

Abstract
Objective
Parkinson disease (PD) is defined by the accumulation of misfolded α‐synuclein (α‐syn) in Lewy bodies and Lewy neurites. It affects multiple cortical and subcortical neuronal populations. The majority of people with PD develop dementia, which is associated with Lewy bodies in neocortex and referred to as Lewy body dementia (LBD). Other neuropathologic changes, including amyloid β (Aβ) and tau accumulation, occur in some LBD cases. We sought to quantify α‐syn, Aβ, and tau accumulation in neocortical, limbic, and basal ganglia regions.
Methods
We isolated insoluble protein from fresh frozen postmortem brain tissue samples for eight brains regions from 15 LBD, seven Alzheimer disease (AD), and six control cases. We measured insoluble α‐syn, Aβ, and tau with recently developed sandwich ELISAs.
Results
We detected a wide range of insoluble α‐syn accumulation in LBD cases. The majority had substantial α‐syn accumulation in most regions, and dementia severity correlated with neocortical α‐syn. However, three cases had low neocortical levels that were indistinguishable from controls. Eight LBD cases had substantial Aβ accumulation, although the mean Aβ level in LBD was lower than in AD. The presence of Aβ was associated with greater α‐syn accumulation. Tau accumulation accompanied Aβ in only one LBD case.
Interpretation
LBD is associated with insoluble α‐syn accumulation in neocortical regions, but the relatively low neocortical levels in some cases suggest that other changes contribute to impaired function, such as loss of neocortical innervation from subcortical regions. The correlation between Aβ and α‐syn accumulation suggests a pathophysiologic relationship between these two processes.
Introduction
The clinical diagnostic features of Parkinson disease (PD) include a combination of tremor, bradykinesia, rigidity, and impaired postural reflexes. PD is defined pathologically by brainstem catecholaminergic neuronal loss, gliosis, and the accumulation of aggregated or misfolded alpha‐synuclein (α‐syn) in neuronal cytoplasmic and neuritic inclusions known as Lewy bodies (LBs) and Lewy neurites (LNs). 1 The identification of dominant mutations in the gene encoding α‐syn (SNCA) in rare familial versions of PD supports the role of α‐syn in the pathogenesis of PD. 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 Dementia occurs frequently in PD. It may begin at approximately the same time as motor symptoms (often referred to as Dementia with Lewy bodies or DLB) or up to 20 years after the onset of motor symptoms (PD with dementia or PDD). The term Lewy body dementia (LBD) encompasses this spectrum of clinical presentations. Immunohistochemistry studies demonstrate that widespread deposition of α‐syn in neocortex, a marker of impaired cortical neuron function, accompanies dementia. 10 , 11 , 12 , 13 , 14
Other neurodegenerative changes occur with cognitive impairment in PD. Aβ plaques and tau‐containing neurofibrillary tangles, neuritic plaques, and dystrophic neurites accumulate to variable extents in cases of PD with dementia. 10 , 11 , 12 , 13 , 15 , 16 , 17 Although substantial neocortical Aβ is common, substantial neocortical tau deposition occurs less often, with pathologic tau accumulation more frequently limited to the medial temporal lobe. Therefore, the combination of Aβ and tau deposition is often not sufficiently extensive to meet NIA‐Reagan criteria for pathologically definite AD or to account independently for dementia according to NIA‐AA criteria. 18 , 19 However, the processes leading to Aβ and tau accumulation likely contribute to limbic and neocortical neuronal dysfunction and therefore may impact cognition.
Cognitive impairment in PD affects multiple domains including executive function, visuospatial function, and memory. 20 , 21 , 22 , 23 Hallucinations and delusions frequently accompany dementia in PD. 23 , 24 An additional characteristic feature of dementia in PD is fluctuations in attention and excessive daytime sleepiness. Possible anatomic and neurochemical correlates of these various cognitive phenotypes include (1) dysfunction in frontal‐striatal circuits and the dopaminergic system relating to executive function 25 , 26 ; (2) medial temporal lobe and basal forebrain cholinergic system dysfunction relating to memory 27 , 28 ; (3) superior parietal, temporal, and visual association cortex dysfunction relating to visuospatial function 29 , 30 , 31 ; (4) amygdala, hippocampus, parietal cortex, and occipital cortex dysfunction relating to hallucinations and delusions 32 , 33 ; and (5) serotonergic and noradrenergic projections, thalamus, and diffuse neocortex dysfunction relating to fluctuating attention. 32 , 33 , 34 , 35 To better understand the pathologic substrates of dementia, we need to better define the relationships between cognitive phenotypes and the deposition of the various protein species in limbic and neocortical regions.
Most previous studies of the neuropathology of PD assessed the deposition of aggregated protein species with immunohistochemistry (IHC). The patterns of protein deposition are complex, encompassing a range of morphology and subcellular localization. In the case of α‐syn, IHC identifies α‐syn accumulation in neuritic processes (LNs) and presynaptic terminals (often referred to as grains) in addition to neuronal cytoplasmic inclusions (LBs). Aβ accumulates in diffuse and cored plaques and in blood vessel walls. Aggregated or misfolded tau accumulates in neurofibrillary tangles (NFTs) within neuronal cytoplasm and proximal dendrites, dystrophic neurites of neuritic plaques, and neuropil threads. Different patterns of tau accumulation may reflect different pathologic processes and have different effects on brain function. IHC has been quantified by image analysis, yielding estimates of densities of pathologic protein deposition in different brain regions. 17 , 36 , 37 Alternatively, biochemical approaches can quantify protein accumulation in frozen tissue samples.
For this study, we developed new methods to quantify the burden of insoluble protein accumulation in frozen tissue samples from individual brain regions. We then applied these methods to postmortem tissue samples collected from a pilot study of 15 clinically and neuropathologically well‐characterized cases of LBD participants who were in a longitudinal clinical study of PD. This enabled our initial analysis of the relationships between misfolded protein accumulation and dementia as well as relationships between the accumulation of different pathologic protein species.
Subjects and Methods
Standard protocol approvals and patient consents
The Human Research Protection Office at Washington University in Saint Louis approved this study. Written informed consent to perform a brain autopsy was obtained from all participants. After death, the immediate next‐of‐kin were contacted and confirmed consent for brain removal and retention of brain tissue for research purposes.
Study participants
The cohort included 15 PD participants with dementia, seven AD participants, and six healthy age‐matched neurologically normal control participants. All PD participants and one control participant (the first 16 to die in this longitudinal study of PD and controls) were recruited through the Movement Disorders Center (MDC) and five control participants were recruited through the Knight Alzheimer Disease Research Center (ADRC), as described previously. 38 Two AD participants were recruited through the MDC and five AD participants were recruited from the Knight ADRC. Both centers are located at Washington University in Saint Louis. The five controls from the ADRC and the seven AD participants were included posthumously based on clinical and neuropathologic features. Postmortem brain tissue was collected between April 2008 and May 2016 and evaluated by IHC staining and neuropathological staging as previously described. 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 Inclusion criteria for the study were (1) PD participants who had a clinical diagnosis of idiopathic PD based on the UK Parkinson Disease Society Brain Bank diagnostic criteria 49 and either Clinical Dementia Rating (CDR global score) 50 ≥1 or a clinical diagnosis of dementia; (2) control participants with CDR ≤ 0.5, Braak Stage 0 LB pathology, Braak stage 0–B Aβ pathology, 41 and Braak stage I‐III tau pathology 41 ; and (3) AD participants with CDR ≥1 or clinical diagnosis of dementia, Braak stage C Aβ pathology, Braak stage V–VI tau pathology.
Motor assessments
MDC clinicians evaluated motor symptoms using the Unified Parkinson Disease Rating Scale motor subscale III (UPDRS‐III). 51 Evaluations were performed OFF antiparkinsonian medications.
