

ABSTRACT
Introduction
Neutralizing antibodies (NAbs) that target key domains of the spike protein in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) may have therapeutic value because of their specificity. Depending on the targeted epitope, single agents may be effective, but combined treatment involving multiple NAbs may be necessary to prevent the emergence of resistant variants.
Areas covered
This article highlights the accelerated regulatory processes established to facilitate the review and approval of potential therapies. An overview of treatment approaches for SARS-CoV-2 infection, with detailed examination of the preclinical and clinical evidence supporting the use of NAbs, is provided. Finally, insights are offered into the potential benefits and challenges associated with the use of these agents.
Expert opinion
NAbs offer an effective, evidence-based therapeutic intervention during the early stages of SARS-CoV-2 infection when viral replication is the primary factor driving disease progression. As the pandemic progresses, appropriate use of NAbs will be important to minimize the risk of escape variants. Ultimately, the availability of effective treatments for COVID-19 will allow the establishment of treatment algorithms for minimizing the substantial rates of hospitalization, morbidity (including long COVID) and mortality currently associated with the disease.
KEYWORDS: Bamlanivimab and etesevimab, casirivimab and imdevimab, COVID-19, neutralizing antibodies, regdanvimab, sotrovimab, SARS-CoV-2
1. Introduction
The coronavirus disease 2019 (COVID-19) pandemic continues to pose an ongoing global public health emergency. Infection rates are high, particularly in the developing world where access to vaccines and treatment can be limited [1].
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection can cause considerable morbidity and mortality. Upon infection, most individuals exhibit mild symptoms and experience swift recovery. However, outcomes can be poor in some patients, particularly the elderly and those with comorbidities. A US study of hospitalized patients estimated mortality rates greater than 20% in the overall hospitalized population, which increased to greater than 70% among those on mechanical ventilation [2]. In the early stages of COVID-19, disease progression is driven by viral replication and infection and represents an early opportunity to effectively change the course of the disease and prevent the development of severe or critical illness and hospitalization. An additional potential benefit of early treatment is a reduction in transmission by decreasing the time that an individual is infectious [3]. Therefore, there is a need for effective treatment options to cover the spectrum of COVID-19, including early-stage disease, and to counteract the emergence of SARS-CoV-2 variants that might confer treatment resistance [4,5].
In the present article, we provide an overview of current and potential treatment approaches for SARS-CoV-2 infection, with a focus on the therapeutic application of neutralizing antibodies (NAbs). A summary of evidence is provided from clinical trials of NAbs currently available for the treatment of patients with SARS-CoV-2 infection. The article concludes with expert commentary on the future prospects for these agents as the pandemic continues to evolve.
2. Regulatory processes during the COVID-19 pandemic
In response to the COVID-19 pandemic, the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) adopted policies for the accelerated review and approval of treatments for COVID-19, as summarized in Figure 1.
Figure 1.
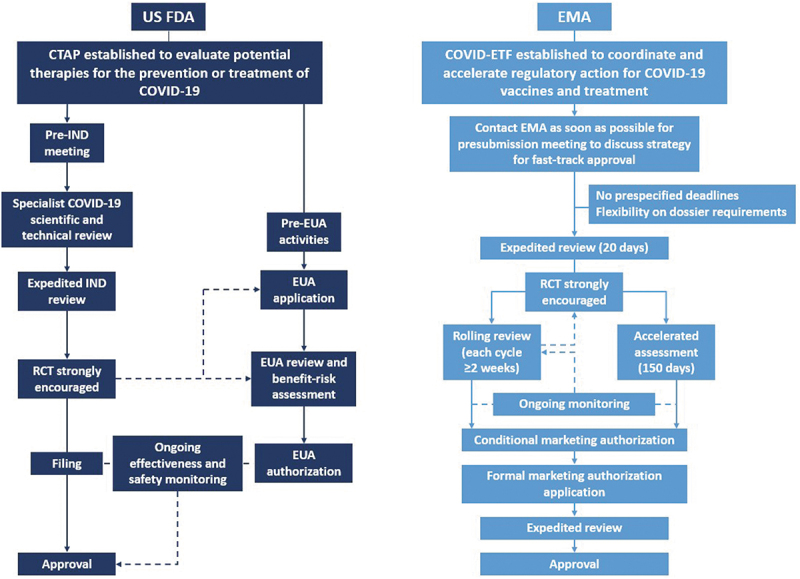
Regulatory pathways adopted by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) during the COVID-19 pandemic. COVID-19, coronavirus disease 2019; COVID-ETF: COVID-19 EMA pandemic Task Force; CTAP: Coronavirus Treatment Acceleration Program; EUA: Emergency Use Authorization; IND: investigational new drug; RCT: randomized controlled trial.
The FDA established the emergency Coronavirus Treatment Acceleration Program (CTAP) to facilitate the timely evaluation of novel treatments for COVID-19, and the FDA’s Center for Drug Evaluation and Research has established a specific team to oversee investigational new drug (IND) applications related to COVID-19 [6]. Additionally, the US Department of Health and Human Services declared on 1 April 2020 that “circumstances justified the authorization of emergency use of drugs and biological products during the COVID-19 pandemic” [7]. Regardless of previous approval status, medical products may be granted Emergency Use Authorization (EUA) in a public health emergency [7]. The EMA also issued guidance to accelerate medicine and vaccine development for COVID-19, and established the COVID-19 EMA pandemic Task Force (COVID-ETF) to coordinate and accelerate regulatory action, for both new products and those already authorized for other conditions.
As of October 2021, more than 640 drug development programs had been planned under the FDA CTAP, more than 470 trials had been reviewed by the FDA, 11 treatments had been authorized for emergency use and one treatment had been approved for COVID-19 [8]. More than 60 studies of NAbs had received safe-to-proceed IND status [8]. In Europe, by October 2021, over 80 COVID-19 treatments had received EMA advice, three treatments were undergoing rolling review, five treatments were under evaluation for marketing authorization extension and one treatment was authorized for use in COVID-19 [9,10]. Five products, including four NAbs or NAb combinations, had been approved for use in the European Union (EU) to treat COVID-19 following Article 5(3) review, which is intended to support national decision-making before formal authorization is issued [11]. In October 2021, the European Commission announced a list of 10 promising candidate therapeutics as part of its EU Strategy on COVID-19 Therapeutics, based on the EMA’s rolling review evaluation. These consisted of six treatments undergoing evaluation for marketing authorization extension or under rolling review and four waiting for rolling review to start [12,13].
3. Overview of treatment approaches for SARS-CoV-2 infection
A broad range of both novel and repurposed therapies have been evaluated as potential treatments for SARS-CoV-2 infection, to be used in combination with supportive care measures. EUAs have permitted clinical use of several agents that have shown promise during early testing. In response to the need for evidence-based treatment of SARS-CoV-2 infection, a number of rapidly evolving guidelines have been published to guide and standardize clinical use of these agents. Recommendations from recognized treatment guidelines, stratified according to patient population and severity of infection, are summarized in Table 1 [14–17].
Table 1.
Treatment guidelines for SARS-CoV-2 infection
| SARS-CoV-2 Patient Population | Recommendations by Organization/Societya |
|---|---|
| Not hospitalized, mild-to-moderate |
|
| Hospitalized and not requiring supplemental oxygen |
|
| Hospitalized and requiring supplemental oxygen |
|
| Hospitalized and requiring oxygen delivery through high-flow device or noninvasive ventilation |
|
| Hospitalized and requiring mechanical ventilation |
|
| Not recommended |
|
| Insufficient data to support a recommendation, or no recommendation |
|
aNIH, National Institutes of Health [14]; ERS, European Respiratory Society [15]; WHO, World Health Organization [16]; Infectious Diseases Society of America (December 2021 update) [17].
bRecommended prior to the occurrence of the dominant omicron variant.
cPatients with elevated markers of systemic inflammation.
dRecommended in combination with remdesivir for patients with a contraindication to corticosteroid therapy.
COVID-19: coronavirus disease 2019; ECMO: extracorporeal mechanical oxygenation; HIV: human immunodeficiency virus; ICU: intensive care unit; N/A: not available.
To date, NAbs have gained EUA and some have received marketing authorization for use in patients with mild-to-moderate disease who are at high risk of progression to severe disease, as described in Table 2 [18–26]. Remdesivir is recommended within some guidelines as a targeted antiviral therapy for hospitalized patients with SARS-CoV-2 infection [27,28] and more recently, with the emergence of omicron as the dominant variant, for ambulatory patients at high risk of progression to severe disease [14,17]. Dexamethasone or other systemic corticosteroids are suggested for hospitalized patients receiving supplemental oxygen or mechanical ventilation [29,30]. In patients with a contraindication to corticosteroid therapy, baricitinib should be used as an add-on to remdesivir instead of remdesivir alone [17,31]. Tocilizumab is recommended in certain guidelines for use in patients with elevated markers of systemic inflammation, generally in combination with corticosteroids [32,33]. Early data from investigation of the IL-1α/β inhibitor anakinra suggested a significant reduction in the risk of disease worsening among patients at risk of respiratory failure, defined by elevated soluble urokinase plasminogen activator receptor levels; in December 2021, the EMA approved anakinra for the treatment of COVID-19 in adult patients with pneumonia requiring supplemental oxygen who are at risk of severe respiratory failure [34,35]. Candidates for oral treatment include the nucleoside analogue molnupiravir, which inhibits SARS-CoV-2 replication via mutagenesis, i.e. by introducing errors into the viral genome [36–38]. Results from the Phase 2/3 MOVe-OUT study demonstrated a reduction in the number of hospitalizations or deaths with molnupiravir compared with placebo when used to treat non-hospitalized patients [39,40]. The EMA has granted molnupiravir conditional authorization for the treatment of adults with COVID-19 who are at increased risk of progressing to severe disease but who do not require supplemental oxygen, and the FDA has issued EUA approval for molnupiravir for the treatment of adults with COVID-19 who are at high risk of progressing to severe disease and are ineligible for other treatments authorized by the FDA [41,42]. In addition, the FDA has issued EUA for nirmatrelvir/ritonavir for the treatment of mild-to-moderate COVID-19 in adults and pediatric patients who are at high risk of progression to severe disease [43]. Other oral treatment candidates include ATR-002, which is being evaluated in a Phase 2 trial in patients with moderate-to-severe COVID-19 requiring hospitalization (NCT04776044), and AT-527, which is currently undergoing a Phase 3 trial having demonstrated antiviral activity in high-risk patients with underlying health conditions at Phase 2 [44,45].
