

Abstract
Objective
To describe the clinical characteristics and outcomes in patients with refractory myasthenia gravis (MG) and to determine the effectiveness and side effects of the drugs used for their treatment.
Methods
This observational retrospective cross‐sectional multicenter study was based on data from the Spanish MG Registry (NMD‐ES). Patients were considered refractory when their MG Foundation of America post‐interventional status (MGFA‐PIS) was unchanged or worse after corticosteroids and two or more other immunosuppressive agents. Clinical and immunologic characteristics of drug‐refractory patients, efficiency and toxicity of drugs used, and outcome (MGFA‐PIS) at end of follow‐up were studied.
Results
We included 990 patients from 15 hospitals. Eighty‐four patients (68 of 842 anti‐acetylcholine receptor [AChR], 5 of 26 anti‐muscle‐specific tyrosine kinase [MusK], 10 of 120 seronegative, and 1 of 2 double‐seropositive patients) were drug refractory. Drug‐refractory patients were more frequently women (p < 0.0001), younger at onset (p < 0.0001), and anti‐MuSK positive (p = 0.037). Moreover, they more frequently presented a generalized form of the disease, bulbar symptoms, and life‐threatening events (p < 0.0001; p = 0.018; and p = 0.002, respectively) than non‐drug‐refractory patients. Mean follow‐up was 9.8 years (SD 4.5). Twenty‐four (50%) refractory patients had side effects to one or more of the drugs. At the end of follow‐up, 42.9% of drug‐refractory patients (42.6% of anti‐AChR, 100% of anti‐MuSK, and 10% of seronegative patients) and 79.8% of non‐drug‐refractory patients (p < 0.0001) achieved remission or had minimal manifestations. Eighty percent of drug‐refractory‐seronegative patients did not respond to any drug tested.
Interpretation
In this study, 8.5% of MG patients were drug‐refractory. New more specific drugs are needed to treat drug‐refractory MG patients.
Introduction
Myasthenia gravis (MG) is an autoimmune disorder caused by antibodies that bind to post‐synaptic proteins at the neuromuscular junction. 1 , 2 The disease is immunologically diverse. 3 Patients usually present antibodies against the acetylcholine receptor (AChR) or the muscle‐specific tyrosine kinase (MuSK). However, around 15% of patients do not have these antibodies and are considered seronegative. 4 Moreover, MG is clinically heterogeneous, and some of its clinical characteristics, such as the presence of thymoma 5 and early disease onset, 6 have been related to greater severity.
Therapeutic strategies developed in recent decades have greatly improved the outcome of MG patients, resulting in significant decreases in mortality and morbidity. Standard treatment of MG consists of acetylcholinesterase inhibitors, immunosuppressive drugs, and/or thymectomy, but it is individualized according to the patient characteristics. In severe weakness cases or exacerbations, immunomodulatory therapies are used. 7 However, around 10–15% of patients are drug refractory. Since 2016, patients are considered drug refractory when their MG Foundation of America post‐intervention status (MGFA‐PIS) is unchanged or worse after corticosteroids and at least two other immunosuppressive agents, used in adequate doses for an adequate duration, with persistent disabling symptoms or side effects, and based on an international experts consensus. 8 Identifying the characteristics of drug‐refractory MG patients could ultimately lead to help the development of new treatments for these patients.
Few articles have specifically studied drug‐refractory MG characteristics and data remain scarce. One recent study found 14.8% of MG patients were treatment refractory. Refractory status was associated with early‐onset, female sex, presence of anti‐MuSK antibodies and thymoma. 9 In another study, around 11% of patients were found to be drug refractory. MG was more severe in these patients and they had more frequent clinical exacerbations. The authors noted that early onset was associated with a higher risk of refractoriness. 10 In both these studies, however, the number of patients was small and the definition of drug‐refractory MG differed from the current definition. In view of the lack of data available regarding the efficacy and side effects of drugs used for the treatment of drug‐refractory MG, further studies on drug‐refractory MG are required.
The aim of this study was to determine the clinical, immunological and therapeutic characteristics in patients with drug refractory MG. Data were extracted from the nationwide neurologist‐driven MG Registry in Spain. 5 , 6 , 11
Methods
Data source: NMD‐ES project
The MG registry is part of the Spanish Neuromuscular Diseases Registry (NMD‐ES). It was founded in 2010 and designed in accordance with current Spanish law on biomedical research and data protection. Patients included must have a confirmed diagnosis of MG based on clinical findings and supported by positive autoantibodies and/or electrophysiological studies. Data are collected by neurologists with expertise in neuromuscular disorders at University hospitals in Spain. The Registry includes 60 MG‐specific items concerning demographic, clinical, immunologic, and therapeutic aspects. Follow‐up information is updated once a year or whenever a significant clinical event occurs. The Registry is reviewed annually to safeguard the quality of the information. Data included at the Registry have been used in previous published studies. 5 , 6 , 11
Patients and clinical evaluation
This is an observational retrospective cross‐sectional multicenter study. We selected all patients in the MG registry with onset of MG between 1 January 2000, and 31 December 2017. We excluded patients lost to follow‐up and patients for whom relevant information was missing. Patients were considered drug refractory as defined by Sanders et al. 8 Follow‐up ended on the date of death or at the end of the study (31 December 2019).
