

Abstract
Introduction
Chronic kidney disease (CKD) is associated with significantly increased morbidity and mortality. No specific treatment of the underlying condition is available for the majority of patients, but ACE-inhibitors (ACE-I) and angiotensin II-receptor blockers (ARB) slows progression in albuminuric CKD. Adding a mineralocorticoid receptor-antagonist (MRA) like spironolactone has an additive effect. However, renin–angiotensin–aldosterone system (RAAS)-blockade increases the risk of hyperkalaemia which is exacerbated by the presence of CKD. Thus, hyperkalaemia may prevent optimal use of RAAS-blockade in some patients.
This project hypothesises that adding a potassium binder (patiromer) allows for improved RAAS-blockade including the use of MRA, thereby reducing albuminuria in patients with albuminuric CKD where full treatment is limited by hyperkalaemia.
If successful, the study may lead to improved treatment of this subgroup of patients with CKD. Furthermore, the study will examine the feasibility of potassium binders in patients with CKD.
Methods and analysis
An open-label, randomised controlled trial including 140 patients with estimated glomerular filtration rate (eGFR) 25–60 mL/min/1.73 m2, a urinary albumin/creatinine ratio (UACR) >500 mg/g (or 200 mg/g if diabetes mellitus) and a current or two previous plasma-potassium >4.5 mmol/L. Patients who develop hyperkaliaemia >5.5 mmol/L during a run-in phase, in which RAAS-blockade is intesified with the possible addition of spironolactone, are randomised to 12-month treatment with maximal tolerated ACE-I/ARB and spironolactone with or without patiromer.
The primary endpoint is the difference in UACR measured at randomisation and 12 months compared between the two groups. Secondary endpoints include CKD progression, episodes of hyperkalaemia, blood pressure, eGFR, markers of cardiovascular disease, diet and quality of life.
Ethics and dissemination
This study is approved by The Central Denmark Region Committees on Health Research Ethics (REFNO 1-10-72-110-20) and is registered in the EudraCT database (REFNO 2020-001595-15). Results will be presented in peer-reviewed journals, at meetings and at international conferences.
Keywords: chronic renal failure, adult nephrology, diabetic nephropathy & vascular disease, nephrology
Strengths and limitations of this study.
This study uses a robust randomised controlled design, investigating if patiromer, through increased renin–angiotensin–aldosterone system (RAAS)-blockade, can reduce albuminuria in patients with chronic kidney disease (CKD) and hyperkalaemia.
The selective run-in phase only allows randomisation of patients where RAAS-blockade is proven to be limited by hyperkalaemia.
A 1-year follow-up will examine long-term tolerability of patiromer in CKD patients, testing if such a treatment regimen is feasible.
The limited sample size and 1-year follow-up that does not allow for evaluation of ‘hard’ endpoints such as time to renal death or decrease in estimated glomerular filtration rate.
Introduction
Chronic kidney disease (CKD) is associated with substantial comorbidity and mortality.1 No curative treatment is currently available for the majority of patients, and current interventions aim to halt or slow the natural progression of the disease. Albuminuria is a well-established predictor of end-stage renal disease (ESRD). The risk of progressing to ESRD is up to 75 times higher among CKD patients with significant albuminuria compared with patients without albuminuria.2 Treatment with inhibitors of the renin–angiotensin–aldosterone system (RAAS) such as ACE inhibitors (ACE-Is) or angiotensin II receptor blockers (ARBs) reduce albuminuria in a dose-dependent manner.3 Furthermore, treatment slows CKD progression in both diabetic and non-diabetic CKD.4 5 RAAS-blockade, in particular the use of ACE-I or ARB, is considered first-line treatment in patients with CKD and albuminuria. Evidence suggests that the change in albuminuria correlates with the protection provided and therefore serves as a surrogate marker of disease progression.6 7 Other studies have shown that reducing albuminuria in CKD patients lowers the cardiovascular risk.8–10 Cardiovascular risk can be assessed from left ventricular hypertrophy, arterial stiffness (pulse wave velocity (PWV) and central blood pressure, BP) and blood biomarkers such as endothelin-1, N-terminal pro b-type natriuretic peptide (NT-pro-BNP) and troponin I (TnI).11–15
Aldosterone is a regulator of BP through fluid and electrolyte homeostasis. Evidence strongly support an additional, direct pathophysiological role for aldosterone in the development of kidney and cardiovascular disease. Mineralocorticoid receptor (MR) activation induces inflammation, oxidative stress and fibrosis16 17 and leads to glomerulosclerosis and cardiac fibrosis.18 19 This increases the risk of kidney function decline, albuminuria and cardiovascular disease. Blood aldosterone levels increase as estimated glomerular filtration rate (eGFR) deteriorates, and CKD is considered a state of relative hyperaldosteronism.20 21 RAAS blockade using ARB/ACE-I insufficiently lowers the aldosterone level, and plasma concentration typically rises after 6–12 months of treatment; a phenomenon known as aldosterone escape.22 MR-antagonists (MRAs) such as spironolactone or eplerenone alone or in combination with ACE-I/ARB reduce albuminuria by 25%–40%.23 24 A large number of studies have suggested renoprotective benefits of treatment with MRA in CKD with persistent albuminuria. This notion was emphasised by the recent FIDELIO-DKD trial. The trial found that the addition of the MRA finerenone to ACE-I/ARB treatment in patients with CKD, type 2 diabetes and albuminuria significantly reduced the risk of renal outcomes and cardiovascular risk.25
Despite inherent benefits, RAAS blockade may be hampered by fear of hyperkalaemia. Severe hyperkalaemia may cause life-threatening cardiac arrhythmias.26 CKD results in a reduced ability to excrete potassium and patients are at significant risk of hyperkalaemia.27 28 Treatment with ACE-Is or ARBs as well as MRAs further inhibits renal potassium excretion, augmenting the risk of hyperkalaemia.29 30 Thus, high potassium levels often limit the optimal use of RAAS blockade in many patients with CKD.28 Novel third generation nonsteroidal selective MRAs such as finerenone has a lower risk of causing hyperkalaemia compared with older generation MRAs, but the number of patients discontinuing treatment due to hyperkalaemia is still 2.5-fold higher compared with placebo.25 Other ongoing clinical trials evaluating the effect of ACE-I or ARB combined with MRAs in CKD exclude patients with hyperkalaemia.16 Thus, patients with CKD with hyperkalaemia may be barred from the potential benefits of complete RAAS blockade including MRA.
