

Abstract
The sphingosine 1-phosphate (S1P) signaling pathway is an attractive target for pharmacological manipulation due to its involvement in cancer progression and immune cell chemotaxis. The synthesis of S1P is catalyzed by the action of sphingosine kinase 1 or 2 (SphK1 or SphK2) on sphingosine and ATP. While potent and selective inhibitors of SphK1 or SphK2 have been reported, development of potent dual SphK1/SphK2 inhibitors are still needed. Towards this end, we report the structure–activity relationship profiling of 2-(hydroxymethyl)pyrrolidine-based inhibitors with 22d being the most potent dual SphK1/SphK2 inhibitor (SphK1 Ki = 0.679 μM, SphK2 Ki = 0.951 μM) reported in this series. 22d inhibited the growth of engineered Saccharomyces cerevisiae and decreased S1P levels in histiocytic lymphoma myeloid cell line (U937 cells), demonstrating inhibition of SphK1 and 2 in vitro. Molecular modeling studies of 22d docked inside the Sph binding pocket of both SphK1 and SphK2 indicate essential hydrogen bond between the 2-(hydroxymethyl)pyrrolidine head to interact with aspartic acid and serine residues near the ATP binding pocket, which provide the basis for dual inhibition. In addition, the dodecyl tail adopts a “J-shape” conformation found in crystal structure of sphingosine bound to SphK1. Collectively, these studies provide insight into the intermolecular interactions in the SphK1 and 2 active sites to achieve maximal dual inhibitory activity.
Keywords: Sphingosine kinase, SphK1, Inhibitor, Sphingosine, Sphingosine 1-phosphate, Lipophilic binding pocket, PF-543, Structure-activity relationship
1. Introduction
In humans and other mammals, sphingolipid signaling has emerged as an attractive candidate for pharmacological intervention. Numerous investigations have focused on the role sphingolipids play in maintaining cellular homeostasis, specially via the sphingosine 1-phosphate (S1P) signaling pathway. Examination of the S1P synthetic pathway has revealed it to be a potential mediator of cellular fate (Fig. 1). In particular, ceramide (Cer) and sphingosine (Sph) promote signaling that result in cell arrest and apoptosis whereas surplus S1P encourages proliferation and survival.1–3 As such, members of the S1P synthetic pathway, particularly S1P, have emerged to be implicated in numerous diseases such as cancer,4–6 fibrosis,7,8 and sickle cell disease (SCD).9,10
Fig. 1.
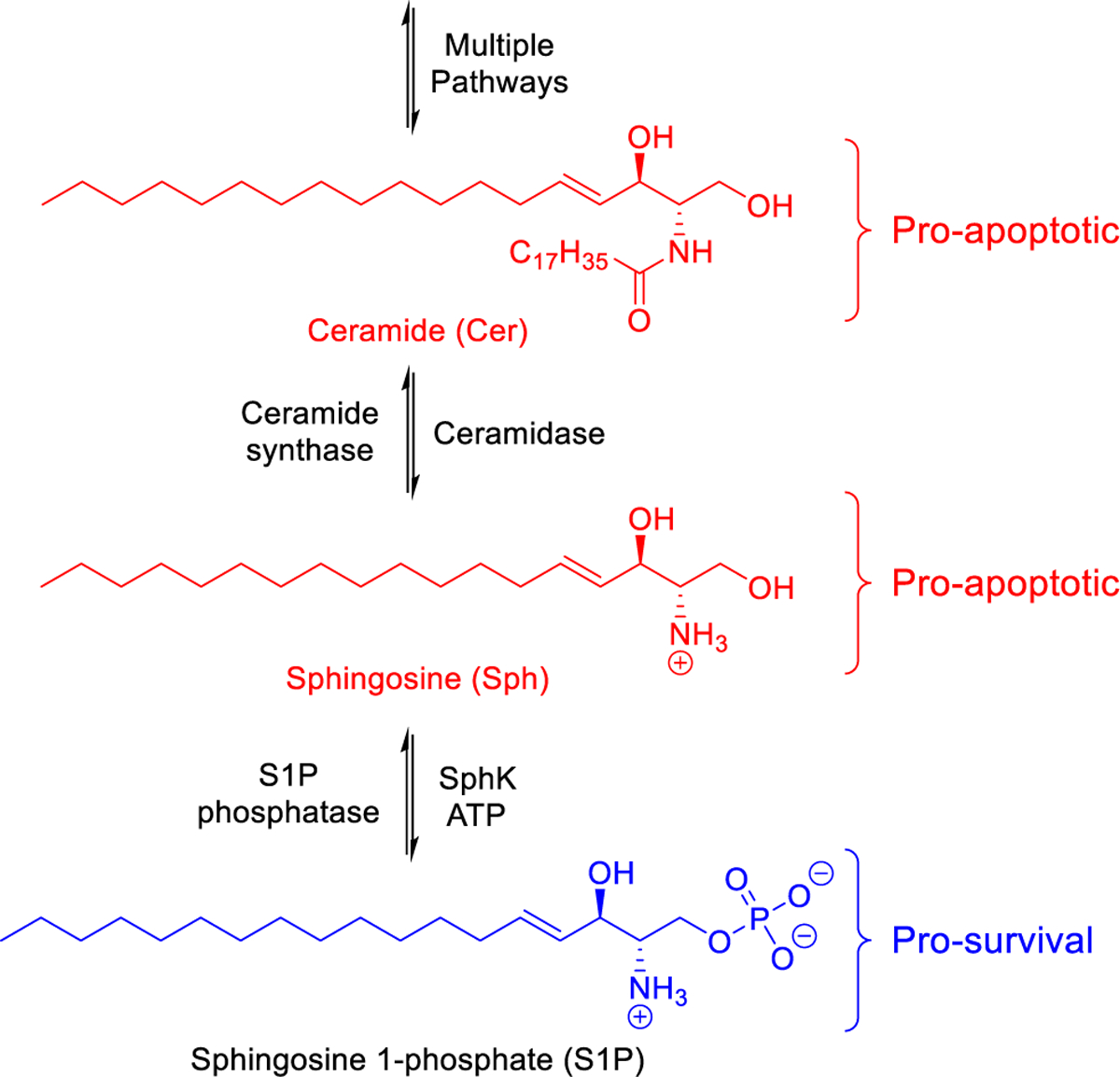
The S1P synthetic pathway and its role in cellular fate.
Functionally, S1P can act as both an intra- and extracellular ligand for multiple targets resulting in various cellular responses. Most notably, extracellular S1P has a high affinity for five G-protein coupled receptors (S1P1–5) that when activated lead to a downstream cascade of pro-survival effects.11–13 Synthetically, the generation of S1P is exclusively due to the ATP-dependent phosphorylation of Sph, mediated by sphingosine kinases (SphK1 and SphK2). The general function of two kinase isoforms is moderately repetitive in that both catalyze the transformation of Sph to S1P. Gene ablation experiments in mice demonstrated that deletion of either isoform was unremarkable, and the animals were viable and fertile. However, elimination of both kinase isoform genes resulted in embryonic death around day E13.5.14,15 There are some differences observed between SphK1 and SphK2 including subcellular distribution, substrate specificity, and active site topography.16–19 One salient difference is the consequence of isoform deficiency, whether accomplished by genetic manipulation or inhibitors, with regard to blood S1P concentrations. Interestingly, rodents deficient in SphK1 displayed circulating S1P levels that were reduced by > 50% whereas SphK2 deficient rodents had blood S1P levels increase nearly 3-fold.20–22 The latter phenomenon is ascribed to be the result of the role SphK2 plays in the clearance of circulating S1P. In short, Sph formed at the hepatocyte membrane is captured by SphK2-mediated phosphorylation and subsequently degraded via S1P lyase. The absence of SphK2 activity thus permits excess Sph to be captured by SphK1-mediated phosphorylation, which is prominent in red blood cells and thereby increasing circulating S1P levels.23 There are suggestions in the literature that inhibiting SphK activity could be beneficial,5,16,24,25 thus prompting a search for reliable SphK1-selective, SphK2-selective, and dual SphK1/SphK2 inhibitors. While potent and selective inhibitors of SphK1 and SphK2 have been reported, potent dual SphK1/SphK2 inhibitors are still needed (Fig. 2A). Among these, 1 (PF-543) is the most potent inhibitor of human SphK1 disclosed. Inhibitor 1 was developed by Schnute and coworkers at Pfizer via a lead hopping strategy of two high-throughput screening hits, 7 and 8 (Fig. 2B).26,27 In short, replacement of the 2-(hydroxymethyl)pyrrolidine head with other hydroxyl pyrrolidine moieties resulted in minimal loss of potency and selectivity for SphK1. Ultimately, it was determined that the 2-(hydroxymethyl)pyrrolidine head of 1 was the most preferred because of its potency in vitro. However, despite its impressive in vitro properties, the need for a continuously administered dose via an implanted osmotic pump device might be an indicator of a short compound half-life, low metabolic stability, or overall poor pharmacokinetic characteristics in vivo. Nonetheless, 1 remains an excellent tool for probing the effects of human SphK1 inhibition in vitro and could serve as a template for the development of potent SphK1/SphK2 dual inhibitors. We herein present the design and synthesis of novel analogues of 1 (PF-543) that retain the 2-(hydroxymethyl)pyrrolidine head while incorporating various lipophilic tail moieties with the interest of transforming SphK1-selective derivatives into potent SphK1/SphK2 dual inhibitors.
Fig. 2.
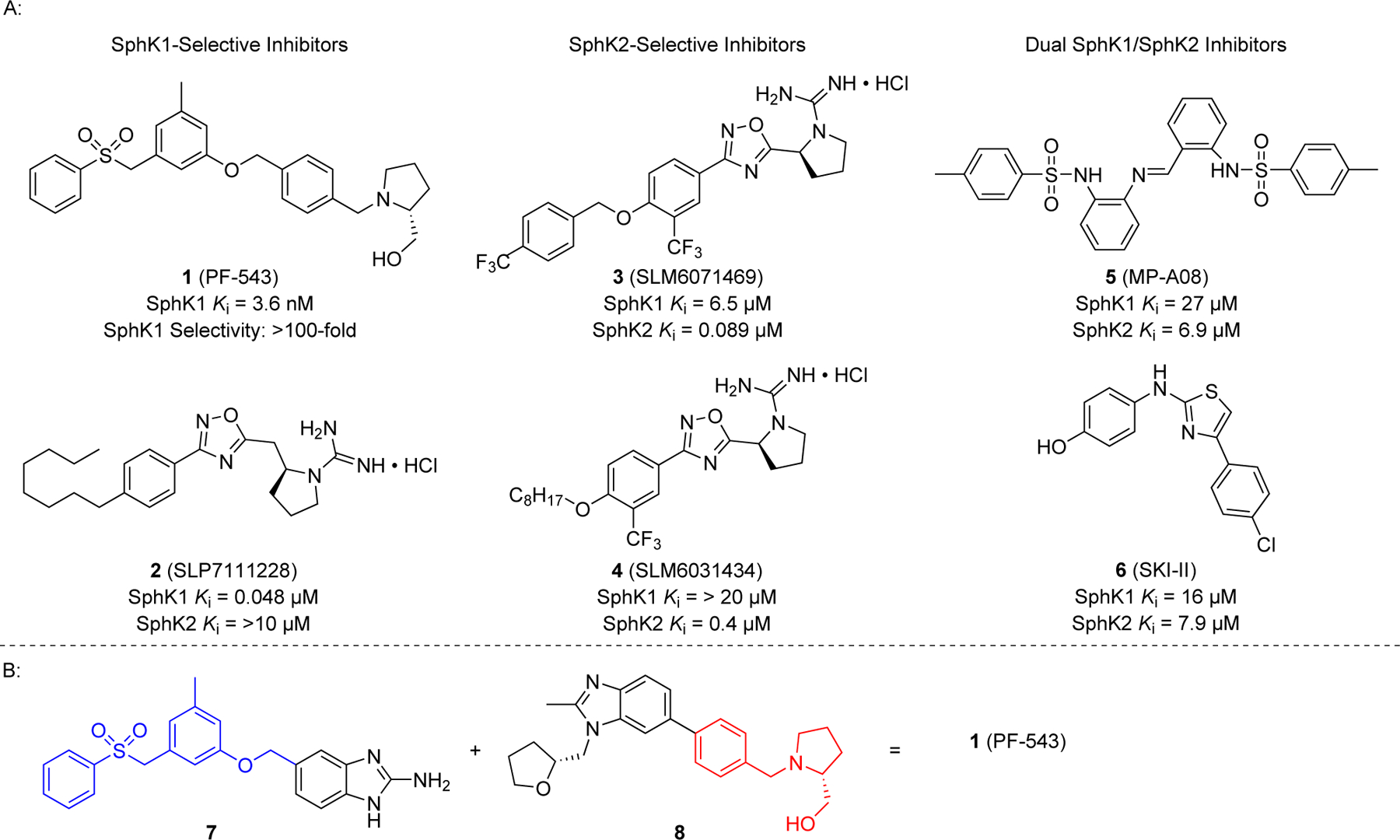
Structures of (A) select SphK inhibitors and (B) lead hopping combination of hit compounds 7 and 8 to generate 1 (PF-543).
2. Results and discussion
2.1. Inhibitor design
Previous work conducted by Schnute and coworkers revealed that replacement of the 2-(hydroxymethyl)pyrrolidine head of 1 resulted in minimal improvements in inhibitor efficacy toward SphK1, with 2-(hydroxymethyl)pyrrolidine being the most preferred.27 Furthermore, molecular modeling studies conducted with 1 docked in the substrate binding pocket of SphK1 revealed that 2-(hydroxymethyl)pyrrolidine head strongly hydrogen bonds with amino acid Asp178, a vital residue for SphK1 substrate recognition, thus explaining its impressive potency and selectivity for the type 1 kinase isoform.28 However, alterations to the aryl sulphonyl tail of 1 resulted in differentiating SphK1/SphK2 inhibitor activity.26,27 Based from these observations, further structural modifications on the scaffold of 1 must retain the 2-(hydroxymethyl) pyrrolidine head to preserve inhibitor SphK1 activity. In this study, various lipophilic substituents (i.e. alkyl, aryl, alkoxy, etc.) were incorporated into the scaffold of 1 in place of the aryl sulphonyl moiety. It was our rational that inclusion of lipophilic moieties that mimic the substrate sphingosine will be tolerated in the enzyme binding pockets of SphK1 and 2. With this approach, we aimed to develop dual SphK1/SphK2 inhibitors.
2.2. Chemistry: Synthesis of 2-(hydroxymethyl)pyrrolidine derivatives
The synthesis of compounds 12a-e, 18a-i, 22a-h, 25a-b, and 30a-c are shown in Schemes 1–3. Compound 9 is available for purchase from commercial sources. As shown in Scheme 1, benzaldehyde intermediates 10a-e were synthesized via a reaction of 9 in the presence of potassium carbonate and various alkyl bromides while refluxed in DMF for 12 h. Subsequently, prolinol intermediates 11a-e were generated by reductive amination utilizing 2-(hydroxymethyl)pyrrolidine. Lastly, a mixture of free amine intermediates 11a-e with HCl in methanol was performed to yield the desired alkoxy analogues 12a-e as HCl salts.
Scheme 1.
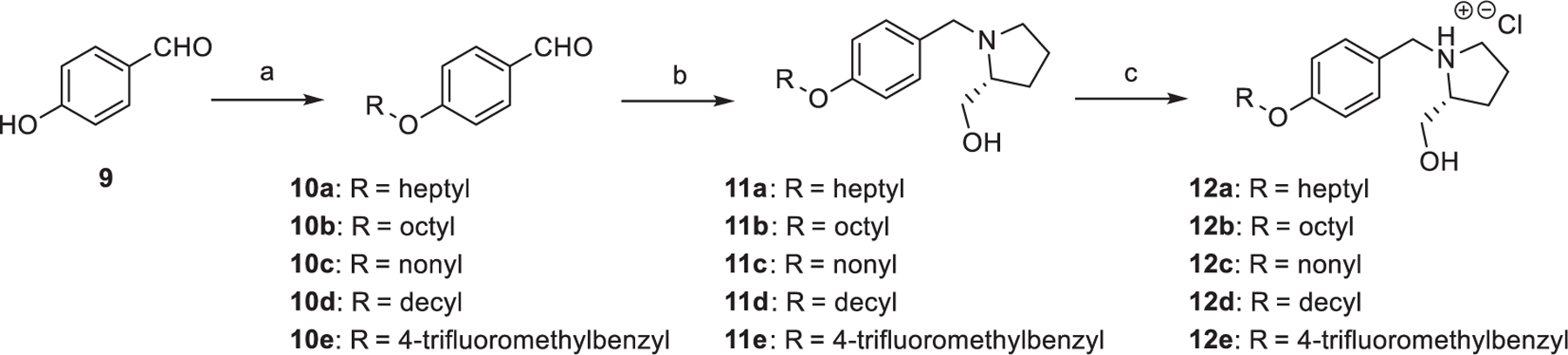
Synthesis of analogues 12a-f. Reagents and conditions: (a) Alkyl bromide derivative, K2CO3, DMF, reflux, 12 h; (b) (R)-Prolinol, TsOH⋅H2O, NaCNBH3, molecular sieves, CH2Cl2, rt, 12 h; (c) HCl (g), MeOH, rt, 10 min.
Scheme 3.
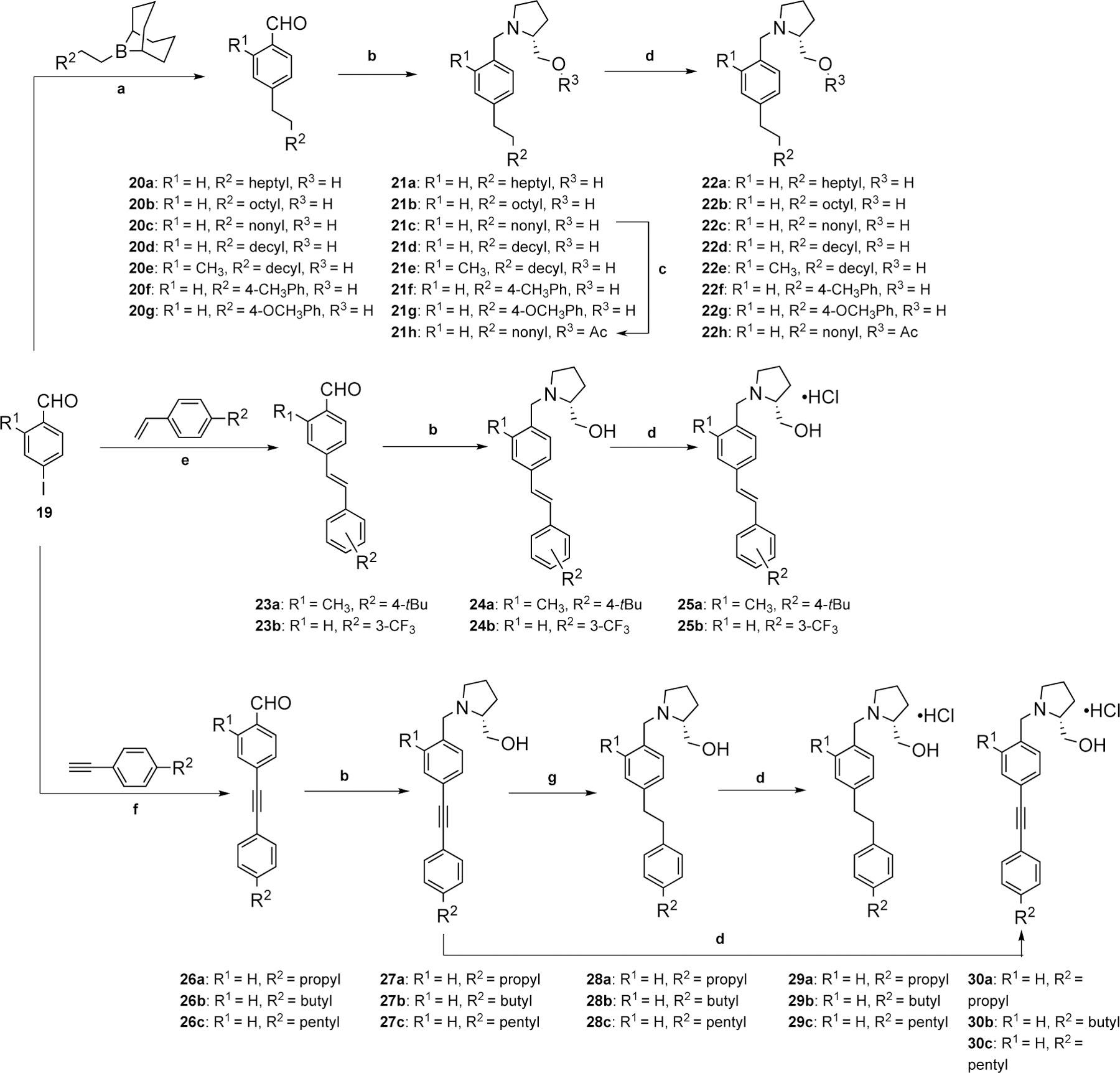
Synthesis of analogues 22a-h, 25a-b, 29a-c, and 30a-c. Reagent and conditions: (a) (i) Alkene derivative, 9-BBN, THF, 100 °C, 30 min, microwave; (ii) Benzaldehyde derivative, Pd(dppf)Cl2, NaOH, THF/H2O, 100 °C, 30 min, microwave; (b) (R)-Prolinol, NaCNBH3, TsOH⋅H2O, MeOH, rt, 12 h; (c) Acetyl chloride, pyridine, rt, 16 h; (d) HCl (g), MeOH, rt, 10 min; (e) Styrene derivative, Pd(PPh3)2Cl2, K2CO3, DMF, reflux, 16 h; (f) Phenylacetylene derivative, Pd(PPh3)2Cl2, CuI, TEA, THF, 50 °C, 1 h, microwave; (g) H2 (g), 10% Pd/C, rt, 1 h.
