

ABSTRACT
Introduction
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has caused the devastating pandemic named coronavirus disease 2019 (COVID-19). Unfortunately, the discovery of antiviral agents to combat COVID-19 is still an unmet need. Transmembrane serine protease 2 (TMPRSS2) is an important mediator in viral infection and thus, TMPRRS2 inhibitors may be attractive agents for COVID-19 treatment.
Areas covered
This review article discusses the role of TMPRSS2 in SARS-CoV-2 cell entry and summarizes the inhibitors of TMPRSS2 and their potential anti-SARS activity. Two known TMPRSS2 inhibitors, namely camostat and nafamostat, approved drugs for the treatment of pancreatitis, are under clinical trials as potential drugs against COVID-19.
Expert opinion
Due to the lack of the crystal structure of TMPRSS2, homology models have been developed to study the interactions of known inhibitors, including repurposed drugs, with the enzyme. However, novel TMPRSS2 inhibitors have been identified through high-throughput screening, and appropriate assays studying their in vitro activity have been set up. The discovery of TMPRSS2ʹs crystal structure will facilitate the rational design of novel inhibitors and in vivo studies and clinical trials will give a clear answer if TMPRSS2 inhibitors could be a new weapon against COVID-19.
KEYWORDS: Clinical trials, COVID-19, inhibitors, SARS-CoV-2, transmembrane serine protease 2 (TMPRSS2)
1. Introduction
In late 2019, a new highly infectious respiratory disease, named coronavirus disease 2019 (COVID-19), emerged in Wuhan, China [1,2]. The causative virus is a member of the Betacoronavirus genus within the Coronaviridae family and has been classified as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), due to its resemblance to SARS-CoV [3]. COVID-19 causes symptoms ranging from mild respiratory illness to pneumonia with acute respiratory distress syndrome that can be fatal [4]. As of 5 December 2021 nearly 265 million confirmed cases have been reported globally, with over 5.2 million confirmed deaths [5]. As the number of SARS-CoV-2 cases has risen critically, new variants of the virus have been identified and some of them have been associated with higher viral load and increased transmissibility [6]. For instance, the latest variant of concern, omicron, which was reported to the World Health Organization (WHO) in November, has a large number of mutations and is characterized by high contagiousness and an increased risk of reinfection [7].
Due to the emergence and severity of this pandemic, there has been a worldwide focus on developing vaccines and repurposing already approved drugs in order to attenuate the infection [8,9]. As of 20 October 2021 at least seven different vaccines have been distributed in numerous countries, with vulnerable populations being prioritized for vaccination [10]. At the same time, various key proteins that mediate SARS-CoV-2 infection and replication have become attractive targets for drug repurposing. Some of these targets are RNA-dependent RNA polymerase (RdRp), SARS-CoV-2 main protease (Mpro) and angiotensin-converting enzyme 2 (ACE2). A number of drugs that displayed promising preliminary results against the disease have been tested in patients with COVID-19, namely remdesivir targeting RdRp [11], hydroxychloroquine [12] and lopinavir [13]. However, recent results from an ongoing clinical trial revealed that these drugs had little or no effect on hospitalized patients with the disease, as indicated by overall mortality, initiation of ventilation, and duration of hospital stay [14]. In particular, Remdesivir exhibited no clinical benefit in hospitalized patients for COVID-19 [15].
The present review focuses on inhibitors of another attractive target against SARS-CoV-2, namely, transmembrane serine protease 2 (TMPRSS2). TMPRSS2 is an important mediator in viral infection and cell entry and it has been associated with influenza viruses and other coronaviruses [16,17]. The finding that SARS-CoV-2 cell entry depends on both ACE2 and TMPRSS2 [16] gave rise to a special attention on TMPRSS2 inhibitors as potential agents to treat COVID-19. Currently, the main drugs targeting TMPRSS2, which are tested against COVID-19, are camostat mesilate and nafamostat mesilate. These inhibitors are approved drugs for the treatment of pancreatitis in Japan and preliminary experiments performed in vitro, confirm their efficacy against SARS-CoV-2 [17]. In the meantime, novel inhibitors of TMPRSS2 have been designed and synthesized, specifically targeting TMPRSS2, employing homology models for modeling and docking studies. The aim of this review article is to summarize the discovery and properties of new compounds and already approved drugs as TMPRSS2 inhibitors and the clinical trials, which are ongoing for testing these drugs as a monotherapy or in combination with other drugs against COVID-19. In addition, computational studies, which have been executed to elucidate the mechanism of TMPRSS2 inhibition by the existing drugs, will be discussed. Due to the lack of TMPRSS2ʹs crystal structure, use of homology models based on other well-known serine protease structures may facilitate target-based drug discovery and design of potential agents to treat COVID-19.
2. TMPRSS2 protein and its role in the proteolytic activation of SARS-CoV-2
2.1. Type II Serine proteases
Trypsin-like serine proteases are proteolytic enzymes of the serine protease family that catalyze the cleavage of peptide bonds, showing specificity for basic amino acids. They have been found to be crucial in physiological processes, such as blood coagulation, immunity, and digestion. The study of serine proteases has led to the identification of a group of enzymes that are anchored to the membrane either by C-terminal (Type I) or N-terminal (Type II) transmembrane domains [18]. The TTSP or Type II subgroup of serine proteases consists of 17 members in humans. The hepsin/TMPRSS (transmembrane protease/serine) subfamily includes 7 members, namely, hepsin, TMPRSS2, TMPRSS3, TMPRSS4, TMPRSS5, MSPL (mosaic serine protease large-form), and enteropeptidase [19].
2.2. Structure and function of TMPRSS2
One important member of the TTSP subgroup is TMPRSS2, which was first discovered in 1997 by Paoloni-Giacobino et al. using exon-trapping [20]. The TMPRSS2 gene maps to human chromosome 21q22.3 (HC21) and is homologous to the human enteropeptidase gene [21], which also maps to HC21. The TMPRSS2 protein consists of 492 amino acids and it is mainly expressed in the epithelial cells of the prostate, breast, bile duct, kidney, colon, small intestine, pancreas, ovary, salivary gland, stomach, and lung [22]. The structure of this protein is characterized by a 70 amino acid N-terminal cytoplasmic domain, a 36 amino acid transmembrane domain, a class A LDL receptor (LDLRA) domain, a scavenger receptor cysteine-rich (SRCR) domain, and an activation domain linked to a serine protease domain via a disulfide bond (Figure 1(a)) [20].
Figure 1.

A. Domain organization of TMPRSS2. B. ACE-2 and TMPRSS2 mediated cell entry of SARS-CoV-2.
TMPRSS2 protein, similar to other TTSPs, is first synthesized as a zymogen, which is activated upon proteolytic cleavage. The in vitro-translated TMPRSS2 zymogen has a molecular mass of 54 kDa, whereas N-linked glycosylation leads to human TMPRSS2 proteins having a molecular mass between 60 and 70 kDa. In cells derived from prostate cancer tissue, a 32-kDa TMPRSS2 fragment has been detected, most likely the protease domain, which may indicate the partial activation of the protease [23]. Afar et al. have studied the proteolytic cleavage and secretion of a fraction (mainly the protease domain) of the TMPRSS2 protein. The LDLRA and the SRCR domains might remain attached to the membrane and potentially function as receptors, independently [24].
In 1999, Lin et al. showed that the mRNA expression of TMPRSS2 could be upregulated in prostate cancer LNCaP cells upon androgen-stimulation [25]. Moreover, it is known that the TMPRSS2 gene is located close to the ERG gene in HC21. There is evidence that TMPRSS2-ERG gene fusions disrupt the normal androgen receptor signaling, thus promoting prostate cancer invasion and progression, and could be found in nearly 50% of prostate cancer samples studied [26]. On the other hand, it has been established that TMPRSS2 is overexpressed in prostate hyperplasia, neoplasia, and metastases in a mouse model of prostate adenocarcinoma (TRAMP), regardless of TMPRSS2-ERG fusion. In particular, it has been proven that TMPRSS2 activity regulates cancer cell invasion and metastasis to other organs, such as the liver and lung. It is evident that TMPRSS2 is a key protease in prostate cancer; however, not many physiological functions for this protein have been discovered. In fact, a study using TMPRSS2 knockout mice showed that they were viable and fertile displaying no apparent abnormalities [27]. Another study suggested that TMPRSS2 could regulate epithelial sodium channels (ENaC), which are expressed in human airways [28].
2.3. Involvement in the proteolytic activation of viruses
Throughout the years, influenza A viruses have been the cause of numerous human epidemics. The infectivity of these viruses has been attributed to the cleavage activation of the influenza virus hemagglutinin (HA) by host cell proteases, which prime the viral protein for low-pH-induced membrane fusion. In 2006, a study showed that in TMPRSS2 transfected MDCK cells, which have been infected with influenza A viruses, multicycle replication of the viruses took place [29]. On the contrary, in cells that were transfected with enzymatically inactive TMPRSS2, multicycle replication of the viruses did not occur. These data led to the suggestion that TMPRSS2 could be connected to the proteolytic activation of influenza A viruses [28]. This suggestion was further supported by the fact that TMPRSS2 was found to be expressed in target cells in the human alveolar epithelium [30]. Finally, two studies confirmed the role of TMPRSS2 in the activation and spread of H1N1, H3N2, and H7N9 influenza A viruses in mice. Specifically, TMPRSS2 knockout mice were able to survive infection with influenza A viruses, which was fatal to wild-type mice [31,32].
TMPRSS2 protein has also been associated with coronaviruses, such as severe acute respiratory syndrome-related coronavirus (SARS-CoV) and Middle East respiratory syndrome-related coronavirus (MERS-CoV), which are highly pathogenic to humans. As a class I viral fusion protein, Coronavirus spike glycoprotein (S) bears a structural and mechanistic resemblance to influenza virus HA. This glucoprotein contains cell receptor-binding domains and virus-cell membrane fusion domains and it requires proteolytic priming in order to be activated [33]. Studies published in 2010 and 2011, indicated that TMPRSS2 protein could activate the S protein, inducing virus-cell membrane fusion at the cell surface in vitro, in Vero E6 and 293 T cells expressing TMPRSS2 [16,34,35]. Eventually, MERS-CoV spike protein was also found to be activated by TMPRSS2, as Vero-TMPRSS2 cells were 100-fold times more susceptible to the virus than parental cells [36]. Finally, animal models were used to confirm the role of TMPRSS2 in coronavirus infections in vivo. In a recent study, Th1-prone C57BL/6 mice were used for SARS-CoV infection, hDPP4-Tg mice were used for MERS-CoV infection and TMPRSS2-knockout mice for each experiment. When infected, the TMPRSS2-knockout mice demonstrated limited body weight loss and milder symptoms in comparison [37].
