

Abstract
Objective
Autologous haematopoietic stem cell transplantation (AHSCT) has the potential to induce sustained periods of disease remission in multiple sclerosis (MS), which is an inflammatory disease of the central nervous system (CNS) characterised by demyelination and axonal degeneration. However, the mechanisms associated with durable treatment responses in MS require further elucidation.
Methods
To characterise the longer term immune reconstitution effects of AHSCT at 24 and 36 months (M) post‐transplant, high‐dimensional immunophenotyping of peripheral blood mononuclear cells from 22 MS patients was performed using two custom‐designed 18‐colour flow cytometry panels.
Results
The higher baseline frequencies of specific pro‐inflammatory immune cells (T‐helper‐17 (Th17) cells, mucosal‐associated invariant T‐cells and CNS‐homing T‐conventional (T‐conv) cells observed in MS patients were decreased post‐AHSCT by 36M. This was accompanied by a post‐AHSCT increase in frequencies and absolute counts of immunoregulatory CD56hi natural killer cells at 24M and terminally differentiated CD8+CD28−CD57+ cells until 36M. A sustained increase in the proportion of naïve B‐cells, with persistent depletion of memory B‐cells and plasmablasts was observed until 36M. Reconstitution of the B‐cell repertoire was accompanied by a reduction in the frequency of circulating T‐follicular helper cells (cTfh) expressing programmed cell death‐1 (PD1+) at 36M. Associations between frequency dynamics and clinical outcomes indicated only responder patients to exhibit a decrease in Th17, CNS‐homing T‐conv and PD1+ cTfh pro‐inflammatory subsets at 36M, and an increase in CD39+ T‐regulatory cells at 24M.
Interpretation
AHSCT induces substantial recalibration of pro‐inflammatory and immunoregulatory components of the immune system of MS patients for up to 36M post‐transplant.
Introduction
Multiple sclerosis (MS) is an immune‐mediated disorder affecting central nervous system (CNS) characterised by demyelination, axonal damage, and neurodegeneration, 1 , 2 for which autologous haematopoietic stem cell transplantation (AHSCT) has been investigated as a treatment option in refractory MS patients. AHSCT is highly effective in suppressing inflammatory activity resulting in remission from clinical relapses and radiological activity in MS patients. 2 , 3 , 4 , 5 , 6 It has been proposed that elimination of the autoreactive immune system and subsequent reconstitution of a renewed immune repertoire underpins the clinical response. 7
We have previously published early lymphocyte reconstitution until 1 year (yr) post‐AHSCT in a well‐characterized clinical cohort of MS patients treated with BEAM (carmustine, etoposide, cytarabine, melphalan) + horse antithymocyte globulin (ATG). 2 Our earlier work demonstrated enrichment of immunoregulatory cells until 1 yr post‐AHSCT, 2 corroborating similar findings from other MS studies 8 , 9 , 10 that extended until 2 yr, 8 , 9 or up to 72M post‐AHSCT, 7 despite using different conditioning regimens. Not all the subsets reported in this current study has been examined earlier in MS post‐AHSCT. For immune subsets that have been previously reported in MS in the peri‐transplant setting, this study examines immune correlates up to extended post‐AHSCT timepoints of 36M. The long‐term timepoint of 36M post‐AHSCT is reflective of actual sustained immunological changes beyond initial post‐chemotherapy lymphopenia.
Subsets that possess CNS‐transmigratory properties were investigated, such as T‐helper‐17 (Th17) 11 , 12 which reduced post‐AHSCT until 15M 13 or remained unchanged at 2 yr, 14 and mucosal‐associated invariant T‐(MAIT) cells which decreased until 1 yr, 2 and 2 yr post‐transplant. 9 Additionally, no study to date has explored the brain‐homing CD49d+ (α4+) lymphocytes post‐AHSCT, despite this being the target of natalizumab.
The role of memory B‐cells in propagating MS pathogenesis is supported by the use of CD20 monoclonal antibody therapy 15 , 16 , 17 in MS. Although a single study 14 described reconstitution of B‐lymphocytes by 12M post‐AHSCT, it was underpowered. As emerging histopathological and clinical evidence indicate role of B‐cells as antigen presenting cells and/or mediators of inflammation in MS, 18 , 19 , 20 a better understanding of B‐cell kinetics post‐AHSCT is imperative. Likewise, plasmablasts, antibody‐secreting B‐cells predominantly present in cerebrospinal fluid and to a lesser degree in circulation, are relevant in MS pathogenesis. 21 , 22 Although plasmablasts are diminished in MS post‐glatiramer acetate treatment, 23 its kinetics post‐AHSCT is unknown. Circulating CD4+ T‐follicular helper (cTfh) cells that regulate B‐cell maturation and memory B‐cell formation, 24 akin to their lymphoid organ counterparts, 25 , 26 , 27 were analysed for subset co‐expressing programmed cell death protein‐1 (PD1). This subset, currently unknown in MS post‐AHSCT, regulates immunological responses in autoimmune diseases, 25 , 28 and PD1 expression may be a surrogate measure of helper function towards humoral response. 25 , 27 , 29
We investigated CD39+ T‐regulatory cells (Tregs) that suppress pro‐inflammatory interleukin‐17 production, 30 CD56hi natural killer (NK) cells, immunoregulatory innate cells that expand following DMTs in MS 31 , 32 and terminally differentiated CD8+CD28−CD57+ T‐cells, whose function although currently unclear, may still assist to limit autoreactivity. 9 , 10
Overall, this study elucidated whether the recalibration of immune balance is maintained beyond 1 yr post‐AHSCT out to 36M and its relationship with disease remission, highlighting possible mechanisms underlying AHSCT‐induced clinical remission. This may enable development of novel targets for cell‐based therapeutics and immunotherapies focused on specific immune populations of interest, which could reduce the need for chemotherapy‐based approaches and their consequent toxicity.
Materials and Methods
Patients
The current study was part of the single‐centre, prospective phase II clinical trial investigating efficacy of AHSCT in MS, approved by the Human Research Ethics Committee (HREC Ref: HREC/10/SVH/135) in December 2010 and registered with the Australian New Zealand Clinical Trial Registry (ACTRN12613000339752). Patients aged 18–60 yr with relapsing‐remitting MS (RRMS) or secondary progressive MS (SPMS), diagnosed according to the McDonald criteria 33 with a Kurtzke Expanded Disability Status Score (EDSS) 34 of 2.0–7.0 at baseline, and refractory to the first‐line disease‐modifying therapies (DMT) were eligible for the trial. Patients were referred by their primary neurologist and had to fulfil the inclusion criteria of failing at least two DMTs prior to AHSCT, with a minimum of one relapse and /or disease progression with evidence of MRI activity in the year prior to referral. Patients with comorbidities precluding AHSCT were excluded from the trial as previously described. 2
As of December 2020, 28 MS patients with RRMS or SPMS who had undergone AHSCT had achieved follow‐up for 36M, from which six patients were excluded from analysis as they were lost to follow‐up due to inter‐state patient relocations or were censored following commencement of an alternate DMT or due to pre‐existing conditions. The remaining 22 MS patients aged 22–55 yrs with 1:1.2 male:female ratio were included in the current study. The patient cohort was also analysed as responders and non‐responders, whereby non‐responders were patients with either a clinical relapse or a new or enlarging T2/FLAIR lesion on MRI and /or gadolinium enhancement of lesions during the study period up to 36M post‐AHSCT. Due to postulated differences in the pathogenesis of relapse versus progression, patients with increasing EDSS score were not considered non‐responders. Eighteen healthy controls (HCs) who were age‐ and sex‐matched (26–55 yr, 1:1 male:female ratio, Table 1) were included as a comparator group. Informed written consent was obtained from MS patients (n = 22) and HCs (n = 18). Demographics of MS patients and HCs, treatments prior to AHSCT, clinical characteristics and AHSCT treatment responses of MS patients are provided in Table 2.
Table 1.
Age and gender comparison of MS patients and HCs.
| Parameters | MS patients | Healthy controls | Statistical comparison |
|---|---|---|---|
| Number of subjects | 22 | 18 | |
| Median age | 34 | 34.5 | |
| Age range | 22–55 | 26–55 | Independent two sample t‐test; p = 0.7339 |
| Gender ratio (male:female) | 1:1.2 | 1:1 | Chi square test, p = 0.7746 |
AHSCT, Autologous haematopoietic stem cell transplantation; ATG, Anti‐thymocyte globulin; BEAM, Carmustine, Etoposide, Cytarabine, Melphalan; cTfh, Circulating T follicular helper; DMT, Disease modifying therapies; HCs, Healthy controls; M, Months; MAIT, Mucosal‐associated invariant T; MS, Multiple Sclerosis; NK, Natural Killer; PD1, Programmed cell death protein 1; T‐conv, T‐conventional; Th17, T helper 17; Tregs, T regulatory cells; yr, Year.
