

ABSTRACT
Background
Renal manifestations of monoclonal gammopathies are of increasing interest among nephrologists. Typical manifestations include light chain cast nephropathy, amyloidosis or renal damage mediated by monoclonal immunoglobulin deposition. Podocytopathies in the setting of an underlying monoclonal gammopathy constitute a rare manifestation of these diseases and, although being described in the literature, remain a challenge since most data derive from case reports.
Methods
A retrospective review of the clinical data of Hospital del Mar and Hospital Vall d’Hebron was performed to identify patients with minimal change disease (MCD) or focal and segmental glomerulosclerosis (FSGS) in the setting of neoplasms that produce monoclonal (M) protein. Additionally, a literature review on this topic was performed. This study aims to describe the clinical characteristics and outcomes of these patients.
Results
Three patients were identified to have podocytopathy and monoclonal gammopathy between the years 2013 and 2020. All three were males and >65 years of age. Two patients were diagnosed with MCD and one patient was diagnosed with FSGS. All patients underwent a kidney biopsy and light and electron microscopic studies were performed. The underlying causes of monoclonal gammopathy were multiple myeloma in two cases and Waldeström macroglobulinemia in one case. Two patients developed nephrotic syndrome during the follow-up. All patients were under active hematological treatment. One patient presented a complete remission of proteinuria whereas the other two presented a partial remission.
Conclusions
Podocytopathies may infrequently be found in patients with monoclonal gammopathies. Patients with overt glomerular proteinuria and hematological disorders with M protein should undergo a kidney biopsy for prompt diagnosis and to specify a prognosis. In addition, further study on this matter must be done to understand the pathophysiology and treat these patients appropriately.
Keywords: focal segmental glomerulosclerosis, MGRS, minimal change disease, multiple myeloma, podocytopathy, Wäldestrom disease
INTRODUCTION
Monoclonal gammopathies constitute a spectrum of clonal cell malignancies or premalignancies, such as multiple myeloma (MM), Waldeström macroglobulinemia (WM) and chronic lymphocytic leukemia (CLL). In 2012, the term monoclonal gammopathy of renal significance (MGRS) was introduced by the International Kidney and Monoclonal Gammopathy Research Group. MGRS was defined as a clonal proliferative disorder that produces nephrotoxic monoclonal immunoglobulins (Igs) and that does not fulfill the hematological criteria for malignancy [1]. These Igs induce diverse clinical manifestations such as renal failure and proteinuria. The most common finding on biopsy is myeloma cast nephropathy (light chain-cast nephropathy), followed by light chain–associated proximal tubulopathy. Other less common findings are amyloidosis and monoclonal Ig deposition diseases, among others [2, 3].
In 2016, Sethi et al. [3] proposed two different mechanisms of renal damage in the setting of a monoclonal gammopathy. First, they described renal lesions directly associated with monoclonal Ig deposition. When this mechanism appears, Ig deposition can result in diseases involving either the tubular or the glomerular compartments. Larger monoclonal (M) proteins will induce glomerular inflammation, while smaller M proteins (low molecular weight chain molecules) are likely to pass through the glomerular barrier and therefore produce tubular direct toxicity, such as proximal tubular light chain–associated toxicity and/or light chain cast nephropathy. Second, an indirect mechanism of glomerular and vascular lesions that could simulate a proliferative glomerulonephritis was also described. The clinical manifestations of this group of patients appear to be similar to a nephritic syndrome (hypertension, impaired renal function, hematuria and proteinuria) [3].
The presence of overt glomerular proteinuria or an overt nephrotic syndrome associated to monoclonal gammopathy has been rarely reported. Observational data of case series reports describe the presence of minimal change disease (MCD) and/or focal segmental glomerulosclerosis (FSGS) in some patients diagnosed as monoclonal gammopathy, although in a low percentage. There are scarce data reporting podocytopathies in patients with monoclonal gammopathies.
Podocytopathies are the most common group of glomerular disorders leading to massive proteinuria. When evaluating pathophysiology, podocytopathies can be divided into MCD, diffuse mesangial sclerosis, FSGS and collapsing glomerulopathy [4].
Herein we present three cases of patients diagnosed with monoclonal gammopathies (MM, WM or MGRS) that developed overt glomerular proteinuria and were diagnosed as MCD or FSGS.