Dementia ratings
Dementia was rated using the CDR scale by experienced raters. The CDR included an assessment of whether cognitive dysfunction was sufficiently severe to impair activities of daily living. PD participants were recruited into the longitudinal study regardless of the presence of cognitive decline and regardless of the time of onset of cognitive decline or dementia (as elicited by history) relative to the onset of motor symptoms. Prior to death, all PD participants had CDR ≥ 1, in accordance with criteria for dementia in PD. 23
Tissue Processing and Extraction
Frozen tissue was dissected from the right hemibrain. Insoluble protein was isolated from frozen tissue samples by sequential extraction and centrifugation. The insoluble fraction was then solubilized in 2% SDS with heating and sonication. Additional details are provided in the supporting information.
ELISA
All ELISAs used the following protocol unless otherwise stated. Costar 96 well, clear, flat bottom, half area, high binding, polystyrene plates (Corning #3690) were used. The capture antibodies (Table S1) were diluted 1 μg/mL in 34 mM sodium bicarbonate, 16 mM sodium carbonate solution with 3 mM sodium azide. The capture antibody solution was added (50 μL/well) and incubated overnight at 4°C. Plates were washed 5X with 150 μL 1XPBS + 0.05% Tween‐20/well. Plates were blocked with 150 μL of 2% BSA (Sigma‐Aldrich #A7906) in PBS at 37°C for 2 h. Prior to standard and sample addition, plates were washed as previously stated. Samples were diluted at 1:33.3 in sample buffer containing no SDS to reach an SDS concentration of 0.06%. Standards and samples were added to the plate 50 μL/well in duplicate wells and incubated overnight at 4°C with slow rotation. Plates were washed as previously stated. The detection antibody (Table S1) solution was diluted 1 μg/mL in PBS + 0.05% BSA + 0.05% Tween. The detection antibody solution was added 50 μL/well and incubated at 37°C for 2 hours. Plates were washed as previously stated. Streptavidin poly‐HRP80 (Fitzgerald # 65R‐S118) was diluted 1:8000 in 1% BSA in 1XPBS+0.05% Tween‐20. The streptavidin solution was added 50 μL/well and incubated for 90 minutes at room temperature in the dark. The plate was washed as described previously. Fifty μL/well of 3,3’,5,5’‐Tetramethylbenzidine Liquid Substrate, Super Slow (Sigma # T5569) was rocked on the rotator until first reading. Readings were done on Synergy 2 (BioTek) at 650 nM at 9, 12, and 15 minutes. The time point with the highest ratio between the top concentration and 0 concentration (background) in the standard curve was chosen for data analysis.
Statistical analyses
Data were analyzed using GraphPad Prism software, version 8 (Graph Pad Software, Inc., La Jolla, CA). Due to unequal group sizes and nonnormal distribution of data points, nonparametric Mann–Whitney U tests were utilized to compare groups. Spearman correlation analysis was used to analyze correlations between different protein measures and between protein and cognitive function measures. All tests were two‐tailed and corrected for multiple comparisons with the Holm–Bonferroni method 52 using a significance level of 0.05.
Results
Demographics and clinical information of participants
Table 1 summarizes the demographics and clinical information of the study participants. Age at death did not significantly differ between control and LBD participants. The AD participants were younger at age of death. The male/female ratio was higher in LBD compared with controls and AD. To analyze insoluble protein accumulation in frozen postmortem tissue samples, we isolated insoluble protein by sequential extraction and centrifugation and then solubilized insoluble protein in 2% SDS. We analyzed samples from eight brain regions: middle frontal gyrus (MFG), anterior cingulate gyrus (ACG), caudate, inferior parietal lobule (IPL), visual association cortex (VAC), precuneus, hippocampus, and amygdala.
Table 1.
Demographic and clinical information of the autopsied cases.
| LBD | Control | AD | |
|---|---|---|---|
| Demographics | |||
| Participants, N | 15 | 6 | 7 |
| Age at Death, y | 81 (71‐93) | 84 (70‐100) | 73 (62‐78) |
| Male/Female, N | 12/3 | 2/4 | 4/3 |
| Clinical characteristics of LBD participants | |||
| Age at LBD diagnosis, y | 63 (54–82) | NA | NA |
| Duration of motor impairment, y | 14 (8–27) | NA | NA |
| UPDRS‐III score (OFF medications) | 44 (35–73.5) | NA | NA |
| LEDD, mg | 800 (0–1350) | NA | NA |
| SSRI medications, N | 10/15 | NA | NA |
| Other antidepressant medications, N a | 2/15 | NA | NA |
| AChE inhibitor medications, N b | 8/15 | NA | NA |
| Neuroleptic medications, N c | 14/15 | NA | NA |
Abbreviations: AChE, acetylcholinesterase. Values are median (range) unless otherwise indicated; LEDD, levodopa equivalent daily dose; NA, not applicable; SSRI, selective serotonin reuptake inhibitors; UPDRS, Unified Parkinson Disease Rating Scale; Y, years.
Mirtazapine and venlafaxine.
Donepezil, rivastigmine, and galantamine.
Quetiapine and clozapine.
Assay performance characteristics of the ELISA assays
We developed and validated sandwich ELISAs for α‐syn, p‐α‐syn, Aβ(1‐42), p‐tau, and tau. Table S1 indicates ELISA antibodies used for α‐syn, p‐α‐syn, Aβ, tau, and p‐tau. Representative standard curves for each ELISA are shown in Fig. S1. The ELISA lower limit of quantification (LLQ) for α‐syn (Syn1/13G5B), p‐α‐syn, Aβ, p‐tau, and tau were 0.15, 0.15, 0.014,17.5, and 0.125 ng/mL, respectively, which corresponded to 0.05, 0.01, 0.94, 1.75, and 0.33 µg/g wet weight tissue when adjusted for the sample dilution used in each ELISA. Since all group comparisons were performed for an individual brain region within the same plate, the coefficients of variation (%CV) for four intraplate controls were the most relevant measures of assay consistency, which are included in Table S2.
Alpha‐synuclein and phospho‐alpha‐synuclein
We used our recently developed α‐syn ELISA (Syn1/13G5B) 53 to measure insoluble α‐syn in LBD, AD, and control samples. The LBD cases had a wide range of α‐syn levels. Three LBD cases had α‐syn levels that were within the range of control cases for MFG, IPL, and Prec, and an additional seven LBD cases overlapped with control cases for IPL, and Prec (Fig. 1A and B). The highest median levels of α‐syn deposition in LBD cases were found in MFG, caudate, and amygdala, whereas, the VAC had very little α‐syn in all LBD cases. We consistently observed quantifiable α‐syn levels in control cases, which may represent a low level of aggregated α‐syn in brain tissue from older controls, or alternatively, it may represent a carryover of the highly abundant soluble α‐syn fraction during the preparation of insoluble fractions.
Figure 1.