The demographic and clinical variables analyzed were: sex, age at onset, AChR and MuSK antibody positivity, severity and distribution of muscle weakness according to the Myasthenia Gravis Foundation of America (MGFA) clinical classification 12 at onset and at maximal worsening, frequency of life‐threatening events (defined as MGFA IVB and V) and myasthenic crises (MGFA V) at onset and at maximal worsening, generalization—defined as patients with a focal ocular form of the disease at onset (MGFA I) but later generalized (MGFA II or higher), thymoma defined by pathologic study in patients undergoing thymectomy and by thoracic CT in the others, thymectomy, follow‐up time defined as the difference between the follow‐up deadline or date of death and the date of diagnosis, and clinical outcome according to the MGFA post‐intervention status (MGFA‐PIS) 12 at the end of follow‐up. We analyzed the following therapeutic variables for drug‐refractory MG patients: immunosuppressive and immunomodulatory agents required and reason for withdrawal classified as inefficacy, side effects, pregnancy, or clinical improvement. The most effective drug was that which achieved the best MGFA‐PIS status (improved or better).
MG standard of care in Spain
The current Standard of care to treat MG in Spain includes: (1) acetylcholinesterase inhibitors as symptomatic therapy; (2) immunosuppressive drugs, being steroids the first‐line immunosuppressive treatment, azathioprine and mycophenolate mofetil as second line immunosuppressors or steroid‐sparing agents, and others (cyclosporine, tacrolimus, rituximab, methotrexate, and cyclophosphamide) in case of side effects or lack of response; (3) thymectomy, performed in patients with thymoma and also in patients younger than 65 years with anti‐AChR antibodies and no thymoma; and (4) immunomodulatory therapies (intravenous immunoglobulin and plasma exchange), predominantly used in acute worsening or MG crisis. However, treatment is individualized according to the patient characteristics. Patients with anti‐MuSK MG are treated with prednisone as first‐line immunosuppressive agent, rituximab if they do not respond favorably to prednisone, and plasma exchange as immunomodulatory therapy for exacerbacions. 7
Standard protocol approvals and patient consents
All participating centers obtained approval from their corresponding ethics committees to participate in the NMD‐ES Registry, and all patients signed an informed consent form for use of their data for scientific purposes.
Data availability statement
The data that support the findings of this study are available upon request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Statistical analysis
A descriptive data analysis was performed. The frequencies of symptoms are reported as percentages. Demographic characteristics are reported as means and SD. Differences in baseline characteristics between patient subgroups were evaluated using the chi‐squared test to compare categorical variables, and Student’s t test to compare quantitative variables. A significant difference was defined as p < 0.05. Missing data were dropped as they were <5% of the sample for the relevant variables. Data analysis was carried out using the STATA 13 software for Windows (StataCorp LP, College Station, TX).
Results
A total of 1168 patients of the 1619 included in the MG Registry at the time of the study had MG onset between 1 January 2000, and 31 December 2017. We excluded 178 patients because relevant information was missing. The sample was therefore made up of 990 patients from 15 hospitals. Mean age at onset was 57.2 years (SD 19.1), 52.2% were men, 842 (85.1%) were anti‐AChR positive, 26 (2.6%) were anti‐MuSK positive, 120 (12.1%) had seronegative MG, and 2 (0.2%) were anti‐AChR and anti‐MuSK double‐positive (Fig. 1). Thymoma was diagnosed in 128 (13.2%) patients.
Figure 1.
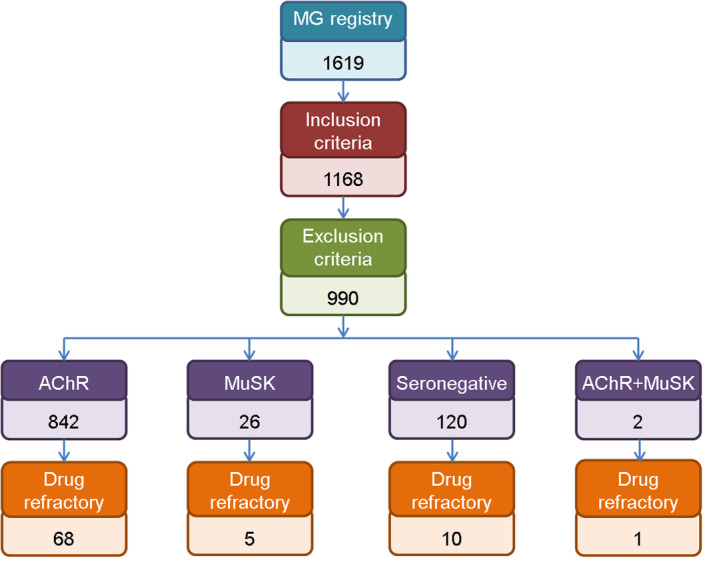
Flowchart shows patients included in the study, immunological characteristics, number of drug‐refractory patients and outcome. MG, myasthenia gravis; anti‐AChR, anti‐acetylcholine receptor; anti‐MuSK, anti‐muscle‐specific tyrosine kinase; MM, minimal manifestations; I, improvement; U, unchanged; W, worsening. [Colour figure can be viewed at wileyonlinelibrary.com]
A total of 84 participants (8.5%) were drug‐refractory. Their mean age at onset was 44.4 years (SD 18.0) and 75% were women. Eighty‐one percent had anti‐AChR antibodies, 6% had anti‐MuSK antibodies, 1.2% had anti‐AChR and anti‐MuSK antibodies, and 11.9% were seronegative (Fig. 1). Among drug‐refractory patients, 18.3% had thymoma. A generalized MG was developed by 92.3% of the patients, 77.5% of them had a generalized MG already at onset. The frequency of bulbar symptoms, life‐threatening events, and myasthenic crises was 47.5%, 13.8%, and 3.8%, respectively, at onset, and 62.8%, 28.2%, and 11.5% at maximal worsening (Table 1). Clinical classification of MGFA at onset and at maximal worsening by immunological subgroup is detailed in Table 2.