Table 2.
Demographics and clinical characteristics of MS patients.
| Patient ID | Sex | Age at Tx | Duration of MS prior to Tx (months) | Relapses in the 24M prior to Tx | No of MRIs with new/enlarging lesions in the 24M prior to Tx | Prior DMTs 1 | EDSS at baseline | Post‐Tx relapses/MRI activity | EDSS at 36M post‐Tx |
|---|---|---|---|---|---|---|---|---|---|
| MS001 | F | 52 | 188 | 0 | 2 | IFNβ1a, MTX, FIN, NTZ | 7 | N | 7.5 |
| MS002 | F | 22 | 62 | 3 | 3 | IFNβ1a, FIN | 3.5 | Relapse with right leg weakness at 13M | 2 |
| MS003 | M | 27 | 130 | 2 | 2 | IFNβ1a, NTZ | 4.5 | N | 6 |
| MS004 | F | 46 | 68 | 3 | 2 | IFNβ1a, GA, DMF | 4.5 | N | 4.5 |
| MS005 | F | 31 | 62 | 1 | 0 | GA, DMF | 4 | Single new non‐enhancing T2/FLAIR hyperintensity on MRI at 36M. No clinical correlate. | 4.5 |
| MS006 | M | 30 | 39 | 2 | 1 | IFNβ1a, FIN, NTZ | 4 | N | 4.5 |
| MS007 | M | 25 | 8 | 4 | 3 | FIN | 2 | N | 2 |
| MS008 | F | 35 | 47 | 2 | 2 | FIN, NTZ, plasma exchange, DMF | 3 | N | 0 |
| MS009 | F | 43 | 36 | 3 | 1 | FIN, NTZ | 5 | Relapse with left leg weakness at 23M | 3 |
| MS010 | M | 33 | 103 | 1 | 2 | IFNβ1a, GA, FIN, MTX | 4.5 | N | 1 |
| MS011 | M | 33 | 115 | 0 | 2 | IFNβ1a, FIN, NTZ, DMF | 7 | N | 8 |
| MS012 | F | 38 | 127 | 3 | 3 | IFNβ1a, FIN, NTZ | 4 | N | 0 |
| MS013 | F | 55 | 259 | 1 | 1 | IFNβ1a, GA, FIN, NTZ, DMF | 6.5 | Right upper limb weakness at 33M | 6.5 |
| MS014 | F | 38 | 142 | 2 | 2 | FIN, NTZ | 6 | N | 6.5 |
| MS015 | M | 31 | 25 | 3 | 4 | IFNβ1a, GA, DMF | 6.5 | N | 7 |
| MS016 | M | 29 | 99 | 2 | 2 | IFNβ1a, GA, DMF | 2 | N | 1 |
| MS017 | M | 28 | 73 | 2 | 1 | IFNβ1a, FIN | 2 | N | 2.5 |
| MS018 | F | 37 | 179 | 1 | 1 | IFNβ1a, GA, MTX, NTZ, DMF | 4 | N | 0 |
| MS019 | F | 37 | 17 | 5 | 6 | GA, NTZ | 6.5 | N | 3 |
| MS020 | F | 35 | 93 | 3 | 3 | IFNβ1a, FIN, NTZ | 6 | N | 3.5 |
| MS021 | M | 44 | 124 | 1 | 4 | IFNβ1a, GA, FIN, NTZ | 3.5 | Sensory disturbance involving right leg at 11M. | 4.5 |
| MS022 | M | 31 | 19 | 2 | 5 | FIN, NTZ | 2.5 | N | 2.5 |
DMF, Dimethyl fumarate; FIN, Fingolimod; GA, Glatiramer acetate; IFN, Interferon; MTX, Mitoxantrone; NTZ, Natalizumab; M, Months; N, No relapse/MRI activity; Tx, Transplant (AHSCT).
Pulse steroid use has not been included in this table.
Transplant regimen
Autologous haematopoietic stem cells were mobilised with intravenous administration of cyclophosphamide 2 g/m2 and subcutaneous administration of granulocyte colony stimulating factor at 10 mcg/kg per day for 10 days. Peripheral blood stem cells were collected by leukapheresis using the Cobe Spectra apheresis system and cryopreserved. Transplant patients received BEAM (carmustine 300 mg/m2/d on day −6, etoposide 200 mg/m2/d on days −5 to −2, cytarabine 200 mg/m2/d on days −5 to −2, melphalan 140 mg/m2/d on day −1) conditioning chemotherapy. The thawed leukapheresis stem cell product was administered on day 0, followed by horse ATG 20 mg/kg/day on days +1 and +2.
Isolation of peripheral blood stem cells from peripheral blood
Peripheral blood samples were collected into ethylenediaminetetraacetic acid tubes from MS patients serially at pre‐AHSCT and post‐AHSCT at 24M and 36M timepoints (±4M), and from HCs as comparator group. Peripheral blood stem cells were isolated using Ficoll‐Paque density‐gradient centrifugation and cryopreserved for subsequent batched immunophenotyping analysis.
Immunophenotyping using polychromatic flow cytometry
Two 18‐colour immunophenotyping panels were used to characterize the post‐AHSCT immunological reconstitution in MS patients via polychromatic flow cytometry. Briefly, cryopreserved peripheral blood stem cell samples were thawed in batches using thawing medium (RPMI + 10% heat‐inactivated foetal bovine serum + 1% GlutaMAX). Pilot experiments conducted in our laboratory has successfully validated the suitability of using cryopreserved specimens for immunophenotyping via comparison with fresh specimens (data not shown). In this study, thawed cells were stained in the dark at room temperature using Near Infrared (NIR) viability dye for 30 min followed by two washes in PBS. Antibody staining was performed using two separate antibody cocktails (each consisting of 17 antibodies). The stained cells were then washed with 1× flow cytometry buffer followed by fixation in 0.5% paraformaldehyde. The multicolour flow cytometry was performed using a 5‐laser 20‐colour BD LSR Fortessa X‐20 Flow Cytometer (BD Biosciences, San Jose, California, USA) and FACSDiva software. The instrument was calibrated using CST beads prior to each run. To correct fluorescence spillover, compensation matrices were generated during acquisition on FACSDiva or post‐acquisition on FlowJo using appropriate single‐stained beads or cells. Unstained and NIR‐only stained cells were also run in addition to the full panel‐stained cells for each patient. The gating and data analysis were performed using FlowJo software (Treestar, Ashland, Oregon, USA). The list of all antibodies used for both immunophenotyping panels are described in Table S1. The examined immune subset phenotypes are described in Table S2 and the gating strategies are shown in Figures S1–S3. All the immune subsets that were examined for changes in frequency dynamics were also analysed for changes in their absolute counts. Only the subsets that showed statistically significant changes in their absolute counts between pre‐ and post‐AHSCT in the whole MS cohort or distinct changes within the responder and non‐responder MS patient groups are shown in Figure S4.
Statistical analysis
The statistical analysis for longitudinal comparison between pre‐AHSCT and post‐AHSCT timepoints within the MS cohort was performed using linear mixed‐effects model (p < 0.05). This maximum likelihood‐based approach can utilise all the observed data points and handles missing datapoints under missing at random (MAR) assumption. Multiple comparisons were adjusted using Holm‐Sidak method. Model checking was performed based on standardised residues. Logarithmic transformations were performed if required after examining the residual analysis. For the longitudinal analysis of the differences within responders and non‐responders, due to a smaller sample size in the non‐responder group, the non‐parametric Friedman’s ANOVA with Dunn’s test with adjustment for multiple comparisons (p < 0.05) was used. The difference between pre‐AHSCT and post‐AHSCT timepoints within the responder group and non‐responder groups were analysed. This analysis only included patients with complete measures available at all timepoints.
The statistical analysis between responders and non‐responders at pre‐transplant timepoint as well as the cross‐sectional statistical analysis between MS samples at specific timepoints and HCs were performed using independent two sample t‐tests to compare continuous outcome, and Mann–Whitney U test was used if the distribution of the continuous measure was highly skewed (p < 0.05). Independent two sample t‐test (p < 0.05) and chi‐square test (p < 0.05) were used to compare the age and sex, respectively between MS cohort and HCs. All analyses were carried out in GraphPad Prism 8 (GraphPad Prism, La Jolla, USA).