In recent years, novel potassium-binding agents have been introduced. These include patiromer (Veltassa), a non-absorbable sodium-free powder for oral use, which binds potassium in the gastrointestinal tract, thereby increasing faecal excretion and lowering plasma (P) potassium.28 Patiromer significantly lowers P-potassium in patients with CKD.31 32 Several studies have proposed the use of patiromer to allow for increased RAAS blockade in patients with hyperkalaemia, CKD and suboptimal RAAS blockade treatment.33 Hyperkalaemia may be addressed through dietary restrictions, but these have limited effect, a profound impact on patient’s lifestyle and freedom and limit the intake of healthy fresh fruits and vegetables.34
Aims and hypotheses
This trial aims to establish if the use of a potassium binding agent (patiromer) in patients with moderate or advanced CKD (eGFR 25–60 mL/min/1.73 m2) leads to a reduction in albuminuria by the concomitant intensified use of RAAS-inhibitors (losartan and/or spironolactone). In secondary analyses, it will examine the effects of this approach on markers of cardiovascular function, dietary habits (including fruit and vegetable intake) and quality-of-life as well as the potential risks; monitoring BP, episodes of hyperkalaemia and renal function.
Thus, the study will address the hypotheses that treatment with patiromer and intensified RAAS-blockade in patients with eGFR 25–60 mL/min, albuminuria and a tendency of high potassium levels leads to:
A significant reduction in albuminuria when compared with patients in maximal RAAS-blockade as allowed by their P-potassium levels without patiromer.
A significant reduction in albuminuria during treatment.
A reduced PWV and left ventricular mass (LVM) along with improvement in blood biomarkers of cardiovascular function.
An increased intake of healthy foods and higher quality of life.
Methods and analysis
The MorphCKD study is an investigator-initiated, multicentre, open-label, parallel group, superiority randomised controlled trial (RCT). Randomisation is performed as a block randomisation with 1:1 allocation.
The study will include patients from the outpatient clinics at the renal departments in Aarhus, Aalborg, Holstebro and Viborg, Denmark. The primary site, Aarhus, will also include patients from the diabetes outpatient clinic and within the hospital public admission area (see under ‘recruitment’).
The study is divided into a run-in phase of 2–8 weeks, followed by randomisation to a treatment phase involving 52 weeks of treatment with or without patiromer (see figure 1). The run-in phase will determine if maximised RAAS-blockade, including treatment with an MRA, leads to clinically significant hyperkalaemia (>5.5 mmol/L) despite dietary counselling, thereby identifying the patients that may benefit from treatment with patiromer who qualify for randomisation to the treatment phase.
Figure 1.
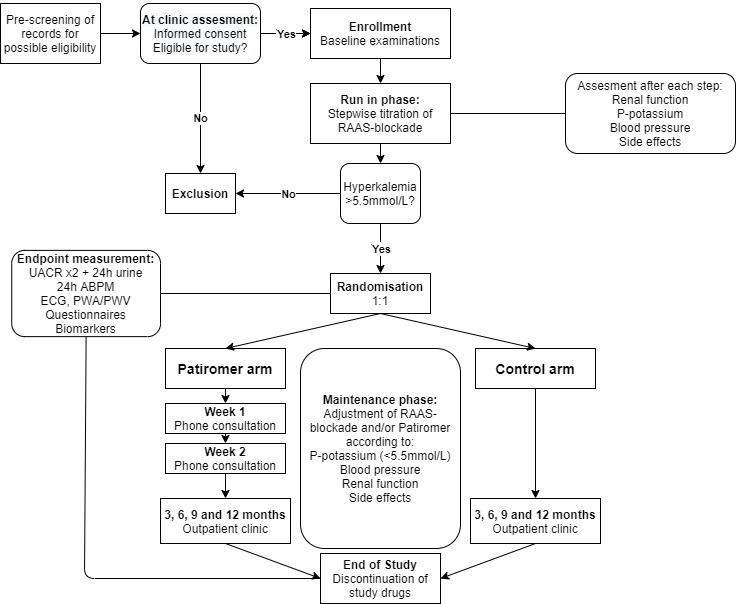
Flow chart of the trial design. ABPM, ambulatory blood pressure monitoring; PWA, pulse wave analysis; PWV, pulse wave velocity; RAAS, renin angiotensin aldosterone system; UACR, urine albumin/creatinine ratio.
Participants
A total of 140 patients fulfilling the eligibility criteria below will be included.
Inclusion criteria
Age 18–80.
eGFR 25–60 mL/min/1.73 m2.
Current P-potassium >4.5 mmol/L or P-potassium >4.5 mmol/L twice within 24 months.
Urine albumin-creatinine ratio (UACR) >500 mg/g or 200 mg/g and diabetes.
A lower UACR threshold for patients with diabetes is based on international guidelines suggesting a more aggressive approach when treating albuminuria in this group of patients,35 in which the renoprotective effects of treating lower levels of albuminuria is well documented.36
Exclusion criteria
Known allergies to both ACE-I and losartan or spironolactone or patiromer.
A history of kidney transplantation or active on the waiting list.
ESRD (defined as the need for dialysis or kidney transplantation).
Any renal disease requiring or being expected to require specific immunosuppressive therapy for the duration of the trial.
Pregnancy or inability to use contraception.
