

Abstract
We describe an engineered violet fluorescent protein from the lancelet Branchiostoma floridae (bfVFP). This is the first example of a GFP‐like fluorescent protein with a stable fluorescent chromophore lacking an imidazolinone ring; instead, it consists of oxidized tyrosine 68 flanked by glycine 67 and alanine 69. bfVFP contains the simplest chromophore reported in fluorescent proteins and was generated from the yellow protein lanFP10A2 by two synergetic mutations, S148H and C166I. The chromophore structure was confirmed crystallographically and by high‐resolution mass spectrometry. The photophysical characteristics of bfVFP (323/430 nm, quantum yield 0.33, and E c 14,300 M−1 cm−1) make it potentially useful for multicolor experiments to expand the excitation range of available FP biomarkers and Förster resonance energy transfer with blue and cyan fluorescent protein acceptors.
Keywords: chromophore, crystal structure, fluorescent protein, gene reporter, lancelet, mutagenesis, protein engineering
Short abstract
1. INTRODUCTION
Fluorescent proteins (FPs) are essential molecular tools widely used to study cellular processes of gene expression, protein localization and trafficking, protein–protein interactions, sensing of intracellular metabolites, and pH, to name a few. 1 These applications rely on the ability of FPs to form a fluorescent chromophore that matures in the course of an autocatalytic posttranslational modification. 2 In the original Aequorea victoria, GFP (avGFP) consists of the chromophore‐forming triad of Ser65, Tyr66, and Gly67. 3 Based on the body of structural research, it is widely accepted that chromophore maturation consists of three consecutive steps: cyclization, dehydration, and oxidation. 4 , 5 , 6 Most of the FPs reported to date emit in the green–red range of 480–700 nm and contain a 4‐(p‐hydroxybenzylidene)‐5‐imidazolinone moiety, where the second chromophore‐forming residue is tyrosine. 7 , 8 The chromophores of blue‐emitting FPs (BFPs, 420–480 nm) are more diverse and can be divided into three groups according to their chromophore structure. 9 The first group contains an aromatic amino acid different from tyrosine as the second chromophore‐forming residue: phenylalanine (e.g., Sirius), 10 histidine (e.g., azurite), 11 or tryptophan (e.g., SCFP3A). 12 The second group has tyrosine‐based chromophores, which, unlike chromophores of avGFP and GFP‐like FPs, are unable to undergo excited state proton transfer when excited with long‐wavelength UV light. 9 The third group, represented by mTagBFP (402/457 nm, quantum yield [QY] 0.63, E C 52,000 M−1 cm−1), contains an unconventional RFP‐like chromophore that skipped Tyr66 oxidation. 13 , 14 In the absence of conjugation between hydroxybenzylidene and imidazolinone rings, electronic conjugation of the chromophore is notably shortened. An mTagBFP‐like chromophore containing leucine as the second chromophore‐forming amino acid was observed in the blue fluorescent protein t‐shBFP derived from the purple chromoprotein of Stichodactyla haddoni. 15 Here, we describe a bright violet‐emitting FP (bfVFP, 323/430 nm) engineered from the previously reported Branchiostoma floridae yellow FP lanFP10A. bfVFP is the second violet‐emitting protein reported since 2009 (Sirius, 355/427 nm, QY 0.24, E C 15,000 M−1 cm−1). 10 X‐ray structural analysis and high‐resolution mass spectrometry revealed that bfVFP contains an unusual chromophore consisting of a single oxidized tyrosine that has never been reported for GFP‐like FPs.
2. RESULTS AND DISCUSSION
2.1. Improving the expression/solubility of lanFP10A
In a previous report, we described the assembly of two synthetic genes coding for the hypothetical proteins lanFP6A and lanFP10A, 16 found along with other GFP‐like proteins in the genome of the lancelet Branchiostoma floridae. 17 These proteins were selected to study the role of alanine as the third chromophore‐forming residue instead of the canonical glycine found in most FPs. 8 To our surprise, lanFP10A turned out to be a poorly yellow FP, tolerating the presence of Ala67 in the GFP‐type chromophore. On the other hand, lanFP6A, which shares 83.9% sequence identity with lanFP10A, did not have a functional chromophore. Following this finding, we demonstrated that the chromophore of lanFP6A undergoes complete autohydrolysis, resulting in a nonfluorescent colorless protein. 18 Western blot analysis of E. coli cells expressing lanFP6A and lanFP10A showed that both were poorly soluble compared to GFP‐like proteins from jellyfish and corals. It also revealed that lanFP6A was more soluble than lanFP10A. 16 At this point, we realized that any attempt to improve or modify the properties of lanFP10A would first require an improvement of its expression/solubility.
In vitro recombination 19 of lanFP10A and lanFP6A, along with replacement of the first two amino acid residues MP‐ with the mNeonGreen N‐termini of MASKEEDNMAS‐, 20 , 21 resulted in lanFP10A2 (Figure 1). Unlike lanFP10A, E. coli colonies expressing lanFP10A2 had an intense yellow color. After protein purification, we determined that the production of lanFP10A2 remarkably improved from 9.7 to 422.9 mg/L. 22 Replacement of the starting sequence of MASKEEDNMAS‐ in lanFP10A2 with the original MP‐ resulted in the lanFP10A3 variant expressed at 75.4 mg/L. This level of expression is 7.7‐fold higher than lanFP10A and 5.6‐fold lower than lanFP10A2, demonstrating the expression tag role of the DNA sequence encoding the starting peptide MASKEEDNMAS. 21
FIGURE 1.