Table 2.
Eligibility criteria for NAb therapy in the treatment of COVID-19
| Use of NAbs: Approved patient populations |
|---|
|
Bamlanivimab/etesevimab [23] and casirivimab/imdevimab [24] have received conditional/emergency use authorization for the treatment of non-hospitalized patients with mild/moderate SARS-CoV-2 infection considered at high risk of progression to severe COVID-19:
|
|
Monoclonal antibodies, such as bamlanivimab plus etesevimab, and casirivimab plus imdevimab, and sotrovimab ‘may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high-flow oxygen or mechanical ventilation’. Therefore, these agents are not authorized for use in patients who:
Sotrovimab has received conditional/emergency use authorization for the treatment of non-hospitalized mild/moderate COVID-19 in adults and pediatric patients (≥12 years of age and weighing ≥40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk of progression to severe COVID-19, including hospitalization or death [74,75]:
The EMA has approved casirivimab/imdevimab for the treatment of COVID-19 in adults and adolescents aged ≥12 years and weighing ≥40 kg who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19 [19]. The EMA has approved regdanvimab for the treatment of adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19, defined as having at least one of the following risk factors [26]:
|
AIDS: acquired immunodeficiency syndrome; BMI: body mass index; CHMP: Committee for Medicinal Products for Human Use; COVID-19: coronavirus disease 2019; HIV: human immunodeficiency virus; NAb: neutralizing antibody.
Mesenchymal stem cells (MSCs) have also been assessed for their potential for treating patients with COVID-19 [46]. Preliminary clinical data from several small studies of patients with COVID-19 (including COVID-19 pneumonia and acute respiratory distress syndrome) support the potential of intravenous administration of MSCs to improve pulmonary function, clinical symptoms and survival [47–49]. Several clinical trials of adipose-derived MSCs for the treatment of COVID-19 are ongoing [50].
4. NAbs as a therapeutic approach to the management of SARS-CoV-2 infection
SARS-CoV-2 has four main structural proteins: spike (S), envelope, membrane and nucleocapsid [51]. S, which forms a glycoprotein, contains S1 and S2 subunits that are responsible for receptor attachment and membrane fusion, respectively [52]. The S protein facilitates viral entry into host cells by binding to angiotensin-converting enzyme 2 (ACE2) receptors and promoting endocytosis. It is therefore a key target for the development of antiviral therapies [52,53]. S1 and S2 protein priming by cellular transmembrane serine protease 2 (TMPRSS2) promotes fusion of viral and host cellular membranes, a process that can be inhibited by anti-SARS-CoV-2 antibodies induced by infection [53].
Antibodies have extremely high specificity for their target epitopes and therefore represent a vital approach to the therapeutic management of SARS-CoV-2 infection. Given the importance of the S protein – and specifically the receptor-binding domain (RBD) – in virus–cell fusion and entry, this represents an ideal target site to disrupt virus–ACE2 interaction and inhibit virus entry into the cell [4] (Figure 2). In addition to blocking the binding of RBD to ACE2, NAbs may target other subunits of the S protein, preventing the conformational changes needed for fusion, and some NAbs may also activate immune-related mechanisms, such as antibody-dependent cellular phagocytosis or antibody-dependent cellular cytotoxicity, via fragment crystallizable (Fc)-mediated effector functions [54,55].
Figure 2.
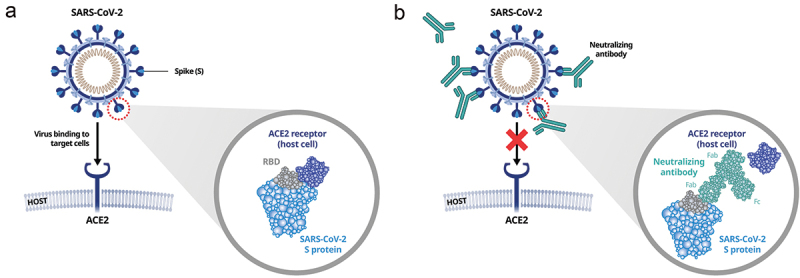
Mechanism of action of anti-receptor-binding domain (RBD) SARS-CoV-2 neutralizing antibodies (NAbs). (a) SARS-CoV-2 infection occurs via binding of the RBD to the angiotensin-converting enzyme 2 (ACE2) receptor on host cells; (b) NAb interferes with binding of the RBD/spike (S) protein to the ACE2 receptor, blocking virus entry into host cells. Fab: fragment antigen binding; Fc: fragment crystallizable.
A number of different NAb structures have been characterized and classified into one of four categories, according to their mode of binding to the S protein [56–58] (Figure 3). Class 1 NAbs (e.g. regdanvimab, etesevimab and imdevimab) are IGHV3-encoded NAbs with short CDRH3 loops and bind to epitopes on the ACE2 binding site (or receptor-binding motif [RBM]) of the RBD in the ‘up’ conformation only. Class 2 NAbs (e.g. bamlanivimab) are also IGHV3-encoded antibodies but with longer CDRH3 loops and bind the RBD in the ‘up’ and ‘down’ conformations [56,58]. Class 1 and Class 2 NAbs exert their neutralizing effect through direct competition with the human ACE2 receptor (i.e. ‘ACE2 blockers’) [57,58]. Class 3 NAbs block the ACE2 binding site, recognize both ‘up’ and ‘down’ RBD conformations and can interact with adjacent RBD protomers. Class 4 NAbs (e.g. casirivimab, sotrovimab) do not overlap with the ACE2 binding site and bind to a highly conserved epitope that is only accessible when the RBD is in the ‘up’ conformation [56,58].
Figure 3.
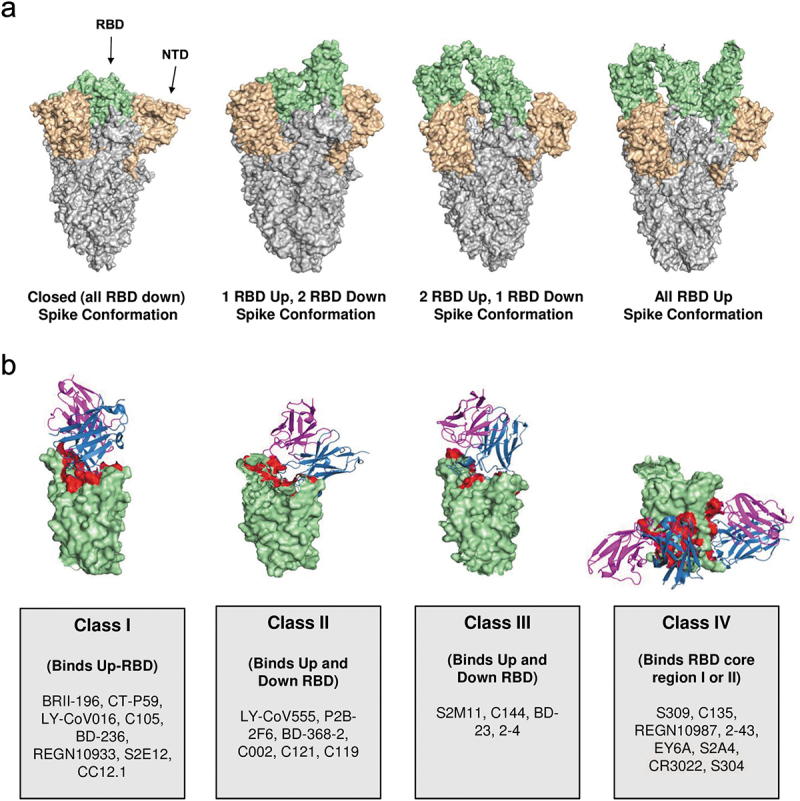
SARS-CoV-2 spike protein structure, conformation and targets of receptor-binding domain (RBD)-dependent neutralizing antibodies (NAbs). (a) Different conformations of spike protein. (b) Representation of four classes of SARS-CoV-2 RBD-dependent NAbs. Figure reproduced with modification from Kumar S, Chandele A, Sharma A. Current status of therapeutic monoclonal antibodies against SARS-CoV-2. PLoS Pathog. 2021;17(9):e1009885 [58] under CC-BY license.
Several NAbs are in development and some have been approved for the treatment of patients with mild-to-moderate COVID-19 who are considered at risk of progression to severe disease [19,26,59]. Patients with mild-to-moderate COVID-19 have been selected as good candidates for treatment with NAbs because early disease is likely virally mediated, whereas in patients with advanced disease, post-viral or peri-viral phenomena (e.g. hypoxia, hyperinflammation) are thought to be the main contributors to disease progression [59–61]. Combinations of NAbs (so-called ‘cocktail therapy’) that target different sites on the S protein can act synergistically, leading to enhanced neutralization [62,63]. In addition, cocktail therapy using combinations of antibodies that bind distinct and non-overlapping regions of the S protein RBD may prevent the emergence of resistant strains, as mutations are unlikely to occur simultaneously at two distinct genetic sites [64]. However, some NAbs may be suitable for use as single agent therapy (i.e. without other NAbs).
Finally, in addition to treating SARS-CoV-2 infection, NAbs may offer protection against infection. Several NAbs that target the N-terminal domain of the S1 subunit have been shown to protect against viral challenge in preclinical models [55,65]. Similarly, RBD-targeting NAbs may also offer prophylactic protection against SARS-CoV-2 infection. Bamlanivimab was shown to protect against SARS-CoV-2 infection in non-human primates [66] and the randomized, placebo-controlled BLAZE-2 study subsequently provided clinical evidence for the preventative potential of this NAb [67]. Casirivimab plus imdevimab prevented COVID-19 and protected against SARS-CoV-2 infection when given to previously uninfected household members of infected individuals [68]. While of interest, comprehensive discussion of NAbs for prophylaxis of SARS-CoV-2 infection is beyond the scope of the present article, which is focused on the application of NAbs for the treatment of SARS-CoV-2 infection.