Table 1.
Clinical characteristics of drug‐refractory and non‐drug‐refractory myasthenia gravis patients and statistical analysis results.
| Drug‐refractory MG (n = 84) | Non‐drug‐refractory MG (n = 906) | p value | |
|---|---|---|---|
| Age at onset | 44.4 (SD 18.0) | 58.4 (SD 18.8) | <0.0001 |
| Age at onset >65 yo (%) | 12 (14.3) | 428 (47.2) | <0.0001 |
| Sex (female %) | 63 (75) | 410 (45.3) | <0.0001 |
| Antibodies (%) | AChR 68 (81) | AChR 774 (85.4) | 0.037 |
| MuSK 5 (6) | MuSK 21 (2.3) | ||
| AChR+MuSK 1 (1.2) | AChR+MuSK 1 (0.1) | ||
| Seronegative 10 (11.9) | Seronegative 110 (12.1) | ||
| Thymoma (%) | 15/82 (18.3) | 113/890 (12.7) | 0.152 |
| MGFA classification at onset (%) | |||
| I | 18 (21.4) | 375 (41.4) | |
| II | 29 (34.5) | 317 (35.0) | |
| III | 19 (22.6) | 125 (13.8) | <0.0001 |
| IV | 11 (13.1) | 49 (5.4) | |
| V | 3 (3.6) | 22 (2.4) | |
| Unknown | 4 (4.8) | 18 (2.0) | |
| MGFA classification at maximal worsening (%) | |||
| I | 6 (7.1) | 247 (27.3) | |
| II | 13 (15.5) | 260 (28.7) | |
| III | 30 (35.7) | 213 (23.5) | <0.0001 |
| IV | 20 (23.8) | 76 (8.4) | |
| V | 9 (10.7) | 61 (6.7) | |
| Unknown | 6 (7.1) | 49 (5.4) | |
| Generalized MG (onset/maximal worsening, %) | 62/80 (77.5) | 513/888 (57.8) | <0.0001 |
| 72/78 (92.3) | 610/857 (71.2) | <0.0001 | |
| Bulbar symptoms (onset/maximal worsening, %) | 38/80 (47.5) | 345/888 (38.9) | 0.130 |
| 49/78 (62.8) | 419/857 (48.9) | 0.018 | |
| Life‐threatening events (onset/maximal worsening, %) | 11/80 (13.8) | 67/888 (7.5) | 0.051 |
| 22/78 (28.2) | 126/857 (14.7) | 0.002 | |
| Myasthenic crisis (onset/maximal worsening, %) | 3/80 (3.8) | 22/888 (2.5) | 0.492 |
| 9/78 (11.5) | 61/857 (7.1) | 0.156 | |
MG, myasthenia gravis; MGFA, Myasthenia Gravis Foundation of America; AChR, acetylcholine receptor; MuSK, muscle‐specific tyrosine kinase.
Table 2.
MGFA clinical classification at onset and at maximal worsening is shown for drug‐refractory myasthenia gravis patients according to immunological subgroup.
| AChR MG | MuSK MG | AChR + MuSK MG | Seronegative MG | |
|---|---|---|---|---|
| MGFA at onset | n = 65 | n = 5 | n = 1 | n = 9 |
| I | 14 | 0 | 0 | 4 |
| IIA | 12 | 0 | 0 | 3 |
| IIB | 12 | 2 | 0 | 0 |
| IIIA | 4 | 1 | 0 | 1 |
| IIIB | 11 | 1 | 0 | 1 |
| IVA | 3 | 0 | 0 | 0 |
| IVB | 7 | 1 | 0 | 0 |
| V | 2 | 0 | 1 | 0 |
| MGFA at maximal worsening | n = 62 | n = 5 | n = 1 | n = 10 |
| I | 4 | 0 | 0 | 2 |
| IIA | 4 | 0 | 0 | 3 |
| IIB | 4 | 1 | 0 | 1 |
| IIIA | 6 | 0 | 0 | 3 |
| IIIB | 19 | 2 | 0 | 0 |
| IVA | 6 | 0 | 0 | 1 |
| IVB | 12 | 1 | 0 | 0 |
| V | 7 | 1 | 1 | 0 |
MG, myasthenia gravis; MGFA, Myasthenia Gravis Foundation of America; AChR, acetylcholine receptor; MuSK, muscle‐specific tyrosine kinase.
