

Abstract
Membrane proteins play essential roles in cellular function and metabolism. Nonetheless, biophysical and structural studies of membrane proteins are impeded by the difficulty of their expression in and purification from heterologous cell‐based systems. As an alternative to these cell‐based systems, cell‐free protein synthesis has proven to be an exquisite method for screening membrane protein targets in a variety of lipidic mimetics. Here we report a high‐throughput screening workflow and apply it to screen 61 eukaryotic membrane protein targets. For each target, we tested its expression in lipidic mimetics: two detergents, two liposomes, and two nanodiscs. We show that 35 membrane proteins (57%) can be expressed in a soluble fraction in at least one of the mimetics with the two detergents performing significantly better than nanodiscs and liposomes, in that order. Using the established cell‐free workflow, we studied the production and biophysical assays for mitochondrial pyruvate carrier (MPC) complexes. Our studies show that the complexes produced in cell‐free are functionally competent in complex formation and substrate binding. Our results highlight the utility of using cell‐free systems for screening and production of eukaryotic membrane proteins.
Keywords: cell‐free expression, complexes, eukaryotic membrane proteins, high throughput, lipidic mimetics
1. INTRODUCTION
Membrane proteins (MPs) make up 20–30% of gene products and are involved in numerous cellular functions and metabolisms. 1 , 2 Membrane proteins are the targets of about half of the known drugs and thus are of great interest for structure–function characterizations. 3 , 4 Relative to their soluble counterparts, only about 3.1% structures deposited in Protein Data Bank (PDB, www.pdb.org) are membrane proteins. The under‐representation of membrane protein structures is mainly due to difficulties associated with their production including low level of expression, toxicity to host cells, sub‐optimal stability in detergent micelle, and mismatches in lipid composition of host cells. 5 , 6 , 7 , 8 , 9 , 10 To overcome these limitations, several approaches have been utilized. On a “genomic” level, one approach is to screen cell‐based expression of large numbers of homologs, orthologs, and other close relatives of genes of interest that may improve the odds of finding a target suitable for structural determination. 11 , 12 Typically, this approach is performed at a dedicated center and is labor‐intensive and expensive. 11 , 13 , 14 , 15 , 16 , 17 The second approach is genetic manipulation of the expression host cells. This has been achieved in particular with various Escherichia coli strains based on T7 RNA polymerase, such as C41, C43, C44, C45, and M56 18 , 19 (see also Ref. 20 for a recent review). The third approach is to screen for host genes that can directly affect the expression of a target. For example, the overexpression of certain Escherichia coli genes has been shown to increase the expression of MP targets. 21 , 22 , 23
Eukaryotic MPs are more successfully expressed using eukaryotic expression systems, mainly insect cells and mammalian cell lines such as HEK293. However, eukaryotic expression systems are labor‐intensive and expensive to support. They are also not particularly conducive to a high‐throughput approach and genetic manipulation of host strains is difficult and not as common as for prokaryotic expression systems (see Refs. 24, 25 for a review).
Complementary to cell‐based approaches, cell‐free systems offer a convenient method in tackling many difficulties such as aggregation, resistance to detergents, and mismatches in lipid composition. 26 , 27 A major advantage of a cell‐free system is the ready inclusion of additives (detergents, lipids, salts, ligands, etc.) to the reaction mixture to assess how they influence protein production and stability. 28 In contrast to cell‐based systems, with the cell‐free systems, MPs can be directly inserted into the mimetic of choice without any intervening extraction steps, which are usually harsh, inefficient, and may lead to aggregates and loss of structure and activity.
In this study, we set out to investigate whether high‐throughput screening of a large set of eukaryotic MP targets using a cell‐free system could be a promising avenue for MP expression. We report the cell‐free expression of 35 out of 61 eukaryotic MP targets in lipidic mimetics. Overall, cell‐free synthesis led to more targets being produced than an E. coli‐based expression system. We also report the cell‐free production and biophysical analysis of mitochondrial pyruvate carrier complexes.
2. RESULTS
2.1. Workflow for cell‐free screening
Starting from genes codon‐optimized for bacterial expression and inserted into a T7 promoter plasmid, our cell‐free screening workflow includes three steps as illustrated in Figure 1. The first step is to prepare cell‐free reaction mixtures containing one of three types of lipidic mimetics: detergents, liposomes, or nanodiscs. The second step is to assemble the reaction mixtures (RMs) and place them in microdialysis cartridges (Pierce™ 96‐well Microdialysis Plate; see also Section 4), which are inserted into 96‐well deep‐well blocks containing a feeding mix (FM) of amino acids, NTPs, energy molecules, and buffer. After overnight reaction, the third step is to detect expressed (or solubilized) targets. For detergent and liposome reactions, both total synthesized protein (T) and solubilized protein (S) are assayed by immunoblot analysis (anti‐His tag). This is achieved by a centrifugation step that cleared the total reaction mixtures from insoluble proteins. The soluble fraction of the produced proteins is then found in the supernatant.
FIGURE 1.
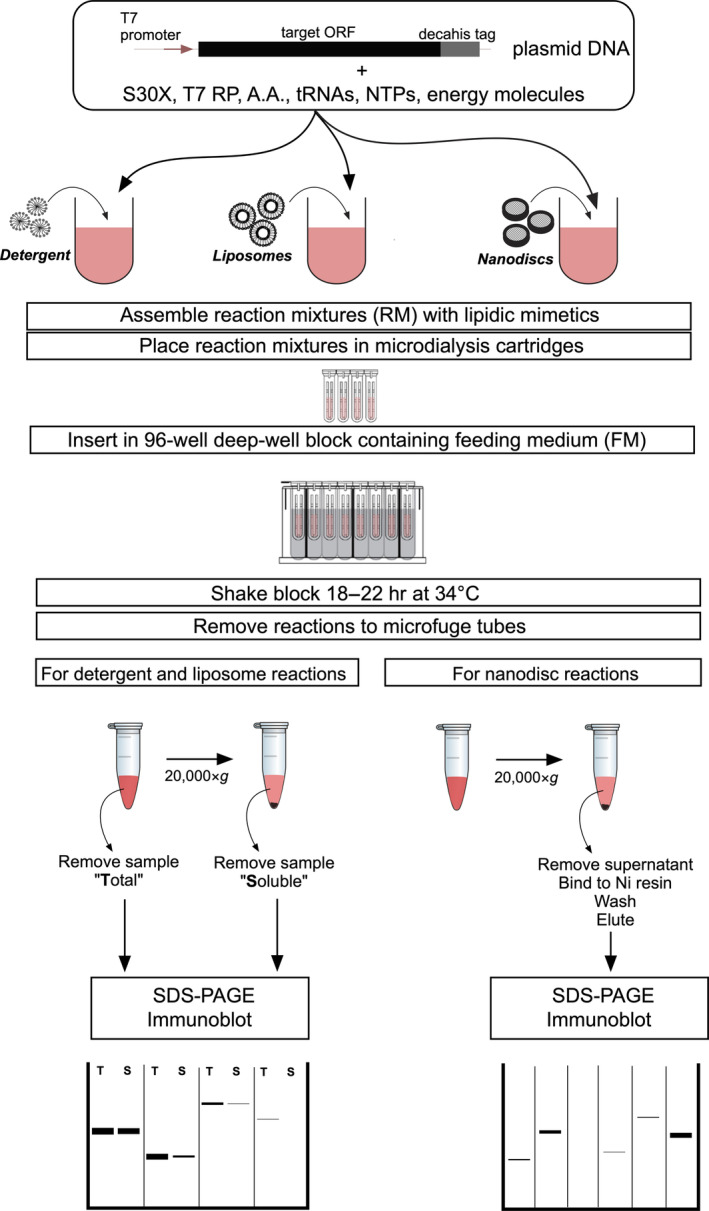
Workflow for high‐throughput cell‐free screening of eukaryotic membrane proteins. For detailed experimental procedures, see Section 4. S30X, E. coli S30 extract; T7 RP, T7 RNA polymerase; A.A., amino acids; NTPs, nucleotide triphosphates
For nanodisc reactions, only solubilized protein (S) was analyzed. This solubilized fraction had to be further purified by Ni2+‐affinity. Without this step, we were not able to analyze the immunoblot assays as lanes looked grossly overloaded and distinct bands were difficult to assess.