MATERIALS AND METHODS
A retrospective review of the clinical data between 2000 and 2020 from Hospital del Mar and Hospital Vall d’Hebron was performed to identify patients with MCD or FSGS in the setting of neoplasms that produce M protein. Three patients that met these criteria were identified: two from Hospital del Mar and one from Hospital Vall d'Hebron. Demographic, clinical, laboratory and histological data from renal biopsies were recorded. The medical records of these patients were reviewed and clinical and analytical data were systematically collected.
Definitions
Nephrotic syndrome was defined by the presence of massive proteinuria [>3.5 g/day and/or a urine protein:creatinine ratio (UPCR) >3.5 g/g], hypoalbuminemia (<3.5 g/dL), dyslipidemia and clinically relevant edema [5, 6]. Monoclonal gammopathies reported in this review were defined according to the International Myeloma Working Group criteria for classification of monoclonal gammopathies and related disorders [1]. MCD was defined as the absence of significant visible glomerular alterations by light microscopy, absence of immune glomerular deposits by immunofluorescent microscopy and the presence of diffuse effacement of podocyte foot processes by electron microscopy in the context of a patient with nephrotic syndrome. Complete remission was defined as <0.3 g/day of proteinuria, while partial remission was defined by a decrease of >50% of initial proteinuria [7]. FSGS was defined as the presence of segmental scars in some glomeruli, with either hyaline or adhesions of the tuft to Bowman's capsule, discarding other glomerulopathy [8].
Methods
We report three cases from two hospitals: Hospital del Mar (Barcelona, Spain) and Hospital Vall d’Hebrón (Barcelona, Spain). We conducted a review on the PubMed database and the terms searched were ‘MM’ or ‘hematological discrasia’ or ‘Waldeström disease’ or ‘WM’ or ‘monoclonal gammopathy’ and ‘nephrotic syndrome’ or ‘MCD’ or ‘FSGS’ or ‘podocytopathy.’ All reviewed articles were searched to identify patients who developed a nephrotic syndrome in the setting of a monoclonal gammopathy.
CASE REPORTS
Patient 1
A 67-year-old male was diagnosed with Waldeström disease in 2017. The patient showed IgM plasma levels of 3345 mg/dL at the moment of diagnosis, with normal renal function [serum creatinine 0.8 mg/dL; glomerular filtration rate (GFR) >60 mL/min/1.72 m2] with no urinary protein excretion (UPCR <30 mg/g). From 2017 to 2020, regular exams were performed in our center. In October 2020 he was referred to nephrology due to a clinical nephrotic syndrome: UPCR 26 000 mg/g, hypoalbuminemia 2.1 g/dL and hypercholesterolemia 292 mg/dL. The patient developed acute kidney injury (serum creatinine 1.31 mg/dL) coinciding with Waldenström disease progression (IgM 7240 mg/dL) with mild excretion of λ Bence-Jones proteinuria of 0.27 mg/dL. Renal biopsy was performed showing three glomeruli with minor morphologic alterations (focal glomerular basal membrane thickening) without sclerosis (Figure 1). Neither immune deposits nor amyloid were identified. Tubules showed acute tubular injury and a diffuse increase of protein reabsorption (Figure 2). There was mild chronic unspecific interstitial inflammation without plasma cells. A single glomerulus was examined with electron microscopy, with diffuse podocyte foot process effacement, in association with foci of microvillus transformation of podocytes (Figure 3). Glomerular basement membrane was normal in thickness and ultrastructure. Mesangial spaces were preserved. No crystalline structures or immune or organized deposits were identified. The biopsy findings were consistent with an MCD, although unsampled FSGS could not be ruled out due to the scant number of examined glomeruli.
Figure 1:

Patient 1. Optical morphology of the glomeruli. H&E: hematoxylin and eosin stain; PAS: periodic acid-Schiff stain; JMS: Jones methenamine stain; TRIM: Masson's trichrome. Magnification ×600.