2.4. Involvement in the proteolytic activation of SARS-CoV-2
As the emerging coronavirus SARS-CoV-2 has caused a pandemic, elucidation of the viral entry mechanism is essential. In two individual studies, it was indicated that the novel virus, which is quite similar to SARS-CoV, enters the host cells through binding of its spike protein to the ACE2 receptor, followed by proteolytic activation by TMPRSS2. SARS-CoV and SARS-CoV-2 spike proteins are almost 76% identical [17,38].
The SARS-CoV-2 spike protein contains two functional domains, S1 and S2 (Figure 1(b)), analogous to other coronaviruses. The S1 domain is responsible for receptor binding and contains an N-terminal domain and a C-terminal domain to serve that purpose. The S2 domain is involved in membrane fusion and contains a transmembrane domain. In most coronaviruses, proteolytic cleavage takes place at the S1/S2 boundary or within the S2 domain. Following cleavage, the S1 and S2 domains remain associated non-covalently [39]. In a recent study, it was found that this proteolytic process takes place in the plasma membrane, where the virus can then easily penetrate the cells in a pH-independent manner. In the absence of TMPRSS2, the virus can be endocytosed and then slowly get activated in a pH-dependent manner via cathepsin L, finally entering the cytosol [40].
3. TMPRSS2 inhibitors – Discovery of novel agents able to inhibit TMPRSS2
In the midst of the pandemic, scientists have focused on finding novel small-molecules able to interact with either SARS-CoV-2 proteins or the host cell proteins. In the case of TMPRSS2, new inhibitors have emerged through virtual screening and molecular docking. Hu et al. reported two new inhibitors after virtual screening and docking of nearly 200,000 compounds and subsequent testing in an enzyme assay [41]. The first, NCGC00378763 (Figure 2), also known as otamixaban, exhibited an IC50 of 0.62 µM against TMPRSS2 and efficient activity in a SARS-Cov-2 pseudotyped particle viral entry assay (IC50 = 15.84 μΜ), while NCGC00386945 (Figure 2) had an IC50 of 1.24 µM, as well as 50% activity in the viral entry assay (IC50 = 0.35 μM) [41]. Otamixaban has also inhibited SARS-Cov-2 infection of precision cut lung slices with a potency comparable to camostat mesilate, a known TMPRSS2 inhibitor. Hempel et al. suggest that though otamixaban exhibited lower potency compared to camostat and nafamostat mesilate in SARS-CoV-2 viral entry in Calu-3 cells, supplementation with camostat or nafamostat in subnanomolar quantities, can enhance its potency drastically in a synergistic manner [42].
Figure 2.

Novel agents that inhibit TMPRSS2 and exhibit anti-SARS-CoV-2 activity and structures of nafamostat and camostat with its metabolites.
Recently, Shapira et al. reported the design and synthesis of ketobenzothiazole-based small-molecule peptidomimetics as TMPRSS2 inhibitors. The most potent compound N-0385 (Figure 2) inhibited TMPRSS2 with an IC50 of 1.9 nM and SARS-CoV-2 infection in Calu-3 cells, with an ED50 of 2.8 nM. Furthermore, a complete inhibition of infection was observed at a concentration of 100 nM in colonoids derived from human donors [43].
Another drug that has shown potential against SARS-CoV-2 is an eight-branched peptide, 8P9R (sequence: NGAICWGPCPTAFRQIGNCGRFRVRCCRIR), with an IC50 of 0.3 μg/mL in Vero-E6 cells. This peptide can cross-link SARS-CoV-2, forming viral clusters that can no longer infect cells through the TMPRSS2-mediated pathway and can also inhibit endosomal acidification to block the endocytic pathway of viral infection. In vivo, 8P9R inhibited SARS-CoV-2 replication in mouse and hamster lungs at a single dose of 0.5 mg/kg the first day and a double dose the second day [44].
Finally, α1-antitrypsin (α1ΑΤ) as indicated by Wettstein et al. inhibited SARS-CoV-2 spike pseudoparticle entry (IC50 ~ 38.5 μΜ) and suppressed viral infection and replication of two SARS-CoV-2 isolates in TMPRSS2-Vero-E6 cells with IC50s of 17.3 and 21.2 μM. Additionally, α1ΑΤ inhibited the replication of SARS-CoV-2 in primary human airway epithelial cells, and it was established that the underlying mechanism of action was the inhibition of TMPRSS2 activity, at first, through computational studies and subsequently, through an enzymatic assay where α1ΑΤ inhibited recombinant TMPRSS2 activity in a dose-dependent manner at concentrations of 5–50 μM [45].
4. Repurposing of TMPRSS2 inhibitors – Camostat and Nafamostat mesilate
TMPRSS2 has emerged as an attractive target for the potential treatment of COVID-19. Therefore, two known TMPRSS2 inhibitors, namely camostat mesilate (4) and nafamostat mesilate (5) (Figure 2), have attracted special interest. These compounds and their derivatives also act as serine protease inhibitors of trypsin, prostasin, matriptase, plasmin, kallikrein, thrombin, and C1 esterases, enzymes connected to the maintenance of normal homeostasis, in particular, digestion, blood circulation, and coagulation [46].
4.1. Camostat mesilate
Camostat mesilate (4) was first synthesized by Ono Pharmaceutical 45 years ago [47,48]. It is currently an approved drug in Japan for the treatment of chronic pancreatitis and postoperative reflux esophagitis, under the trade name FOIPAN® [49]. Supporting this application, it has been demonstrated that camostat mesilate administration prevented the progression of pancreatic fibrosis in rats [50]. Furthermore, when given to hypertensive rats, camostat mesilate causes reduction of blood pressure, and overall improvement of kidney function, potentially by inhibiting prostasin, a protein implicated in epithelial sodium channel regulation [51]. When applied to airway mucous membranes, camostat inhibits the airway epithelial sodium channel (ENaC) function in Guinea pig trachea and enhances the mucociliary clearance in sheep bronchi [52]. In addition, it reduces sodium transport in the airway of humans with cystic fibrosis [53]. Intriguingly, an association between the use of camostat mesilate and reduction of hematuria and/or proteinuria in children has been observed [54]. Finally, in a study using genetically obese and diabetic rats, administration of camostat mesilate was able to reverse hyperglycemia and obesity and improve insulin resistance [55].
As an inhibitor of TMPRSS2, camostat mesilate has exhibited promising antiviral activity. Starting with influenza viruses, camostat mesilate administration ameliorated influenza A/Taiwan/1/86 virus pathology in mice and was effective against both influenza type A and B virus infections in vitro, in MDCK cells [56] and human tracheal surface epithelial cells [57]. Concerning coronaviruses, camostat mesilate mediated inhibition of TMPRSS2 led to a 10-fold reduction in SARS-CoV virus titer in vitro [36] and increased the survival rate of infected mice to 60% [58]. Additionally, it has been shown that camostat can partially block infection by SARS-CoV and human coronavirus NL63 in HeLa cells expressing TMPRSS2 and can prevent viral entry and growth in human Calu-3 airway epithelial cells, when paired with a cathepsin inhibitor [59]. Its action against MERS-CoV has been also demonstrated. In fact, when camostat mesilate was used at a concentration of 10 μM, it impaired viral entry in Vero-TMPRSS2 cells by 15-fold, while the viral RNA present in culture supernatant of Calu3 cells was reduced 270-fold after addition of 100 mM camostat on the third day post MERS-CoV infection [38]. These results led to the suggestion of camostat mesilate as a possible treatment for SARS-CoV-2. Hoffman et al. reported that camostat mesilate partially blocked SARS-2-S-driven entry into Caco-2 and Vero-TMPRSS2 cells [17], and thus, clinical trials with camostat mesilate for the treatment of COVID-19 have been initiated. To specify, 22 clinical trials have been registered to test camostat mesylate against COVID-19 (Table 1). The majority of the trials are randomized, double, triple, or quadruple, blinded studies with outcome measures including viral load, disease progression, and survival rate. Thirteen trials are testing camostat mesilate as a single therapy on hospitalized patients or outpatients and nine trials are using camostat mesilate in combination with other drugs. These clinical trials are underway in various countries and are either recruiting or not yet recruiting patients with no results announced at this stage. It is worth mentioning that in a small retrospective observational case series with eleven patients with COVID-19, treatment with camostat mesilate resulted in a decreased severity of the disease. In comparison to the patients that were treated with hydroxychloroquine, patients in the camostat group exhibited a decline in inflammatory markers and improvement of oxygenation, as well as a decreased Sepsis-related Organ Failure Assessment score [60]. In fact, a recent study has shown that the inhibitory effect of hydroxychloroquine on SARS-CoV-2 entry is attenuated by TMPRSS2, which explains its inefficacy in clinical trials. It is also stated that SARS-CoV-2 is more dependent on TMPRSS2 than SARS-CoV-1 [61].
Table 1.