With all components ready, the entire cell‐free screening workflow may be completed in 2–3 days.
2.2. Membrane protein targets
To get a better understanding of whether cell‐free systems (CF) can be used for screening membrane proteins with different lipidic mimetics, we compiled a set of MP targets. Initially, we identified 61 targets from the UniProt database (www.uniprot.org) for which no structures had been reported to the Protein Data Bank. The list includes 42 targets from the mouse‐ear cress Arabidopsis thaliana, two from the poplar tree Populus trichocarpa, six from green algae Chlamydomonas reinhardtii, and 11 from H. sapiens (Table S1). The DNA sequence for all these targets was codon‐optimized for expression in E. coli, synthesized, and cloned into a pET23 derivative containing a C‐terminal deca‐histidine tag. These expression plasmids were then used in cell‐free mini‐scale reactions using E. coli. S30 extract and in‐house purified T7 RNA polymerase. 29 Figure 1 shows the workflow of our high‐throughput screening for these eukaryotic MP targets. Immunoblot assay results are reported in Tables S1 and S2.
2.3. Cell‐free screening in detergents Brij‐35 and DDM
Dodecylmaltoside (DDM) is a widely used detergent for membrane protein extraction and purification. It is relatively mild and has good tolerance in CF (Refs. 28, 30, 31 and references therein). From our CF screening, we found that 15 targets were expressed and soluble to various degrees in DDM as assessed by immunoblotting (see Tables S1 and S2). This translates to an expression rate of about ~25%, which is in the range of what we have found to be the rate when expressing prokaryotic membrane protein targets in E. coli hosts (Refs. 32, 33 and data not shown). For the combined plant and plant‐like targets (Arabidopsis thaliana, Populus trichocarpa, and Chlamydomonas reinhardtii), the rate came out to 26%.
Although DDM is widely used, we asked whether other detergents could perform as well or even better for cell‐free screening. It is known that many detergents are too harsh for cell‐free experiments, such as phosphocholine, n‐octyl‐β‐d‐glucopyranoside (β‐OG), 3‐cholamidopropyl dimethylammonio 1‐propanesulfonate (CHAPS), but that polyoxyethylene alkyl‐ether (or Brij), although mild for extraction, seems very well suited for cell‐free use (Refs. 28, 34, 35, 36 and references therein). We thus used Brij‐35 in our cell‐free screening and found that 29 targets (48%) were expressed, including 24 of the 50 plant targets (48%), a significant improvement over DDM (Tables S1 and S2). We noticed that several targets were synthesized both in the presence of DDM or Brij‐35. Interestingly, they were not soluble in DDM, whereas they were solubilized with Brij‐35 (Figure 2a–d and data not shown). Only two targets (Bax Inhibitor 1 from poplar tree and A. thaliana fatty acid export 3) were produced and soluble in DDM but not with Brij‐35 (Figure 2d,e, Table S1). We did not detect any targets that were synthesized but not soluble in the presence of Brij‐35.
FIGURE 2.
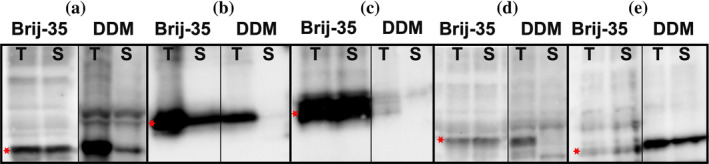
Cell‐free expression in detergents Brij‐35 and DDM. Anti‐His immunoblot of total cell‐free reactions (T) and soluble fractions (S) of selected targets expressed in the presence of Brij‐35 and DDM. (a) Fatty acid desaturase 4‐like 2 (34 kDa). (b) Mitochondrial pyruvate carrier 1 (15 kDa). (c) Transmembrane protein 126A (24 kDa). (d) β‐Carotene‐3‐hydroxylase 2 (36 kDa). (e) Fatty acid export 3 (40 kDa). Each lane contains 10% of total or soluble fractions. Relevant protein bands are indicated by a red star (*). Molecular weights include His tag. The band in the DDM soluble fraction of panel a is a background band
The phenomenon of enhanced solubilization by Brij‐35 was especially evident with the three mitochondrial pyruvate carriers (MPCs) from A. thaliana (Figures 2b and 3a). When the solubilized fractions were further purified by Ni2+‐affinity chromatography, only trace amounts of purified MPC1 and 2 could be detected with DDM, most likely due to a concentration effect of the purification and over‐exposure of the immunoblot (Figure 3b). Interestingly, we were able to detect higher molecular weight bands for MPC1 and 2 in Brij‐35 indicative of oligomerization, which is consistent with demonstrations that MPCs do oligomerize 37 , 38 , 39 and with the preservation of tertiary structure during Brij‐35 solubilization.
FIGURE 3.
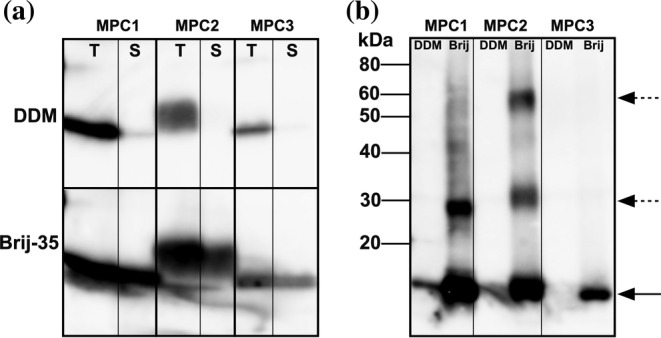
Cell‐free expression of mitochondrial pyruvate carriers (MPCs) in detergents Brij‐35 and DDM. Anti‐His immunoblot of total cell‐free reactions (T) and soluble fractions (S) (a), and of Nickel‐affinity purified proteins (b) of the A. thaliana MPC1, 2, and 3. The solid arrow in panel B indicates monomers and the dashed arrows oligomers. For total or soluble fractions, 10% was loaded on gel. For Ni‐purified, the amount was 25% of purified protein
2.4. Comparison between cell‐free and E. coli‐based expression
When the same set of targets was used for expression in E. coli BL21(DE3)‐pLysS cells using a 1‐ml mini‐scale expression system, 33 we found that 18 and 14 targets expressed when cells were extracted with DDM and Brij‐3, respectively (Tables S1 and S2). As mentioned above, the rates of expression, 30 and 23%, are what we have observed for expression of bacterial targets in our mini‐scale platform. In the cell‐based system, DDM performed somewhat better than Brij‐35, the reverse of what was observed in the cell‐free system. This is expected as DDM is considered a stronger detergent and thus better at extracting proteins from membranes. Although the rates for DDM in the cell‐free versus the cell‐based system were comparable (25% vs. 30%, Table S2), the number of targets expressed in the cell‐based expression using Brij‐35 was only half of that with Brij‐35 in the cell‐free system (48% vs. 23%, Table S2). For the plant targets, the rate of expression with DDM was about the same in both cell‐free and cell‐based systems (21% and 24%, respectively, Table S2); however, the rate with Brij‐35 was significantly higher for cell‐free than for cell‐based (39% vs. 18%, Table S2). A major contributor to this drop was the Chlamydomonas targets as none of them were expressed in E. coli.