Figure 2:
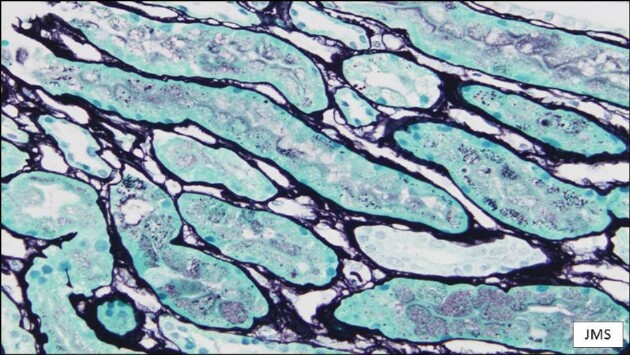
Patient 1. Cortical tubules showing a diffuse increase in protein reabsorption. Absence of abnormal casts. Jones methenamine stain; magnification ×400.
Figure 3:

Patient 1. Electron microscopy findings showing podocyte foot process effacement (black arrows) and microvillus transformation (white arrows).
The patient was treated with plasma exchange for hyperviscosity (five treatments) and with chemotherapy based on bendamustin and rituximab. At the same time, 1 mg/kg/day of prednisone was started for nephrotic syndrome treatment. To date, the patient has gone through five R-bendamustin cycles. The patient presented with complete remission 8 weeks after treatment with prednisone and currently shows <300 mg/mg UPCR and normal renal function. Serum IgM levels decreased to <1000 mg/dL and the patient shows no symptomatology attributable to WM.
Patient 2
An 86-year-old male was referred to nephrology due to detection of proteinuria in September 2013. Initial UPCR determination was 3565 mg/g with normal renal function (creatinine 1.16 mg/dL and GFR 60 mL/min/1.72 m2), urinary immunofixation evidenced κ Bence-Jones proteinuria (657 mg/dL). Serum immunosubtraction proved the presence of an IgG κ monoclonal gammopathy and thus the patient was referred to hematology. Bone marrow aspiration was consistent with IgG κ MM. Renal biopsy was performed, showing 16 glomeruli, some of them globally sclerosed (12.5%), and none of them with segmental sclerosis. Evaluable glomeruli showed minor optical alterations (mild glomerular basement membrane thickening). Tubules showed focal tubular acute injury and arteries showed moderate arteriosclerosis and moderate arteriolar hyalinosis. Immunofluorescence microscopy showed no immune deposits. Two glomeruli were available for electron microscopy with mild and segmental collapse of capillary lumens. Focal and segmental podocyte foot process effacement was detected. Podocyte cytoplasm showed edematous change and focal microvillus transformation, one of them containing one cytoplasmic pseudocyst (Figure 4). Glomerular basement membranes presented normal thickness and no ultrastructural alterations. Mesangial spaces were within the normal limits. With these results in correlation with light and immunofluorescent microscopy data, MCD was suggested.
Figure 4:

Patient 2. Electron microscopy showing podocyte foot process effacement (black arrows), microvillus transformation (white arrows) and occasional cytoplasmic pseudocysts (red arrow).
The patient was first treated with a chemotherapy regimen based on melphalan and prednisone, completing nine cycles, until 2016, when progression of the disease was detected and a second-line treatment based on lenalidomide and dexamethasone was started. In August 2018, new progression of the disease was detected and a third-line treatment was started with daratumumab and bortezomib, completing nine cycles. To date, the patient is under treatment with daratumumab and dexamethasone. Tumoral progressions in 2016 and 2018 were associated with a relapse of proteinuria, responding to the different treatment lines. Initially prednisone was started at a dose of 1 mg/kg/day (60 mg maximum per day) for MCD treatment. In February 2015 the patient presented a relapse of proteinuria, so prednisone treatment was reintroduced with progressive decrease in proteinuria. To date, the patient presents a partial remission of MCD, with 24-hour proteinuria of 500 mg/day and normal renal function (creatinine 0.89 mg/dL, GFR 77 mL/min/1.72 m2).