Regular need for trimethoprim or NSAIDs.
Current treatment with aliskiren.
Disseminated cancer disease.
Addison’s disease.
HF defined as ejection fraction <40% or active treatment at a HF clinic or similar.
Porphyria.
Severe constipation with a regular use of laxatives or previous recurrent ileus.
Fructose/galactose-intolerance.
Severe liver insufficiency (Child-Pugh Score B-C).
Clinically significant severe renal artery stenosis.
Investigator’s evaluation that participation in the trial may cause serious harm to the patient (eg, previous severe acute kidney injury (AKI) in relation to RAAS-blockade).
Initiation of an SGLT2-inhibitor within 30 days prior to inclusion.
Interventions and randomisation
Dietary counselling to limit potassium intake is provided at inclusion. Patients not treated with ACE-I/ARB at inclusion will commence losartan 50 mg/day for the run-in phase (step 1 below). Patients already treated with ACE-I/ARB will continue this treatment at the current dose with the addition of an MRA (starting at step 3 below). Based on tolerability, RAAS-blockade is increased in four steps:
Losartan 50 mg/day.
Losartan 100 mg/day.
Losartan 100 mg/day or current ACE-I/ARB+spironolactone 25 mg/day.
Losartan 100 mg/day or current ACE-I/ARB+spironolactone 50 mg/day.
Blood samples and home BP monitoring will be performed 1–2 weeks after each dose change and the patient is contacted by phone to record home BP and to inform about blood results. Tolerability is evaluated by P-potassium, creatinine, BP and side effects. The dose of losartan or spironolactone is reduced to the previous step and the patient proceeds to randomisation if P-potassium is >5.5 mmol/L.
Patients that reach step four without significant hyperkalaemia are excluded from the study.
Patients completing the run-in phase with an episode of significant hyperkalaemia (>5.5 mmol/L), a UACR >300 mg/g or 150 mg/g and diabetes, a most recent P-potassium >4.0 mmol/L and no other contraindications (eg, AKI) to continued and increased RAAS-blockade are randomised to open-label treatment in one of two regimens:
Patiromer with stepwise dose-increase/dose-decrease with increased RAAS-blockade in addition to standard clinical care and dietary counselling. Patiromer will be dosed based on P-potassium and tolerability until maximal RAAS-blockade with P-potassium ≤5.5 mmol/L.
No patiromer (control group) with standard clinical care, dietary counselling and maximal RAAS-blockade with P-potassium ≤5.5 mmol/L.
The lower threshold for albuminuria at randomisation compared with baseline is used to allow for some reduction of albuminuria resulting from the increase in RAAS-blockade during the run-in phase. Permuted block randomisation with random varying block sizes of 2, 4 and 6 is used to allocate patients to the patiromer- or control group at a 1:1 ratio, stratified by albuminuria >1000 mg/g (yes/no) and diabetes (yes/no). These stratifications have been included to minimise imbalances in patients with diabetes or severe albuminuria as such may have a different pathophysiology and thereby response to treatment compared with non-diabetics and patients with moderate albuminuria. The random allocation list is generated and uploaded to REDCap, a secure online database used for the electronic case report form, by an independent service provider (Clinical Trial Unit, Dept. of Clinical Medicine, Aarhus University) maintaining proper allocation concealment of randomisation.
After randomisation, patients are followed for up to 52 weeks with blood sampling and outpatient visits every 3 months and allowing for additional visits if considered clinically required based on the assessment of the local investigator.
The dose of study drugs (RAAS-blockade) is determined by the four steps previously described, aiming at the possible highest step with a P-potassium <5.5 mmol/L. Dose increases are only allowed on planned consultations (phone or outpatient clinic), but decreases may be introduced at any point depending on the results of blood test, BP or other adverse effects. In the patiromer group, patiromer is prescribed as tolerated at a daily dose of 8.4 g, 16.8 g or 25.2 g in order to maintain P-potassium <5.5 mmol/L. The dosing of patiromer is increased concomitantly with any increase in RAAS-blockade, unless P-potassium is ≤4.6 mmol/L. RAAS-blockade is decreased if hyperkalaemia >5.5 mmol/L is recorded at the highest tolerated patiromer dose.
All study drugs are stopped at the last outpatient visit after 52 weeks. Patients entering the study on ACE-I/ARB will continue this treatment without patiromer. Blood and urine samples are collected 4 weeks later for the evaluation of eGFR, albuminuria and P-potassium after discontinuation of study drugs.
Additional medication
Any inhibitors of the RAAS-system other than those mentioned above as well as additional potassium binders are not allowed during the study period. Hypertension is treated aiming at a systolic BP between 110 and 130 mm Hg in both groups. All antihypertensives, except additional inhibitors of the RAAS-system, may be used as per the discretion of the treating physician. Loop and thiazide diuretics may be prescribed for hypertension and/or fluid retention. Hypomagnesaemia is treated with oral magnesium supplements. Study drugs including losartan, spironolactone and patiromer can be reduced or suspended depending on BP, hyperkalaemia as per protocol or any side effects deemed to outweigh the benefit of the treatment (eg, AKI, gastrointestinal intolerance, biochemical abnormalities). SGLT2-inhibitors may not be prescribed during the trial.
Endpoints
The primary endpoint is:
The difference in UACR from randomisation to end of treatment compared between the two groups. This is measured as an average of two morning spot UACR samples collected both at randomisation and at the last study visit.
Secondary endpoints include:
The difference in 24 hours urine albumin from randomisation to end of treatment compared between the two groups.
The difference in albuminuria (evaluated by morning spot UACR and 24 hours urine collection) at the end of treatment between the two groups.
The difference in the extent of RAAS-blockade (ACE-I/ARB and MRA) at the end of treatment between the two groups.
The difference in kidney function (eGFR and urine creatinine clearance) at the end of treatment and the changes in eGFR from randomisation to end of treatment between the two groups.