The synthesis of diaryl ether analogues 18a-i was completed as shown in Scheme 2. Execution of a nucleophilic aromatic substitution reaction of commercially available 4-fluorobenzonitrile with assorted aryl alcohols (13a-e) and cesium carbonate was completed to afford benzonitrile intermediates 14a-e. Next, nitrile reduction was facilitated with DIBAL-H to afford the corresponding benzaldehyde intermediates 15a-e. Subsequently, 15a-c were carried forward to a reductive amination reaction to afford free amine intermediates 17a-c. In contrast, benzaldehyde intermediates 15d and 15e were utilized for Suzuki-Miyaura coupling reactions with various aryl boronic acids to yield intermediates 16d-i. Afterward, compounds 16d-i were employed for a reductive amination reaction to afford free amine intermediates 17d-i. Finally, analogues 17a-i were combined with HCl in methanol to afford the corresponding diaryl ether derivatives 18a-i as HCl salts.
Scheme 2.
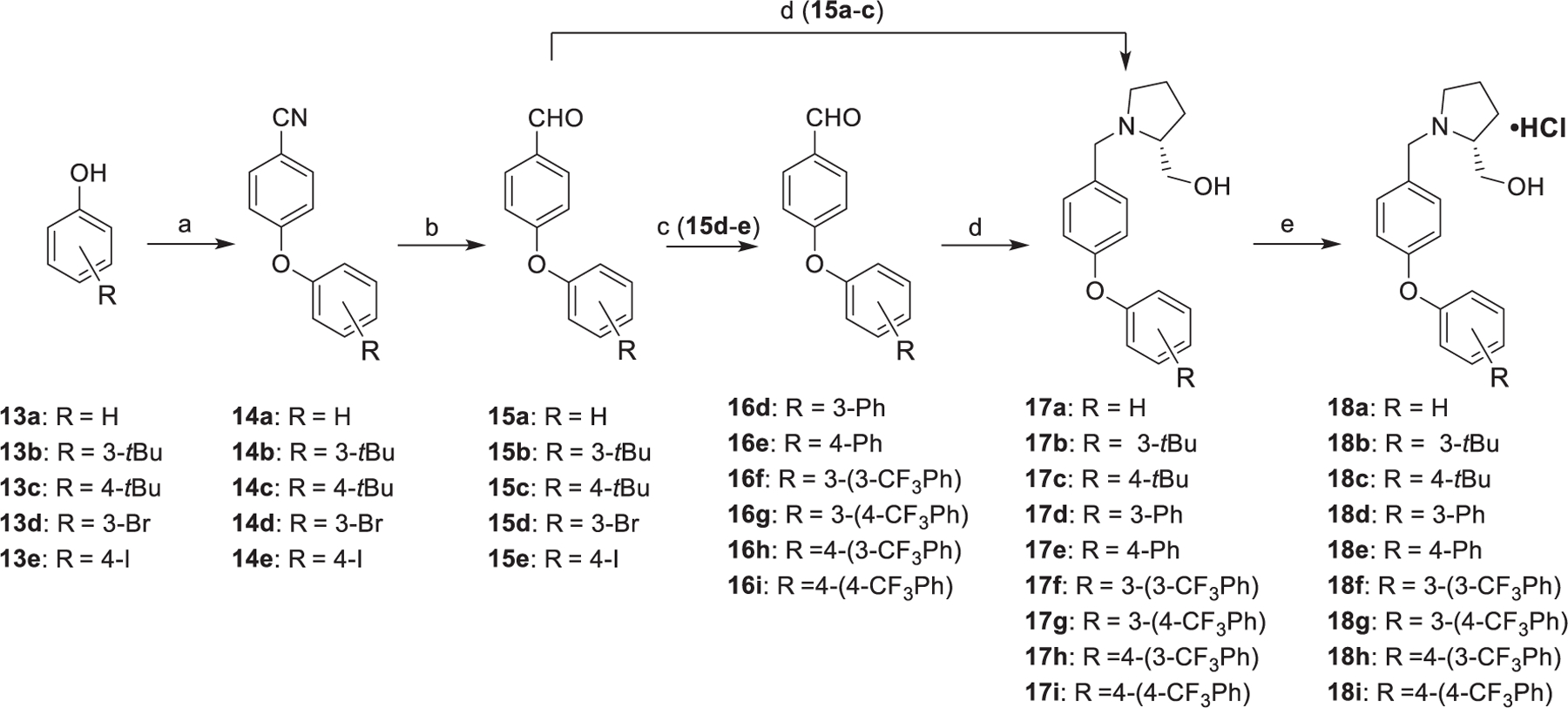
Synthesis of analogues 18a-i. Reagent and conditions: (a) 4-fluorobenzonitrile, Cs2CO3, DMF, reflux, 12 h; (b) DIBAL-H, CH2Cl2, − 70 °C to rt, 4 h; (c) Arylboronic acid derivative, Pd(dppf)Cl2, NaOH, THF/H2O, 100 °C, 30 min, microwave; (d) (R)-Prolinol, NaCNBH3, TsOH⋅H2O, MeOH, rt, 12 h; (e) HCl (g), MeOH, rt, 10 min.
The synthesis of alkyl analogues 22a-h, alkenyl analogues 25a-b, and alkynyl analogues 29a-c, and 30a-c is outlined in Scheme 3. Compound 19 is available for purchase from commercial sources. A Suzuki-Miyaura coupling reaction with various substituted borane derivatives was completed to generate benzaldehyde intermediates 20a-g. The production of 2-(hydroxymethyl)pyrrolidine intermediates 21a-g was accomplished via reductive amination. Completion of an acylation reaction with free amine 21c and acetyl chloride was performed to produce acetoxy derivative 21 h. Lastly, intermediates 21a-h were added to a mixture of HCl and methanol to produce alkyl derivatives 22a-h as HCl salts. The synthesis of alkenyl benzaldehyde intermediates 23a-b was completed via a Heck coupling reaction with 19 and various substituted styrene derivatives. Then, installation of the pyrrolidine head was facilitated via reductive amination to produce 24a and 24b. Lastly, intermediates 24a and 24b were added to a mixture of HCl and methanol to produce alkenyl analogues 25a and 25b as HCl salts. The production of alkynyl benzaldehyde intermediates 26a-c was accomplished via a Sonogashira coupling reaction with 19 and various substituted phenylacetylene derivatives. Reductive amination of the benzaldehyde was completed to produce pyrrolidine intermediates 27a-c, which upon catalytic hydrogenation of the internal triple bond generated 28a-c. Intermediates 27a-c were also added to a mixture of HCl and methanol to produce alkynyl analogues 30a-c as HCl salts. Likewise, reduced intermediates 28a-c were added to a mixture of HCl and methanol to produce alkane analogues 29a-c as HCl salts.
2.3. Biological evaluation of derivatives
This goal of this investigation was to develop a potent dual SphK1/SphK2 inhibitor whose design was inspired from key structural components of 1 and Sph. A library of analogues that contain an 2-(hydroxymethyl)pyrrolidine head moiety and various lipophilic tail substituents (i.e. alkyl, aryl, alkoxy, etc.) were synthesized and evaluated according to a previously described protocol.29,30 In short, this assay utilizes recombinant SphK1 or SphK2 and γ-[32P]ATP to screen and rapidly identify inhibitors suitable for further evaluation. All compounds were assayed at 1.0 μM for SphK1 and 0.3 μM for SphK2. Previous inhibitors developed in our group bearing an alkoxy moiety have shown success in inhibiting SphKs but with a guanidine head group.31,32 Thus, alkylether compounds bearing heptyloxy (12a), octyloxy (12b), nonyloxy (12c), and decyloxy (12d) tails were synthesized and assayed against SphK1 and 2 (Table 1). Incorporation of a heptyloxy (12a) tail resulted in poor SphK1 and 2 inhibition. Further extension of the alkoxy tail (12b-d) displayed an alkyl chain length effect on dual SphK1/SphK2 inhibition, with the nonyloxy (12c) variant indicating to be the optimal length and inhibiting SphK1 and SphK2 activity by 84% and 44% respectively. To further explore tolerated changes on this region, we synthesized a 4-trifluoromethylbenzoxy tail (12e) based on our previous investigations.18 However, 12e displayed weak SphK inhibition. Next, the effect of substitution with a much bulkier aryl substituent (18a-i) was explored to probe the size of the ligand docking space as well as potential π-π stacking or hydrophobic interactions with nearby active site residues. Compared to alkoxy derivative 12c, the inclusion of a phenoxy tail (18a) resulted in poor SphK1 and 2 inhibition. Analogues bearing a tert-butyl (18b) and phenyl groups (18d) at the meta-position also gave minimal enzyme inhibition. This is thought to be the result of the acute bond angle caused by the 1,3-disubstitution pattern most likely resulting in steric clash inside the binding pocket. When tert-butyl (18c) and phenyl (18e) moieties were migrated to the para-position, both compounds displayed a moderate increase in potency towards both kinase isoforms; however, they were still roughly half as potent as 12c. The effect of an additional trifluoromethyl group on the terminal phenyl ring was explored with analogues 18f-i. Of these compounds, only 18 h displayed significant inhibitory activity (SphK1 = 70%, SphK2 = 52%), indicating that meta-CF3 substitution on the terminal phenyl ring plays an important role for favorable binding. Next, to mimic the lipophilic tail of Sph, analogues with alkyl tails were synthesized in varying lengths (22a-e, and 22 h). Interestingly, elongation of the alkyl tail had a positive impact on SphK1 activity with 22b-22d having 76% inhibition. 22b and 22d had strong inhibitory activity against SphK2. We also introduced a methyl group on the internal phenyl ring (22e) but without improvement in activity. We next investigated inserting a phenyl ring onto the alkyl chain with compounds 22f, 22 g, and 29a-c. These analogs showed no improvement in activity. Analogues with rigid tail groups bearing internal double or triple bonds were next evaluated (25a-b, 30a-c). These were likewise ineffective inhibitors of SphKs.
Table 1.
Activity of sphingosine kinase inhibitors.a
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Cmpd | R1 | R2 | R3 | % SphK1 Inhibition | % SphK2 Inhibition | Cmpd | R1 | R2 | R3 | % SphK1 Inhibition | % SphK2 Inhibition |
| 12a |
|
H | H | 5 ± 2 | 0 ± 4 | 22a |
|
H | H | 62 ± 4 | 44 ± 3 |
| 12b |
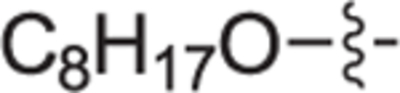
|
H | H | 36 ± 1 | 1 ± 2 | 22b |
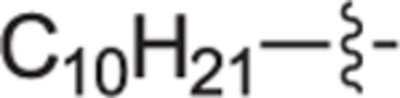
|
H | H | 76 ± 2 | 54 ± 1 |
| 12c |
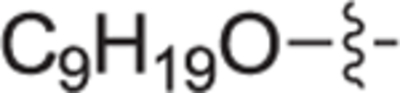
|
H | H | 84 ± 2 | 44 ± 3 | 22c |
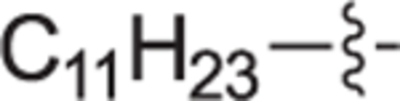
|
H | H | 76 ± 3 | 18 ± 7 |
| 12d |
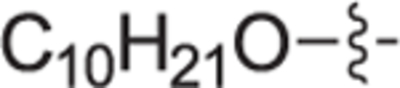
|
H | H | 62 ± 6 | 11 ± 1 | 22d |
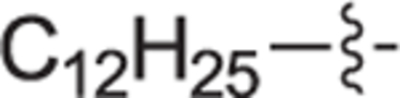
|
H | H | 78 ± 2 | 37 ± 5 |
| 12e |
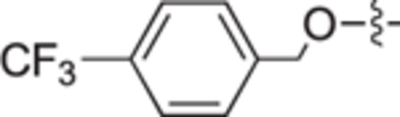
|
H | H | 14 ± 4 | 0 ± 7 | 22e |
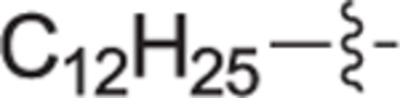
|
CH3 | H | 67 ± 2 | 38 ± 3 |
| 18a |
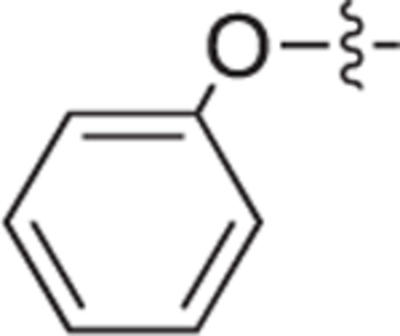
|
H | H | 4 ± 7 | 13 ± 4 | 22h |
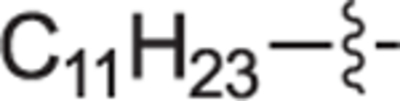
|
H | Ac | 47 ± 6 | 26 ± 6 |
| 18b |
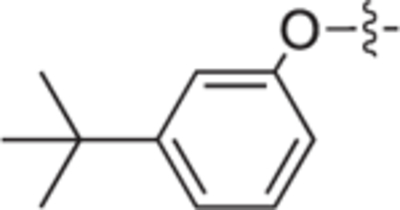
|
H | H | 0 ± 4 | 11 ± 5 | 22f |
|
H | H | 4 ± 4 | 3 ± 5 |
| 18c |
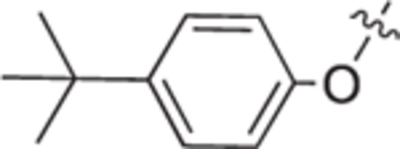
|
H | H | 42 ± 4 | 22 ± 1 | 22g |
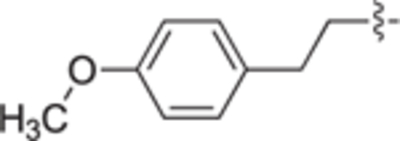
|
H | H | 8 ± 7 | 2 ± 2 |
| 18d |
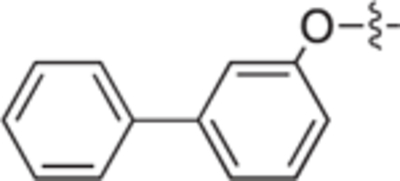
|
H | H | 7 ± 5 | 0 ± 1 | 29a |
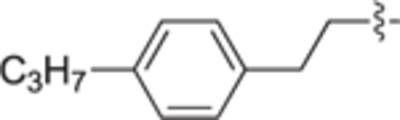
|
H | H | 29 ± 8 | 3 ± 4 |
| 18e |
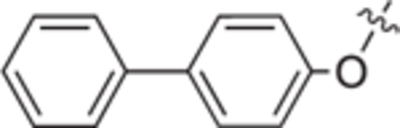
|
H | H | 35 ± 3 | 9 ± 6 | 29b |
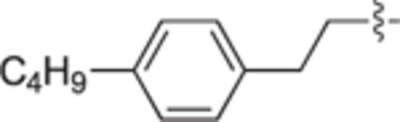
|
H | H | 31 ± 4 | 0 ± 3 |
| 18f |
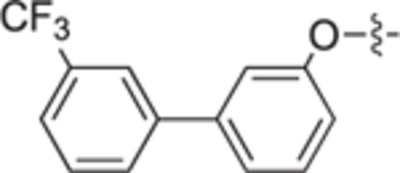
|
H | H | 19 ± 8 | 11 ± 2 | 29c |
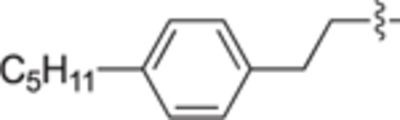
|
H | H | 36 ± 3 | 15 ± 5 |
| 18g |
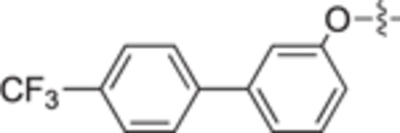
|
H | H | 12 ± 1 | 1 ± 1 | 25a |
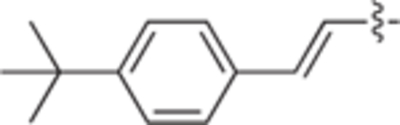
|
CH3 | H | 23 ± 3 | 0 ± 1 |
| 18h |
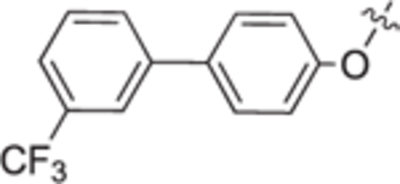
|
H | H | 70 ± 5 | 52 ± 2 | 25b |
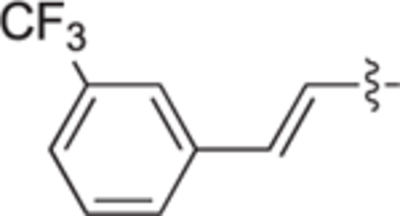
|
H | H | 45 ± 1 | 13 ± 5 |
| 18i |
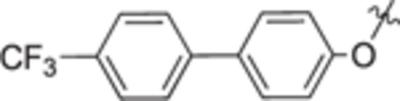
|
H | H | 7 ± 7 | 0 ± 5 | 30a |
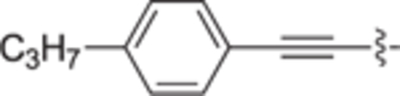
|
H | H | 16 ± 1 | 0 ± 4 |
| 30b |
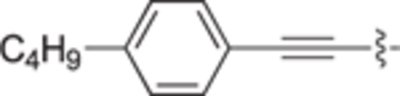
|
H | H | 53 ± 4 | 25 ± 1 | ||||||
| 30c |
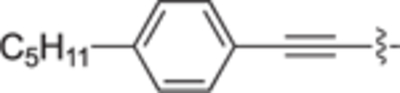
|
H | H | 49 ± 1 | 19 ± 2 | ||||||
SphK inhibition is presented as % of control (no inhibitor added). Recombinant human SphK1 and SphK2 were isolated from cell lysates. Enzyme activity was measured for SphK1 and SphK2 with 10 μM and 5 μM sphingosine respectively, and 250 μM γ-[32P] ATP. Compounds were assayed at 1.0 μM for SphK1 and 0.3 μM for SphK2 in triplicate.
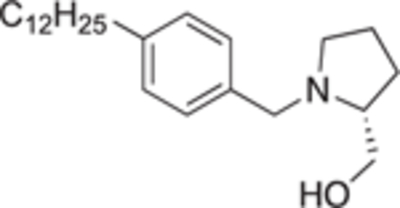
Because the initial screen utilized 0.3 μM for SphK2 and 1.0 μM for SphK1 inhibitor concentration, the apparent low activity with SphK2 is expected to increase when extrapolated to 1 μM. To confirm SphK inhibition, select compounds that displayed the greatest dual SphK1 (>70%) and SphK2 (>18%) inhibitory activity were subjected to a cell-based assay that uses the budding yeast Saccharomyces cerevisiae as a platform for assessing inhibitors of human SphK1 and 2 (hSphK1 and 2).33 In short, this assay exploits the observed toxicity of increased concentrations of phosphorylated long chain bases (LCBs), such as S1P, towards S. cerevisiae. Rescue of a genetically modified (to enhance phospho-LCB toxicity) yeast cell strain harboring plasmids expressing either hSphK1 or 2 in the presence of select inhibitors can be accomplished in a dose dependent fashion by measuring the growth of yeast culture. As shown in Table 2, top performing compounds 18 h and 22b-d were assayed with S. cerevisiae and EC50 values towards SphK1 and SphK2 were determined. After consideration of metabolic stability, compound 12c was abandoned because of a structural alert for the formation of iminomethide. With regard to hSphK1 in yeast cells, the dodecyl (22d) analogue was the most potent inhibitor with an EC50 of 154 nM, followed by decyl (22b), undecyl (22c), and diaryl ether (18 h), respectively, with EC50 values ranging from 202 nM to 299 nM. When tested against hSphK2, undecyl analogue 22c and 22d had EC50 values within experimental error of 246 ± 21 and 290 ± 21 nM, respectively. Taken altogether, inhibitor 22d was determined to be the most effective dual SphK1/SphK2 inhibitor tested. Subsequently, an inhibitory constant (Ki) was determined for the compound 22d for both SphK1 and 2 (Table 2).21 Dodecyl analogue 22d displayed good potency (SphK1 Ki = 679 nM, SphK2 Ki = 951 nM) towards both enzyme isoforms with a slight selectivity (1.4-fold) for SphK1.
Table 2.
| Cmpd | Structure | hSphK1 EC50 (nM) | hSphK2 EC50 (nM) | hSphK1 KI (nM) | hSphK2 KI (nM) |
|---|---|---|---|---|---|
| 18h |
|
299 ± 19 | 488 ± 36 | – | – |
| 22b |
|
202 ± 31 | 401 ± 50 | – | – |
| 22c |
|
276 ± 22 | 246 ± 21 | – | – |
| 22d |
|
154 ± 11 | 290 ± 30 | 679 ± 12 | 951 ± 32 |
EC50 values of various compounds were calculated from the growth curves of yeast strain KYA1 encoding either hSphK1 or hSphK2 in the presence of inhibitor over 24 h. By way of comparison, the EC50 at hSphK1 for 1 (PF-543) in this assay is 5700 nM23. For detailed assay conditions, see the experimental section.