Overall, three targets were detected in the cell‐based system that was not detected in the cell‐free method, whereas 20 total targets could be detected in the cell‐free system but not in E. coli cells, albeit that most of them were with Brij‐35 and in the low to medium range of expression (Table S1).
2.5. Cell‐free screening in liposomes
Liposomes are lipid‐based mimetics forming a cell‐like compartment that mimics native membrane bilayers. 40 , 41 We thus investigated how liposomes may be utilized in our cell‐free based screening of membrane proteins. We first used phosphatidyl‐choline (PC), a widely used lipid in liposomes, 28 , 42 in combination with phosphatidyl‐ethanolamine (PE; ratio of 95:5 of PC:PE). Surprisingly, only two targets (a lipid phosphate phosphatase and a molybdate transporter, both from A. thaliana) were soluble (Tables S1 and S2). Other targets were synthesized but were not soluble (Figure 4, “PC–PE” panels). Although PC and PE constitute the majority of phospholipids in Arabidopsis tissues such as seeds, roots, leaves, and extra‐chloroplastid organelles, 43 in chloroplasts there is no PE present and the two major phospholipids are PC and phosphatidyl‐glycerol (PG, ratio of PC:PG of 56:44; see Ref. 43). As about half of our targets are predicted to localize in chloroplasts (Table S1), we explored whether liposomes composed of PC and PG would perform better than PC–PE liposomes. As expected, we could identify a total of nine targets (15%) soluble in PC–PG liposomes including the two previously detected with PC–PE liposomes (Tables S1 and S2). Of those seven new targets, five are localized in chloroplasts, one is a human mitochondrial protein, and one was a Chlamydomonas sugar nucleotide transporter for which the subcellular localization remains unclear. Notably, these seven targets were also produced in the presence of PC–PE liposomes but could not be solubilized. Two examples are shown in Figure 4a,b for mitochondrial fission process protein 1 and fatty acid export 3. As with PC–PE some targets were synthesized in the presence PC–PG but were not soluble (Figure 4c,d).
FIGURE 4.
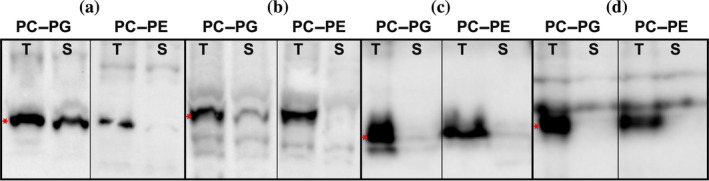
Cell‐free expression in liposomes. Anti‐His immunoblot of total cell‐free reactions (T) and soluble fractions (S) of selected targets expressed in the presence of PC–PE and PC–PG liposomes. (a) Mitochondrial fission process protein 1 (20 kDa). (b) Fatty acid export 3 (40 kDa). (c) Mitochondrial pyruvate carrier 1 (15 kDa). (d) Transmembrane protein 126A (24 kDa). Each lane contains 10% of total or soluble fractions. Relevant protein bands are indicated by a red star (*). Molecular weights include His tag
2.6. Cell‐free screening in MSP nanodiscs
Nanodiscs are also lipid‐based mimetics that can be used in cell‐free systems (Refs. 28, 44 and references therein). Nanodiscs circumscribe a patch of phospholipid bilayer by a membrane scaffold protein (MSP). For our cell‐free screening, we first used the MSP1E3D1 nanodiscs 45 , 46 , 47 (~12–13 nm in diameter) prefilled with DMPC (1,2‐dimyristoyl‐sn‐glycero‐3‐phosphocholine). We purified the reaction mixtures by Ni2+‐affinity chromatography, and we were able to identify five targets by immunoblots (8% expression rate; Tables S1 and S2).
As with the liposomes, we wanted to test the performance with a phospho‐glycerol. We, therefore, repeated the experiment but using nanodiscs prefilled with DMPG (1,2‐dimyristoyl‐sn‐glycero‐3‐phospho‐glycerol). Using DMPG nanodiscs, we were able to detect a total of 10 targets (16% expression rate). These included almost all targets that can be expressed in DMPC nanodiscs (Figure 5, Tables S1 and S2). It thus appears that DMPG is a more suitable lipid to form nanodiscs for use with cell‐free systems.
FIGURE 5.
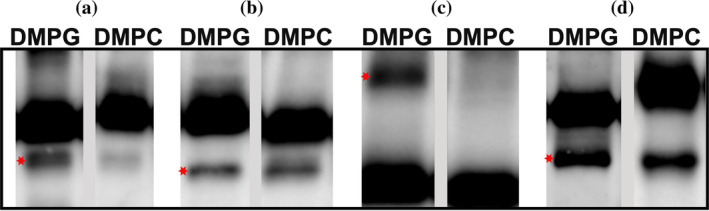
Cell‐free expression in MSP nanodiscs. Anti‐His immunoblot of Nickel‐purified cell‐free reactions of selected targets expressed in nanodiscs filled with DMPG or DMPC. (a) C. reinhardtii Bax Inhibitor 1 (30 kDa). (b) Lipid phosphate phosphatase γ (28 kDa). (c) Phosphate/phosphoenolpyruvate translocator PPT2 (46 kDa). (d) Mitochondrial fission process protein 1 (20 kDa). About one‐quarter of the purified protein was loaded on gel. Relevant protein bands are indicated by a red star (*). Molecular weights include His tag
2.7. Cell‐free production of functional mitochondrial pyruvate carrier complexes
In our cell‐free screening, we found that MPCs oligomerize (Figure 3). In fact, it is suggested that the functional carrier is a heterodimer of either MPC1/2 or MPC1/3. 38 , 39 To assess whether we could produce MPC complexes using our cell‐free set‐up, we performed reactions that contained two plasmids, each encoding an MPC with either a His‐tag or Strep‐tag at its C‐terminus. After reactions were completed, proteins were purified by Ni2+‐affinity resins followed by immunoblotting using antibodies against Strep‐tag. As shown in Figure 6, we can detect three associated complexes (MPC1/2, MPC1/3, and MPC2/3). The assembly of these complexes was not due to nonspecific binding of Strep‐tagged proteins to the Ni2+‐NTA resin (Figure 6, Lanes d and e). We were also able to detect the binding events of His‐tagged MPC1 to Strep‐tagged MPC2 or MPC3 using microscale thermophoresis (MST) 48 (Figure S1).
FIGURE 6.

Coexpression of mitochondrial pyruvate carrier complexes in Brij‐35. Anti‐Strep immunoblot of reaction mixture (lanes a–c) and of Ni2+‐purified proteins (lanes d–i) of cell‐free reactions containing the following plasmid combinations: (a) MPC1‐His. (b) MPC2‐His/MPC3‐Strep. (c) MPC3‐His/MPC2‐Strep. (d) MPC2‐Strep. (e) MPC3‐Strep. (f) MPC1‐His/MPC2‐Strep. (g) MPC1‐His/MPC3‐Strep. (h) MPC2‐His/MPC3‐Strep. (i) MPC3‐His/MPC2‐Strep. For lanes a–c, 10% of soluble fraction was assessed. For lanes d–i, 25% of purified protein was tested
With the cell‐free produced MPC complexes, we asked whether substrate pyruvate might enhance the complex formation. Indeed, the addition of sodium pyruvate increased expression of MPC1 and MPC3 significantly, but to a lesser extent for MPC2 (Figure 7 and data not shown). The optimal concentration of sodium pyruvate is about 100 μM for MPC1 and 250 μM for MPC3. This phenomenon is specific to sodium pyruvate as the lithium salt of pyruvate and sodium succinate had little to no effect (Figure 7).