Results
Immune reconstitution of primary lymphocyte populations out to 36M in MS patients
Investigating the long‐term pattern of immune reconstitution of MS patients after AHSCT is critical in determining the immunological recalibration occurring beyond the initial period of chemotherapy‐induced lymphopenia. The frequencies of CD3+, CD4+, and CD8+ T‐cell, and CD19+ B‐cell lymphocyte subsets in MS patients pre‐ and post‐AHSCT are shown in Figure 1. By 24M, the frequencies (Fig. 1A and B) and absolute counts (data not shown) of CD3+ T‐cells and CD4+ T‐cells had returned to pre‐AHSCT levels, whereas frequencies of CD8+ T‐cells and CD19+ B‐cells exceeded baseline at 24M and 36M post‐AHSCT (Fig. 1C and E). The absolute counts of CD8+ T‐cells had returned to baseline by 36M while CD19+ B‐cell numbers remained higher at 24M and 36M (Fig. S4A and C). The CD4:CD8 ratio remained below baseline at 24M and started to normalise by 36M post‐AHSCT (Fig. 1D, Fig. S4B).
Figure 1.
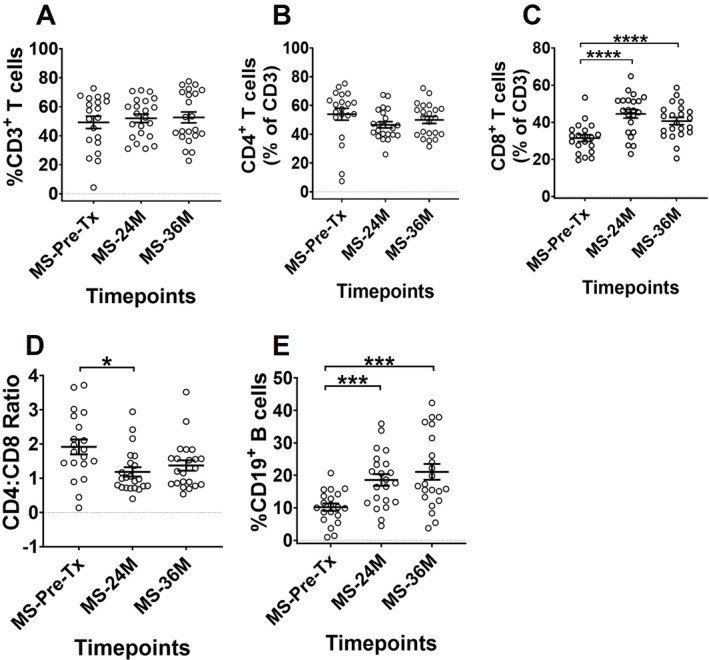
Comparison of lymphocyte reconstitution between pre‐AHSCT and post‐AHSCT timepoints in MS patients. The frequencies of (A) CD3+ T cells, (B) CD4+ T cells, (C) CD8+ T cells, (D) CD4:CD8 ratio and (E) CD19+ B cells. The frequencies in graphs (A) and (E) are shown as percentage of live lymphocytes, and (B) and (C) as percentage of CD3+. Gating strategy can be obtained in Figure S1. Statistical analysis was performed using linear mixed‐effects model (p < 0.05) and multiple comparisons adjusted using Holm‐Sidak method. Logarithmic transformations were performed for analysing difference between pre‐AHSCT and post‐AHSCT timepoint in CD4:CD8 ratio. Pre‐AHSCT (Pre‐Tx) n = 20, 24 months (24M) n = 22, 36 months (36M) n = 22. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Black bars and asterisks indicate longitudinal comparison between pre‐ and post‐AHSCT timepoints within MS cohort.
Persistent reduction in pro‐inflammatory immune subsets at 36M post‐transplant
We investigated the frequencies of pro‐inflammatory cells like Th17, MAIT and CD49d+ CNS‐homing T‐conventional (T‐conv) due to their involvement in MS pathogenesis. We also performed a cross‐sectional comparison between MS patients and HCs to examine the extent of immune dysregulation in MS patients pre‐AHSCT and whether this normalised by 36M after AHSCT. The frequency of various pro‐inflammatory cells that may be involved in the pathogenesis of the disease is shown in Figure 2. We observed that Th17 cells were significantly elevated in MS patients prior to AHSCT compared to HCs, likely reflective of active disease necessitating inclusion in the trial. A sustained decrease was observed in the frequency of Th17 subset at both 24M and 36M timepoints as compared to pre‐AHSCT frequency, lower than HCs at 36M (Fig. 2A). No significant changes were observed in the Th17 absolute cell numbers (data not shown).
Figure 2.
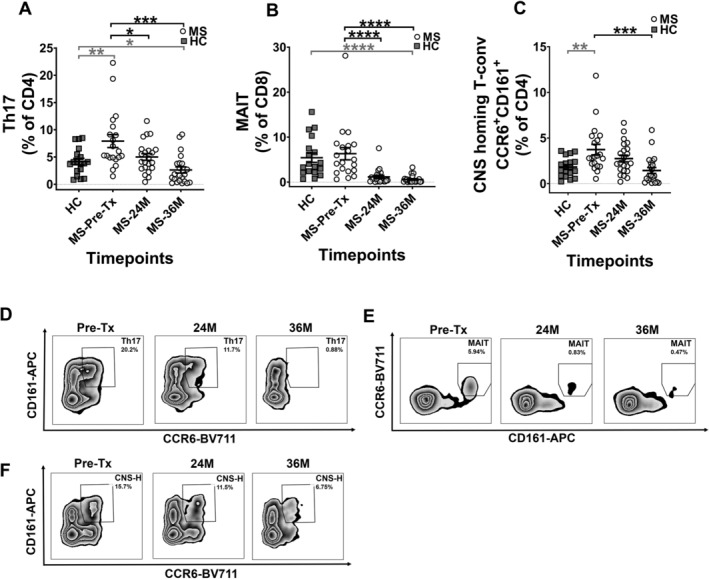
Decrease in frequency of pro‐inflammatory immune subsets in MS patients. The frequencies of (A) Th17, (B) MAIT and (C) CNS‐homing T‐conventional (T‐conv) cells. The frequencies in graphs (A) and (C) are shown as percentage of CD4+, and (B) as percentage of CD8+. Representative flow cytometry plots for subsets at each timepoint are shown in (D–F). (D) Zebra plots showing Th17 subset, with numbers on the plots indicating memory Th17 percentage gated from CD45RA− parent population. (E) Zebra plots showing MAIT subset, with numbers on the plots indicating MAIT cell percentage gated from CD8+ parent population. (F) Zebra plots showing CNS‐homing T‐conv subset (CNS‐H), with numbers on the plots indicating CNS‐H subset percentage gated from CD49d+β7− T‐conv cells parent population. Gating strategy can be obtained in Figure S1. Statistical analysis was performed using linear mixed‐effects model (p < 0.05) and multiple comparisons adjusted using Holm‐Sidak method. Logarithmic transformations were performed for analysing difference between pre‐AHSCT and post‐AHSCT timepoint in Th17, MAIT and CNS‐homing T‐conv cell populations. Statistical analysis between MS at pre‐/36M post‐AHSCT and HCs was performed using independent two‐sample t‐tests (p < 0.05). As normality assumption failed for Th17, MAIT and CNS‐homing populations in MS cohort at 36M post‐AHSCT, Mann–Whitney U test was used to compare between MS at 36M pre‐AHSCT against HCs for these subsets. MS Pre‐AHSCT (Pre‐Tx) n = 20, 24 months (24M) n = 22, 36 months (36M) n = 22, HCs n = 18. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Grey bars and asterisks indicate cross‐sectional comparison between MS at pre‐/36M post‐AHSCT and HC cohorts, whereas black bars and asterisks indicate longitudinal comparison between pre‐ and post‐AHSCT timepoints within MS cohort.
MAIT cells exhibited comparable frequencies in MS patients at pre‐AHSCT timepoint to that of HCs, and were significantly reduced in their frequency and absolute cell numbers at both the 24M and 36M post‐AHSCT timepoints (Fig. 2B, Fig. S4D) to frequencies below HCs at 36M.
We next evaluated a putative CNS‐homing T‐conv cell population, whose proportion were significantly elevated in MS patients pre‐AHSCT as compared to HCs and was significantly reduced at 36M post‐AHSCT timepoints when compared to pre‐AHSCT (Fig. 2C). By 36M the frequency of this subset in MS patients was comparable to HCs. No changes were observed post‐AHSCT in their absolute numbers (data not shown).