Inhibitory constants for recombinant enzymes were obtained by kinetic analysis of S1P production using variable concentration of sphingosine and a fixed concentration of ATP in the presence or absence of compounds.
To further validate the SphK inhibitory activity, compound 22d was incubated with mammalian U937 cells to determine the effect on S1P synthesis (Fig. 3). U937 cells are a histiocytic lymphoma myeloid cell line that express both SphK1 and SphK2. In this assay, cells were incubated with inhibitor, lysed, and S1P levels were determined by LC-MS/MS. To our delight, administration of 22d from 0.1 μM to 1.0 μM resulted in a concentration-dependent reduction of S1P levels, thus indicating dose-dependent SphK inhibition. Taken together, the activity of 22d in both the yeast assay as well as in U937 cells provide strong support for dual SphK inhibition.
Fig. 3.
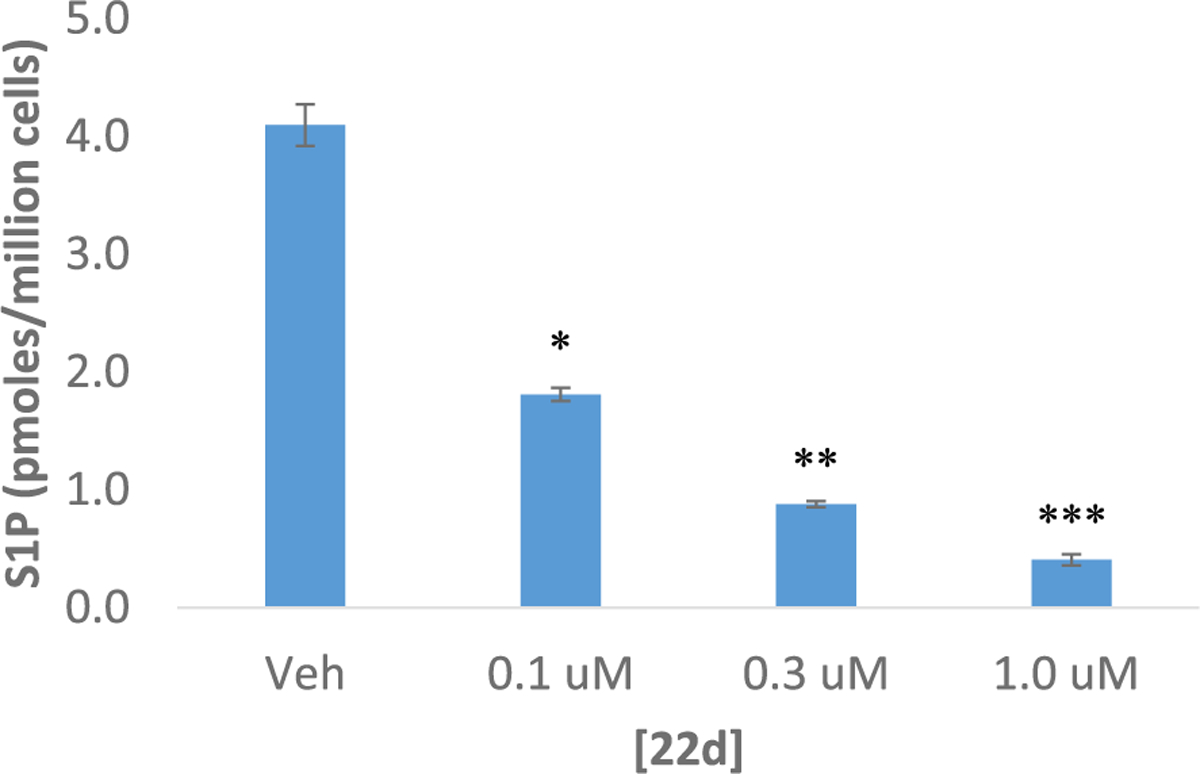
The effect of 22d on S1P concentrations in U937 cells. After a 2.0 h incubation, U937 cells were harvested by centrifugation and lysed, and levels of S1P were measured using LC-MS/MS. Amounts associated with cells are expressed as the number of picomoles per 106 cells. The experiment was performed in duplicate. The level of significance is indicated for each experiment (*P < 0.05, **P < 0.005, ***P < 0.001) using an unpaired t-test (compared to control).
2.4. Molecular modeling of 22d in the active site of hSphK1 and hSphK2
Molecular docking of 22d into the Sph binding site of SphK1 and SphK2 was performed to provide insight into the intermolecular interactions within the substrate binding pocket (Fig. 4). Compound 22d fits in the Sph binding pocket of SphK1 that favors strong hydrogen bond formation between the 2-(hydroxymethyl)pyrrolidine moiety: (i) the primary alcohol hydrogen bond (3.0 Å) with Ser168 and (ii) the tertiary nitrogen hydrogen bond (3.6 Å) with Asp178 (Fig. 4A). This is in contrast with the co-crystal structure of PF-573 bound to SphK128 where the Asp178 carboxylate moiety to participates in hydrogen bonding with both the primary alcohol and tertiary nitrogen. The change in the positioning between 22d and PF573 likely results from the rearrangement of the tail groups within the binding pocket. Nonetheless, this data suggests the 2-(hydroxymethyl)pyrrolidine head group within our scaffold is vital for compound binding affinity and its corresponding observed inhibitory activity towards SphK1. As shown in Fig. 4C, docking of 22d into the homology model of SphK218,19 reveals a similar hydrogen bond between the pyrrolidine nitrogen to Asp308 and hydroxyl group to Ser298. We hypothesize that the similar binding pose around this region is the structural basis for the dual inhibitory of 22d. In addition to the 2-(hydroxymethyl)pyrrolidine head, hydrophobic interaction is observed between 22d and Ile174 of SphK1 (Fig. 4A). The lack of a large, bulky moiety in the central region of our scaffold contributes to the slight observed SphK1 selectivity given that bulkier substituents have demonstrated to be better tolerated in the hydrophobic core of the Sph binding cavity of SphK2 rather than SphK1. This is due to the presence of the smaller Val304 residue in SphK2 (Ile174 in SphK1) that allows inhibitors bearing larger moieties on this portion of the scaffold to migrate deeper into the binding pocket of SphK2.18 Indeed, the dodecyl alkyl tail of compound 22d is posed in a “J-shape” conformation that has been observed in SphK1 structures crystallized with various inhibitors.34,28 Fig. 4B and D show the Van der Waals radii of 22d within the SphK1 and SphK2 binding pocket wherein a larger volume for SphK2 accommodates the aliphatic tail.
Fig. 4.
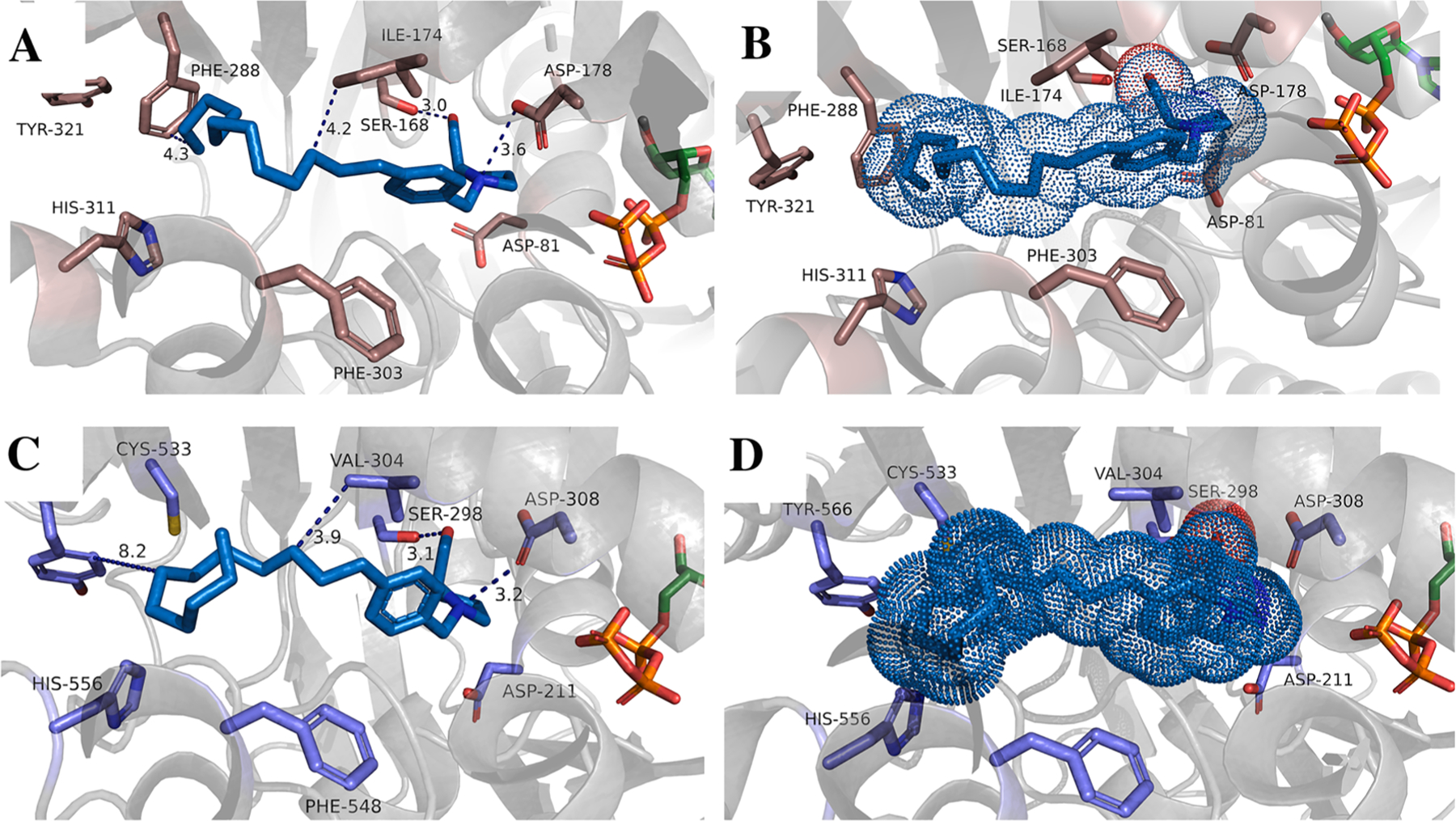
Docking of inhibitor 22d into hSphK1 (A, B) and hSphK2 (C, D). Key residues in the binding pocket are represented by grey sticks and are labeled. ATP is shown in orange and colored by element. The SphK1 and SphK2 protein structures are depicted in grey cartoon. Distances between interacting atoms are shown as dashed lines. Inhibitors are shown as stick and colored by element. Panels B and D illustrate the inhibitor as Van der Waals radii in dots to represent volume occupancy of the inhibitor.
3. Conclusions
The S1P pathway remains an intriguing target for pharmacological intervention. The biologic role S1P and its generative enzymes SphK1 and SphK2 play in various diseases states have been the subject of intense investigation over the last two decades. While potent and selective inhibitors of SphK1 and SphK2 have been reported, development of potent dual SphK1/SphK2 inhibitors are still needed. Particularly regarding SphK1, many biologically active inhibitors have been developed with the 2-(hydroxymethyl)pyrrolidine-based inhibitor 1 being the most potent (SphK1 Ki = 3.6 nM) to date. Towards that end, we utilized compound 1 as a template and performed a structure–activity relationship profiling of 2-(hydroxymethyl)pyrrolidine-based dual inhibitors of SphK1 and 2 with 22d being the most effective dual SphK1/SphK2 inhibitor (SphK1 Ki = 679 nM, SphK2 Ki = 951 nM) reported in this series. Biological assessment of 22d demonstrated success in reducing measured concentrations of S1P in both S. cerevisiae and U937 cell lines. Furthermore, molecular modeling studies suggest that hydrogen bonding of Ser298 and Asp278 with the 2-(hydroxymethyl) pyrrolidine moiety provides the structural basis for dual inhibitory activity at SphK1 and SphK2. Collectively, these studies give insight into the role 2-(hydroxymethyl)pyrrolidine-based inhibitors play inside the SphK1 and SphK2 active sites and provide a foundation for the development of future dual SphK1/SphK2 inhibitors designed to target and manipulate the S1P signaling pathway.
4. Experimental
4.1. Biological assays and molecular modeling
4.1.1. Recombinant hSphK1 and hSphK2 cell lysate assay
The percent inhibition of SphK1 from synthesized compounds was carried forward using a previously described protocol.30,35 Recombinant human SphK1 (10 μM) and human SphK2 (5 μM) was isolated from a SF9 cell lysate expressing respective plasmid DNAs and incubated with (1.0 μM for SphK1 and 0.3 μM for SphK2) or without compound, sphingosine, and 250 μM γ-[32P] ATP via scintillation counting. SphK activity was determined by the amount of γ-[32P]-S1P as a function of inhibitor concentration. Compounds were assayed in triplicate.
4.1.2. Recombinant yeast assay
The growth of recombinant yeast (Saccharomyces cerevisiae) cells encoding either hSphK1 or hSphK2 was performed according to our previously described protocol.33 Briefly, yeast strain KYA1 harboring plasmid pGAL-HsSPHK1 encoding hSphK1, or plasmid pGAL-HsSPHK2 encoding hSphK2 was selected, maintained and grown in SC-URA media with 2% glucose overnight at 30 °C. Following overnight growth media was diluted 1:100 into SC-URA media supplemented with 2% galactose and various concentrations of test inhibitor. After another incubation period of 24–48 h at 30 °C, cellular growth was quantified by measuring absorbance at 600 nm and EC50 values were calculated from the inhibitor concentration-absorbance curve.
4.1.3. U937 cell culture assay
The growth of U937 cells was conducted in RPMI 1640 media supplemented with (L)-glutamate, 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C in an atmosphere containing 5% CO2. Growth media was replaced with media containing 0.5% FBS 24 h prior to inhibitor introduction. After, cells were treated with compound 22d for 2 h before being harvested and lipids extracted for LC-MS/MS analysis as described previously.29
4.1.4. Molecular modeling
To visualize and rationalize the observed biological efficacy of 22d, molecular docking was utilized to predict position and ligand–protein interactions of the inhibitor in the sphingosine binding pocket of SphK1 and SphK2. The SphK1 and 2 model, with ATP and Mg2+ bound, was generated with Molecular Operating Environment (MOE) and energy minimized as previously described.32,19 In order to draw, display, and characterize chemical structures, substructures, and reactions for preparation in docking programs, Marvin 17.3.13, 2017 was used (ChemAxon; http://www.chemaxon.com) to create structure files that were cleaned in 3D. AutoDock Tools was used to prepare the protein and ligand files.36 In order to perform the docking, AutoDock Vina was utilized to dock the inhibitor to SphK1 or 2, with 9 poses being created for each inhibitor.37 The grid box was set to 20 X 20 X 28 Å3 with grid spacing of 1.000 Å. To include all known key residues in the sphingosine binding cavity, the grid box was positioned at the approximate center of the ligand-binding cavity based on the position of sphingosine in the crystal structure (PDB ID: 3VZB).34,32 The lowest energy docked pose for the inhibitor was then used to visualize and rationalize the observed biological efficacy towards SphK1 and SphK2.
4.2. Chemistry
4.2.1. General materials and methods
All solvents were dried using a PureSolv solvent drying system prior to use. All chemical reagents were purchased from commercial sources and used without further purification. Flash column chromatography was performed using a Combiflash Rf purification system with flash grade 40–63 μm silica gel. Where indicated, all reactions utilizing a microwave reactor were conducted with a Discover SP microwave synthesizer (CEM Corporation). 1H NMR spectra were obtained with either a Bruker Advance-II 500 MHz or Agilent 400-MR 400 MHz spectrometer. Chemical shifts are reported in ppm with the solvent resonance as the internal standard (CDCl3:7.26 ppm, CD3OD: 3:31 ppm). 13C NMR spectra were obtained with either a Bruker Advance II 500 MHz or Agilent 400-MR 400 MHz spectrometer with complete proton decoupling. 13C NMR chemical shifts are reported in ppm with the solvent resonance as the internal standard (CDCl3: 77.2 ppm, CD3OD: 49.0 ppm). High-resolution mass spectrometry (HRMS) was performed on an LC-MS time-of-flight mass spectrometer by electrospray ionization (ESI).
4.2.2. General procedure for the synthesis of compounds 10a-e
To a round bottom flask 4-hydroxybenzaldehyde (1.0 equiv.) was added to a solution of DMF, potassium carbonate (5.0 equiv.) and the appropriate alkyl halide (1.2 equiv.) followed by flushing the reaction container with nitrogen gas. The reaction mixture was refluxed at 100 °C for 16 h until TLC indicated the starting material had been fully consumed. Subsequently, the resulting solution was partitioned between EtOAc and LiBr aqueous solution. Using additional EtOAc, the aqueous LiBr solution was washed three times and the combined organic layers were dried over Na2SO4, filtered, and concentrated via vacuum. The resulting concentrate was purified by silica gel chromatography.
4.2.2.1. 4-(heptyloxy)benzaldehyde (10a).
Clear oil, yield 87%. 1H NMR (400 MHz, CDCl3) δ: 9.85 (s, 1H), 7.80 (d, J = 8.9 Hz, 2H), 6.96 (d, J = 8.9 Hz, 2H), 4.01 (t, J = 6.6 Hz, 2H), 1.83 –1.73 (m, 2H), 1.48 – 1.39 (m, 2H), 1.39 – 1.23 (m, 6H), 0.90 – 0.84 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 190.9, 164.4, 132.1, 129.8, 114.8, 68.5, 31.9, 29.2, 29.1, 26.0, 22.7, 14.2. HRMS (ESI + ): calcd for C14H21O2 [M + H]+ 221.1536; found, 221.1548.
4.2.2.2. 4-(octyloxy)benzaldehyde (10b).
Clear oil, yield 74%. 1H NMR (400 MHz, CDCl3) δ: 9.88 (s, 1H), 7.82 (d, J = 8.9 Hz, 1H), 6.99 (d, J = 8.7 Hz, 2H), 4.04 (t, J = 6.6 Hz, 2H), 1.87 –1.75 (m, 2H), 1.47 (dt, J = 15.2, 6.8 Hz, 2H), 1.40 – 1.21(m, 8H), 0.91 – 0.86(m, 3H). 13C NMR (101 MHz, CDCl3) δ: 190.9, 164.4, 132.1, 129.9, 68.6, 31.9, 29.4, 29.4, 29.2, 26.1, 22.8, 14.2. HRMS (ESI + ): calcd for C15H23O2 [M + H]+ 235.1693; found: 235.1707.
4.2.2.3. 4-(nonyloxy)benzaldehyde (10c).
Clear oil, yield 80%. 1H NMR (400 MHz, CDCl3) δ: 9.87 (s, 1H), 7.82 (d, J = 8.9 Hz, 2H), 6.98 (d, J = 8.8 Hz, 2H), 4.03 (t, J = 6.6 Hz, 2H), 1.86 – 1.75 (m, 2H), 1.50 – 1.40 (m, 2H), 1.40 – 1.21 (m, 10H), 0.92 – 0.83 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 190.9, 164.4, 132.1, 129.8, 114.9, 68.6, 32.0, 29.6, 29.5, 29.4, 29.2, 26.1, 22.8, 14.2. HRMS (ESI + ): calcd for C16H25O2 [M + H]+ 249.1849; found: 249.1865.
4.2.2.4. 4-(decyloxy)benzaldehyde (10d).
Clear oil, yield 72%. 1H NMR (400 MHz, CDCl3) δ: 9.87 (s, 1H), 7.82 (d, J = 8.9 Hz, 2H), 6.99 (d, J = 8.6 Hz, 2H), 4.03 (t, J = 6.6 Hz, 2H), 1.86 –1.76 (m, 2H), 1.51 – 1.41 (m, 2H), 1.29 (dd, J = 14.2, 5.5 Hz, 12H), 0.91 – 0.84 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 191.0, 164.4, 132.1, 129.9, 114.9, 68.6, 32.0, 29.7, 29.7, 29.5, 29.5, 29.2, 26.1, 22.8, 14.3. HRMS (ESI + ): calcd for C17H27O2 [M + H]+ 263.2006; found: 263.2029.
4.2.2.5. 4-((4-(trifluoromethyl)benzyl)oxy)benzaldehyde (10e).
White solid, yield 65%. 1H NMR (400 MHz, CDCl3) δ: 9.90 (s, 1H), 7.86 (d, J = 8.8 Hz, 2H), 7.67 (d, J = 8.1 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 7.08 (d, J = 8.8 Hz, 2H), 5.22 (s, 2H). 13C NMR (101 MHz, CDCl3) δ: 190.8, 163.4, 140.1, 132.2, 130.6, 127.5, 125.9, 125.9, 125.8, 125.8, 115.2, 69.5. HRMS(ESI + ): calcd for C30H22F6KO4 [2 M + K]+ 599.1054, found: 599.1084.