FIGURE 7.
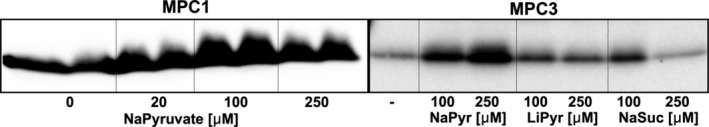
Cell‐free production of MPCs in presence of various ligands. Anti‐His immunoblot of soluble fraction of His‐tagged MPC1 or MPC3. Cell‐free reactions were performed in the presence of Brij‐35 (0.2% final) and indicated concentrations of ligands. Ligands were added in both reaction and feeding mixture. Pyr, pyruvate; Suc, succinate. 10% of the soluble fraction was assessed
We next used MST to evaluate whether the cell‐free produced MPCs could bind pyruvate. We used MPC1, MPC3, or MPC1/3 for titration with a series concentrations of sodium pyruvate, and we observed MST responses indicating binding events with the MPC1/3 complex exhibiting a significantly lower K d than MPC1 or MPC3 alone (Figure 8). Therefore, our cell‐free produced MPCs are capable of substrate binding.
FIGURE 8.
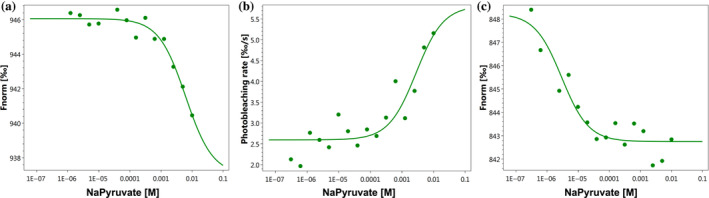
Microscale thermophoresis (MST) analysis of interactions between MPCs and pyruvate. Unlabeled MPC1‐His (500 nM, a), MPC3‐Strep (250 nM, b), or labeled His‐tagged MPC1 (50 nM) and unlabeled MPC3‐Strep (100 nM, c) was titrated with the indicated concentration of sodium pyruvate. MST was performed on Nanotemper Monolith NT.LabelFree at medium MST power and 20% excitation power (a and b) or on a Monolith NT.115 at medium MST power and 60% excitation power (c). Output for a and c was normalized fluorescence (1,000 units = relative fluorescence before MST power) and photobleaching rate for B (0/00% s−1 decline from −3 s to 0 s). Calculated K d s were ~6 mM (a), ~3 mM (b), and ~3 μM (c)
3. DISCUSSION
In this work, we present a cell‐free based screening workflow for expression of eukaryotic membrane proteins, which remains a bottleneck for structure–function characterizations. Our workflow features a parallel screening of six lipid mimetics using microdialysis devices and a 96‐well plate, thus providing a high‐throughput system for screening many eukaryotic membrane protein targets within 48–72 hr. To make use of E. coli extracts for eukaryotic membrane protein production, we optimized codons for all genes.
In our screening for 61 MPs, we have used six lipidic mimetics, which we found to be a good combination to cover various conditions of detergents, lipids, and nanodiscs. Not surprisingly, we found that detergents performed better than liposomes or nanodiscs. In our hands, Brij‐35 was significantly better than DDM in the cell‐free system. It has been shown that essentially only very mild surfactants such as Brij or digitonin work best (see Ref. 28 and references therein). In fact, when we used GDN, a synthetic derivative of digitonin, 49 on a select subset targets we found it to perform well, but no better than Brij‐35 (data not shown).
DDM performed comparably with Brij‐35 in the cell‐based system; however, in the cell‐free system, the rate of expression with DDM was only about half of that with Brij‐35. Overall, the two detergents covered 31 of the 61 targets in the cell‐free setup (51%), but only 18 (30%) in the cell‐based system, a rate that corresponds to what we routinely see for prokaryotic MP expression in our laboratory. 32 , 33 It should be noted that in the cell‐based expression the detergents are used to extract the proteins from the membranes whereas in the cell‐free methods that is not the case. Not surprisingly then a mild detergent such as Brij‐35 does not perform as well when used to extract proteins but is superior in the cell‐free method.
Although adding liposomes and nanodiscs to the cell‐free reactions did not increase the expression coverage by significant amounts (from 31 to 35), we envision that these lipidic mimetics are still worthwhile: first, combinations of different lipids might result in expression (or solubility) of targets previously not detected. This was shown with the liposomes where substituting PG for PE increased the solubility of targets significantly. Second, liposomes and nanodiscs more closely represent a natural environment for membrane proteins than detergents. Lipids play a vital role in the folding process, topological organization, and modulation of functionality of membrane proteins. 50 , 51 This becomes particularly important when studying biophysical and functional characterizations of MPs. 27 Our own data with pyruvate binding to the mitochondrial pyruvate carriers (MPCs) shows that although the MPCs produced with Brij‐35 did show binding of pyruvate, the K d s were relatively high, especially for the single MPC proteins, and the binding affinity can probably be improved by synthesizing the MPCs in a lipid bilayer.
It is important to note that the screening platform described here can be expanded easily to include additional lipidic mimetics, be they detergents such as additional members of the Brij family or other maltosides, 52 , 53 , 54 liposomes with different lipid compositions or blended with diblock polymers, 55 , 56 , 57 lipid‐detergent mixtures (i.e., bicelles, 27 , 58 , 59 ), improved nanodiscs derivatives 60 or other amphipathic copolymers such as amphipols. 61 It should also be noted that only non‐ionic amphipols (NAPols or Nvoy) appear to work in cell‐free systems and that ionic amphipols, such as A8‐35, and SMAs are inhibitory. 61 , 62 , 63 Our own limited data however suggest that even NAPols might be inhibitory in cell‐free reactions (data not shown).
Therefore, it becomes imperative to screen a wide range of lipidic mimetics for any particular membrane protein to find a suitable environment, for both structural and functional analysis, for the target. For instance, 23 targets could not be at all expressed and solubilized in any of our cell‐free or cell‐based assays (Table S1). These so‐called “recalcitrant” targets are a major impediment in the membrane protein field and more innovative ways need to be developed for them (Section 1). However, it is also likely that some of these targets could be expressed in our cell‐free setup with mimetics that have not been tested as described above. For example, Blesneac et al. 34 found that the production of soluble rat uncoupling protein UCP1 could be improved significantly in the presence of fluorinated surfactants and cardiolipin.
Another approach is to add additives to improve expression and/or stabilize the target in a conducive environment as was shown with pyruvate and the mitochondrial pyruvate carriers. For instance, Kai et al. 64 tested more than 20 chemical “chaperones” that can potentially stabilize membrane proteins. Those included sugars, modified amino acids, alcohols, and PEGs of different sizes.