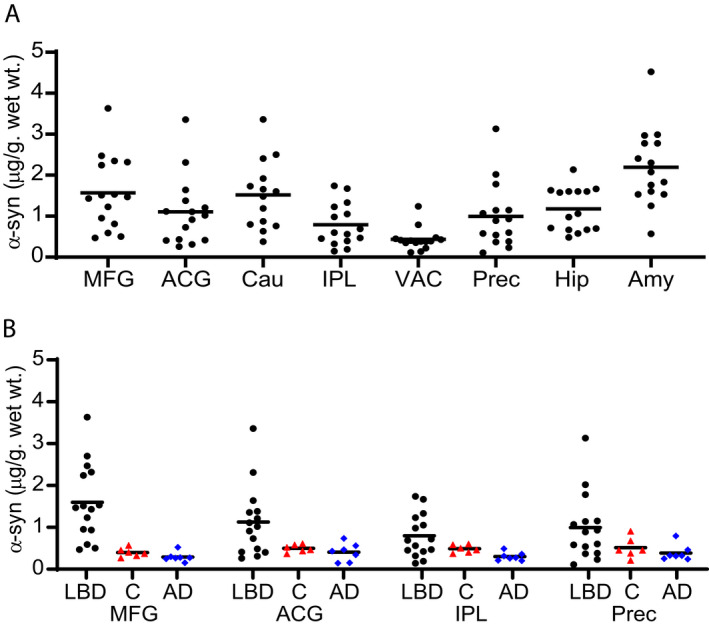
Regional distribution of insoluble α‐syn concentration in LBD and Control cases (Syn1‐13G5B ELISA). Levels of insoluble α‐syn were measured by sandwich ELISA in eight brain regions from LBD (black circles) cases (A) and four brain regions from control (red triangles) and AD (blue diamonds) cases (B). There was a wide range of α‐syn accumulation among LBD cases, including some cases in which α‐syn accumulation overlapped with levels measured in control cases. The highest median levels of α‐syn deposition were in MFG, caudate, and amygdala. Control and AD cases had low levels of insoluble α‐syn in all brain regions tested. The lower limit of quantification (LLQ) for the ELISA was 0.15 ng/mL or 0.05 µg/g wet wt tissue. All samples were in the quantifiable range. The α‐syn level in MFG was significantly higher in LBD compared with control cases. Data were analyzed with the two‐tailed Mann–Whitney test using a significance level of 0.05 and corrected for multiple comparisons with the Holm‐Bonferroni method. 52 Mid‐Frontal Gyrus (MFG), Anterior Cingulate Gyrus (ACG), Caudate (Cau), Inferior Parietal Lobule (IPL), Visual Association cortex (VAC), Precuneus (Prec), Hippocampus (Hip), and Amygdala (Amy).
Since immunohistochemistry produces highly selective staining for pathologic α‐syn when phospho‐serine 129 α‐syn antibodies are utilized, we also developed an ELISA utilizing the anti‐phospho‐serine 129 α‐syn antibody psyn81a. p‐α‐syn was below LLQ in all Control brain samples with the exception of one MFG and one ACG sample (Fig. 2). Consistent with the α‐syn measurements, p‐α‐syn levels in LBD were below the LLQ in MFG for three cases and were below LLQ in IPL for seven cases and precuneus for six cases (Fig. 2A). The highest median levels of p‐α‐syn accumulation were found in ACG, caudate, and amygdala. Levels were below the LLQ in all but two LBD cases for VAC. α‐syn and p‐α‐syn were significantly correlated in all regions, but correlations were much stronger in regions such as caudate, hippocampus, and amygdala compared with other regions with weaker correlations such as ACG, MFG, and IPL (Fig. 3). We did not observe detectable p‐α‐syn deposition in samples from AD cases (Fig. 2B).
Figure 2.
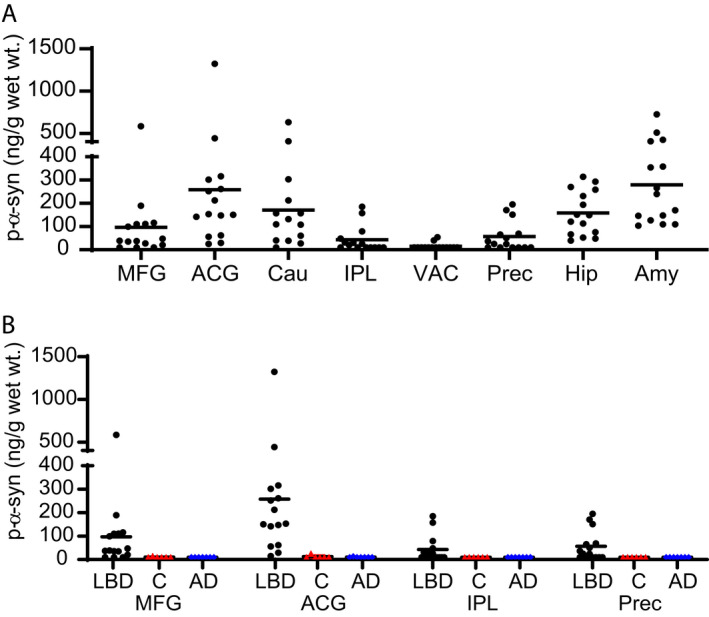
Regional distribution of p‐α‐syn concentration in LBD and Control cases (pSyn/81A‐Syn1B ELISA). Levels of insoluble p‐α‐syn were measured by sandwich ELISA in eight brain regions from LBD (black circles) cases (A) and four brain regions from control (red triangles) and AD (blue diamonds) cases (B). The highest median levels of p‐α‐syn in LBD cases were in ACG, caudate, and amygdala. The LLQ for the ELISAs was 0.15 ng/mL or 9.99 ng/g wet wt tissue. All samples from control and AD cases were below the lower limit of quantification for p‐α‐syn except one control MFG sample and one control ACG sample. The α‐syn level in ACG was significantly higher in LBD compared with control cases. Data were analyzed with the two‐tailed Mann–Whitney test using a significance level of 0.05 and corrected for multiple comparisons with the Holm‐Bonferroni method. 52
Figure 3.
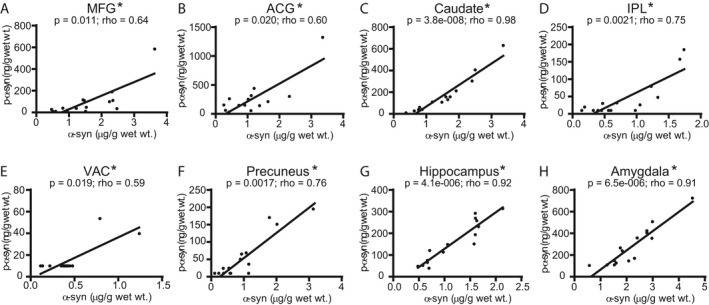
Correlations between insoluble α‐syn (Syn1‐13G5B ELISA) and p‐α‐syn (pSyn/81A‐Syn1B ELISA) concentration measurements in LBD cases. We observed significant correlations (Spearman) between α‐syn and p‐α‐syn in all eight brain regions: (A) MFG, (B) ACG, (C) caudate, (D) IPL, (E) VAC, (F) precuneus, (G) hippocampus, (H) amygdala, with the strongest correlations (Spearman rho values > 0.9) in caudate, hippocampus, and amygdala. Data were corrected for multiple comparisons with the Holm‐Bonferroni method. 52 Lines were generated by linear regression.
We also analyzed the LBD cases but not Control or AD cases with our previously published α‐syn ELISA that uses Syn211 and FL140 antibodies (Fig. S2). This ELISA is significantly less sensitive than the Syn1/13G5 ELISA and is also not available for continued use because the polyclonal FL140 antibody has been discontinued by the commercial supplier. We analyzed correlations between the two α‐syn ELISAs and observed significant correlations in all brain regions except ACG (Fig. S3). The highest median levels of α‐syn measured by the Syn211/FL140 ELISA were in ACG, caudate, hippocampus, and amygdala. There was a significant correlation between α‐syn Syn211/FL140 ELISA and p‐α‐syn in all brain regions (Fig. S4). The strongest correlations were found in MFG, caudate, and precuneus.
Aβ (1‐42)
Accumulation of Aβ is a hallmark of AD and has also been implicated in the development of dementia in LBD patients. 10 , 12 , 41 , 42 , 45 , 54 , 55 , 56 , 57 We used a well‐characterized ELISA that is selective for Aβ(1‐42), the predominant Aβ species in plaques. 58 We detected insoluble Aβ(1‐42), in 8 of the 15 (53%) LBD cases studied (Fig. 4A), and these LBD cases had widespread Aβ accumulation by IHC (Table S3). The highest levels by ELISA of Aβ for LBD were found in MFG, ACG, and IPL. These eight LBD cases with quantifiable Aβ levels were compared with AD cases (Fig. 4B), and the mean level of Aβ was lower in LBD cases compared with AD cases in all four regions. Levels in LBD and AD were compared using the Mann–Whitney test and corrected for multiple comparisons with the Holm‐Bonferroni method. Two control cases had elevated levels of Aβ, one with substantially elevated levels in all four brain regions analyzed (Fig. 4B). Both these control cases had Braak stage B Aβ pathology by IHC.