4.2.3. General procedure for the synthesis of compounds 11a-e, 17a-i, 21a-h, 24a-b, 27a-c
To a round bottom flask containing CH2Cl2 the appropriate aldehyde (1.0 equiv.), (R)-prolinol (1.3 equiv.) and 100 mg 4.0 Å molecular sieves were added followed by flushing the reaction container with nitrogen gas. The solution was allowed to stir at room temperature for 8 h before the addition of p-toluenesulfonic acid monohydrate (0.05 equiv.) and sodium cyanoborohydride (1.3 equiv.). The solution was stirred at room temperature under nitrogen for an additional 12 h. After, the solution was filtered with solids removed and partitioned between ethyl acetate and water. The organic layers were washed with saturated sodium bicarbonate solution and brine, dried over sodium sulfate, and concentrated via vacuum. The resulting concentrate was purified by silica gel chromatography and carried forward without characterization. Following silica gel chromatography, the corresponding ester (1.0 equiv.) was added to a round bottom flask containing THF and put under nitrogen gas. Next, the mixture was cooled in an ice bath for 5 min before the addition of lithium aluminum hydride (0.7 equiv.). The mixture was allowed to rise to room temperature and then refluxed for 10 h. Subsequently, the reaction was cooled in ice bath and quenched with 1 mL ethyl acetate and 20 mL saturated sodium hydroxide solution. The mixture was then extracted with ethyl acetate, washed with saturated sodium bicarbonate solution and brine, dried over sodium sulfate, and concentrated via vacuum. The concentrate was purified using silica gel column chromatography to yield the desired product.
4.2.3.1. (R)-(1-(4-(heptyloxy)benzyl)pyrrolidin-2-yl)methanol (11a).
White solid, yield 60%. 1H NMR (400 MHz, CDCl3) δ: 7.21 (d, J = 8.7 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H), 3.95 – 3.89 (m, 3H), 3.64 (dd, J = 11.0, 3.6 Hz, 1H), 3.46 (dd, J = 11.0, 2.8 Hz, 1H), 3.39 (d, J = 12.9 Hz, 1H), 3.00 (dt, J = 9.5, 4.6 Hz, 1H), 2.79 (td, J = 6.0, 3.1 Hz, 1H), 2.35 (q, J = 8.5 Hz, 1H), 1.99 – 1.87 (m, 1H), 1.86 – 1.79 (m, 1H), 1.79 – 1.66 (m, 4H), 1.49 – 1.39 (m, 2H), 1.38 – 1.24 (m, 6H), 0.93 – 0.85 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 158.6, 130.2, 130.0, 114.4, 68.0, 64.6, 61.8, 58.0, 54.3, 31.8, 29.1, 27.7, 26.1, 23.4, 22.7, 14.2. HRMS (ESI + ): calcd for C19H32NO2 [M + H]+ 306.2428, found: 306.2425.
4.2.3.2. (R)-(1-(4-(octyloxy)benzyl)pyrrolidin-2-yl)methanol (11b).
White solid, yield 61%. 1H NMR (400 MHz, CDCl3) δ: 7.19 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H), 3.93 (t, J = 6.6 Hz, 2H), 3.89 (d, J = 12.8 Hz, 1H), 3.64 (dd, J = 10.7, 3.5 Hz, 1H), 3.41 (dd, J = 10.7, 2.2 Hz, 1H), 3.31 (d, J = 12.8 Hz, 1H), 3.00 – 2.92 (m, 2H), 2.75 – 2.68 (m, 2H), 2.34 – 2.23 (m, 1H), 1.98 –1.85 (m, 1H), 1.85 – 1.73 (m, 3H), 1.73 – 1.60 (m, 2H), 1.45 (dt, J = 15.0, 6.9 Hz, 2H), 1.38 – 1.23 (m, 8H), 0.93 – 0.84 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 158.4, 131.2, 130.0, 114.4, 68.1, 64.2, 61.8, 58.0, 54.5, 32.0, 29.5, 29.5, 29.4, 28.0, 26.2, 23.6, 22.8. HRMS (ESI + ): calcd for C20H34NO2 [M + H]+ 320.2584, found: 320.2589.
4.2.3.3. (R)-(1-(4-(nonyloxy)benzyl)pyrrolidin-2-yl)methanol (11c).
White solid, yield 59%. 1H NMR (400 MHz, CDCl3) δ: 7.19 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H), 3.93 (t, J = 6.6 Hz, 2H), 3.88 (d, J = 12.8 Hz, 1H), 3.64 (dd, J = 10.7, 3.5 Hz, 1H), 3.42 (dd, J = 10.7, 2.2 Hz, 1H), 3.30 (d, J = 12.8 Hz, 1H), 3.00 – 2.91 (m, 1H), 2.75 – 2.67 (m, 1H), 2.28 (td, J = 9.4, 7.6 Hz, 1H), 1.99 – 1.82 (m, 1H), 1.85 – 1.61 (m, 3H), 1.69 (s, 2H), 1.45 (dt, J = 15.0, 7.1 Hz, 2H), 1.39 – 1.21 (m, 10H), 0.93 – 0.84 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 158.4, 131.1, 129.9, 114.4, 68.1, 64.2, 61.9, 58.0, 54.4, 32.0, 29.7, 29.5, 29.4, 29.4, 28.0, 26.2, 23.5, 22.8, 14.2. HRMS (ESI + ): calcd for C21H36NO2 [M + H]+ 334.2741, found: 334.2764.
4.2.3.4. (R)-(1-(4-(decyloxy)benzyl)pyrrolidin-2-yl)methanol (11d).
White solid, yield 58%. 1H NMR (400 MHz, CDCl3) δ: 7.36 (d, J = 8.7 Hz, 2H), 6.91 (d, J = 8.7 Hz, 2H), 4.39 (d, J = 13.1 Hz, 1H), 4.12 (d, J = 13.1 Hz, 1H), 3.92 (t, J = 6.6 Hz, 2H), 3.86 – 3.75 (m, 2H), 3.60 (m, 1H), 3.42 (m, 1H), 3.01 (m, 1H), 2.19 – 1.86 (m, 4H), 1.80 – 1.70 (m, 1H), 1.41 (m, 2H), 1.37 – 1.19 (m, 12H), 0.89 – 0.82 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 160.6, 132.4, 120.9, 115.4, 68.3, 68.0, 60.9, 53.9, 32.0, 29.7, 29.7, 29.5, 29.5, 29.3, 26.6, 26.2, 23.0, 22.8, 14.3. HRMS (ESI + ): calcd for C22H38NO2 [M + H]+ 348.2897, found: 348.2902.
4.2.3.5. (R)-(1-(4-((4-(trifluoromethyl)benzyl)oxy)benzyl)pyrrolidin-2-yl)methanol (11e).
White solid, yield 60%. 1H NMR (400 MHz, CDCl3) δ: 7.64 (d, J = 8.1 Hz, 2H), 7.54 (d, J = 8.7 Hz, 2H), 7.39 (d, J = 8.8 Hz, 2H), 7.00 (d, J = 8.8 Hz, 2H), 5.12 (s, 2H), 4.34 (d, J = 13.1 Hz, 1H), 3.97 (d, J = 13.1 Hz, 1H), 3.78 (d, J = 4.8 Hz, 2H), 3.45 (dq, J = 10.0, 4.9 Hz, 1H), 3.36 (dt, J = 11.0, 5.8 Hz, 1H), 2.88 (dt, J = 11.0, 7.3 Hz, 1H), 2.19 – 2.04 (m, 1H), 2.06 – 1.85 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 159.4, 140.7, 132.1, 127.6, 125.7, 115.6, 69.3, 67.6, 61.1, 58.8, 54.1. HRMS (ESI + ): calcd for C20H23F3NO2 [M + H]+ 366.1675, found: 366.1688.
4.2.3.6. (R)-(1-(4-phenoxybenzyl)pyrrolidin-2-yl)methanol (17a).
Clear oil, yield 53%. 1H NMR (400 MHz, CDCl3) δ: 7.33 (dd, J = 8.6, 7.4 Hz, 2H), 7.28 (d, J = 8.5 Hz, 2H), 7.13 – 7.07 (m, 1H), 7.01 (dd, J = 8.7, 1.1 Hz, 2H), 6.97 (d, J = 8.6 Hz, 2H), 3.98 (d, J = 13.0 Hz, 1H), 3.68 (dd, J = 10.9, 3.5 Hz, 1H), 3.47 (dd, J = 11.0, 2.6 Hz, 1H), 3.40 (d, J = 13.0 Hz, 1H), 3.03 (ddd, J = 9.5, 5.6, 3.8 Hz, 1H), 2.80 (ddt, J = 9.2, 6.1, 3.0 Hz, 1H), 2.35 (td, J = 9.2, 7.8 Hz, 1H), 2.01 – 1.90 (m, 1H), 1.89 – 1.80 (m, 1H), 1.78 – 1.70 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 157.3, 156.6, 133.4, 130.4, 129.9, 123.4, 119.0, 118.9, 64.7, 61.8, 58.1, 54.5, 27.8, 23.6. HRMS (ESI + ): calcd for C18H22NO2 [M + H]+ 284.1645, found: 284.1656.
4.2.3.7. (R)-(1-(4-(3-(tert-butyl)phenoxy)benzyl)pyrrolidin-2-yl)methanol (17b).
Clear oil, yield 61%. 1H NMR (400 MHz, CDCl3) δ: 7.32 (d, J = 8.5 Hz, 2H), 7.30 – 7.27 (m, 1H), 7.19 – 7.15 (m, 1H), 7.11 (dd, J = 2.4, 1.9 Hz, 1H), 6.99 (d, J = 8.5 Hz, 2H), 6.81 (dddd, J = 8.0, 2.4, 1.0, 0.4 Hz, 1H), 4.04 (d, J = 13.0 Hz, 1H), 3.72 (dd, J = 11.1, 3.4 Hz, 1H), 3.56 – 3.46 (m, 1H), 3.14 – 3.05 (m, 2H), 2.89 (td, J = 5.9, 3.0 Hz, 1H), 2.43 (dt, J = 9.5, 8.2 Hz, 1H), 2.03 – 1.94 (m, 1H), 1.91 – 1.86 (m, 1H), 1.82 – 1.75 (m, 2H), 1.33 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 157.1, 156.8, 153.7, 130.5, 129.3, 120.6, 118.5, 116.7, 115.9, 110.2, 65.0, 61.8, 58.2, 54.4, 34.9, 31.4, 27.7, 23.6. HRMS (ESI + ): calcd for C22H30NO2 [M + H]+ 340.2271, found: 340.2278.
4.2.3.8. (R)-(1-(4-(4-(tert-butyl)phenoxy)benzyl)pyrrolidin-2-yl)methanol (17c).
Clear oil, yield 56%. 1H NMR (400 MHz, CDCl3) δ: 7.37 (d, J = 2.1 Hz, 2H), 7.35 (d, J = 2.3 Hz, 2H), 6.97 (d, J = 8.6 Hz, 2H), 6.94 (d, J = 9.0 Hz, 2H), 4.30 (d, J = 13.0 Hz, 1H), 3.89 (d, J = 13.1 Hz, 1H), 3.78 (dd, J = 12.1, 3.6 Hz, 1H), 3.72 (dd, J = 12.1, 5.3 Hz, 1H), 3.38 – 3.27 (m, 2H), 2.84 – 2.73 (m, 2H), 2.13 – 2.05 (m, 1H), 1.98 – 1.86 (m, 3H), 1.32 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 158.7, 153.9, 147.0, 131.8, 126.9, 119.1, 118.5, 67.0, 61.2, 58.4, 54.1, 34.5, 31.6, 27.0, 23.2. HRMS (ESI + ): calcd for C22H30NO2 [M + H]+ 340.2271, found: 340.2275.
4.2.3.9. (R)-(1-(4-([1,1′-biphenyl]-3-yloxy)benzyl)pyrrolidin-2-yl)methanol (17d).
Clear oil, yield 36%. 1H NMR (400 MHz, CDCl3) δ: 7.56 (d, J = 7.1 Hz, 1H), 7.26 – 7.25 (m, 1H), 7.03 (d, J = 8.2 Hz, 2H), 7.01 – 6.97 (m, 1H), 4.28 – 4.18 (m, 1H), 3.82 – 3.73 (m, 2H), 3.70 – 3.64 (m, 1H), 3.29 – 3.17 (m, 1H), 2.72 – 2.64 (m, 1H), 2.09 – 2.05 (m, 1H), 1.94 – 1.83 (m, 2H). 13C NMR (101 MHz, CDCl3) δ: 157.9, 157.1, 143.4, 140.4, 131.6, 130.3, 128.9, 127.8, 127.2, 122.7, 118.9, 118.2, 76.8, 61.4, 58.3, 54.3, 29.8, 27.2, 23.2, 14.3. HRMS (ESI + ): calcd for C24H26NO2 [M + H]+ 360.1958, found: 360.1963.
4.2.3.10. (R)-(1-(4-([1,1′-biphenyl]-4-yloxy)benzyl)pyrrolidin-2-yl)methanol (17e).
Clear oil, yield 53%. 1H NMR (400 MHz, CDCl3) δ 7.58 – 7.52 (m, 4H), 7.42 (t, J = 7.6 Hz, 1H), 7.35 – 7.28 (m, 3H), 7.06 (d, J = 8.7 Hz, 1H), 7.01 (d, J = 8.5 Hz, 1H), 4.02 (d, J = 13.0 Hz, 1H), 3.68 (dd, J = 11.1, 3.4 Hz, 1H), 3.54 – 3.44 (m, 2H), 3.07 (dt, J = 9.6, 4.4 Hz, 1H), 2.85 (td, J = 6.0, 3.0 Hz, 1H), 2.39 (q, J = 8.4 Hz, 1H), 2.01 – 1.90 (m, 1H), 1.89 – 1.81 (m, 1H), 1.81 – 1.71 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 159.9, 159.1, 145.4, 142.4, 133.6, 132.3, 130.9, 129.8, 129.2, 124.7, 120.9, 120.2, 63.4, 60.3, 56.3, 31.8, 29.2, 25.2, 16.3. HRMS (ESI + ): calcd for C24H26NO2 [M + H]+ 360.1958, found: 360.1967.
4.2.3.11. (R)-(1-(4-((3′ -(trifluoromethyl)-[1,1′ -biphenyl]-3-yl)oxy) benzyl)pyrrolidin-2-yl)methanol (17f).
Clear oil, yield 40%. 1H NMR (500 MHz, CDCl3) δ: 7.79 (s, 1H), 7.73 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.54 (t, J = 7.7 Hz, 1H), 7.44 (t, J = 7.9 Hz, 1H), 7.38 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 8.2 Hz, 1H), 7.25 (t, J = 2.1 Hz, 1H), 7.03 (d, J = 8.6 Hz, 3H), 4.21 (d, J = 13.1 Hz, 1H), 3.77 – 3.70 (m, 2H), 3.65 (dd, J = 11.7, 4.5 Hz, 1H), 3.21 (td, J = 6.6, 2.8 Hz, 1H), 3.15 (q, J = 5.0 Hz, 1H), 2.66 – 2.60 (m, 1H), 2.06 – 2.01 (m, 1H), 1.91 – 1.83 (m, 3H). 13C NMR (126 MHz, CDCl3) δ: 157.3, 157.3, 141.7, 141.2, 131.6, 131.3, 131.3, 131.0, 130.8, 130.4, 129.6, 129.3, 127.3, 125.2, 124.3, 124.3, 124.3, 124.3, 123.9, 123.9, 123.9, 123.8, 123.0, 122.5, 120.8, 118.8, 118.6, 118.0, 66.1, 61.4, 58.2, 54.1, 27.1, 23.2. HRMS (ESI + ): calcd for C25H25F3NO2 [M + H]+: 428.1832, found: 428.1840.
4.2.3.12. (R)-(1-(4-((4′ -(trifluoromethyl)-[1,1′ -biphenyl]-3-yl)oxy) benzyl)pyrrolidin-2-yl)methanol (17 g).
Clear oil, yield 47%. 1H NMR (500 MHz, CDCl3) δ: 7.71 – 7.63 (m, 4H), 7.43 (t, J = 7.9 Hz, 1H), 7.39 – 7.32 (m, 3H), 7.25 (t, J = 2.1 Hz, 1H), 7.06 – 7.00 (m, 3H), 4.15 – 4.09 (m, 1H), 3.72 (dd, J = 11.5, 3.4 Hz, 1H), 3.63 – 3.55 (m, 2H), 3.16 (ddd, J = 10.4, 6.7, 3.7 Hz, 1H), 3.00 (ddt, J = 9.5, 6.5, 3.6 Hz, 1H), 2.55 –2.46 (m, 1H), 2.03 – 1.97 (m, 1H), 1.92 – 1.77 (m, 3H). 13C NMR (126 MHz, CDCl3) δ: 157.5, 156.8, 143.9, 143.9, 141.7, 131.0, 130.4, 130.0, 129.8, 129.5, 129.3, 127.4, 125.8, 125.8, 125.7, 125.7, 125.3, 123.1, 122.4, 121.0, 118.9, 118.5, 117.8, 77.3, 65.6, 61.1, 58.2, 54.2, 27.3, 23.41. HRMS (ESI + ): calcd for C25H25F3NO2 [M + H]+: 428.1832, found: 428.1844.
4.2.3.13. (R)-(1-(4-((3′ -(trifluoromethyl)-[1,1′ -biphenyl]-4-yl)oxy) benzyl)pyrrolidin-2-yl)methanol (17 h).
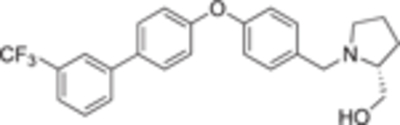
Clear oil, yield 68%. 1H NMR (500 MHz, CDCl3) δ: 7.79 (s, 1H), 7.73 (d, J = 7.5 Hz, 1H), 7.61 – 7.51 (m, 4H), 7.40 (d, J = 8.1 Hz, 2H), 7.10 (d, J = 8.3 Hz, 2H), 7.04 (d, J = 8.2 Hz, 2H), 4.26 (d, J = 13.0 Hz, 1H), 3.82 – 3.75 (m, 2H), 3.69 (dd, J = 11.8, 4.8 Hz, 1H), 3.30 – 3.21 (m, 2H), 2.74 – 2.67 (m, 1H), 2.11 – 2.03 (m, 1H), 1.95 – 1.83 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 157.4, 156.8, 141.2, 135.2, 131.5, 131.3, 131.0, 130.8, 130.2, 129.3, 129.0, 128.7, 127.4, 125.2, 123.8, 123.8, 123.8, 123.7, 123.7, 123.7, 123.6, 123.6, 123.1, 120.9, 66.5, 61.3, 58.3, 54.1, 27.1, 23.1. HRMS (ESI + ): calcd for C25H25F3NO2 [M + H]+: 428.1832, found: 428.1839.
4.2.3.14. (R)-(1-(4-((4′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)oxy) benzyl)pyrrolidin-2-yl)methanol (17i).
Clear oil, yield 44%. 1H NMR (500 MHz, CDCl3) δ: 7.71 – 7.64 (m, 4H), 7.57 (d, J = 8.6 Hz, 2H), 7.36 (d, J = 8.5 Hz, 2H), 7.10 (d, J = 8.7 Hz, 2H), 7.04 (d, J = 8.5 Hz, 2H), 4.07 (d, J = 13.0 Hz, 1H), 3.71 (dd, J = 11.3, 3.4 Hz, 1H), 3.57 – 3.51 (m, 2H), 3.13 (dd, J = 6.8, 3.6 Hz, 1H), 2.93 (dt, J = 8.0, 4.3 Hz, 1H), 2.46 (q, J = 8.8 Hz, 1H), 2.05 – 1.95 (m, 1H), 1.88 (dt, J = 12.4, 6.5 Hz, 1H), 1.84 – 1.76 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 157.5, 156.4, 144.0, 144.0, 134.8, 130.7, 129.5, 129.2, 129.0, 128.7, 128.7, 127.5, 127.1, 125.8, 125.8, 125.7, 125.7, 125.4, 123.2, 121.0, 119.1, 119.1, 65.3, 61.6, 58.2, 54.3, 27.5, 23.5. HRMS (ESI + ): calcd for C25H25F3NO2 [M + H]+: 428.1832, found:428.1834.
4.2.3.15. (R)-(1-(4-nonylbenzyl)pyrrolidin-2-yl)methanol (21a).
Clear oil, yield 70%. 1H NMR (400 MHz, CDCl3) δ: 7.22 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 8.0 Hz, 2H), 3.96 (d, J = 12.9 Hz, 1H), 3.67 (dd, J = 10.9, 3.5 Hz, 1H), 3.46 (dd, J = 10.9, 2.5 Hz, 1H), 3.39 (d, J = 12.9 Hz, 1H), 3.05 – 2.98 (m, 1H), 2.78 (ddt, J = 9.1, 6.1, 3.0 Hz, 1H), 2.61 – 2.55 (m, 2H), 2.34 (td, J = 9.1, 7.9 Hz, 1H), 1.99 – 1.88 (m, 1H), 1.88 – 1.77 (m, 1H), 1.76 – 1.66 (m, 2H), 1.65 – 1.54 (m, 2H), 1.36 – 1.22 (m, 12H), 0.91 – 0.85 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 142.1, 135.8, 128.9, 128.5, 64.6, 61.8, 58.4, 54.5, 35.7, 32.0, 31.6, 29.7, 29.6, 29.5, 29.4, 27.8, 23.5, 22.8, 14.2. HRMS (ESI + ): calcd for C21H36NO [M + H]+ 318.2791, found: 318.2802.