Regulation of immune subsets that facilitate humoral immune response at 36M post‐transplant
The frequency of CD27+ memory B‐cell populations was significantly depleted at 24M and 36M post‐AHSCT. This change was accompanied by significantly increased CD27− naïve B‐cell frequencies, resulting in a decreased memory:naïve B‐cell ratio, below that of HCs at 36M (Fig. 3A–C). Further, a significant decline was observed in the CD27hiCD38hi plasmablast frequency at both 24M and 36M post‐AHSCT timepoints, rendering this population significantly lower than HCs at 36M (Fig. 3D). The absolute cell numbers of naïve B‐cells (Fig. S4E) remained significantly elevated, whereas the memory:naive B‐cell ratio was significantly reduced (Fig. S4F) in MS patients at both 24M and 36M post‐AHSCT, with no change in absolute numbers of memory B‐cells and plasmablasts (data not shown).
Figure 3.
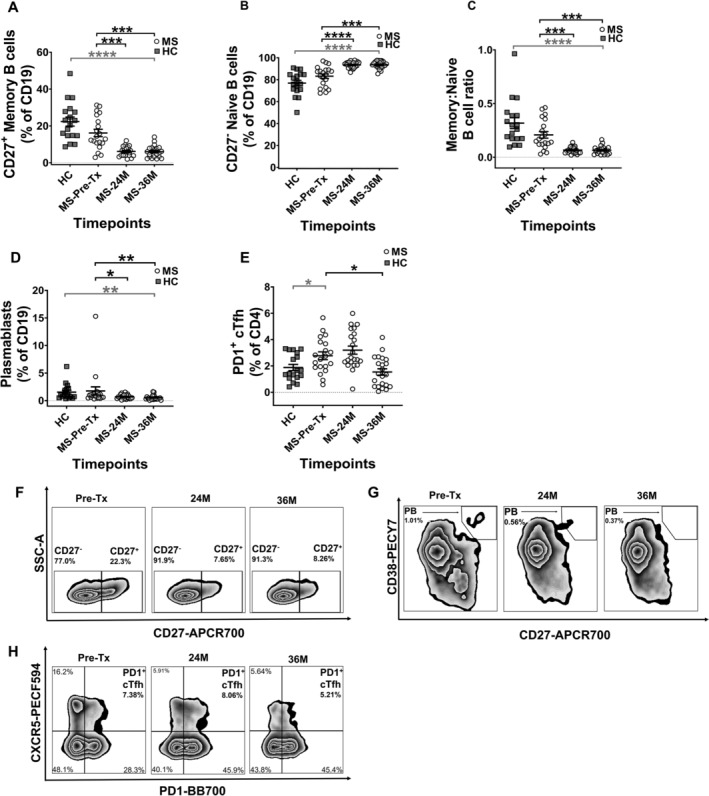
Regulation of frequency in immune subsets associated with humoral immune response. The frequencies of (A) Memory B cells, (B) Naïve B cells, (C) Memory:Naïve B cell ratio, (D) Plasmablasts and (E) Circulating PD1+ cTfh. The frequencies in (A), (B), and (D) are shown as percentage of CD19+ and (E) as percentage of CD4+. Representative flow cytometry plots for subsets at each timepoint are shown in (F–H). (F) Zebra plots showing naïve and memory B cell subsets, with numbers on the plots indicating their percentages gated from CD19+ parent population. (G) Zebra plots showing plasmablast (PB) subset, with numbers on the plots indicating PB percentage gated from CD19+ parent population. (H) Zebra plots showing PD1+ cTfh subset, with numbers on the plots indicating PD1+ cTfh percentages gated from CD45RA− parent population. Gating strategy can be obtained in Figure S2. Statistical analysis was performed using linear mixed‐effects model (p < 0.05) and multiple comparisons adjusted using Holm‐Sidak method. Logarithmic transformations were performed for analysing difference between pre‐AHSCT and post‐AHSCT timepoint in Memory B cells, Memory:Naïve B cell ratio and Plasmablasts. Statistical analysis between MS at pre‐/36M post‐AHSCT and HCs was performed using independent two‐sample t‐tests (p < 0.05). MS Pre‐AHSCT (Pre‐Tx) n = 20, 24 months (24M) n = 22, 36 months (36M) n = 22, HCs n = 18. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Grey bars and asterisks indicate cross‐sectional comparison between MS at pre‐/36M post‐AHSCT and HC cohorts, whereas black bars and asterisks indicate longitudinal comparison between pre‐ and post‐AHSCT timepoints within MS cohort.
Based on previous studies associating B‐cell response and PD1‐expressing cTfh lymphocytes, 25 , 27 we next investigated the frequency of PD1+ cTfh in MS patients. Pre‐transplant, MS patients demonstrated significantly elevated frequencies of PD1+ cTfh cells, putative facilitators of B cell‐mediated immune response, as compared to HCs (Fig. 3E). At 36M post‐AHSCT, a significant decrease in the PD1+ cTfh population was observed, resulting in frequency comparable to HCs (Fig. 3E). There were no changes observed in their absolute cell numbers between pre‐AHSCT and post‐AHSCT timepoint in MS patients (data not shown).
Frequency of key immunoregulatory and immunosenescent subsets at 36M post‐AHSCT
Changes in immunoregulatory subsets following AHSCT are shown in Figure 4. At baseline, MS patients exhibited similar CD4+ Treg frequencies as HCs, and higher CD39+ Treg frequencies than HCs. Post‐AHSCT no changes were observed in CD4+ Treg and CD39+ Treg frequencies (Fig. 4A and B) or absolute numbers (data not shown), however by 36M MS patients exhibited lower frequency of CD4+ Tregs than HCs.
Figure 4.
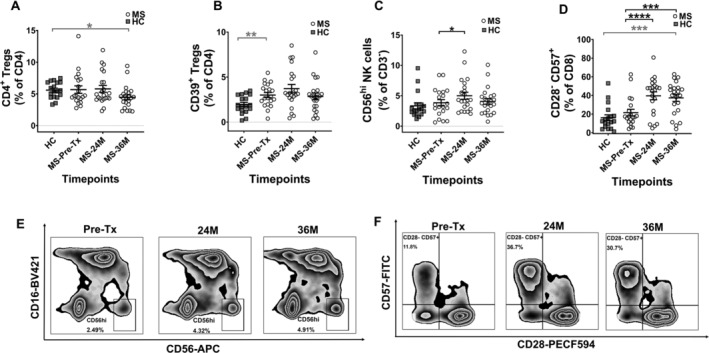
Increase in immunoregulatory and immunosenescent subsets at post‐AHSCT timepoints in MS patients. The frequencies of (A) CD4+ Tregs, (B) CD39+ Tregs, (C) CD56hi NK cells and (D) CD8+CD28−CD57+ T cells. The frequencies in graphs (A) and (B) are shown as a percentage of CD4+, (C) as percentage of CD3−, and (D) as percentage of CD8+. (E) Zebra plots showing CD56hi NK cell subset, with numbers on the plots indicating their percentage gated from CD3– parent population. (F) Zebra plots showing CD28–CD57+ subset, with numbers on the plots indicating their percentage gated from CD8+ parent population. Gating strategy can be obtained in Figure S3. Statistical analysis was performed using linear mixed‐effects model (p < 0.05) and multiple comparisons adjusted using Holm‐Sidak method. Logarithmic transformations were performed for analysing difference between pre‐AHSCT and post‐AHSCT timepoint in CD4+ Tregs, CD39+ Tregs and CD56hi NK cells. Statistical analysis between MS at pre‐/36M post‐AHSCT and HCs was performed using independent two‐sample t‐tests (p < 0.05). Pre‐AHSCT (Pre‐Tx) n = 20, 24 months (24M) n = 22, 36 months (36M) n = 22, HCs n = 18. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Grey bars and asterisks indicate cross‐sectional comparison between MS at pre‐/36M post‐AHSCT and HC cohorts, whereas black bars and asterisks indicate longitudinal comparison between pre‐ and post‐AHSCT timepoints within MS cohort.
MS patients showed comparable frequencies of the immunoregulatory CD56hi NK cell subset pre‐AHSCT compared to HCs. The frequency (Fig. 4C) and absolute cell numbers (Fig. S4G) of CD56hi NK cells were increased at 24M post‐AHSCT, however by 36M post‐AHSCT returned to frequencies comparable with HCs.
MS patients showed comparable frequencies of the terminally differentiated CD8+CD28−CD57+ T‐cells to HCs pre‐AHSCT. The frequency (Fig. 4D) and absolute cell numbers (Fig. S4H) of the CD8+CD28−CD57+ T‐cell subset were significantly increased at both 24M post‐AHSCT and 36M post‐AHSCT, exceeding baseline frequencies of HCs at 36M.
