safety study of ASO5-2 in cynomolgus monkeys. Twenty-eight normal monkeys averaging 2.5 kg in weight were treated with 0 [n=8], 1.5 [n=12] or 6.0 [n=8] mg of ASO5-2. Necropsy was performed at 29 or 90 days (half the animals at each timepoint). Treatment was by intrathecal injection on Days 1, 14, 28 for animals assigned to the Day 29 and Day 90 necropsies and additionally on Days 57 and 85 for animals assigned to the Day 90 necropsy. No behavioral or neurological deficits were observed out to 90 days at either dose; the necropsies did not show pathological findings attributable to ASO5-2 (data not shown).
ASO5-2 suppresses polyGP levels in a C9ORF72 patient
Further encouraged by the large animal studies, we administered ASO5-2, which we have designated afinersen, in ascending doses to a single individual patient with atypical motor neuron dysfunction who harbored a C9ORF72 mutation, with appropriate authorization by the Western Institutional Review board and the FDA (IND141673). This individual was a 60-year-old male with a C9ORF72 G4C2 expansion of approximately 2,400 repeats. He was assessed neurologically because of recurrent left foot-drop induced by distance running followed by slowly progressive weakness in the feet and hands. On examination he demonstrated distal weakness in the legs, diffuse hyporeflexia and subtle sensory loss in the distal legs and feet. Physiological studies documented focally enlarged motor units with evidence of scattered foci of active denervation. CSF analysis revealed elevated levels of polyGP dipeptide repeats (Table 2) but was otherwise benign. As summarized in Fig. 3, on August 29, 2019, he was treated with intrathecal ASO5-2 at 0.5 mg/kg, followed by 1.0 mg/kg two weeks later. On January 26, 2020, he received a third dose at 1.5 mg/kg, increased to 2.0 mg/kg on February 13; 2.0 mg/kg was subsequently administered four more times (Fig. 3). He experienced no medically or neurologically adverse effects from these interventions; his laboratory safety studies were unremarkable. In multiple CSF evaluations (Table 2), his CSF cell counts, protein and glucose levels were largely unremarkable. At the time of the 4th and 5th CSF analyses, he demonstrated very mild pleocytosis (but not in both CSF tubes on a given day). He also showed a progressive increase in CSF phosphorylated neurofilament heavy and neurofilament light chains (Table 2), which peaked after the 4th 2.0 mg/kg dose of ASO5-2 and then partially subsided as the interval between doses was extended.
Table 2.
Cerebrospinal Fluid Parameters in Relation to Dosing of ASO5-1 in Patient
| Chems1 | ASO | ASO | GP | Tube 1 | Tube n | Differential Tube 1 | Differential Tube n | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Date | Weeks | Glu | Pro | Dose2 | Level3 | Rel4 | pNFH5 | NFL6 | WBC7 | RBC8 | WBC | RBC | n | cells | lymphs | monos | polys | cells | lymphs | monos | polys |
| 05/9/16 | 71 | 56 | 0 | 1.00 | 2 | 37 | 1 | 5 | 3 | 16 | 31 | 63 | 6 | 100 | 45 | 54 | 1 | ||||
| 8/24/19 | 0 | 59 | 0.5 | 0 | 1.00 | 253.8 | 2,249.8 | 0 | 0 | 4 | 5 | 100 | |||||||||