4.2.3.16. (R)-(1-(4-decylbenzyl)pyrrolidin-2-yl)methanol (21b).
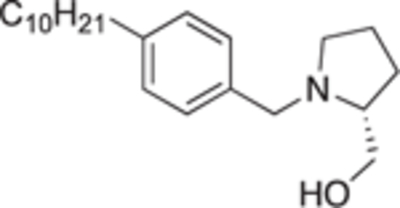
Clear oil, yield 72%. 1H NMR (400 MHz, CDCl3) δ: 7.38 (d, J = 8.2 Hz, 2H), 7.16 (d, J = 8.0 Hz, 2H), 4.18 (d, J = 13.1 Hz, 1H), 3.85 (d, J = 13.4 Hz, 1H), 3.76 (dd, J = 12.3, 3.1 Hz, 1H), 3.66 (dd, J = 12.3, 4.6 Hz, 1H), 3.34 – 3.27 (m, 1H), 3.26 – 3.20 (m, 1H), 2.69 (dd, J = 8.1, 3.7 Hz, 1H), 2.60 – 2.54 (m, 2H), 2.03 – 1.89 (m, 3H), 1.88 – 1.79 (m, 1H), 1.56 (q, J = 7.2 Hz, 2H), 1.32 – 1.20 (m, 14H), 0.88 – 0.83 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 143.9, 130.3, 129.0, 126.1, 67.0, 61.2, 58.6, 54.0, 35.7, 32.0, 31.4, 29.8, 29.7, 29.7, 29.7, 29.6, 29.4, 27.0, 23.5, 22.8, 14.2. HRMS (ESI + ): calcd for C22H38NO [M + H]+ 332.2948, found: 332.2955.
4.2.3.17. (R)-(1-(4-undecylbenzyl)pyrrolidin-2-yl)methanol (21c).
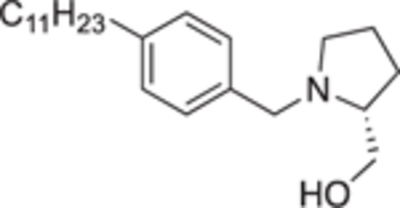
Clear oil, yield 65%. 1H NMR (400 MHz, CDCl3) δ: 7.20 (d, J = 8.0 Hz, 2H), 7.12 (d, J = 7.9 Hz, 2H), 3.92 (d, J = 12.9 Hz, 1H), 3.65 (dd, J = 10.7, 3.4 Hz, 1H), 3.42 (dd, J = 10.7, 2.1 Hz, 1H), 3.32 (d, J = 12.9 Hz, 1H), 2.98 (dq, J = 9.3, 5.6, 4.4 Hz, 1H), 2.77 – 2.67 (m, 1H), 2.62 – 2.53 (m, 2H), 2.29 (td, J = 9.3, 7.6 Hz, 1H), 1.93 (dq, J = 12.6, 8.8 Hz, 1H), 1.87 – 1.78 (m, 1H), 1.69 (td, J = 8.9, 5.7 Hz, 2H), 1.59 (dt, J = 15.0, 6.2 Hz, 2H), 1.34 – 1.19 (m, 16H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 141.9, 136.6, 128.8, 128.5, 64.3, 61.8, 58.3, 54.6, 35.8, 32.1, 31.7, 29.8, 29.8, 29.7, 29.7, 29.5, 29.5, 28.0, 23.6, 22.8, 14.3. HRMS (ESI + ): calcd for C23H40NO [M + H]+ 346.3104, found: 346.3132.
4.2.3.18. (R)-(1-(4-dodecylbenzyl)pyrrolidin-2-yl)methanol (21d).
Clear oil, yield 69%. 1H NMR (400 MHz, CDCl3) δ: 7.25 (d, J = 8.3 Hz, 2H), 7.16 (d, J = 8.0 Hz, 2H), 4.07 (d, J = 13.0 Hz, 1H), 3.74 – 3.66 (m, 1H), 3.55 (dd, J = 11.7, 4.2 Hz, 1H), 3.12 (dt, J = 10.0, 4.9 Hz, 1H), 2.97 (m, 1H), 2.63 – 2.55 (m, 2H), 2.50 (m, 1H), 2.06 – 1.92 (m, 2H), 1.92 – 1.74 (m, 2H), 1.59 (m, 2H), 1.37 – 1.18 (m, 18H), 0.91 – 0.81 (m, 3H). HRMS (ESI + ): calcd. for C24H42NO [M + H]+ 360.3261, found: 360.3266.
4.2.3.19. (R)-(1-(4-dodecyl-2-methylbenzyl)pyrrolidin-2-yl)methanol (21e).
Clear oil, yield 33%. 1H NMR (400 MHz, CDCl3) δ: 7.38 (d, J = 8.2 Hz, 2H), 7.23 (d, J = 8.2 Hz, 2H), 7.08 (d, J = 7.9 Hz, 2H), 7.03 (d, J = 8.2 Hz, 2H), 4.45 (d, J = 13.0 Hz, 1H), 4.15 (d, J = 13.1 Hz, 1H), 3.88 – 3.74 (m, 2H), 3.66 – 3.58 (m, 1H), 3.43 (ddd, J = 11.7, 6.8, 4.9 Hz, 1H), 3.02 (ddd, J = 11.3, 8.3, 7.2 Hz, 1H), 2.93 – 2.82 (m, 4H), 2.31 (s, 3H), 2.21 – 2.12 (m, 1H), 2.09 – 1.89 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 144.2, 138.2, 135.6, 130.9, 129.6, 129.2, 128.4, 127.2, 68.2, 60.9, 58.9, 54.1, 37.8, 37.2, 26.6, 23.0, 21.2. HRMS (ESI + ): calcd for C25H44NO [M + H]+ 374.3417, found: 374.3412.
4.2.3.20. (R)-(1-(4-(4-methylphenethyl)benzyl)pyrrolidin-2-yl)methanol (21f).
Clear oil, yield 42%; 1H NMR (400 MHz, CDCl3) δ: 7.30 (d, J = 7.8 Hz, 2H), 7.17 (d, J = 8.1 Hz, 2H), 7.11 – 7.04 (m, 4H), 4.05 (d, J = 13.0 Hz, 1H), 3.70 (dd, J = 11.6, 3.3 Hz, 1H), 3.65 – 3.51 (m, 2H), 3.17 (s, 1H), 2.99 (s, 1H), 2.92 – 2.83 (m, 4H), 2.54 – 2.46 (m, 1H), 2.32 (s, 3H), 2.02 – 1.73 (m, 5H). 13C NMR (101 MHz, CDCl3) δ: 138.5, 135.4, 129.6, 129.5, 129.0, 128.7, 128.3, 65.7, 61.4, 58.5, 54.2, 37.7, 37.4, 27.3, 23.5, 21.0. HRMS (ESI + ): calcd for C21H28NO [M + H]+ 310.2165, found: 310.2170.
4.2.3.21. (R)-(1-(4-(4-methoxyphenethyl)benzyl)pyrrolidin-2-yl)methanol (21 g).
Clear oil, yield 48%. 1H NMR (400 MHz, CDCl3) δ: 7.45 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.2 Hz, 2H), 7.07 (d, J = 16.3 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 4.09 (d, J = 13.0 Hz, 1H), 3.83 (s, 3H), 3.73 (dd, J = 11.6, 3.3 Hz, 1H), 3.67 – 3.55 (m, 2H), 3.17 (d, J = 6.4 Hz, 1H), 3.00 (s, 1H), 2.52 (q, J = 8.4 Hz, 9H), 2.04 – 1.74 (m, 1H). 13C NMR (101 MHz, CDCl3) δ: 159.5, 130.1, 130.0, 128.8, 127.9, 126.6, 126.1, 114.3, 65.8, 61.6, 58.7, 55.5, 54.4, 32.1, 29.8, 27.5, 23.6. HRMS (ESI + ): calcd for C21H28NO2 [M + H]+ 326.2115, found: 326.2119.
4.2.3.22. (R,E)-(1-(4-(4-(tert-butyl)styryl)-2-methylbenzyl)pyrrolidin-2-yl)methanol (24a).
Clear oil, yield 42%. 1H NMR (400 MHz, CDCl3) δ: 7.18 (d, J = 7.5 Hz, 1H), 6.96 (d, J = 7.4 Hz, 2H), 3.94 (d, J = 13.0 Hz, 1H), 3.64 (dd, J = 10.9, 3.4 Hz, 1H), 3.46 – 3.34 (m, 2H), 3.03 – 2.94 (m, 1H), 2.76 (s, 1H), 2.54 (t, J = 8.9, 6.7 Hz, 2H), 2.34 (s, 3H), 2.33 – 2.27 (m, 1H), 2.01 – 1.89 (m, 1H), 1.89 – 1.79 (m, 1H), 1.76 – 1.64 (m, 2H), 1.58 (p, J = 10.6, 4.8 Hz, 2H), 1.37 – 1.20 (m, 19H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 136.7, 136.4, 130.8, 130.4, 130.0, 126.0, 65.5, 62.0, 56.5, 54.7, 35.7, 32.1, 31.7, 29.8, 29.8, 29.8, 29.7, 29.6, 29.5, 27.8, 23.8, 22.9, 19.5, 14.3, 14.2. HRMS (ESI + ): calcd for C25H34NO [M + H]+ 364.2635, found: 364.2622.
4.2.3.23. (R,E)-(1-(4-(3-(trifluoromethyl)styryl)benzyl)pyrrolidin-2-yl) methanol (24b).
Clear oil, yield 51%. 1H NMR (400 MHz, CDCl3) δ: 7.75 (s, 1H), 7.66 (d, J = 7.3 Hz, 1H), 7.54 – 7.43 (m, 4H), 7.32 (d, J = 7.9 Hz, 2H), 7.13 (q, J = 16.4 Hz, 2H), 3.98 (d, J = 13.1 Hz, 1H), 3.68 (dd, J = 10.8, 3.5 Hz, 1H), 3.46 (dd, J = 10.8, 2.3 Hz, 1H), 3.39 (d, J = 13.2 Hz, 1H), 3.04 – 2.97 (m, 1H), 2.80 – 2.72 (m, 1H), 2.71 (s, 1H), 2.37 – 2.26 (m, 1H), 2.01 – 1.90 (m, 1H), 1.90 – 1.80 (m, 1H), 1.77 – 1.66 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 140.1, 139.6, 132.4, 132.0, 131.2, 131.0, 131.0, 131.0, 131.0, 130.6, 130.6, 129.8, 128.6, 125.4, 125.4, 125.3, 125.3, 124.4, 124.3, 124.3, 124.3, 70.0, 60.7, 59.3, 55.6, 27.3, 23.2. HRMS (ESI + ): calcd for C21H23F3NO [M + H]+ 362.1726, found: 362.1712.
4.2.3.24. (R)-(1-(4-((4-propylphenyl)ethynyl)benzyl)pyrrolidin-2-yl) methanol (27a).
Clear oil, yield 55%. 1H NMR (400 MHz, CDCl3) δ: 7.48 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 7.16 (d, J = 8.3 Hz, 2H), 3.98 (d, J = 13.2 Hz, 1H), 3.66 (dd, J = 10.8, 3.5 Hz, 1H), 3.45 (dd, J = 10.8, 2.2 Hz, 1H), 3.37 (d, J = 13.2 Hz, 1H), 3.00 – 2.94 (m, 1H), 2.77 – 2.70 (m, 1H), 2.62 – 2.57 (m, 2H), 2.32 – 2.24 (m, 1H), 1.99 – 1.89 (m, 1H), 1.85 (dt, J = 13.0, 6.4 Hz, 1H), 1.75 – 1.68 (m, 2H), 1.64 (dt, J = 14.8, 7.4 Hz, 3H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 143.2, 139.5, 131.7, 131.6, 128.8, 128.6, 122.4, 120.6, 89.6, 88.8, 64.5, 62.0, 58.5, 54.6, 38.1, 27.9, 24.5, 23.6, 13.9. HRMS (ESI + ): calcd for C23H28NO [M + H]+ 334.2165, found: 334.2142.
4.2.3.25. (R)-(1-(4-((4-butylphenyl)ethynyl)benzyl)pyrrolidin-2-yl)methanol (27b).
Clear oil, yield 36%; 1H NMR (400 MHz, CDCl3) δ: 7.48 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.1 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 7.16 (d, J = 8.0 Hz, 2H), 3.98 (d, J = 13.2 Hz, 1H), 3.66 (dd, J = 10.8, 3.5 Hz, 1H), 3.45 (dd, J = 10.8, 2.3 Hz, 1H), 3.37 (d, J = 13.2 Hz, 1H), 2.98 (dt, J = 9.3, 4.4 Hz, 1H), 2.74 (ddt, J = 9.1, 5.9, 2.9 Hz, 1H), 2.66 – 2.57 (m, 2H), 2.34 – 2.23 (m, 1H), 1.95 (dt, J = 12.6, 8.7 Hz, 1H), 1.84 (td, J = 13.0, 12.6, 7.1 Hz, 1H), 1.71 (tt, J = 8.6, 3.8 Hz, 2H), 1.66 – 1.54 (m, 2H), 1.36 (dq, J = 14.6, 7.3 Hz, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 143.5, 139.4, 131.7, 131.6, 128.8, 128.6, 122.3, 120.5, 89.6, 88.7, 64.5, 61.9, 58.5, 54.6, 35.7, 33.5, 27.8, 23.6, 22.4, 14.1. HRMS (ESI + ): calcd for C24H30NO [M + H]+ 348.2322, found: 348.2342.
4.2.3.26. (R)-(1-(4-((4-pentylphenyl)ethynyl)benzyl)pyrrolidin-2-yl) methanol (27c).
Clear oil, yield 47%. 1H NMR (400 MHz, CDCl3) δ: 7.48 (d, J = 8.3 Hz, 2H), 7.44 (d, J = 8.3 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 7.16 (d, J = 8.4 Hz, 2H), 3.98 (d, J = 13.2 Hz, 1H), 3.66 (dd, J = 10.8, 3.5 Hz, 1H), 3.45 (dd, J = 10.8, 2.2 Hz, 1H), 3.37 (d, J = 13.2 Hz, 1H), 3.03 – 2.93 (m, 1H), 2.80 – 2.69 (m, 1H), 2.66 – 2.57 (m, 2H), 2.35 – 2.23 (m, 1H), 2.02 – 1.78 (m, 2H), 1.77 – 1.66 (m, 2H), 1.62 (dt, J = 15.2, 7.6 Hz, 2H), 1.33 (td, J = 7.8, 6.5, 4.5 Hz, 4H), 0.94 – 0.85 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 143.5, 139.5, 131.7, 131.6, 128.8, 128.6, 122.3, 120.5, 89.6, 88.7, 64.5, 61.9, 58.5, 54.6, 36.0, 31.6, 31.1, 27.9, 23.6, 22.7, 14.2. HRMS (ESI + ): calcd for C25H32NO [M + H]+ 362.2478, found: 362.2456.
4.2.4. General procedure for the synthesis of compounds 12a-e, 18a-i, 22a-h, 25a-b, 29a-c, 30a-c
To a round bottom flask containing methanol (0.2 M), the appropriate tertiary amine analogue (1.0 equiv.) was added and stirred. Next, the mixture was bubbled with HCl gas for 1 min and then stirred for 5 – 10 min. The reaction progress was monitored by TLC. Lastly, the organic mixture was concentrated via vacuum and triturated with diethyl ether to afford the corresponding analogue as an HCl salt.
4.2.4.1. (R)-(1-(4-(heptyloxy)benzyl)pyrrolidin-2-yl)methanol (12a).
White solid, yield 95%. 1H NMR (400 MHz, CD3OD) δ: 7.48 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 4.58 (d, J = 13.0 Hz, 1H), 4.24 (d, J = 13.0 Hz, 1H), 4.01 (t, J = 6.4 Hz, 2H), 3.79 – 3.68 (m, 3H), 3.42 – 3.36 (m, 1H), 3.30 – 3.23 (m, 1H), 2.30 – 2.20 (m, 1H), 2.14 – 2.05 (m, 1H), 1.83 – 1.74 (m, 2H), 1.53 – 1.44 (m, 2H), 1.39 – 1.31 (m, 6H), 0.95 – 0.89 (m, 3H). 13C NMR (101 MHz, CD3OD) δ: 161.8, 133.4, 123.3, 116.0, 69.5, 69.1, 60.8, 59.0, 55.2, 32.9, 30.3, 30.1, 27.3, 27.1, 23.6, 23.1, 14.4. HRMS (ESI + ): calcd for C19H32NO2 [M + H]+ 306.2428, found: 306.2438.
4.2.4.2. (R)-(1-(4-(octyloxy)benzyl)pyrrolidin-2-yl)methanol (12b).
White solid, yield 97%. 1H NMR (400 MHz, CD3OD) δ: 7.37 (d, J = 8.7 Hz, 2H), 6.93 (d, J = 8.7 Hz, 2H), 4.49 (d, J = 13.0 Hz, 1H), 4.13 (d, J = 13.0 Hz, 1H), 3.93 (t, J = 6.4 Hz, 2H), 3.72 – 3.57 (m, 3H), 3.34 – 3.26 (m, 1H), 3.21 – 3.14 (m, 1H), 2.17 (ddt, J = 10.5, 6.3, 3.4 Hz, 1H), 2.07 – 1.98 (m, 1H), 1.93 – 1.83 (m, 2H), 1.77 – 1.69 (m, 2H), 1.41 (t, J = 7.7 Hz, 2H), 1.31 – 1.23 (m, 8H), 0.87 – 0.81 (m, 3H). 13C NMR (101 MHz, CD3OD) δ: 161.9, 133.3, 123.3, 116.1, 69.4, 69.1, 60.8, 59.1, 55.3, 33.0, 30.4, 30.4, 30.3, 27.3, 27.1, 23.7, 23.1, 14.4. HRMS (ESI + ): calcd for C20H34NO2 [M + H]+ 320.2584, found: 320.2582.
4.2.4.3. (R)-(1-(4-(nonyloxy)benzyl)pyrrolidin-2-yl)methanol (12c).
White solid, yield 97%. 1H NMR (400 MHz, CD3OD) δ: 7.42 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 4.54 (d, J = 12.9 Hz, 1H), 4.18 (d, J = 13.0 Hz, 1H), 4.00 (t, J = 6.4 Hz, 2H), 3.80 – 3.70 (m, 1H), 3.71 – 3.61 (m, 2H), 3.41 – 3.33 (m, 1H), 3.27 – 3.20 (m, 1H), 2.29 – 2.16 (m, 1H), 2.14 – 2.02 (m, 1H), 1.99 – 1.85 (m, 2H), 1.77 (dt, J = 14.6, 6.5 Hz, 2H), 1.53 – 1.41 (m, 2H), 1.40 – 1.24 (m, 10H), 0.94 – 0.86 (m, 3H). 13C NMR (101 MHz, CD3OD) δ: 146.3, 131.9, 130.3, 129.0, 69.8, 60.8, 59.4, 55.5, 36.6, 33.0, 32.5, 30.7, 30.6, 30.4, 30.3, 27.3, 23.7, 23.1, 14.4. HRMS (ESI + ): calcd for C21H36NO2 [M + H]+ 334.2741, found: 334.2762.
4.2.4.4. (R)-(1-(4-(decyloxy)benzyl)pyrrolidin-2-yl)methanol (12d).
White solid, yield 91%. 1H NMR (400 MHz, CD3OD) δ: 7.44 (d, J = 8.6 Hz, 2H), 6.99 (d, J = 8.7 Hz, 2H), 4.55 (d, J = 13.0 Hz, 1H), 4.20 (d, J = 13.0 Hz, 1H), 4.00 (t, J = 6.4 Hz, 2H), 3.78 – 3.66 (m, 3H), 3.40 – 3.35 (m, 1H), 3.27 – 3.21 (m, 1H), 2.28 – 2.19 (m, 1H), 2.12 – 2.04 (m, 1H), 1.97 – 1.87 (m, 2H), 1.82 – 1.72 (m, 2H), 1.47 (p, J = 6.9 Hz, 2H), 1.40 – 1.25 (m, 12H), 0.92 – 0.86 (m, 3H). 13C NMR (101 MHz, CD3OD) δ: 161.9, 133.4, 123.3, 116.1, 69.5, 69.2, 60.8, 59.1, 55.3, 33.1, 30.7, 30.7, 30.5, 30.5, 30.3, 27.3, 27.1, 23.7, 23.1, 14.4. HRMS (ESI + ): calcd for C22H38NO2 [M + H]+ 348.2897, found: 348.2917.
4.2.4.5. (R)-(1-(4-((4-(trifluoromethyl)benzyl)oxy)benzyl)pyrrolidin-2-yl)methanol (12e).
White solid, yield 90%. 1H NMR (400 MHz, CD3OD) δ: 7.53 (s, 2H), 6.91 (s, 2H), 4.26 (m, 4H), 3.89 (d, J = 19.7 Hz, 2H), 3.60 (s, 3H), 3.00 (s, 1H), 2.07 (s, 3H), 1.76 (s, 2H), 1.34 (m, 10H), 0.85 (s, 3H). 13C NMR (101 MHz, CD3OD) δ: 160.6, 133.1, 120.6, 115.2, 68.8, 68.3, 61.0, 59.1, 53.8, 50.8, 32.0, 29.6, 29.5, 29.3, 29.2, 26.6, 26.1, 23.7, 22.8, 14.2. HRMS (ESI + ): calcd for C20H23F3NO2 [M + H]+ 366.1675, found: 366.1680.