AHSCT‐induced changes in key immunological subsets associated with clinical response in MS patients
The described immune subsets were further analysed based on patient’s clinical response. The MS patient cohort (n = 22) was divided into responders (n = 17) and non‐responders (n = 5) based on the clinical relapse or new MRI activity during the study period out to 36M post‐AHSCT. Only responder (n = 15) and non‐responder (n = 5) patients with complete datapoints were included for the longitudinal subgroup analysis of immune subset frequencies. The frequencies of pro‐inflammatory subsets including Th17, putative CNS‐homing T‐conv and PD1+ cTfh were demonstrated to be significantly reduced at 36M post‐AHSCT within the responder, but not the non‐responder cohort (Fig. 5A–C). Furthermore, the frequency of immunoregulatory CD39+ Tregs was found to be increased at 24M timepoint in responder patients, whereas it remained unchanged in non‐responder patients (Fig. 5D). Additionally, upon comparison of pre‐AHSCT frequencies of Th17, putative CNS‐homing T‐conv, PD1+ cTfh and CD39+ Tregs between the responder and non‐responder patient cohorts, no statistically significant differences were observed (Fig. S5A–D). No discernable longitudinal differences were detected in the frequencies of other immune subsets within responder and non‐responder patients (data not shown). Only the absolute numbers of immune subsets where discernible changes were observed within responder and non‐responder patients’ post‐AHSCT are shown in Figure S4I–L.
Figure 5.
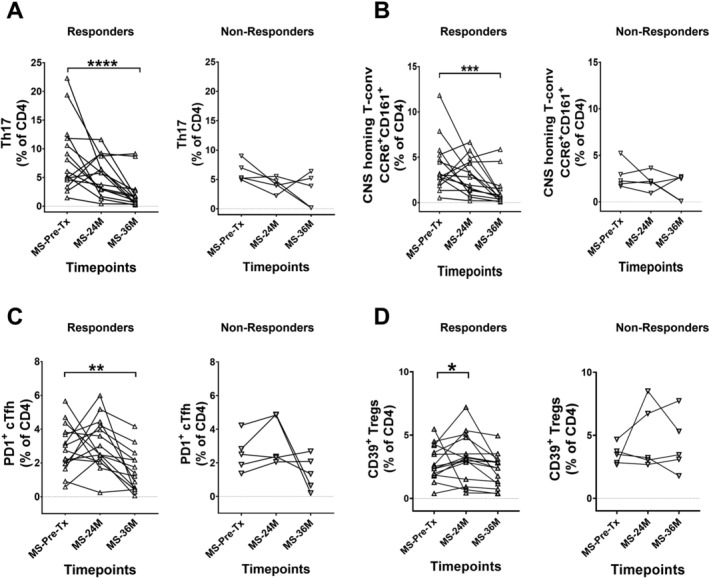
AHSCT‐induced changes in key immune subsets are associated with clinical response in MS patients. The MS patient cohort was split into responders (▵) and non‐responders (▽) based on clinical relapse and MRI activity. The frequencies of (A) Th17 cells in responders and non‐responders, (B) CNS‐homing populations in responders and non‐responders, (C) PD1+ cTfh in responders and non‐responders and (D) CD39+ Tregs in responders and non‐responders. Statistical analysis was performed using parametric Friedman’s ANOVA with Dunn’s test (p < 0.05). Linear mixed‐effects model was not used for this analysis due to smaller sample size in non‐responder group. As the Friedman’s ANOVA analyses complete datasets, two patients from responder group that had missing timepoints were excluded for statistical comparisons. In responder group, pre‐AHSCT (Pre‐Tx) n = 15, 24 months (24M) n = 15, 36 months (36M) n = 15. In non‐responder group, pre‐AHSCT (Pre‐Tx) n = 5, 24 months (24M) n = 5, 36 months (36M) n = 5. **p < 0.01, ****p < 0.0001. Black bars and asterisks indicate longitudinal comparison within responder and non‐responder patient subgroups in MS cohort.
Discussion
The substantial immunological changes evidenced in this study highlight the potential of AHSCT to shift the immune balance of MS patients from an autoreactive nature to a more tolerant phenotype. The study findings persist until 36M post‐transplant, well beyond the resolution of lymphopenia, thereby complementing our previous study that examined early immune reconstitution until 1 yr post‐AHSCT. 2 Dysregulations in immune balance have been noted in MS 35 and this is the first study to report some of the described immune subsets up to 36M post‐AHSCT in MS patients undergoing BEAM + ATG myeloablative conditioning. In addition to immune subsets identified in our previous study, like MAIT cells, CD8+CD28−CD57+ T‐cells and CD56hi NK cells, that demonstrated durable recalibration, we also identified immunological shifts in putative CNS‐homing T‐conv, and PD1+ cTfh subsets, a novel finding in MS patients post‐AHSCT. T‐helper‐1 subset was not examined as Darlington et al. 13 demonstrated reduction of Th17 and not T‐helper‐1 cells underlie abrogation of MS post‐AHSCT, where T‐helper‐1 cells remained unchanged post‐AHSCT.
Higher frequencies of Th17 lymphocytes, key mediators of neuroinflammation, 11 , 12 is likely a reflection of an active inflammatory state in MS patients’ pre‐AHSCT. Persistent reduction of Th17 frequency post‐AHSCT indicates a more tolerant post‐transplant immune milieu, extending a previous study demonstrating early post‐AHSCT (12M–15M) reduction in Th17 frequency in MS patients who underwent high‐intensity immunoablative conditioning. 13 Post‐AHSCT Th17 frequencies in MS patients with BEAM + ATG conditioning remained similar to baseline at 2 yr 14 or akin to HCs, despite without longitudinal comparisons. 36 In the current study the residual proportion of Th17 cells at 36M is significantly lower than HCs, indicating a profound suppression of inflammatory activity post‐AHSCT. When correlated with clinical outcome, the significant reduction of Th17 cells was observed longitudinally only within responder patients (Fig. 5A). However it is noted that the responder patient cohort had higher baseline Th17 frequencies than non‐responder cohort but their difference was not statistically significant (Fig. 5A, Fig. S5A). Corroborating previous studies, Durelli et al. 37 showed that Th17 frequencies closely paralleled active MS, whilst another study indicated a close association between Th17 frequency reduction and abrogation of new disease activity in MS patients. 13 Overall, our findings indicate dysregulated Th17 frequencies existing in MS patients pre‐AHSCT, which are subsequently corrected post‐AHSCT, maintained until 36M.
MAIT cells that comprise a predominant fraction of the CD8+ T‐cells producing interleukin‐17 or co‐producing interleukin‐17/interferon‐γ pro‐inflammatory cytokines 9 were significantly depleted post‐AHSCT, although observed in similar frequencies in MS at pre‐AHSCT and HCs. The CD161 and CCR6 expression on T‐cells have been demonstrated to enable transmigration into CNS. 9 , 38 Previous reports have confirmed transmigratory property of MAIT cells into brain by identifying CD8+CD161+ and CD161+TCRVα7.2+ cells in the white matter and inflammatory infiltrates of MS post‐mortem brain tissues, 9 suggesting their involvement in MS pathogenesis. The further reduction of MAIT cell frequency in MS patients by 36M post‐AHSCT below HCs, further highlights the substantial downregulation of inflammatory activity post‐AHSCT.
The putative CNS‐homing T‐conv was identified using upstream markers denoting non‐gut homing integrin expression (α4+β7−), and CNS‐transmigratory markers CCR6 and CD161. α4β1 (very late antigen‐4) regulates lymphocyte trafficking into CNS, bone marrow and skin by interacting with vascular cell adhesion protein‐1 on endothelial cells. 39 Further, α4β7 integrin mediates T‐cell migration to gut‐associated lymphoid tissue by interacting with mucosal vascular addressin cell adhesion molecule‐1. 39 , 40 Natalizumab targets integrin α4 on both α4β1 and α4β7, 41 and inhibits lymphocyte migration into CNS by blocking α4β1 integrin and vascular cell adhesion protein‐1 interaction. 42 , 43 It has been reported that α4 integrin dichotomously binds to either β1 or β7, 39 , 41 , 44 which was previously determined in our laboratory using a different HC cohort and flow cytometry panel (data not shown). We have therefore been able to use α4+β7− cells as a surrogate for α4+β1+ cells, allowing further investigation of CNS‐migratory potential by using CCR6 and CD161, whose frequency reduced in MS patients post‐AHSCT, particularly within the responder group.