Clinical trials testing the efficacy of camostat mesilate for the treatment of COVID-19
| NCT number (clinicaltrials.gov) |
Title | Status | Interventions | Phase | Location |
|---|---|---|---|---|---|
| NCT04353284 | Camostat Mesylate in COVID-19 Outpatients | Recruiting | Drug: Camostat mesilate Placebo controlled |
Phase 2 | United States |
| NCT04583592 | Camostat Efficacy vs. Placebo for Outpatient Treatment of COVID-19 (CAMELOT) | Active, not recruiting | Drug: Camostat mesilate Placebo controlled |
Phase 2 | United States |
| NCT04524663 | Oral Camostat Compared With Standard Supportive Care in Mild-Moderate COVID-19 Patients | Recruiting | Drug: Camostat mesilate Placebo controlled |
Phase 2 | United States |
| NCT04608266 | CAMOVID: Evaluation of Efficacy and Safety of Camostat Mesylate for the Treatment of SARS-CoV-2 Infection – COVID-19 in Ambulatory Adult Patients | Recruiting | Drug: Camostat mesilate Placebo controlled |
Phase 3 | France |
| NCT04625114 | The Potential of Oral Camostat in Early COVID-19 Disease in an Ambulatory Setting to Reduce Viral Load and Disease Burden | Recruiting | Drug: Camostat mesilate Placebo controlled |
Phase 2 | Belgium |
| NCT04455815 | A Trial Looking at the Use of Camostat to Reduce Progression of Symptoms of Coronavirus (COVID-19) in People Who Have Tested Positive But Are Able to Stay at Home | Recruiting | Drug: Camostat mesilate | Phase 2 Phase 3 | UK |
| NCT04662073 | COVID-19 Outpatient Pragmatic Platform Study (COPPS) – Camostat Sub-Protocol | Not yet recruiting | Drug: Camostat mesilate Placebo controlled |
Phase 2 | United States |
| NCT04652765 | Camostat With Bicalutamide for COVID-19 | Recruiting | Drug: Camostat Mesilate Drug: Bicalutamide | Phase 1 | United States |
| NCT04730206 | The DAWN Camostat Trial for Ambulatory COVID-19 Patients | Not yet recruiting | Drug: Camostat mesilate Placebo controlled |
Phase 3 | Belgium |
| NCT04750759 | Safety and Preliminary Efficacy of the Combination of Niclosamide and Camostat | Recruiting | Drug: Niclosamide + Camostat mesilate Placebo controlled |
Phase 2 | Germany |
| NCT04657497 | A Study of FOY-305 in Patients With SARS-Cov-2 Infection (COVID-19) | Recruiting | Drug: FOY-305 Placebo controlled |
Phase 3 | Japan |
| NCT04355052 | Open Label Study to Compare Efficacy, Safety and Tolerability of Hydroxychloroquine Combined With Azithromycin Compared to Hydroxychloroquine Combined With Camostat Mesylate and to ‘no Treatment’ in SARS CoV 2 Virus | Recruiting | Drug: hydroxychloroquine with camostat mesilate Drug: Hydroxychloroquine with Azithromycin Placebo controlled |
Phase 3 | Israel |
| NCT04530617 | Camostat and Artemisia Annua vs Placebo in COVID-19 Outpatients | Recruiting | Drug: Camostat Mesilate Drug: Artemisia Annua Leaf Placebo controlled |
Phase 2 | Mexico |
| NCT04681430 | Reconvalescent Plasma/Camostat Mesylate Early in SARS-CoV-2 Q-PCR (COVID-19) Positive High-risk Individuals | Recruiting | Biological: Convalescent plasma Drug: Camostat Mesilate Placebo controlled |
Phase 2 | Germany |
| NCT04662086 | COVID-19 Outpatient Pragmatic Platform Study (COPPS) – Master Protocol | Not yet recruiting | Drug: Acebilustat Drug: Camostat Mesilate Placebo controlled |
Phase 2 | United States |
| NCT04321096 | The Impact of Camostat Mesilate on COVID-19 Infection | Active, not recruiting | Drug: Camostat Mesilate Placebo controlled |
Phase 1 Phase 2 | Denmark |
| NCT04374019 | Novel Agents for Treatment of High-risk COVID-19 Positive Patients | Recruiting | Drug: Ivermectin Drug: Camostat Mesilate Drug: Artesunate |
Phase 2 | United States |
| NCT04470544 | Camostat Mesilate Treating Patients With Hospitalized Patients With COVID-19 | Recruiting | Drug: Camostat Mesilate Placebo controlled |
Phase 2 | United States |
| NCT04644705 | Safety and Pharmacokinetics of a Novel Niclosamide Solution in Combination With Camostat | Recruiting | Drug: Niclosamide with Camostat Mesilate Drug: Placebo controlled |
Phase 1 | Germany |
| NCT04721535 | A Study of DWJ1248 in Prevention of COVID-19 Infection After the Exposure of SARS-COV-2 | Not yet recruiting | Drug: DWJ1248 Placebo controlled |
Phase 3 | South Korea |
| NCT04521296 | Efficacy and Safety of DWJ1248 in Patients With Mild to Moderate COVID-19 Compared to the Placebo | Not yet recruiting | Drug: DWJ1248 Placebo controlled |
Phase 2 | South Korea |
| NCT04713176 | Efficacy and Safety of DWJ1248 With Remdesivir in Severe COVID-19 Patients | Recruiting | Drug: DWJ1248 with Remdesivir Placebo controlled |
Phase 3 | South Korea |
Camostat mesilate acts as a prodrug with a short plasma half-life. The acetamide side-chain ester group is easily hydrolyzed to produce GBPA (4-(4-guanidino-benzoyloxy)phenylacetic acid, Figure 2) in vivo, in the gut or after systemic administration. GPBA, also known as FOY-251, potently inhibits TMPRSS2 with an estimated half-life of 1 hour after intravenous infusion of camostat mesilate. This compound is further metabolized to GBA (Figure 2), which is inactive against TMPRSS2 [62]. Hoffman et al. demonstrated that GBPA was able to inhibit SARS-CoV-2 infection of Calu-3 cells in vitro and human precision-cut-lung slices (PCLS) ex vivo, with an efficiency that was similar to that of camostat mesilate [63]. On the other hand, its ability to inhibit the enzymatic activity of recombinant TMPRSS2 was reduced compared to camostat mesilate.
4.2. Nafamostat mesilate
Another serine protease inhibitor that could potentially be repurposed against SARS-CoV-2 is nafamostat mesilate (5), also known as FUT-175. It was synthesized by Fujii et al. in 1981 [64]. This drug is used for the treatment of pancreatitis, intravascular coagulation, and systemic inflammatory response syndrome by suppressing enzymes like thrombin, plasmin, kallikrein, trypsin, and Cl esterase in the complement system [65], as well as factors VIIa, Xa, and XIIa in the coagulation cascade [66]. In addition, it has been shown that nafamostat mesilate can inhibit proliferation, migration, and invasion of colorectal cancer cells, by disrupting NF-κB signaling in vitro [67]. It is also effective against other types of cancer, namely pancreatic cancer [68], lung cancer [69], gallbladder cancer [70], gastric cancer [71] and breast cancer [72]. The fat mass and obesity-associated protein (FTO), a demethylase that plays an important role in physiological processes, is associated with various disease processes, such as type II diabetes, Alzheimer’s disease, cardiovascular diseases, acute myeloid leukemia, and lung cancer and seems to be inhibited by nafamostat [73]. Furthermore, it has been proven that nafamostat and its derivative sepimostat exhibit potent neuroprotective effects in vitro and in vivo. They act as neuroprotective agents antagonizing the NMDA receptors through binding to the NR2B subunit [74].
Nafamostat has been recognized for its antimicrobial and antiviral properties. Specifically, nafamostat mesilate exhibited in vitro inhibition of Chlamydia proliferation and proved to be effective against Chlamydia-induced arthritis [75]. In the case of viruses, nafamostat mesilate has been effective against influenza virus type A and B in MDCK cells, with EC50s of 0.44 and 1.5 μg/mL, respectively. Also, the inhibitor caused a reduction of viral hemagglutinin titer in influenza viruses A and B and parainfluenza virus type 1 in ovo [76]. Additionally, nafamostat mesilate potently inhibited the membrane fusion of MERS-CoV spike protein in 293 FT cells with an IC50 value of 0.1 μM and Calu-3 cells with an IC50 value of 1 nM [77]. Moreover, a study testing the effect of nafamostat mesilate in the activation of different coronaviruses, namely SARS-CoV, MERS-CoV, and SARS-CoV-2, indicated that nafamostat inhibited the infection of lung cells (Calu-3), with EC50 values of 1.4 nM (SARS-CoV), 5.9 nM (MERS-CoV) and 5 nM (SARS-CoV-2), respectively, compared to camostat mesilate with 198 nM (SARS-CoV), 444 nM (MERS-CoV), and 87 nM (SARS-CoV-2), respectively [78]. Furthermore, a different study testing the inhibition of the novel coronavirus in infected Vero E6 cells showed that nafamostat inhibited the infection (EC50 22.50 μM) [79]. Finally, nafamostat and camostat were able to inhibit SARS-CoV-2 pseudovirus entry in human induced pluripotent stem cell (iPSC)-derived lung organoids (LORGs) [80]. These findings were further supported by Inoue and coworkers, showing that nafamostat mesilate blocked the infection of Calu-3 cells with an effective concentration of 10 nM, as well as VeroE6/TMPRSS2 with an effective concentration of 30 μΜ, highlighting a cell-type-dependence [81]. Importantly, both camostat and nafamostat have also been tested against three different variants of SARS-CoV-2 in Calu-3 cells and have exhibited the same effectiveness against all variants, ranging from 0.102 to 0.170 μΜ for camostat and 0.016 to 0.024 μΜ for nafamostat, respectively [82]. In vivo, it has been reported that nafamostat potently inhibited SARS-CoV-2 infection in a mouse model of COVID-19 when administered intranasally at a dose of 3 mg/kg. In detail, K18-hACE2 mice that were pretreated with nafamostat for 2 h and then infected with SARS-CoV-2 exhibited reduced weight loss, mortality, and viral loads [83]. Currently, there are seven different clinical trials, recruiting or not yet recruiting patients that will test the efficacy of nafamostat mesilate for COVID-19 and one completed trial with no results announced to date. Most of these trials will be randomized, open-label, phase II or III trials, while one of them will be double blind and placebo controlled. These trials are running in various countries and are summarized in Table 2.
Table 2.
Clinical trials testing the efficacy of nafamostat mesilate for the treatment of COVID-19
| NCT number (clinicaltrials.gov) |
Title | Status | Interventions | Phase | Location |
|---|---|---|---|---|---|
| NCT04390594 | Efficacy and Safety Evaluation of Treatment Regimens in Adult COVID-19 Patients in Senegal | Recruiting | Drug: Nafamostat Mesilate | Phase 3 | Senegal |
| NCT04418128 | Clinical Efficacy of Nafamostat Mesylate for COVID-19 Pneumonia | Not yet recruiting | Drug: Nafamostat Mesilate | Phase 2 Phase 3 | South Korea |
| NCT04352400 | Efficacy of Nafamostat in Covid-19 Patients (RACONA Study) | Not yet recruiting | Drug: Nafamostat Mesilate Placebo controlled | Phase 2 Phase 3 | Italy |
| NCT04628143 | A Study Evaluating the Efficacy and Safety of CKD-314 in Hospitalized Adult Patients Diagnosed With COVID-19 Pneumonia | Not yet recruiting | Drug: Nafamostat Mesilate | Phase 2 | South Korea |
| NCT04623021 | A Study Evaluating the Efficacy and Safety of CKD-314 (Nafabelltan) in Hospitalized Adult Patients Diagnosed With COVID-19 Pneumonia | Completed | Drug: Nafamostat Mesilate | Phase 2 | Russia |
| NCT04473053 | Rapid Experimental Medicine for COVID-19 | Recruiting | Drug: Nafamostat Mesilate Drug: TD139 |
Phase 2 Phase 3 | UK |
| NCT04483960 | Australasian COVID-19 Trial (ASCOT) ADAptive Platform Trial | Recruiting | Drug: Nafamostat Mesilate Biological: Convalescent plasma Drug: Enoxaparin Drug: Dalteparin Drug: Tinzaparin Drug: Aspirin |
Phase 3 | Australia |
Nafamostat mesilate is administered by IV infusion and has a plasma half-life of approximately 23 minutes. This compound is mainly metabolized to 6-amidino-2-naphthol and 4-guanidinobenzoic acid, which do not inhibit the trypsin-like serine protease, prostasin, contrary to nafamostat [84]. The activity of these metabolites against TMPRSS2 has not been investigated.