Patient 3
A 72-year-old male was diagnosed with MM in April 2007. Plasma immunosubtraction found the presence of a monoclonal peak of IgG λ. Since diagnosis the patient has required different lines of treatment for different relapses. In July 2018, proteinuria of 1580 mg/day was detected for the first time, coinciding with a peak of λ levels of 2700 mg/L. Proteinuria diminished at the same time as λ levels after the new treatment was started. Afterwards, in July 2019, proteinuria of 1638 mg/24 h was again detected and a new increase of λ levels was observed. Proteinuria increased to a maximum of 4300 mg/24 h and his chemotherapy was changed to carfilzomib/dexamethasone with very good partial remission. However, proteinuria had a slow decline, with the presence of 50% of albuminuria. Thus the patient was referred to nephrology for persistent proteinuria (1624 mg/24 h) and a renal biopsy was performed. The renal biopsy showed 20 glomeruli, 1 of them globally sclerosed and 3 of them with segmental sclerosis. The rest of the glomeruli did not show optical alterations. Tubules showed mild atrophy and moderate intimal thickening and mild arteriolar hyalinosis were observed in the arteries. Immunofluorescence microscopy showed no immune deposits. Two glomeruli were available for electron microscopy, one of them with sclerosis, the viable glomerulus with diffuse podocyte foot process effacement (>50%), in association with foci of microvillus and vacuolization transformation of podocytes. Glomerular basement membranes had preserved architecture and some were thickened, ranging between 416 and 589 nm with an average thickness of 485 nm. Neither crystalline structures or immune or organized deposits were identified. Mesangial spaces were within the normal limits. With these results, FSGS was suggested. The patient did not fulfill clinical or analytical criteria for nephrotic syndrome and renal function was preserved. To date, proteinuria has decreased to 910 mg/day with only chemotherapy.
Literature review
In the past 2 decades, nine different cases have been reported of patients with monoclonal gammopathy developing a primary podocytopathy. These cases are summarized in Table 2. Most of the reports correspond to male patients (77.7%). A total of four WM, four MM and one single case of monoclonal gammopathy of uncertain significance (MGUS) were reported. Most of them (66.6%) showed κ M protein, whereas in 33.3% of cases these data were not available. We found three cases of MCD, while the rest consisted of FSGS, two of which presented as collapsing FSGS variant. These data are consistent with other reported case series, such as the one by Kofman et al. in 2014 [16]. They conducted a retrospective study of 18 patients with MCD in the context of non-Hodgkin lymphoma (NHL). They found that 33.3% of the patients had an underlying diagnosis of WM, 5.5% MM and 22.22% B-type CLL. The remaining patients were diagnosed as mantle cell lymphoma (MCL), peripheral T-cell lymphoma (PTCL) or marginal zone B-cell lymphoma. According to this study, MCD was preferentially associated with B-cell neoplasms. The majority of cases (55%) had κ M protein and a single patient had a λ monoclonal component associated to WM. No electron microscopy was performed.
Table 2.
Review of reported cases in the literature since 2000
| Year [Ref.] | Age-sex | Hematological disease | Nephrotic syndrome (yes/no) | Proteinuria (g/24 h)/ | Bence–Jones protenuria | Serum albumine diagnose (g/dL) | M-onoclonal protein (mg/dL) | Serum creatinine at diagnose (mg/dL) | Renal disease | Renal disease treatment | Remission (CR/NR/PR) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2012 [18] | 67-M | WM | Yes | 9.4 | No | 1.4 | IgM ĸ (2460) | 1.8 | MCD | DXMb | CR |
| 2020 [19] | 68-F | WM | Yes | 16.2a | No | 3.9 | IgM ĸ (1693) | 0.6 | FSGS | PDN | PR |
| 2013 [20] | 67-M | WM | Yes | 12.7 | No | – | IgM ĸ | 0.9 | MCD | PDN | CR |
| 2019 [21] | 52-M | WM | Yes | – | No | 1.99 | IgM (4190) | 2.38 | MCD | DXMb | CR |
| 2014 [22] | 62-M | MM | Yes | 4.3 | No | 3.2 | IgG ĸ (1500) | 0.8 | FSGS | PDN/CP | PR |
| 2018 [23] | 48-M | MM | No | 8 | No | 3.1 | – | 2.71 | FSGS | PDN | PR |
| 2010 [24] | 72-M | MGUSd | No | 4.8a | – | – | IgM | 5 | FSGSb | – | – |
| 2004 [25] | 51-F | MM | No | 32.6 | Yes | 2.5 | IgA ĸ (4400) | 3.5 | FSGSb | DXMb | CR |
| 2001[26] | 36-M | MM | No | 9.5 | Yes | – | IgG ĸ | 1.7 | FSGS | DXMb | NR |
CP: cyclophosphamide; CR: complete remission; DXM: dexamethasone; NR: no remission; PDN: prednisone; PR: partial remission.
Proteinuria was obtained by urinary protein:creatinine ratio.