| 9/9/19 | 2 | 65 | 48 | 1.0 | 11.7 | 1.05 | 309.2 | 2,478.2 | 1 | 2 | 4 | 50 | 36 | 64 | |||||||
| 10/7/19 | 6 | 62 | 52 | 19.9 | 1.08 | 444.8 | 2,840.3 | 1 | 0 | 4 | 18 | 67 | 33 | ||||||||
| 1/16/20 | 20.4 | 69 | 54 | 1.5 | 1.7 | 0.62 | 353.4 | 2,922.0 | 1 | 3 | 2 | 0 | 4 | 9 | 67 | 33 | 4 | 50 | 50 | ||
| 1/30/20 | 22.4 | 71 | 54 | 20.5 | 0.62 | 299.4 | 3,212.4 | 3 | 211 | 1 | 2 | 4 | 100 | 58 | 36 | 6 | 36 | 67 | 33 | ||
| 2/13/20 | 24.4 | 59 | 55 | 2.0 | 15.1 | 0.53 | 350.2 | 3,078.4 | 1 | 3 | 1 | 1 | 3 | 27 | 74 | 26 | 11 | 55 | 45 | ||
| 2/26/20 | 26.3 | 65 | 65 | 42.5 | 0.37 | 516.7 | 3,687.9 | 1 | 1 | 1 | 1 | 3 | 42 | 45 | 55 | 50 | 44 | 56 | |||
| 3/9/20 | 28.0 | 64 | 52 | 2.0 | 26.1 | 0.38 | 442.4 | 4,137.3 | 3 | 1 | 1 | 1 | 3 | 66 | 58 | 42 | 81 | 62 | 38 | ||
| 4/23/20 | 34.4 | 67 | 62 | 17.7 | 0.24 | 810.8 | 11,269.3 | 4 | 173 | 8 | 1 | 3 | 100 | 41 | 58 | 1 | 100 | 54 | 46 | ||
| 5/4/20 | 36.0 | 65 | 66 | 2.0 | 14.4 | 0.21 | 1,156.3 | 13,626.6 | 11 | 0 | 3 | 2 | 3 | 100 | 58 | 42 | 6 | 6 | 67 | 33 | |
| 7/27/20 | 48.0 | 67 | 49 | 2.0 | 9.33 | 0.23 | 1,337.9 | 2,790.5 | 8 | 4 | 3 | 7 | 2 | 23 | 78 | 22 | 100 | 65 | 33 | 2 | |
| 10/20/20 | 60.1 | 66 | 40 | 2.0 | 8.9 | 0.30 | 966.2 | 10,361.5 | 2 | 1 | 1 | 0 | 3 | 19 | 47 | 33 | 58 | 62 | 48 | 3 | |
mg/dl;
mg/dl;
ng/ml;
ng/ml (baseline 0.01–0.03 ng/ml);
pg/ml (normal 2–300, ALS 250–12,000)
pg/ml (normal 100–1,500, ALS 1,000–20,000)
cells/ml
cells/ml
Glu – glucose; Pro – protein; GP Rel – poly(GP) level relative to baseline
pNFH – phosphorylated neurofilament heavy chain; NFL – neurofilament light chain
WBC – white blood cell count; RBC red blood cell count; cells - #/ml
lymphs – lymphocytes; monos – monocytes; polys – polymorphonuclear cells
Figure 3. Clinical summary and afinersen (ASO5-2) dosing.

A single patient received multiple doses of ASO5-2 as indicated with arrows below the graph. The patient’s ALSFRS-R score before and during treatment is shown as a blue line and points. The patient’s poly(GP) DPR level, relative to the baseline (100%) is shown as a red line and points.
The toxic polydipeptides translated from the expanded hexanucleotide repeat are detectable in the CSF of ALS patients. We monitored polyGP levels as a biomarker to ascertain the efficacy of silencing of the repeat-containing transcripts. The patient’s CSF polyGP levels in two samples prior to dosing were in the range 0.01–0.03 ng/ml. After sequential doses of 2.0 mg/kg, the relative CSF polyGP levels dropped by approximately 80%, correlating with increasing CNS levels of ASO5-2 (Fig. 3, Table 2). During the months of treatment, the patient’s ALS functional rating score (ALSFRS-R) was largely stable. Initially it was 38 (48 is normal), decreasing to a nadir of 33, but then improving to 38 concomitantly with the higher doses of ASO5-2.
We also monitored levels of ASO5-2 within the patient’s CSF (Fig. S6). The data showed an excellent fit to a two-phase exponential curve, reflecting a fast phase presumably due to initial clearance from CSF (t1/2 2.6 days), followed by slow clearance (t1/2 7.8 weeks). A dose of 2 mg/kg every three months showed stable levels of ASO in CSF and stable silencing of polyGP.