Figure 4.
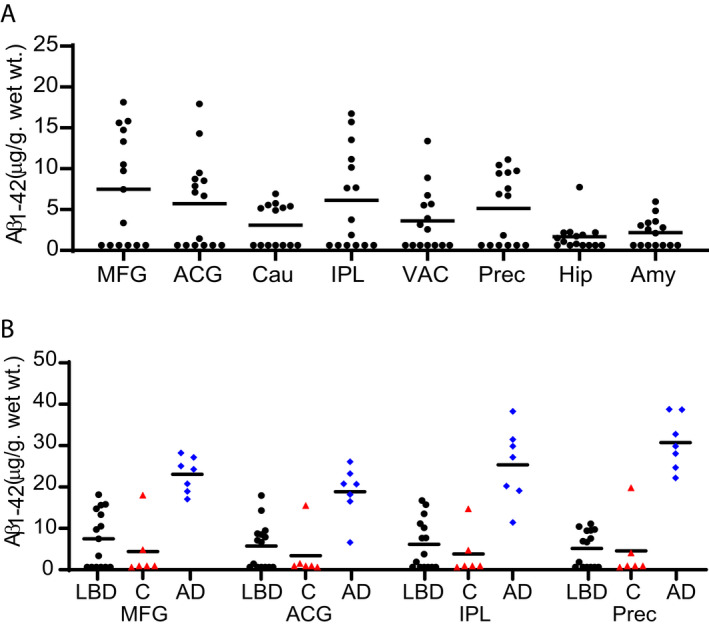
Regional distribution of insoluble Aβ(1‐42) concentration in LBD, Control, and AD cases. Levels of insoluble Aβ(1‐42) were measured by sandwich ELISA. (A) Eight of the 15 cases had measurable Aβ levels in most brain regions. For LBD cases with measurable Aβ, the highest levels of Aβ were found in MFG, ACG, IPL, and Precuneus. (B) Aβ levels were measurable in two of the six control (red triangles) cases. One control case had high levels of Aβ in all brain regions. AD (blue diamonds) cases had significantly higher Aβ levels compared with the subgroup of LBD (black circles) cases that had detectable Aβ levels in all four brain regions examined. The LLQ for the ELISAs was 14.17 pg/mL or 0.63 µg/g wet wt tissue. Data were analyzed with the two‐tailed Mann–Whitney test using a significance level of 0.05 and corrected for multiple comparisons with the Holm‐Bonferroni method. 52
Previous histologic studies observed correlations between LB and Aβ plaque density in LBD. We compared insoluble α‐syn in subgroups defined by the presence or absence of Aβ accumulation. The LBD with Aβ accumulation subgroup had significantly higher α‐syn levels in ACG and amygdala (Fig. 5). In addition, there was a consistent trend toward higher mean α‐syn levels in the subgroup with Aβ accumulation for all regions. Similar results were observed for the comparison of p‐α‐syn levels between the two subgroups (Fig. S6). We also analyzed correlations between insoluble α‐syn and Aβ measured by the ELISAs. Significant correlations between α‐syn (Syn1/13G5B ELISA) and Aβ in LBD cases were found in ACG, IPL, precuneus, and amygdala (Fig. 6). We found correlations between Aβ and α‐syn measured by syn211/FL140 ELISA (Fig. S5) in four of the eight brains regions (amygdala, precuneus, MFG, and IPL). Three of the four brains regions with significant correlations (IPL, precuneus, and amygdala) were common between the two α‐syn ELISAs. Three brain regions (MFG, amygdala, and precuneus) had significant correlations between p‐α‐syn and Aβ (Fig. S7).
Figure 5.
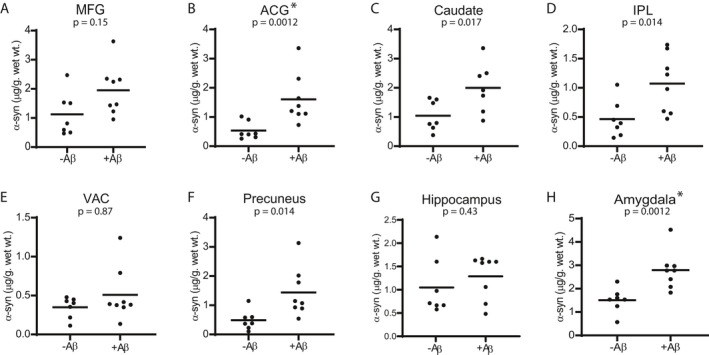
Comparison of insoluble α‐syn concentrations (Syn1‐13G5B) between LBD with measurable Aβ and LBD cases without measurable Aβ. The subgroup with Aβ accumulation had significantly higher α‐syn concentrations in ACG and amygdala, compared with the subgroup without measurable Aβ. Mean α‐syn levels were 18% higher in the PD with Aβ accumulation subgroup in the hippocampus and were 32%–73% in other regions. Data were analyzed with the two‐tailed Mann–Whitney test using a significance level of 0.05 and corrected for multiple comparisons with the Holm‐Bonferroni method. 52
Figure 6.
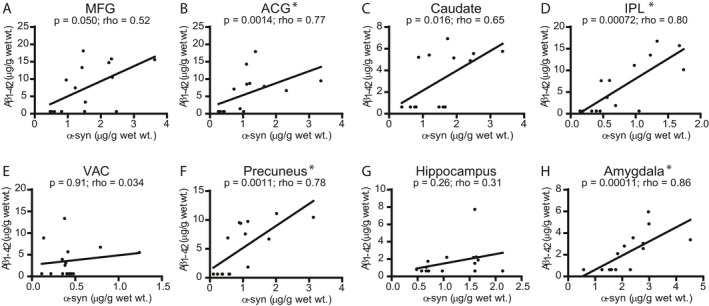
Correlations between α‐syn (Syn1‐13G5B ELISA) and Aβ concentrations in LBD cases. We observed significant correlations (Spearman) between α‐syn and Aβ in four (ACG (B), IPL (D), precuneus (F), and amygdala (H)) of the eight brain regions. Data were corrected for multiple comparisons with the Holm‐Bonferroni method. 52 Lines were generated by linear regression.
Tau and phospho‐tau
Tauopathy is strongly associated with the development of dementia in AD. 39 , 41 , 59 Immunohistochemistry studies have identified hyperphosphorylated tau accumulation in LBD cases. 10 , 11 , 17 , 37 , 54 , 57 , 60 We utilized an ELISA to measure total insoluble tau and also developed an ELISA for p‐tau based on the PHF‐1 antibody. PHF‐1 is specific for tau phosphorylation at serine 396/404 and is commonly used for histolopathologic analysis of tau accumulation. One LBD case had elevated p‐tau accumulation relative to other LBD cases in all regions, although it was still within the range of the levels found in Control cases and lower than most AD cases (Fig. 7A and B). This case also had increased deposition of total tau relative to the other LBD cases (Fig. S8A). AD cases had elevated levels of tau compared with LBD cases (Fig. S8B). We observed tau and p‐tau correlations in caudate and VAC, but not in other regions for LBD cases (Fig. S9). We detected a significant correlation between total tau and α‐syn (Syn1‐13G5B) in VAC and amygdala and detected a correlation between total tau and Aβ in MFG, but we did not identify significant correlations between p‐tau and either α‐syn or Aβ in LBD cases (data not shown).
Figure 7.