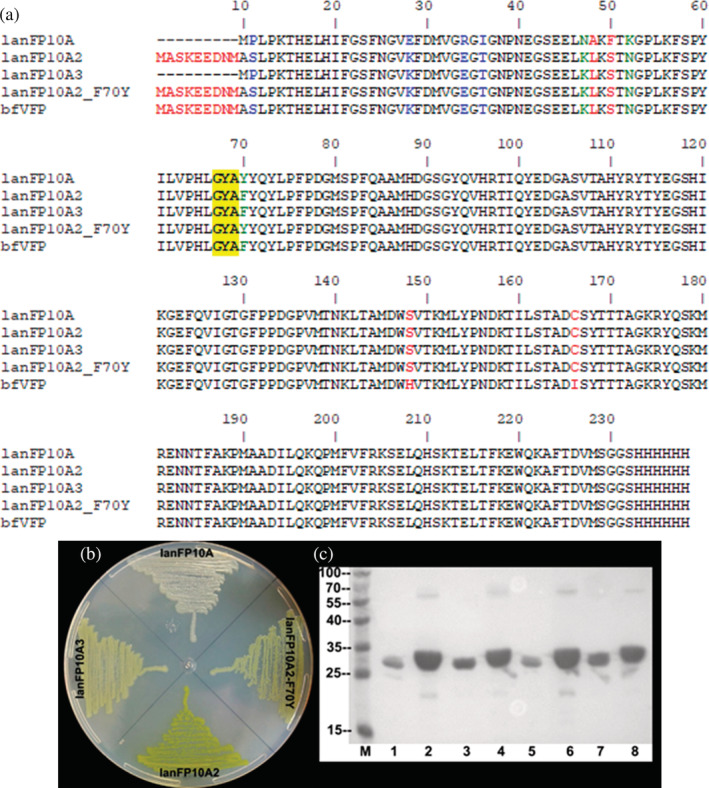
Improving the solubility of lanFP10A. (a) Amino acid sequence of proteins generated during improvement of solubility of lanFP10A; the chromophore‐forming triad is highlighted in yellow. bfVFP was added to the sequence alignment. (b) Phenotype of E. coli cells expressing the proteins whose sequence is shown above. (c) SDS–PAGE of cells expressing the proteins shown above revealed by Western blot using anti‐His antibody coupled to alkaline phosphatase. M: protein marker, 1 and 2: lanFP10A, 3 and 4 lanFP10A2, 5 and 6: lanFP10A3, 7 and 8: lanFP10A2‐F70Y. Samples 1, 3, 5, and 7: soluble fractions; samples 2, 4, 6, and 8: total extracts
Similar to lanFP10A, 16 the absorption spectrum of lanFP10A2 did not overlap with its excitation spectrum, suggesting the coexistence of two protein species in different proportions: the major one contained a nonfluorescent chromophore displaying maximum absorption at 463 nm, and the minor one contained a fluorescent chromophore with excitation at 492 nm and emission at 503 nm (Figure S1). Determination of the extinction coefficient (E c) and QY for lanFP10A2 (42,068 M−1 cm−1 and 0.003, respectively) confirmed the poor fluorescence of the protein, which resembled chromoproteins. 23 , 24
2.2. Generation and analysis of bfVFP
Once the solubility issue of lanFP10A was resolved, we focused on improving its fluorescence. For this goal, we used a similar approach to the pioneering work reported by Bulina et al., where mutagenesis of the internal amino acids Ala144 and Ser159 of the nonfluorescent chromoprotein Anemonia sulcata (asCP) converted the protein into some fluorescent variants. 25 Additionally, mutagenesis of the equivalent amino acids Ser146, Ile161, and Lys163 of the fluorescent protein DsRed converted the protein into a nonfluorescent chromoprotein. These experiments demonstrated that not only chromophore‐forming amino acids but also amino acids located in the spatially closest vicinity of the chromophore determine the photophysical properties of fluorescent proteins. As Chudakov et al. accurately cited, residues located at positions equivalent to His148, Phe165, and Ile167 in avGFP are the primary determinants of the protonation state of the chromophore, its polarization, its spatial cis or trans configuration, and its rotational freedom. 26 Therefore, variations in these amino acids can modify the absorption, excitation and emission spectra, converting proteins into fluorescent or nonfluorescent forms.
An alignment of the amino acid sequences of asCP and DsRed versus lanFP10A2 (Figure S2) revealed that Ser148, Ala164, and Cys166 were the equivalent amino acid residues in our target protein lanFP10A2. Combinatorial saturation mutagenesis of these three amino acid residues via the oligonucleotide method previously reported by our group 27 yielded an experimental library of 5 × 104 transformants encompassing 8,000 different protein variants (203). The library was spread on several LB plates and incubated at 37°C for 24 hr. The subsequent screening of the library revealed some greenish fluorescent colonies when the dishes were irradiated with blue light and visualized through an orange filter. We carried out DNA sequencing of some of these colonies and purified and characterized the encoded FPs (Figure S3 and Table S1). As expected, these variants were clearly more fluorescent than the parental protein lanFP10A2. Because all of them were poorly fluorescent compared with the classical enhanced green fluorescent protein (EGFP), nothing else was done with these variants. EGFP is an improved variant of avGFP bearing the mutations F64L and S65T. 28
However, when the same plates were visualized under 302 nm UV transillumination, we discovered that some colonies displayed interesting violet fluorescence. DNA sequencing of the recovered plasmids from four colonies showed that they all contained the same lanFP10A2‐S148H/C166I mutant. The new protein was named Branchiostoma floridae Violet Fluorescent Protein (bfVFP) to honor the organism that encoded the parental protein lanFP10A. 16 , 17 Structural comparison of lanFP10A (PDB ID: 6M9X) 18 and EGFP (PDB ID: 2Y0G) 29 demonstrated that His148 and Ile166 in bfVFP are equivalent to His148 and Ile167 in EGFP. However, the mutation G67A in the third chromophore‐forming residue of EGFP chromophore produced an unstable nonfluorescent protein as previously described, 16 , 30 contrary to bfVFP stability. BfVFP is so stable that has remained fluorescent after 3 years of storage at 4°C and at least 3 hr of exposition over a 302 nm transilluminator.
BfVFP has an excellent level of expression and solubility and was purified at 328 mg/L of culture. It shows higher expression levels than two previously reported BFPs, mTagBFP2 14 and Sirius, 10 obtained at 174.3 and 30.7 mg/L, respectively. Under irradiation with 302, 365, and 400 nm light, bfVFP, Sirius, and mTagBFP2 produced a distinct fluorescence pattern (Figure 2). bfVFP displayed a maximum absorption/excitation at 323 nm and maximum emission at 430 nm. It has a Stokes shift of 107 nm and the shortest excitation wavelength ever reported for FPs.
FIGURE 2.
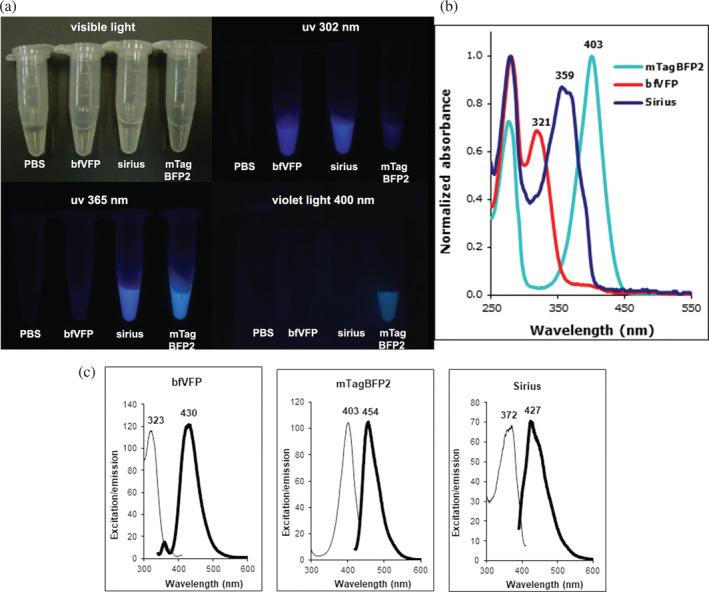
bfVFP compared with two other blue fluorescent proteins: Sirius and mTagBFP2. (a) Visualization of the pure proteins at 1 mg/ml under irradiation at different lights. (b) UV–Vis absorbance of the three BFPs. (c) Excitation and emission spectra of the three BFPs
The photophysical characteristics of bfVFP suggested that its chromophore had a different structure than other reported FPs, 7 including blue‐emitting mTagBFP2 and Sirius. The E c and QY of bfVFP, Sirius, and mTagBFP2 determined under the same experimental conditions show that bfVFP is slightly brighter than Sirius and dimmer than mTagBFP2 (Table 1). Due to 10.7‐fold higher expression, E. coli cells expressing bfVFP appear 15‐fold brighter than those transformed with Sirius. Size exclusion chromatography (SEC) suggests that bfVFP is a dimer, whereas lanFP10A2 is a tetramer (Figure S4). bfVFP is stable in the pH range of 4–9. Exposure to basic pH (10–12) results in the rise of a small 487 nm absorbance peak, indicating that a fraction of the protein undergoes the formation of the GFP‐type chromophore (Figure S5).