The difference in BP (ambulatory and 24 hours) at the end of treatment and the changes in BP from randomisation to end of treatment between the two groups.
The difference in PWV/pulse wave analysis (PWA) at the end of treatment and the changes in PWV/PWA from randomisation to end of treatment between the two groups.
The difference in LVM (ECG) at the end of treatment and the changes in LVM from randomisation to end of treatment between the two groups.
The difference in cardiac biomarkers at the end of treatment and the changes in cardiac biomarkers from randomisation to end of treatment between the two groups.
The difference in P-potassium at the end of treatment between the two groups.
The difference in the number of episodes with severe hyperkalaemia (>6.2 mmol/L) from randomisation to end of treatment between the two groups.
The difference in the number of episodes with AKI (KDIGO stage 1–3) from randomisation to end of treatment between the two groups.
The difference in the questionnaire base assessment of the consumption of fruits and vegetables at the end of treatment and the changes in fruits and vegetables consumption from randomisation to end of treatment between the two groups.
The difference in QoL at the end of treatment and the changes in QoL from randomisation to end of treatment between the two groups.
Sample size and power calculation
The study will include 140 patients under the assumption that 30% will not meet randomisation criteria, leaving 98 participants (49 in each group) for randomisation. This will provide 80% power to detect a clinically relevant 1.5-fold greater reduction in the amount of albuminuria in the patiromer group compared with the control group, with a risk of type 1 error of 0.05 assuming an 80% coefficient of variation in the change of UACR.37 The study will continue as long as the number of randomised participants is expected to be no less than 55% (54 patients), which will provide power to detect a 1.75-fold greater reduction in the amount of albuminuria.
Recruitment
All patients from the renal outpatient clinics at the three centres and all patients serviced by Aarhus University Hospital (covering a population of approx. 900 000) who have provided a blood sample within the last 2 years are prescreened. The prescreening algorithm uses the LABKA II database containing the result of all blood samples analysed within the relevant regions and identifies patients aged 18–80 with a history (within 2 years) of P-potassium >4.5 mmol/L, UACR >200 mg/g and eGFR 25–60 mL/min/1.73 m2. The resulting patient records including blood samples are manually screened by a local investigator. Potentially eligible patients are contacted with the participant information by letter. In addition, patients in the nephrology outpatient clinics in Aarhus, Aalborg, Holstebro and Viborg and the diabetes outpatient clinic in Aarhus are contacted in person at their next appointment. After written informed consent they are screened for inclusion including blood samples to ensure that they meet the inclusion criteria at the time of inclusion.
Data collection
The patient’s medical history including current treatment is registered at inclusion. Patient height is measured at inclusion and weight at every visit. A physical examination including vital signs are performed at inclusion, randomisation and final visit. A urine sample for UACR is collected at inclusion, twice at randomisation and final visit for the primary outcome and at every 3-month visit during the treatment phase. If possible, a morning sample is preferred. Timed 24 hours urine samples are collected at randomisation and 52 weeks. P- potassium, creatinine, eGFR and sodium are measured at inclusion, weekly during the run-in phase, during titration of RAAS-blockade and every 3 months in the outpatient clinic. P-total CO2 and ionised calcium are measured at inclusion, randomisation and every 3 month visit. P-magnesium is measured at all visits during the maintenance phase in the patiromer group. Cardiac biomarkers (Endothelin-1, NT-proBNP and TnI), ECG, blood samples evaluating biomarkers of fruit and vegetable consumption (lutein, β-cryptoxanthin, α-carotene and β-carotene, lycopene and vitamin C38), PWV and PWA as a measure of arteriosclerosis, 36-item short form health survey (SF-3639) measuring QoL, MyFood2440 food diaries and 24 hours ABPM are all performed at randomisation and 52 weeks. Please refer to table 1 for a timeline of data collection.
Table 1.
Study timeline and visits
| Timeline (weeks) | Inclusion −8 |
-6 | −4* | −2* | t0 Randomisation/ (exclusion) |
2* | 4* | 13 | 26 | 39 | 52 | 56 |
| Study phase | Run-in phase | Maintenance phase | ||||||||||
| Interventions | ||||||||||||
| Increased RAAS-blockade |

|
|||||||||||
| Patiromer-treatment (intervention group) and/or RAAS-blockade titration |
|
|||||||||||
| Assesments | ||||||||||||
| Eligibility screen | x | |||||||||||
| Informed consent | x | |||||||||||
| Pregnancy test (if fertile woman) | x | |||||||||||
| Medical record | x | |||||||||||
| Physical examination | x | x | x | |||||||||
| Weight | x | x | x | x | X | x | ||||||
| Dietary counselling | x | |||||||||||
| P-Creatinine, eGFR, P-potassium, P-sodium | x | X | x | x | x | x | x | x | x | X | x | x |
| P-magnesium (patiromer group) | x | x | x | x | x | X | x | |||||
| P-total CO2, P-ionised Calcium | x | x | x | x | X | x | x | |||||
| BpTRU or similar local ABPM | x | x | x | x | X | x | ||||||
| 24 hours ABPM | x | x | ||||||||||
| At-home ABPM | x | x | x | x | x | |||||||
| UACR | x | x | x | x | xx | x | x | xx | x | |||
| Questionnaire—side effects | x | x | x | x | X | x | ||||||
| Questionnaire—SF-3639 | x | x | ||||||||||
| Questionnaire—MyFood2440 | x | x | ||||||||||
| Carotenoids and C-vitamin | x | x | ||||||||||
| 24 hours urine collection | x | x | ||||||||||
| PWA/PWV | x | x | ||||||||||
| ECG | x | x | ||||||||||
| Endothelin-1, NT-pro-BNP and TnI | x | x | ||||||||||
| Pill count | x (x) | x | x | X | x | |||||||
| Type of visit | ||||||||||||
| Phone consultation | x | x | x | x | x | |||||||
| Outpatient clinic | x | x (x) | x | x | X | x | ||||||
*Only If RAAS-blockade has been increased. During the run-in phase, the patient proceeds directly to randomisation if P-potassium >5.5 mmol/L.