4.2.4.6. (R)-(1-(4-phenoxybenzyl)pyrrolidin-2-yl)methanol (18a).
White solid, yield 90%. 1H NMR (400 MHz, CD3OD) δ: 7.56 – 7.51 (m, 2H), 7.42 – 7.36 (m, 2H), 7.20 – 7.14 (m, 1H), 7.06 – 7.01 (m, 4H), 4.62 (d, J = 13.0 Hz, 1H), 4.26 (d, J = 13.0 Hz, 1H), 3.84 – 3.72 (m, 3H), 3.48 – 3.40 (m, 1H), 3.29 – 3.24 (m, 1H), 2.28 – 2.19 (m, 1H), 2.15 – 2.07 (m, 1H), 2.04 – 1.88 (m, 2H). 13C NMR (101 MHz, CD3OD) δ: 160.5, 157.6, 133.8, 131.1, 126.1, 125.3, 120.6, 119.6, 69.7, 67.0, 58.9, 55.4, 27.3, 23.1. HRMS (ESI + ): calcd for C18H22NO2 [M + H]+ 284.1645, found: 284.1656.
4.2.4.7. (R)-(1-(4-(3-(tert-butyl)phenoxy)benzyl)pyrrolidin-2-yl)methanol (18b).
White solid, yield 92%. 1H NMR (400 MHz, CD3OD) δ: 7.54 – 7.49 (m, 2H), 7.31 (td, J = 7.9, 0.4 Hz, 1H), 7.23 (ddd, J = 7.9, 1.8, 1.1 Hz, 1H), 7.07 (ddd, J = 2.4, 1.9, 0.4 Hz, 1H), 7.04 – 7.00 (m, 2H), 6.81 (ddd, J = 8.0, 2.4, 1.1 Hz, 1H), 4.61 (d, J = 13.0 Hz, 1H), 4.25 (d, J = 13.0 Hz, 1H), 3.80 – 3.68 (m, 3H), 3.40 (ddd, J = 11.4, 7.5, 4.8 Hz, 1H), 3.29 – 3.23 (m, 1H), 2.28 – 2.20 (m, 1H), 2.13 – 2.07 (m, 1H), 1.99 – 1.89 (m, 2H), 1.30 (s, 9H). 13C NMR (101 MHz, CD3OD) δ: 160.8, 157.2, 155.0, 133.7, 130.6, 125.8, 122.4, 119.3, 118.0, 117.7, 69.7, 60.8, 58.9, 55.4, 35.6, 31.6, 27.3, 23.1. HRMS (ESI + ): calcd for C22H30NO2 [M + H]+ 340.2271, found: 340.2278.
4.2.4.8. (R)-(1-(4-(4-(tert-butyl)phenoxy)benzyl)pyrrolidin-2-yl)methanol (18c).
White solid, yield 94%. 1H NMR (400 MHz, CD3OD) δ: 7.72 (d, J = 8.3 Hz, 2H), 7.66 – 7.62 (m, 2H), 7.47 – 7.35 (m, 4H), 4.70 (d, J = 12.9 Hz, 1H), 4.32 (d, J = 12.9 Hz, 1H), 3.83 – 3.72 (m, 3H), 3.43 (ddd, J = 11.6, 6.9, 4.2 Hz, 1H), 3.31 (dt, J = 3.4, 1.7 Hz, 2H), 2.27 (dtd, J = 14.3, 8.3, 7.4, 4.3 Hz, 1H), 2.17 – 2.09 (m, 1H), 2.02 – 1.89 (m, 2H), 1.38 (s, 9H). 13C NMR (101 MHz, CD3OD) δ: 153.1, 144.8, 141.1, 132.5, 129.8, 128.9, 126.0, 125.1, 69.9, 60.8, 59.2, 55.5, 35.7, 31.8, 27.3, 23.1. HRMS (ESI + ): calcd for C22H30NO2 [M + H]+ 340.2271, found: 340.2275.
4.2.4.9. (R)-(1-(4-([1,1′-biphenyl]-3-yloxy)benzyl)pyrrolidin-2-yl)methanol (18d).
White solid, 97%. 1H NMR (400 MHz, CD3OD) δ: 7.58 – 7.52 (m, 4H), 7.49 – 7.39 (m, 4H), 7.36 – 7.31 (m, 1H), 7.26 (ddd, J = 2.3, 1.6, 0.5 Hz, 1H), 7.13 – 7.08 (m, 2H), 7.01 (ddd, J = 7.5, 2.4, 1.6 Hz, 1H), 4.62 (d, J = 12.9 Hz, 1H), 4.25 (d, J = 13.0 Hz, 1H), 3.81 – 3.68 (m, 3H), 3.44 – 3.37 (m, 1H), 3.29 – 3.22 (m, 1H), 2.28 – 2.21 (m, 1H), 2.14 – 2.07 (m, 1H), 1.99 – 1.88 (m, 2H). 13C NMR (101 MHz, CD3OD) δ: 159.1, 156.8, 143.4, 140.1, 132.4, 130.1, 128.6, 127.4, 126.6, 124.9, 122.5, 118.4, 117.9, 117.7, 68.4, 59.4, 57.5, 54.0, 25.9, 21.8. HRMS (ESI + ): calcd for C24H26NO2 [M + H]+ 360.1958, found: 360.1963.
4.2.4.10. (R)-(1-(4-([1,1′ -biphenyl]-4-yloxy)benzyl)pyrrolidin-2-yl)methanol (18e).
White solid, yield 90%. 1H NMR (400 MHz, CD3OD) δ: 7.66 – 7.63 (m, 2H), 7.62 – 7.58 (m, 2H), 7.57 – 7.53 (m, 2H), 7.45 – 7.40 (m, 2H), 7.35 – 7.30 (m, 1H), 7.13 – 7.08 (m, 4H), 4.63 (d, J = 13.0 Hz, 1H), 4.27 (d, J = 13.0 Hz, 1H), 3.82 – 3.69 (m, 3H), 3.45 – 3.40 (m, 1H), 3.30 – 3.24 (m, 1H), 2.31 – 2.21 (m, 1H), 2.18 – 2.07 (m, 1H), 2.01 – 1.88 (m, 2H). 13C NMR (101 MHz, CD3OD) δ: 160.4, 157.1, 141.5, 138.6, 133.8, 129.9, 129.6, 128.3, 127.8, 126.2, 120.9, 119.7, 69.7, 60.8, 58.9, 55.4, 27.3, 23.1. HRMS (ESI + ): calcd for C24H26NO2 [M + H]+ 360.1958, found: 360.1968.
4.2.4.11. (R)-(1-(4-((3′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)oxy) benzyl)pyrrolidin-2-yl)methanol (18f).
White solid, yield 98%. 1H NMR (500 MHz, CD3OD) δ: 7.84 – 7.76 (m, 2H), 7.61 (t, J = 5.2 Hz, 2H), 7.55 – 7.52 (m, 2H), 7.50 – 7.45 (m, 1H), 7.44 – 7.41 (m, 1H), 7.26 (d, J = 2.0 Hz, 1H), 7.10 – 7.06 (m, 2H), 7.05 – 7.02 (m, 1H), 4.60 (d, J = 13.0 Hz, 1H), 4.24 (d, J = 12.9 Hz, 1H), 3.78 – 3.73 (m, 1H), 3.71 – 3.66 (m, 2H), 3.37 (ddd, J = 11.6, 7.3, 4.5 Hz, 1H), 3.27 – 3.22 (m, 2H), 2.25 – 2.18 (m, 1H), 2.07 (qd, J = 8.0, 3.1 Hz, 1H), 1.98 – 1.83 (m, 2H). 13C NMR (126 MHz, CD3OD) δ 188.4, 186.6, 171.2, 170.7, 162.1, 160.5, 160.3, 160.0, 160.0, 159.0, 154.8, 154.7, 153.6, 152.7, 152.1, 148.2, 148.1, 147.3, 98.0, 89.0, 87.0, 83.6, 55.5, 51.3. HRMS (ESI + ): calcd for C25H25F3NO2 [M + H]+ 428.1832, found: 428.1840.
4.2.4.12. (R)-(1-(4-((4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)oxy) benzyl)pyrrolidin-2-yl)methanol (18 g).
White solid, yield 95%. 1H NMR (500 MHz, CD3OD) δ: 7.81 (d, J = 8.0 Hz, 2H), 7.73 (dd, J = 8.4, 4.5 Hz, 4H), 7.55 (d, J = 8.6 Hz, 2H), 7.17 – 7.11 (m, 4H), 4.63 (d, J = 13.0 Hz, 1H), 4.26 (d, J = 13.0 Hz, 1H), 3.81 – 3.77 (m, 1H), 3.74 – 3.68 (m, 1H), 3.41 (td, J = 7.2, 3.7 Hz, 1H), 3.30 – 3.25 (m, 2H), 2.25 (dddd, J = 12.9, 8.7, 6.2, 3.1 Hz, 1H), 2.15 – 2.09 (m, 1H), 2.00 – 1.89 (m, 2H). 13C NMR (126 MHz, CD3OD) δ 160.1, 158.1, 145.3, 136.7, 133.8, 130.0, 128.3, 126.8, 126.8, 126.8, 126.8, 126.5, 124.7, 120.9, 120.1, 69.8, 60.7, 58.9, 55.4, 27.3, 23.1. HRMS (ESI + ): calcd for C25H25F3NO2 [M + H]+ 428.1832, found: 428.1837.
4.2.4.13. (R)-(1-(4-((3′ -(trifluoromethyl)-[1,1′ -biphenyl]-4-yl)oxy) benzyl)pyrrolidin-2-yl)methanol (18 h).
White solid, yield 97%. 1H NMR (500 MHz, CD3OD) δ: δ 7.79 (s, 1H), 7.73 (d, J = 7.5 Hz, 1H), 7.61 – 7.51 (m, 4H), 7.40 (d, J = 8.1 Hz, 1H), 7.10 (d, J = 8.3 Hz, 2H), 7.04 (d, J = 8.2 Hz, 2H), 4.26 (d, J = 13.0 Hz, 1H), 3.82 – 3.75 (m, 2H), 3.69 (dd, J = 11.8, 4.8 Hz, 1H), 3.30 – 3.21 (m, 2H), 2.74 – 2.67 (m, 1H), 2.11 – 2.03 (m, 1H), 1.95 – 1.83 (m, 3H). 13C NMR (126 MHz, CD3OD) δ 157.4, 156.8, 141.2, 135.2, 131.5, 131.3, 131.0, 130.8, 130.2, 129.3, 129.0, 128.7, 127.4, 125.2, 123.8, 123.8, 123.8, 123.7, 123.7, 123.7, 123.6, 123.6, 123.1, 120.9, 66.5, 61.3, 58.3, 54.1, 27.1, 23.1. HRMS (ESI + ): calcd for C25H25F3NO2 [M + H]+ 428.1832, found: 428.1839.
4.2.4.14. (R)-(1-(4-((4′ -(trifluoromethyl)-[1,1′ -biphenyl]-4-yl)oxy) benzyl)pyrrolidin-2-yl)methanol (18i).
White solid, yield 91%. 1H NMR (500 MHz, CD3OD) δ: 7.80 (s, 2H), 7.63 (d, J = 8.6 Hz, 2H), 7.57 (d, J = 4.5 Hz, 2H), 7.52 (d, J = 8.2 Hz, 2H), 7.07 (dd, J = 13.8, 8.4 Hz, 4H), 4.59 (d, J = 13.0 Hz, 1H), 4.23 (d, J = 13.0 Hz, 1H), 3.77 – 3.71 (m, 1H), 3.70 – 3.65 (m, 1H), 3.39 – 3.33 (m, 1H), 3.26 – 3.20 (m, 2H), 2.24 – 2.17 (m, 1H), 2.09 – 2.02 (m, 1H), 1.95 – 1.83 (m, 2H). 13C NMR (126 MHz, CD3OD) δ 162.6, 160.5, 145.2, 139.2, 136.5, 135.2, 134.9, 134.6, 134.4, 134.1, 133.4, 132.4, 131.5, 129.3, 127.4, 127.4, 127.4, 127.3, 127.2, 126.9, 126.9, 126.9, 125.0, 123.5, 122.6, 72.4, 63.4, 61.4, 58.0, 29.9, 25.7. HRMS (ESI + ): calcd for C25H25F3NO2 [M + H]+ 428.1832, found: 428.1834.
4.2.4.15. (R)-(1-(4-nonylbenzyl)pyrrolidin-2-yl)methanol (22a).
White solid, yield 92%. 1H NMR (400 MHz, CD3OD) δ: 7.45 (d, J = 8.1 Hz, 2H), 7.34 – 7.27 (m, 2H), 4.60 (d, J = 12.9 Hz, 1H), 4.24 (d, J = 12.9 Hz, 1H), 3.80 – 3.67 (m, 3H), 3.41 – 3.29 (m, 1H), 3.30 – 3.22 (m, 1H), 2.68 – 2.62 (m, 2H), 2.24 (qd, J = 6.0, 2.9 Hz, 1H), 2.14 – 2.06 (m, 1H), 1.98 – 1.86 (m, 2H), 1.66 – 1.58 (m, 2H), 1.37 – 1.25 (m, 12H), 0.93 – 0.86 (m, 3H). 13C NMR (101 MHz, CD3OD) δ: 144.9, 130.5, 128.9, 127.6, 68.4, 59.4, 57.9, 54.1, 35.2, 31.6, 31.1, 29.3, 29.2, 29.0, 28.9, 22.3, 21.7, 13.0. HRMS (ESI + ): calcd for C21H36NO [M + H]+ 318.2791, found: 318.2802.
4.2.4.16. (R)-(1-(4-decylbenzyl)pyrrolidin-2-yl)methanol (22b).
White solid, yield 90%. 1H NMR (400 MHz, CD3OD) δ: 8.02 (d, J = 8.2 Hz, 2H), 7.80 (d, J = 8.0 Hz, 2H), 4.83 (d, J = 13.1 Hz, 1H), 4.49 (d, J = 13.4 Hz, 1H), 4.40 (dd, J = 12.3, 3.1 Hz, 1H), 4.30 (dd, J = 12.3, 4.6 Hz, 1H), 3.99 – 3.91 (m, 1H), 3.90 – 3.84 (m, 1H), 3.33 (dd, J = 8.1, 3.7 Hz, 2H), 3.24 – 3.18 (m, 2H), 2.67 – 2.53 (m, 3H), 2.52 – 2.43 (m, 1H), 2.20 (q, J = 7.2 Hz, 2H), 1.96 – 1.84 (m, 16H), 1.52 – 1.47 (m, 3H). 13C NMR (101 MHz, CD3OD) δ: 145.9, 132.3, 131.0, 128.1, 69.0, 63.2, 60.6, 56.0, 37.7, 34.0, 33.4, 31.8, 31.7, 31.7, 31.7, 31.6, 31.4, 29.0, 25.5, 24.8, 16.2. HRMS (ESI + ): calcd for C22H38NO [M + H]+ 332.2948, found: 332.2955.
4.2.4.17. (R)-(1-(4-undecylbenzyl)pyrrolidin-2-yl)methanol (22c).
White solid, yield 91%. 1H NMR (400 MHz, CD3OD) δ: 7.27 (d, J = 7.5 Hz, 2H), 7.15 (d, J = 7.8 Hz, 2H), 4.02 (d, J = 13.0 Hz, 1H), 3.70 (dd, J = 11.3, 3.3 Hz, 1H), 3.55 – 3.49 (m, 2H), 3.11 (ddd, J = 9.9, 6.6, 3.4 Hz, 1H), 2.92 (td, J = 6.0, 3.0 Hz, 1H), 2.60 – 2.57 (m, 2H), 2.49 – 2.40 (m, 1H), 2.00 – 1.91 (m, 1H), 1.90 – 1.84 (m, 1H), 1.83 – 1.72 (m, 2H), 1.59 (p, J = 7.2 Hz, 2H), 1.35 – 1.22 (m, 16H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ: 142.8, 134.1, 129.4, 128.7, 65.4, 61.6, 58.5, 54.3, 35.8, 32.0, 31.6, 29.8, 29.8, 29.7, 29.6, 29.5, 27.6, 23.6, 22.8, 14.3. HRMS (ESI + ): calcd for C23H40NO [M + H]+ 346.3104, found: 346.3132.
4.2.4.18. (R)-(1-(4-dodecylbenzyl)pyrrolidin-2-yl)methanol (22d).
White solid, yield 89%. 1H NMR (400 MHz, CD3OD) δ: 7.43 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 8.5 Hz, 2H), 4.59 (d, J = 12.7 Hz, 1H), 4.21 (d, J = 12.9 Hz, 1H), 3.79 – 3.71 (m, 1H), 3.71 – 3.64 (m, 2H), 3.40 – 3.35 (m, 1H), 3.27 – 3.21 (m, 1H), 2.69 – 2.61 (m, 2H), 2.27 – 2.18 (m, 1H), 2.15 – 2.03 (m, 1H), 2.00 – 1.84 (m, 2H), 1.62 (p, J = 7.7 Hz, 2H), 1.35 – 1.23 (m, 18H), 0.92 – 0.86 (m, 3H). 13C NMR (101 MHz, CD3OD) δ: 146.4, 131.9, 130.4, 129.0, 69.8, 60.7, 59.4, 55.5, 36.6, 33.1, 32.6, 30.8, 30.8, 30.7, 30.7, 30.6, 30.5, 30.3, 27.3, 23.7, 23.1, 14.4. HRMS (ESI + ): calcd for C24H42NO [M + H]+ 360.3261, found: 360.3266.
4.2.4.19. (R)-(1-(4-dodecyl-2-methylbenzyl)pyrrolidin-2-yl)methanol (22e).
White solid, yield 98%. 1H NMR (400 MHz, CD3OD) δ: 7.36 (d, J = 7.8 Hz, 1H), 7.16 (s, 1H), 7.11 (dd, J = 7.8, 1.8 Hz, 1H), 4.77 (d, J = 13.2 Hz, 1H), 4.15 (d, J = 13.2 Hz, 1H), 3.96 – 3.89 (m, 1H), 3.76 – 3.69 (m, 2H), 3.41 – 3.33 (m, 1H), 2.61 (t, J = 7.7 Hz, 2H), 2.47 (s, 3H), 2.34 – 2.21 (m, 1H), 2.18 – 2.06 (m, 1H), 2.00 – 1.86 (m, 2H), 1.65 – 1.56 (m, 2H), 1.37 – 1.23 (m, 19H), 0.90 (t, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ: 146.5, 139.5, 132.8, 132.5, 127.8, 127.6, 70.5, 60.4, 57.0, 55.6, 36.5, 33.1, 32.5, 30.8, 30.7, 30.7, 30.7, 30.6, 30.5, 30.3, 27.0, 23.7, 23.0, 19.5, 14.4. HRMS (ESI + ): calcd for C25H44NO [M + H]+ 374.3417, found: 374.3427.
4.2.4.20. (R)-(1-(4-(4-methylphenethyl)benzyl)pyrrolidin-2-yl)methanol (22f).
White solid, yield 93%. 1H NMR (400 MHz, CD3OD) δ: 7.40 (d, J = 8.1 Hz, 2H), 7.23 (d, J = 8.1 Hz, 2H), 7.03 – 6.96 (m, 4H), 4.56 (d, J = 12.9 Hz, 1H), 4.21 (d, J = 12.9 Hz, 1H), 3.74 – 3.63 (m, 3H), 3.37 – 3.32 (m, 1H), 3.26 – 3.19 (m, 1H), 2.94 – 2.81 (m, 4H), 2.24 (s, 3H), 2.20 (d, J = 3.2 Hz, 1H), 2.13 – 2.02 (m, 1H), 1.97 – 1.85 (m, 2H). 13C NMR (101 MHz, CD3OD) δ: 143.9, 138.0, 135.0, 130.4, 129.2, 128.5, 128.0, 127.8, 68.4, 59.4, 58.0, 54.1, 37.3, 36.8, 25.9, 21.8, 19.7. HRMS (ESI + ): calcd for C21H28NO [M + H]+ 310.2165, found: 310.2176.
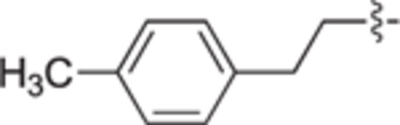
4.2.4.21. (R)-(1-(4-(4-methoxyphenethyl)benzyl)pyrrolidin-2-yl)methanol (22 g).
White solid, yield 94%. 1H NMR (400 MHz, CD3OD) δ: 7.24 (d, J = 8.0 Hz, 2H), 7.14 (d, J = 8.0 Hz, 2H), 7.09 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 3.98 (d, J = 12.9 Hz, 1H), 3.79 (s, 3H), 3.67 (dd, J = 11.1, 3.4 Hz, 1H), 3.50 – 3.42 (m, 2H), 3.05 (ddd, J = 9.7, 5.8, 4.0 Hz, 1H), 2.89 – 2.82 (m, 5H), 2.42 – 2.34 (m, 1H), 1.98 – 1.92 (m, 1H), 1.90 – 1.83 (m, 1H), 1.78 – 1.71 (m, 2H). 13C NMR (101 MHz, CD3OD) δ: 158.0, 141.3, 135.6, 133.9, 129.5, 129.2, 128.7, 113.9, 64.9, 61.7, 58.5, 55.4, 54.5, 38.0, 37.1, 27.8, 23.6. HRMS (ESI + ): calcd for C21H28NO2 [M + H]+ 326.2115, found: 326.2135.