TABLE 1.
Fluorescent properties of bfVFP compared with Sirius and mTagBFP2
| FP | Ex (nm) | Em (nm) | EC (M−1 cm−1) | QY | Brightness | Relative expression | Oligomeric state | Maturation time (hr) a |
|---|---|---|---|---|---|---|---|---|
| bfVFP | 323 | 430 | 14,500 | 0.33 | 4.72 | 10.7 | Dimer | 20 |
| Sirius | 372 | 427 | 18,900 | 0.18 | 3.4 | 1 | Monomer | 20 |
| mTagBFP | 403 | 454 | 27,900 | 0.64 | 17.7 | 5.7 | Monomer | 16 |
Time when 5 ml cultures reached their maximum fluorescence at 37°C and agitation at 200 rpm.
2.3. Structural determination of lanFP10A2 and bfVFP
The X‐ray crystal structures of lanFP10A2 (PDB ID: 7FSA) and bfVFP (PDB ID: 7FS9) were solved at 1.65 and 1.8 Å resolution, respectively (Tables S2 and S3). Both proteins have a typical GFP‐like FP fold: an 11‐stranded β‐barrel with an α‐helix twined around the central axis of the barrel and the chromophore located in the center of the α‐helix.
LanFP10A2 was prepared from lanFP10A, which was the first example of a successful GFP‐like FP with alanine as the third chromophore‐forming residue. 16 , 18 LanFP10A2 differs from lanFP10A by 10 positions: M10A/P11S/E28K/R34E/I36T/N47K/A48L/F50S/K52N/Y70F (lanFP10A2 numbering in Figure 1). All but Y70F mutations are located on the surface of the protein (Figure 3). Substitutions P11S, E28K, R34E, I36T, and N47K alter the surface charge, likely improving the solubility of lanFP10A2. A48L contributes to the giant hydrophobic cluster comprising Leu18, Ile20, Phe24, Phe29, Met31, Leu46, Leu48, Leu55, Leu57, Leu62, Leu66, Phe124, Val126, and Leu215, which isolates the chromophore from the solvent (Figure S6a). F50S and K52N change the conformation of the polypeptide chain in loop 46–57, shifting it ~2 Å closer to the β‐strand formed by residues 27–38 (Figure S6b). Phe70 removes a steric conflict with Leu18 present in parental lanFP10A, where the distances between the Tyr70 hydroxyl and Leu18 Cα and Cβ atoms are 3.2 and 3.1 Å, respectively (Figure S6c). In lanFP10A2, this conflict is resolved, and the respective distances between the Phe70 Cz atom and Leu18 Cα and Cβ atoms are 4.7 and 4.2 Å (Figure S6d). The importance of the mutation Y70F in lanFP10A2 was confirmed when this mutation was reverted to the original amino acid, giving rise to the mutant lanFP10A2‐F70Y. This protein was produced at 168.3 mg/L, demonstrating a 2.5‐fold drop in expression efficiency compared with LanFP10A2 (Figure 1). Both lanFP10A and lanFP10A2 have tyrosines at positions 71 and 111, which are necessary for the successful maturation of the Gly—Tyr—Ala chromophore. 18 The chromophore position is stabilized by four direct H‐bonds between the chromophore and its immediate environment: 2.6 Å H‐bond between the hydroxyl of p‐hydroxyphenyl ring and Ser148; 2.6 Å H‐bond between N2 and Glu219; 3.2 Å H‐bond between the carbonyl oxygen of imidazolone ring and Arg97; and 2.5 Å H‐bond between C═O of Ala69 and Tyr113 (Figure S6e).
FIGURE 3.
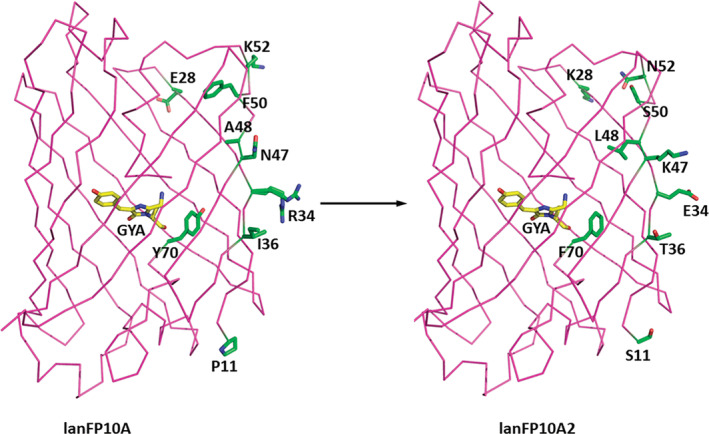
Crystal structure of lanFP10A2 (PDB ID: 7FSA) showing the amino acid changes acquired during the recombination PCR process to improve the solubility of lanFP10A (PDB ID: 6M9X)
As mentioned above, bfVFP differs from lanFP10A2 in two positions: 148 and 166. These mutations, S148H and C166I, caused a dramatic change in the spatial position of the Gly—Tyr—Ala tripeptide that resulted in multiple conformational changes for many bulky residues in the nearest chromophore environment seen in the overlap of lanFP10A2 and bfVFP structures (Figure S7a). Replacement of C166I resulted in a domino‐effect conformational change for the following residues in the nearest chromophore environment: Met180, Arg97, Tyr111, Tyr113, Phe124, and Tyr115 (Figure S7b). The same effect derived from S148H replacement impacted Tyr68, Phe70, and Tyr71 (Figure S7c). Interestingly, bfVFP has a unique chromophore that lacks an imidazolone ring (Figure 4a). It is an oxidized Tyr68 containing a double bond between the carbons Cα–Cβ, enabling conjugation of its aromatic ring and its own carbonyl group. The chromophore adopts a cis configuration, similar to most fluorescent chromophores. 26 The uniform occupancy of the chromophore moiety unequivocally indicates that oxidized tyrosine is the exclusive product of the posttranslational modification of bfVFP.
FIGURE 4.