5. NAbs in the treatment of SARS-CoV-2 infection
In this section, the primary and key secondary efficacy endpoints, together with safety information, are presented for each study reviewed.
5.1. Bamlanivimab plus etesevimab
Bamlanivimab (LY-CoV555) is a recombinant, neutralizing human immunoglobulin G1 monoclonal antibody (mAb) [66] and etesevimab (LY-CoV016, also known as JS016) is a recombinant human monoclonal NAb [69]. Both specifically bind to the SARS-CoV-2 S protein, inhibiting viral attachment and entry into host cells.
The randomized, Phase 2/3 BLAZE-1 clinical trial [18,21] included 613 patients with SARS-CoV-2 infection, and those participating in Phase 2 had to have at least one symptom of mild or moderate disease. Bamlanivimab monotherapy (doses of 700 mg, 2800 mg or 7000 mg) was not associated with statistically significant reductions in viral load at day 11 compared with placebo. However, statistically significant changes in viral load were seen in patients receiving combination therapy (2800 mg each of bamlanivimab and etesevimab; Table 3) [18,70]. The difference (95% confidence interval [CI]) from placebo in change from baseline in log viral load at day 11 was −0.27 (−0.71 to 0.16; p = 0.21) with bamlanivimab 2800 mg and −0.57 (−1.00 to −0.14; p = 0.01) with combination treatment [18]. COVID-19-related hospitalizations or emergency department visits were reported in nine patients receiving placebo (5.8%) and one patient (0.9%) receiving combination therapy. Data from the Phase 3 cohort of the study (n = 1035) demonstrated reduced COVID-19-related hospitalization or death from any cause (primary endpoint) in 11/518 (2.1%) patients in the bamlanivimab and etesevimab group versus 36/517 (7.0%) of patients in the placebo group. No deaths were reported in the bamlanivimab and etesevimab group, whereas 10 were reported in the placebo group (nine of which were deemed COVID-19 related). By day 7, a greater reduction from baseline in log viral load was observed among patients in the bamlanivimab and etesevimab group compared with placebo [70]. The BLAZE-4 trial (NCT04634409) has evaluated treatment of symptomatic SARS-CoV-2 infection with bamlanivimab in combination with other NAbs, including etesevimab and sotrovimab.
Table 3.
Pivotal clinical trials with bamlanivimab/etesevimab, casirivimab/imdevimab and regdanvimab [18,22,84]
| Study | Patients | Treatments | Virologic Outcomes | Clinical Outcomes | Treatment-Emergent AEs | Hypersensitivity/Infusion Reactions | Serious AEs |
|---|---|---|---|---|---|---|---|
| BLAZE-1 [18,70](Phase 2/3) | Ambulatory patients with SARS-CoV-2 infection and ≥1 mild/moderate symptom(s) | Bamlanivimab 700 mg (n = 101) [18] |
Mean difference in change from baseline log viral load versus placebo Day 11: 0.09 log10 cp/mL 95% CI −0.35 to 0.52 |
Hospitalizations or ED visits within 29 days: 1.0% (1 event) |
27 (26.7%) Most common: nausea, diarrhea |
6 events across all bamlanivimab monotherapy doses (1.9%)a | 0 |
| Bamlanivimab 2800 mg (n = 107) [18] |
Mean difference in change from baseline log viral load versus placebo Day 11: −0.27 log10 cp/mL 95% CI −0.71 to 0.16 |
Hospitalizations or ED visits within 29 days: 1.9% (2 events) |
26 (24.3%) Most common: nausea, diarrhea |
0 | |||
| Bamlanivimab 7000 mg (n = 101) [18] |
Mean difference in change from baseline log viral load versus placebo Day 11: 0.31 log10 cp/mL 95% CI −0.13 to 0.76; p = 0.16 |
Hospitalizations or ED visits within 29 days: 2.0% (2 events) |
22 (21.8%) Most common: diarrhea, nausea |
0 | |||
| Bamlanivimab 2800 mg/etesevimab 2800 mg (n = 112) [18] |
Mean difference in change from baseline log viral load versus placebo Day 11: −0.57 log10 cp/mL 95% CI −1.00 to −0.14; p = 0.01 |
Hospitalizations or ED visits within 29 days: 0.9% (1 event) |
19 (17.0%) Most common: nausea, diarrhea |
2 (1.8%)a | 1 (0.9%)b | ||
| Placebo (n = 156) [18] |
Hospitalizations or ED visits within 29 days: 5.8% (9 events) |
42 (26.9%) Most common: diarrhea, nausea |
1 (0.6%)a | 1 (0.6%)c | |||
| Bamlanivimab 2800 mg/etesevimab 2800 mg (n = 518) [70] |
Mean difference in change from baseline log viral load versus placebo Day 7: −1.20 log10 cp/mL 95% CI −1.46 to −0.94; p < 0.001 |
Hospitalizations or death within 29 days: 2.1% (11 patients) |
69 (13.3%) Most common: rash, nausea |
Not reported | 7 (1.4%) | ||
| Placebo (n = 517) [70] |
Hospitalizations or death within 29 days: 7.0% (36 patients) |
60 (11.6%) Most common: rash, nausea, dizziness |
Not reported | 5 (1.0%) | |||
| Weinreich, et al. [22] (Phase 1–2) |
Non-hospitalized patients with SARS-CoV-2 infection | Casirivimab and imdevimab 2.4 g (n = 92) |
LS mean difference in change from baseline log viral load versus placebo Day 7: −0.25 log10 cp/mL 95% CI −0.60 to 0.10 |
Medically attended visit within 29 days: 3% (3 patients) |
Overall incidence not reported Most common AEs of special interest: vomiting, nausea |
0d | 1 (1%) |
| Casirivimab and imdevimab 8.0 g (n = 90) |
LS mean difference in change from baseline log viral load versus placebo Day 7: −0.56 log10 cp/mL 95% CI −0.91 to −0.21 |
Medically attended visit within 29 days: 3% (3 patients) |
Overall incidence not reported Most common AEs of special interest: abdominal pain, pruritus, urticaria, chills, flushing |
2 (2%)d | 0 | ||
| Placebo (n = 93) |
Medically attended visit within 29 days: 6% (6 patients) |
Overall incidence not reported Most common AEs of special interest: hypertension, hypoxia, vomiting, nausea, rash, dizziness, headache |
1 (1%)d | 2 (2%) | |||
| Ison, et al. [84](Phase 2/3 – Part 1) | Non-hospitalized patients with mild-to-moderate SARS-CoV-2 infection | Regdanvimab 40 mg/kg (n = 100) |
Median (95% CI) time to negativef RT-qPCR, days All: 12.8 (9.00–12.87) Mild infection: 12.72 (8.90–12.89) Moderate infection: 12.75 (8.84–15.78) |
Median (95% CI) time to clinical recovery, days: All: 5.4 (3.97–6.78) Mild infection: 4.37 (2.15–7.67) Moderate infection: 5.73 (4.13–7.33) |
31 (29.5%) Most common treatment-emergent AE related to study drug: hypertriglyceridemia |
1 (1.0%) | 0e |
| Regdanvimab 80 mg/kg (n = 103) |
Median (95% CI) time to negativef RT-qPCR, days All: 11.9 (8.94–12.91) Mild infection: 9.05 (8.85–12.92) Moderate infection: 12.72 (8.89–13.82) |
Median (95% CI) time to clinical recovery, days: All: 6.2 (5.53–7.85) Mild infection: 5.49 (3.15–7.60) Moderate infection: 7.30 (5.58–10.72) |
27 (24.5%) No treatment-emergent AE related to study drug reported in >1 patient |
0 | 0e | ||
| Placebo (n = 104) |
Median (95% CI) time to negativef RT-qPCR, days All: 12.9 (12.69–13.89) Mild infection: 12.95 (8.96–15.84) Moderate infection: 12.87 (10.83–15.83) |
Median (95% CI) time to clinical recovery, days: All: 8.8 (6.72–11.73) Mild infection: 6.88 (4.80–8.78) Moderate infection: 10.81 (6.81–n.c.) |
34 (30.9) Most common treatment-emergent AEs related to study drug: hypertriglyceridemia, infusion-related reaction |
2 (1.8%) | 0e | ||
| Ison, et al. [84](Phase 2/3 – Part 2) | Non-hospitalized patients with mild-to-moderate SARS-CoV-2 infection | Regdanvimab 40 mg/kg (n = 656) |
[Exploratory] Mean change from baseline in viral load titer Day 7: –2.770 log10 cp/mL |
Patients progressing to severe COVID-19 up to day 28 (hospitalization, oxygen therapy or mortality): High-risk patients: 3.1% All patients: 2.4% |
198 (30.4%) Specific AEs not reported |
4 (0.6%) | 4 (0.6%)e |
| Placebo (n = 659) |
[Exploratory] Mean change from baseline in viral load titer Day 7: –2.236 log10 cp/mL |
Patients progressing to severe COVID-19 up to day 28 (hospitalization, oxygen therapy or mortality): High-risk patients: 11.1% All patients: 8.0% |
202 (31.1%) Specific AEs not reported |
7 (1.1%) | 1 (0.2%)e |
All trials conducted prior to the current dominance of the omicron variant.
aMost occurred during infusion, were mild in severity and unrelated to dose.
bUrinary tract infection considered unrelated to study medication.
cUpper abdominal pain considered unrelated to study medication.
dGrade ≥2 within 4 days of infusion.
eTreatment-emergent serious AEs.
fThreshold <2.33 log10 cp/mL.
AE: adverse event; CI: confidence interval; COVID-19: coronavirus disease 2019; cp: copies; ED: emergency department; LS: least-squares; n.c.: not calculable; RT-qPCR: quantitative reverse transcription–polymerase chain reaction.