Compared to non‐drug‐refractory patients, patients with drug‐refractory MG were more frequently women (p < 0.0001), younger at onset (p < 0.0001), and anti‐MuSK positive (p = 0.0001). They also more frequently had generalized MG (p = 0.001 at onset and p < 0.0001 at maximal worsening), bulbar symptoms (p = 0.018), and life‐threatening events (p = 0.002) at maximal worsening. Table 1 shows clinical data of drug‐refractory and non‐drug‐refractory patients.
Drug‐refractory patients were treated with a mean of 3.2 (minimum 2–maximum 6) other‐than‐prednisone immunosuppressive agents. Anti‐AChR‐positive drug‐refractory patients were treated with a mean of 3.6 (min 2–max 5 drugs), anti‐MuSK‐positive patients with a mean of 3.3 (min 2–max 6), and seronegative patients with a mean of 2.3 (min 2–max 3). The most frequently used immunosuppressive therapies for drug‐refractory and non‐drug‐refractory MG patients are shown in Figure S1A. Drug‐refractory patients needed intravenous immunoglobulin (86.9% vs. 23.7%, p < 0.0001) and plasma exchange (19% vs. 4.4%, p < 0.0001) more frequently than non‐drug‐refractory patients at some point of the disease (Fig. S1B). Drug‐refractory patients were more frequently thymectomized than non‐drug‐refractory patients (44.0% vs. 23.4%, p < 0.0001). At the last follow‐up, complete stable remission was achieved in 1 of the 37 thymectomized drug‐refractory patients and pharmacological remission was achieved in another. Eight patients had minimal manifestations, 7 improved, 18 were unchanged, and 2 worsen.
The drug that most improved MGFA‐PIS status for each patient is detailed in Figure 2. Anti‐AChR MG patients improved with a variety of drugs, the most frequent being rituximab (15%) and tacrolimus (15%). Thirty‐two percent of patients did not respond to any of the drugs administered. Anti‐MuSK patients mainly responded favorably to rituximab (80%). The only patient with both anti‐AChR‐ and anti‐MuSK‐positive antibodies also responded favorably to rituximab. Eighty percent of seronegative MG patients did not respond to any drug tested.
Figure 2.
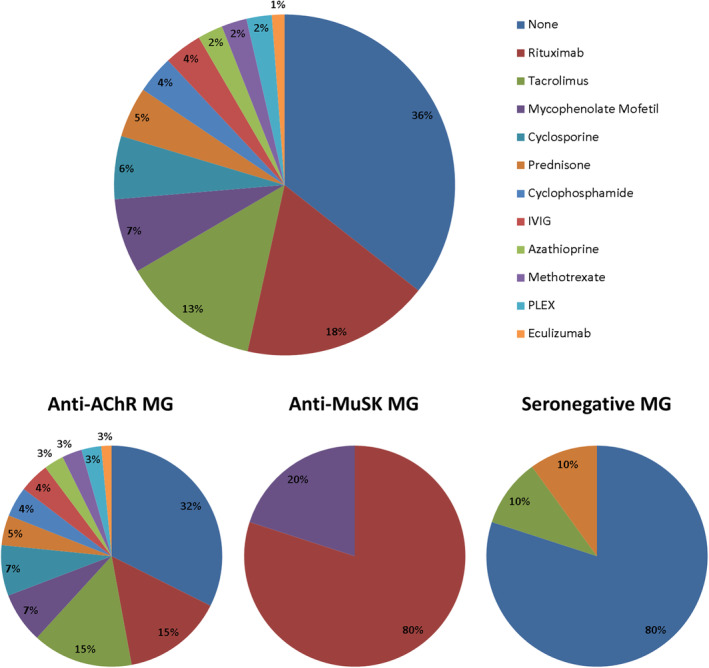
The drug that achieved the best MGFA‐PIS status (improved, minimal manifestations, or remission) for each patient is represented in percentages for the total of drug‐refractory patients, and broken down based on the immunological profile (anti‐acetylcholine receptor‐positive patients, anti‐muscle‐specific tyrosine kinase‐positive patients, and seronegative patients). MG, myasthenia gravis; anti‐AChR, anti‐acetylcholine receptor; anti‐MuSK, anti‐muscle‐specific tyrosine kinase. [Colour figure can be viewed at wileyonlinelibrary.com]
Forty‐two patients (50%) presented side effects that led to drug withdrawal; 24 of them (28.6%) with 1 drug, 11 patients (13.1%) with 2 drugs, 5 patients (6%) with 3 drugs, and 2 patients (2.4%) with 4 drugs. Azathioprine and mycophenolate mofetil were usually withdrawn because of inefficacy rather than because of side effects. However, in the case of cyclosporine and tacrolimus, withdrawal because of inefficacy and withdrawal because of side effects were both main reasons, and in similar frequencies. The most frequent side effects for each immunosuppressor are detailed in Table 3.
Table 3.