Dexamethasone was part of the chemotherapy for hematological dyscrasia.
FSGS collapsing variant was found on kidney biopsy.
Diagnosis of cell dyscrasia was made 2 years prior to renal disease.
Table 1.
Clinical and analytical characteristics of three reported cases
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Hematological disease | IgM Κ WM | IgG Κ MM | IgG λ MM |
| Age at diagnosis (years) | 67 | 86 | 72 |
| Renal disease onset after hematological diagnosis (months) | 52 | 0 | 312 |
| Renal function at diagnosis [GFR (MDRD; mL/min/1.72 m2)] | 55 | 60 | 83 |
| Protein:creatinine ratio (mg/g) | 26 350 | 3565 | 1849 |
| Urine monoclonal component (%) | 2.81 | 64.8 | 0 |
| Serum monoclonal component (%) | 50.9 | 4 | – |
| Serum albumin at renal disease diagnosis (g/dL) | 2.1 | 3.2 | 4.1 |
| Nephrotic syndrome (yes/no) | Yes | Yes | No |
| Renal pathology diagnosis | Podocytopathy | MCD | FSGS |
| Renal replacement therapy | No | No | No |
| Hematological remission | UT | UT | UT |
| Renal remission | CR | PR | PR |
| Renal replacement therapy | No | No | No |
| Renal disease's specific treatment | Steroids | Steroids | No |
| Other treatments | PEX | – | – |
CR: complete remission; PEX: plasmapheresis; PR: partial remission; UT: under treatment.
In 2005, Dingli et al. [17] reported a cohort from the Mayo Clinic database of 13 patients with idiopathic FSGS and a monoclonal cell disorder. In this cohort, most of the patients were males (85%) and the majority were diagnosed with an MGUS (69%). The median age at onset was 65 years, except two patients who were diagnossed at <50 years of age. Presentation with nephrotic syndrome was not unanimous. In our review, of nine patients, 55.5% presented with a nephrotic syndrome, while in Dingli et al. [17] it was only 23% presented like this. Patients diagnosed with MCD are more frequently associated with nephrotic syndrome. In the current literature review, all MCD patients initially presented as nephrotic syndrome. In the study by Kofman et al. [16], all 18 patients exhibited typical features of nephrotic syndrome (all of them with light microscopy studies consistent with MCD).
DISCUSSION
Herein we present three cases of patients that presented a monoclonal gammopathy and developed a podocytopathy (either MCD or FSGS). It is not common to find these two entities in the same patient and the mechanism by which this occurs still needs to be elucidated.
Renal manifestations of hematological diseases have grown in importance over the past few years. It should be noted that since the term MGRS was coined in 2012 by the International Kidney and Monoclonal Gammopathy Research Group, interest among nephrologists concerning monoclonal gammopathies has become more evident. Podocytopathies represent a challenge since etiological mechanisms are not fully understood. MCD is a clinical condition characterized by heavy proteinuria and nephrotic syndrome. Immunofluorescence staining is typically negative and podocyte foot process effacement can be detected only by electron microscopy. FSGS typically refers to a description of glomerular damage and is typically associated with heavy proteinuria and nephrotic syndrome, but kidney biopsies typically show sclerotic lesions that involve focal segments of glomeruli and are associated with variable podocyte foot process effacement [9–11].
Podocytopathies can be primary or secondary to an underlying disease. Multiple studies have hypothesized a direct relationship with a definite trigger or cause. MCD has been reported in association with major blood disorders such as Hodgkin lymphoma and NHL, as described by Kofman et al. [16] Other lymphoproliferative disorders such as MM and epithelial malignant neoplasms have also been described in patients presenting with heavy proteinuria and MCD. It is interesting to note that since these relationships exist, the hypothesis of a circulating factor that can trigger podocyte damage has been discussed in recent years.
MCD and FSGS have historically been described as different entities. Nowadays this separation is under discussion, since both entities have a similar clinical behavior. Several authors propose these two entities to be different histological manifestations of the same underlying disease. In the past few years the term idiopathic nephrotic syndrome (INS) has gained popularity [11]. To date, no determinant circulating factor has been identified for either form of INS.