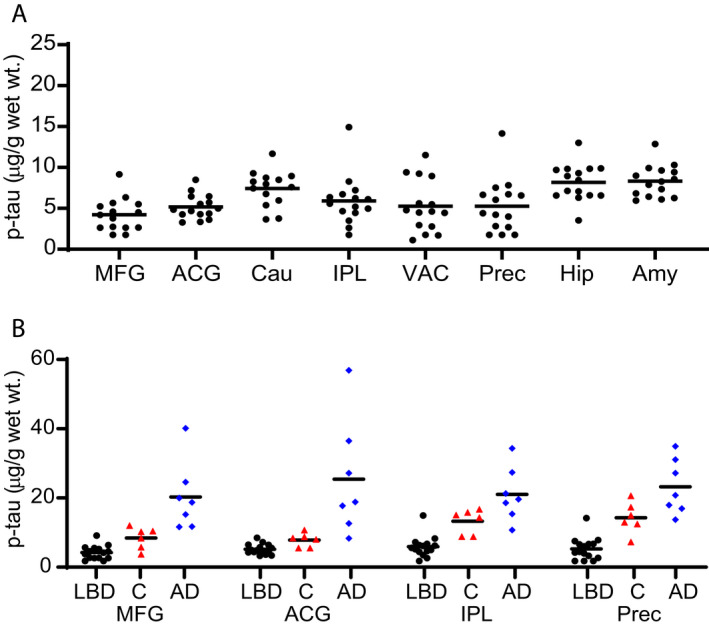
Regional distribution of phospho‐tau concentration in LBD, Control, and AD cases. Levels of insoluble phospho‐tau were measured by sandwich ELISA in eight brain regions from LBD cases (A), four brain regions from control (red triangles) cases, and four regions from AD cases (blue diamonds) (B). The LBD cases (black circles) had low levels of p‐tau. One LBD case had elevated p‐tau accumulation relative to other LBD cases in all regions, although it was still within the range of the levels found in control cases. The LBD cases were significantly lower than the control and AD cases. The highest levels of p‐tau in LBD cases were found in the caudate, hippocampus, and amygdala. The LLQ for the ELISAs was 17.5 ng/mL or 1.75 µg/g wet weight tissue. Data were analyzed with the two‐tailed Mann–Whitney test using a significance level of 0.05 and corrected for multiple comparisons with the Holm‐Bonferroni method. 52
Histology vs ELISA
We used immunohistochemistry to analyze formalin‐fixed paraffin‐embedded tissue samples from the contralateral (left) hemibrain of each LBD case, according to our diagnostic neuropathology protocols. 43 Six of the eight brain regions analyzed by ELISA also were scored for pathologic burden of LBs identified by p‐α‐syn staining; diffuse plaques (DP) and cored plaques (CP) identified by Aβ staining; and NFTs and neuritic plaques (NP) identified by p‐tau staining. We compared histopathology scores to the ELISA results for p‐α‐syn, Aβ, and p‐tau (Fig. S10‐S11). We performed an exploratory Spearman correlation analysis, which was limited by the poor distribution of data points for most analyses. DP and CP scores correlated to some degree with ELISA measures of Aβ for most regions, but there was no indication of correlations between LB scores and ELISA measures or between NFT or NP scores and ELISA measures.
Dementia severity vs ELISA
Since all the LBD cases were diagnosed with dementia, we compared the CDR sum of boxes score and MMSE scores closest to the time of death with the mean level of p‐α‐syn detected in the neocortical regions of MFG, IPL, VAC, and precuneus (Fig. 8). Dementia severity as measured by CDR sum of boxes correlated with higher p‐α‐syn concentration. There was also a significant correlation between worse MMSE and higher p‐α‐syn concentration. There were no correlations between these cognitive measures and either mean neocortical insoluble Aβ or mean insoluble p‐α‐syn concentration in limbic regions (data not shown). There were also no significant correlations when we analyzed for correlations between each protein measure and time from PD onset to dementia onset, age of dementia onset, dementia duration, age at death, and sex (data not shown).
Figure 8.
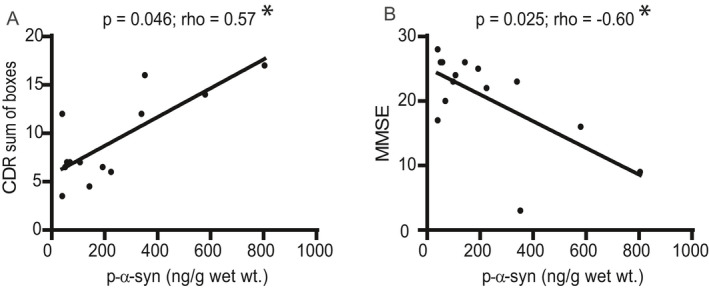
Correlations between cognitive measures (CDR sum of boxes and MMSE) and neo‐cortical phospho‐alpha‐synuclein levels in LBD cases. We analyzed correlations (Spearman) between cognitive measures (CDR sum of boxes, MMSE) and total aggregated p‐α‐syn in the neocortical regions of MFG, IPL, VAC, and precuneus. (A) There was a significant correlation between CDR sum of boxes score and neocortical p‐α‐syn (p = 0.046, rho = 0.57). (B) There was also a significant negative correlation between MMSE and neocortical p‐α‐syn (p = 0.025; rho = −0.60). Lines were generated by linear regression.
Discussion
We report the results of a new approach to analyze the accumulation of insoluble α‐syn, Aβ, and tau in Lewy body dementia. Our approach relies on the isolation of insoluble protein from frozen postmortem tissue, followed by solubilization in SDS and quantification by sandwich ELISAs. We applied these methods to 15 cases of LBD, measuring pathologic accumulation of α‐syn, Aβ42 and tau in eight brain areas; we also applied them to seven AD cases and six controls, in four of these brain areas. While all LBD cases had Braak stage 6 LB pathology as determined by standard immunohistochemistry analysis, we observed a wide range of insoluble α‐syn levels in basal ganglia, limbic and cortical regions, with a subset of cases having low neocortical levels that were not clearly distinguished from controls. Aβ accumulated in 53% of the LBD cases, in line with previous immunohistochemistry studies. The LBD plus Aβ subgroup had higher α‐syn accumulation compared with the LBD only group in the majority of brain regions examined, and levels of Aβ correlated with levels of α‐syn. However, in contrast to the pathologic picture of symptomatic AD, widespread pathologic Aβ accumulation in LBD was not accompanied by widespread tau accumulation. These results provide new insight into pathophysiologic processes in PD, including the relationship between regional α‐syn accumulation and dementia as well as relationships between different accumulating protein species.
The wide range of insoluble α‐syn levels measured in cortical, limbic, and basal ganglia regions indicates that complex causes of brain dysfunction, beyond the level of α‐syn accumulation alone, underlie the cognitive impairment and dementia in LBD. Most consistent among the 15 cases is the high α‐syn and p‐α‐syn accumulation in ACG, caudate, amygdala, and hippocampus. In neocortical regions, many cases had high levels of insoluble α‐syn. However, three cases had low cortical α‐syn concentrations that were not clearly distinguished from levels measured in control cases. Multiple studies have observed that neocortical LBs are associated with dementia in PD 10 , 11 , 12 , 13 and these cases all had LBs in neocortical regions that were sufficient to meet Braak stage 6 neuropathologic criteria. However, for a subset of cases, the total burden of α‐syn fibrils present in a combination of LBs, LNs, and grain pathology is relatively low and difficult to distinguish from Control cases. It is not clear whether the low level of insoluble α‐syn measured by our α‐syn ELISA in Control cases corresponds to the presence of α‐syn fibrils. It may instead represent the carryover of a small fraction of soluble α‐syn during multiple rounds of homogenization and centrifugation. Our p‐α‐syn ELISA appears to be more specific for pathologic α‐syn accumulation in LBD cases since p‐α‐syn levels were not quantifiable in all but two control tissue samples.