Despite these promising findings, there are also data pointing to limitations regarding treatment with NAbs. In the ACTIV-3/TICO LY-CoV555 study, the addition of bamlanivimab to remdesivir did not improve the likelihood of a favorable pulmonary outcome in hospitalized patients without end-organ failure [20]. The FDA revoked the EUA for bamlanivimab monotherapy in April 2021 due to concerns regarding an increase in circulating viral variants that were resistant to bamlanivimab alone [70,71]. Combination therapy with bamlanivimab plus etesevimab has been granted an EUA by the FDA and a positive scientific opinion by the EMA’s Committee for Medicinal Products for Human Use [60], for use in adult and pediatric patients (aged ≥12 years and weighing ≥40 kg) with mild-to-moderate disease, a positive SARS-CoV-2 viral test result and high risk of progression to severe COVID-19 and/or hospitalization (Table 2). However, some SARS-CoV-2 variants may be unsusceptible to treatment with bamlanivimab plus etesevimab [72,73]. Both of these antibodies lack neutralization activity against B.1.351 (beta variant) and P.1 (gamma variant). For this reason, the US National Institutes of Health (NIH) recommended that use of bamlanivimab plus etesevimab be stopped temporarily; however, with activity against the recently predominant delta variant, its use has again been considered acceptable where the combined frequency of potentially resistant variants is low [23,76]. Neutralization assays have shown smaller reductions in susceptibility with other variants, such as epsilon, iota and kappa, and the effects on clinical outcomes are not yet known [23]. The omicron variant is predicted to have markedly reduced susceptibility to bamlanivimab plus etesevimab [77].
5.2. Casirivimab plus imdevimab
Casirivimab (REGN10987) and imdevimab (REGN10933), collectively known as REGN-COV2, are two non-competing NAbs that target non-overlapping epitopes on the SARS-CoV-2 S protein [78]. Preclinical studies in rodents and non-human primates have shown that this cocktail of NAbs can reduce viral load in the upper and lower respiratory tract and also reduce virus-induced pathological sequelae, such as interstitial pneumonia [78].
In a randomized, double-blind, placebo-controlled Phase 1–3 clinical trial, data from the first 275 non-hospitalized patients with SARS-CoV-2 infection indicate that REGN-COV2 (at a dose of 2400 mg or 8000 mg intravenously) is associated with a significant reduction in viral titer, when compared with placebo 7 days after treatment initiation (Table 3) [22]. Among patients who were serum-antibody negative at baseline, the difference versus placebo in the change in viral load from day 1 through day 7 was −0.41 log10 copies/mL ± 0.15 (95% CI −0.71 to −0.10) in the overall population (both doses of REGN-COV2) and −0.56 log10 copies/mL ± 0.18 (95% CI −0.91 to −0.21) in those receiving the 8000 mg dose of REGN-COV2. In total, 6% of patients receiving placebo and 3% of those receiving the REGN-COV2 cocktail required one or more medically attended visits (absolute difference vs placebo: −3%; 95% CI −16% to 9%). More recent information from a Phase 3 trial including 4567 patients suggests that casirivimab plus imdevimab may reduce the risk of hospitalization or death [79]. The number of patients who were hospitalized or died was lower with casirivimab plus imdevimab 1200 mg (7 [1%]) versus placebo (24 [3.2%]), corresponding to a risk reduction of 70%. The corresponding risk reduction with the 2400 mg dose was 71%. Median time to COVID-19 symptom resolution was 10 days in patients receiving casirivimab plus imdevimab at doses of 1200 mg or 2400 mg, compared with 14 days in patients receiving placebo [79].
The FDA has issued an EUA for casirivimab plus imdevimab for the treatment of adult and pediatric patients (aged ≥12 years) with mild-to-moderate COVID-19 infection who are at high risk of progressing to severe COVID-19 and/or hospitalization (Table 2) [80]. Casirivimab plus imdevimab was granted marketing authorization by the EMA and is indicated for treatment of COVID-19 in adults and adolescents aged ≥12 years and weighing ≥40 kg who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19 (Table 2) [19].
Data from the R10933-10987-COV-2067 casirivimab plus imdevimab clinical trial indicate that the G446V mutation, which had a 135-fold reduced susceptibility to imdevimab compared with wild-type in a neutralization assay, but which retained susceptibility to casirivimab and the casirivimab plus imdevimab combination, was detected at an allele fraction of ≥15% [24]. The clinical impact of this mutation is currently unknown; thus far it has not been associated with any of the currently identified variants of concern (VOCs) [81]. According to US NIH COVID-19 treatment guidelines, the alpha, beta, gamma and delta VOCs remain susceptible to casirivimab plus imdevimab based on in vitro studies, and no change in clinical activity against these variants is anticipated [14]. The omicron variant is predicted to have markedly reduced susceptibility to casirivimab plus imdevimab [77].
5.3. Regdanvimab
Regdanvimab (CT-P59) is a NAb with activity against various SARS-CoV-2 isolates, including those containing the D614G mutation that is associated with all currently identified VOCs [81]. Complex crystal structure analyses indicate that regdanvimab blocks the interaction regions of the SARS-CoV-2 RBD for the ACE2 receptor. In animal models of SARS-CoV-2 infection, administration of regdanvimab was associated with a reduction in viral load and alleviation of symptoms [82].
Regdanvimab was shown to be well tolerated in Phase 1 studies [83]. In a Phase 1 study in 32 healthy volunteers, adverse events of headache, elevated C-reactive protein level and pyrexia (all grade 1) were reported in two participants within the first 14 days after regdanvimab intravenous infusion [83]. In a Phase 1 study of 18 patients with mild SARS-CoV-2 infection, reduction in viral load was greater with regdanvimab than with placebo among patients with maximum viral loads >105 copies/mL [83]. However, there was no difference in viral load reduction between regdanvimab and placebo in patients with lower viral loads (<105 copies/mL). The mean time to recovery was 3.39 days in patients receiving regdanvimab (three dose-groups combined), compared with 5.25 days among those receiving placebo. The mean times to recovery with regdanvimab 20, 40 and 80 mg/kg were 4.43, 3.21 and 2.52 days, respectively [83].
Data up to 28 days are also available from a two-part, randomized, placebo-controlled, double-blind study that enrolled outpatients with mild-to-moderate COVID-19 [84]. In part 1 of the study, patients received a single dose of regdanvimab 40 mg/kg (n = 101), regdanvimab 80 mg/kg (n = 103) or placebo (n = 103) [84]. For these treatment groups, respectively, median time to undetectable viral load was 12.8, 11.9 and 12.9 days; median time to clinical recovery was 5.35, 6.23 and 8.77 days; and the proportion of patients requiring hospitalization or oxygen therapy was 4.0%, 4.9% and 8.7% (Table 3) [84]. Among the subgroup of patients with moderate SARS-CoV-2 infection aged ≥50 years, 7.5%, 10.0% and 23.7% of those receiving regdanvimab 40 mg/kg, regdanvimab 80 mg/kg and placebo, respectively, required hospitalization or oxygen therapy due to SARS-CoV-2 infection. Based on the results of part 1 of this study, the 40 mg/kg dose was selected. Part 2 of the study involved 1315 patients, of whom 656 were treated with regdanvimab 40 mg/kg and 659 received placebo [84]. In line with results from part 1, regdanvimab significantly reduced the risk of hospitalization, oxygen therapy and mortality due to SARS-CoV-2 infection (Table 3). These events occurred in 3.1% of patients at high risk of progressing to severe COVID-19 who had received regdanvimab 40 mg/kg, compared with 11.1% in the placebo group (risk difference 8.0%; 95% CI 4.5% to 11.7%; p < 0.0001 [primary study endpoint]) [84]. Corresponding results in the overall study cohort were 2.4% and 8.0% (risk difference 5.9%; 95% CI 3.3% to 8.5%; p < 0.0001). The risk reduction was 72% for high-risk patients and 70% for all patients. The median time to clinical recovery in high-risk patients was significantly shorter in the regdanvimab 40 mg/kg group than in the placebo group (9.27 vs >14 days; between-group difference ≥4.73 days; p < 0.0001). In the overall cohort, the median clinical recovery times were 8.38 and 13.25 days, respectively (between-group difference 4.87 days; p < 0.0001). Incidence rates for treatment-emergent adverse events (TEAEs) related to study drug were similar across the treatment groups in both parts of the study [84]. One patient (a recipient of regdanvimab 40 mg/kg in part 2 of the study) experienced a serious TEAE, which did not result in discontinuation.
Regdanvimab received its first full approval on 17 September 2021 in South Korea for the treatment of COVID-19 in adult patients aged >50 years with at least one underlying medical condition (obesity, cardiovascular disease, chronic lung disease, diabetes, chronic kidney disease, chronic liver disease, and patients on immunosuppressive agents) and mild symptoms of COVID-19, and in adult patients with moderate symptoms of COVID-19 [85]. Regdanvimab was granted marketing authorization by the EMA in November 2021 and is indicated for the treatment of patients with COVID-19 who do not require supplemental oxygen therapy and who are at increased risk of progressing to severe COVID-19.
Results from recent in vitro studies show that regdanvimab has reduced binding affinity and reduced potency in pseudovirus neutralization assays against a SARS-CoV-2 RBD triple mutant containing the three mutations that characterize the beta VOC (B.1.351). However, the clinically relevant dose (accounting for differences in human and ferret pharmacokinetics) of regdanvimab effectively decreased the viral load of this variant in the upper and lower respiratory tract in a ferret challenge model, to an extent similar to that seen when using the wild-type virus [86]. In vitro studies assessing the activity of regdanvimab against the delta, epsilon, gamma and kappa variants of SARS-CoV-2 have also been performed, along with delta and gamma variant challenge experiments in mice [87]. Results show that, similar to the beta variant, regdanvimab has neutralizing potency against the delta, epsilon, gamma and kappa variants despite being less active against these variants versus the wild-type virus [87]. Moreover, in a mouse study, regdanvimab reduced mortality, weight loss and respiratory tract viral load associated with the delta and gamma variants [87]. An in vitro examination that specifically focused on the delta variant compared 50% inhibitory concentration (IC50) values of clinical-stage NAbs, including regdanvimab, and demonstrated similar reductions in susceptibility against the delta variant for NAbs, except casirivimab [88]. Despite these discrepancies in neutralizing activity for regdanvimab versus casirivimab, differences in posology (regdanvimab 40 mg/kg [2400 mg for an individual weighing 60 kg] and casirivimab 600 mg for an individual weighing ≥40 kg) may result in minimal effective clinical difference.