4.2.4.22. (R)-(1-(4-undecylbenzyl)pyrrolidin-2-yl)methyl acetate (22 h).
White solid, 90%. 1H NMR (400 MHz, CD3OD) δ 7.49 (d, J = 8.0 Hz, 2H), 7.32 (d, J = 7.9 Hz, 2H), 4.57 (d, J = 12.9 Hz, 1H), 4.36 – 4.31 (m, 2H), 4.20 (dd, J = 12.8, 6.8 Hz, 1H), 3.98 – 3.93 (m, 1H), 3.49 (ddd, J = 12.0, 7.3, 5.3 Hz, 1H), 3.41 – 3.34 (m, 1H), 2.66 (dd, J = 8.5, 6.8 Hz, 2H), 2.41 – 2.31 (m, 1H), 2.19 – 2.12 (m, 1H), 2.08 (s, 3H), 2.05 – 1.91 (m, 2H), 1.67 – 1.59 (m, 2H), 1.31 (d, J = 17.3 Hz, 16H), 0.92 – 0.87 (m, 3H). 13C NMR (101 MHz, CD3OD) δ: 171.8, 146.5, 132.0, 130.5, 128.8, 67.2, 63.1, 59.6, 56.0, 36.6, 33.1, 32.5, 30.8, 30.7, 30.7, 30.6, 30.5, 30.3, 27.6, 23.7, 23.1, 20.6, 14.5. HRMS (ESI + ): calcd for C25H42NO2 [M + H]+ 388.3210, found: 388.3240.
4.2.4.23. (R,E)-(1-(4-(4-(tert-butyl)styryl)-2-methylbenzyl)pyrrolidin-2-yl)methanol (25a).
White solid, yield 94%. 1H NMR (400 MHz, CD3OD) δ: 7.54 – 7.47 (m, 3H), 7.47 – 7.43 (m, 1H), 7.44 – 7.38 (m, 2H), 7.18 (q, J = 16.3 Hz, 3H), 4.20 (d, J = 13.2 Hz, 1H), 4.00 – 3.91 (m, 1H), 3.81 – 3.70 (m, 2H), 2.53 (s, 3H), 2.36 – 2.24 (m, 2H), 2.21 – 2.09 (m, 2H), 2.03 – 1.89 (m, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, CD3OD) δ: 152.3, 141.0, 140.0, 135.6, 133.2, 131.2, 130.3, 129.1, 127.7, 127.5, 126.7, 125.4, 70.5, 60.4, 56.9, 55.7, 35.5, 31.7, 27.0, 23.0, 19.5. HRMS (ESI + ): calcd for C25H33NO [M + H]+ 363.2562, found: 363.2564.
4.2.4.24. (R,E)-(1-(4-(3-(trifluoromethyl)styryl)benzyl)pyrrolidin-2-yl) methanol (25b).
White solid, yield 94%. 1H NMR (400 MHz, CD3OD) δ: 7.86 (s, 2H), 7.73 (d, J = 7.5 Hz, 2H), 7.61 – 7.53 (m, 4H), 7.36 (s, 2H), 4.67 (d, J = 12.9 Hz, 1H), 4.29 (d, J = 13.0 Hz, 1H), 3.86 – 3.68 (m, 3H), 3.46 – 3.37 (m, 1H), 3.30 – 3.22 (m, 1H), 2.33 – 2.20 (m, 1H), 2.19 – 2.06 (m, 1H), 2.05 – 1.87 (m, 2H). 13C NMR (101 MHz, CD3OD) δ: 140.1, 139.6, 132.4, 132.0, 131.2, 131.0, 131.0, 131.0, 131.0, 130.6, 130.6, 129.8, 128.6, 125.4, 125.4, 125.3, 125.3, 124.4, 124.3, 124.3, 124.3, 70.0, 60.7, 59.3, 55.6, 27.3, 23.2. HRMS (ESI + ): calcd for C21H22F3NO [M + H]+ 361.1653, found: 361.1663.
4.2.4.25. (R)-(1-(4-(4-propylphenethyl)benzyl)pyrrolidin-2-yl)methanol (29a).
White solid, yield 90%. 1H NMR (400 MHz, CD3OD) δ: 7.41 (d, J = 8.1 Hz, 2H), 7.29 – 7.24 (m, 2H), 7.04 (s, 4H), 4.58 (d, J = 12.8 Hz, 1H), 4.21 (d, J = 12.9 Hz, 1H), 3.69 (dt, J = 13.4, 7.2 Hz, 2H), 3.36 (ddd, J = 6.7, 4.8, 2.4 Hz, 1H), 3.24 (dt, J = 11.4, 7.9 Hz, 1H), 2.98 – 2.85 (m, 4H), 2.55 – 2.50 (m, 2H), 2.26 – 2.19 (m, 1H), 2.12 – 2.05 (m, 1H), 1.93 (dq, J = 12.9, 6.9, 6.2 Hz, 2H), 1.65 – 1.55 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ: 145.4, 141.3, 139.7, 131.8, 130.6, 129.4, 129.4, 127.7, 69.7, 60.7, 59.4, 55.5, 38.7, 38.6, 38.3, 27.3, 25.8, 23.1, 14.1. HRMS (ESI + ): calcd for C23H32NO [M + H]+ 338.2478, found: 338.2498.
4.2.4.26. (R)-(1-(4-(4-butylphenethyl)benzyl)pyrrolidin-2-yl)methanol (29b).
White solid, yield 94%. 1H NMR (400 MHz, CDCl3) δ: 7.31 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H), 7.16 (s, 4H), 4.06 (d, J = 13.0 Hz, 1H), 3.74 (dd, J = 11.1, 3.4 Hz, 1H), 3.59 – 3.49 (m, 2H), 3.13 (dd, J = 9.7, 5.5 Hz, 1H), 2.99 – 2.87 (m, 4H), 2.68 – 2.59 (m, 2H), 2.51 – 2.40 (m, 1H), 2.06 – 1.85 (m, 2H), 1.87 – 1.74 (m, 2H), 1.66 (dt, J = 15.2, 7.5 Hz, 2H), 1.42 – 1.34 (m, 2H), 1.00 – 0.91 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 141.7, 140.9, 139.3, 132.5, 129.5, 128.9, 128.8, 128.7, 65.2, 62.0, 58.8, 54.7, 38.1, 37.9, 36.0, 31.7, 28.0, 23.9, 23.0, 14.5. HRMS (ESI + ): calcd for C24H34NO [M + H]+ 352.2635, found: 352.2638.
4.2.4.27. (R)-(1-(4-(4-pentylphenethyl)benzyl)pyrrolidin-2-yl)methanol (29c).
White solid, yield 98%. 1H NMR (400 MHz, CDCl3) δ: 7.30 (d, J = 8.3 Hz, 2H), 7.21 (d, J = 8.1 Hz, 2H), 7.15 (s, 4H), 4.05 (d, J = 12.8 Hz, 1H), 3.73 (dd, J = 11.0, 3.3 Hz, 1H), 3.58 – 3.48 (m, 2H), 3.12 (dd, J = 9.7, 5.6 Hz, 1H), 2.98 – 2.86 (m, 4H), 2.67 – 2.58 (m, 2H), 2.50 – 2.39 (m, 1H), 2.05 – 1.84 (m, 2H), 1.86 – 1.73 (m, 2H), 1.65 (dt, J = 15.0, 7.6 Hz, 2H), 1.41 – 1.33 (m, 2H), 0.99 – 0.90 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 141.9, 141.1, 139.5, 132.7, 129.7, 129.1, 129.0, 128.9, 65.4, 62.2, 59.0, 54.9, 38.3, 38.1, 36.2, 32.2, 31.9, 28.2, 24.1, 23.2, 14.7. HRMS (ESI + ): calcd for C25H36NO [M + H]+ 366.2791, found: 366.2814.
4.2.4.28. (R)-(1-(4-((4-propylphenyl)ethynyl)benzyl)pyrrolidin-2-yl) methanol (30a).
Yellow solid, yield 98%. 1H NMR (400 MHz, CD3CD) δ: 7.62 – 7.59 (m, 2H), 7.57 – 7.54 (m, 2H), 7.44 – 7.41 (m, 2H), 7.23 – 7.18 (m, 2H), 4.70 – 4.63 (m, 1H), 4.29 (d, J = 13.0 Hz, 1H), 3.82 – 3.69 (m, 3H), 3.43 – 3.36 (m, 1H), 3.28 – 3.24 (m, 1H), 2.61 (dd, J = 8.4, 6.8 Hz, 2H), 2.29 – 2.23 (m, 1H), 2.16 – 2.09 (m, 1H), 2.01 – 1.88 (m, 2H), 1.71 – 1.59 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CD3CD) δ: 143.6, 131.7, 131.2, 130.7, 130.1, 128.4, 125.2, 119.9, 90.7, 87.1, 68.7, 59.4, 57.8, 54.2, 37.5, 25.9, 24.1, 21.8, 12.7. HRMS (ESI + ): calcd for C23H27NO [M + H]+ 333.2093, found: 333.2103.
4.2.4.29. (R)-(1-(4-((4-butylphenyl)ethynyl)benzyl)pyrrolidin-2-yl)methanol (30b).
Yellow solid, yield 93%. 1H NMR (400 MHz, CD3CD) δ: 7.63 – 7.59 (m, 2H), 7.56 – 7.52 (m, 2H), 7.44 – 7.40 (m, 2H), 7.23 – 7.19 (m, 2H), 4.66 (d, J = 12.9 Hz, 1H), 4.27 (d, J = 12.9 Hz, 1H), 3.82 – 3.68 (m, 3H), 3.42 – 3.35 (m, 1H), 3.29 – 3.23 (m, 1H), 2.66 – 2.61 (m, 2H), 2.29 – 2.22 (m, 1H), 2.17 – 2.09 (m, 1H), 2.00 – 1.90 (m, 2H), 1.65 – 1.57 (m, 2H), 1.41 – 1.31 (m, 2H), 0.94 (s, 3H). 13C NMR (101 MHz, CD3CD) δ: 145.3, 133.1, 132.6, 132.1, 131.5, 129.7, 126.7, 121.2, 92.2, 88.4, 70.1, 60.7, 59.2, 55.6, 36.5, 34.7, 27.2, 23.3, 23.1, 14.2. HRMS (ESI + ): calcd for C24H29NO [M + H]+ 347.2249, found: 347.2269.
4.2.4.30. (R)-(1-(4-((4-pentylphenyl)ethynyl)benzyl)pyrrolidin-2-yl) methanol (30c).
Yellow solid, yield 99%. 1H NMR (400 MHz, CD3CD) δ: 7.63 – 7.59 (m, 2H), 7.57 – 7.53 (m, 2H), 7.44 – 7.40 (m, 2H), 7.22 – 7.17 (m, 2H), 4.66 (d, J = 12.9 Hz, 1H), 4.28 (d, J = 13.0 Hz, 1H), 3.84 – 3.68 (m, 3H), 3.40 (ddd, J = 11.5, 7.3, 4.5 Hz, 1H), 3.29 – 3.23 (m, 1H), 2.65 – 2.60 (m, 2H), 2.29 – 2.22 (m, 1H), 2.16 – 2.08 (m, 1H), 2.00 – 1.87 (m, 2H), 1.67 – 1.58 (m, 2H), 1.39 – 1.30 (m, 4H), 0.93 – 0.88 (m, 3H). 13C NMR (101 MHz, CD3CD) δ: 145.3, 133.1, 132.6, 132.1, 131.5, 129.7, 126.7, 121.2, 92.1, 88.5, 70.1, 60.7, 59.2, 55.6, 36.8, 32.5, 32.1, 27.2, 23.5, 23.1, 14.4. HRMS (ESI + ): calcd for C25H31NO [M + H]+ 361.2406, found: 361.2408.
4.2.5. General procedure for the synthesis of compounds 14a-e
To a round bottom flask containing DMF the appropriate phenol analogue (1.5 equiv.), sodium bicarbonate (5.0 equiv.) and 4-fluorobenzonitrile (1.0 equiv.) were added and refluxed under nitrogen gas for 12 h. After, the resulting solution was partitioned between EtOAc and LiBr aqueous solution. Using additional EtOAc, the aqueous LiBr solution was washed three times and the combined organic layers were dried over Na2SO4, filtered, and concentrated via vacuum. The resulting concentrate was purified by silica gel chromatography.
4.2.5.1. 4-phenoxybenzonitrile (14a).
White solid, yield 80%. 1H NMR (400 MHz, CDCl3) δ: 7.57 (d, J = 9.0 Hz, 2H), 7.40 (dd, J = 8.5, 7.6 Hz, 2H), 7.22 (t, J = 7.4 Hz, 1H), 7.06 (dd, J = 8.6, 1.1 Hz, 2H), 6.99 (d, J = 9.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 161.6, 154.7, 134.1, 130.2, 125.1, 120.4, 118.8, 117.9, 105.7. HRMS (ESI + ): calcd for C13H10NO [M + H]+ 196.0757, found: 196.0744.
4.2.5.2. 4-(3-(tert-butyl)phenoxy)benzonitrile (14b).
White solid, yield 70%. 1H NMR (400 MHz, CDCl3) δ: 7.58 (d, J = 9.0 Hz, 2H), 7.32 (t, J = 7.9 Hz, 1H), 7.26 – 7.22 (m, 1H), 7.09 (t, J = 2.1 Hz, 1H), 6.98 (d, J = 8.8 Hz, 2H), 6.84 (ddd, J = 7.9, 2.4, 1.1 Hz, 1H), 1.30 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 162.0, 154.6, 154.3, 134.2, 129.8, 122.3, 119.0, 117.8, 117.8, 117.4, 105.6, 35.0, 31.4. HRMS (ESI + ): calcd for C17H18NO [M + H]+ 252.1383, found: 252.1363.
4.2.5.3. 4-(4-(tert-butyl)phenoxy)benzonitrile (14c).
White solid, yield 73%. 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 8.9 Hz, 2H), 7.42 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 2.3 Hz, 2H), 6.98 (d, J = 2.2 Hz, 2H), 1.35 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 162.0, 152.3, 148.2, 134.1, 127.1, 120.0, 119.0, 117.7, 105.5, 34.5, 31.5. HRMS (ESI + ): calcd for C17H18NO: [M + H]+ 252.1383, found: 252.1366.
4.2.5.4. 4-(3-bromophenoxy)benzonitrile (14d).
White solid, yield 79%. 1H NMR (400 MHz, CDCl3) δ: 7.62 (d, J = 8.9 Hz, 2H), 7.35 (ddd, J = 8.0, 1.8, 1.1 Hz, 1H), 7.27 (t, J = 8.1 Hz, 1H), 7.23 – 7.20 (m, 1H), 7.04 – 6.98(m, 3H). 13C NMR (101 MHz, CDCl3) δ: 160.9, 155.8, 134.4, 131.4, 128.2, 123.6, 118.9, 118.7, 118.5, 106.7. HRMS (ESI + ): calcd for C13H9BrNO [M + H]+ 273.9862, found: 273.9860.
4.2.5.5. 4-(4-iodophenoxy)benzonitrile (14e).
White solid, yield 82%. 1H NMR (400 MHz, CDCl3) δ: 7.68 (d, J = 5.9 Hz, 2H), 7.60 (dd, J = 5.0, 2.3 Hz, 2H), 7.00 (d, J = 8.9 Hz, 2H), 6.82 (d, J = 8.9 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 161.0, 155.0, 139.4, 134.4, 122.5, 118.7, 118.3, 106.5. HRMS (ESI + ): calcd for C13H9INO [M + H]+ 321.9723, found: 321.9751.
4.2.6. General procedure for the synthesis of compounds 15a-e
To a round bottom flask containing THF, the appropriate aryl nitrile analogue (1.0 equiv.) was added followed by the addition of nitrogen gas. Next, the solution was cooled to − 70 °C before the addition of a DIBAL-H (1.1 equiv.) solution (1.0 M in THF). After, the mixture was stirred at − 40 °C for 5 h under nitrogen until TLC indicated the starting material had been fully consumed. Subsequently, the reaction vessel was placed in an ice bath and quenched with 1 mL ethyl acetate and 20 mL HCl (1.0 M). After, the resulting solution was partitioned between EtOAc and brine. Using additional EtOAc, the aqueous brine solution was washed three times and the combined organic layers were dried over Na2SO4, filtered, and concentrated via vacuum. The resulting concentrate was purified by silica gel chromatography.
4.2.6.1. 4-phenoxybenzaldehyde (15a).
White solid, yield 82%. 1H NMR (400 MHz, CDCl3) δ: 9.92 (s, 1H), 7.84 (d, J = 8.8 Hz, 2H), 7.46 – 7.37 (m, 2H), 7.23 (t, J = 7.4 Hz, 1H), 7.09 (dd, J = 8.7, 1.1 Hz, 2H), 7.06 (d, J = 8.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 190.9, 163.3, 155.2, 132.1, 131.4, 130.3, 125.1, 120.5, 117.7. HRMS (ESI + ) [M + H]+ calcd for C13H11O2: 199.0754, found: 199.0753.
4.2.6.2. 4-(3-(tert-butyl)phenoxy)benzaldehyde (15b).
White solid, yield 65%. 1H NMR (400 MHz, CDCl3) δ: 9.91 (s, 1H), 7.84 (d, J = 8.8 Hz, 2H), 7.33 (t, J = 7.9 Hz, 1H), 7.29 – 7.22 (m, 1H), 7.14 – 7.10 (m, 1H), 7.05 (d, J = 8.7 Hz, 1H), 6.91 – 6.83 (m, 1H), 1.32 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 190.9, 163.6, 154.9, 154.1, 132.1, 131.2, 129.7, 122.1, 117.8, 117.5, 117.4, 31.4. HRMS (ESI + ) [M + H]+ calcd for C17H19O2: 255.1380, found: 255.1369.
4.2.6.3. 4-(4-(tert-butyl)phenoxy)benzaldehyde (15c).
White solid, yield 69%. 1H NMR (400 MHz, CDCl3) δ: 9.91 (s, 1H), 7.83 (d, J = 8.8 Hz, 2H), 7.42 (d, J = 8.8 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H), 7.01 (d, J = 8.8 Hz, 2H), 1.35 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 190.9, 163.7, 152.7, 148.1, 132.1, 131.2, 127.1, 120.1, 117.5, 34.6, 31.6. HRMS (ESI + ) [M + H]+ calcd for C17H19O2: 255.1380, found: 255.1372.
4.2.6.4. 4-(3-bromophenoxy)benzaldehyde (15d).
White solid, yield 66%. 1H NMR (400 MHz, CDCl3) δ: 9.91 (s, 1H), 7.84 (d, J = 8.6 Hz, 2H), 7.34 – 7.30 (m, 1H), 7.23 (d, J = 5.2 Hz, 1H), 7.21 (dd, J = 3.6, 1.5 Hz, 1H), 7.05 (d, J = 8.7 Hz, 2H), 7.01 – 6.97 (m, 1H). 13C NMR (101 MHz, CDCl3) δ: 190.8, 162.4, 156.2, 132.2, 132.0, 131.3, 128.1, 123.6, 123.3, 119.0, 118.2. HRMS (ESI + ) calcd for C13H10BrO2 [M + H]+ 276.9859, found: 276.9860.
4.2.6.5. 4-(4-iodophenoxy)benzaldehyde (15e).
White solid, yield 69%. 1H NMR (400 MHz, CDCl3) δ: 9.93 (s, 1H), 7.86 (d, J = 8.9 Hz, 2H), 7.71 (d, J = 8.9 Hz, 2H), 7.07 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.9 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 190.8, 162.6, 155.4, 139.3, 132.1, 131.8, 122.5, 118.0, 88.3. HRMS (ESI + ): calcd for C13H10IO2 [M + H]+ 324.9720, found: 324.9701.
4.2.7. General procedure for the synthesis of compounds 16d-i
To a microwave reaction tube, the appropriate aryl halide (1.0 equiv.) and aryl boronic acid (1.3 equiv.) were dissolved in THF. Next, the resulting solution was purged with argon gas for 5 min followed by the addition of sodium hydroxide (aq.) (3.0 equiv.) to form a THF/H2O = 5:1 mixture. After, the mixture was further purged with argon gas for 5 min before the addition of Pd(dppf)Cl2 (0.1 equiv.). The resulting mixture was then heated in a microwave reactor at 100 °C for 10 min. After, the solid was removed via filtration and the resulting solution was partitioned between ethyl acetate and water. The organic layer was washed with a saturated sodium bicarbonate solution and brine, dried over sodium sulfate, and concentrated via vacuum. The resulting mixture was then purified by silica gel chromatography to yield the desired product.
4.2.7.1. 4-([1,1′-biphenyl]-3-yloxy)benzaldehyde (16d).
Off-white solid, yield 53%. 1H NMR (400 MHz, CDCl3) δ: 9.93 (t, J = 0.4 Hz, 1H), 7.87 (d, J = 8.8 Hz, 2H), 7.58 (dd, J = 8.3, 1.4 Hz, 2H), 7.51 – 7.41 (m, 4H), 7.39 – 7.34 (m, 1H), 7.33 (ddd, J = 2.3, 1.4, 0.7 Hz, 1H), 7.12 (d, J = 8.4 Hz, 2H), 7.07 (dt, J = 6.8, 2.5 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ: 190.9, 163.3, 155.7, 143.7, 140.2, 132.1, 131.5, 130.6, 129.0, 128.0, 127.2, 123.8, 119.2, 117.8. HRMS (ESI + ): calcd for C19H15O2 [M + H]+ 275.1067, found: 275.1077.