The mean neocortical p‐α‐syn concentration correlated with dementia severity, as measured by CDR sum of boxes score and MMSE. However, the wide range of insoluble α‐syn burden in neocortical regions, including a subset of cases with low levels, indicates that changes other than cortical neuron synucleinopathy may contribute to cognitive impairment in at least some cases. Our observations overlap with other studies in which a subset of LBD cases, classified as limbic or transitional Lewy body disease, 37 , 45 , 61 have high LB density in the brainstem and limbic regions but low or absent LB pathology in neocortical regions. We observe consistently high levels of α‐syn accumulation in the hippocampus, amygdala, and caudate regions in LBD cases. We also previously reported the substantial loss of dopaminergic, serotonergic, and noradrenergic innervation in neocortical, limbic, and basal ganglia regions for these same 15 LBD cases. 38 Specifically, cases that had a low α‐syn burden in this study had substantial deficits in dopaminergic, serotonergic, and noradrenergic innervation of neocortical, limbic, and basal ganglia regions. These findings support the idea that loss of projections from subcortical nuclei, including substantia nigra, raphe nuclei, and locus coeruleus, also may contribute to the development of dementia in LBD. Further studies of both pathologic protein accumulation and neurotransmitter deficits in additional cases, including cases without dementia, will help to better define the relative contributions of these different pathologic substrates of dementia. Furthermore, analysis of additional cases may also reveal relationships to deficits in specific cognitive domains.
The correlation between insoluble α‐syn and Aβ accumulation in LBD indicates a pathophysiologic relationship that has implications for previously observed associations of Aβ with shorter survival 13 and increased rate of cognitive decline in LBD. 10 , 62 , 63 Previous studies have observed α‐syn–Aβ correlations based on immunohistochemistry analysis of LBs and plaques. 10 , 17 , 37 , 64 , 65 In PD, where CSF levels of α‐syn are lower compared with control cases, levels of CSF α‐syn and CSF Aβ1‐42 are correlated. 66 These correlations may arise from pathophysiologic mechanisms, such as impaired protein homeostasis, where impaired protein turnover pathways affect both proteins. Alternatively, metabolism changes, neuroinflammation, or impaired synaptic function in PD may contribute to both α‐syn and Aβ accumulation. We previously observed shorter survival in LBD cases with Aβ compared with those without Aβ accumulation. 13 Others have observed shorter time to develop cognitive impairment associated with low CSF Aβ levels. 10 , 62 , 63 While these observations may be explained by effects of Aβ independent of α‐syn, the faster rate of disease progression may also relate to increased α‐syn deposition associated with Aβ accumulation or the underlying disease mechanisms driving the accumulation of both proteins.
We did not see substantial levels of insoluble phospho‐tau in the LBD cases with Aβ accumulation. Only a single LBD case overlapped with the range of phospho‐tau levels quantified in AD cases with Braak stage V and VI tau pathology. This observation, which is similar to our previous observation that widespread tau deposition is rare in PD based on immunohistochemistry analysis, indicates that Aβ accumulation is associated with distinct pathophysiologic processes in LBD. Tau accumulation, or pathways leading to tau accumulation, may be less involved in LBD or at least less involved in neocortical regions compared with symptomatic AD. Other studies have observed an association between Aβ and tau accumulation based on IHC scores and staging, 10 , 17 and although tau accumulation is limited primarily to temporal lobe regions in most cases, it may still contribute to cognitive dysfunction in PD. 17 , 60 It is also possible that differences in the patterns of pathologic protein deposition among different centers may reflect differences in case selection; for example, individuals with α‐syn accumulation only may be more likely to present to movement disorder centers, and those with co‐occurring AD neuropathic changes may be more likely to present to centers focusing on cognitive disorders. 10 , 11 , 17 , 37 , 54 , 57 , 60
The development of an α‐syn PET imaging agent would provide more detailed information on longitudinal patterns of α‐syn accumulation and their relationship to clinical features of disease progression. 67 One limitation in interpreting our postmortem analysis is that we do not know whether cell death late in the disease course may lead to clearance of pathologic protein and a decline in levels measured at autopsy, a question that can be addressed by longitudinal PET studies. However, our autopsy results provide guidance for the initial clinical development of α‐syn imaging agents. The α‐syn ELISA results highlight ACG, caudate, amygdala, and hippocampus as having the highest levels of α‐syn accumulation in LBD at autopsy among the brain areas sampled here. Some, but not all, cases have high neocortical levels of α‐syn. IHC studies also endorse brainstem nuclei, olfactory bulb, basal forebrain, and olfactory cortex as sites of dense α‐syn accumulation. The generally high variance in α‐syn accumulation among LBD cases predicts that variable signal will be observed among individuals in initial clinical studies of α‐syn PET tracers for PD and LBD. Further ELISA analyses of PD cases without dementia, which are less common at autopsy, will provide more information about expected levels across the disease spectrum.
Our study has several limitations with respect to the goal of understanding relationships between insoluble protein accumulation and clinical features of LBD. Our sandwich ELISA approach improves throughput and precision for quantifying insoluble protein accumulation, but throughput is still limited by the time required to dissect and process frozen tissue samples. Quantification by the ELISA method is applied to a relatively small sample of frozen tissue. For quantification of caudate, ACG, and neocortical regions, the values represent concentrations present in approximately 1 g samples of gray matter, whereas for hippocampus and amygdala, quantification is based on 0.1 g samples, which are selected from cryostat sections produced from brain regions where smaller volumes are available for analysis. The approach is not easily applied to small subcortical nuclei, such as nucleus basalis, raphe nuclei, and locus coeruleus, where the tissue volumes are very small, and regions of interest are more difficult to isolate from frozen tissue. Because the analysis relies on the isolation of an insoluble fraction, the burden of soluble oligomeric species is not assessed. However, our recent study using a similar biochemical fractionation method indicates that levels of soluble oligomeric α‐syn species are very low, relative to insoluble α‐syn, in PD tissue. 68 The choice of antibodies for sandwich ELISAs may bias toward detection of specific conformations and posttranslational modifications recognized with the highest affinity by these antibodies. We utilized phosphorylation‐specific antibodies to complement the analysis of “total” α‐syn and tau, which may improve specificity given the preferential phosphorylation of insoluble species. However, antibodies used to measure total α‐syn and tau also may have biases toward specific species, which may explain the imperfect correlations we observe between sandwich ELISA values obtained with different α‐syn antibodies.
Additional studies are needed to better understand the relationship between pathologic protein accumulation and dementia, including specific cognitive phenotypes associated with PD. The inclusion of cases without dementia will be particularly helpful for future studies, which is challenging given the relatively low number of cases at autopsy. Analysis of additional markers, including markers to identify the loss of innervation from subcortical nuclei, as well as the loss of cortical neurons, and their connections, may also be helpful to understand the relative contributions of these pathologic changes and their relationship to pathologic protein accumulation.
Author Contributions
Study concept and organization: Drs. Kotzbauer, Perlmutter, and Campbell; Execution and acquisition of data: Miller, Dhavale, O’Shea, Andruska, Liu, Buddhala, Loftin, Franklin, Cairns, Perrin; Analysis and interpretation of data: Miller, Dhavale, Cairns, Perrin, Campbell, Perlmutter, Kotzbauer; Drafting of the manuscript: Drs. Miller and Kotzbauer; Critical revision of the manuscript for important intellectual content: All authors; Statistical analysis: Drs. Miller, Kotzbauer, and Campbell; Obtained funding: Drs. Kotzbauer, Campbell, and Perlmutter.
Supporting information
Data S1. Materials and methods.
Figure S1. Representative standard curves.
Figure S2. Regional distribution of insoluble α‐syn concentrations in LBD cases as measured by the Syn211‐FL140 ELISA.
Figure S3. Correlation between α‐syn concentration measurements obtained by the Syn1‐13G5B ELISA and the Syn211‐FL140 ELISA in LBD cases.
Figure S4. Correlations between α‐syn (Syn211‐FL140 ELISA) and p‐α‐syn measurements in LBD cases.
Figure S5. Correlations between α‐syn (Syn211‐FL140 ELISA) and Aβ concentrations in LBD cases.
Figure S6. Comparison of insoluble phospho‐α‐syn concentrations (pSyn/81A‐13G5B) between LBD with measurable Aβ and LBD cases without measurable Aβ.