ABPM, ambulatory blood pressure monitoring; BpTRU, An automatic blood pressure monitor set to average the last 5 of 6 blinded blood pressures with 2 minute intervals; eGFR, estimated glomerular filtration rate; NT-pro-BNP, N-terminal pro b-type natriuretic peptide; PWA, pulse wave analysis; PWV, pulse wave velocity; RAAS, renin–angiotensin–aldosterone system; SF-36, 36-Item short form health survey; TnI, troponin I; UACR, urine albumin/creatinine ratio.
Questionnaires and food diaries may be filled in online prior to the visit using REDCap or the Myfood24 website. An invitation with a unique link is sent to the patient via email. Alternatively, they are completed with the assistance of the investigator on the day of the visit. Vital signs are measured using BpTRU, an automatic blood pressure monitor set to average the last 5 of 6 blinded blood pressures, or equivalent at every visit and with an automatic digital BP monitor at home for phone consultations. Pill counts are done at every visit to assess compliance. Patients are asked if and to what extent they have taken the study drugs at all contacts. Adherence techniques such as morning routines and medication in relation to meals are discussed if required.
Blood and urine biochemical analyses are performed at local biochemical laboratories using standard automated assay. Reference intervals have been standardised on a national level and all Danish laboratories use these for reference. The UACR at randomisation and final visit is calculated as an average of 2 measurements over 2 days at each time point to minimise variability.41 The patient is carefully instructed to provide morning urine samples. Urine collection, including 24 hours urine collection, is performed by the patient at home prior to the randomisation and 12-month-visit. PWV and PWA are measured using the Sphygmocor system. Applanation tonometry is applied on the carotid, femoral, and radial artery. A minimum operator index of 85 is used. Length is measured as 80% of the distance from the carotid artery to the femoral artery. Home BP is measured three times in the morning and evening for 3 days. An average of day 2 and 3 is reported. Twenty-4 hour ABPM is recorded with measurements every 30 min. ECG is recorded by an experienced nurse. Each centres’ personnel will be trained and instructed in all study procedures by the study principal investigator (PI).
Data management
Study data, including adverse events (AE) and serious AEs/reaction, will be collected and managed using REDCap electronic data capture tools hosted at Aarhus University.42 43 All data are entered electronically by the local investigator at each site. Original data is stored in the patients’ electronic records or in a participant file. Participant files are stored in a secure place and kept for 15 years after end of study. Data will be exported from RedCap for final analysis using a suitable statistical software package.
Statistical methods
The primary endpoint is analysed using a t-test comparing the differences in UACR between randomisation and 12 months between the two groups. The ratio between groups with confidence intervals and the p value will be reported. A secondary two-way repeated measures ANOVA including UACR at randomisation, 3, 6, 9 and 12 months is performed and the P value is reported. The data are analysed as intention to treat with a secondary treated-as analysis. A separate t-test will compare the difference in UACR from randomisation to the time patiromer is discontinued. Data from patients discontinuing treatment before 12 months of follow-up is included using carry-over of the last available dataset before stopping. Missing data for the primary endpoint will be replaced by the most recent observation carried over. Previous studies have shown that most of the effect of increased RAAS-blockade on albuminuria is seen early after treatment initiation with little change thereafter, suggesting that the UACR at the closest possible time point is a fair proxy measure of the 12 month value. Imputations may be applied for secondary analyses if feasible. All variables are analysed for normal distribution and skewed data are log-transformed when appropriate. Non-normally distributed variables on both the standard-scale and log-scale are analysed using non-parametric testing. Repeated measurements are analysed by a linear model when feasible.
Safety measures
AEs, defined as any medical occurrence in a trial participant without regard to the possible cause, are collected from when the consent has been signed and until final visit. Participants are asked about any new such events at each contact and will fill out a questionnaire at each outpatient clinic visit. Serious AEs will be reported directly to the PI and sponsor. Investigators will evaluate any AEs possible relation to study drugs based on temporal relationship, known mechanism of action and known side effects for classification of adverse reactions.
The following individual safety outcomes are evaluated by the investigator at each contact:
A decline in eGFR >30% from inclusion, 20% from previous visit or 5 mL/min/1.73 m2 from previous visit if eGFR <25 mL/min/1.73 m2 should lead to a temporary reduction or discontinuation of spironolactone and/or ACE-I/ARB (Losartan or other).
An increase in P-creatinine >100% from the previous visit, the possible need for acute dialysis or other findings suggesting severe AKI leads to admittance for treatment.
If P-potassium is >5.5 mmol/L on maximal patiromer dose, RAAS-blockade is reduced by 50% or spironolactone is discontinued. P-potassium is repeated within 2 days or as soon as possible.
If P-potassium is >5.9 mmol/L, ACE-I/ARB and spironolactone are temporarily discontinued. P-potassium is repeated within 1 day.
If P-potassium is >6.2 mmol/L, the patient is admitted and treated in accordance with local guidelines. ACE-I/ARB and spironolactone are temporarily discontinued.
If P-magnesium is <0.6 mmol/L, the patient is treated with oral magnesium supplements as per discretion of the local investigator and P-magnesium is repeated within 7–10 days.
If P-magnesium is <0.5 mmol/L despite maximal tolerated magnesium supplement treatment, patiromer is discontinued and p-magnesium is repeated within 2 days.
In case of events 1–5 above, RAAS-blockade may the reinitiated at the previous dosage if and when kidney function is restored and/or P-potassium <5.4 mmol/L following discontinuation or dose reduction of the RAAS inhibitor.
The study is halted if at any point a significant higher number of the following events are observed in patiromer group compared with the control group:
Events with hyperkalaemia >6.2 mmol/L.