Discussion
Ten years after the identification of the G4C2 repeat expansion in the gene C9ORF72 as the most common genetic cause of ALS and FTD, multiple investigations have defined potential disease mechanisms. Partial loss of function of C9ORF72 likely contributes to the neurotoxicity of hexanucleotide expansions but does not by itself recapitulate motor neuron degeneration5,35. Forced expression of mutant human C9ORF72 in mice also fails to reproduce frank motor neuron pathology but does replicate important molecular signatures, including intranuclear RNA foci and cytoplasmic polydipeptides. These are generated selectively by the two of the three C9ORF72 transcripts, V1 and V3, that harbor hexanucleotide expansions. Together, these observations suggest that an effective therapeutic strategy will be to suppress expression of V1 and V3 to reduce levels of repeat-containing RNA and polydipeptides while maintaining baseline levels of V2, which produces the full-length C9ORF72 protein and thereby avoids haploinsufficiency. Here, we report that ASOs targeting the 5’junction of the expanded repeat selectively reduce C9ORF72 repeat-containing transcripts V1 and V3 in a dose dependent manner after ICV administration in C9BAC transgenic mice, without reducing the overall level of C9ORF72 V2 transcripts. This intervention significantly reduces numbers of intranuclear RNA foci and depresses level of polyGP dipeptides, which are predicted to be produced from both sense and anti-sense strands of the hexanucleotide repeat. Moreover, in a single patient this also reduced polyGP dipeptide levels by approximately 80%.
This report confirms earlier reports that ASOs can suppress V1 and V3 transcripts in C9-ALS/FTD20–26 and extends our understanding of this therapeutic approach in several important ways. Importantly, we have examined the impact of modifications of the backbone on both the potency and toxicity of the ASOs. Reducing the degree of PS modification substantially increased the acute tolerability in the CNS without impairing its potent biological activity. While there are published examples of mixed-backbone ASOs used in the CNS31–33,36–39, to the best of our knowledge this is the first presentation of comparative data on the efficacy and tolerability of fully vs partially PS-modified ASOs and the first to highlight the critical importance of the position of PS modifications in maintaining efficacy while reducing toxicity.
In the course of these studies of ribose and backbone modifications, we compared the efficacy of ICV-bolus vs chronically administered ASOs. It appears that, for comparable doses, delivery by bolus is more effective than by pump. For example, V1-V3 transcripts were reduced ~80% by ASO5 at 30 nmol delivered by IV bolus, while pump delivery of 50 nmol failed to reduce levels of these transcripts. An analogous difference was observed in ASO therapy in a murine model of spinal muscular atrophy29, in which bolus administration proved more effective than chronic infusion.
The observed efficacy in mice and safety in both mice and primates prompted our study of repeated dosing of ASO5-2 via intrathecal delivery in a single patient. This was well tolerated as gauged by clinical examination and the safety profile in blood and CSF (Table 2). With repeated dosing of ASO5-2, we observed a progressive but transient elevation of two CSF biomarkers, phosphorylated neurofilament and neurofilament light chains. We ascribe this to some degree of nerve root irritation provoked by the intrathecal ASO or to some component of the disease not influenced by ASO5-2. This phenomenon is described in the literature in humans but fortunately has not been associated with major neurological consequences40. In our case, these biomarkers subsided somewhat in the later CSFs, as the interval between dosing was extended, suggesting a dose-response relation. The patient’s clinical metrics improved across the course of therapy, despite the increment in the CSF neurofilament biomarkers.
Most importantly, our study provides proof-of-concept that ASO therapy in a human can effectively and safely suppress levels of the C9ORF72 transcript that harbor expansions (V1 and V3) without significantly affecting the predominant V2 transcript. In our models in vitro and in vivo, and in an individual with a C9ORF72 expansion, we observed an 80% reduction in levels of polyGP dipeptide. Suppression of CSF levels of a C9ORF72 polydipeptide has not previously been reported in humans. There is substantial evidence that significant pathology is derived from the C9ORF72 polydipeptides12,15,41–45, and directly toxic effects of the expanded RNA transcript have also been described46. Our findings strongly encourage the view that suppressing the expression of the mutant alleles of C9ORF72 may be clinically beneficial, regardless of whether the primary benefit is mediated by reduction in mutant transcripts or polydipeptide levels. Larger clinical trials now underway to assess the clinical impact of therapies that silence C9ORF72 will likely illuminate this hypothesis.
METHODS
Design of Mouse and Human Studies
The goal of this study was to develop an antisense oligonucleotide (ASO) as a potential clinical therapeutic candidate to treat C9-ALS/FTD. Our experimental approach combined C9 patient derived samples to evaluate selective efficacy in vitro and C9BAC transgenic mice to assess safety, efficacy and duration of effect in vivo. Per experiment, all mice were aged-matched and randomly assigned to control or experimental group. Samples and data were blindly collected, processed and quantified. Molecular and physiological readouts include expression level of V1V3 repeat containing transcripts, all transcripts and peptides, weight of body and organs and motor functions. All outliers were included in data analysis. Exploratory experiments were performed on at least five mice per genotype. Sample size was calculated using the G-power analysis method based on previously defined effect size and standard deviation measuring variability within the sample.