Immunosuppressor therapies used for the treatment of drug‐refractory MG, percentage of withdrawal, reasons for withdrawal, and reported side effects.
| Immunosuppressor (number of patients) | Withdrawal (%) | Reasons for withdrawal (%) | Side effects |
|---|---|---|---|
| Prednisone (84) | 18 (21.4) |
6 (33.3) inefficacy 6 (33.3) side effects 6 (33.3) improvement or remission |
1 High blood pressure 1 Diabetes mellitus 1 High blood pressure + diabetes mellitus 1 Osteopenia 1 Compulsion 1 Weight gain |
| Azathioprine (70) | 63 (90) |
40 (63.5) inefficacy 22 (34.9) side effects 1 (1.6) inefficacy and side effects |
5 Hepatotoxicity 5 Abdominal pain 4 Anemia 3 Allergy 1 Pancytopenia 1 Leucopenia 1 Arthralgia 3 Unknown |
| Mycophenolate Mofetil (57) | 43 (75.4) |
32 (74.4) inefficacy 8 (18.6) side effects 3 (7.0) desire for pregnancy |
2 Abdominal pain 2 Hepatotoxicity 1 Diarrhea 1 Abdominal pain and diarrhea 1 Pancytopenia 1 Unknown |
| Cyclosporine (56) | 45 (80.4) |
21 (46.7) inefficacy 21 (46.7) side effects 2 (4.4) inefficacy and side effects 1 (2.2) desire for pregnancy |
10 Nephrotoxicity 4 Abdominal pain 2 High blood pressure and headache 1 Hepatotoxicity 1 Hypertrichosis 1 Gum hyperplasia 1 Tremor 1 Anemia and leucopenia 2 Unknown |
| Tacrolimus (33) | 17 (51.5) |
8 (47.1) inefficacy 7 (41.2) side effects 2 (11.8) remission |
2 Nausea and vomits 1 Skin rash 1 Alopecia 1 Hyperglycemia 1 Vision loss 1 Unknown |
| Rituximab (40) | 35 (87.5) |
21 (60.0) inefficacy 2 (5.7) inefficacy and side effects 13 (37.1) remission |
1 Psoriasis exacerbation 1 Progressive multifocal leukoencephalopathy |
| Methotrexate (6) | 5 (83.3) |
2 (40) inefficacy 1 (20) side effects 2 (40) remission |
1 Abdominal pain |
| Cyclophosphamide (7) | 7 (100) |
3 (42.9) inefficacy 1 (14.3) inefficacy and side effects 3 (42.9) remission |
1 Toxic organizing pneumonia |
MG, myasthenia gravis.
Patients were followed for a mean of 9.8 years (SD 4.5). At the end of follow‐up, fewer drug‐refractory patients achieved remission or had minimal manifestations than non‐drug‐refractory patients (42.9% vs. 79.8%, p < 0.0001) (Fig. S2A). After breaking down the sample based on immunological subtype, we found that 42.6% of anti‐AChR‐positive MG patients achieved remission or had minimal manifestations at the end of follow‐up. This MGFA‐PIS status was achieved in 10% of seronegative patients and 100% of anti‐MuSK‐positive patients (Fig. S2B).
Discussion
In our series, 8.5% of patients with MG were drug‐refractory despite the numerous therapeutic options available. These patients were more likely to be female, to present early‐age at onset, and to have anti‐MuSK antibodies. We also observed that drug‐refractory MG patients had more severe forms of the disease than their non‐drug‐refractory counterparts, specifically in terms of generalized MG, bulbar symptoms, and life‐threatening events during the course of the disease. Furthermore, they more frequently needed rescue treatments with intravenous immunoglobulin and plasma exchange.
The frequency of drug‐refractory MG patients was lower than that reported in previous studies where around 10–15% of patients were drug‐refractory. 9 , 10 These differences may be due to the variation of definitions of drug‐refractoriness over time. However, it may also be because we have learned to treat some subgroups of patients more efficiently in recent years. For instance, in our series, anti‐MuSK patients diagnosed after rituximab effectiveness was described were not considered drug‐refractory because immediately after prednisone failure they were treated with rituximab rather than other immunosuppressive drugs that we now know are rarely effective. 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 Previous studies suggested thymoma is more frequent in drug‐refractory MG patients 9 but we did not find any statistically significant difference compared to the frequency in non‐drug‐refractory MG patients. Maybe a more precise thymoma staging and a more accurate treatment strategy following recent international standards 22 had a beneficial impact on our thymoma‐associated MG patients outcome.
The clinical and immunological characteristics associated with drug‐refractory MG may indicate that distinctive immunopathologic pathways or biological mechanisms are involved in the development of drug‐refractory MG. Studying these underlying molecular processes should be a priority as they may lead to the identification of therapeutic targets that could help treat drug‐refractory MG more efficiently. The anti‐CD20 agent rituximab, for instance, has extensively proven to be highly effective for the treatment of anti‐MuSK‐positive patients, 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 as for other IgG4‐related diseases. 23 Our results regarding rituximab efficacy in treating anti‐MuSK‐positive patients are in agreement with previously reported studies. Other emerging drugs target important molecules involved in MG. The terminal complement activation inhibitor eculizumab merits consideration in the treatment of drug‐refractory anti‐AChR‐positive MG, 13 , 14 , 24 although we have scarce data about this drug as it is not still available in our country and the only patient that responded was part of a clinical trial. Other anti‐complement strategies 25 , 26 and neonatal Fc receptor (FcRn) antagonists 26 , 27 are currently on trial but unfortunately none of the patients received these therapies during the study period. The development of these new molecules highlights the importance of studying the molecular pathophysiology underlying disease development and progression.