Regdanvimab is under investigation as part of a cocktail therapy with the investigational NAb CT-P63. Positive Phase 1 results from CT-P63 included strong neutralizing activity against the omicron variant based on X-ray crystallography and pseudo-virus testing (unpublished data) [89].
5.4. Sotrovimab
Sotrovimab (VIR-7831) is an investigational dual-action SARS-CoV-2 NAb with the potential to both block viral entry into healthy cells and clear infected cells [74]. VIR-7831 was derived from a parent antibody isolated from a recovered patient who had been infected with SARS-CoV in 2003.
The randomized, double-blind, placebo-controlled Phase 3 study (COMET-ICE; NCT04545060), evaluated sotrovimab in adults with SARS-CoV-2 infection at high risk of hospitalization [90]. The primary efficacy outcome was hospitalization for >24 hours for any cause or death within 29 days after randomization. In a prespecified interim analysis of 583 patients, 3 patients (1%) in the sotrovimab group and 21 patients (7%) in the placebo group had disease progression leading to hospitalization or death (relative risk reduction 85%, 97.24% CI: 44 to 96; p = 0.002). All five patients who were admitted to intensive care were in the placebo group. Additionally, fewer patients in the sotrovimab group required emergency department visits without hospitalization (or with hospitalization for <24 hours) compared with the placebo group. Adverse events were reported in 17% of sotrovimab-treated patients and 19% of placebo-treated patients, and serious adverse events were more common in the placebo group (6%) than in the sotrovimab group (2%) [90].
The EMA has completed a pre-authorization review of sotrovimab and recommended that, prior to marketing authorization, sotrovimab can be used to treat confirmed COVID-19 in adults and adolescents (aged ≥12 years and weighing ≥40 kg) who do not require supplemental oxygen therapy and are at risk of progressing to severe COVID-19 [12].
The FDA has issued an EUA for sotrovimab for the treatment of mild-to-moderate COVID-19 in adults and pediatric patients (aged ≥12 years and weighing ≥40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk of progression to severe COVID-19, including hospitalization or death [74].
In vitro studies have shown sotrovimab to retain activity against variants of interest and concern, including the alpha, beta, delta, gamma and lambda variants [14,90]. Sotrovimab is the only currently available NAb anticipated to have activity against the omicron variant [77].
6. Pharmacoeconomics
Pharmacoeconomic studies have begun to evaluate the cost of treating COVID-19. The cost of treating patients with severe disease who require admission to the intensive care unit (ICU) or invasive mechanical ventilation (IMV) is substantially higher than for patients with mild-to-moderate disease who do not require ICU admission or IMV [91–93]. Moreover, a recent analysis showed that therapies reducing the length of hospital stay, COVID-related mortality and incidence of mechanical ventilation are likely to be cost-effective [94]. Despite a lack of pharmacoeconomic data specific for NAbs, clinical trials have consistently shown a lower risk of hospitalization or death and a shorter time to symptom resolution and/or clinical recovery in patients treated with NAbs versus controls [79,83,84]. Thus, NAbs may contribute to reducing healthcare costs associated with hospitalization and intensive care admission, as well as the considerable burden on the healthcare system posed by severe and critical cases of COVID-19.
7. Discussion
Therapeutic intervention during the early stages of the disease – during which viral replication and infection are the primary factors driving disease progression – represents an early opportunity to alter the course of the disease prior to the onset of post-viral inflammatory and immune responses. Impacts of the post-acute phase of COVID-19 (commonly known as long COVID) can also be considerable. Estimates of the proportion of patients who experience persistent symptoms vary; it may range between 33% and 87% in hospitalized patients, and as many as two-thirds of patients with non-critical disease may be affected [95]. This significant health burden might be reduced by effectively treating early disease. As summarized in the present article, NAbs are well suited to early therapeutic intervention and will likely play an increasingly important role as the pandemic continues to evolve.
Concerns around the potential for SARS-CoV-2 variants to have an increased ability to spread and escape from immunity have prompted researchers to investigate ways to combat the evolution of viral variants.
RNA viruses such as SARS-CoV-2 tend to acquire mutations quickly because of the poorer proofreading ability of the RNA polymerase enzymes in comparison with that seen in DNA viruses [96,97]. Coronaviruses mutate more slowly than most other RNA viruses, likely because of the proofreading function of the exonuclease nsp14 that corrects potentially fatal RNA copying mistakes [96,98]. Nevertheless, by April 2021, researchers had described more than 1 million SARS-CoV-2 genome sequences, with mutations occurring particularly in the S protein RBD [99–101]. Although many mutations will not influence the ability of the virus to spread or cause disease [96], mutations that affect the RBD of SARS-CoV-2 are of particular concern, as this RBD is the molecular target for many vaccines and antibody-based treatments for SARS-CoV-2 infection [102].
Several of the World Health Organization (WHO)-defined VOCs have mutations in the RBD, including the alpha variant, which originated in the UK; the beta variant, which originated in South Africa; the gamma variant, which originated in Brazil; the delta variant, which originated in India; and the most recently identified omicron variant, which originated in multiple countries [100,103]. Similarly, RBD mutations are also seen in WHO-defined variants of interest (VOIs), lambda (originating in Peru) and mu (Colombia) [103]. The SARS-CoV-2 D614G mutation, which affects S1 and has now spread globally, is associated with increased infectivity and transmission, and its incidence has increased over time. As of December 2021, the D614G mutation is present in all WHO VOCs (alpha, beta, gamma, delta and omicron) and in both VOIs (lambda and mu) [5,99,104]. The NAbs currently approved or authorized for emergency use for the treatment of COVID-19 show variability with respect to their activity against the different VOCs, as summarized in section 5.
In addition to naturally occurring VOCs, the development of drug-resistant variants of SARS-CoV-2 may be promoted by selective pressure from the treatment of COVID-19 with NAbs [59]. Clinical trials of bamlanivimab monitored SARS-CoV-2 strains for S protein variations potentially associated with drug resistance (as previously identified in non-clinical serial resistance and directed evolution studies) [18,59]. In the BLAZE-1 study, known bamlanivimab-resistant variants were identified at baseline [59], and an exploratory analysis of ongoing viral sequencing identified putative bamlanivimab-resistant variants in all treatment groups, including placebo. However, the clinical significance of these resistant variants remains unclear, as several clinical outcomes were comparable in the combination and monotherapy groups despite a larger reduction in viral load being observed in the combination group. Notably, the bamlanivimab monotherapy groups had a higher proportion of patients with a variant detected at more than one time point during the viral time course (4.1–7.2%) than did the placebo or combination bamlanivimab and etesevimab group (both 0%) [18]. The latter finding supports the notion that using NAbs in combination (cocktail therapy) has the potential to thwart the substantial threat posed by antibody resistance if combinations that bind non-overlapping epitopes are used, because mutations are unlikely to occur simultaneously at two distinct genetic sites [64]. A recent study mapped mutations to the S protein RBD that escape antibody recognition [105]. Interestingly, the findings implied that antibody cocktails may not have to target distinct regions of the RBD to resist viral escape, as several cocktails of antibodies that compete for binding on the S protein but have different escape mutations remained resistant to viral escape. Furthermore, the authors postulated that such cocktails may even be preferable to those targeting distinct regions, because the acquisition of multiple different escape mutations for the ACE2-binding interface could impose important loss of receptor binding on the virus [105].
The epitopes for casirivimab and imdevimab are non-overlapping, providing rationale for their combination, and the binding affinities of these NAbs to SARS-CoV-2 RBD are concentration dependent, with 50% effective concentration (EC50) values in the sub-nanomolar range [106]. In contrast, bamlanivimab and etesevimab bind two different but overlapping epitopes of the SARS-CoV-2 RBD and consequently compete with one another for binding of the S protein [107]. Binding affinities of bamlanivimab and etesevimab for the SARS-CoV-2 RBD were in the low nanomolar range, with reported equilibrium dissociation constants (KD) of 1.5 nM and 46.5 nM, respectively [107]. Regdanvimab binds with picomolar affinity (EC50 4.4 ng/mL corresponding to 3.0 × 10–11 M) to regions of the SARS-CoV-2 RBD that interact with the ACE2 receptor [108]. However, the binding orientation of regdanvimab to the RBD is different from that of other NAbs, suggesting that the epitopes recognized by regdanvimab and the steric hindrance afforded by binding are distinct to those of other NAbs [82]. Finally, sotrovimab binds with high affinity (KD 0.21 nM) to a highly conserved epitope in the SARS-CoV-2 RBD that is outside of the RBM [109]. These differences between NAbs could offer potential alternative treatment options in the event of resistance to any single agent or combination of agents.
In addition to the potential development of drug-resistant variants, there has been a theoretical concern that antibody-based therapies may exacerbate COVID-19 severity via antibody-dependent enhancement (ADE) [110], another area of focus for researchers. ADE in viral infections can occur through two mechanisms: by increasing viral uptake and replication in Fcγ receptor-expressing immune cells, and through increased immune activation by Fc-mediated effector function or immune complex formation [59,110]. ADE can be mediated by both pathways when antibodies bind to viral antigens without blocking or clearing infection (e.g. with non-NAbs or antibodies at subneutralizing levels) [110]. There is no evidence of ADE with SARS-CoV-2 infection and NAb treatment, but measures to mitigate the theoretical risk are possible (e.g. modifying the Fc region of the antibody so that it cannot elicit effector immune responses) [59,110].
While all NAbs currently authorized for the treatment of COVID-19 can be administered intravenously, several can be administered via subcutaneous injection [62,63], and others currently under development are administered intramuscularly [111]. Early studies are also showing promising results using nebulized NAb formulations [112]. As we have postulated, NAb therapy is likely to have the greatest effect early during infection, when viral replication and infection drive disease progression [59–61]. Simplifying administration of these therapies raises the intriguing possibility of earlier treatment availability in less specialized healthcare facilities, general practitioner offices or maybe even in the community setting, as self-administration.
Given the constantly changing situation regarding SARS-CoV-2 variants, it is important for physicians to refer to the most up-to-date information issued by organizations such as the WHO, EMEA, FDA and US Centers for Disease Control and Prevention when making prescribing decisions.