Incidentally, none of the 23 recalcitrant targets can actually be synthesized in the total reaction mixtures (Figure 1 and Section 4). It is worthwhile noting that the average and median molecular weights of this subset of targets are higher than for the total set (p < .01) and significantly higher than any of the expressed ones (p < .001; see Table S3). It is thus possible that expression of these targets is just a matter of an improved transcriptional/translational system and that could be achieved by optimizing several parameters including reaction temperature, potassium, and magnesium concentration, as well as modifying regulatory elements to improve RNA transcription and protein synthesis. 64 , 65 , 66 , 67 , 68
Many membrane proteins are stabilized and function as oligomers. Cell‐free expression is applicable to the formation of homooligomers; however, attention is needed to screen and optimization of MP production for heterodimers. With our MPC complexes as an example, we found the highest yield when MPC1/2 or MPC1/3 were co‐expressed. Incidentally, binding of pyruvate as measured using MST was highest for the MPC1/3 complex as compared to MPC1 or MPC3 alone (Figure 8). This is not surprising as it has been shown that the functional MPC is most likely a heterodimer. 37 , 38 , 69 Although we were not able to find any K d s for MPCs in the literature, the K d we measured for the A. thaliana MPC1/3 complex (~ 3 μM) seems to be lower than Michaelis constants (K M) for rat or mouse liver MPC activity, 70 but higher than IC50s found for some MPC inhibitors. 71
In addition, substrate sodium pyruvate seems to further enhance the MPC production in a concentration‐dependent manner (Figure 7). Therefore, cell‐free systems offer a convenient way to titrate the formation of hetero‐complexes that are challenging, if not impossible, to produce in cell‐based systems. With the further tuning of the ratio of added MPC1/2 and MPC1/3 plasmids, one can even tune the ratio of hetero‐complex components.
To the best of our knowledge, this is the first instance of screening such a large number of eukaryotic membrane proteins using a cell‐free system and three types of lipidic mimetics. Shinoda et al. tested 19 human membrane proteins using mostly liposomes and bicelles 27 and Isaksson et al. tested 24 mostly human membrane proteins although 18 of those were aquaporins and UCPs. 35 Other high throughput systems used soluble (i.e., cytoplasmic) targets or very few membrane proteins among them or used exclusively prokaryotic membrane proteins. 42 , 72 , 73 Our results highlight the highly efficient use of cell‐free systems for screening and production of eukaryotic membrane proteins.
4. MATERIALS AND METHODS
4.1. Reagents
Amino acids, nucleotides, pyruvate kinase (PK), phospho(enol) pyruvate (PEP), acetyl phosphate (AcP), and folinic acid were all from Sigma‐Aldrich (St. Louis, MO). RiboLock RNase inhibitor was from Thermofisher. E. coli tRNA and complete protease inhibitor cocktail tablets were from Roche. n‐Dodecyl‐β‐d‐maltopyranoside (DDM), GDN, NAPol, were from Anatrace and Brij‐35 from Millipore (Burlington, MA). l‐α‐Phosphatidylcholine (PC) and l‐α‐phosphatidylethanolamine (PE) were from Sigma‐Aldrich, and 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phospho‐glycerol (POPG), 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphocholine (DMPC), and 1,2‐dimyristoyl‐sn‐glycero‐3‐phospho‐glycerol (DMPG) were from Avanti Polar Lipids (Alabaster, AL).
The mouse anti‐HIS antibody was from Takara Bio (Kusatsu, Japan). The mouse anti‐STREP antibody was from IBA Lifesciences (Göttingen, Germany). Goat anti‐mouse IgG‐Peroxidase antibody was from Sigma‐Aldrich. SDS protein gels were from BioRad (Hercules, CA; Criterion 4–20% Tris‐Cl). Blocking reagent (SuperBlock T20), PBS‐Tween, and chemiluminescence reagent (SuperSignal™ West Femto Maximum Sensitivity Substrate) were from Thermofisher (Waltham, MA).
Isopropyl‐beta‐d‐thiogalactoside (IPTG), 4‐(2‐Aminoethyl)‐benzenesulfonylfluoride hydrochloride (AEBSF), Tris (2‐Carboxyethyl) phosphine (TCEP), and Nickel chelating resin were from Gold Biotechnology (St. Louis, MO).
4.2. E. coli strains and DNAs
The E. coli A19 strain [Hfr, rna‐19, gdhA2, his‐95, relA1, spoT1, metB1] was from the E. coli Genetic Stock Center at Yale University (cgsc.biology.yale.edu). The expression strain BL21(DE3)‐pLysS [F− ompT, hsdSB(rB −, mB −), gal, dcm, (DE3), pLysS‐(CamR)], and the plasmid pAR1219 were from Sigma (St. Louis, MO). All target DNAs were codon‐optimized, synthesized, and cloned into a pET23 derivative containing a C‐terminal TEV cleavage site followed by a deca‐histidine tag.
4.3. S30 E. coli extract and T7 RNA polymerase preparation
For S30 extract preparation, the A19 strain was grown in Terrific Broth (TB) to an OD600 of 3–4. Cells were collected by centrifugation and washed three times in S30 buffer A (10 mM Tris‐Acetate, pH 8.2, 14 mM Mg[OAc]2, 60 mM KCl, and 6 mM β‐Mercaptoethanol). Cells were then thoroughly resuspended with a volume, corresponding 1.1 × the pellet, of S30 buffer B (buffer A plus 1 mM DTT and 1 mM AEBSF) and broken up with a micro‐fluidizer. The resulting extract was clarified by centrifugation (30,000×g, 30 min), adjusted with NaCl to 400 mM, incubated at 42°C for 45 min and then dialyzed against S30 buffer C (10 mM Tris‐Acetate, pH 8.2, 14 mM Mg(OAc)2, 60 mM K(OAc), and 0.5 mM DDT). The dialyzed lysate was then cleared again by centrifugation, aliquoted, and stored at −80°C.
For T7 RNA polymerase production, pAR1219 plasmid was transformed in BL21 Star (Novagen). Cells were grown in LB to an OD600 of about 0.7–0.8, collected by centrifugation, resuspended in Resuspension Buffer A (30 mM Tris‐Cl, pH = 8, 50 mM NaCl, 10 mM EDTA, 10 mM β‐mercaptoethanol, 1 mM AEBSF, and 5% glycerol), broken up by sonication and debris was removed by centrifugation (20,000×g, 30 min). Genomic DNA was removed by adding streptomycin sulfate to the clarified lysate stepwise to a final concentration of 2% and the resulting precipitate was spun down by centrifugation (20,000×g, 30 min). The lysate was then purified over an anion exchange column (HiPrepQ FF 16 10, GE) using a NaCl gradient from 50–500 mM in Resuspension Buffer A. Fractions containing the T7 RNA polymerase protein (as judged by protein gel electrophoresis) were pooled and dialyzed against a buffer containing 10 mM K2HPO4, pH = 8, 100 mM NaCl, 0.5 mM EDTA, 1 mM DTT, and 5% glycerol. The dialyzed lysate was adjusted to a final glycerol concentration of 10% and concentrated to about 4 mg/ml. Finally, glycerol concentration was adjusted to 50% and the preparation was aliquoted and stored at −20°C.