This is the first paper to report the reconstitution of specific B‐cell sub‐populations until 36M post‐transplant in MS. A relative increase in naïve B‐cell population drives an expansion of CD19+ cells more generally, whilst memory B‐cells and plasmablasts remained suppressed for 36M. Akin to a multitude of studies 10 , 13 , 14 , 45 demonstrating re‐emergence of a diversified naïve T‐cell repertoire in MS patients following AHSCT, a similar regeneration of the B‐cell pool appears evident, providing a repopulated humoral immune system capable of mounting immune response to novel antigenic challenges post‐transplant. Antibody‐secreting plasmablasts, 22 , 46 although circulating at similar frequencies in MS and HCs, their reduction post‐AHSCT suggests suppressed humoral activity. Due to limitations in the existing flow cytometry panels this study did not examine other B‐cell subsets such as regulatory B‐cells. A study by Cencioni et al. 47 has demonstrated transitional B‐cells (CD19+CD24hiCD38hi) in MS patients to possess a defective function when compared to HCs.
In patients with autoimmune conditions like juvenile dermatomyositis and systemic lupus erythematosus (SLE), cTfh frequency correlated with disease activity and humoral response indicators. 26 , 28 Further, PD1 expression on cTfh cells is considered to be a surrogate measure of their helper function on B‐cells, 25 , 27 , 29 and SLE and rheumatoid arthritis patients exhibited higher CCR7loPD1hi cTfh subset frequency associated with elevated autoantibody levels and disease activity scores. 25 No studies to date have explored PD1+ cTfh subset in MS, despite high interleukin‐21 expression, the main cytokine secreted by cTfh cells, found in MS lesions. 48 Our study findings demonstrating the correction of high PD1+ cTfh frequencies in MS patients by 36M post‐AHSCT, being particularly relevant within the responder group, may imply a potential PD1+ cTfh‐mediated regulation of inflammatory immune response in these MS patients.
Our findings show comparable CD4+ Tregs frequencies between MS patients at pre‐AHSCT and HCs, in agreement with previous studies. 30 The frequencies of CD39+ Tregs were elevated in our MS cohort as compared to HCs, suggesting it is a qualitative not quantitative deficiency of Tregs that contributes to MS pathogenesis. Our previous study has shown an early increase in both CD4+ Tregs and CD39+ Treg percentage post‐AHSCT. 2 In the current study we observed no significant changes in CD4+ Tregs and CD39+ Treg frequencies post‐AHSCT with a modest increase in CD39+ Tregs within responder patients at 24M. Nevertheless, the impact on early surges of Tregs in MS patients post‐AHSCT should be assessed by their functional properties.
Previous studies have demonstrated CD56hi NK cells to possess impaired immunoregulatory function in MS patients, 49 and mediates an immunomodulatory effect on T‐cell survival following daclizumab treatment. 33 CD56hi NK cells expanded following daclizumab 33 and interferon‐β 34 treatment and positively correlated with clinical response. Previous study on MS patients who underwent non‐myeloablative conditioning followed by AHSCT has shown transient increase in the CD56hi NK cell frequency up to 6M post‐AHSCT 9 and our prior work demonstrated an increase in this population until 1 yr post‐AHSCT. 2 Here, the modest increase in CD56hi NK cell populations at 24M returned to baseline frequencies by 36M.
Terminally differentiated CD8+CD28−CD57+ cells increased until 36M post‐AHSCT in this study, were demonstrated to expand until 2 yr post‐AHSCT following a high‐intensity myeloablative conditioning by Muraro et al. 10 and until 4 yr post‐AHSCT by Arruda et al. 8 albeit following a lymphoablative conditioning regimen. This heterogeneous terminally differentiated subset is characterised by loss of CD28 and gain of CD57 expression after several rounds of proliferation. 50 , 51 While its precise function remains debated and unclear, it is proposed to contribute towards limiting autoreactivity via space attrition or soluble immunoregulatory factors by previous MS studies 9 and possess cytotoxic function. 50 , 51 One MS study also demonstrated expansion of CD8+CD57+ T‐cells until 2 yr post‐AHSCT, which exhibited immunosuppressive activity against CD4+ T‐cells. 9
This study has a number of limitations. In an attempt to counter the impact on baseline populations by previous DMTs, an average of 2‐month interval was used between last DMT and baseline blood sample. Notably, no patients in this cohort were treated in the immediate pre‐transplant period with an immune reconstitution therapy (alemtuzumab, cladribine) nor long‐acting B‐cell depleting therapy (Ocrelizumab), therefore we expect the effect of prior DMT to be minimal. Results presented here were not analysed in regard to MS disability progression post‐AHSCT, given this neurodegenerative component of the disease is likely distinct from the altered immune state that drives relapsing‐remitting CNS inflammation. Outcomes were analysed in terms of patients who relapsed post‐AHSCT, either clinically or with a new MRI lesion. In MS studies, ‘no evidence of disease activity’ is never complete as it is only assessed at pre‐specified intervals, and clinical relapse and recurrent MRI activity could still be indicative of a reduced inflammatory response compared to pre‐AHSCT inflammatory activity levels. Although novel sustained shifts were observed in circulatory immune subset frequencies, we cannot comment on the functional corollary of cellular phenotype in the absence of further ex vivo work. Being a major referral centre for treating MS with AHSCT in Australia, patient recruitments occur from across the country. It was difficult to collect blood samples from all patients at the exact relapse timepoint due to logistic constraints. Therefore, we could not perform additional correlations between immunological changes and disease activity in non‐responders. Furthermore, due to the fortunately high response rate in MS patients to AHSCT, the number of non‐responder patients was small. Thus, making robust conclusions about the association of certain subtype alterations with response is problematic. Larger prospective and multi‐centre studies in the future would be required to confirm this finding and to analyse correlations between immunological findings and clinical response.
In conclusion, our study finds several immune populations altered in proportion post‐AHSCT, although absolute counts of few remained unchanged. We postulate that following the period of lymphopenia, AHSCT serves to recalibrate the balance of lymphocyte subsets which may be dysregulated in active MS. Our findings indicate these changes persist out to 36M post‐AHSCT, comprised of a profound reduction in pro‐inflammatory subsets and sustained expansion of subsets that may possess immunoregulatory potential.
Author Contributions
JJM, IJS and DDFM designed clinical trial. MV, KH, JCM, MLK, CDF, JJZ, JJM and DDFM designed research methodology. MV, JJZ, KH and CDF set up flow cytometry panels. MV, KH and JCM prepared experimental samples. MV acquired flow cytometry data. MV, KH, JCM, MLK, CDF, JJZ, BW, IJS, DDFM and JJM analysed and interpreted data. JCM assessed patients’ clinical response. MV, KH, JCM, MLK and CDF performed biobank storage. JJM and DDFM provided funding for the study. MV generated drafts for manuscript and all authors contributed to manuscript revisions. All authors reviewed and edited the final manuscript.
Conflict of Interest
Nothing to report.
Supporting information
Table S1. List of antibodies used for flow cytometry panels.
Table S2. Phenotypes and gating hierarchy of immune subsets.
Figure S1. Gating strategy of pro‐inflammatory T cells. Gating strategy for (A) Th17 cells, (B) MAIT cells and (C) CNS‐homing T‐conv (CNS‐H) cells are indicated.
Figure S2. Gating strategy of immune subsets regulatory humoral response cells. Gating strategy for (A) Naïve and memory B cells, (B) Plasmablasts (PB) and (C) PD1+ cTfh cells are indicated.
Figure S3. Gating strategy of immunoregulatory and immunosenescent cells. Gating strategy for (A) CD4+ Tregs, (B) CD39+ Tregs, (C) CD56hi NK cells, (D) CD8+CD28−CD57+ cells are indicated.