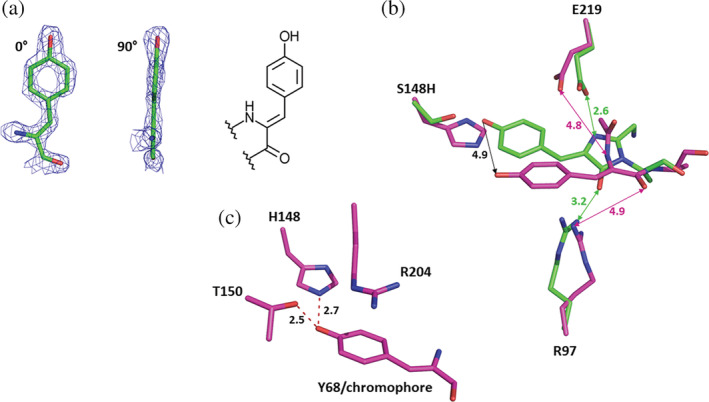
Structural analysis of bfVFP crystal. (a) Chemical structure of the chromophore found in bfVFP: oxidized tyrosine 68 in cis conformation. (b) Contrary to lanFP10A2 (shown in green), the chromophore‐forming triad in bfVFP (shown in purple) forms no H‐bonds with either Arg97 or Glu219. (c) The position of the bfVFP chromophore is stabilized by two H‐bonds between the p‐hydroxyphenyl ring and the NH group of His148 (2.8 Å) and the C═O of Thr150 (2.5 Å)
The absence of the imidazolone ring significantly shortens the conjugation system of the chromophore. It manifests in a 72 nm hypsochromic shift of bfVFP fluorescence (430 vs. 502 nm for lanFP10A2), making it one of the most blueshifted GFP‐like proteins. The residue responsible for the absence of the imidazolone ring is presumably His148, which pushes the p‐hydroxyphenyl ring of the chromophore ~4.9 Å away from its conventional position in lanFP10A2 (Figure 4b). This moves the entire Gly—Tyr—Ala tripeptide away from catalytic Glu219 and Arg97 sites: the distance between the Glu219 carboxylic group and N67 (Gly67) becomes 4.7 Å (2.6 Å in lanFP10A2), and between Arg97 and the carbonyl oxygen of Ala69 is 4.9 Å (3.2 Å in lanFP10A2). The displacement is accompanied by distortion of the Gly—Tyr—Ala tripeptide, resulting in a 4.4 Å distance between the carbonyl group of Gly67 and the amino group of Ala69, which makes cyclization of the imidazolone ring unlikely. The position of the chromophore is stabilized by two H‐bonds between the p‐hydroxyphenyl ring and the NH group of His148 (2.8 Å) and C═O of T150 (2.5 Å) and cation–π stacking interactions with Arg204 (Figure 4c). The most surprising feature of the resulting chromophore is not the lack of an imidazolone ring but the double bond between Cα and Cβ atoms of Tyr68 (Figure 4a), suggesting that while the maturation Gly—Tyr—Ala chromophore bypassed cyclization and dehydration steps, it did not avoid oxidation, which is even more intriguing since the chromophore‐forming triad in bfVFP forms no H‐bonds with either Arg97 or Glu219.
Analysis of mature lanFP10A2 by high‐performance electrospray ionization mass spectrometry (ESI‐MS) yielded a value of 26,678 Da, whereas the calculated molecular weight of its unmodified polypeptide chain was 26,698 Da (Figure S8). The loss of 20 Da by mature lanFP10A2 during posttranslational modification confirms the GFP‐type chromophore revealed by the X‐ray crystal structure described above. ESI‐MS of pure bfVFP gave a value of 26,755 Da for the protein (Figure S8). This result is in good agreement with the calculated value of 26,757 for the unmodified polypeptide. The loss of two Daltons during the posttranslational reaction corresponds to dehydrogenation and confirms an oxidized Tyr68 chromophore.
2.4. Amino acid determinants for the bfVFP phenotype
In our earlier works, 16 , 18 we demonstrated that the lanFP10 scaffold could support the maturation of GFP‐type chromophores with both Gly69 and Ala69 as the third chromophore‐forming residue. bfVFP differs from lanFP10A2 by two positions, His148 and Ile166, causing a dramatic change in its posttranslational modification. To verify whether Ala69 also contributes to this change, we considered 12 mutants probing positions 69, 148, and 166. Reverse single point mutations A69G, H148S, and I166C all resulted in the formation of the GFP‐type chromophore with a variable ratio of anionic and neutral species (Figure 5). Single mutants bfVFP‐H148F, bfVFP‐H148W, bfVFP‐H148Y, and the double mutant bfVFP‐Y68H/H148Y turned out to be nonfluorescent, despite a notably higher solubility than bfVFP (Figure S9). The absorbance spectra of bfVFP mutants I166L, I166V, and I166M indicated the presence of a mixture of bfVFP‐ and GFP‐type chromophores (Figure 5). In bfVFP‐I166F, bulky Phe166 inhibited maturation of the GFP‐like chromophore and yielded only a tiny fraction of the bfVFP‐like species, despite the high expression levels (Figure S9). BfVFP‐I166A had an improved level of expression but was completely insoluble and unavailable for characterization.
FIGURE 5.
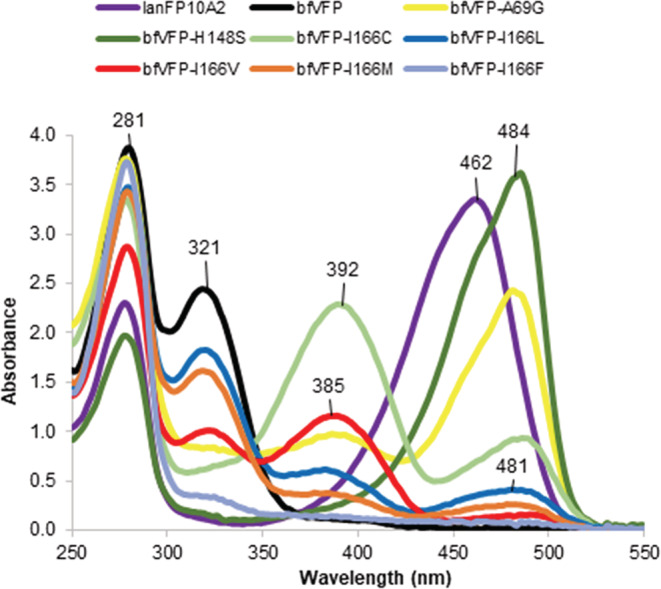
Absorbance spectra of lanFP10A2, bfVFP, and its mutants
The driving force of the posttranslational modification of GFP‐like proteins is a steric conflict between the unmodified chromophore‐forming tripeptide in a constrained kinked conformation and the tight confinement of the β‐barrel. 31 , 32 Typically, the conflict is resolved by the maturation or hydrolysis of the chromophore accompanied by the tripeptide shortening. 16 , 18 , 32 , 33 In bfVFP, the conformation of GYA tripeptide is affected by His148 and Ile166 on the one side, and by Ala69 that is bulkier than canonical Gly on the other side. Although in the crystal structure of bfVFP we see the final result of GYA oxidation, having uncyclized tripeptide, we argue that a combination of His148, Ala69, and Ile166 shifts GYA away from the catalytic residues Glu219 and Arg97 (Figure 4), hampering the conventional chromophore maturation. Replacement of His148 and Ala69 with the original Ser and Gly residues yielded anionic GFP‐like chromophore and made catalytic residues available for interactions with GYA tripeptide. 30 , 34 The contribution of Ile166 in the formation of bfVFP chromophore is more complex. Reverse mutation bfVFP‐I161C produced a 3:1 mixture of protonated and deprotonated GFP‐like chromophores. Val, Leu, and Met in position 166 yielded a mixture of bfVFP and GFP‐like chromophores. We speculate that less bulky residues 166 present GYA tripeptide with two alternative maturation pathways: cyclization and formation of GFP‐like chromophore or oxidation and formation of bfVFP‐like species (Scheme 1). The simultaneous presence of the two chromophore species, the ratio of which does not change with time (the spectra were recorded several days after purification of mutant proteins), suggests that once oxidized, Tyr68 cannot participate in cyclization for production of a GFP‐like chromophore, ruling out the oxidized mechanism proposed by density functional calculations. 35
SCHEME 1.