4.4. Cell‐free reactions
Cell‐free miniscale reactions were in a total volume of 110 μl and were performed in Pierce 96‐well microdialysis devices (10 kDa cut‐off; Thermofisher). The reaction mixture (RM) consisted of 35% of S30 extract, 0.015 mg/ml of DNA, 0.3 U/μl RiboLock, 5–6 μl of T7 RNA polymerase, 0.5 mg/ml tRNA and 0.1 mg/ml of PK in a feeding mixture (FM) containing amino acids at 1 mM each, PEP and AcP at 20 mM each, GTP, CTP, and UTP (0.8 mM each), ATP (1.2 mM), DTT (2 mM), folinic acid (0.1 mg/ml), complete protease inhibitor cocktail (0.05 tablet/ml), 100 mM HEPES‐KOH pH = 8, 20 mM EDTA, 11 mM Mg(OAc)2, 150 mM K(OAc), 2% PEG 8000, and 0.05% NaN3. The microdialysis devices were placed in 96‐well deep‐block plates containing ≈1.9 ml of 0.4× of FM. For detergent screening, DDM and Brij‐35 were included in both RM and FM at 0.2% (w/v) final. GDN and NAPol were at final concentrations of 9.3 and 3 mg/ml, respectively, in both RM and FM. PC–PE (95%:5%, w/w) and PC–POPG (56%:44%, w/w) liposomes were added to RM only at a final concentration of 2.7 mg/ml. Nanodiscs (MSP1E3) with DMPC or DMPG were added to RM only at a final concentration of 10 μM. Reactions were performed at 34°C overnight in a SHEL LAB shaking incubator (Sheldon Manufacturing, Cornelius, OR) at 700 rpm.
4.5. Purification of cell‐free reactions
Reactions were transferred to 1.5 ml Eppendorf tubes and 10 μl was removed for assessing total protein synthesis (T). For the soluble fractions (S), tubes were spun at 20,000×g at 4°C for 30 min and 10 μl were removed from the supernatant. For Ni2+‐affinity purification, soluble fractions were placed into a 96‐well filter plate deep‐well block (Thomson Instrument Company, Oceanside, CA) and 35 μl of a 50% Ni‐Sepharose slurry was added. The mixtures were then diluted with PBS to about 500 μl and shaken for 1–2 hr at 4°C. Ni2+‐sepharose beads were washed twice and proteins were eluted in a total of 40 μl as described previously. 33
4.6. Immunoblotting assay
For detergent and liposome experiments, equal amounts of T and S fractions (5–10 μl) were separated on Criterion gels. For nanodisc experiments, 10 μl of Ni2+‐affinity‐purified fraction was used. Gels were run in Tris‐Glycine‐SDS buffer in a Dodeca Cell chamber (BioRad) following manufacturer's manual. Proteins were transferred to PVDF membranes using an iBlot 2 transferring machine (Thermofisher). Membranes were blocked for 1 hr in blocking buffer and then incubated overnight with the primary antibody diluted in blocking buffer (1:5,000 for the anti‐His and 1:1,000 for the anti‐STREP). Membranes were then washed three times in PBS‐Tween, incubated for 1 hr with a 1:70,000 dilution of anti‐mouse IgG‐Peroxidase antibody, and then washed again three times. Finally, membranes were incubated for 1 min in enhanced chemiluminescence (ECL) reagent and imaged using a ChemiDoc imaging system (BioRad). Only immunoblot bands in the range (±20%) of the expected molecular weight were considered. Bands had to be clearly above background to be included as expressed. For each lipidic mimetic, the strongest bands were assigned three stars (***; see also Table S1). All other bands were categorized as either medium or weak and given either two (**) or one (*) star, respectively.
4.7. BL21‐pLysS mini‐scale expression
Expression and purification of targets in BL21‐pLysS were performed essentially as described previously. 33 In short, 30 μl of an overnight saturated culture was added to 1 ml of 2xTY (plus carbenicillin and chloramphenicol) in a 96‐well deep‐well block, and cells were grown for 75 min at 37°C at which point IPTG was added to a final concentration of 0.4 mM and cells were grown for an additional 4 hr at 37°C. Cells were recovered by centrifugation and pellets were resuspended in 0.5 ml of Resuspension buffer (50 mM HEPES, pH 7.8, 300 mM NaCl, 20 mM imidazole, pH 7.8, 1 mM MgCl2, 1.2 mg/ml AEBSF, and 0.5 mM TCEP). After two rounds of sonication, DDM or Brij‐35 was added to a final 2% and extraction was done in the cold for at least 1 hr on a shaking platform. Lysate was then centrifuged at 4,500×g and 10 μl of soluble supernatant was removed and assayed by immunoblotting as described above.
4.8. Microscale thermophoresis
MST was performed on Nanotemper Technologies' Monolith NT.115 for labeled targets and Monolith NT.LabelFree for unlabeled targets following the manual from the manufacturer (Nanotemper Technologies, München, Germany). For labeled His‐tagged targets, protein (100 nM final) was incubated with 50 nM of RED‐tris‐NTA label for 30 min at RT before binding assays. Binding assays for both NT.115 and NT.LabelFree were performed by mixing an equal volume of labeled or unlabeled protein and 1:1 serially diluted ligand in assay buffer (40 mM HEPES, pH 7.8, 200 mM NaCl, 0.2% Brij‐35). Samples were then loaded into capillaries (Nanotemper Technologies) and measured using the manufacturer's recommended settings. Results were monitored by using MO. Control software and K d s were determined with MO. Affinity (Nanotemper Technologies).
CONFLICT OF INTEREST
Renato Bruni, Aisha Laguerre, Anna‐Maria Kaminska, Sean McSweeney, Wayne A. Hendrickson, and Qun Liu declare no competing interests.
AUTHOR CONTRIBUTIONS
Renato Bruni: Conceptualization (equal); data curation (lead); formal analysis (equal); methodology (equal); project administration (lead); supervision (equal); validation (equal); visualization (equal); writing – original draft (lead); writing – review and editing (equal). Aisha Laguerre: Conceptualization (supporting); methodology (supporting). Anna‐Maria Kaminska: Data curation (supporting). Sean McSweeney: Conceptualization (supporting); funding acquisition (supporting). Wayne A. Hendrickson: Conceptualization (supporting); formal analysis (supporting); funding acquisition (lead); project administration (supporting); resources (supporting); writing – original draft (supporting); writing – review and editing (supporting). Qun Liu: Conceptualization (equal); data curation (supporting); formal analysis (equal); investigation (equal); methodology (equal); project administration (supporting); supervision (equal); validation (equal); visualization (equal); writing – original draft (supporting); writing – review and editing (equal).
Supporting information
Appendix S1 Supporting Information
ACKNOWLEDGMENTS
This work was supported by NIH NIGMS grant 5P41GM116799 through the Center on Membrane Protein Production and Analysis. Qun Liu was supported by the U.S. Department of Energy, Office of Science, Biological and Environmental Research, as part of the Quantitative Plant Science Initiative at BNL.
Bruni R, Laguerre A, Kaminska A‐M, McSweeney S, Hendrickson WA, Liu Q. High‐throughput cell‐free screening of eukaryotic membrane protein expression in lipidic mimetics. Protein Science. 2022;31:639–651. 10.1002/pro.4259
Funding information National Institute of General Medical Sciences, Grant/Award Number: 5P41GM116799‐05; U.S. Department of Energy, Office of Science, Biological and Environmental Research
Contributor Information
Renato Bruni, Email: rbruni@nysbc.org.
Qun Liu, Email: qunliu@bnl.gov.