Figure S7. Correlations between p‐α‐syn (pSyn/81A‐Syn1B ELISA) and Aβ fibril concentrations in LBD cases.
Figure S8. Regional distribution of insoluble tau concentrations.
Figure S9. Correlations between phospho‐tau and tau levels in LBD cases.
Figure S10. Relationship between ELISA measurements and ICH‐based scoring of pathological α‐syn, Aβ, and tau accumulation.
Figure S11. Representative photomicrographs of immunohistochemistry.
Table S1. Capture and detection antibodies used in the ELISA assays.
Table S2. Assay performance characteristics of ELISA assays.
Table S3. Clinical and Pathologic Information for Study Participants.
Table S4. Insoluble protein data from ELISAs.
Acknowledgments
Support for this work was provided by grants from the Michael J. Fox Foundation; NIH grants NS097799, NS075321, NS097437, and NS110436 from the National Institute of Neurological Disorders and Stroke; NIH grants AG003991, AG066444, and AG026276 from the National Institute of Aging; the American Parkinson Disease Association (APDA) Advanced Research Center for Parkinson Disease at Washington University in St Louis; the Greater St Louis Chapter of the APDA; and the Barnes Jewish Hospital Foundation (Elliot Stein Family Fund and Parkinson Disease Research Fund). We are grateful for the technical support of the Betty Martz Laboratory for Neurodegenerative Research at Washington University in Saint Louis.
Funding Information
The sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Funding Statement
This work was funded by Michael J. Fox Foundation for Parkinson's Research ; American Parkinson Disease Association (APDA) Advanced Research Center for Parkinson Disease at Washington University in St Louis; Barnes Jewish Hospital Foundation (Elliot Stein Family Fund and Parkinson Disease Research Fund); National Institute of Aging grants AG003991, AG026276, and AG066444; National Institute of Neurological Disorders and Stroke grants NS075321, NS097437, NS097799, and NS110436; Greater St Louis Chapter of the APDA.
REFERENCES
- 1. Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol. 1996;55(3):259‐272. [DOI] [PubMed] [Google Scholar]
- 2. Appel‐Cresswell S, Vilarino‐Guell C, Encarnacion M, et al. Alpha‐synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord. 2013;28(6):811‐813. [DOI] [PubMed] [Google Scholar]
- 3. Golbe LI, Di Iorio G, Bonavita V, Miller DC, Duvoisin RC. A large kindred with autosomal dominant Parkinson's disease. Ann Neurol. 1990;27(3):276‐282. [DOI] [PubMed] [Google Scholar]
- 4. Kruger R, Kuhn W, Muller T, et al. Ala30Pro mutation in the gene encoding alpha‐synuclein in Parkinson's disease. Nat Genet. 1998;18(2):106‐108. [DOI] [PubMed] [Google Scholar]
- 5. Lesage S, Anheim M, Letournel F, et al. G51D alpha‐synuclein mutation causes a novel parkinsonian‐pyramidal syndrome. Ann Neurol. 2013;73(4):459‐471. [DOI] [PubMed] [Google Scholar]
- 6. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science. 1997;276(5321):2045‐2047. [DOI] [PubMed] [Google Scholar]
- 7. Proukakis C, Dudzik CG, Brier T, et al. A novel alpha‐synuclein missense mutation in Parkinson disease. Neurology. 2013;80(11):1062‐1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singleton AB, Farrer M, Johnson J, et al. alpha‐Synuclein locus triplication causes Parkinson's disease. Science. 2003;302(5646):841. [DOI] [PubMed] [Google Scholar]
- 9. Zarranz JJ, Alegre J, Gomez‐Esteban JC, et al. The new mutation, E46K, of alpha‐synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55(2):164‐173. [DOI] [PubMed] [Google Scholar]
- 10. Compta Y, Parkkinen L, O'Sullivan SS, et al. Lewy‐ and Alzheimer‐type pathologies in Parkinson's disease dementia: which is more important? Brain. 2011;134(Pt 5):1493‐1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov Disord. 2008;23(6):837‐844. [DOI] [PubMed] [Google Scholar]
- 12. Hurtig HI, Trojanowski JQ, Galvin J, et al. Alpha‐synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology. 2000;54(10):1916‐1921. [DOI] [PubMed] [Google Scholar]
- 13. Kotzbauer PT, Cairns NJ, Campbell MC, et al. Pathologic accumulation of alpha‐synuclein and Abeta in Parkinson disease patients with dementia. Arch Neurol. 2012;69(10):1326‐1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012;72(4):587‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jellinger KA. Dementia with Lewy bodies and Parkinson's disease‐dementia: current concepts and controversies. J Neural Transm. 2018;125(4):615‐650. [DOI] [PubMed] [Google Scholar]
- 16. Braak H, Rüb U, Jansen Steur EN, Del Tredici K, de Vos RA. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology. 2005;64(8):1404‐1410. [DOI] [PubMed] [Google Scholar]
- 17. Coughlin D, Xie SX, Liang M, et al. Cognitive and pathological influences of tau pathology in lewy body disorders. Ann Neurol. 2019;85(2):259‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18(4 Suppl):S1‐S2. [PubMed] [Google Scholar]
- 19. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goetz CG, Emre M, Dubois B. Parkinson's disease dementia: definitions, guidelines, and research perspectives in diagnosis. Ann Neurol. 2008;64(Suppl 2):S81‐S92. [DOI] [PubMed] [Google Scholar]
- 21. Janvin CC, Larsen JP, Salmon DP, Galasko D, Hugdahl K, Aarsland D. Cognitive profiles of individual patients with Parkinson's disease and dementia: comparison with dementia with lewy bodies and Alzheimer's disease. Mov Disord. 2006;21(3):337‐342. [DOI] [PubMed] [Google Scholar]
- 22. Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson's disease. Lancet Neurol. 2010;9(12):1200‐1213. [DOI] [PubMed] [Google Scholar]
- 23. Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord. 2007;22(12):1689‐1707; quiz 837. [DOI] [PubMed] [Google Scholar]
- 24. Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson's disease: prevalence, phenomenology and risk factors. Brain. 2000r;123(Pt 4):733‐745. [DOI] [PubMed] [Google Scholar]
- 25. Bosboom JL, Stoffers D, Wolters E. Cognitive dysfunction and dementia in Parkinson's disease. J Neural Transm. 2004;111(10–11):1303‐1315. [DOI] [PubMed] [Google Scholar]
- 26. Troster AI. Neuropsychological characteristics of dementia with Lewy bodies and Parkinson's disease with dementia: differentiation, early detection, and implications for "mild cognitive impairment" and biomarkers. Neuropsychol Rev. 2008;18(1):103‐119. [DOI] [PubMed] [Google Scholar]
- 27. Hall H, Reyes S, Landeck N, et al. Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson's disease. Brain. 2014;137(Pt 9):2493‐2508. [DOI] [PubMed] [Google Scholar]
- 28. Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337‐351. [DOI] [PubMed] [Google Scholar]
- 29. Firbank MJ, Colloby SJ, Burn DJ, McKeith IG, O'Brien JT. Regional cerebral blood flow in Parkinson's disease with and without dementia. NeuroImage. 2003;20(2):1309‐1319. [DOI] [PubMed] [Google Scholar]
- 30. Mentis MJ, McIntosh AR, Perrine K, et al. Relationships among the metabolic patterns that correlate with mnemonic, visuospatial, and mood symptoms in Parkinson's disease. Am J Psychiatry. 2002;159(5):746‐754. [DOI] [PubMed] [Google Scholar]
- 31. Pereira JB, Junque C, Marti MJ, Ramirez‐Ruiz B, Bargallo N, Tolosa E. Neuroanatomical substrate of visuospatial and visuoperceptual impairment in Parkinson's disease. Mov Disord. 2009;24(8):1193‐1199. [DOI] [PubMed] [Google Scholar]
- 32. Junque C, Ramirez‐Ruiz B, Tolosa E, et al. Amygdalar and hippocampal MRI volumetric reductions in Parkinson's disease with dementia. Mov Disord. 2005;20(5):540‐544. [DOI] [PubMed] [Google Scholar]