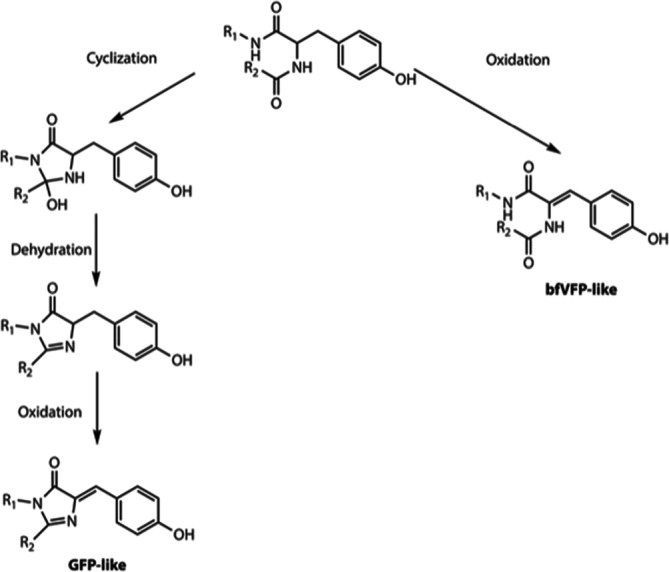
Alternative pathways for maturation of the GYA tripeptide in bfVFP and its mutants
Many groups studied chromophore maturation; these attempts predominantly resulted in the comparison of cyclized, uncyclized, and hydrolyzed chromophore‐forming tripeptide. Direct structural evidence for the chromophore in the state preceding the oxidation is scarce, and little is known about what it requires except for the presence of oxygen. 13 , 34 , 35 , 36 , 37 , 38 For the traditional mechanism of cyclization–dehydration–oxidation, oxidation is considered the rate‐limiting step, and it was argued whether it precedes dehydration or follows it. In our earlier work in aceGFP‐G222E variant, we captured the chromophore in the native unoxidized form as a noncoplanar, nonconjugated two‐ring structure (avGFP numbering), showing that cyclization–dehydration–oxidation is a preferred route of GFP‐like chromophore maturation. 39 The immature aceGFP‐G222E could be irreversibly oxidized to a green fluorescent state by UV‐irradiation, while the nearest chromophore environment did not change in any meaningful way. The overall consensus was that the brightly fluorescing chromophores of GFP‐like proteins result from an oxidative tyrosine modification mediated by peptide cyclization reaction. 40
Our findings present a unique example where, skipping cyclization and dehydration, GYA tripeptide of bfVFP undergoes oxidation, yielding a new violet‐emitting chromophore. Autocatalytic oxidation of Tyr68 without the typically observed imidazolinone or interactions with the catalytic Glu219 and Arg97 is unprecedented and has not been reported to date. The case of bfVFP and its mutants exploring position 166 demonstrates that once oxidized, Tyr68 cannot participate in the cyclization and formation of GFP‐like chromophore.
Currently, bfVFP surpasses Sirius as the most blueshifted FP; it has higher QY, brightness, and expression levels. bfVFP can be potentially used in multicolor experiments to expand the excitation range of available FP biomarkers and as a donor in Förster resonance energy transfer with blue and cyan fluorescent protein acceptors. Undoubtedly, the photophysical characteristics of bfVFP can be further improved by directed evolution, as was done for other FPs. 7 , 26 , 41
3. CONCLUSIONS
We described an engineered violet fluorescent protein from Branchiostoma floridae (bfVFP) with the chromophore of an oxidized tyrosine lacking the imidazolinone ring. bfVFP was generated from the highly soluble yellow protein lanFP10A2 by two synergetic mutations, S148H and C166I. Extensive mutagenesis along with X‐ray crystal structure analysis suggests that a tight environment created by His148 and Ala69 distorts and shifts the chromophore‐forming tripeptide away from catalytic Glu219. To our surprise, the GYA tripeptide forgoes cyclization and dehydration but undergoes oxidation of the posttranslational modification, yielding a new violet‐emitting chromophore. The photophysical characteristics of bfVFP are superior to those of the violet‐emitting fluorescent protein Sirius reported to date. They make bfVFP potentially useful for multicolor experiments to expand the excitation range of available FP biomarkers and Förster resonance energy transfer with blue and cyan fluorescent protein acceptors.
4. MATERIALS AND METHODS
4.1. LanFP10A solubility
An equimolar mixture of plasmids containing the genes encoding lanFP6A and lanFP10A (20 ng each) was subjected to limited PCR amplification using 50 pmol of the forward primer ACACAGGAACAGCATATGGCTAGCAAAGAAGAAgataacatggcgagcCTGCCAAAAACCCATGAAC‐TTCACATTTT and 50 pmol of the reverse primer ACTCACTATAGGGCGAATTG (named pJOQrv) under the following modified PCR conditions. Twenty‐five cycles were performed: denaturing at 94°C for 60 s, annealing at 58°C for 60 s, and extension at 72°C for 10 s instead of 60 s. This modification of the amplification time allows recombination between similar genes. 19 The ancillary reagents for the PCR were from New England Biolabs, including vent DNA polymerase. The forward primer was designed to anneal 21nt at the beginning of both genes, as well as to encode the GFP‐type amino termini MASKEEDNMAS‐ and the NdeI restriction site. The reverse primer was designed to anneal to the plasmid sequence at some nucleotides downstream from the stop codon of both genes, which also amplified the terminal XhoI restriction site. The library of recombined genes was cloned as an NdeI/XhoI insert in our previously described pJOQ plasmid, which expresses genes under constitutive conditions under the control of the trc promoter. 42 The library was transformed into E. coli electrocompetent cells, strain MC1061, and the transformants were cultured on LB plates supplemented with kanamycin at 30 μg/ml. The plates were incubated at 37°C for 20 hr. After this time, plasmids from yellow colonies were isolated and submitted for DNA sequencing to reveal the nature of the new genes. In this screening, LanFP10A2 was found.
4.2. Combinatorial mutagenesis of the amino acid residues Ser148, Ala164, and Cys166
Using the gene coding for lanFP10A2 as a template, the amino acid residues Ser148, Ala164, and Cys166 were subjected to combinatorial mutagenesis as previously reported using two mutagenic primers assembled by the resin‐splitting method. 27 Mutations in Ser148 were performed by partial PCR with the forward primer pJOQfw (CGGTTCTGGCAAATATTCTG), which pairs some bases upstream of the starting methionine codon, and the reverse mutagenic primer GGTTTTATCGTTCGGATACAGCATCTTCGTCACZZZCCAATCCATCGC‐GGTCAGTTTATT. In this nucleotide sequence, ZZZ represents a mixture of 20 anticodons coding for the 20 different natural amino acid residues. The second partial PCR was performed with the pJOQrv primer and the forward mutagenic primer ATGCTGTATCCGAACGATAAAACCATTCTGAGCACGXXXGACXXX‐TCTTATACCACGACGGCGGGT, where XXX represents a mixture of 20 direct codons for the 20 different amino acid residues. Both PCRs were purified by agarose gel electrophoresis, mixed and used as templates in a third PCR with the external primers pJOQfw and pJOQrv to assemble the complete mutant genes. The gene library was cloned and transformed as previously described for improvement of lanFP10A.