Another important observation from our study is that around 40% of drug‐refractory patients did not achieve remission or minimal manifestations MGFA‐PIS status at the end of a long follow‐up. And importantly, a significant percentage of these participants were treatment intolerant to some of the drugs received. Fifty percent of drug‐refractory patients had adverse events with one or more of the drugs tested. Some of these side effects were severe and could have increased the disease burden. Also, some of the treatments we currently use are potentially teratogenic, a risk that cannot be overlooked when a vast majority of drug‐refractory patients are young females. This point again supports the need to develop more specific, well tolerated drugs to treat MG.
In the present study, the most effective drugs to treat drug‐refractory MG patients were rituximab and tacrolimus and we found the same results when we analyzed separately anti‐AChR positive patients. We highlight that two of five patients treated with methotrexate achieved remission, although a recent clinical trial of oral weekly methotrexate in anti‐AChR generalized MG patients showed that it is not a useful drug to spare steroids with class I level of evidence. 28 Regarding thymectomy, drug‐refractory patients were more frequently thymectomized, but they were younger at onset and consequently more likely to have undergone thymic removal. Furthermore, most patients in the study were diagnosed of MG before the clinical trial that demonstrated the efficacy of thymectomy in non‐thymomatous anti‐AChR positive patients was published. 29 The lack of such evidence at this stage may have impacted on the percentage of patients who were eventually thymectomized. We know that 10 of the 37 thymectomized drug‐refractory patients achieved a MGFA‐PIS status of remission or minimal manifestations at last follow‐up. However, we have no specific data about the clinical response to thymectomy in these patients. The retrospective nature of the study and the fact that patients were simultaneously treated with other agents prevented an accurate evaluation. Overall, these results show the heterogeneous response of drug‐refractory anti‐AChR MG patients and highlight the importance of the search for treatment response biomarkers.
Another important finding in our study is that 80% of seronegative drug‐refractory MG patients did not respond to any drug used and 90% did not achieve a minimal manifestations or remission MGFA‐PIS status. The accuracy of diagnosis in these patients might be questioned here, but we should emphasize that all patients included in the MG registry had a diagnosis of confirmed MG based on both clinical features and electrophysiological studies. Moreover, these results are in line with previously published findings that also suggested a poorer outcome for seronegative patients. 30 In this study, drug‐refractory‐seronegative MG patients received fewer other‐than‐prednisone immunosuppressor agents than anti‐AChR and MuSK positive patients. Maybe the lack of a detectable antibody or the fact that they had mainly mild forms of the disease prevented us from adopting a more aggressive pharmacological approach. Another hypothesis is that the unknown underlying physiopathology of seronegative MG prevents us from finding a specific treatment, as was the case with anti‐MuSK positive patients before the antibody was discovered and the efficacy of rituximab was demonstrated. 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 Research focusing on finding the antibodies involved in seronegative MG is of high significance.
Although some of our observations are in accordance with those in previously published studies, 9 , 10 it would be inaccurate to compare results because the definition of drug‐refractoriness varies. A main strength of this study is that it is the first to classify patients based on the current universally accepted definition. 8 Furthermore, we used standardized, updated information from a large number of patients from 15 hospitals around the country, and the series of drug‐refractory MG patients is the widest reported to date. The main limitation is that the sample may be biased in terms of disease severity and non‐response to commonly used drugs. This is because data were mainly collected at tertiary university hospitals, which may lead to overestimation of the percentage of drug‐refractory patients. Also, our results may be not transferable to smaller hospitals or other settings in other countries where not all treatments are available. However, the design of the study allowed us to review the clinical and immunological characteristics and the efficacy and side effects of the drugs used to treat many drug‐refractory MG patients, which was the aim of this article. An epidemiological study is needed to determine the exact prevalence of drug‐refractory MG patients.
Conclusions
In conclusion, we demonstrate that a subgroup of MG patients with distinct clinical features is drug‐refractory to available therapies. Many of these patients never achieve the therapeutic goal and drug side effects are common. Seronegative drug‐refractory patients specifically need more effective drugs, as most patients do not respond favorably to any treatment. These findings highlight the need for more specific therapies to treat MG.
Conflict of Interest
Authors report no conflict of interests.
Supporting information
Figure S1. (A) Percentages of use of immunosuppressive therapies are represented in bars for drug‐refractory and non‐drug‐refractory myasthenia gravis patients. (B) Percentages of use of immunomodulatory therapies are represented in bars for drug‐refractory and non‐drug‐refractory myasthenia gravis patients. MG, myasthenia gravis; IVIG, intravenous immunoglobulin; PLEX, plasma exchange.