As commented above, the fact that MCD and FSGS can be related to underlying diseases contributes to the hypothesis of an underlying circulating factor that provokes podocyte injury, disturbing its function and increasing glomerular permeability. Several factors have been pointed to as potential candidates. FSGS permeability factors include cardiotrophin-like cytokine 1 and soluble urokinase receptor, among others. MCD studies have focused on cytokines. Nonetheless, to date, studies have failed to identify a single permeability factor for both entities [11]. In the cases we reported, the link of a hematological underlying disease with the onset of a renal disease such as MCD and FSGS strengthens, as far as we are concerned, the possibility of a circulating factor when a B-cell neoplasm has to be found. This hypothesis seems to be supported by the fact that sometimes progression of the hematological neoplasm worsens proteinuria (as described in patients 2 and 3). In our cases, the treatment of monoclonal gammopathies with renal manifestations is not decided by the renal histology, but rather by the nature of the clone producing the M protein. Therefore, attacking the monoclonal gammopathy would diminish the production of a circulating factor that causes podocyte injury [12].
In MM, Igs, or light chains have been described to present structural changes that alter their molecular charge. This happens because it seems there is an alteration in the cationization and glycosylation processes [13]. This is important because, as previously known, the glomerular filtration barrier not only discriminates on a size basis, but as Brenner et al. [14] described in 1978, for a given molecular size there is different clearance of a substance if there is a change in its molecular charge. Thus charge shifts in plasma proteins seen in lymphoproliferative disorders as described in MM could become potential circulating effectors for podocyte injury. For all this, the particular role of Igs in the setting of an MCD, and therefore in FSGS, needs to be studied.
Some observational studies have tried to elucidate an epidemiological link between a monoclonal gammopathy and the presence of a primary podocytopathy. Bergón and Miravalles [15] described a prevalence of monoclonal gammopathies of 0.19% in a Spanish cohort. The prevalence increased with patient age and for patients >60 years of age, such as our cohort, the prevalence was 1.86%.
Between 2010 and 2020 FSGS and/or MCD was reported in 163 renal biopsies. This represents a prevalence of 1.84%. Using the prevalence observed by Bergón and Miravalles [15] and assuming a Poisson distribution, the Poisson probability of observing at least three cases with an expected mean of 1.86 is 0.3.
In our three reported cases, as seen in Figure 1, the onset of renal disease was not necessarily simultaneous to the hematological diagnosis. Patient 1 began with an overt glomerular proteinuria 52 months (4.3 years) after the onset of the hematological disease (WM), noticing that MCD appeared simultaneously with severe progression of WM that began with a nephrotic syndrome and hyperviscosity symptomatology and was eligible to start treatment. Patient 2 was simultaneously diagnosed as both MM and overt proteinuria and patient 3 was diagnosed a decade after the onset of MM and after being treated with multiple treatment lines since the diagnosis.
The natural history of INS in the context of a monoclonal gammopathy remains unknown and represents a subject of study. The 18 cases of MCD associated with NHL reported by Kofman et al. [16] had different onset timing. They differentiated three different groups. In group 1, MCD preceded NHL by at least 3 months (4–20 months); in group 2, the onset of both entities was simultaneously; and in group 3, NHL preceded MCD by at least 3 months (3–26 months). In this study, the largest group was group 2, with a total of 10 patients. This was the group in which a greater heterogeneity of lymphoid disorders was found: WM (n = 3), marginal zone lymphoma (MZL; n = 4), CLL (n = 1), MCL (n = 1) and PTCL (n = 1). No MM was found in that group. This differs from our patient 2, who was diagnosed as MM at the same time as MCD. In the previously mentioned study, group 3 is interesting from our point of view since all four patients were diagnosed with WM (n = 3) and MM (n = 1). On the other hand, group 1 had four patients diagnosed as CLL or MZL. It is not possible to draw conclusions from a retrospective study like the one mentioned, but still it is striking that our three reported cases do not totally escape from the description and group differentiation that Kofman et al. [16] described.