4.3. Site‐directed mutagenesis
The mutants lanFP10A2‐F70Y, bfVFP‐A69G, bfVFP‐H148S, bfVFP‐H148F, bfVFP‐H148W, bfVFP‐H148Y, bfVFP‐Y68H/H148Y, bfVFP‐I166C, bfVFP‐I166L, bfVFP‐I166V, bfVFP‐I166M, bfVFP‐I166A, and bfVFP‐I166F were constructed by site‐directed mutagenesis using the overlapping PCR approach in a similar mode as that for construction of the combinatorial library described above. 43 For each mutation, a pair of specific complimentary oligonucleotides was used to perform the two partial PCRs.
4.4. Protein analysis by electrophoresis
One colony of E. coli MC1061 cells expressing each target protein was used to inoculate 5 ml of LB supplemented with kanamycin. The cultures were incubated at 37°C for 24 hr, agitated at 200 rpm and then centrifuged at 13,000 rpm for 5 min. The supernatant was discarded, and the pellet was washed with 1 ml of PBS and resuspended in 500 μl of the same buffer. Twenty microliters of this suspension were withdrawn and centrifuged, the supernatant was removed with a micropipette, and the remaining pellet was treated with 20 μl of denaturing loading buffer and boiled for 5 min to constitute the total protein extract. The remaining suspension of cells was sonicated for 5 min to constitute the protein soluble fraction after centrifugation. For SDS–PAGE analysis of the soluble fraction or purified proteins, the samples were prepared by mixing 2.5 μl of each fraction, 2.5 μl of denaturing loading buffer and 5 μl of water. For denaturing conditions, the samples were boiled for 5 min before being loaded on the gel, whereas for semidenaturing conditions, the samples were loaded without boiling. Purified proteins analyzed by SDS–PAGE were visualized by staining with Coomassie blue, whereas total extracts and soluble fractions were analyzed by western blotting using alkaline phosphatase‐conjugated anti‐His to specifically detect histidine‐tagged proteins, which were revealed by the addition of the blue liquid substrate system BClP®/NBT.
4.5. Protein purification
Proteins expressed in E. coli strain MC1061 were produced in 1 L LB/km cultures grown at 37°C under agitation at 200 rpm for 24 hr. After this process, the cells were recovered by centrifugation, resuspended in 30 ml of PBS and lysed by sonication in two pulses of 4 min, with intermediate cooling over water ice for 30 min. After centrifugation at 13,000 rpm, the clear supernatant was loaded on three HisTrap HP/1 ml columns collocated in tandem (GE Health care), and the proteins were purified using an imidazole gradient from 0 to 300 mM in 30 min at a flow rate of 1 ml/min, with detection at 280 nm. Imidazole solutions were prepared in 100 mM phosphate buffer (pH 7.2) containing 0.5 M NaCl. Fractions containing the pure protein were combined, concentrated on centrifugal filter devices (Amicon Ultra4 10k, Millipore) and washed with PBS (2 × 4 ml) to remove the residual imidazole. The pure proteins were redissolved in 1.5 ml of PBS, and their concentration was determined by the bicinchoninic acid assay using the commercial QuantiPro™ kit from Sigma–Aldrich, as well as BSA standards from the same supplier. Protein concentration was determined by the procedure recommended by the supplier, measuring the absorbance at 562 nm on a DU 730 UV/Vis spectrophotometer from Beckman Coulter.
4.6. Protein analysis by SEC
Forty microliters of each purified protein, ranging in concentration from 2 to 5 mg/ml, was independently analyzed on a Superdex 200 10/300 GL column from Pharmacia using 0.1 M NaCl in 0.1 M phosphate buffer pH 7.2 as the mobile phase at a flow rate of 0.75 ml/min. Detection was achieved by UV absorption at 280 nm. The samples were also analyzed on a Superdex increase 75 10/300 GL column under the same running conditions.
4.7. Absorbance spectroscopy
The UV–visible spectra of pure proteins in PBS were obtained by recording the absorbance in the range 260–650 nm using silica quartz cuvettes and the same spectrophotometer mentioned above.
4.8. Fluorescence characterization
The excitation and emission spectra of the pure mutant proteins were recorded on an LS‐55 fluorescence spectrometer (Perkin Elmer). The emission spectrum of each protein was obtained by fixing the excitation wavelength at the maximum absorbance wavelength obtained by UV/Vis spectrophotometry and recording the emission in the range “excitation wavelength + 20 nm” up to 750 nm using 5‐nm emission and excitation slits. The excitation spectra were recorded in a similar mode by fixing the emission wavelength at 20 nm above its maximum and recording excitation from 350 nm to the maximum emission wavelength.
4.9. Extinction coefficient and quantum yield
The E c of pure proteins at their maximum absorbance peak was determined spectrophotometrically by measuring the absorbance of six duplicate dilutions. Plotting concentration (M) versus absorbance produced a linear graph whose slope was E c in units M−1 cm−1. Each of the dilutions was further diluted 25‐fold, and fluorescence was measured at the maximum emission wavelength with excitation at the maximum absorbance wavelength. The plot of absorbance versus fluorescence produced a linear graph with a particular slope.
As a standard to determine the QY of the mutants, we determined the relationship between absorbance (488 nm) and fluorescence (507 nm) for the pure protein EGFP, whose reported value is 0.60. 44 Dividing the sample's slope by EGFP's slope and multiplying by 0.60 gives the sample's QY.
4.10. Protein crystallization
For crystallization, lanFP10A2 and bfVFP were dialyzed against phosphate‐buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM K2HPO4 pH 7.4) and concentrated to 30.7 and 20.3 mg/ml, respectively. Initial crystallization conditions were found using a Mosquito drop setter (SPT Labtech). In its default setup, Mosquito mixed 0.4 μl of the protein solution with an equal amount of reservoir solution, and the drops were incubated against 140 μl of the same reservoir at 20°C for 3 days. Successful hits were further optimized manually. The crystals suitable for X‐ray study were grown by a hanging drop vapor diffusion method. Typically, 2 μl of protein was mixed with an equal volume of well solution and incubated against 500 μl of the same solution at 20°C for 2 weeks. The best lanFP10A2 and bfVFP crystals were grown from 0.08 M TRIS pH 8.0, 24% PEG 2000 MME and 1.35 M LiSO4, 0.09 M TRIS pH 8.5, respectively.