Figure S2. (A) Percentage of Myasthenia Gravis Foundation of America post‐intervention status at end of follow‐up are represented in bars for drug‐refractory and non‐drug‐refractory myasthenia gravis patients. Curly brackets show the percentage of drug‐refractory and non‐drug‐refractory myasthenia gravis patients that achieved the treatment goal. (B) Percentage of Myasthenia Gravis Foundation of America post‐intervention status at end of follow‐up are represented in bars for seronegative, double seropositive, anti‐muscle‐specific tyrosine kinase‐positive and anti‐acetylcholine receptor‐positive drug‐refractory MG patients. MG, myasthenia gravis; CSR, complete stable remission; PR, pharmacologic remission; MM, minimal manifestations; I, improvement; U, unchanged; W, worsening; anti‐AChR, anti‐acetylcholine receptor; Anti‐MuSK, anti‐muscle‐specific tyrosine kinase.
Acknowledgments
The authors thank the patients for contributing their data to this registry and Carolyn Newey for language support. E. Cortés‐Vicente, R. Álvarez‐Velasco, R. Rojas‐Garcia, J. Turon‐Sans, E. Pascual‐Goñi, E. Gallardo, I. Illa, C. Paradas and T. Sevilla are members of the European Reference Network for Neuromuscular Diseases. E. Cortés‐Vicente, R. Álvarez‐Velasco, R. Rojas‐Garcia, J. Turon‐Sans, E. Pascual‐Goñi, E. Gallardo, I. Illa, C. Paradas, T. Sevilla, C. Casasnovas work on a CSUR (centro, servicio, unidad de referencia) on rare neuromuscular diseases. E. Cortés‐Vicente, R. Álvarez‐Velasco, R. Rojas‐Garcia, J. Turon‐Sans, E. Pascual‐Goñi, E. Gallardo, I. Illa, C. Casasnovas and A. Ramos‐Fransi are members of XUECs (Xarxes d'unitats d'expertesa clínica en malalties minoritàries).
Funding Information This work was funded by the Instituto de Salud Carlos III through the project PI19/01774 (cofunded by the European Union ERDF), PI I. Illa and E. Gallardo. E. Cortés‐Vicente was supported by a Juan Rodés grant (JR19/00037) from the Fondo de Investigación en Salud, Instituto de Salud Carlos III and co‐funded by European Union (ERDF/ESF, “A way to make Europe”/“Investing in your future”), Ministry of Health (Spain). R. Álvarez‐Velasco was supported by grant SLT008/18/00207 from the Health Research and Innovation Strategic Plan (PERIS). The NMD‐ES Project and F. Pla‐Junca (data curator) are partially funded by the Centro de Investigación Biomédica en Red de Enfsermedades Raras (CIBERER).
Funding Statement
This work was funded by Centro de Investigacion Biomedica en Red de Enfermedades Raras (CIBERER); Health Research and Innovation Strategic Plan (PERIS) grant SLT008/18/00207; Instituto de Salud Carlos III grants JR19/00037 and PI19/01774; Ministry of Health ; European Union .
References
- 1. Gilhus NE. Myasthenia gravis. N Engl J Med. 2016;375:2570‐2581. [DOI] [PubMed] [Google Scholar]
- 2. Querol L, Illa I. Myasthenia gravis and the neuromuscular junction. Curr Opin Neurol. 2013;26:459‐465. [DOI] [PubMed] [Google Scholar]
- 3. Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8:475‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berrih‐Aknin S, Frenkian‐Cuvelier M, Eymard B. Diagnostic and clinical classification of autoimmune myasthenia gravis. J Autoimmun. 2014;48‐49:143‐148. [DOI] [PubMed] [Google Scholar]
- 5. Álvarez‐Velasco R, Gutiérrez‐Gutiérrez G, Trujillo JC, et al. Clinical characteristics and outcomes of thymoma associated myasthenia gravis. Eur J Neurol. 2021;28(6):2083–2091. 10.1111/ene.14820 [DOI] [PubMed] [Google Scholar]
- 6. Cortés‐Vicente E, Álvarez‐Velasco R, Segovia S, et al. Clinical and therapeutic features of myasthenia gravis in adults based on age at onset. Neurology. 2020;94(11):e1171‐e1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cortés‐Vicente E, Gallardo E, Álvarez‐Velasco R, Illa I. Myasthenia gravis treatment updates. Curr Treat Options Neurol. 2020;22:24. [Google Scholar]
- 8. Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis executive summary. Neurology. 2016;87:419‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suh J, Goldstein JM, Nowak RJ. Clinical characteristics of refractory myasthenia gravis patients. Yale J Biol Med. 2013;86(2):255‐260. [PMC free article] [PubMed] [Google Scholar]
- 10. Rath J, Brunner I, Tomschik M, et al. Frequency and clinical features of treatment‐refractory myasthenia gravis. J Neurol. 2020;267(4):1004‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ramos‐Fransi A, Rojas‐García R, Segovia S, et al. Myasthenia gravis: descriptive analysis of life‐threatening events in a recent nationwide registry. Eur J Neurol. 2015;22:1056‐1061. [DOI] [PubMed] [Google Scholar]
- 12. Jaretzki A, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Neurology. 2000;55:16‐23. [DOI] [PubMed] [Google Scholar]
- 13. Mantegazza R, Antozzi C. When myasthenia gravis is deemed refractory: clinical signposts and treatment strategies. Ther Adv Neurol Disord. 2018;11:1756285617749134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dalakas MC. Immunotherapy in myasthenia gravis in the era of biologics. Nat Rev Neurol. 2019;15(2):113‐124. [DOI] [PubMed] [Google Scholar]