Figure S4. Analysis of absolute counts of immune subsets within whole MS cohort, and within responder and non‐responder patient groups (expressed as cells/μL). First two rows show absolute counts of immune subsets that showed significant changes between pre‐ and post‐AHSCT comparison. (A) CD8+ T cells (B) CD4:CD8 ratio, (C) CD19+ B cells, (D) MAIT cells, (E) Naïve B cells, (F) Memory:Naïve B cells, (G) CD56hi NK cells and (H) CD8+CD28−CD57+ cells. Statistical analysis was performed using linear mixed‐effects model (p < 0.05) and multiple comparisons adjusted using Holm‐Sidak method. Logarithmic transformations were performed for analysing difference between pre‐AHSCT and post‐AHSCT timepoint in CD4:CD8 ratio, CD19+ B cells, MAIT, Naïve B cells, Memory:Naïve B cell ratio and CD8+CD28−CD57+ cells. Pre‐AHSCT (Pre‐Tx) n = 19, 24 months (24M) n = 22, 36 months (36M) n = 22. Last two rows show absolute counts of immune subsets that demonstrated distinct responses within responder and non‐responder patient groups. Absolute counts of (I) Th17 in responder and non‐responder patients, (J) CNS‐homing T‐conv cells in responder and non‐responder patients, (K) Naïve B cells in responder and non‐responder patients and (L) CD56hi NK cells in responder and non‐responder patients. Statistical analysis was performed using parametric Friedman’s ANOVA with Dunn’s test (p < 0.05). Linear mixed‐effects model was not used for this analysis due to smaller sample size in non‐responder group. As the Friedman’s ANOVA analyses complete datasets, three patients from responder group that had missing timepoints of absolute count values were excluded for statistical comparisons. In responder group, pre‐AHSCT (Pre‐Tx) n = 14, 24 months (24M) n = 14, 36 months (36M) n = 14. In non‐responder group, pre‐AHSCT (Pre‐Tx) n = 5, 24 months (24M) n = 5, 36 months (36M) n = 5. For all graphs, *p < 0.05, **p < 0.01, ***p < 0.001. Black bars and asterisks indicate longitudinal comparison between pre‐ and post‐AHSCT timepoints within responder or non‐responder cohort.
Figure S5. Comparison of immune subset frequencies between responder and non‐responder MS patients at pre‐AHSCT. (A) Th17, (B) CNS‐homing T‐conv, (C) PD1+ cTfh and (D) CD39+ Tregs. Statistical analysis between responders (▵) (n = 15) and non‐responders (▽) (n = 5) at pre‐AHSCT were performed using independent two‐sample t‐tests (p < 0.05).
Acknowledgements
The funding for this study was provided by research scholarships and grants from Multiple Sclerosis Research Australia, Medich Family Foundation, NHMRC, St Vincent’s Clinic Research Foundation, Maple‐Brown Family Charitable Foundation, Reset Australia, John Kirkpatrick Medical Foundation. The authors would like to thank Dr Zhixin Liu from the Mark Wainwright Analytical Centre, University of New South Wales for assistance with statistical analysis for the current study.
Funding InformationThe funding for this study was provided by research scholarships and grants from Multiple Sclerosis Research Australia, Medich Family Foundation, NHMRC, St Vincent’s Clinic Research Foundation, Maple‐Brown Family Charitable Foundation, Reset Australia, John Kirkpatrick Medical Foundation.
Funding Statement
This work was funded by John Kirkpatrick Medical Foundation; Maple‐Brown Family Charitable Foundation; Medich Family Foundation; Multiple Sclerosis Research Australia ; National Health and Medical Research Council ; Reset Australia; St Vincent’s Clinic Research Foundation.
References
- 1. Baecher‐Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron. 2018;97(4):742‐768. [DOI] [PubMed] [Google Scholar]
- 2. Moore JJ, Massey JC, Ford CD, et al. Prospective phase II clinical trial of autologous haematopoietic stem cell transplant for treatment refractory multiple sclerosis. J Neurol Neurosurg Psychiatry. 2019;90(5):514‐521. [DOI] [PubMed] [Google Scholar]
- 3. Burt RK, Balabanov R, Burman J, et al. Effect of nonmyeloablative hematopoietic stem cell transplantation vs continued disease‐modifying therapy on disease progression in patients with relapsing‐remitting multiple sclerosis: a randomized clinical trial. JAMA. 2019;321(2):165‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muraro PA, Pasquini M, Atkins HL, et al. Long‐term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol. 2017;74(4):459‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nash RA, Hutton GJ, Racke MK, et al. High‐dose immunosuppressive therapy and autologous HCT for relapsing‐remitting MS. Neurology. 2017;88(9):842‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boffa G, Massacesi L, Inglese M, et al. Long‐term clinical outcomes of hematopoietic stem cell transplantation in multiple sclerosis. Neurology. 2021;96(8):e1215‐e1226. doi: 10.1212/wnl.0000000000011461 [DOI] [PubMed] [Google Scholar]
- 7. Muraro PA, Martin R, Mancardi GL, Nicholas R, Sormani MP, Saccardi R. Autologous haematopoietic stem cell transplantation for treatment of multiple sclerosis. Nat Rev Neurol. 2017;13(7):391‐405. [DOI] [PubMed] [Google Scholar]
- 8. Arruda LCM, de Azevedo JTC, de Oliveira GLV, et al. Immunological correlates of favorable long‐term clinical outcome in multiple sclerosis patients after autologous hematopoietic stem cell transplantation. Clin Immunol. 2016;169:47‐57. [DOI] [PubMed] [Google Scholar]
- 9. Abrahamsson SV, Angelini DF, Dubinsky AN, et al. Non‐myeloablative autologous haematopoietic stem cell transplantation expands regulatory cells and depletes IL‐17 producing mucosal‐associated invariant T cells in multiple sclerosis. Brain. 2013;136(Pt 9):2888‐2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muraro PA, Douek DC, Packer A, et al. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J Exp Med. 2005;201(5):805‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jadidi‐Niaragh F, Mirshafiey A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand J Immunol. 2011;74(1):1‐13. [DOI] [PubMed] [Google Scholar]
- 12. Kebir H, Kreymborg K, Ifergan I, et al. Human TH17 lymphocytes promote blood‐brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13(10):1173‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Darlington PJ, Touil T, Doucet JS, et al. Diminished Th17 (not Th1) responses underlie multiple sclerosis disease abrogation after hematopoietic stem cell transplantation. Ann Neurol. 2013;73(3):341‐354. [DOI] [PubMed] [Google Scholar]
- 14. Karnell FG, Lin D, Motley S, et al. Reconstitution of immune cell populations in multiple sclerosis patients after autologous stem cell transplantation. Clin Exp Immunol. 2017;189(3):268‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hauser SL, Waubant E, Arnold DL, et al. B‐cell depletion with rituximab in relapsing‐remitting multiple sclerosis. N Engl J Med. 2008;358(7):676‐688. [DOI] [PubMed] [Google Scholar]
- 16. Hauser SL, Bar‐Or A, Comi G, et al. Ocrelizumab versus interferon beta‐1a in relapsing multiple sclerosis. N Engl J Med. 2017;376(3):221‐234. [DOI] [PubMed] [Google Scholar]
- 17. Hauser SL, Bar‐Or A, Cohen JA, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. 2020;383(6):546‐557. [DOI] [PubMed] [Google Scholar]
- 18. Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118(10):3420‐3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen P, Roch T, Lampropoulou V, et al. IL‐35‐producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507(7492):366‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cencioni MT, Mattoscio M, Magliozzi R, Bar‐Or A, Muraro PA. B cells in multiple sclerosis—from targeted depletion to immune reconstitution therapies. Nat Rev Neurol. 2021;17(7):399‐414. [DOI] [PubMed] [Google Scholar]
- 21. Cepok S, Rosche B, Grummel V, et al. Short‐lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain. 2005;128(Pt 7):1667‐1676. [DOI] [PubMed] [Google Scholar]
- 22. Rivas JR, Ireland SJ, Chkheidze R, et al. Peripheral VH4+ plasmablasts demonstrate autoreactive B cell expansion toward brain antigens in early multiple sclerosis patients. Acta Neuropathol. 2017;133(1):43‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ireland SJ, Guzman AA, O’Brien DE, et al. The effect of glatiramer acetate therapy on functional properties of B cells from patients with relapsing‐remitting multiple sclerosis. JAMA Neurol. 2014;71(11):1421‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. 2019;50(5):1132‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He J, Tsai LM, Leong YA, et al. Circulating precursor CCR7(lo)PD‐1(hi) CXCR5+ CD4+ T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. 2013;39(4):770‐781. [DOI] [PubMed] [Google Scholar]