The flow chart for the clinical study is presented in Figure 3. This study was conducted with approval from the UMass Medical School and Western Institutional Review Boards (WIRB # 20183136) and the FDA (IND 141673). Our initial spinal fluid evaluation was performed under the auspices of a Longitudinal Biomarker Study, UMass. Medical School IRB docket #14341. Patient consent was obtained for these investigations as approved by the IRB.
Antisense Oligonucleotides
LNA phosphoramidites were synthesized in house using standard methods47 from the 3’-hydroxyl precursors. All other phosphoramidites were purchased from ChemGenes. 0.1M DDTT (ChemGenes) was used as the sulfurizing reagent and 0.25M BTT (AIC) as the activator. ASOs were synthesized on a Dr. Oligo 48, ABI394, AKTA Oligopilot10 or AKTA Oligopilot 100 synthesizers, according to the required scale. LNA and MOE phosphoramidites were coupled for 8 minutes. Oligonucleotides were deprotected in concentrated aqueous ammonia at 55°C for 18 h and purified using ion-exchange chromatography (eluting with 30% acetonitrile in water containing increasing gradients of NaClO4). Final purification, desalting, concentration and pH adjustment were effected by diafiltration in an Amicon centrifugal filter. All oligonucleotides were characterized by LCMS.
Dual Luciferase Reporter Assay System
C9-intron1 containing two G4C2 motifs (334 nucleotides 5’ of the G4C2 repeat motif and 769 nucleotides in the 3’ end) was amplified from blood genomic DNA using the following forward and reverse primers 5’-acgtatgcggccgcacgtaacctacggtgtc-3’, 5’-atacgtgcggccgctaccatcagtcaagtgatg-3’, and was cloned into the psiCHECK™-2 vector (Promega). C9intron1 expression was measured with the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer instructions.
Cell culture
HEK293T cells
HEK293T cells were transfected with 6.5μg of psiCHECK™-2 vector (Promega) containing C9 intron1 using Lipofectamine 3000 with P3000 reagent according to the manufacturer instructions (ThermoScientific) in T25 flasks. One day after transfection, cells were plated into a 96-well plate in DMEM supplemented with 10% FBS and treated with the indicated dose of ASO the next day using Lipofectamine RNAiMax Reagent (ThermoScientific). Cells were lysed 48hours after ASO treatment and luciferase signals were quantified.
C9 Patient Derived Fibroblasts.
Skin biopsies obtained from two unrelated C9 carriers were cut into small pieces and placed on a culture dish with DMEM supplemented with 15% fetal bovine serum to allow fibroblasts to expand. ASO treatment was performed on cells plated in 10 cm dish using Lipofectamine RNAiMax Reagent (ThermoScientific). Total RNA was isolated 72 hours after treatment.
C9BAC Derived Primary cortical neurons.
Embryos were removed at E15.5 from pregnant wild-type C57BL/6 females crossed with homozygous C9BAC males. Cortical tissue of each embryo was dissected on ice-cold Hank’s Balanced Salt Solution (ThermoScientific). Pooled tissue was minced and digested with 0.05% Trypsin-EDTA (Life Technology) at 37°C for 12 min. Digestion was halted by addition of 10% FBS/DMEM. Cells were triturated, resuspended in neurobasal media supplemented with Glutamax (ThermoScientific), 2% penicillin/streptomyocin and B27 supplement (ThermoScientific) and seeded at 0.5 × 10E6 cells/well in 6-well plates pre-coated with poly-ornithine (Sigma). Neurons were treated with ASO at the indicated dose five days after culture and collected 15 days after treatment.
C9ORF72 Bac Transgenic Mice
C9BAC mice were generated as previously described18 and backcrossed to C57BL/6. All experimental protocols and procedures were approved by the University of Massachusetts Medical School Institutional Animal Care and Use Committee.
Stereotaxic Pump Implantation and Bolus Injection of ASO in the Mouse Brain.