4.11. X‐ray data collection, structure solution, and crystallographic refinement
X‐ray diffraction data were collected at SER‐CAT beamline 22‐BM (Advanced Photon Source, Argonne National Laboratory, Argonne, IL). Prior to data collection, the crystals were briefly soaked in a cryoprotectant solution containing 20% glycerol and 80% reservoir solution and were flash‐cooled in a 100 K nitrogen stream. To minimize radiation damage, a helical data collection strategy was applied. Diffraction images were processed with HKL2000. 45 Crystal structures were solved by the molecular replacement method with MOLREP 46 using the coordinates of lanFP10A (PDB ID: 6M9X) as a search model. 18 Crystallographic refinement was performed with REFMAC5, 47 alternating with manual revision of the model with COOT. 48 Localization of water molecules was performed with COOT. Structure validation was performed with COOT and PROCHECK. 49 Crystallographic data and refinement statistics are given in Tables S2 and S3, respectively. The coordinates and structure factors of lanFP10A2 and bfVFP were deposited in the Protein Data Bank under accession codes 7SFA and 7SF9, respectively.
AUTHOR CONTRIBUTIONS
Paul Gaytán: Conceptualization (lead); data curation (equal); formal analysis (equal); funding acquisition (equal); investigation (equal); methodology (equal); supervision (equal); writing – original draft (equal); writing – review and editing (equal). Abigail Roldán‐Salgado: Conceptualization (supporting); data curation (equal); investigation (equal); methodology (equal). Liya Muslinkina: Formal analysis (equal); investigation (equal); methodology (equal); supervision (equal); writing – review and editing (equal). Sergei Pletnev: Formal analysis (equal); funding acquisition (equal); investigation (equal); supervision (equal); writing – review and editing (equal). Nadya Pletneva: Supervision (equal). Vladimir Pletnev: Data curation (equal); formal analysis (equal); funding acquisition (equal); writing – review and editing (equal).
Supporting information
TABLE S1: Characteristics of green fluorescent mutants of lanFP10A2 probing positions 148, 164, and 166
Table S2: Data collection statistics
Table S3: Refinement statistics
Figure S1: Absorption, excitation, and emission spectra of the pure fluorescent protein lanFP10A2
Figure S2: Amino acid sequence alignment of lanFP10A2 vs. asCP and DsRed
Figure S3: Purified fluorescent mutants of lanFP10A2
Figure S4: Experiments of size exclusion chromatography
Figure S5: Absorbance spectra of bfVFP at different pH values
Figure S6: Structural details observed in lanFP10A2 after improving the solubility of the parental protein lanFP10A
Figure S7: Overlap of lanFP10A2 and bfVFP structures
Figure S8. Mass spectra of the pure, mature proteins lanFP10A2 and bfVFP
Figure S9: SDS–PAGE analysis
ACKNOWLEDGMENTS
This project was supported by the Russian Foundation for Basic Research (grant 19‐04‐00107), the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases, National Institutes of Health, and internal resources from the DNA synthesis core facility of the Institute of Biotechnology, Universidad Nacional Autónoma de México. Diffraction experiments were carried out at synchrotron beamline 22‐BM of the Southeast Regional Collaborative Access Team (SER‐CAT), located at the Advanced Photon Source, Argonne National Laboratory. Use of the APS was supported by the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences under Contract No. W‐31‐109‐Eng‐38. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government. Technical assistance by Jorge Arturo Yáñez, Eugenio López‐Bustos, Santiago Becerra‐Ramírez, Ana Yanci Alarcón, and Leopoldo Güereca is highly appreciated. We are especially grateful to Dr Takuya Nishigaki and Dr Chris Wood who reviewed our manuscript kindly and thoroughly.
Roldán‐Salgado A, Muslinkina L, Pletnev S, Pletneva N, Pletnev V, Gaytán P. A novel violet fluorescent protein contains a unique oxidized tyrosine as the simplest chromophore ever reported in fluorescent proteins. Protein Science. 2022;31:688–700. 10.1002/pro.4265
Funding information Division of Intramural Research, National Institute of Allergy and Infectious Diseases; Russian Foundation for Basic Research, Grant/Award Number: 19‐04‐00107; Universidad Nacional Autónoma de México; National Institutes of Health
REFERENCES
- 1. Lippincott‐Schwartz J, Patterson GH. Development and use of fluorescent protein markers in living cells. Science. 2003;300:87–91. [DOI] [PubMed] [Google Scholar]
- 2. Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. [DOI] [PubMed] [Google Scholar]
- 3. Heim R, Prasher DC, Tsien RY. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci U S A. 1994;91:12501–12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang F, Moss LG, Phillips GN Jr. The molecular structure of green fluorescent protein. Nat Biotechnol. 1996;14:1246–1251. [DOI] [PubMed] [Google Scholar]
- 5. Bravaya KB, Subach OM, Korovina N, Verkhusha VV, Krylov AI. Insight into the common mechanism of the chromophore formation in the red fluorescent proteins: The elusive blue intermediate revealed. J Am Chem Soc. 2012;134:2807–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pletnev S, Subach FV, Dauter Z, Wlodawer A, Verkhusha VV. Understanding blue‐to‐red conversion in monomeric fluorescent timers and hydrolytic degradation of their chromophores. J Am Chem Soc. 2010;132:2243–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Olenych SG, Claxton NS, Ottenberg GK, Davidson MW. The fluorescent protein color palette. Curr Protoc Cell Biol Chapter 21, Unit 21 5. 2007;33:21.5.1–21.5.34. [DOI] [PubMed] [Google Scholar]
- 8. Subach FV, Verkhusha VV. Chromophore transformations in red fluorescent proteins. Chem Rev. 2012;112:4308–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ai HW, Shaner NC, Cheng Z, Tsien RY, Campbell RE. Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochemistry. 2007;46:5904–5910. [DOI] [PubMed] [Google Scholar]
- 10. Tomosugi W, Matsuda T, Tani T, et al. An ultramarine fluorescent protein with increased photostability and pH insensitivity. Nat Methods. 2009;6:351–353. [DOI] [PubMed] [Google Scholar]
- 11. Mena MA, Treynor TP, Mayo SL, Daugherty PS. Blue fluorescent proteins with enhanced brightness and photostability from a structurally targeted library. Nat Biotechnol. 2006;24:1569–1571. [DOI] [PubMed] [Google Scholar]
- 12. Goedhart J, von Stetten D, Noirclerc‐Savoye M, et al. Structure‐guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun. 2012;3:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Subach OM, Malashkevich VN, Zencheck WD, et al. Structural characterization of acylimine‐containing blue and red chromophores in mTagBFP and TagRFP fluorescent proteins. Chem Biol. 2010;17:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Subach OM, Gundorov IS, Yoshimura M, et al. Conversion of red fluorescent protein into a bright blue probe. Chem Biol. 2008;15:1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang HY, Ko TP, Chang YC, et al. Crystal structure of the blue fluorescent protein with a Leu‐Leu‐Gly tri‐peptide chromophore derived from the purple chromoprotein of Stichodactyla haddoni . Int J Biol Macromol. 2019;130:675–684. [DOI] [PubMed] [Google Scholar]