REFERENCES
- 1. Tan S, Tan HT, Chung MC. Membrane proteins and membrane proteomics. Proteomics. 2008;8(19):3924–3932. [DOI] [PubMed] [Google Scholar]
- 2. Wallin E, von Heijne G. Genome‐wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7(4):1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bakheet TM, Doig AJ. Properties and identification of human protein drug targets. Bioinformatics. 2009;25(4):451–457. [DOI] [PubMed] [Google Scholar]
- 4. Yildirim MA, Goh KI, Cusick ME, Barabási AL, Vidal M. Drug‐target network. Nat Biotechnol. 2007;25(10):1119–1126. [DOI] [PubMed] [Google Scholar]
- 5. Opekarova M, Tanner W. Specific lipid requirements of membrane proteins—A putative bottleneck in heterologous expression. Biochim Biophys Acta. 2003;1610(1):11–22. [DOI] [PubMed] [Google Scholar]
- 6. Wagner S, Baars L, Ytterberg AJ, et al. Consequences of membrane protein overexpression in Escherichia coli . Mol Cell Proteomics. 2007;6(9):1527–1550. [DOI] [PubMed] [Google Scholar]
- 7. Wagner S, Bader ML, Drew D, de Gier JW. Rationalizing membrane protein overexpression. Trends Biotechnol. 2006;24(8):364–371. [DOI] [PubMed] [Google Scholar]
- 8. Sonoda Y, Cameron A, Newstead S, et al. Tricks of the trade used to accelerate high‐resolution structure determination of membrane proteins. FEBS Lett. 2010;584(12):2539–2547. [DOI] [PubMed] [Google Scholar]
- 9. Sonoda Y, Newstead S, Hu NJ, et al. Benchmarking membrane protein detergent stability for improving throughput of high‐resolution X‐ray structures. Structure. 2011;19(1):17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kang HJ, Lee C, Drew D. Breaking the barriers in membrane protein crystallography. Int J Biochem Cell Biol. 2013;45(3):636–644. [DOI] [PubMed] [Google Scholar]
- 11. Kloppmann E, Punta M, Rost B. Structural genomics plucks high‐hanging membrane proteins. Curr Opin Struct Biol. 2012;22(3):326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Almo SC, Garforth SJ, Hillerich BS, Love JD, Seidel RD, Burley SK. Protein production from the structural genomics perspective: Achievements and future needs. Curr Opin Struct Biol. 2013;23(3):335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lundstrom K. Structural genomics for membrane proteins. Cell Mol Life Sci. 2006;63(22):2597–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Markley JL, Aceti DJ, Bingman CA, et al. The center for eukaryotic structural genomics. J Struct Funct Genomics. 2009;10(2):165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sauder MJ, Rutter ME, Bain K, et al. High throughput protein production and crystallization at NYSGXRC. Methods Mol Biol. 2008;426:561–575. [DOI] [PubMed] [Google Scholar]
- 16. Kim Y, Babnigg G, Jedrzejczak R, et al. High‐throughput protein purification and quality assessment for crystallization. Methods. 2011;55(1):12–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elsliger MA, Deacon AM, Godzik A, et al. The JCSG high‐throughput structural biology pipeline. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66(Pt 10):1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miroux B, Walker JE. Over‐production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260(3):289–298. [DOI] [PubMed] [Google Scholar]
- 19. Angius F, Ilioaia O, Amrani A, et al. A novel regulation mechanism of the T7 RNA polymerase based expression system improves overproduction and folding of membrane proteins. Sci Rep. 2018;8(1):8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karyolaimos A, Ampah‐Korsah H, Zhang Z, de Gier JW. Shaping Escherichia coli for recombinant membrane protein production. FEMS Microbiol Lett. 2018;365(15):fny250. [DOI] [PubMed] [Google Scholar]
- 21. Michou M, Kapsalis C, Pliotas C, Skretas G. Optimization of recombinant membrane protein production in the engineered Escherichia coli strains SuptoxD and SuptoxR. ACS Synth Biol. 2019;8(7):1631–1641. [DOI] [PubMed] [Google Scholar]
- 22. Skretas G, Georgiou G. Simple genetic selection protocol for isolation of overexpressed genes that enhance accumulation of membrane‐integrated human G protein‐coupled receptors in Escherichia coli . Appl Environ Microbiol. 2010;76(17):5852–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Skretas G, Makino T, Varadarajan N, Pogson M, Georgiou G. Multi‐copy genes that enhance the yield of mammalian G protein‐coupled receptors in Escherichia coli . Metab Eng. 2012;14(5):591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McKenzie EA, Abbott WM. Expression of recombinant proteins in insect and mammalian cells. Methods. 2018;147:40–49. [DOI] [PubMed] [Google Scholar]
- 25. Martinez‐Solis M, Herrero S, Targovnik AM. Engineering of the baculovirus expression system for optimized protein production. Appl Microbiol Biotechnol. 2019;103(1):113–123. [DOI] [PubMed] [Google Scholar]
- 26. Henrich E, Hein C, Dötsch V, Bernhard F. Membrane protein production in Escherichia coli cell‐free lysates. FEBS Lett. 2015;589(15):1713–1722. [DOI] [PubMed] [Google Scholar]
- 27. Shinoda T, Shinya N, Ito K, et al. Cell‐free methods to produce structurally intact mammalian membrane proteins. Sci Rep. 2016;6:30442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hein C, Henrich E, Orban E, Dötsch V, Bernhard F. Hydrophobic supplements in cell‐free systems: Designing artificial environments for membrane proteins. Eng Life Sci. 2014;14:365–379. [Google Scholar]
- 29. Rues RB, Henrich E, Boland C, Caffrey M, Bernhard F. Cell‐free production of membrane proteins in Escherichia coli lysates for functional and structural studies. Methods Mol Biol. 2016;1432:1–21. [DOI] [PubMed] [Google Scholar]
- 30. Schwarz D, Junge F, Durst F, et al. Preparative scale expression of membrane proteins in Escherichia coli‐based continuous exchange cell‐free systems. Nat Protoc. 2007;2(11):2945–2957. [DOI] [PubMed] [Google Scholar]
- 31. Schwarz D, Klammt C, Koglin A, et al. Preparative scale cell‐free expression systems: New tools for the large scale preparation of integral membrane proteins for functional and structural studies. Methods. 2007;41(4):355–369. [DOI] [PubMed] [Google Scholar]
- 32. Punta M, Love J, Handelman S, et al. Structural genomics target selection for the New York consortium on membrane protein structure. J Struct Funct Genomics. 2009;10(4):255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bruni R, Kloss B. High‐throughput cloning and expression of integral membrane proteins in Escherichia coli . Curr Protoc Protein Sci. 2013;74:29.6.1–29.6.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blesneac I, Ravaud S, Juillan‐Binard C, et al. Production of UCP1 a membrane protein from the inner mitochondrial membrane using the cell free expression system in the presence of a fluorinated surfactant. Biochim Biophys Acta. 2012;1818(3):798–805. [DOI] [PubMed] [Google Scholar]
- 35. Isaksson L, Enberg J, Neutze R, Göran Karlsson B, Pedersen A. Expression screening of membrane proteins with cell‐free protein synthesis. Protein Expr Purif. 2012;82(1):218–225. [DOI] [PubMed] [Google Scholar]
- 36. Rebuffet E, Frick A, Järvå M, Törnroth‐Horsefield S. Cell‐free production and characterisation of human uncoupling protein 1‐3. Biochem Biophys Rep. 2017;10:276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bricker DK, Taylor EB, Schell JC, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, drosophila, and humans. Science. 2012;337(6090):96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. He L, Jing Y, Shen J, et al. Mitochondrial pyruvate carriers prevent cadmium toxicity by sustaining the TCA cycle and glutathione synthesis. Plant Physiol. 2019;180(1):198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shen JL, Li CL, Wang M, et al. Mitochondrial pyruvate carrier 1 mediates abscisic acid‐regulated stomatal closure and the drought response by affecting cellular pyruvate content in Arabidopsis thaliana . BMC Plant Biol. 2017;17(1):217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Verchere A, Broutin I, Picard M. Reconstitution of membrane proteins in liposomes. Methods Mol Biol. 2017;1635:259–282. [DOI] [PubMed] [Google Scholar]