For intracerebroventricular (ICV) infusion of ASO or PBS vehicle through a micro-osmotic pump (Alzet pump model 1007D attached to Alzet brain infusion kit 3), wild-type C57BL/6 or C9BAC transgenic mice were anesthetized and maintained on 2.5% isoflurane via a nose cone under a stereotaxic frame. Implantation procedure was performed as previously described48, with a 3mm cannula implantation 0.2mm posterior and 1.0mm lateral to the right of Bregma.
For ICV bolus injection mice were anesthetized with isoflurane and placed into a stereotaxic frame. 10μL of sterile PBS or ASO was injected into the right lateral ventricle using the following coordinates: 0.2 mm posterior and 1.0 mm lateral to the right from the Bregma and lowered to a depth of 3 mm.
Mouse Behavior Monitoring
Over the course of treatment and immediately prior to sacrifice, each animal was blindly weighed and evaluated weekly by a trained observer for adverse events, defined as any behavior not typical in a naïve matched control animal, including, but not limited to: limb clasping, tremors, abnormal respiration, paralysis, spasticity, impaired reflex, hyperactivity and lethargy.
Rotarod
Coordinated motor functions were assessed in control and treated mice using the rotarod test as previously described18. Briefly, mice were tested weekly beginning two weeks prior to ASO/vehicle administration and ending at the week of sacrifice. Each animal was given three trials on a 4–40rpm accelerating rotarod for 5 minutes with a one minute inter trial interval. Latencies to fall for each animal was automatically recorded by a computer and plotted as a mean +/− SEM.
Blood Biochemistry
Whole blood samples were collected after cardiac puncture (terminal procedure). Blood biochemistry was performed using the VetScan Comprehensive diagnostic profile (Abaxis, Union City, CA).
Southern Blot
Southern blot was performed on 10 μg genomic DNA isolated using Gentra Puregene Tissue kit (Qiagen). DNA was digested overnight with AluI and DdeI at 37°C and separated by electrophoresis on a 0.6% agarose gel, transferred to a positively charged nylon membrane (Roche Applied Science), cross-linked by UV, and hybridized overnight at 55°C with a digoxigenin-labeled G2C4 DNA probe in hybridization mix buffer (EasyHyb, Roche). The digoxigenin-labeled probe was detected with anti-digoxigenin antibody and CDP-Star reagent as recommended by the manufacturer (Roche).
RNA Extraction and Quantitative RealTime-PCR
Total RNA was isolated from snap frozen cortex or spinal cord tissue using Trizol (ThermoScientific) and subsequently treated with DNase I (Qiagen). One μg of total RNA was reverse transcribed into cDNA using random hexamers and MultiScribe reverse transcriptase (ThermoScientific) following the manufacturer’s instructions. Quantitative PCR was performed on a StepOnePlus Real-Time PCR system using SYBR Green Master Mix (Applied Biosystems) and 0.2 μM of forward and reverse primers as described in41. Ct values for each sample and gene were normalized to GAPDH. The 2exp (−ΔΔCt) method was used to determine the relative expression of each target gene.
Fluorescence in situ Hybridization (FISH)
FISH was performed as previously described49 using a 5’ end Cy3-conjugated (G2C4)4 oligonucleotide DNA probe at 55°C in hybridization buffer containing formamide 40%/2XSSC/0.1% Tween-20/DNA salmon sperm. Samples were then washed twice in pre-warmed wash buffer (formamide 40%/2XSSC/0.1% Tween-20) and in stringency wash buffer (0.2XSSC/0.1%Tween20) at 55°C. Samples were then mounted in Prolong Gold Antifade reagent with DAPI (ThermoScientific). Confocal images were taken with a Leica TCS SP5 II laser scanning confocal microscope and processed with Leica LAS AF software.
Detection of poly(GP) and poly(PR)
Rabbit polyclonal anti-poly GP and PR antibodies were generated to the repeat motif (GP8, PR8) by New England Peptide. Poly(GP) or poly(PR) levels in lysates were measured using a sandwich immunoassay that utilizes Meso Scale Discovery (MSD) electrochemiluminescence detection technology. Tissue samples were lysed in RIPA buffer supplemented with 1× Roche complete protease inhibitor and 1X Halt Phosphatase Inhibitor Cocktail (ThermoScientific) using TissueLyserII (Qiagen) followed by sonication on ice. Samples were gently homogenized on a rocker at 4°C for 30 min. Debris were removed by centrifugation (15 min, 14 000 g, 4°C) and the supernatant collected. Total protein concentration was determined using the BCA Protein Assay Kit (Thermo Scientific). 50 μg of total protein diluted in PBS-Tween supplemented with 10% fetal bovine serum was loaded per well in duplicate wells. Serial dilutions of recombinant (GP)8 or (PR)8 spiked in wild-type C57Bl/6 brain protein extracts were used to prepare the standard curve. Response values corresponding to the intensity of emitted light upon electrochemical stimulation of the assay plate using the MSD QUICKPLEX SQ120 were acquired and background corrected using the average response from lysates obtained from wild-type C57Bl/6 brain extract.