5. Other small-molecule TMPRSS2 inhibitors
Bromhexine hydrochloride (6, Figure 3) is an orally bioavailable drug, which is used as a mucolytic cough suppressant, with few to no adverse effects [85]. This drug is further metabolized to ambroxol (7, Figure 3) by removal of a methyl group and hydroxylation on the cyclohexyl ring. Ambroxol is also a mucoactive agent, which is in use in cough syrup [86]. Bromhexine hydrochloride selectively inhibits TMPRSS2 activity with an IC50 value of 0.75 μM, while it is less active against hepsin, matriptase, trypsin and thrombin [87]. Furthermore, the efficacy of bromhexine against the cytopathic effect (CPE) of SARS-CoV-2 infection has been demonstrated by the National Institutes of Health (NIH), with an AC50 of 8.9 μΜ [88].
Figure 3.
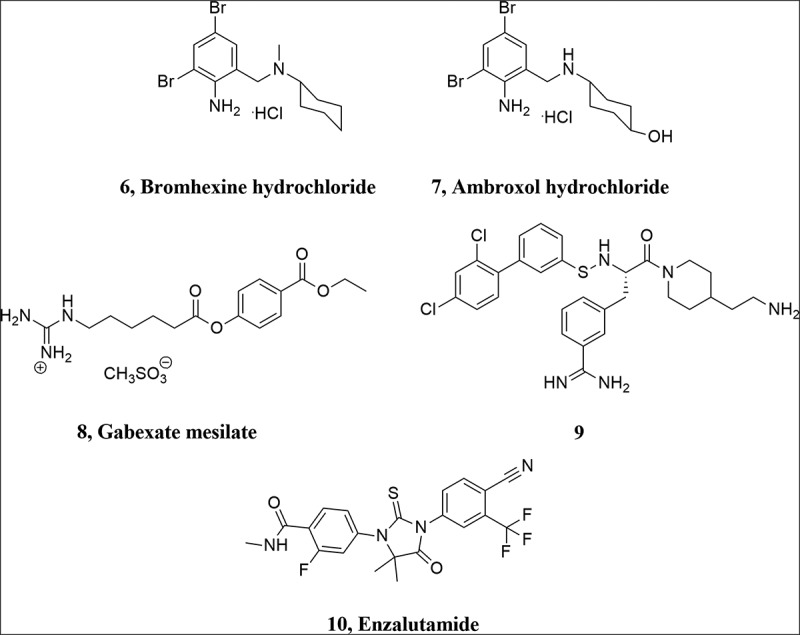
Structure of bromhexine hydrochloride, ambroxol hydrochloride, gabexate mesylate, enzalutamide and compound 9.
Currently, bromhexine hydrochloride is one of the already approved drugs that are being tested in clinical trials for COVID-19. There are six clinical trials testing this drug (Table 3), two of them are completed and three are recruiting patients. Preliminary results in one of these trials, conducted in China, suggest that bromhexine hydrochloride might be helpful in alleviating hepatic or lung injury caused by SARS-CoV-2 [89]. In another trial conducted in Iran in a group of 78 patients, it was reported that early treatment with bromhexine at doses of 8 mg three times a day alleviated respiratory symptoms and led to a reduced rate of intensive care unit admissions, mechanical ventilation, and mortality. While these results are encouraging, subsequent large-scale clinical trials for validation should ensue [90].
Table 3.
Clinical trials testing the efficacy of bromhexine hydrochloride for the treatment of COVID-19
| NCT number (clinicaltrials.gov) |
Title | Status | Interventions | Phase | Location |
|---|---|---|---|---|---|
| NCT04355026 | Use of Bromhexine and Hydroxychloroquine for Treatment of COVID-19 Pneumonia | Recruiting | Drug: Bromhexine Oral Tablet and/or hydroxychloroquine tablet | Phase 4 | Slovenia |
| NCT04273763 | Evaluating the Efficacy and Safety of Bromhexine Hydrochloride Tablets Combined With Standard Treatment/ Standard Treatment in Patients With Suspected and Mild Novel Coronavirus Pneumonia (COVID-19) | Active, not recruiting | Drug: Bromhexine Hydrochloride Drug: Arbidol Hydrochloride Drug: Recombinant Human Interferon α2b |
- | China |
| NCT04405999 | Prevention of Infection and Incidence of COVID-19 in Medical Personnel Assisting Patients With New Coronavirus Disease | Completed | Drug: Bromhexine Hydrochloride | Phase 4 | Russia |
| NCT04424134 | BromhexIne And Spironolactone For CoronаVirUs Infection Requiring HospiTalization | Recruiting | Drug: Bromhexine and Spironolactone Drug: Base therapy |
Phase 3 | Russia |
| NCT04340349 | Low-dose Hydroxychloroquine and Bromhexine: a Novel Regimen for COVID-19 Prophylaxis in Healthcare Professionals | Enrolling by invitation | Drug: Hydroxychloroquine Sulfate Drug: Bromhexine Placebo controlled |
Early Phase 1 | Mexico |
| IRCT20200317046797N4 | Effect of Bromhexine Hydrochloride on clinical improvement and outcome of COVID-19-induced pneumonia | Completed | Drug: Bromhexine Drug: Base therapy |
Phase 3 | Iran |
Gabexate mesilate (8, Figure 3) is a TMPRSS2 inhibitor, which has been approved for use in Italy and Japan for the treatment of pancreatitis and disseminated intravascular coagulation [91]. It has been established that gabexate mesilate can inhibit influenza virus A and B in vitro in MDCK and human tracheal epithelial cells [57,76], and in vivo in a murine model [92]. This inhibitor is similar to camostat and nafamostat in structure, but less effective in most cases. Recently, in an enzymatic TMPRSS2 assay, Shrimp et al. reported that gabexate mesilate inhibited TMPRSS2 with an IC50 value of 130 nM and was the least potent compound compared to nafamostat mesilate (IC50 0.27 nM), camostat mesilate (IC50 6.2 nM) and FOY-251 (IC50 33.3 nM), while bromhexine hydrochloride showed no inhibition of TMPRSS2 [93].
In addition, in an effort to find new potent inhibitors of TMPRSS2 for the treatment of influenza virus, a series of 3-amidinophenylalanyl-derived inhibitors was synthesized and the new compounds were tested against TMPRSS2. The most potent compound 9 inhibited TMPRSS2 with a Ki value of 0.9 nM and efficiently blocked influenza virus propagation in human airway epithelial cells [94].
On a final note, TMPRSS2 expression and enzymatic activity have been found to be arguably higher in male mice and male human oral tissue, which suggests that androgen regulation of TMPRSS2 may possibly affect protease activity [95]. Also, not only TMPRSS2 and ACE-2 seem to be co-expressed in oral tissues, their expression levels were particularly higher in the elderly group [96]. In a different study, treatment with enzalutamide (10), a nonsteroidal antiadrogen drug that has been used against prostate cancer, reportedly reduced TMPRSS2 levels in human lung cells and in mouse lung, as well as SARS-CoV-2 entry and infection [97]. These findings seem to be contradicted in other recent studies, where no evidence for increased TMPRSS2 expression was found in male human or mouse lungs. Moreover, treatment with enzalutamide did not decrease pulmonary TMPRSS2 in male mice [98]. In another study, enzalutamide was able to inhibit SARS-CoV-2 infection in human prostate cells, but not in human lung cells and organoids [99].
6. Molecular modeling studies
The crystal structure of TMPRSS2 has not been elucidated so far (October 2021) and thus homology modeling has been used for the construction of its 3D structure. Rahman et al. [100] used SWISS-MODEL to create the 3D model of TMPRSS2 and TMPRSS1 was used as the template (PDB ID: 5CE1). Evaluation of the structure was accomplished, and MOE was used to identify the active site residues (Asn146, Arg147, Cys148, Val149, Arg150, Leu151, Asp187, Met188, Tyr190, Ile221, Tyr222, Lys223, Asn368, Pro369, Gly370, Met371, Lys449, Asn450, Ile452, and Trp454). The authors docked 2,140 compounds in TMPRSS2, which resulted in 85 compounds with a considerable better docking score than camostat mesylate, employed as a reference compound. Docking pose of camostat revealed its interaction with Asn146, Cys148, Asn450, Asp187 and the formation of seven H-bonds. Some natural compounds, along with geniposide, demonstrated some promising docking results, which need further investigation.
To identify a protein template for building the homology model, Elmezayen et al. [101] used the BLAST tool to retrieve proteins with high similarity in sequence. According to their calculations, human transmembrane protease serine (DESC1) demonstrated 41% similarity and was used as the template to build the homology model of TMPRSS2 in MODELER. Minimization and validation of the model was carried out using online tools. Authors conducted virtual screening with AutoDock vina and among the top hits were the compounds Rubitecan, Loprazolam, ZINC000000702323 and ZINC000012481889 (ZINC15). The interactions formed by these molecules with the active site include residues His296, Lys300, Lys 342, Lys340, Lys390, Ser339, Tyr337, Asp435, Ser436, Gln438, Gly439, Ser460, Gly462, Ser441. Although Elmezayen et al. do not mention the key residues and the method they used for defining the active site, it seems that they differ from the active site that Rahman et al. established and therefore further investigation is required.
Kishk et al. identified proteins with known X-ray structure and high homology for TMPRSS2 using SWISS-MODEL and NCBI blastp [102]. These proteins included transmembrane protease serine 1 and 13 and a group of human plasma kallikrein. Homology models were built with SWISS-MODEL, evaluated, and subjected to molecular dynamics simulations to measure their energetic stability. Docking of nafamostat in the TMPRSS2 active site with MOE program revealed the position of the phenyl guanidine group in the S1 pocket and its hydrogen bond interactions with residues Asp435, Ser436, Ser463 and Gly462. The hydrophobic pocket, which consists of Thr459, Ser460, Trp461 and Val473, accommodates the phenyl ring. The active site was determined by site finder and positioned to contain the catalytic Ser441 and S1 pocket amino acids, including Glu389, Tyr416, Asp435, Ser436, Cys437, Gln438, Thr459, Ser460, Trp461, Gly462, Ser463, Gly464, Cys465, Ala466, Arg470, Pro471, Gly472 and Val473.