8. Conclusion
The magnitude of the global COVID-19 pandemic, coupled with the expansive disease manifestation of this infection, mandates broadening the scope of treatment approaches. Experience has shown that appropriate management in the early stages of disease is crucial, because in later stages the inflammatory response predominates and the evolution of disease becomes unpredictable. An increasing body of evidence suggests that NAbs represent an effective treatment option that may be used alongside other therapies in the management of SARS-CoV-2 infection. Further clinical evaluation and real-world evaluation of these agents is needed to optimize their clinical use, particularly among high-risk patients, and to help reduce the global burden of COVID-19.
9. Expert opinion
There is an ongoing, pressing need, recognized by regulators, to bolster the range of treatments available for the prevention and treatment of SARS-CoV-2 infection, with emerging evidence pointing to the value of NAbs. Indeed, as experience with treating COVID-19 grows, so does confidence in being able to predict which patients are most likely to progress to severe disease and, therefore, are most likely to benefit from NAbs. In the same way, recognizing and proactively treating immunocompromised patients will likely be essential to avoid the development of serious and persistent disease with successive acute viral episodes.
As the pandemic continues, the risk of resistant variants increases; over the coming years, combination therapy with NAbs is likely to play a role in minimizing this risk. However, a number of important questions remain to be answered.
Most of the clinical trials on which regulatory submissions and subsequent EUAs and approvals were based specifically excluded vaccinated individuals. From a pathophysiological standpoint, administering anti-S protein-targeted NAbs to previously vaccinated individuals is not anticipated to pose major risks. Furthermore, given the increasing number of breakthrough infections [113], it will be important to evaluate the use of NAbs in vaccinated individuals. Therefore, it is imperative that data on the safety and efficacy of NAbs in this patient population are collected from real-world studies based on routine clinical practice.
Early indications suggest that several mAb therapies have reduced effectiveness against the omicron variant [114]. This has most likely emerged as part of natural evolutionary pressure, and not as drug-induced resistance, since the clinical trials would have been conducted prior to the circulation of the omicron variant. As such, the same evolutionary pressure could allow reversion of at least part of the S protein mutations that now cause reduced binding affinity. Genetic surveillance of the dominant variants circulating locally is becoming paramount to allow tailored treatment decisions for each setting and, potentially, for each patient. This is particularly relevant when variant replacement is occurring, and when both the previous and current dominant variants are co-circulating, especially if the circulating variants display different susceptibilities to the available NAbs (as is the case with the omicron and delta variants). Prompt serology and sequencing are also becoming increasingly important when deciding on fast and effective NAb treatments for unvaccinated or severely immunocompromised patients [115].
Recurrent infection with SARS-CoV-2 has been reported in a number of patients, although recurrent infection rates were relatively low (estimated to occur in 0.04% of all patients with COVID-19 in one US study [116]) prior to the emergence of the omicron variant. NAbs may play a role in treating patients with recurrent infections; however, treatment algorithms remain to be established. NAbs could also provide a therapeutic option for immunocompromised patients who fail to respond to vaccination and are at risk of recurrent infections and prolonged infections [117].
The potential role of NAbs in preventing post-acute phase COVID-19 (i.e. long COVID) also remains to be elucidated. The UK ZOE symptom study estimated that approximately 15% of people experiencing symptomatic COVID-19 went on to suffer long COVID with a duration of ≥4 weeks, and a smaller proportion continued to experience symptoms for longer durations (e.g. 2.2% for ≥12-weeks) [118]. The strongest predictors for long COVID of duration ≥4 weeks were increasing age and the number of symptoms experienced during the first week [118].
Reflecting on the immense scientific gains achieved in the first 2 years of the COVID era in terms of our knowledge base and array of available interventions, we anticipate that in the next 5 years, the range of effective treatment options will continue to grow, and early treatment of infected patients in whom disease progression is likely will become commonplace. Optimal use of NAbs alongside other interventions for COVID-19 will be informed by future clinical trial data and real-world evidence as this become available.
Acknowledgments
Medical writing support (including development of a draft outline and subsequent drafts in consultation with the authors, assembling tables and figures, collating author comments, copyediting, fact checking and referencing) was provided by Duncan Campbell, PhD, CMPP, at Aspire Scientific (Bollington, UK), and was funded by Celltrion Healthcare Co., Ltd. (Incheon, Republic of Korea).
Funding Statement
This paper was not funded.
Article highlights
With an urgent need for effective treatments for COVID-19, regulatory agencies have adopted policies for the accelerated review and approval of COVID-19 treatments.
Neutralizing antibodies (NAbs) bind specifically to important components of SARS-CoV-2, and clinical trials have shown NAbs to have therapeutic potential.
In a randomized, placebo-controlled Phase 3 study, regdanvimab reduced the risk of hospitalization, oxygen therapy and mortality by 72% versus placebo in patients at high risk of progression to severe COVID-19.
In a randomized, placebo-controlled Phase 3 study, casirivimab plus imdevimab 1200 mg reduced the risk of hospitalization or death by 70% versus placebo in high-risk outpatients; the corresponding risk reduction with casirivimab plus imdevimab 2400 mg was 71%.
The randomized, placebo-controlled Phase 3 COMET-ICE study demonstrated an 85% relative risk reduction for hospitalization or death with sotrovimab versus placebo in adults with SARS-CoV-2 infection at high risk of hospitalization.
Data from the Phase 3 cohort of the BLAZE-1 study demonstrated reduced rates of COVID-19-related hospitalization or death from any cause with bamlanivimab plus etesevimab (2.1%) compared with placebo (7.0%) in patients with SARS-CoV-2 infection.
This box summarizes key points contained in the article.
Declaration of interest
EMR has been an investigator in clinical trials with Celltrion, Inc. and Pfizer, and has made presentations for Merck Sharp & Dohme and Pfizer. DC, SK and SY are employees of Celltrion Healthcare Co., Ltd. OS has been an investigator in COVID-19 clinical trials by Algernon Pharmaceuticals, Atea Pharmaceuticals, Diffusion Pharmaceuticals, Regeneron Pharmaceuticals, PureTech and Celltrion, Inc.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in, or financial conflict with, the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.World Health Organization . WHO Coronavirus (COVID-19) dashboard. 2021. [cited 2021 May 23]; Available from: https://covid19.who.int/
- 2.Fried MW, Crawford JM, Mospan AR, et al. Patient characteristics and outcomes of 11,721 patients with coronavirus disease 2019 (COVID-19) hospitalized across the United States. Clin Infect Dis. 2021;72(10):e558–e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim PS, Read SW, Fauci AS.. Therapy for early COVID-19: a critical need. JAMA. 2020;324(21):2149–2150. [DOI] [PubMed] [Google Scholar]
- 4.Liu CH, Lu CH, Wong SH, et al. Update on antiviral strategies against COVID-19: unmet needs and prospects. Front Immunol. 2020;11:616595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization . SARS-CoV-2 variants. 2020. [cited 2021 May 23]; Available from: https://www.who.int/emergencies/disease-outbreak-news/item/2020-DON305
- 6.U.S. Food and Drug Administration . COVID-19 public health emergency: general considerations for pre-IND meeting requests for COVID-19 related drugs and biological products. Guidance for industry and investigators. 2020 [cited 2021 May 26]; Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/covid-19-public-health-emergency-general-considerations-pre-ind-meeting-requests-covid-19-related
- 7.Department of Health and Human Services . Emergency use authorization declaration. A Notice by the Health and Human Services Department on 04/01/2020. 2020. [cited 2021 May 26]; Available from: https://www.federalregister.gov/documents/2020/04/01/2020-06905/emergency-use-authorization-declaration
- 8.U.S. Food and Drug Administration . Coronavirus Treatment Acceleration Program (CTAP). 2021. [cited 2021 Oct 26]; Available from: https://www.fda.gov/drugs/coronavirus-covid-19-drugs/coronavirus-treatment-acceleration-program-ctap; • FDA-accelerated program designed for expedited review.
- 9.European Medicines Agency . COVID-19 treatments: research and development 2021 [cited 2021 Nov 8]; Available from: https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines/treatments-covid-19/covid-19-treatments-research-development
- 10.European Medicines Agency . COVID-19 treatments: under evaluation. 2021. [cited 2021 Nov 8]; Available from: https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines/treatments-covid-19/covid-19-treatments-under-evaluation; • EMA task force for rolling review of SARS-CoV-2 therapeutics.
- 11.European Medicines Agency . COVID-19 treatments: article 5 (3)reviews. 2021. [cited 2021 Jul 10]; Available from: https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines/treatments-covid-19/covid-19-treatments-article-53-reviews
- 12.European Commission . European Health Union: commission establishes portfolio of 10 most promising treatments for COVID-19. 2021. [cited 2021 Dec 13]; Available from: https://cyprus.representation.ec.europa.eu/news/european-health-union-commission-establishes-portfolio-10-most-promising-treatments-covid-19-2021-10-22_en
- 13.European Commission . Questions and answers on the list of 10 candidate COVID-19 therapeutics. 2021. [cited 2021 Dec 13]; Available from: https://ec.europa.eu/commission/presscorner/detail/en/qanda_21_5367
- 14.National Institutes of Health . Coronavirus disease 2019 (COVID-19) treatment guidelines. 2022. [cited 2021 Jan 10]; Available from: https://www.covid19treatmentguidelines.nih.gov/ [PubMed]
- 15.Chalmers JD, Crichton ML, Goeminne PC, et al. Management of hospitalised adults with coronavirus disease 2019 (COVID-19): a European Respiratory Society living guideline. Eur Respir J. 2021;57(4):2100048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.World Health Organization . Therapeutics and COVID-19: living guideline. 2021 Jul 6 [cited 2021 Aug 31]; Available from: https://www.who.int/publications/i/item/WHO-2019-nCoV-therapeutics-2021.2 [PubMed]
- 17.Bhimraj A, Morgan R, Hirsch Shumaker A, et al. IDSA guidelines on the treatment and management of patients with COVID-19. Version 6.0.0. 2021 [cited 2022 Jan 25]; Available from: https://www.idsociety.org/practice-guideline/covid-19-guideline-treatment-and-management/
- 18.Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA. 2021;325(7):632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Pivotal trial of bamlanivimab as monotherapy.