When it comes to treatment options among these patients, different strategies have been described in the literature depending on clinical features and monoclonal gammopathy, mainly based on chemotherapy. For instance, Dingli et al. [17] reported that all four patients with MM and FSGS responded to chemotherapy with a decrease in proteinuria (two presented a partial remission and two presented a complete remission). In our review, FSGS was most commonly associated with partial remission or no remission. Although FSGS may respond to a corticosteroid-based therapy, it is usually associated with a low response. It is worth mentioning that steroid therapy received in idiopathic FSGS may differ from the steroid therapy associated with chemotherapy [16, 17]. Treatment for neoplasms is intermittent while a steroid regimen in FSGS or MCD is usually daily. From the literature review we conducted, all three patients presenting with MCD had a complete remission. All received corticosteroids; one as treatment of the nephrotic syndrome and the other two associated with chemotherapy for the underlying neoplasm. On the other hand, patient 2 presented a partial remission despite treatment with prednisone for the underlying MCD. It is interesting to note that patient 2 had a difficult control of its proteinuria when neoplasm progressed. Kofman et al. [16] described that in their study all patients received steroids, either alone or associated with chemotherapy. In this study, 14 of 18 patients were treated with standard steroid protocol (in the case of idiopathic MCNS). All of them presented with complete remission and the mean duration of steroid therapy for groups 1 and 2 was 6.1–6.9 months while for group 3 (NHLS precedes MCNS by at least 3 months) was 13 months. They also suggested that relapse was more common when MCD and NHL did not occur simultaneously (group 1 and 3).
In conclusion, there is information showing that podocytopathies may be found in the setting of a monoclonal gammopathy with an inherent link between both entities. It seems that some of monoclonal gammopathies (MM, WM, MGUS) might be promoters of podocyte injury and therefore help in the development of MCD or FSGS. This happens more often when a κ monoclonal component is found. The idea that these entities share a pathophysiologic mechanism is supported by the temporal relationships that we described and by the fact that chemotherapy seems to have a positive effect on proteinuria.
Contributor Information
Andrés Ribas, Nephrology Department, Hospital del Mar, Barcelona, Spain.
Adrián Puche, Pathology Department, Hospital del Mar, Barcelona, Spain.
Javier Gimeno, Pathology Department, Hospital del Mar, Barcelona, Spain.
Laia Sans, Nephrology Department, Hospital del Mar, Barcelona, Spain.
Clara Barrios, Nephrology Department, Hospital del Mar, Barcelona, Spain.
Eva Márquez, Nephrology Department, Hospital del Mar, Barcelona, Spain.
Dolores Naranjo, Pathology Department, Hospital del Mar, Barcelona, Spain.
Belén Lloveras, Pathology Department, Hospital del Mar, Barcelona, Spain.
Joan Lop, Pathology Department, Hospital del Mar, Barcelona, Spain.
Natàlia Ramos, Nephrology Department, Hospital Vall d'Hebrón, Barcelona, Spain.
Maria José Soler, Nephrology Department, Hospital Vall d'Hebrón, Barcelona, Spain.
Alejandra Gabaldon, Pathology Department, Hospital Vall d'Hebrón, Barcelona, Spain.
Marta Crespo, Nephrology Department, Hospital del Mar, Barcelona, Spain.
Eva Rodríguez, Nephrology Department, Hospital del Mar, Barcelona, Spain.
CONFLICT OF INTEREST STATEMENT
None declared. All co-authors have seen and agree with the contents of the manuscript and there are no financial interests to report. We certify that the submission is original work and is not under review at any other publication. M.J.S. is the CKJ Editor-in-Chief Elect.