Their findings document the inhibitory activity of nafamostat, in which the carbonyl oxygen of the ester moiety interacts with the catalytic Ser441 via a water molecule. It is the guanidinobenzoate moiety of nafamostat, which mimics the natural substrate resulting in inhibitory activity. In addition, according to their docking results, camostat, and gabexate follow a similar binding mode, while sivelestat did not demonstrate a strong binding, as it was moved away from the binding site during their molecular dynamics simulations.
Baby et al. used the human plasma kallikrein (PDB ID: 5TJX), identified with BLAST tool, as the template to build the homology model of TMPRSS2 protein. Schrödinger Prime Module was employed for the construction of the model, which further modified with Refine Loops software and evaluated accordingly [103]. Schrödinger’s High Throughput Virtual Screening was employed to dock ~2800 FDA-acquiesced molecules in TMPRSS2 and ACE2, for which the receptor grid was created based on the co-crystalized ligand. Their calculations resulted in a number of drugs that demonstrated a promising binding mode, including valrubicin, lopinavir, fleroxacin, alvimopan, arbekacin, and dequalinium. For these compounds, molecular dynamics in Maestro Desmond was accomplished and suggested a superiority of valrubicin and lopinavir in their formation of a stable complex with the proteins. The main interactions formed in these complexes were hydrophobic and hydrophilic with residues such as Trp461, Trp461, Lys300.
Pooja et al. used the PSI-BLAST tool to search for high homology proteins in RCSB PDB. SWISS-MODEL server was used for building the model, which was further evaluated with a number of tools [104]. HADDOCK, CLUSPRO, and dog site server used to determine the binding site in order to dock seven marketed drugs and eighteen natural compounds with Autodock VINA in TMPRSS2. The S1 domain was identified to consist of the following amino acids: His296, Ser441, Asp345, Gly496, Gln438, Gln498, Gln506, Tyr505 and His296, Ser441, Asp345, Asn437 Gly404, Asp405, Gly496 and Gln498. For the evaluation of the docking results, camostat was used as the reference compound. Based on their docking results, camostat forms hydrogen bonds with His296, Cys281, Glu389, Lys390 and Gln438, while the formation of van der Waals interactions with Trp461, Gly462, Ser460, Gly385, His279, Thr393, Lys392 and Gly391 help to stabilize the complex. The authors also mention the carbon–hydrogen interactions with the catalytic Ser441 and Gly439, Cys297 and Leu302. Meloxicam, nafamostat, ganodermanontriol, columbin, myricetin, proanthocyanidin A2, jatrorrhizine and baicalein were the compounds with the highest docking scores. For nafamostat, the authors described its van der Waals interactions with the catalytic Ser441 and His296 and the formation of three hydrogen bonds with Ser436, Asp435 and Gly461. Interestingly, based on their docking results, meloxicam forms three hydrogen bonds with Gly462, Ser441, and Ser460, while several van der Waals interactions stabilize the complex.
A slightly different approach for identifying a template protein was described by Huggins [105]. He aligned the sequence of the human members of the TMPRSS2 family and found that TMPRSS15 was suitable as a template to build TMPRSS2 protein. After applying several modifications on TMPRSS15 sequence to build TMPRSS2, structural modifications of the protein followed in Schrödinger’s Prime and then energy minimization and molecular dynamics using the Desmond package applied in the system. Then, structural data for 250 compounds, which are targeting S1A proteases and they are approved, investigational, or experimental drugs, were aligned with the homology model and Embrace minimization with GBSA solvation was used to produce MMGBSA energy scores for the complexes. This procedure led to the identification of several drugs (Drugbank ID: DB04442, DB03082, DB08697, DB07247, DB03782, DB02398, DB03865, DB01725), which could effectively bind to TMPRSS2. In addition, the main common characteristic of these structures is the positively charged warhead they contain, which is similar to nafamostat, camostat, and gabexate.
Escalante et al. used the plasma kallikrein A (PDB: 2ANY) as the template for building the homology model in Prime [106]. They studied the interactions of the residues close to the catalytic center and they documented a triad of His296, Ser441 and Asp345 residues, which forms a network of hydrogen bonds, contributing to the activation of the serine for a nucleophilic attack to the substrate. As soon as His296 is positively charged, Asp45 can stabilize it via the formation of a hydrogen bond. The Glide algorithm and then the cdock algorithm were used to simulate the nucleophilic attack of Ser441 to the ligand, and based on the results, it is more likely that Ser441 attacks the carbonyl group from below and it is not a top side attack. The authors used quantum mechanics/molecular mechanics (QM/MM) molecular dynamics simulations to study the apo structure as well as complexes of protein-camostat and protein-GBPA. QM was used to model the residues of the catalytic triad in the apo structure and included all atoms of the ligand in the complex protein-camostat. A strength of the Ser441 to His296 H-bonding interaction was measured upon insertion of the ligand. Hydrogen bonds between the ligand, especially the guanidino donor, and Asp435, Ser436, Ser463 and Gly464 in combination with polar interactions contribute to the stabilization of the complex. In the GBPA complex, the benzoyl guanidino moiety behaves similar to camostat, while a number of π-π interactions formed and could guide the design of new inhibitors toward an enhancement of their binding affinity against TMPRSS2.
Two additional studies on TMPRSS2 homology modeling are pre-prints on ChemRxiv until today. Although pre-prints should not be mentioned as established information, we include them in this review due to the limited information existing on TMPRSS2 related to SARS-CoV-2 and due to the emergency of the situation.
The first report describes the construction of seven homology models of TMPRSS2 in Schrodinger Advanced Homology Modeling Interface, using human plasma kallikrein, Factor XIa and Hepsin as protein templates [107]. Rensi et al. chose these proteins using the BLAST tool for identifying high sequence similarity. They used SiteMap tool to define the active site and they validated the results by manually checking if the catalytic triad Ser-Asp-His was included. Finally, they used Glide to dock their ligands library. Their results for the known inhibitors (nafamostat, camostat) are in a good alignment with their experimental inhibitory activity, which adds positively to their predictions, including otamixaban, edoxaban, and argatroban. Otamixaban is predicted to have the best binding score and forms interactions with His 41, Asp180, and Ser186.
The second report describes the construction of the homology model using human plasma kallikrein as template in PRIMO online tool [108]. The binding pocket was identified by CASTp and residues His296, Asp345 and Ser441 were marked as the catalytic triad. Three inhibitors, camostat mesylate, nafamostat, and bromhexine hydrochloride, were docked in the created model. Docking of camostat mesilate seems to form interactions with Ser441, His296, Glu299, Asp435, Val473 as well as with Val28, Asp440, Thr459, Ser460, Trp461, and Tyr474. Nafamostat also maintains the formation of hydrogen bonds with Asp435, Gly464 and Ser441, while bromhexine hydrochloride interacts with Gln438, His279, Val280, Cys281 and His296.
In 2009, it has been shown that camostat mesilate or its metabolite GBPA bind in the active site of the enzyme prostasin [109]. As demonstrated by X-ray crystallography (PDB 3FVF), GBA formed a covalent bond with Ser238 after cleavage of camostat mesylate or GBPA by the triad His85-Asp134-Ser238, which corresponds to His296-Asp345-Ser441 in human TMPRSS2.
Most recently, Hempel et al. reported a detailed study on the molecular mechanism of TMPRSS2 inhibition by camostat and nafamostat mesilate [110]. First, they confirmed that camostat and nafamostat inhibited TMPRSS2 by employing a cell-based assay established by them [111]. The estimated IC50 values for camostat and nafamostat were 142 nM and 55 nM, respectively. For their simulations, they used homology models [107] of TMPRSS2 domain with either camostat or nafamostat docked to them. According to the general mechanism of hydrolysis by serine proteases, the non-covalent substrate-enzyme complex, initially formed, is cleaved and catalyzed by the conserved catalytic triad Asp345, His296, and Ser441, resulting in the formation of covalent acyl-enzyme intermediate between the inhibitor and Ser441. The ‘oxyanion hole,’ consisting of the backbone NH of Gly439 and Ser441, stabilizes the carbonyl of the scissile bond. The Michaelis complex initially formed, as well as the covalent complex generated by the action of either camostat or nafamostat, are shown in Figure 4A. Figure 4B shows the interaction of camostat with the active site of TMPRSS2 [112]. The studies of Hempel et al. showed that the non-covalent complexes of camostat and nafamostat are relatively short-lived, while the main inhibitory effect has to be attributed to the formation of the long-lived covalent acyl–enzyme complex between Ser441 and the guanidinobenzoyl moiety of the drugs.
Figure 4.
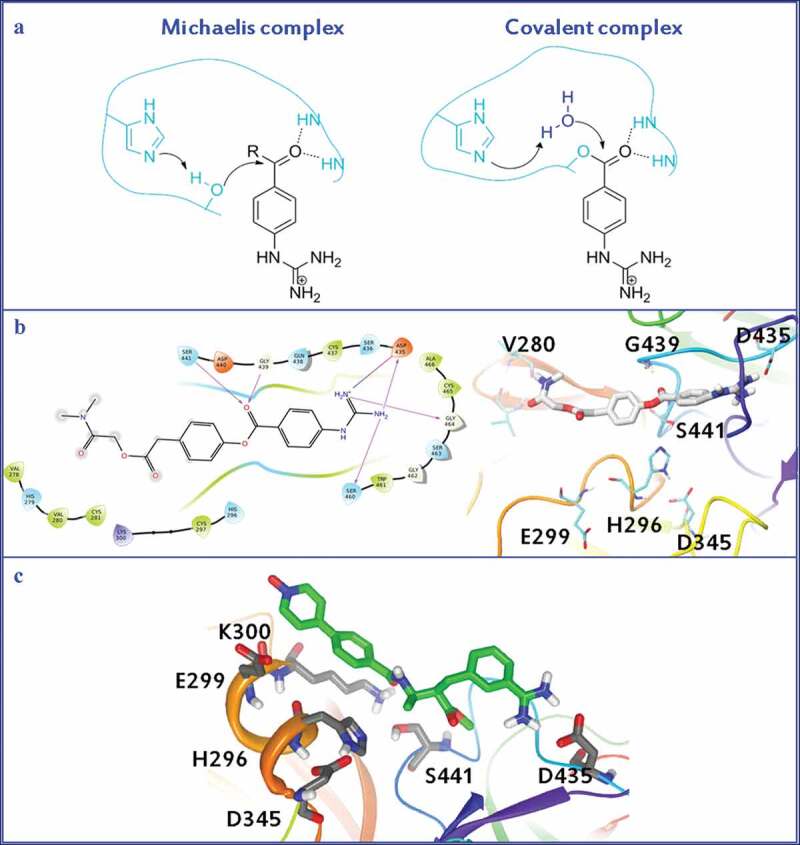
A. Michaelis complex and covalent complex generated by the action of either camostat or nafamostat mesilate. B. Interaction of camostat mesilate with the active site of TMPRSS2. C. Interaction of otamixaban with the active site of TMPRSS2.