Detection of pNFH and NFL
Neurofilament light chain (NFL) and phosphorylated neurofilament heavy chain (pNFH) were detected in CSF samples using sandwich immunoassays and the MSD QUICKPLEX SQ120. For pNFH measures, the Iron Horse Diagnostics clinically validated assay was used with a monoclonal anti-pNFH antibody as capture and sulfo-tagged polyclonal anti-pNFH antibody for detection. Purified pNFH was used to generate a calibration curve. All assays were performed in triplicate with a coefficient of variation (CV) < 5% for all samples. The lower limits of quantification of the pNFH assay is 1.2 pg/ml in CSF. For NFL measures, a R-PLEX human NFL MSD assay kit was used (MSD catalog # F217X) following manufacturer specifications. All assays were performed in triplicate with a CV < 5% for all samples. The lower limits of quantification of the NFL assay is 2.0 pg/ml.
Immunohistochemistry
Brains were rapidly removed from euthanized animals. The contralateral hemibrain was post-fixed in 10% formalin. Paraffin-embedded or cryoprotected blocks were cut in 10μm thick sagittal sections. Slides were permeabilized with Triton 0.1% for 10 min. Non-specific antibody binding was blocked by incubation with 10% goat serum in PBS/Tween0.01% for one hour. Primary antibodies were diluted in blocking solution and sections were incubated overnight at 4°C. After three washes in PBS/Tween 0.01%, sections were incubated with Alexa fluor-488 or −546 conjugated secondary antibodies diluted in PBS for one hour at room temperature. Autofluorescence was quenched by slide immersion in 0.5% Sudan BlackB in 70% ethanol and cell nuclei were stained with DAPI. Primary antibodies used: mouse anti-NeuN (1:500, Millipore), rabbit anti-P/S ASO (1:500, in house). Briefly, a rabbit polyclonal antibody raised in house by inoculating two female New Zealand White rabbits with a fully-PS-modified, KLH-conjugated ASO. Boosts and bleeds were carried out at regular intervals over one year, and antisera were used for histology (Moazami and Watts, in preparation).
Haematoxylin and eosin (HE) staining
8 to 10 μm thick sections of mouse liver and kidney were cut from formalin-fixed, paraffin embedded blocks. Standard HE staining was performed.
Mass spectrometric characterization of clinical antisense oligonucleotide
The sequence and modification pattern of ASO5-2 was confirmed by mass spectrometric (MS) analysis performed by nanoflow electrospray ionization (i.e., nanospray) on a Fourier transform ion cyclotron resonance (FTICR) mass spectrometer (vide infra for conditions). The multiply charged ions observed in negative ion mode (Figure S4a) provided a monoisotopic molecular mass of 6461.1894 u, which matched very closely the expected value of 6461.1888 Da calculated from sequence. In subsequent experiments, the [M - 4H]4− molecular ion at 1615 m/z was submitted to tandem mass spectrometry (MS2) to achieve sequence confirmation (vide infra). Upon isolation in the mass selective quadrupole (Q) and activation by collisions with Ar in the collision cell (q), the precursor ion exhibited typical fragment series corresponding to the central region of the construct, which confirmed the sequence spanning from A7 to C13 (Fig. S5b). Distinctive signals were additionally detected at 1179 and 1971 m/z, which were assigned to fragments corresponding to the entire G1:T6 and G14:C18 moieties, respectively. In turn, each of these first-generation fragments were individually isolated and activated in the FTICR cell to complete MS3 determinations (Fig. S5c–d). These experiments provided abundant second-generation fragments that confirmed the sequences of the construct’s terminal regions.