Patients with eGFR <15 mL/min/1.73 m2 or requiring dialysis for >3 months.
Deaths.
Admissions (except events due to hyperkalaemia, covered in point 1).
A combined endpoint of the four above.
These outcomes will be evaluated by the PI after each such event using Fischer’s exact test.
Study oversight and monitoring
The study is monitored by the Good Clinical Practice unit at Aarhus and Aalborg University Hospitals. It does not include an independent data monitoring committee due to the open label design, limited number of sites and continuous monitoring of significant safety outcomes as described above. Any systematic or serious risk to the participants will be immediately apparent to the PI and sponsor. The study may be audited by the Danish Medicines Agency. Safety reports are forwarded to the Danish Medicines Agency and The Central Denmark Region Committees on Health Research Ethics annually.
Patient and public involvement statement
Participants were involved in changes to the design of the study. They preferred less transportation and fewer hospital visits. From their feedback, some visits were replaced by phone consultations and blood sampling prior to most visits was made possible at 28 local sites across the Central Denmark Region. Once the trial has been published, participants will be informed of the results via email using the REDCap distribution tool.
Current trial status
The first participant was included in the study late August 2020 and is planned to continue until March 2022. At the time of writing (August 2021), 56 participants have been included and 14 have been randomised to the treatment phase. Enrolment was halted from December 2020 due to the lockdown following COVID-19 in Denmark but was resumed in March 2021.
Ethics and dissemination
The study protocol was initially approved by The Central Denmark Region Committees on Health Research Ethics (REFNO 1-10-72-110-20) on 23 June 2020 with the latest version being approved on 1 July 2021 and by the Danish Medicines Agency on 10 June 2021. The research will be conducted in accordance with the Helsinki Declaration and Good Clinical Practice. All protocol amendments will be approved by the Ethics Committee and Danish Medicines Agency before implementation when required and all investigators will be notified directly.
A local investigator will obtain a written, informed consent from all participants prior to inclusion. The consent form follows the standards and template from the Danish National Committee on Health Research Ethics.
All principal investigators and sponsor will have access to the cleaned dataset.
Trial results, positive as well as negative, will be submitted for publication in peer reviewed, international journals and presented at conferences and meetings.
Discussion
This study investigates the feasibility of daily treatment with an established potassium-binding agent in moderate and severe CKD patients with albuminuria. It will examine if treatment enables increased RAAS-blockade and leads to a greater decline in albuminuria. Previous studies have shown that patiromer allows for the use of spironolactone in patients with CKD with hyperkalaemia44; however, it is unknown if the approach leads to an effect on albuminuria in this distinct group of patients. The study aims to fill this gap in current knowledge.
The open-label study design should closely mimic the clinical decision making and the delicate task of balancing hyperkalaemia and renoprotection. This will provide information on the practicability and potential benefits of such an approach in patients with hyperkaliemia otherwise barred from full pharmaceutical blockage of the RAAS-system. Of note, the study includes a 1-year follow-up to examine potential complications to long-term treatment including non-adherence, hypotension, AKI and other adverse effects. Additional strengths of the study include the extensive list of outcomes and the RCT design. Furthermore, the unique and selective run-in phase only allows randomisation of patients that are proven to potentially benefit from treatment with a potassium binder, which should be in accordance with clinical practice. In addition, since all included interventions involves established and approved drugs, the road to implementation ought to be short.
There are some potential limitations and challenges. First, the small sample size and 1-year follow-up does not allow for evaluation of harder renal endpoints such as progression to ESRD or a 50% reduction in eGFR. However, albuminuria is a widely accepted surrogate marker of disease progression in albuminuric CKD. In addition, it is closely correlated to the protective effects of RAAS-blockade. Second, the study is not powered to detect minor differences in the change in albuminuria; however, it will be able to identify a 1.5 times greater reduction in UACR. Third, the 1-year follow-up may challenge patient adherence to treatment. Fourth, patients already treated with ACE-I or ARB may be on a submaximal dose of these medications when Spironolactone is added as per protocol to their current treatment. This was considered necessary to avoid the complexity of either having to manage a large number of different ACE-Is or ARBs as potential study drugs or requiring an initial switch to for example, Losartan, which may increase the duration of the run-in phase significantly and thus, the risk of early participant drop out. Fifth, the open-label design may introduce selection bias in physicians’ use of non-investigational drugs. Standard operating procedures on concomitant treatment, including instructions for the use of diuretics, are established to mitigate such bias. The open-label design does provide some potential benefits, allowing for a setup that closely resembles clinical practice and for a more practical safety algorithm to prevent potentially life-threatening hyperkalaemia. The primary outcome is based on biochemical findings. It is very unlikely that this is affected by the open-label design.
The power calculations are based on the number of randomised patients after the run-in phase. It is assumed that 30% of the included patient will not be eligible for randomisation; however, the accuracy of this number has not been established and thus, the number of actual randomised patients may be different. The extensive screening algorithm, which is based on results from blood samples in all four outpatient clinics and the entire Aarhus University public admission area, will however ensure that all eligible candidates are invited to the study. This is particularly important as the number of patients with an eGFR between 25 and 60 mL/min/1.73 m2, concomitant and significant albuminuria, and previous or current P-potassium >4.5 mmol/L may be limited.
The study is partly based on the assumption that adding MRA to ACE-I/ARB treatment in this subgroup of patients is beneficial if hyperkalaemia can be controlled, supported by the recent results of the FIDELIO-DKD trial25; however, the underlying principle is also applicable to patients in which maximal dosing of ACE-I or ARBs is barred by hyperkalaemia. If this study establishes the feasibility of such approach, it should pave the way for larger studies with hard endpoints to corroborate the use of potassium binders in patients currently excluded from maximal RAAS-inhibition.