- 16. Roldan‐Salgado A, Sanchez‐Barreto C, Gaytan P. LanFP10‐A, first functional fluorescent protein whose chromophore contains the elusive mutation G67A. Gene. 2016;592, 281–290. [DOI] [PubMed] [Google Scholar]
- 17. Baumann D, Cook M, Ma L, et al. A family of GFP‐like proteins with different spectral properties in lancelet Branchiostoma floridae. Biol Direct. 2008;3:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muslinkina L, Roldán‐Salgado A, Gaytán P, et al. Structural factors enabling successful GFP‐like proteins with alanine as the third chromophore‐forming residue. J Mol Biol. 2019;431:1397–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao H, Giver L, Shao Z, Affholter JA, Arnold FH. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat Biotechnol. 1998;16:258–261. [DOI] [PubMed] [Google Scholar]
- 20. Rodriguez‐Mejia JL, Roldan‐Salgado A, Osuna J, Merino E, Gaytan P. A codon deletion at the beginning of green fluorescent protein genes enhances protein expression. J Mol Microbiol Biotechnol. 2017;27:1–10. [DOI] [PubMed] [Google Scholar]
- 21. Shaner NC, Lambert GG, Chammas A, et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods. 2013;10:407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. [DOI] [PubMed] [Google Scholar]
- 23. Wiedenmann J, Elke C, Spindler KD, Funke W. Cracks in the beta‐can: Fluorescent proteins from Anemonia sulcata (Anthozoa, Actinaria). Proc Natl Acad Sci U S A. 2000;97:14091–14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lukyanov KA, Fradkov AF, Gurskaya NG, et al. Natural animal coloration can be determined by a nonfluorescent green fluorescent protein homolog. J Biol Chem. 2000;275:25879–25882. [DOI] [PubMed] [Google Scholar]
- 25. Bulina ME, Chudakov DM, Mudrik NN, Lukyanov KA. Interconversion of Anthozoa GFP‐like fluorescent and non‐fluorescent proteins by mutagenesis. BMC Biochem. 2002;3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90:1103–1163. [DOI] [PubMed] [Google Scholar]
- 27. Gaytan P, Roldan‐Salgado A. Elimination of redundant and stop codons during the chemical synthesis of degenerate oligonucleotides. Combinatorial testing on the chromophore region of the red fluorescent protein mKate. Acs Synthetic Biology. 2013;2:453–462. [DOI] [PubMed] [Google Scholar]
- 28. Cormack BP, Valdivia RH, Falkow S. FACS‐optimized mutants of the green fluorescent protein (GFP). Gene. 1996;173:33–38. [DOI] [PubMed] [Google Scholar]
- 29. Royant A, Noirclerc‐Savoye M. Stabilizing role of glutamic acid 222 in the structure of enhanced green fluorescent protein. J Struct Biol. 2011;174:385–390. [DOI] [PubMed] [Google Scholar]
- 30. Sniegowski JA, Phail ME, Wachter RM. Maturation efficiency, trypsin sensitivity, and optical properties of Arg96, Glu222, and Gly67 variants of green fluorescent protein. Biochem Biophys Res Commun. 2005;332:657–663. [DOI] [PubMed] [Google Scholar]
- 31. Lemay NP, Morgan AL, Archer EJ, Dickson LA, Megley CM, Zimmer M. The role of the tight‐turn, broken hydrogen bonding, Glu222 and Arg96 in the post‐translational green fluorescent protein chromophore formation. Chemical Physics. 2008;348:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Branchini BR, Nemser AR, Zimmer M. A computational analysis of the unique protein‐induced tight turn that results in posttranslational chromophore formation in green fluorescent protein. J Am Chem Soc. 1998;120:1–6. [Google Scholar]
- 33. Barondeau DP, Kassmann CJ, Tainer JA, Getzoff ED. Understanding GFP posttranslational chemistry: Structures of designed variants that achieve backbone fragmentation, hydrolysis, and decarboxylation. J Am Chem Soc. 2006;128:4685–4693. [DOI] [PubMed] [Google Scholar]
- 34. Wood TI, Barondeau DP, Hitomi C, Kassmann CJ, Tainer JA, Getzoff ED. Defining the role of arginine 96 in green fluorescent protein fluorophore biosynthesis. Biochemistry. 2005;44:16211–16220. [DOI] [PubMed] [Google Scholar]
- 35. Donnelly M, Fedeles F, Wirstam M, Siegbahn PE, Zimmer M. Computational analysis of the autocatalytic posttranslational cyclization observed in histidine ammonia‐lyase. A comparison with green fluorescent protein. J Am Chem Soc. 2001;123:4679–4686. [DOI] [PubMed] [Google Scholar]
- 36. Barondeau DP, Tainer JA, Getzoff ED. Structural evidence for an enolate intermediate in GFP fluorophore biosynthesis. J Am Chem Soc. 2006;128:3166–3168. [DOI] [PubMed] [Google Scholar]
- 37. Pouwels LJ, Zhang L, Chan NH, Dorrestein PC, Wachter RM. Kinetic isotope effect studies on the de novo rate of chromophore formation in fast‐ and slow‐maturing GFP variants. Biochemistry. 2008;47:10111–10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grigorenko BL, Krylov AI, Nemukhin AV. Molecular modeling clarifies the mechanism of Chromophore maturation in the green fluorescent protein. J Am Chem Soc. 2017;139:10239–10249. [DOI] [PubMed] [Google Scholar]
- 39. Pletneva NV, Pletnev VZ, Lukyanov KA, et al. Structural evidence for a dehydrated intermediate in green fluorescent protein chromophore biosynthesis. J Biol Chem. 2010;285:15978–15984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wachter RM. The family of GFP‐like proteins: Structure, function, photophysics and biosensor applications. Introduction and perspective. Photochem Photobiol. 2006;82:339–344. [DOI] [PubMed] [Google Scholar]
- 41. Kremers GJ, Gilbert SG, Cranfill PJ, Davidson MW, Piston DW. Fluorescent proteins at a glance. J Cell Sci. 2011;124:157–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tejerizo GT, Bañuelos LA, Cervantes L, et al. Development of molecular tools to monitor conjugative transfer in rhizobia. J Microbiol Methods. 2015;117:155–163. [DOI] [PubMed] [Google Scholar]
- 43. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site‐directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. [DOI] [PubMed] [Google Scholar]
- 44. Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys J. 1997;73:2782–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Otwinowski Z, Minor W. Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. [DOI] [PubMed] [Google Scholar]
- 46. Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66:22–25. [DOI] [PubMed] [Google Scholar]
- 47. Murshudov GN, Skubák P, Lebedev AA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J App Cryst. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1: Characteristics of green fluorescent mutants of lanFP10A2 probing positions 148, 164, and 166
Table S2: Data collection statistics
Table S3: Refinement statistics
Figure S1: Absorption, excitation, and emission spectra of the pure fluorescent protein lanFP10A2
Figure S2: Amino acid sequence alignment of lanFP10A2 vs. asCP and DsRed
Figure S3: Purified fluorescent mutants of lanFP10A2
Figure S4: Experiments of size exclusion chromatography
Figure S5: Absorbance spectra of bfVFP at different pH values
Figure S6: Structural details observed in lanFP10A2 after improving the solubility of the parental protein lanFP10A
Figure S7: Overlap of lanFP10A2 and bfVFP structures
Figure S8. Mass spectra of the pure, mature proteins lanFP10A2 and bfVFP
Figure S9: SDS–PAGE analysis