- 19.European Medicines Agency . Casirivimab plus imdevimab: Summary of Product Characteristics. 2021. [cited 2021 Nov 30]; Available from: https://www.ema.europa.eu/en/documents/product-information/ronapreve-epar-product-information_en.pdf
- 20.ACTIV-3/TICO LY-CoV555 Study Group ; Lundgren JD, Grund B, Barkauskas CE, et al. A neutralizing monoclonal antibody for hospitalized patients with Covid-19. N Engl J Med. 2021;384(10):905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen P, Nirula A, Heller B, et al. SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with Covid-19. N Engl J Med. 2021;384(3):229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinreich DM, Sivapalasingam S, Norton T, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with Covid-19. N Engl J Med. 2021;384(3):238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Pivotal trial of casirivimab/imdevimab combination.
- 23.Lilly E and Company . Fact sheet for health care providers. Emergency use authorization (EUA) of bamlanivimab and etesevimab. 2021. [cited 2021 Jul 07]; Available from: https://www.fda.gov/media/145802/download
- 24.Regeneron . Fact sheet for health care providers. Emergency use authorization (EUA) of REGEN-COVTM (casivirimab and imdevimab). 2021 [cited 2021 Sep 16]; Available from: https://www.fda.gov/media/145611/download
- 25.European Medicines Agency . Conditions of use, conditions for distribution and patients targeted and conditions for safety monitoring addressed to member states for unauthorised product Regkirona (regdanvimab). 2021. [cited 2021 Jul 11]; Available from: https://www.ema.europa.eu/en/documents/referral/celltrion-use-regdanvimab-treatment-covid-19-article-53-procedure-conditions-use-conditions_en.pdf
- 26.European Medicines Agency . Regdanvimab: Summary of Product Characteristics. 2021. [cited 2021 Nov 30]; Available from: https://www.ema.europa.eu/en/documents/product-information/regkirona-epar-product-information_en.pdf
- 27.Spinner CD, Gottlieb RL, Criner GJ, et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. JAMA. 2020;324(11):1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldman JD, Lye DCB, Hui DS, et al. Remdesivir for 5 or 10 days in patients with severe Covid-19. N Engl J Med. 2020;383(19):1827–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.RECOVERY Collaborative Group; Horby P, Lim WS, Emberson JR, et al. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. 2021;384(8):693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomazini BM, Maia IS, Cavalcanti AB, et al. Effect of dexamethasone on days alive and ventilator-free in patients with moderate or severe acute respiratory distress syndrome and COVID-19: the CoDEX randomized clinical trial. JAMA. 2020;324(13):1307–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalil AC, Patterson TF, Mehta AK, et al. Baricitinib plus remdesivir for hospitalized adults with Covid-19. N Engl J Med. 2021;384(9):795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soin AS, Kumar K, Choudhary NS, et al. Tocilizumab plus standard care versus standard care in patients in India with moderate to severe COVID-19-associated cytokine release syndrome (COVINTOC): an open-label, multicentre, randomised, controlled, phase 3 trial. Lancet Respir Med. 2021;9(5):511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosas IO, Brau N, Waters M, et al. Tocilizumab in hospitalized patients with severe Covid-19 pneumonia. N Engl J Med. 2021;384(16):1503–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kyriazopoulou E, Poulakou G, Milionis H, et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat Med. 2021;27(10):1752–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.European Medicines Agency . Anakinra: Summary of Product Characteristics. 2021. [cited 2022 Jan 4]; Available from: https://www.ema.europa.eu/en/documents/product-information/kineret-epar-product-information_en.pdf
- 36.Gordon CJ, Tchesnokov EP, Schinazi RF, et al. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J Biol Chem. 2021;297(1):100770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou S, Hill CS, Sarkar S, et al. beta-d-N4-hydroxycytidine inhibits SARS-CoV-2 through lethal mutagenesis but is also mutagenic to mammalian cells. J Infect Dis. 2021;224(3):415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabinger F, Stiller C, Schmitzova J, et al. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat Struct Mol Biol. 2021;28(9):740–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caraco Y, Crofoot GE, Moncada PA, et al. Phase 2/3 trial of molnupiravir for treatment of Covid-19 in nonhospitalized adults. NEJM Evid. 2021. Dec 16. DOI: 10.1056/EVIDoa2100043. [DOI] [PubMed] [Google Scholar]
- 40.Jayk Bernal A, Gomes da Silva MM, Musungaie DB, et al. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N Engl J Med. 2021;20211216. DOI: 10.1056/NEJMoa2116044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.European Medicines Agency . EMA issues advice on use of Lagevrio (molnupiravir) for the treatment of COVID-19. 2021. [cited 2021 Dec 13]; Available from: https://www.ema.europa.eu/en/news/ema-issues-advice-use-lagevrio-molnupiravir-treatment-covid-19
- 42.U.S. Food and Drug Administration . Fact sheet for healthcare providers: emergency use authorization for molnupiravir. 2021. [cited 2022 Jan 4]; Available from: https://www.fda.gov/media/155054/download
- 43.U.S. Food and Drug Administration . Fact sheet for healthcare providers: emergency use authorization for Paxlovid. 2021. [cited 2022 Jan 4]; Available from: https://www.fda.gov/media/155050/download
- 44.Pfizer . Pfizer’s novel COVID-19 oral antiviral treatment candidate reduced risk of hospitalization or death by 89% in interim analysis of phase 2/3 EPIC-HR study. 2021. [cited 2021 Nov 8]; Available from: https://www.pfizer.com/news/press-release/press-release-detail/pfizers-novel-covid-19-oral-antiviral-treatment-candidate
- 45.Atea Pharmaceuticals . Atea pharmaceuticals provides update and topline results for phase 2 MOONSONG trial evaluating AT-527 in the outpatient setting. 2021. [cited 2021 Nov 8]; Available from: https://ir.ateapharma.com/news-releases/news-release-details/atea-pharmaceuticals-provides-update-and-topline-results-phase-2
- 46.Gentile P, Sterodimas A. Adipose-derived stromal stem cells (ASCs) as a new regenerative immediate therapy combating coronavirus (COVID-19)-induced pneumonia. Expert Opin Biol Ther. 2020;20(7):711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leng Z, Zhu R, Hou W, et al. Transplantation of ACE2(-) mesenchymal stem cells improves the outcome of patients with COVID-19 pneumonia. Aging Dis. 2020;11(2):216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu R, Yan T, Feng Y, et al. Mesenchymal stem cell treatment improves outcome of COVID-19 patients via multiple immunomodulatory mechanisms. Cell Res. 2021;20211026. DOI: 10.1038/s41422-021-00573-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lanzoni G, Linetsky E, Correa D, et al. Umbilical cord mesenchymal stem cells for COVID-19 acute respiratory distress syndrome: a double-blind, phase 1/2a, randomized controlled trial. Stem Cells Transl Med. 2021;10(5):660–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gentile P, Sterodimas A, Pizzicannella J, et al. Research progress on mesenchymal stem cells (MSCs), adipose-derived mesenchymal stem cells (AD-MSCs), drugs, and vaccines in inhibiting COVID-19 disease. Aging Dis. 2020;11(5):1191–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Satarker S, Nampoothiri M. Structural proteins in severe acute respiratory syndrome coronavirus-2. Arch Med Res. 2020;51(6):482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11(1):1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abraham J. Passive antibody therapy in COVID-19. Nat Rev Immunol. 2020;20(7):401–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Du L, Yang Y, Zhang X. Neutralizing antibodies for the prevention and treatment of COVID-19. Cell Mol Immunol. 2021;18(10):2293–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barnes CO, Jette CA, Abernathy ME, et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588(7839):682–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Valdez-Cruz NA, Garcia-Hernandez E, Espitia C, et al. Integrative overview of antibodies against SARS-CoV-2 and their possible applications in COVID-19 prophylaxis and treatment. Microb Cell Fact. 2021;20(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar S, Chandele A, Sharma A. Current status of therapeutic monoclonal antibodies against SARS-CoV-2. PLoS Pathog. 2021;17(9):e1009885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taylor PC, Adams AC, Hufford MM, et al. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat Rev Immunol. 2021;21(6):382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boechat JL, Chora I, Morais A, et al. The immune response to SARS-CoV-2 and COVID-19 immunopathology - Current perspectives. Pulmonology. 2021;27(5):423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khadke S, Ahmed N, Ahmed N, et al. Harnessing the immune system to overcome cytokine storm and reduce viral load in COVID-19: a review of the phases of illness and therapeutic agents. Virol J. 2020;17(1):154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu H, Yuan M, Huang D, et al. A combination of cross-neutralizing antibodies synergizes to prevent SARS-CoV-2 and SARS-CoV pseudovirus infection. Cell Host Microbe. 2021;29(5):806–818.e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pandey M, Ozberk V, Eskandari S, et al. Antibodies to neutralising epitopes synergistically block the interaction of the receptor-binding domain of SARS-CoV-2 to ACE 2. Clin Transl Immunol. 2021;10(3):e1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baum A, Fulton BO, Wloga E, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369(6506):1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suryadevara N, Shrihari S, Gilchuk P, et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell. 2021;184(9):2316–2331.e2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jones BE, Brown-Augsburger PL, Corbett KS, et al. The neutralizing antibody, LY-CoV555, protects against SARS-CoV-2 infection in nonhuman primates. Sci Transl Med. 2021;13(593):eabf1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cohen MS, Nirula A, Mulligan MJ, et al. Effect of bamlanivimab vs placebo on incidence of COVID-19 among residents and staff of skilled nursing and assisted living facilities: a randomized clinical trial. JAMA. 2021;326(1):46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Brien MP, Forleo-Neto E, Musser BJ, et al. Subcutaneous REGEN-COV antibody combination to prevent Covid-19. N Engl J Med. 2021;385(13):1184–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu X, Li N, Wang G, et al. Tolerability, safety, pharmacokinetics, and immunogenicity of a novel SARS-CoV-2 neutralizing antibody, etesevimab in Chinese healthy adults: a randomized, double-blind, placebo-controlled, first-in-human phase 1 study. Antimicrob Agents Chemother. 2021;65(8):e0035021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dougan M, Nirula A, Azizad M, et al. Bamlanivimab plus etesevimab in mild or moderate Covid-19. N Engl J Med. 2021;385(15):1382–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.U.S. Food and Drug Administration . Coronavirus (COVID-19) update: FDA revokes emergency use authorization for monoclonal antibody bamlanivimab. 2021 [cited 2021 May 23]; Available from: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-revokes-emergency-use-authorization-monoclonal-antibody-bamlanivimab