Supplementary Material
Footnotes
Contributors: HB conceived the study and had editorial rights on the protocol. FHM was first author of the protocol, primary investigator in Aarhus and implemented the study on all sites as the coordinating investigator. CDP held an advisory role in the writing of the protocol. JHC was the primary investigator in Aalborg. All authors contributed to refinement of the study protocol including the design and approved the final manuscript.
Funding: This work is supported by Aarhus University Graduate School and by Vifor Pharma. The latter includes free of charge patiromer. Study administration, financial statement and conflicts of interestNeither the study sponsor (HB), PI (FHM) nor any other investigators have any financial involvement with the primary funder (Vifor Pharma) or the primary study drug (patiromer). HB has previously participated in advisory boards and meetings hosted by Vifor Pharma. These have all been reported to the Danish Medicines Agency. The authors have no further conflicts of interest to declare.
Competing interests: None declared.
Patient and public involvement: Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review: Not commissioned; externally peer reviewed.
Ethics statements
Patient consent for publication
Not applicable.
References
- 1.Chen TK, Knicely DH, Grams ME. Chronic kidney disease diagnosis and management: a review. JAMA 2019;322:1294–304. 10.1001/jama.2019.14745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iseki K, Kinjo K, Iseki C, et al. Relationship between predicted creatinine clearance and proteinuria and the risk of developing ESRD in Okinawa, Japan. Am J Kidney Dis 2004;44:806–14. 10.1016/S0272-6386(04)01080-7 [DOI] [PubMed] [Google Scholar]
- 3.Weinberg MS, Kaperonis N, Bakris GL. How high should an ACE inhibitor or angiotensin receptor blocker be dosed in patients with diabetic nephropathy? Curr Hypertens Rep 2003;5:418–25. 10.1007/s11906-003-0088-8 [DOI] [PubMed] [Google Scholar]
- 4.Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001;345:861–9. 10.1056/NEJMoa011161 [DOI] [PubMed] [Google Scholar]
- 5.Lewis EJ, Hunsicker LG, Bain RP, et al. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med Overseas Ed 1993;329:1456–62. 10.1056/NEJM199311113292004 [DOI] [PubMed] [Google Scholar]
- 6.Levey AS, Gansevoort RT, Coresh J, et al. Change in albuminuria and GFR as end points for clinical trials in early stages of CKD: a scientific workshop sponsored by the National kidney Foundation in collaboration with the US food and drug administration and European medicines Agency. American Journal of Kidney Diseases 2020;75:84–104. 10.1053/j.ajkd.2019.06.009 [DOI] [PubMed] [Google Scholar]
- 7.Waijer SW, Gansevoort RT, Heerspink HJL. Change in albuminuria as a surrogate endpoint. Curr Opin Nephrol Hypertens 2019;28:519–26. 10.1097/MNH.0000000000000541 [DOI] [PubMed] [Google Scholar]
- 8.Balamuthusamy S, Srinivasan L, Verma M, et al. Renin angiotensin system blockade and cardiovascular outcomes in patients with chronic kidney disease and proteinuria: a meta-analysis. Am Heart J 2008;155:791–805. 10.1016/j.ahj.2008.01.031 [DOI] [PubMed] [Google Scholar]
- 9.Stenvinkel P, Ketteler M, Johnson RJ, et al. IL-10, IL-6, and TNF-alpha: central factors in the altered cytokine network of uremia--the good, the bad, and the ugly. Kidney Int 2005;67:1216–33. 10.1111/j.1523-1755.2005.00200.x [DOI] [PubMed] [Google Scholar]
- 10.Navalkar S, Parthasarathy S, Santanam N, et al. Irbesartan, an angiotensin type 1 receptor inhibitor, regulates markers of inflammation in patients with premature atherosclerosis. J Am Coll Cardiol 2001;37:440–4. 10.1016/S0735-1097(00)01138-4 [DOI] [PubMed] [Google Scholar]
- 11.Peters CD, Kjaergaard KD, Jensen JD, et al. No significant effect of angiotensin II receptor blockade on intermediate cardiovascular end points in hemodialysis patients. Kidney Int 2014;86:625–37. 10.1038/ki.2014.69 [DOI] [PubMed] [Google Scholar]
- 12.Jankowich M, Choudhary G. Endothelin-1 levels and cardiovascular events. Trends Cardiovasc Med 2020;30:1-8. 10.1016/j.tcm.2019.01.007 [DOI] [PubMed] [Google Scholar]
- 13.Barton M. Reversal of proteinuric renal disease and the emerging role of endothelin. Nat Clin Pract Nephrol 2008;4:490–501. 10.1038/ncpneph0891 [DOI] [PubMed] [Google Scholar]
- 14.Dahlöf B, Devereux RB, Kjeldsen SE, et al. Cardiovascular morbidity and mortality in the losartan intervention for endpoint reduction in hypertension study (life): a randomised trial against atenolol. Lancet 2002;359:995–1003. 10.1016/S0140-6736(02)08089-3 [DOI] [PubMed] [Google Scholar]
- 15.Malmqvist K, Kahan T, Edner M, et al. Regression of left ventricular hypertrophy in human hypertension with irbesartan. J Hypertens 2001;19:1167–76. 10.1097/00004872-200106000-00023 [DOI] [PubMed] [Google Scholar]
- 16.Barrera-Chimal J, Girerd S, Jaisser F. Mineralocorticoid receptor antagonists and kidney diseases: pathophysiological basis. Kidney Int 2019;96:302–19. 10.1016/j.kint.2019.02.030 [DOI] [PubMed] [Google Scholar]
- 17.Gilbert KC, Brown NJ. Aldosterone and inflammation. Current Opinion in Endocrinology, Diabetes and Obesity 2010;17:199–204. 10.1097/MED.0b013e3283391989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shavit L, Lifschitz MD, Epstein M. Aldosterone blockade and the mineralocorticoid receptor in the management of chronic kidney disease: current concepts and emerging treatment paradigms. Kidney Int 2012;81:955–68. 10.1038/ki.2011.505 [DOI] [PubMed] [Google Scholar]