Finally, Hempel et al. suggested that when administration of otamixaban is combined with supplemental camostat or nafamostat, otamixaban’s potency can be significantly enhanced against TMPRSS2 activity and SARS-CoV-2 infection of a human lung cell line [42]. The authors produced 109 ms of atomistic MD simulations to describe the interactions formed between TMPRSS2 and otamixaban. For the computational calculations, the TMPRSS2 structure from Hempel et al. was used [110]. Autodock Vina was employed for the docking calculations and OpenMM 7.4.0 was used for the MD production. Figure 4C shows the interactions of otamixaban with TMPRSS2ʹs active site, as it was produced by MD simulations. The long, hydrophobic pyridine-N-oxide moiety of the molecule interacts with Glu299, Lys300 and His296 and the benzamidine moiety with Asp435. The authors suggest that otamixaban can act as a stable, non-covalent reversible inhibitor.
It is crucial that authors developing homology models should make them available to public providing the pdb files of their docking results, as Hempel et al. did. This will allow for a direct comparison between predicted binding modes and active sites from different research groups and will help to proceed with the development of novel inhibitors of TMPRSS2.
In conclusion, in the absence of crystallographic data for TMPRSS2, the 3D structure of the protein has been constructed based on homology models. At least eight studies have been reported on building the 3D structure of the enzyme. Literature and computational tools have been employed to identify proteins that could be used as models for the homology modeling and these proteins include TMPRSS1, TMPRSS13, TMPRSS15 and a group of human plasma kallikrein. A number of compounds have been docked resulting in some new candidates such as rubitecan, loprazolam, valrubicin, and lopinavir. In addition, the mechanism of TMPRSS2 inhibition by camostat and nafamostat mesylate has been proposed. This includes the formation of a Michaelis complex, which includes the conserved catalytic triad Asp345, His296 and Ser441 and the backbone NH of Gly439.
7. Conclusion
Due to the COVID-19 pandemic, SARS-CoV-2 has become the subject of intense research, with a focus on the elucidation of the viral entry mechanism and the development of efficient antiviral drugs. The key role of certain enzymes in SARS-CoV-2 infection has been elucidated and specific enzymes that facilitate viral entry have been targeted, such as TMPRSS2, a type II serine protease that is responsible for the proteolytic activation of the virus in the cell surface. With the use of virtual screening and molecular docking, new agents have been developed that could readily inhibit TMPRSS2 and SARS-CoV-2 infection in vitro, namely, otamixaban, ketobenzothiazole-based small-molecule peptidomimetics, 8P9R and α1-antitrypsin (Table 4). At the same time, a great effort for drug repurposing has been undertaken. Camostat mesilate and nafamostat mesilate potently attenuated SAR-CoV-2 infection in vitro and in vivo and a plethora of phase II and III clinical trials for COVID-19, employing these drugs, are underway (Table 4). Other repurposed drugs are bromhexine and ambroxol hydrochloride and gabexate mesilate that also act as TMPRSS2 inhibitors. In addition, treatment with enzalutamide, has been associated with reduced TMPRSS2 levels in human lung cells and SARS-CoV-2 infection but these findings are disputed in other studies. Finally, as the crystal structure of TMPRSS2 remains unknown, numerous homology models have been established that have provided essential insight into the development of TMPRSS2 inhibitors and their mode of action.
Table 4.
Summary of reported compounds and approved drugs
| Compound/drug | TMPRSS2 inhibition | SARS-CoV-2 antiviral potency | Clinical status | Ref |
|---|---|---|---|---|
| NCGC00378763 (Otamixaban) |
IC50 = 0.62 μM | EC50 = 15.84 μΜ (SARS-Cov-2 PP viral entry assay) |
- | [41] |
| NCGC00386945 | IC50 = 1.24 μM | EC50 = 0.35 μΜ (SARS-Cov-2 PP viral entry assay) |
- | [41] |
| N-0385 | IC50 = 1.9 nM | ED50 = 2.8 nM (Calu-3 cells) |
- | [43] |
| 8P9R | - | IC50 = 0.3 μg/ml (Vero-E6 cells) |
- | [44] |
| α1ΑΤ | Dose-dependent inhibition at 5–50 μM | IC50 = 17.3 μM IC50 = 21.2 μM (2 different SARS-CoV-2 isolates in Vero-E6 cells) |
- | [45] |
| Camostat mesilate | IC50 = 6.2 nM | EC50 = 87 nM (Calu-3 cells) EC50 = 0.102–0.170 μM (3 different variants in Calu-3 cells) |
Multiple Phase 1, 2, 3 clinical trials (see Table 1) |
[78,82,93] |
| Nafamostat mesilate | IC50 = 0.27 nM | EC50 = 5 nM (Calu-3 cells) EC50 = 22.50 μΜ (Vero-E6 cells) EC50 = 0.016–0.024 μM (3 different variants in Calu-3 cells) EC50 = 10 nM (Calu-3 cells) |
Multiple Phase 1, 2, 3 clinical trials (see Table 2) |
[78,79,81,82,93] |
| Bromhexine hydrochloride | IC50 = 0.75 μM | AC50 = 8.9 μM (CPE of SARS-CoV-2) |
Phase 1, 3, 4 clinical trials (see Table 3) |
[87,88] |
| Gabexate mesilate | IC50 = 130 nM | - | - | [93] |
| Compound 9 | Ki = 0.9 nM | - | - | [94] |
| Enzalutamide | TMPRSS2 downregulation at 10 μM | 50% inhibition at 10 μΜ (SARS-Cov-2 PP viral entry assay) |
- | [97] |
8. Expert opinion
The new beta-coronavirus SARS-CoV-2, which appeared first in China in December 2019, has caused the devastating COVID-19 pandemic, resulting in the loss of more than 5 million human lives so far and unmeasurable economic consequences. Although several efficient vaccines have been developed and approved for use, a lot of people globally either do not have access to them or are not willing to be vaccinated. In addition, it seems that vaccination offers reduced protection for the very recently emerged omicron variant. Unfortunately, up to now no clinical effective small-molecule drug has been found for the treatment of COVID-19. Thus, there is an urgent need for the development of novel antiviral agents able to treat COVID-19.
The scientific community has directed its attention to various enzymes involved in the replication of SARS-Cov-2, such as the RNA-dependent RNA polymerase (RdRp) and the SARS-CoV-2 main protease (Mpro or 3CLpro), which are highly important targets for the discovery of novel anti-SARS-Cov-2 agents. Equally important and attractive targets are the host cell proteins involved in the viral entry into human lung cells. In addition to ACE2, which has been recognized as a key target for the cell entry mechanism, TMPRSS2 is an important mediator in viral infection and cell entry. In view of the urgency caused by the pandemic and the emerging new variants, the development or repurposing of an array of drugs, targeting different enzymes affiliated with SARS-CoV-2, is of utmost importance. Furthermore, it is essential to develop treatments for different stages and symptoms of COVID-19 to encompass all aspects of the disease. In light of this, drug repurposing could lead to effective therapeutic solutions, potentially overcoming high drug prices and the slow pace of drug discovery and development.
The discovery and development of TMPRSS2 inhibitors has emerged as a promising approach for the identification of new antiviral agents. Two TMPRSS2 inhibitors already approved drugs for the treatment of pancreatitis, camostat and nafamostat, were the first agents that attracted the interest and entered clinical trials as candidate drugs against COVID-19. Various clinical trials have started; however, the outcome of such studies will prove or not their clinical usefulness. Up to now, there are no results of clinical trials, and the efforts of the scientific community in this field have to be intensified.
In addition to the efforts for repurposing camostat and nafamostat, the design, identification, and development of novel TMPRSS2 inhibitors is of paramount importance. Employment of high-throughput screening approaches may orient our studies, and already a few chemical entities have been identified as novel TMPRSS2 inhibitors by adopting such approaches. A major drawback for the rational design of TMPRSS2 inhibitors is the lack of the crystal structure of TMPRSS2. To overcome this lack, homology models have been already developed, and indeed have helped in identifying novel chemical motifs able to inhibit TMPRSS2. Computational approaches play a predominant role in the identification of new agents inhibiting TMPRSS2, providing thus chemical motifs for the further development of drug candidates against COVID-19. Molecular docking and molecular dynamics simulations may lead to pharmacophore-based virtual screening. Most recently, Hu et al. successfully demonstrated such a strategy [41]. In any case, the determination of the crystal structure of TMPRSS2 may greatly facilitate the rational design of novel TMPRRS2 inhibitors. We encourage researchers to develop homology models and to make them available to the public in order to facilitate drug development.
Another interesting topic of research that needs clarification is the involvement of TMPRSS2 in gender and age differences related to the severity of COVID-19. A few reports have very recently appeared on this topic [95,96,98], however further research is required to shed light on the role of TMPRSS2 in age and sex differences, in order to understand how the regulation of this enzyme may influence the COVID-19 outcomes.
All in all, in our perspective, TMPRSS2 is a key target for drug development and repositioning for the treatment of COVID-19. Also, the repositioning of drugs, such as camostat and nafamostat mesilate, could substantially improve the chances of finding temporary cost-efficient therapeutic solutions affording us time to develop new antiviral agents as potential weapons for therapy or prevention of any future epidemics or pandemics.
Article highlights
• Transmembrane serine protease 2 (TMPRSS2) is an enzyme that plays an important role in cell entry and viral infection by SARS-CoV-2, and thus, it is an attractive target for the development of inhibitors as new antiviral agents.
• Adopting high-throughput strategies and homology model studies, novel agents able to inhibit TMPRRS2 have been already identified.
• Camostat and nafamostat, known TMPRRS2 inhibitors and approved drugs for the treatment of pancreatitis, are under clinical trials against COVID-19.
• The determination of TMPRSS2 crystal structure may facilitate the rational design of novel inhibitors.
• Inhibitors of TMPRRS2 may be new therapeutic agents for the treatment of COVID-19.
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China. N Engl J Med. 2019;382(8):727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying . 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5(4): 536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lake MA. What we know so far: COVID-19 current clinical knowledge and research. Clin Med. 2020;20(2):124–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The World Health Organization, Weekly epidemiological update, Coronavirus disease 2019 (COVID-19); 2021. Nov 2. [cited 2021 Dec 30]. Available from https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---7-december-2021
- 6.The World Health Organization . Tracking SARS-CoV-2 variants; 2021. Oct 29. [cited 2021 Dec 30].Available from: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/
- 7.The World Health Organization . Classification of Omicron (B.1.1.529): SARS-CoV-2 variant of concern; 2021. Nov 26. [cited 2021 Dec 30]. Available from: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern
- 8.Liu C, Zhou Q, Li Y, et al. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent Sci. 2020;6(3):315–331. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Report about agents known to be effective against other RNA viruses.