- 41. Routledge SJ, Linney JA, Goddard AD. Liposomes as models for membrane integrity. Biochem Soc Trans. 2019;47(3):919–932. [DOI] [PubMed] [Google Scholar]
- 42. Niwa T, Sasaki Y, Uemura E, et al. Comprehensive study of liposome‐assisted synthesis of membrane proteins using a reconstituted cell‐free translation system. Sci Rep. 2015;5:18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li‐Beisson Y, Shorrosh B, Beisson F, et al. Acyl‐lipid metabolism. Arabidopsis Book. 2013;11:e0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rues RB, Gräwe A, Henrich E, Bernhard F. Membrane protein production in E. coli lysates in presence of preassembled nanodiscs. Methods Mol Biol. 2017;1586:291–312. [DOI] [PubMed] [Google Scholar]
- 45. Bayburt TH, Carlson JW, Sligar SG. Reconstitution and imaging of a membrane protein in a nanometer‐size phospholipid bilayer. J Struct Biol. 1998;123(1):37–44. [DOI] [PubMed] [Google Scholar]
- 46. Schuler MA, Denisov IG, Sligar SG. Nanodiscs as a new tool to examine lipid‐protein interactions. Methods Mol Biol. 2013;974:415–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Denisov IG, Schuler MA, Sligar SG. Nanodiscs as a new tool to examine lipid‐protein interactions. Methods Mol Biol. 2019;2003:645–671. [DOI] [PubMed] [Google Scholar]
- 48. Jerabek‐Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol. 2011;9(4):342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chae PS, Rasmussen SGF, Rana RR, et al. A new class of amphiphiles bearing rigid hydrophobic groups for solubilization and stabilization of membrane proteins. Chemistry. 2012;18(31):9485–9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Corin K, Bowie JU. How bilayer properties influence membrane protein folding. Protein Sci. 2020;29(12):2348–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dowhan W, Vitrac H, Bogdanov M. Lipid‐assisted membrane protein folding and topogenesis. Protein J. 2019;38(3):274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Junge F, Luh LM, Proverbio D, et al. Modulation of G‐protein coupled receptor sample quality by modified cell‐free expression protocols: A case study of the human endothelin a receptor. J Struct Biol. 2010;172(1):94–106. [DOI] [PubMed] [Google Scholar]
- 53. Rath P, Demange P, Saurel O, et al. Functional expression of the PorAH channel from Corynebacterium glutamicum in cell‐free expression systems: Implications for the role of the naturally occurring mycolic acid modification. J Biol Chem. 2011;286(37):32525–32532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Klammt C, Schwarz D, Fendler K, Haase W, Dötsch V, Bernhard F. Evaluation of detergents for the soluble expression of alpha‐helical and beta‐barrel‐type integral membrane proteins by a preparative scale individual cell‐free expression system. FEBS J. 2005;272(23):6024–6038. [DOI] [PubMed] [Google Scholar]
- 55. Jacobs ML, Boyd MA, Kamat NP. Diblock copolymers enhance folding of a mechanosensitive membrane protein during cell‐free expression. Proc Natl Acad Sci USA. 2019;116(10):4031–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Beales PA, Khan S, Muench SP, Jeuken LJC. Durable vesicles for reconstitution of membrane proteins in biotechnology. Biochem Soc Trans. 2017;45(1):15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Seneviratne R, Khan S, Moscrop E, et al. A reconstitution method for integral membrane proteins in hybrid lipid‐polymer vesicles for enhanced functional durability. Methods. 2018;147:142–149. [DOI] [PubMed] [Google Scholar]
- 58. Wada T, Shimono K, Kikukawa T, et al. Crystal structure of the eukaryotic light‐driven proton‐pumping rhodopsin, Acetabularia rhodopsin II, from marine alga. J Mol Biol. 2011;411(5):986–998. [DOI] [PubMed] [Google Scholar]
- 59. Lyukmanova EN, Shenkarev ZO, Khabibullina NF, et al. Lipid‐protein nanodiscs for cell‐free production of integral membrane proteins in a soluble and folded state: Comparison with detergent micelles, bicelles and liposomes. Biochim Biophys Acta. 2012;1818(3):349–358. [DOI] [PubMed] [Google Scholar]
- 60. Johansen NT, Tidemand FG, Nguyen TTTN, Rand KD, Pedersen MC, Arleth L. Circularized and solubility‐enhanced MSPs facilitate simple and high‐yield production of stable nanodiscs for studies of membrane proteins in solution. FEBS J. 2019;286(9):1734–1751. [DOI] [PubMed] [Google Scholar]
- 61. Bazzacco P, Billon‐Denis E, Sharma KS, et al. Nonionic homopolymeric amphipols: Application to membrane protein folding, cell‐free synthesis, and solution nuclear magnetic resonance. Biochemistry. 2012;51(7):1416–1430. [DOI] [PubMed] [Google Scholar]
- 62. Klammt C, Perrin MH, Maslennikov I, et al. Polymer‐based cell‐free expression of ligand‐binding family B G‐protein coupled receptors without detergents. Protein Sci. 2011;20(6):1030–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Periasamy A, Shadiac N, Amalraj A, et al. Cell‐free protein synthesis of membrane (1,3)‐beta‐d‐glucan (curdlan) synthase: Co‐translational insertion in liposomes and reconstitution in nanodiscs. Biochim Biophys Acta. 2013;1828(2):743–757. [DOI] [PubMed] [Google Scholar]
- 64. Kai L, Orbán E, Henrich E, Proverbio D, Dötsch V, Bernhard F. Co‐translational stabilization of insoluble proteins in cell‐free expression systems. Methods Mol Biol. 2015;1258:125–143. [DOI] [PubMed] [Google Scholar]
- 65. Caschera F, Noireaux V. Preparation of amino acid mixtures for cell‐free expression systems. Biotechniques. 2015;58(1):40–43. [DOI] [PubMed] [Google Scholar]
- 66. Garamella J, Marshall R, Rustad M, Noireaux V. The all E. coli TX‐TL toolbox 2.0: A platform for cell‐free synthetic biology. ACS Synth Biol. 2016;5(4):344–355. [DOI] [PubMed] [Google Scholar]
- 67. Marshall R, Noireaux V. Synthetic biology with an all E. coli TXTL system: Quantitative characterization of regulatory elements and gene circuits. Methods Mol Biol. 2018;1772:61–93. [DOI] [PubMed] [Google Scholar]
- 68. Haberstock S, Roos C, Hoevels Y, et al. A systematic approach to increase the efficiency of membrane protein production in cell‐free expression systems. Protein Expr Purif. 2012;82(2):308–316. [DOI] [PubMed] [Google Scholar]
- 69. Herzig S, Raemy E, Montessuit S, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337(6090):93–96. [DOI] [PubMed] [Google Scholar]
- 70. Gray LR, Rauckhorst AJ, Taylor EB. A method for multiplexed measurement of mitochondrial pyruvate carrier activity. J Biol Chem. 2016;291(14):7409–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McCommis KS, Finck BN. Mitochondrial pyruvate transport: A historical perspective and future research directions. Biochem J. 2015;466(3):443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Langlais C, Guilleaume B, Wermke N, et al. A systematic approach for testing expression of human full‐length proteins in cell‐free expression systems. BMC Biotechnol. 2007;7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Savage DF, Anderson CL, Robles‐Colmenares Y, Newby ZE, Stroud RM. Cell‐free complements in vivo expression of the E. coli membrane proteome. Protein Sci. 2007;16(5):966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information