All analyses were carried out on a Bruker (Billerica, MA) 12T solariX FTICR equipped with a 12-tesla superconducting magnet and a home-built ion source that enabled static nanospray operations. No sample desalting or chromatographic steps were employed. All samples consisted of a 4 μM solution of ASO5-2 in 150 mM ammonium acetate (pH 7.4) and 10% volume of 2-propanol. In each analysis, a 5 mL aliquot was loaded onto a quartz emitter prepared in-house. A stainless steel wire was inserted from the back end to provide the voltage necessary to achieve a stable spray. The instrument was calibrated by using a 1 mg/mL CsI solution that provided an accuracy of 87 ppb. Detection was accomplished in broadband mode, which afforded a typical 230,000 resolution. The first activation step was carried out in the collision cell (q) of the instrument, which was flooded with a low pressure of Ar and subjected to an 18.5 V activation voltage. The second activation step was carried out in the FTICR cell by irradiating selected ions with a frequency that was 250 Hz off-resonance and 0.65–1.29% power. The data were processed by using the DataAnalysis package provided by Bruker. The results were interpreted by using the Mongo Oligo Mass Calculator v2.08 available at https://mods.rna.albany.edu/masspec/Mongo-Oligo.
Oligonucleotide pharmacokinetics
Patient CSF was stored at −80°C until analysis. ASO levels in patient CSF were quantified using an assay based on splint ligation and qPCR as described50 CSF from the same patient prior to treatment was added to the hybridization mixture and spiked with a known concentration series of ASO to generate a standard curve. Sequences of oligonucleotides used for detection of ASO 5–2 were as follows: Probe A: 5′ CTCGACCTCTCTATGGGCAGTCACGACAGGAGTCGCGCG 3′; Probe B: 5′ pCTAGGGGCCGCTGAGTCGGAGACACGCAGGGCTTAA 3′; Forward primer: 5′ GCTCGACCTCTCTATGGGC 3′; Reverse primer: 5′ TTAAGCCCTGCGTGTCTCC 3′; Double-quenched probe: 5′ /FAM/CTAGCGCGC/ZEN/GACTCCGTCGTG/IABkFQ/ 3′. Fitting ASO levels to a two-stage exponential clearance model allowed an excellent fit with empirically measured ASO levels (Fig. S6).
Statistical Analysis
All data were graphed as mean ± SEM, showing data points, and statistics analyzed using GraphPad Prism Software (Version 9.1.1). Tests between multiple groups (comparisons indicated in the corresponding figure legends) used one-way analysis of variance (ANOVA) corrected with Dunnett’s multiple comparisons post-hoc test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns not significant.
Supplementary Material
Acknowledgments:
The authors thank the Brown and Watts labs, Wave Life Sciences, the Animal Medicine and DERC Morphology Cores and F. Ladam for advice, technical support and manuscript review.
Funding:
This work was funded by the NIH (R01 NS111990 to RHB and JKW), the Angel Fund for ALS Research, and the Ono Pharmaceutical Foundation (Breakthrough Science Award to JKW). RHB also acknowledges funding from ALSOne, ALS Finding a Cure, the Cellucci Fund for ALS Research and the Max Rosenfeld Fund. We also acknowledge the NEALS Biorepository for providing all or part of the biofluids from the ALS, healthy controls and non-ALS neurological controls used in this study. The project described in this publication was supported in part by the University of Massachusetts CTSA award number UL1TR001453 from the National Center for Advancing Translational Sciences of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
LIST OF ABBREVIATIONS
- ALS
Amyotrophic Lateral Sclerosis
- ASO
Antisense Oligonucleotide
- C9BAC
Chromosome 9 Bacterial Artificial Chromosome
- C9ORF72
Chromosome 9 Open Reading Frame 72
- DPR
dipeptide repeat
- FTD
Frontotemporal Dementia
- HRE
hexanucleotide repeat expansion
- ICV
Intracerebroventricular
- LNA
Locked Nucleic Acid
- MOE, 2’-O-MOE
2’-O-2-methoxyethyl
- MTD
Maximum Tolerated Dose
- nmol
nanomole
- PO
Phosphodiester
- PS
Phosphorothioate
- V1, V2, V3
transcript variants 1, 2, 3
Footnotes
Competing Interests Statement
The authors have filed a patent related to this research. RHB is co-founder of Apic Bio.
Data and materials availability
The data supporting the findings of this study are available within the main text and the supplementary information. The full clinical trial protocol for this study is available upon request to the corresponding authors. C9BAC mice and PS-targeted polyclonal antibody are available by contacting the investigators.