- 15. Díaz‐Manera J, Martínez‐Hernández E, Querol L, et al. Long‐lasting treatment effect of rituximab in MuSK myasthenia. Neurology. 2012;78:189‐193. [DOI] [PubMed] [Google Scholar]
- 16. Illa I, Díaz‐Manera J, Rojas‐García R, et al. Sustained response to rituximab in anti‐ AChR and anti‐ MuSK positive myasthenia gravis patients. J Neuroimmunol. 2008;201‐202:90‐94. [DOI] [PubMed] [Google Scholar]
- 17. Nowak RJ, Dicapua DB, Zebardast N, Goldstein JM. Response of patients with refractory myasthenia gravis to rituximab: a retrospective study. Ther Adv Neurol Disord. 2011;4:259‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iorio R, Damato V, Alboini PE, Evoli A. Efficacy and safety of rituximab for myasthenia gravis: a systematic review and meta‐analysis. J Neurol. 2015;262:1115‐1119. [DOI] [PubMed] [Google Scholar]
- 19. Tandan R, Hehir MK, Waheed W, Howard DB. Rituximab treatment of myasthenia gravis: a systematic review. Muscle Nerve. 2017;56:185‐196. [DOI] [PubMed] [Google Scholar]
- 20. Hehir MK, Hobson‐Webb LD, Benatar M, et al. Rituximab as treatment for anti‐MuSK myasthenia gravis: multicenter blinded prospective review. Neurology. 2017;9:1069‐1077. [DOI] [PubMed] [Google Scholar]
- 21. Cortés‐Vicente E, Rojas‐García R, Díaz‐Manera J, et al. The impact of rituximab infusion protocol on the long‐term outcome in anti‐MuSK myasthenia gravis. Ann Clin Transl Neurol. 2018;5:710‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lamarca A, Moreno V, Feliu J. Thymoma and thymic carcinoma in the target therapies era. Cancer Treat Rev. 2013;39(5):413‐420. [DOI] [PubMed] [Google Scholar]
- 23. Huijbers MG, Querol LA, Niks EH, et al. The expanding field of IgG4‐mediated neurological autoimmune disorders. Eur J Neurol. 2015;22(8):1151‐1161. doi: 10.1111/ene.12758 [DOI] [PubMed] [Google Scholar]
- 24. Howard JF Jr, Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in anti‐ acetylcholine receptor antibody‐ positive refractory generalized myasthenia gravis (REGAIN): a phase 3, randomised, double‐blind, placebo‐controlled, multicentre study. Lancet Neurol. 2017;16:976‐986. [DOI] [PubMed] [Google Scholar]
- 25. Howard JF Jr, Nowak RJ, Wolfe GI, et al. Clinical effects of the self‐administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis: results of a phase 2 randomized, double‐blind, placebo‐controlled, multicenter clinical trial. JAMA Neurol. 2020;77(5):582‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dalakas M. Progress in the therapy of myasthenia gravis: getting closer to effective targeted immunotherapies. Curr Opin Neurol. 2020;33(5):545‐552. [DOI] [PubMed] [Google Scholar]
- 27. Howard JF Jr, Bril V, Burns TM, et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology. 2019;92(23):e2661‐e2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pasnoor M, He J, Herbelin L, et al. A randomized controlled trial of methotrexate for patients with generalized myasthenia gravis. Neurology. 2016;87(1):57‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med. 2016;375(6):511‐522. doi: 10.1056/NEJMoa1602489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tomschik M, Hilger E, Rath J, et al. Subgroup stratification and outcome in recently diagnosed generalized myasthenia gravis. Neurology. 2020;95(10):e1426‐e1436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (A) Percentages of use of immunosuppressive therapies are represented in bars for drug‐refractory and non‐drug‐refractory myasthenia gravis patients. (B) Percentages of use of immunomodulatory therapies are represented in bars for drug‐refractory and non‐drug‐refractory myasthenia gravis patients. MG, myasthenia gravis; IVIG, intravenous immunoglobulin; PLEX, plasma exchange.
Figure S2. (A) Percentage of Myasthenia Gravis Foundation of America post‐intervention status at end of follow‐up are represented in bars for drug‐refractory and non‐drug‐refractory myasthenia gravis patients. Curly brackets show the percentage of drug‐refractory and non‐drug‐refractory myasthenia gravis patients that achieved the treatment goal. (B) Percentage of Myasthenia Gravis Foundation of America post‐intervention status at end of follow‐up are represented in bars for seronegative, double seropositive, anti‐muscle‐specific tyrosine kinase‐positive and anti‐acetylcholine receptor‐positive drug‐refractory MG patients. MG, myasthenia gravis; CSR, complete stable remission; PR, pharmacologic remission; MM, minimal manifestations; I, improvement; U, unchanged; W, worsening; anti‐AChR, anti‐acetylcholine receptor; Anti‐MuSK, anti‐muscle‐specific tyrosine kinase.
Data Availability Statement
The data that support the findings of this study are available upon request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.