- 19.Remuzzi G, Cattaneo D, Perico N. The aggravating mechanisms of aldosterone on kidney fibrosis. J Am Soc Nephrol 2008;19:1459–62. 10.1681/ASN.2007101079 [DOI] [PubMed] [Google Scholar]
- 20.Berl T, Katz FH, Henrich WL, et al. Role of aldosterone in the control of sodium excretion in patients with advanced chronic renal failure. Kidney Int 1978;14:228–35. 10.1038/ki.1978.114 [DOI] [PubMed] [Google Scholar]
- 21.Hené RJ, Boer P, Koomans HA, et al. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982;21:98–101. 10.1038/ki.1982.14 [DOI] [PubMed] [Google Scholar]
- 22.Bomback AS, Klemmer PJ. The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol 2007;3:486–92. 10.1038/ncpneph0575 [DOI] [PubMed] [Google Scholar]
- 23.Alexandrou M-E, Papagianni A, Tsapas A, et al. Effects of mineralocorticoid receptor antagonists in proteinuric kidney disease. J Hypertens 2019;37:2307–24. 10.1097/HJH.0000000000002187 [DOI] [PubMed] [Google Scholar]
- 24.Currie G, Taylor AHM, Fujita T, et al. Effect of mineralocorticoid receptor antagonists on proteinuria and progression of chronic kidney disease: a systematic review and meta-analysis. BMC Nephrol 2016;17:127. 10.1186/s12882-016-0337-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bakris GL, Agarwal R, Anker SD, et al. Effect of Finerenone on chronic kidney disease outcomes in type 2 diabetes. N Engl J Med Overseas Ed 2020;383:2219–29. 10.1056/NEJMoa2025845 [DOI] [PubMed] [Google Scholar]
- 26.Weiss JN, Qu Z, Shivkumar K. Electrophysiology of hypokalemia and hyperkalemia. In: Circulation: arrhythmia and electrophysiology. vol 10, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovesdy CP. Management of hyperkalaemia in chronic kidney disease. Nat Rev Nephrol 2014;10:653–62. 10.1038/nrneph.2014.168 [DOI] [PubMed] [Google Scholar]
- 28.Georgianos PI, Agarwal R. Revisiting RAAS blockade in CKD with newer potassium-binding drugs. Kidney Int 2018;93:325–34. 10.1016/j.kint.2017.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raebel MA. Hyperkalemia associated with use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. Cardiovasc Ther 2012;30:e156–66. 10.1111/j.1755-5922.2010.00258.x [DOI] [PubMed] [Google Scholar]
- 30.Kumar R, Kanev L, Woods SD, et al. Managing hyperkalemia in high-risk patients in long-term care. Am J Manag Care 2017;23:S27–36. [PubMed] [Google Scholar]
- 31.Bakris GL, Pitt B, Weir MR, et al. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015;314:151–61. 10.1001/jama.2015.7446 [DOI] [PubMed] [Google Scholar]
- 32.Weir MR, Bakris GL, Bushinsky DA, et al. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015;372:211–21. 10.1056/NEJMoa1410853 [DOI] [PubMed] [Google Scholar]
- 33.Sutherland CS, Braunhofer PG, Vrouchou P, et al. A cost-utility analysis of Raasi Enabling-Patiromer in patients with hyperkalemia. Value in Health 2017;20:A490. 10.1016/j.jval.2017.08.519 [DOI] [Google Scholar]
- 34.Cupisti A, Kovesdy C, D’Alessandro C, et al. Dietary approach to recurrent or chronic hyperkalaemia in patients with decreased kidney function. Nutrients 2018;10:261. 10.3390/nu10030261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Official Journal of the internatiOnal Society of nephrology KDIGO . Clinical practice guideline for the management of blood pressure in chronic kidney disease, 2012. Available: http://www.kidney-international.org [Accessed 10 Jan 2022].
- 36.Karalliedde J, Viberti G. Evidence for renoprotection by blockade of the renin-angiotensin-aldosterone system in hypertension and diabetes. J Hum Hypertens 2006;20:239–53. 10.1038/sj.jhh.1001982 [DOI] [PubMed] [Google Scholar]
- 37.Belle G, Martin DC. Sample size as a function of coefficient of variation and ratio of means. American Statistician 1993;47:165–7. [Google Scholar]
- 38.Woodside JV, Draper J, Lloyd A, et al. Use of biomarkers to assess fruit and vegetable intake. In: Proceedings of the nutrition Society. 76. Cambridge University Press, 2017: 308–15. 10.1017/S0029665117000325 [DOI] [PubMed] [Google Scholar]
- 39.Lins L, Carvalho FM. SF-36 total score as a single measure of health-related quality of life: Scoping review. SAGE Open Med 2016;4:205031211667172. 10.1177/2050312116671725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wark PA, Hardie LJ, Frost GS, et al. Validity of an online 24-h recall tool (myfood24) for dietary assessment in population studies: comparison with biomarkers and standard interviews. BMC Med 2018;16:136. 10.1186/s12916-018-1113-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naresh CN, Hayen A, Weening A, et al. Day-to-day variability in spot urine albumin-creatinine ratio. Am J Kidney Dis 2013;62:1095–101. 10.1053/j.ajkd.2013.06.016 [DOI] [PubMed] [Google Scholar]
- 42.Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009;42:377–81. 10.1016/j.jbi.2008.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harris PA, Taylor R, Minor BL, et al. The REDCap consortium: building an international community of software platform partners. J Biomed Inform 2019;95:103208. 10.1016/j.jbi.2019.103208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agarwal R, Rossignol P, Romero A, et al. Patiromer versus placebo to enable spironolactone use in patients with resistant hypertension and chronic kidney disease (amber): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2019;394:1540–50. 10.1016/S0140-6736(19)32135-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.