- 9.Gil C, Ginex T, Maestro I, et al. COVID-19 Drug targets and potential treatments. J Med Chem. 2020;63(21):12359–12386. [DOI] [PubMed] [Google Scholar]
- 10.The World Health Organization . COVID-19 vaccines; 2021. Oct 20. [cited 2021 Dec 30]. Available from: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/covid-19-vaccines
- 11.Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19 - Final report. N Engl J Med. 2020;383(19):1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Cao R, Xu M, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020;6(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choy KT, Wong AY, Kaewpreedee P, et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Res. 2020;178:104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan H, Peto R, Henao-Restrepo AM, et al. WHO Solidarity Trial Consortium . Repurposed antiviral drugs for Covid-19 - interim WHO solidarity trial results. N Engl J Med. 2021;384(6):497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ader F, Bouscambert-Duchamp M, Hites M, et al. Remdesivir plus standard of care versus standard of care alone for the treatment of patients admitted to hospital with COVID-19 (DisCoVeRy): a phase 3, randomised, controlled, open-label trial. Lancet Infect Dis. 2021. S1473-3099(21)00485-0 . DOI: 10.1016/S1473-3099(21)00485-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsuyama S, Nagata N, Shirato K, et al. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol. 2010;84(24):12658–12664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181(2):271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Publication on the SARS-CoV-2 cell entry mechanism and the role of TMPRSS2.
- 18.Antalis TM, Bugge TH, Wu Q. Membrane-anchored serine proteases in health and disease. Prog Mol Biol Transl Sci. 2011;99:1–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabo R, Bugge TH. Type II transmembrane serine proteases in development and disease. Int J Biochem Cell Biol. 2008;40(6–7):1297–1316. [DOI] [PubMed] [Google Scholar]
- 20.Paoloni-Giacobino A, Chen H, Peitsch MC, et al. Cloning of the TMPRSS2 gene, which encodes a novel serine protease with transmembrane, LDLRA, and SRCR domains and maps to 21q22.3. Genomics. 1997;44(3):309–320. [DOI] [PubMed] [Google Scholar]
- 21.Kitamoto Y, Yuan X, Wu Q, et al. Enterokinase, the initiator of intestinal digestion, is a mosaic protease composed of a distinctive assortment of domains. Proc Natl Acad Sci USA. 1994;91(16):7588–7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bugge TH, Antalis TM, Wu Q. Type II transmembrane serine proteases. J Biol Chem. 2009;284(35):23177–23181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YW, Lee MS, Lucht A, et al. TMPRSS2, a serine protease expressed in the prostate on the apical surface of luminal epithelial cells and released into semen in prostasomes, is misregulated in prostate cancer cells. Am J Pathol. 2010;176(6):2986–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Afar DE, Vivanco I, Hubert RS, et al. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res. 2001;61(4):1686–1692. [PubMed] [Google Scholar]
- 25.Lin B, Ferguson C, White JT, et al. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Res. 1999;59(17):4180–4184. [PubMed] [Google Scholar]
- 26.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. [DOI] [PubMed] [Google Scholar]
- 27.Kim TS, Heinlein C, Hackman RC, et al. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol Cell Biol. 2006;26(3):965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donaldson SH, Hirsh A, Li DC, et al. Regulation of the epithelial sodium channel by serine proteases in human airways. J Biol Chem. 2002;277(10):8338–8345. [DOI] [PubMed] [Google Scholar]
- 29.Böttcher E, Matrosovich T, Beyerle M, et al. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol. 2006;80(19):9896–9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bertram S, Glowacka I, Blazejewska P, et al. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J Virol. 2010;84(19):10016–10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hatesuer B, Bertram S, Mehnert N, et al. TMPRSS2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 2013;9(12):e1003774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakai K, Ami Y, Tahara M, et al. The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J Virol. 2014;88(10):5608–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Millet JK, Whittaker GR. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015;202:120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shulla A, Heald-Sargent T, Subramanya G, et al. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol. 2011;85(2):873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glowacka I, Bertram S, Müller MA, et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. 2011;85(9):4122–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shirato K, Kawase M, Matsuyama S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol. 2013;87(23):12552–12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iwata-Yoshikawa N, Okamura T, Shimizu Y, et al. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol. 2019;93(6):e01815–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11(1):1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koch J, Uckeley ZM, Doldan P, et al. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 2021;40(16):e107821. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Paper discussing the role of TMPRSS2 in SARS-CoV-2 viral entry.
- 41.Hu X, Shrimp JH, Guo H, et al. Discovery of TMPRSS2 inhibitors from virtual screening as a potential treatment of COVID-19. ACS Pharmacol Transl Sci. 2021;4(3):1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Publication focusing on the development of new TMPRSS2 inhibitors.
- 42.Hempel T, Elez K, Krüger N, et al. Synergistic inhibition of SARS-CoV-2 cell entry by otamixaban and covalent protease inhibitors: pre-clinical assessment of pharmacological and molecular properties. Chem Sci. 2021;12(38):12600–12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shapira T, Monreal IA, Dion SP, et al. A novel highly potent inhibitor of TMPRSS2-like proteases blocks SARS-CoV-2 variants of concern and is broadly protective against infection and mortality in mice. Preprint. bioRxiv 2021. DOI: 10.1101/2021.05.03.442520 [DOI] [Google Scholar]
- 44.Zhao H, To KKW, Lam H, et al. Cross-linking peptide and repurposed drugs inhibit both entry pathways of SARS-CoV-2. Nat Commun. 2021;12(1):1517. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Article presenting a new cross-linking peptide that can attenuate SARS-CoV-2 infection in vitro.
- 45.Wettstein L, Weil T, Conzelmann C, et al. Alpha-1 antitrypsin inhibits TMPRSS2 protease activity and SARS-CoV-2 infection. Nat Commun. 2021;12(1):1726. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Paper identifying alpha-1 antitrypsin as a TMPRSS2 inhibitor.
- 46.Nimishakavi S, Raymond WW, Gruenert DC, et al. Divergent inhibitor susceptibility among airway lumen-accessible tryptic proteases. PLoS One. 2015;10(10):e0141169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujii S, Uegai Y, Watanabe T, et al. Guanidinobenzoic acid derivatives, US4021472A . 1975.
- 48.Fujii S, Watanabe T, Shiota M, et al. Guanidinobenzoic acid compounds and process for preparing the same, US4224342A 1977.
- 49.Kanoh M, Ibata H, Miyagawa M, et al. Clinical effects of camostat in chronic pancreatitis. Biomed Res. 1989;10:145–150. [Google Scholar]
- 50.Gibo J, Ito T, Kawabe K, et al. Camostat mesilate attenuates pancreatic fibrosis via inhibition of monocytes and pancreatic stellate cells activity. Lab Invest. 2005;85(1):75–89. [DOI] [PubMed] [Google Scholar]
- 51.Maekawa A, Kakizoe Y, Miyoshi T, et al. Camostat mesilate inhibits prostasin activity and reduces blood pressure and renal injury in salt-sensitive hypertension. J Hypertens. 2009;27(1):181–189. [DOI] [PubMed] [Google Scholar]
- 52.Coote K, HC A-W, Sugar R, et al. Camostat attenuates airway epithelial sodium channel function in vivo through the inhibition of a channel-activating protease. J Pharmacol Exp Ther. 2009;329(2):764–774. [DOI] [PubMed] [Google Scholar]
- 53.Rowe SM, Reeves G, Hathorne H, et al. Reduced sodium transport with nasal administration of the prostasin inhibitor camostat in subjects with cystic fibrosis. Chest. 2013;144(1):200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asami T, Tomisawa S, Uchiyama M. Effect of oral camostat mesilate on hematuria and/or proteinuria in children. Pediatr Nephrol. 2004;19(3):313–316. [DOI] [PubMed] [Google Scholar]
- 55.Jia D, Taguchi M, Otsuki M. Synthetic protease inhibitor camostat prevents and reverses dyslipidemia, insulin secretory defects, and histological abnormalities of the pancreas in genetically obese and diabetic rats. Metabolism. 2005;54(5):619–627. [DOI] [PubMed] [Google Scholar]
- 56.Lee MG, Kim KH, Park KY, et al. Evaluation of anti-influenza effects of camostat in mice infected with non-adapted human influenza viruses. Arch Virol. 1996;141(10):1979–1989. [DOI] [PubMed] [Google Scholar]
- 57.Yamaya M, Shimotai Y, Hatachi Y, et al. The serine protease inhibitor camostat inhibits influenza virus replication and cytokine production in primary cultures of human tracheal epithelial cells. Pulm Pharmacol Ther. 2015;33:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou Y, Vedantham P, Lu K, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015;116:76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawase M, Shirato K, van der Hoek L, et al. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. 2012;86(12):6537–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hofmann-Winkler H, Moerer O, Alt-Epping S, et al. Camostat mesylate may reduce severity of coronavirus disease 2019 sepsis: a first observation. Crit Care Explor. 2020;2(11):e0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ou T, Mou H, Zhang L, et al. Hydroxychloroquine-mediated inhibition of SARS-CoV-2 entry is attenuated by TMPRSS2. PLoS Pathog. 2021;17(1):e1009212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Midgley I, Hood AJ, Proctor P, et al. Metabolic fate of 14C-camostat mesylate in man, rat and dog after intravenous administration. Xenobiotica. 1994;24(1):79–92. [DOI] [PubMed] [Google Scholar]
- 63.Hoffmann M, Hofmann-Winkler H, Smith JC, et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. bioRxiv. 2020. 2020.08.05.237651 DOI: 10.1101/2020.08.05.237651 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Preprint reporting camostat mesilate’s antiviral activity against SARS-CoV-2.