- 33. Kalaitzakis ME, Christian LM, Moran LB, Graeber MB, Pearce RK, Gentleman SM. Dementia and visual hallucinations associated with limbic pathology in Parkinson's disease. Parkinsonism Relat Disord. 2009;15(3):196‐204. [DOI] [PubMed] [Google Scholar]
- 34. Ballard CG, Aarsland D, McKeith I, et al. Fluctuations in attention: PD dementia vs DLB with parkinsonism. Neurology. 2002;59(11):1714‐1720. [DOI] [PubMed] [Google Scholar]
- 35. Boulougouris V, Tsaltas E. Serotonergic and dopaminergic modulation of attentional processes. Prog Brain Res. 2008;172:517‐542. [DOI] [PubMed] [Google Scholar]
- 36. Armstrong RA, Kotzbauer PT, Perlmutter JS, et al. A quantitative study of alpha‐synuclein pathology in fifteen cases of dementia associated with Parkinson disease. J Neural Transm. 2014;121(2):171‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferman TJ, Aoki N, Crook JE, et al. The limbic and neocortical contribution of α‐synuclein, tau, and amyloid β to disease duration in dementia with Lewy bodies. Alzheimer's Dementia. 2018;14(3):330‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buddhala C, Loftin SK, Kuley BM, et al. Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Ann Clin Transl Neurol. 2015;2(10):949‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18(4 Suppl):S1‐S2. [PubMed] [Google Scholar]
- 40. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82(4):239‐259. [DOI] [PubMed] [Google Scholar]
- 42. Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease‐related pathology. Cell Tissue Res. 2004;318(1):121‐134. [DOI] [PubMed] [Google Scholar]
- 43. Cairns NJ, Taylor‐Reinwald L, Morris JC, Alzheimer's Disease Neuroimaging I . Autopsy consent, brain collection, and standardized neuropathologic assessment of ADNI participants: the essential role of the neuropathology core. Alzheimers Dement. 2010;6(3):274‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Khachaturian ZS. Diagnosis of Alzheimer's disease. ArchNeurol. 1985;42(11):1097‐1105. [DOI] [PubMed] [Google Scholar]
- 45. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863‐1872. [DOI] [PubMed] [Google Scholar]
- 46. McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113‐1124. [DOI] [PubMed] [Google Scholar]
- 47. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41(4):479‐486. [DOI] [PubMed] [Google Scholar]
- 48. Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56(1):33‐39. [DOI] [PubMed] [Google Scholar]
- 49. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico‐pathological study of 100 cases. J Neurol Neurosur Psychiatry. 1992;55(3):181‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Morris JC. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr. 1997;9(Suppl 1):173‐176; discussion 7–8. [DOI] [PubMed] [Google Scholar]
- 51. Fahn S, Elton RL, UPDRS Program Members . Unified Parkinson's disease rating scale. In: Marsden CD, Goldstein M, Caine DB, editor. Recent developments in Parkinson's disease. Florham Park, NJ: Macmillan HealthCare Information; 1987. p. 153‐163, 293‐304. [Google Scholar]
- 52. Holm S. A Simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6(2):65‐70. [Google Scholar]
- 53. Davis AA, Inman CE, Wargel ZM, et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med. 2020;12(529). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ballard C, Ziabreva I, Perry R, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology. 2006;67(11):1931‐1934. [DOI] [PubMed] [Google Scholar]
- 55. Braak H, Rub U, Jansen Steur EN, Del TK, de Vos RA. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology. 2005;64(8):1404‐1410. [DOI] [PubMed] [Google Scholar]
- 56. Jellinger KA, Attems J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol. 2008;115(4):427‐436. [DOI] [PubMed] [Google Scholar]
- 57. Sabbagh MN, Adler CH, Lahti TJ, et al. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord. 2009;23(3):295‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cirrito JR, Wallace CE, Yan P, et al. Effect of escitalopram on Abeta levels and plaque load in an Alzheimer mouse model. Neurology. 2020;95(19):e2666‐e2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Price JL, Morris JC. Tangles and plaques in nondemented aging and "preclinical" Alzheimer's disease. AnnNeurol. 1999;45(3):358‐368. [DOI] [PubMed] [Google Scholar]
- 60. Spotorno N, Coughlin DG, Olm CA, et al. Tau pathology associates with in vivo cortical thinning in Lewy body disorders. Ann Clin Transl Neurol. 2020;7(12):2342‐2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fujishiro H, Ferman TJ, Boeve BF, et al. Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol. 2008;67(7):649‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Siderowf A, Xie SX, Hurtig H, et al. CSF amyloid beta 1–42 predicts cognitive decline in Parkinson disease. Neurology. 2010;75(12):1055‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shahid M, Kim J, Leaver K, et al. An increased rate of longitudinal cognitive decline is observed in Parkinson's disease patients with low CSF Aß42 and an APOE ε4 allele. Neurobiol Dis. 2019;127:278‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lashley T, Holton JL, Gray E, et al. Cortical alpha‐synuclein load is associated with amyloid‐beta plaque burden in a subset of Parkinson's disease patients. Acta Neuropathol. 2008;115(4):417‐425. [DOI] [PubMed] [Google Scholar]
- 65. Pletnikova O, West N, Lee MK, et al. Abeta deposition is associated with enhanced cortical alpha‐synuclein lesions in Lewy body diseases. Neurobiol Aging. 2005;26(8):1183‐1192. [DOI] [PubMed] [Google Scholar]
- 66. Buddhala C, Campbell MC, Perlmutter JS, Kotzbauer PT. Correlation between decreased CSF alpha‐synuclein and Abeta(1)(‐)(4)(2) in Parkinson disease. Neurobiol Aging. 2015;36(1):476‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bagchi DP, Yu L, Perlmutter JS, et al. Binding of the Radioligand SIL23 to alpha‐Synuclein Fibrils in Parkinson Disease Brain Tissue Establishes Feasibility and Screening Approaches for Developing a Parkinson Disease Imaging Agent. PLoS One. 2013;8(2):e55031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yamasaki TR, Holmes BB, Furman JL, et al. Parkinson's disease and multiple system atrophy have distinct alpha‐synuclein seed characteristics. J Biol Chem. 2019;294(3):1045‐1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Materials and methods.
Figure S1. Representative standard curves.
Figure S2. Regional distribution of insoluble α‐syn concentrations in LBD cases as measured by the Syn211‐FL140 ELISA.
Figure S3. Correlation between α‐syn concentration measurements obtained by the Syn1‐13G5B ELISA and the Syn211‐FL140 ELISA in LBD cases.
Figure S4. Correlations between α‐syn (Syn211‐FL140 ELISA) and p‐α‐syn measurements in LBD cases.
Figure S5. Correlations between α‐syn (Syn211‐FL140 ELISA) and Aβ concentrations in LBD cases.
Figure S6. Comparison of insoluble phospho‐α‐syn concentrations (pSyn/81A‐13G5B) between LBD with measurable Aβ and LBD cases without measurable Aβ.
Figure S7. Correlations between p‐α‐syn (pSyn/81A‐Syn1B ELISA) and Aβ fibril concentrations in LBD cases.
Figure S8. Regional distribution of insoluble tau concentrations.
Figure S9. Correlations between phospho‐tau and tau levels in LBD cases.
Figure S10. Relationship between ELISA measurements and ICH‐based scoring of pathological α‐syn, Aβ, and tau accumulation.
Figure S11. Representative photomicrographs of immunohistochemistry.
Table S1. Capture and detection antibodies used in the ELISA assays.
Table S2. Assay performance characteristics of ELISA assays.
Table S3. Clinical and Pathologic Information for Study Participants.
Table S4. Insoluble protein data from ELISAs.