- 26. Morita R, Schmitt N, Bentebibel S‐E, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34(1):108‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Locci M, Havenar‐Daughton C, Landais E, et al. Human circulating PD‐1+CXCR3−CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. 2013;39(4):758‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simpson N, Gatenby PA, Wilson A, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010;62(1):234‐244. [DOI] [PubMed] [Google Scholar]
- 29. Byford ET, Carr M, Ladikou E, Ahearne MJ, Wagner SD. Circulating Tfh1 (cTfh1) cell numbers and PD1 expression are elevated in low‐grade B‐cell non‐Hodgkin's lymphoma and cTfh gene expression is perturbed in marginal zone lymphoma. PLoS One. 2018;13(1):e0190468‐e0190468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fletcher JM, Lonergan R, Costelloe L, et al. CD39+Foxp3+ regulatory T cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol. 2009;183(11):7602‐7610. [DOI] [PubMed] [Google Scholar]
- 31. Bielekova B, Catalfamo M, Reichert‐Scrivner S, et al. Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL‐2Rα‐targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci. 2006;103(15):5941‐5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vandenbark AA, Huan J, Agotsch M, et al. Interferon‐beta‐1a treatment increases CD56bright natural killer cells and CD4+CD25+ Foxp3 expression in subjects with multiple sclerosis. J Neuroimmunol. 2009;215(1):125‐128. [DOI] [PubMed] [Google Scholar]
- 33. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 Revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kurtzke JF. Rating neurologic impairment in multiple sclerosis. An expanded disability status scale (EDSS). Neurology. 1983;33(11):1444‐1444. [DOI] [PubMed] [Google Scholar]
- 35. Bar‐Or A, Li R. Cellular immunology of relapsing multiple sclerosis: interactions, checks, and balances. Lancet Neurol. 2021;20(6):470‐483. [DOI] [PubMed] [Google Scholar]
- 36. Burman J, Fransson M, Tötterman TH, Fagius J, Mangsbo SM, Loskog AS. T‐cell responses after haematopoietic stem cell transplantation for aggressive relapsing‐remitting multiple sclerosis. Immunology. 2013;140(2):211‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Durelli L, Conti L, Clerico M, et al. T‐helper 17 cells expand in multiple sclerosis and are inhibited by interferon‐β. Ann Neurol. 2009;65(5):499‐509. [DOI] [PubMed] [Google Scholar]
- 38. Reboldi A, Coisne C, Baumjohann D, et al. C‐C chemokine receptor 6‐regulated entry of TH‐17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10(5):514‐523. [DOI] [PubMed] [Google Scholar]
- 39. von Andrian UH, Engelhardt B. Alpha4 integrins as therapeutic targets in autoimmune disease. N Engl J Med. 2003;348(1):68‐72. [DOI] [PubMed] [Google Scholar]
- 40. Erle DJ, Briskin MJ, Butcher EC, Garcia‐Pardo A, Lazarovits AI, Tidswell M. Expression and function of the MAdCAM‐1 receptor, integrin alpha 4 beta 7, on human leukocytes. J Immunol. 1994;153(2):517‐528. [PubMed] [Google Scholar]
- 41. Engelhardt B, Kappos L. Natalizumab: targeting α4‐integrins in multiple sclerosis. Neurodegener Dis. 2008;5(1):16‐22. [DOI] [PubMed] [Google Scholar]
- 42. Coisne C, Mao W, Engelhardt B. Cutting edge: Natalizumab blocks adhesion but not initial contact of human T cells to the blood‐brain barrier in vivo in an animal model of multiple sclerosis. J Immunol. 2009;182(10):5909‐5913. [DOI] [PubMed] [Google Scholar]
- 43. Miller DH, Khan OA, Sheremata WA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348(1):15‐23. [DOI] [PubMed] [Google Scholar]
- 44. DeNucci CC, Pagán AJ, Mitchell JS, Shimizu Y. Control of alpha4beta7 integrin expression and CD4 T cell homing by the beta1 integrin subunit. J Immunol. 2010;184(5):2458‐2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muraro PA, Robins H, Malhotra S, et al. T cell repertoire following autologous stem cell transplantation for multiple sclerosis. J Clin Invest. 2014;124(3):1168‐1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Claes N, Fraussen J, Stinissen P, Hupperts R, Somers V. B cells are multifunctional players in multiple sclerosis pathogenesis: insights from therapeutic interventions. Front Immunol. 2015;6:642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cencioni MT, Ali R, Nicholas R, Muraro PA. Defective CD19+CD24(hi)CD38(hi) transitional B‐cell function in patients with relapsing‐remitting MS. Mult Scler. 2021;27(8):1187‐1197. doi: 10.1177/1352458520951536 [DOI] [PubMed] [Google Scholar]
- 48. Tzartos JS, Craner MJ, Friese MA, et al. IL‐21 and IL‐21 receptor expression in lymphocytes and neurons in multiple sclerosis brain. Am J Pathol. 2011;178(2):794‐802. doi: 10.1016/j.ajpath.2010.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laroni A, Armentani E, Kerlero de Rosbo N, et al. Dysregulation of regulatory CD56(bright) NK cells/T cells interactions in multiple sclerosis. J Autoimmun. 2016;72:8‐18. doi: 10.1016/j.jaut.2016.04.003 [DOI] [PubMed] [Google Scholar]
- 50. Strioga M, Pasukoniene V, Characiejus D. CD8+ CD28− and CD8+ CD57+ T cells and their role in health and disease. Immunology. 2011;134(1):17‐32. doi: 10.1111/j.1365-2567.2011.03470.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pangrazzi L, Reidla J, Carmona Arana JA, et al. CD28 and CD57 define four populations with distinct phenotypic properties within human CD8+ T cells. Eur J Immunol. 2020;50(3):363‐379. doi: 10.1002/eji.201948362 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of antibodies used for flow cytometry panels.
Table S2. Phenotypes and gating hierarchy of immune subsets.
Figure S1. Gating strategy of pro‐inflammatory T cells. Gating strategy for (A) Th17 cells, (B) MAIT cells and (C) CNS‐homing T‐conv (CNS‐H) cells are indicated.
Figure S2. Gating strategy of immune subsets regulatory humoral response cells. Gating strategy for (A) Naïve and memory B cells, (B) Plasmablasts (PB) and (C) PD1+ cTfh cells are indicated.
Figure S3. Gating strategy of immunoregulatory and immunosenescent cells. Gating strategy for (A) CD4+ Tregs, (B) CD39+ Tregs, (C) CD56hi NK cells, (D) CD8+CD28−CD57+ cells are indicated.
Figure S4. Analysis of absolute counts of immune subsets within whole MS cohort, and within responder and non‐responder patient groups (expressed as cells/μL). First two rows show absolute counts of immune subsets that showed significant changes between pre‐ and post‐AHSCT comparison. (A) CD8+ T cells (B) CD4:CD8 ratio, (C) CD19+ B cells, (D) MAIT cells, (E) Naïve B cells, (F) Memory:Naïve B cells, (G) CD56hi NK cells and (H) CD8+CD28−CD57+ cells. Statistical analysis was performed using linear mixed‐effects model (p < 0.05) and multiple comparisons adjusted using Holm‐Sidak method. Logarithmic transformations were performed for analysing difference between pre‐AHSCT and post‐AHSCT timepoint in CD4:CD8 ratio, CD19+ B cells, MAIT, Naïve B cells, Memory:Naïve B cell ratio and CD8+CD28−CD57+ cells. Pre‐AHSCT (Pre‐Tx) n = 19, 24 months (24M) n = 22, 36 months (36M) n = 22. Last two rows show absolute counts of immune subsets that demonstrated distinct responses within responder and non‐responder patient groups. Absolute counts of (I) Th17 in responder and non‐responder patients, (J) CNS‐homing T‐conv cells in responder and non‐responder patients, (K) Naïve B cells in responder and non‐responder patients and (L) CD56hi NK cells in responder and non‐responder patients. Statistical analysis was performed using parametric Friedman’s ANOVA with Dunn’s test (p < 0.05). Linear mixed‐effects model was not used for this analysis due to smaller sample size in non‐responder group. As the Friedman’s ANOVA analyses complete datasets, three patients from responder group that had missing timepoints of absolute count values were excluded for statistical comparisons. In responder group, pre‐AHSCT (Pre‐Tx) n = 14, 24 months (24M) n = 14, 36 months (36M) n = 14. In non‐responder group, pre‐AHSCT (Pre‐Tx) n = 5, 24 months (24M) n = 5, 36 months (36M) n = 5. For all graphs, *p < 0.05, **p < 0.01, ***p < 0.001. Black bars and asterisks indicate longitudinal comparison between pre‐ and post‐AHSCT timepoints within responder or non‐responder cohort.
Figure S5. Comparison of immune subset frequencies between responder and non‐responder MS patients at pre‐AHSCT. (A) Th17, (B) CNS‐homing T‐conv, (C) PD1+ cTfh and (D) CD39+ Tregs. Statistical analysis between responders (▵) (n = 15) and non‐responders (▽) (n = 5) at pre‐AHSCT were performed using independent two‐sample t‐tests (p < 0.05).