REFERENCES
- 1. International Myeloma Working Group . Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121: 749–757 [PubMed] [Google Scholar]
- 2. Boudhabhay I, Titah C, Talbot Aet al. Multiple myeloma with crystal-storing histiocytosis, crystalline podocytopathy, and light chain proximal tubulopathy, revealed by retinal abnormalities: a case report. Medicine (Baltimore) 2018; 97: e13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sethi S, Fervenza FC, Rajkumar SV. Spectrum of manifestations of monoclonal gammopathy-associated renal lesions. Curr Opin Nephrol Hypertens 2016; 25: 127–137 [DOI] [PubMed] [Google Scholar]
- 4. van den Berg JG, Weening JJ.. Role of the immune system in the pathogenesis of idiopathic nephrotic syndrome. Clin Sci (Lond) 2004; 107: 125–136 [DOI] [PubMed] [Google Scholar]
- 5. Chen YT, Hsu HJ, Hsu CKet al. Correlation between spot and 24h proteinuria: derivation and validation of equation to estimate daily proteinuria. PLoS One 2019; 14: e0214614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rajkumar SV, Dimopoulos MA, Palumbo Aet al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15: e538–e548 [DOI] [PubMed] [Google Scholar]
- 7. Vivarelli M, Massella L, Ruggiero Bet al. Minimal change disease. Clin J Am Soc Nephrol 2017; 12: 332–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rosenberg AZ, Kopp JB. Focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2017; 12: 502–517[published correction appears in Clin J Am Soc Nephrol 2018; 13:1889] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leung N, Bridoux F, Batuman Vet al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol 2019; 15: 45–59[published correction appears in Nat Rev Nephrol. 2019; 15:121] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bertelli R, Bonanni A, Caridi Get al. Molecular and cellular mechanisms for proteinuria in minimal change disease. Front Med (Lausanne). 2018; 5: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maas RJ, Deegens JK, Smeets Bet al. Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol 2016; 12: 768–776 [DOI] [PubMed] [Google Scholar]
- 12. Leung N, Drosou ME, Nasr SH. Dysproteinemias and glomerular disease. Clin J Am Soc Nephrol 2018; 13: 128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garg P. A review of podocyte biology. Am J Nephrol 2018; 47: 3–13 [DOI] [PubMed] [Google Scholar]
- 14. Brenner BM, Hostetter TH, Humes HD. Molecular basis of proteinuria of glomerular origin. N Engl J Med 1978; 298: 826–833 [DOI] [PubMed] [Google Scholar]
- 15. Bergón E, Miravalles E. Retrospective study of monoclonal gammopathies detected in the clinical laboratory of a Spanish healthcare district: 14-year series. Clin Chem Lab Med 2007; 45: 190–196 [DOI] [PubMed] [Google Scholar]
- 16. Kofman T, Zhang SY, Copie-Bergman Cet al. Minimal change nephrotic syndrome associated with non-Hodgkin lymphoid disorders: a retrospective study of 18 cases. Medicine (Baltimore) 2014; 93: 350–358[published correction appears in Medicine (Baltimore). 2014; 93: 414] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dingli D, Larson DR, Plevak MFet al. Focal and segmental glomerulosclerosis and plasma cell proliferative disorders. Am J Kidney Dis 2005; 46: 278–282 [DOI] [PubMed] [Google Scholar]
- 18. Salviani C, Guido G, Serriello Iet al. Renal involvement in Waldenström's macroglobulinemia: case report and review of literature. Ren Fail 2014; 36: 114–118 [DOI] [PubMed] [Google Scholar]
- 19. Batra A, Herz Allah S, Thajudeen B. Focal segmental glomerulosclerosis in Waldenström's macroglobulinemia. Case Rep Nephrol 2020; 2020: 8895705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grabe DW, Li B, Haqqie SS. A case of nephrotic syndrome with minimal-change disease and Waldenstrom's macroglobulinemia. J Clin Med Res 2013; 5: 481–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mwamunyi MJ, Zhu HY, Zhang Cet al. Pseudothrombus deposition accompanied with minimal change nephrotic syndrome and chronic kidney disease in a patient with Waldenström's macroglobulinemia: a case report. World J Clin Cases 2019; 7: 2393–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shah R, Shah N, Shah Aet al. Steroid-resistant nephrotic syndrome secondary to primary focal segmental glomerulosclerosis and smoldering multiple myeloma. Proc (Bayl Univ Med Cent) 2014; 27: 19–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oweis AO, Al Shelleh SA, Aldaoud Net al. Multiple myeloma in a patient with focal segmental glomerulosclerosis: a case report. Am J Case Rep 2018; 19: 946–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jain N, Tantoco MR, Thomas B, Laut J. 138: Monoclonal gammopathy of uncertain significance (MGUS) and focal segmental glomerulosclerosis (FSGS)–a case report. Natl Kidney Found 2010 Spring Clin Meet Abstr Glomerular Dis 2010; 55; PB66. [Google Scholar]
- 25. Shah S, Cavenagh J, Sheaf Met al. Remission of collapsing focal segmental glomerulosclerosis following chemotherapy for myeloma. Am J Kidney Dis 2004; 43: e10–e12 [DOI] [PubMed] [Google Scholar]
- 26. Charney DA, Wasser W. A 36-year-old man with a monoclonal gammopathy and nephrotic syndrome. Am J Kidney Dis 2003; 42: 1097–1101 [DOI] [PubMed] [Google Scholar]