- 64.Fujii S, Hitomi Y. New synthetic inhibitors of C1r, C1 esterase, thrombin, plasmin, kallikrein and trypsin. Biochim Biophys Acta. 1981;661(2):342–345. [DOI] [PubMed] [Google Scholar]
- 65.Ueda N, Midorikawa A, Ino Y, et al. Inhibitory effects of newly synthesized active center-directed trypsin-like serine protease inhibitors on the complement system. Inflamm Res. 2000;49(1):42–46. [DOI] [PubMed] [Google Scholar]
- 66.Aoyama T, Ino Y, Ozeki M, et al. Pharmacological studies of FUT-175, nafamostat mesilate. I. Inhibition of protease activity in in vitro and in vivo experiments. Jpn J Pharmacol. 1984;35(3):203–227. [DOI] [PubMed] [Google Scholar]
- 67.Lu YX, Ju HQ, Wang F, et al. Inhibition of the NF-κB pathway by nafamostat mesilate suppresses colorectal cancer growth and metastasis. Cancer Lett. 2016;380(1):87–97. [DOI] [PubMed] [Google Scholar]
- 68.Lai PC, Chiu TH, Huang YT. Overexpression of BDNF and TrkB in human bladder cancer specimens. Oncol Rep. 2010;24(5):1265–1270. [DOI] [PubMed] [Google Scholar]
- 69.Homma S, Hayashi K, Yoshida K, et al. Nafamostat mesilate, a serine protease inhibitor, suppresses interferon-gamma-induced up-regulation of programmed cell death ligand 1 in human cancer cells. Int Immunopharmacol. 2018;54:39–45. [DOI] [PubMed] [Google Scholar]
- 70.Iwase R, Haruki K, Fujiwara Y, et al. Combination chemotherapy of nafamostat mesylate with gemcitabine for gallbladder cancer targeting nuclear factor-κB activation. J Surg Res. 2013;184(1):605–612. [DOI] [PubMed] [Google Scholar]
- 71.Haruki K, Shiba H, Fujiwara Y, et al. Inhibition of nuclear factor-κB enhances the antitumor effect of paclitaxel against gastric cancer with peritoneal dissemination in mice. Dig Dis Sci. 2013;58(1):123–131. [DOI] [PubMed] [Google Scholar]
- 72.Mander S, You DJ, Park S, et al. Nafamostat mesilate negatively regulates the metastasis of triple-negative breast cancer cells. Arch Pharm Res. 2018;41(2):229–242. [DOI] [PubMed] [Google Scholar]
- 73.Han X, Wang N, Li J, et al. Identification of nafamostat mesilate as an inhibitor of the fat mass and obesity-associated protein (FTO) demethylase activity. Chem Biol Interact. 2019;297:80–84. [DOI] [PubMed] [Google Scholar]
- 74.Fuwa M, Kageyama M, Ohashi K, et al. Nafamostat and sepimostat identified as novel neuroprotective agents via NR2B N-methyl-D-aspartate receptor antagonism using a rat retinal excitotoxicity model. Sci Rep. 2019;9(1):20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Inman RD, Chiu B. Nafamostat mesylate, a serine protease inhibitor, demonstrates novel antimicrobial properties and effectiveness in chlamydia-induced arthritis. Arthritis Res Ther. 2012;14(3):R150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hosoya M, Matsuyama S, Baba M, et al. Effects of protease inhibitors on replication of various myxoviruses. Antimicrob Agents Chemother. 1992;36(7):1432–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamamoto M, Matsuyama S, Li X, et al. Identification of nafamostat as a potent inhibitor of middle east respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother. 2016;60(11):6532–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hoffmann M, Schroeder S, Kleine-Weber H, et al. Nafamostat mesylate blocks activation of SARS-CoV-2: new treatment option for COVID-19. Antimicrob Agents Chemother. 2020;64(6):e00754–20. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Letter reporting nafamostat’s inhibitory potency against SARS-CoV-2 infection of human lung cells.
- 79.Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tiwari SK, Wang S, Smith D, et al. Revealing tissue-specific SARS-CoV-2 infection and host responses using human stem cell-derived lung and cerebral organoids. Stem Cell Reports. 2021;16(3):437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yamamoto M, Kiso M, Sakai-Tagawa Y, et al. The anticoagulant nafamostat potently inhibits SARS-CoV-2 S protein-mediated fusion in a cell fusion assay system and viral infection in vitro in a cell-type-dependent manner. Viruses. 2020;12(6):629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee J, Lee J, Kim HJ, et al. TMPRSS2 and RNA-dependent RNA polymerase are effective targets of therapeutic intervention for treatment of COVID-19 caused by SARS-CoV-2 variants (B.1.1.7 and B.1.351). Microbiol Spectr. 2021;9(1):e0047221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li K, Meyerholz DK, Bartlett JA, et al. The TMPRSS2 inhibitor nafamostat reduces SARS-CoV-2 pulmonary infection in mouse models of COVID-19. mBio. 2021;12(4):e0097021. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• In vivo effect of nafamostat mesilate in mouse models of COVID-19.
- 84.Tsukagoshi S. [Pharmacokinetics studies of nafamostat mesilate (FUT), a synthetic protease inhibitor, which has been used for the treatments of DIC and acute pancreatitis, and as an anticoagulant in extracorporeal circulation]. Gan To Kagaku Ryoho. 2000;27:767–774. Japanese. PMID: 10832450 [PubMed] [Google Scholar]
- 85.Fischer J, Ganellin CR. Analogue-based drug discovery. Weinheim, Germany: John Wiley & Sons; 2006. p. 544. [Google Scholar]
- 86.Malerba M, Ragnoli B. Ambroxol in the 21st century: pharmacological and clinical update. Expert Opin Drug Metab Toxicol. 2008;4(8):1119–1129. [DOI] [PubMed] [Google Scholar]
- 87.Lucas JM, Heinlein C, Kim T, et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014;4(11):1310–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.OpenData COVID-19 . 2020. [cited 30 Dec 2021]. Available from: https://opendata.ncats.nih.gov/covid19/databrowser?q=bromhexine
- 89.Li T, Sun L, Zhang W, et al. Bromhexine hydrochloride tablets for the treatment of moderate COVID-19: an open-label randomized controlled pilot study. Clin Transl Sci. 2020;13(6):1096–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ansarin K, Tolouian R, Ardalan M, et al. Effect of bromhexine on clinical outcomes and mortality in COVID-19 patients: a randomized clinical trial. Bioimpacts. 2020;10(4):209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cavallini G, Tittobello A, Frulloni L, et al. Gabexate for the prevention of pancreatic damage related to endoscopic retrograde cholangiopancreatography. gabexate in digestive endoscopy--Italian group. N Engl J Med. 1996;335(13):919–923. [DOI] [PubMed] [Google Scholar]
- 92.Kosai K, Seki M, Yanagihara K, et al. Gabexate mesilate suppresses influenza pneumonia in mice through inhibition of cytokines. J Int Med Res. 2008;36(2):322–328. [DOI] [PubMed] [Google Scholar]
- 93.Shrimp JH, Kales SC, Sanderson PE, et al. An enzymatic TMPRSS2 assay for assessment of clinical candidates and discovery of inhibitors as potential treatment of COVID-19. ACS Pharmacol Transl Sci. 2020;3(5):997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Meyer D, Sielaff F, Hammami M, et al. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem J. 2013;452(2):331–343. [DOI] [PubMed] [Google Scholar]
- 95.Okwan-Duodu D, Lim EC, You S, et al. TMPRSS2 activity may mediate sex differences in COVID-19 severity. Signal Transduct Target Ther. 2021;6(1):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peng J, Sun J, Zhao J, et al. Age and gender differences in ACE2 and TMPRSS2 expressions in oral epithelial cells. J Transl Med. 2021;19(1):358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Leach DA, Mohr A, Giotis ES, et al. The antiandrogen enzalutamide downregulates TMPRSS2 and reduces cellular entry of SARS-CoV-2 in human lung cells. Nat Commun. 2021;12(1):4068. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Article reporting enzalutamide’s inhibitory potency against SARS-CoV-2 infection of human lung cells.
- 98.Baratchian M, McManus JM, Berk MP, et al. Androgen regulation of pulmonary AR, TMPRSS2 and ACE2 with implications for sex-discordant COVID-19 outcomes. Sci Rep. 2021;11(1):11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li F, Han M, Dai P, et al. Distinct mechanisms for TMPRSS2 expression explain organ-specific inhibition of SARS-CoV-2 infection by enzalutamide. Nat Commun. 2021;12(1):866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rahman N, Basharat Z, Yousuf M, et al. Virtual screening of natural products against type II transmembrane serine protease (TMPRSS2), the priming agent of coronavirus 2 (SARS-CoV-2). Molecules. 2020;25(10):2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Elmezayen AD, Al-Obaidi A, Şahin AT, et al. Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J Biomol Struct Dyn. 2021;39(8):2980–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kishk SM, Kishk RM, Yassen ASA, et al. Molecular insights into human transmembrane protease serine-2 (TMPS2) inhibitors against SARS-CoV2: homology modelling, molecular dynamics, and docking studies. Molecules. 2020;25(21):5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baby K, Maity S, Mehta CH, et al. SARS-CoV-2 entry inhibitors by dual targeting TMPRSS2 and ACE2: an in silico drug repurposing study. Eur J Pharmacol. 2021;896:173922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pooja M, Reddy GJ, Hema K, et al. Unravelling high-affinity binding compounds towards transmembrane protease serine 2 enzyme in treating SARS-CoV-2 infection using molecular modelling and docking studies. Eur J Pharmacol. 2021;890:173688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huggins DJ. Structural analysis of experimental drugs binding to the SARS-CoV-2 target TMPRSS2. J Mol Graph Model. 2020;100:107710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Escalante DE, Ferguson DM. Structural modeling and analysis of the SARS-CoV-2 cell entry inhibitor camostat bound to the trypsin-like protease TMPRSS2. Med Chem Res. 2021;1–11. DOI: 10.1007/s00044-021-02708-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rensi S, Altman RB, and Liu T, et al. Homology modeling of TMPRSS2 yields candidate drugs that may inhibit entry of SARS-CoV-2 into human cells. ChemRxiv. 2020. . doi: 10.26434/chemrxiv.12009582. [DOI] [Google Scholar]
- 108.Sonawane KD, Barale SS, Dhanavade MJ, et al. Structural insights and inhibition mechanism of TMPRSS2 by experimentally known inhibitors camostat mesylate, nafamostat and bromhexine hydrochloride to control SARS-coronavirus-2: a molecular modeling approach. Inform Med Unlocked. 2021;24:100597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spraggon G, Hornsby M, Shipway A, et al. Active site conformational changes of prostasin provide a new mechanism of protease regulation by divalent cations. Protein Sci. 2009;18(5):1081–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hempel T, Raich L, Olsson S, et al. Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat. Chem Sci. 2021;12:983–992. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Molecular docking studies of TMPRSS2 with camostat and nafamostat mesilate.
- 111.Azouz NP, Klingler AM, Callahan V, et al. Alpha 1 antitrypsin is an inhibitor of the SARS-CoV-2-priming protease TMPRSS2. bioRxiv. 2020. 2020.05.04.077826 DOI: 10.1101/2020.05.04.077826 Preprint [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.TMPRSS2 structural ensembles. [cited 30 Dec 2021]. Available from: https://github.com/noegroup/tmprss2_structures