- 72.Hoffmann M, Arora P, Gross R, et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell. 2021;184(9):2384–2393.e2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Starr TN, Greaney AJ, Dingens AS, et al. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep Med. 2021;2(4):100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.European Medicines Agency . EMA issues advice on use of sotrovimab (VIR-7831) for treating COVID-19. 2021. [cited 2021 May 26]; Available from: https://www.ema.europa.eu/en/news/ema-issues-advice-use-sotrovimab-vir-7831-treating-covid-19
- 75.U.S. Food and Drug Administration . Emergency use authorization (EUA) for emergency use of sotrovimab. 2021 [cited 2021; Available from: https://www.fda.gov/media/149534/download
- 76.National Institutes of Health . The COVID-19 Treatment Guidelines Panel’s statement on bamlanivimab plus etesevimab for the treatment of mild to moderate COVID-19 in nonhospitalized patients. 2021. [cited 2021 Sep 20]; Available from: https://www.covid19treatmentguidelines.nih.gov/therapies/statement-on-bamlanivimab-plus-etesevimab/
- 77.National Institutes of Health . The COVID-19 Treatment Guidelines Panel’s statement on therapies for high-risk, nonhospitalized patients with mild to moderate COVID-19. 2021. [cited 2021 Dec 10]; Available from: https://www.covid19treatmentguidelines.nih.gov/therapies/statement-on-therapies-for-high-risk-nonhospitalized-patients/
- 78.Baum A, Ajithdoss D, Copin R, et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science. 2020;370(6520):1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weinreich DM, Sivapalasingam S, Norton T, et al. REGEN-COV antibody combination and outcomes in outpatients with Covid-19. N Engl J Med. 2021;385(23):e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.U.S. Food and Drug Administration . Emergency use authorization (EUA) for emergency use of casirivimab and imdevimab. 2020 [cited 2021 May 26]; Available from: https://www.fda.gov/media/143891/download
- 81.Centers for Disease Control and Prevention . SARS-CoV-2 variant classifications and definitions. 2021. [cited 2021 Aug 31]; Available from: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html#
- 82.Kim C, Ryu DK, Lee J, et al. A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021;12(1):288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim JY, Jang YR, Hong JH, et al. Safety, virologic efficacy, and pharmacokinetics of CT-P59, a neutralizing monoclonal antibody against SARS-CoV-2 spike receptor-binding protein: two randomized, placebo-controlled, phase i studies in healthy individuals and patients with mild SARS-CoV-2 infection. Clin Ther. 2021;43(10):1706–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ison MG, Kim JY, Sandulescu O, et al. Therapeutic effect of regdanvimab in patients with mild to moderate COVID-19: day 28 results from a multicentre, randomised, controlled pivotal trial. Presented at: the 31st European Congress of Clinical Microbiology and Infectious Diseases (ECCMID); 2021 Jul 9–12; Vienna, Austria, Abstract 4650. [Google Scholar]; •• Pivotal trial for regdanvimab.
- 85.Syed YY. Regdanvimab: first approval. Drugs. 2021;81(18):2133–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ryu DK, Song R, Kim M, et al. Therapeutic effect of CT-P59 against SARS-CoV-2 South African variant. Biochem Biophys Res Commun. 2021;566:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ryu DK, Kang B, Noh H, et al. The in vitro and in vivo efficacy of CT-P59 against Gamma, Delta and its associated variants of SARS-CoV-2. Biochem Biophys Res Commun. 2021;578:91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Efficacy results for regdanvimab against delta and gamma variants.
- 88.Mlcochova P, Kemp S, Dhar MS, et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature. 2021;599:114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Sensitivity of delta variant to clinical-stage neutralizing antibodies.
- 89.Celltrion Healthcare [press release] . Celltrion announces positive results for its cocktail therapy candidates including neutralisation data against Omicron variant. 2022. [cited 2022 Jan 10]; Available from: https://www.celltrionhealthcare.com/en-us/board/newsdetail?modify_key=556&pagenumber=1&keyword=&keyword_type=
- 90.Gupta A, Gonzalez-Rojas Y, Juarez E, et al. Early treatment for Covid-19 with SARS-CoV-2 neutralizing antibody sotrovimab. N Engl J Med. 2021;385(21):1941–1950. [DOI] [PubMed] [Google Scholar]; •• Pivotal trial of sotrovimab.
- 91.Di Fusco M, Shea KM, Lin J, et al. Health outcomes and economic burden of hospitalized COVID-19 patients in the United States. J Med Econ. 2021;24(1):308–317. [DOI] [PubMed] [Google Scholar]
- 92.Pulse . COVID-19 treatment in Korea cost $57,562 per critically-ill, bill charged on state. 2020 [cited 2021 Jun 25]; Available from: https://pulsenews.co.kr/view.php?year=2020&no=471089
- 93.Lee D, Choi B. Policies and innovations to battle Covid-19 - A case study of South Korea. Health Policy Technol. 2020;9(4):587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sheinson D, Dang J, Shah A, et al. A cost-effectiveness framework for COVID-19 treatments for hospitalized patients in the United States. Adv Ther. 2021;38(4):1811–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nalbandian A, Sehgal K, Gupta A, et al. Post-acute COVID-19 syndrome. Nat Med. 2021;27(4):601–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Callaway E. The coronavirus is mutating - does it matter? Nature. 2020;585(7824):174–177. [DOI] [PubMed] [Google Scholar]
- 97.Carey LB. RNA polymerase errors cause splicing defects and can be regulated by differential expression of RNA polymerase subunits. elife. 2015;4:e09945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Robson F, Khan KS, Le TK, et al. Coronavirus RNA proofreading: molecular basis and therapeutic targeting. Mol Cell. 2020;79(5):710–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Troyano-Hernáez P, Reinosa R, Holguín Á. Evolution of SARS-CoV-2 envelope, membrane, nucleocapsid, and spike structural proteins from the beginning of the pandemic to September 2020: a global and regional approach by epidemiological week. Viruses. 2021;13(2):243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Plante JA, Mitchell BM, Plante KS, et al. The variant gambit: COVID-19ʹs next move. Cell Host Microbe. 2021;29(4):508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tai W, He L, Zhang X, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol. 2020;17(6):613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dai L, Gao GF. Viral targets for vaccines against COVID-19. Nat Rev Immunol. 2021;21(2):73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.World Health Organization . Tracking SARS-CoV-2 variants. 2021. [cited 2021 Dec 14]; Available from: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/
- 104.Stanford University Coronavirus Antiviral & Resistance Database . SARS-CoV-2 variants. 2021. [cited 2021 Sep 16]; Available from: https://covdb.stanford.edu/page/mutation-viewer/
- 105.Greaney AJ, Starr TN, Gilchuk P, et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe. 2021;29(1):44–57. e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.European Medicines Agency . Casirivimab and imdevimab: assessment report. 2021. [cited 2021 Nov 30]; Available from: https://www.ema.europa.eu/en/documents/referral/regn-cov2-antibody-combination-casirivimab/imdevimab-covid19-article-53-procedure-assessment-report_en.pdf
- 107.European Medicines Agency . Bamlanivimab and etesevimab: assessment report. 2021. [cited 2021 Nov 30]; Available from: https://www.ema.europa.eu/en/documents/referral/eli-lilly-company-limited-antibody-combination-bamlanivimab/etesevimab-covid19-article-53-procedure-assessment-report_en.pdf
- 108.European Medicines Agency . Regdanvimab: assessment report. 2021. [cited 2021 Nov 30]; Available from: https://www.ema.europa.eu/en/documents/referral/regdanvimab-treatment-covid-19-celltrion-covid-19-article-53-procedure-assessment-report_en.pdf
- 109.European Medicines Agency . Sotrovimab: assessment report. 2021. [cited 2021 Nov 30]; Available from: https://www.ema.europa.eu/en/documents/referral/sotrovimab-also-known-vir-7831-gsk4182136-covid19-article-53-procedure-assessment-report_en.pdf
- 110.Lee WS, Wheatley AK, Kent SJ, et al. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat Microbiol. 2020;5(10):1185–1191. [DOI] [PubMed] [Google Scholar]
- 111.Dolgin E. ‘Super-antibodies’ could curb COVID-19 and help avert future pandemics. Nat Biotechnol. 2021;39(7):783–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Piepenbrink MS, Park JG, Oladunni FS, et al. Therapeutic activity of an inhaled potent SARS-CoV-2 neutralizing human monoclonal antibody in hamsters. Cell Rep Med. 2021;2(3):100218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.CDC COVID-19 Vaccine Breakthrough Case Investigations Team . COVID-19 vaccine breakthrough infections reported to CDC - United States, January 1-April 30, 2021. MMWR Morb Mortal Wkly Rep. 2021;70(21):792–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kozlov M. Omicron overpowers key COVID antibody treatments in early tests. Nature. 2021;20211221. DOI: 10.1038/d41586-021-03829-0 [DOI] [PubMed] [Google Scholar]
- 115.Planas D, Saunders N, Maes P, et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2021. Dec 23. DOI: 10.1038/s41586-021-04389-z. [DOI] [PubMed] [Google Scholar]
- 116.Peltan ID, Beesley SJ, Webb BJ, et al. Evaluation of potential COVID-19 recurrence in patients with late repeat positive SARS-CoV-2 testing. PLoS One. 2021;16(5):e0251214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Corey L, Beyrer C, Cohen MS, et al. SARS-CoV-2 variants in patients with immunosuppression. N Engl J Med. 2021;385(6):562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sudre CH, Murray B, Varsavsky T, et al. Attributes and predictors of long COVID. Nat Med. 2021;27(4):626–631. [DOI] [PMC free article] [PubMed] [Google Scholar]