REFERENCES
- 1.Dejesus-Hernandez M, et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renton AE, et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21- Linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cook C & Petrucelli L Genetic Convergence Brings Clarity to the Enigmatic Red Line in ALS. Neuron 101, 1057–1069 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Amick J & Ferguson SM C9orf72: At the intersection of lysosome cell biology and neurodegenerative disease. Traffic 18, 267–276 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belzil VV, et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol 126, 895–905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu Q, et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci 23, 615–624 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Rourke JG, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burberry A, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med 8, 347ra393 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi Y, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 24, 313–325 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ash PE, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mori K, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Mizielinska S, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tao Z, et al. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum Mol Genet (2015). [DOI] [PubMed] [Google Scholar]
- 14.Freibaum BD, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loveland AB, et al. Ribosome inhibition by C9orf72-ALS/FTD poly-PR and poly-GR proteins revealed by cryo-E<. BioRxiv (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khvorova A & Watts JK The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol 35, 238–248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jafar-Nejad P, et al. The atlas of RNase H antisense oligonucleotide distribution and activity in the CNS of rodents and non-human primates following central administration. Nucleic Acids Res (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peters OM, et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 88, 902–909 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Rourke JG, et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 88, 892–901 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donnelly CJ, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sareen D, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 5, 208ra149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lagier-Tourenne C, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A 110, E4530–4539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang J, et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 90, 535–550 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gendron TF, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci Transl Med 9(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cook CN, et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci Transl Med 12(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang K, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Almeida S, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol 126, 385–399 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zu T, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 110, E4968–4977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rigo F, et al. Pharmacology of a central nervous system delivered 2’-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther 350, 46–55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eckstein F Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther 24, 374–387 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Sztainberg Y, et al. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 528, 123–126 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Becker LA, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tabrizi SJ, et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N Engl J Med 380, 2307–2316 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Moazami MP, et al. Quantifying and mitigating motor phenotypes induced by antisense oligonucleotides in the central nervous system. BioRxiv (2021). [Google Scholar]
- 35.Waite AJ, et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 35, 1779 e1775–1779 e1713 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meng L, et al. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 518, 409–412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao HT, et al. LRRK2 Antisense Oligonucleotides Ameliorate alpha-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol Ther Nucleic Acids 8, 508–519 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohan A, et al. Antisense oligonucleotides selectively suppress target RNA in nociceptive neurons of the pain system and can ameliorate mechanical pain. Pain 159, 139–149 (2018). [DOI] [PubMed] [Google Scholar]
- 39.McCampbell A, et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest 128, 3558–3567 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabrizi SJ, Smith AV & Bennett CF Targeting Huntingtin in Patients with Huntington’s Disease - Reply. N Engl J Med 381, 1181–1182 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Aditi, Folkmann AW & Wente SR Cytoplasmic hGle1A regulates stress granules by modulation of translation. Mol Biol Cell 26, 1476–1490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jovicic A, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18, 1226–1229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang YJ, et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med 24, 1136–1142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuva-Aydemir Y, Almeida S, Krishnan G, Gendron TF & Gao FB Transcription elongation factor AFF2/FMR2 regulates expression of expanded GGGGCC repeat-containing C9ORF72 allele in ALS/FTD. Nat Commun 10, 5466 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moens TG, et al. C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol 137, 487–500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coyne AN, et al. G4C2 Repeat RNA Initiates a POM121-Mediated Reduction in Specific Nucleoporins in C9orf72 ALS/FTD. Neuron (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar R, et al. The first analogues of LNA (locked nucleic acids): phosphorothioate-LNA and 2’-thio-LNA. Bioorg Med Chem Lett 8, 2219–2222 (1998). [DOI] [PubMed] [Google Scholar]
- 48.DeVos SL & Miller TM Direct intraventricular delivery of drugs to the rodent central nervous system. J Vis Exp, e50326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran H, et al. Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron 87, 1207–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shin M, Krishnamurthy PM & Watts JK Quantification of Antisense Oligonucleotides by Splint Ligation and Quantitative Polymerase Chain Reaction. BioRxiv (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the main text and the supplementary information. The full clinical trial protocol for this study is available upon request to the corresponding authors. C9BAC mice and PS-targeted polyclonal antibody are available by contacting the investigators.