4.2.7.2. 4-([1,1′-biphenyl]-4-yloxy)benzaldehyde (16e).
White solid, yield 61%. 1H NMR (400 MHz, CDCl3) δ: 9.94 (s, 1H), 7.87 (d, J = 8.8 Hz, 2H), 7.63 (d, J = 8.7 Hz, 2H), 7.60 (dd, J = 8.4, 1.3 Hz, 2H), 7.46 (t, J = 7.5 Hz, 2H), 7.41 – 7.32 (m, 2H), 7.16 (d, J = 8.7 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 190.9, 163.3, 154.7, 140.3, 138.2, 132.1, 131.5, 129.0, 128.9, 127.5, 127.1, 120.8, 117.8. HRMS (ESI + ): calcd for C19H15O2 [M + H]+ 275.1067, found: 275.1057.
4.2.7.3. 4-((3′ -(trifluoromethyl)-[1,1′ -biphenyl]-3-yl)oxy)benzaldehyde (16f).
Off-white solid, yield 58%. 1H NMR (400 MHz, CDCl3) δ: 9.94 (s, 1H), 7.88 (d, J = 8.7 Hz, 2H), 7.82 (s, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.61 – 7.51 (m, 1H), 7.51 (t, J = 7.9 Hz, 1H), 7.46 (dt, J = 7.7, 1.6 Hz, 1H), 7.37 – 7.31 (m, 1H), 7.12 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 190.9, 163.1, 155.9, 142.2, 141.0, 132.2, 131.9 (q, 2J CF3 = 33.6 Hz), 130.9, 130.5, 130.5, 129.5, 124.6 (q, 3J CF3 = 3.8 Hz), 124.0 (q, 4J CF = 3.9 Hz), 123.8, 122.6, 121.2 (q, 1J CF3 = 277.1 Hz), 120.0, 119.3, 117.8. HRMS (ESI + ): calcd for C20H14F3O2 [M + H]+ 343.0940, found: 343.0943.
4.2.7.4. 4-((4′ -(trifluoromethyl)-[1,1′ -biphenyl]-3-yl)oxy)benzaldehyde (16 g).
Off-white solid, yield 50%. 1H NMR (400 MHz, CDCl3) δ: 9.94 (s, 1H), 7.88 (d, J = 8.8 Hz, 2H), 7.74 – 7.63 (m, 4H), 7.52 (t, J = 7.8 Hz, 1H), 7.50 – 7.42 (m, 1H), 7.36 – 7.30 (m, 1H), 7.15 – 7.09 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 190.8, 163.0, 155.9, 143.6, 142.2, 132.1, 131.7, 130.9, 127.5, 126.0, 123.8, 120.0, 119.3, 117.9. HRMS (ESI + ): calcd for C20H14F3O2 [M + H]+ 343.0940, found: 343.0949.
4.2.7.5. 4-((3′ -(trifluoromethyl)-[1,1′ -biphenyl]-4-yl)oxy)benzaldehyde (16 h).
Off-white solid, yield 59%. 1H NMR (400 MHz, CDCl3) δ: 9.95 (s, 1H), 7.88 (d, J = 8.7 Hz, 2H), 7.83 (s, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.68 – 7.53 (m, 4H), 7.19 (d, J = 8.6 Hz, 2H), 7.13 (d, J = 8.7 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 190.9, 163.0, 155.4, 141.1, 136.6, 132.1, 131.7, 130.4, 129.5, 129.1, 124.2, 120.9, 118.0. HRMS (ESI + ): calcd for C20H14F3O2 [M + H]+ 343.0940, found: 343.0946.
4.2.7.6. 4-((4′ -(trifluoromethyl)-[1,1′ -biphenyl]-4-yl)oxy)benzaldehyde (16i).
Off-white solid, yield 63%. 1H NMR (400 MHz, CDCl3) δ: 9.95 (s, 1H), 7.88 (d, J = 8.7 Hz, 2H), 7.70 (d, J = 2.3 Hz, 4H), 7.64 (d, J = 8.6 Hz, 2H), 7.19 (d, J = 8.6 Hz, 2H), 7.13 (d, J = 8.6 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 190.8, 162.9, 155.6, 143.8, 136.5, 132.1, 131.7, 129.1, 127.4, 125.9, 120.8, 118.1. HRMS (ESI + ): calcd for C20H14F3O2 [M + H]+ 343.0940, found: 343.0935.
4.2.8. General procedure for the synthesis of compounds 20a-g
To a microwave reaction tube containing THF (0.2 M), the appropriate alkene (1.3 equiv.) and 9-BBN (1.6 equiv.) were dissolved followed by addition of argon gas. Next, the resulting mixture was heated in a microwave reactor at 100 °C for 10 min. After heating, the solution was again purged with argon for 5 min followed by the addition of sodium hydroxide (aq.) (3.0 equiv.) to form a THF/H2O = 5:1 mixture. Once more, the mixture was further purged with argon for 5 min followed by the addition of 4-iodobenzaldehyde (1.0 equiv.) then Pd(dppf)Cl2 (0.1 equiv.). The resulting mixture was heated in a microwave reactor at 100 °C for 10 min. After, the solid was removed via filtration and solution was partitioned between ethyl acetate and water. The resulting organic layer was washed with saturated sodium bicarbonate solution and brine, dried over sodium sulfate, and concentrated via vacuum. The resulting mixture was then purified by silica gel chromatography to yield the desired product.
4.2.8.1. 4-nonylbenzaldehyde (20a).
Tan oil, yield 54%. 1H NMR (400 MHz, CDCl3) δ: 9.97 (s, 1H), 7.79 (d, J = 8.3 Hz, 2H), 7.33 (d, J = 7.9 Hz, 2H), 2.72 – 2.64 (m, 2H), 1.64 (p, J = 7.5 Hz, 2H), 1.36 – 1.22 (m, 12H), 0.97 – 0.81 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 192.2, 150.7, 134.5, 130.0, 129.2, 36.4, 32.0, 31.2, 29.7, 29.6, 29.4, 29.4, 22.8, 14.3. HRMS (ESI + ): calcd for C16H25O [M + H]+ 233.1900, found: 233.1905.
4.2.8.2. 4-decylbenzaldehyde (20b).
Tan oil, yield 51%. 1H NMR (400 MHz, CDCl3) δ: 9.97 (s, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 2.95 – 2.43 (m, 2H), 1.63 (m, 2H), 1.28 (d, J = 20.3 Hz, 14H), 1.15 – 0.61 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 192.2, 150.7, 134.5, 130.0, 129.2, 36.4, 32.0, 31.2, 29.7, 29.7, 29.6, 29.5, 29.4, 22.8, 14.3. HRMS (ESI + ): calcd for C17H27O [M + H]+247.2056, found: 247.2059.
4.2.8.3. 4-undecylbenzaldehyde (20c).
Tan oil, yield 59%. 1H NMR (400 MHz, CDCl3) δ: 9.97 (s, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 3.00 – 2.46 (m, 2H), 1.64 (p, J = 7.4 Hz, 2H), 1.45 – 1.05 (m, 16H), 1.15 – 0.56 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 192.2, 150.7, 134.5, 130.0, 129.2, 36.4, 32.1, 31.2, 29.8, 29.8, 29.7, 29.6, 29.5, 29.4, 22.8, 14.3. HRMS (ESI + ): calcd for C18H29O [M + H]+ 261.2213, found: 261.2214.
4.2.8.4. 4-dodecylbenzaldehyde (20d).
Brown oil, yield 61%. 1H NMR (400 MHz, CDCl3) δ: 9.97 (s, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 2.72 – 2.64 (m, 2H), 1.64 (dt, J = 15.0, 7.4 Hz, 2H), 1.37 – 1.21 (m, 18H), 0.91 – 0.84 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 192.2, 150.7, 134.5, 130.0, 129.2, 36.4, 32.1, 31.2, 29.8, 29.8, 29.7, 29.6, 29.5, 29.4, 22.8, 14.3. HRMS (ESI + ): calcd for C19H31O [M + H]+ 275.2369, found: 275.2371.
4.2.8.5. 4-dodecyl-2-methylbenzaldehyde (20e).
Clear oil, yield 59%. 1H NMR (400 MHz, CDCl3) δ: 9.98 (s, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 7.9 Hz, 2H), 7.09 (d, J = 7.8 Hz, 2H), 7.04 (d, J = 8.1 Hz, 2H), 3.02 – 2.96 (m, 2H), 2.91 (ddd, J = 8.7, 6.5, 2.0 Hz, 2H), 2.32 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 192.2, 149.4, 138.0, 135.8, 134.7, 130.0, 129.3, 128.4, 38.3, 37.1, 21.2. HRMS (ESI + ): calcd for C20H33O [M + H]+ 289.2526, found: 289.2545.
4.2.8.6. 4-(4-methylphenethyl)benzaldehyde (20f).
Yellow oil, yield 58%. 1H NMR (400 MHz, CDCl3) δ 9.98 (s, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 7.9 Hz, 2H), 7.09 (d, J = 7.8 Hz, 2H), 7.04 (d, J = 8.1 Hz, 2H), 3.02 – 2.96 (m, 2H), 2.91 (ddd, J = 8.7, 6.5, 2.0 Hz, 2H), 2.32 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 192.2, 149.4, 138.0, 135.8, 134.7, 130.0, 129.3, 128.4, 38.3, 37.1, 21.2. HRMS (ESI + ): calcd for C16H17O [M + H]+ 225.1274, found: 225.1285.
4.2.8.7. 4-(4-methoxyphenethyl)benzaldehyde (20 g).
Tan oil, yield 52%. 1H NMR (400 MHz, CDCl3) δ 9.97 (s, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 7.9 Hz, 2H), 7.05 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 3.79 (s, 3H), 2.97 (ddd, J = 9.0, 6.7, 2.0 Hz, 2H), 2.89 (ddd, J = 8.5, 6.6, 2.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 192.2, 158.1, 149.3, 134.7, 133.1, 130.0, 129.5, 129.4, 114.0. HRMS (ESI + ): calcd for C16H17O2 [M + H]+ 241.1223, found: 241.1233.
4.2.9. General procedure for the synthesis of compound 21 h
To a round bottom flask containing CH2Cl2 (0.2 M), compound 21c (1 equiv.) was dissolved and then cooled in an ice bath for 5 min. After, pyridine (3.0 equiv.) was added slowly followed by acetic anhydride (2.0 equiv.). The mixture was allowed to slowly rise to room temperature and stirred overnight. After, the resulting solution was partitioned between EtOAc and brine. Using additional EtOAc, the aqueous brine solution was washed three times and the combined organic layers were dried over Na2SO4, filtered, and concentrated via vacuum. The resulting concentrate was purified by silica gel chromatography to yield the final product.
4.2.9.1. (R)-(1-(4-undecylbenzyl)pyrrolidin-2-yl)methyl acetate (21 h).
Tan oil, yield 72%. 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J = 8.0 Hz, 2H), 7.11 (d, J = 8.2 Hz, 2H), 4.11 (dd, J = 11.0, 5.2 Hz, 1H), 4.05 – 4.00 (m, 2H), 3.38 (d, J = 12.9 Hz, 1H), 2.93 (ddd, J = 9.4, 6.9, 3.0 Hz, 1H), 2.80 (dq, J = 8.5, 5.7 Hz, 1H), 2.61 – 2.54 (m, 2H), 2.31 – 2.18 (m, 1H), 2.06 (s, 3H), 1.98 – 1.88 (m, 1H), 1.76 – 1.55 (m, 5H), 1.26 (m, 16H), 0.87 (t, 3H). 13C NMR (101 MHz, CDCl3) δ 171.2, 141.7, 136.7, 67.3, 61.9, 59.3, 54.5, 35.8, 32.1, 31.7, 29.8, 29.8, 29.7, 29.7, 29.5, 28.6, 23.0, 22.8, 21.2, 14.3. HRMS (ESI + ): calcd for C25H42NO2 [M + H]+ 388.3210, found: 388.3204.
4.2.10. General procedure for the synthesis of compounds 23a-b
Aryl halide (1 equiv.), aryl alkene (1.3 equiv.) and potassium carbonate (3 equiv.) were added into microwave reaction tube containing DMF. The mixture was purged with argon for 5 min before the addition of Pd(PPh)3Cl2 (0.1 equiv.) and was heated to 140 °C for 2 h in the microwave. The reaction was extracted with ethyl acetate and saturated LiBr solution. The combined organic layers were washed with brine and dried over sodium sulfate. After filtration and concentration via reduced pressure, the resulting oil residue was purified on a silica column with hexane and ethyl acetate as the eluent to yield pure product. The residue was purified using silica gel column chromatography (5% ethyl acetate/hexanes) to yield the product.
4.2.10.1. (E)-4-(4-(tert-butyl)styryl)-2-methylbenzaldehyde (23a).
White solid, yield 65%. 1H NMR (400 MHz, CDCl3) δ: 10.21 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.19 – 7.14 (m, 1H), 7.09 – 7.04 (m, 1H), 2.65 (s, 3H), 2.63 – 2.60 (m, 2H), 1.66 – 1.58 (m, 2H), 1.35 – 1.22 (m, 18H), 0.88 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 192.6, 149.7, 140.8, 132.6, 132.3, 132.1, 126.6, 36.2, 36.0, 32.1, 31.2, 31.2, 29.8, 29.8, 29.7, 29.6, 29.6, 29.5, 29.5, 22.8, 19.8, 14.3. HRMS (ESI + ): calcd for C20H23O [M + H]+ 279.1743, found: 279.1733.
4.2.10.2. (E)-4-(3-(trifluoromethyl)styryl)benzaldehyde (23b).
White solid, yield 70%. 1H NMR (400 MHz, CDCl3) δ: 10.02 (s, 1H), 7.94 – 7.86 (m, 2H), 7.79 (s, 1H), 7.73 – 7.69 (m, 1H), 7.70 – 7.66 (m, 2H), 7.59 – 7.48 (m, 2H), 7.24 (q, J = 16.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 191.5, 142.6, 137.3, 135.7, 131.1, 130.5, 130.3, 129.9, 129.9, 129.9, 129.9, 129.3, 129.2, 127.1, 124.9, 124.9, 124.8, 124.8, 123.5, 123.4, 123.4, 123.4, 110.0. HRMS (ESI + ): calcd for C16H12F3O [M + H]+ 277.0835, found: 277.0846.
4.2.11. General procedure for the synthesis of compounds 26a-c
To a microwave reaction tube containing THF (0.2 M), the appropriate 4-iodobenzaldehyde derivative (1.0 equiv.), phenylacetylene derivative (1.3 equiv.) and triethylamine (3.0 equiv.) were added and then purged with argon gas for 5 min. After, Pd(PPh)3Cl2 (0.1 equiv.) was added and the solution was further purged with argon gas for an additional 5 min before the addition of CuI (0.05 equiv.). The mixture was heated to 50 °C for 30 min in the microwave. The resulting mixture was heated in a microwave reactor at 50 °C for 30 min. After, the resulting mixture was concentrated via vacuum and then purified by silica gel chromatography to yield the desired product.
4.2.11.1. 4-((4-propylphenyl)ethynyl)benzaldehyde (26a).
Yellow oil, yield 59%. 1H NMR (400 MHz, CDCl3) δ: 10.02 (s, 1H), 7.86 (d, J = 8.5 Hz, 2H), 7.66 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 8.3 Hz, 2H), 7.19 (d, J = 8.4 Hz, 2H), 2.61 (t, J = 7.6 Hz, 2H), 1.73 – 1.58 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 191.6, 144.2, 135.4, 132.2, 131.9, 130.1, 129.7, 128.8, 119.8, 94.0, 88.1, 38.2, 24.5, 13.9. HRMS (ESI + ): calcd for C18H17O [M + H]+ 249.1274, found: 249.1267.
4.2.11.2. 4-((4-butylphenyl)ethynyl)benzaldehyde (26b).
Yellow oil, yield 55%. 1H NMR (400 MHz, CDCl3) δ 10.01 (s, 1H), 7.85 (d, J = 8.5 Hz, 2H), 7.66 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.5 Hz, 2H), 2.66 – 2.60 (m, 2H), 1.66 – 1.55 (m, 2H), 1.36 (dq, J = 14.5, 7.3 Hz, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 191.5, 144.4, 135.4, 132.1, 131.8, 130.0, 129.7, 128.7, 119.7, 94.0, 88.1, 35.8, 33.5, 22.4, 14.1. HRMS (ESI + ): calcd for C19H19O [M + H]+ 263.1430, found: 263.1426.
4.2.11.3. 4-((4-pentylphenyl)ethynyl)benzaldehyde (26c).
Yellow oil, yield 61%. 1H NMR (400 MHz, CDCl3) δ: 10.01 (s, 1H), 7.86 (d, J = 8.3 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.2 Hz, 2H), 7.19 (d, J = 8.1 Hz, 2H), 2.67 – 2.58 (m, 2H), 1.62 (p, J = 7.6 Hz, 2H), 1.40 – 1.25 (m, 4H), 0.90 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 191.6, 144.4, 135.4, 132.1, 131.8, 130.0, 129.7, 128.7, 119.7, 94.0, 88.1, 36.1, 31.6, 31.0, 22.7, 14.2. HRMS (ESI + ): calcd for C20H21O [M + H]+ 277.1587, found: 277.1590.
4.2.12. General procedure for the synthesis of compounds 28a-c
To a round bottom flask containing methanol (0.2 M), the appropriate alkyne derivative (1.0 equiv.) was added followed by addition of 10% Pd/C (0.1 equiv.) and then put under an argon gas atmosphere. After, while stirring at room temperature, the solution was subjected to a vacuum followed by a subsequent purge of hydrogen gas. This process of vacuum followed by hydrogen gas purge was repeated twice more. Next, while under a hydrogen gas atmosphere, the mixture was stirred for 2 h. Subsequently, the hydrogen gas atmosphere was replaced by argon gas. Afterward, the solid was removed via filtration upon celite and the solvent was removed via vacuum. The resulting concentrate was then purified by silica gel chromatography to yield the final product.
4.2.12.1. (R)-(1-(4-(4-propylphenethyl)benzyl)pyrrolidin-2-yl)methanol (28a).
Clear oil, yield 55%. 1H NMR (400 MHz, CDCl3) δ: 7.22 (d, J = 8.0 Hz, 2H), 7.16 (d, J = 8.0 Hz, 2H), 7.11 (s, 4H), 3.94 (d, J = 12.9 Hz, 1H), 3.66 (dd, J = 10.7, 3.5 Hz, 1H), 3.43 (dd, J = 10.7, 2.1 Hz, 1H), 3.35 (d, J = 12.9 Hz, 1H), 3.03 – 2.94 (m, 1H), 2.90 (s, 4H), 2.79 – 2.68 (m, 1H), 2.62 – 2.50 (m, 1H), 2.33 – 2.27 (m, 1H), 2.02 – 1.77 (m, 2H), 1.76 – 1.57 (m, 4H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 140.9, 140.4, 139.1, 136.9, 128.8, 128.5, 128.5, 128.4, 64.3, 61.9, 58.3, 54.6, 37.8, 37.8, 37.7, 28.0, 24.8, 23.6, 14.0. HRMS (ESI + ): calcd for C23H32NO [M + H]+ 338.2478, found: 338.2480.
4.2.12.2. (R)-(1-(4-(4-butylphenethyl)benzyl)pyrrolidin-2-yl)methanol (28b).
Clear oil, yield 82%. 1H NMR (400 MHz, CDCl3) δ: 7.24 (d, J = 8.1 Hz, 1H), 7.15 (d, J = 8.2 Hz, 2H), 7.09 (s, 4H), 4.00 (d, J = 13.1 Hz, 1H), 3.67 (dd, J = 11.1, 3.4 Hz, 1H), 3.53 – 3.42 (m, 2H), 3.06 (dd, J = 9.6, 5.3 Hz, 1H), 2.92 – 2.80 (m, 4H), 2.61 – 2.53 (m, 2H), 2.45 – 2.33 (m, 1H), 2.00 – 1.78 (m, 2H), 1.80 – 1.68 (m, 2H), 1.60 (dt, J = 15.2, 7.5 Hz, 2H), 1.35 – 1.28 (m, 2H), 0.93 – 0.85 (m, 3H). 13C NMR (101 MHz, CDCl3) δ:140.4, 139.7, 138.0, 131.2, 128.3, 127.7, 127.5, 127.4, 64.0, 60.8, 57.5, 53.4, 36.8, 36.6, 34.7, 30.4, 26.7, 22.6, 21.7, 13.2. HRMS (ESI + ): calcd for C24H34NO [M + H]+ 352.2635, found: 352.3633.
4.2.12.3. (R)-(1-(4-(4-pentylphenethyl)benzyl)pyrrolidin-2-yl)methanol (28c).
Clear oil, yield 51%. 1H NMR (400 MHz, CDCl3) δ: 7.25 (d, J = 8.3 Hz, 2H), 7.16 (d, J = 8.1 Hz, 2H), 7.10 (s, 4H), 4.00 (d, J = 13.0 Hz, 1H), 3.68 (dd, J = 11.1, 3.4 Hz, 1H), 3.53 – 3.43 (m, 2H), 3.07 (dd, J = 9.7, 5.5 Hz, 1H), 2.93 – 2.81 (m, 5H), 2.62 – 2.53 (m, 2H), 2.45 – 2.34 (m, 1H), 2.00 – 1.79 (m, 2H), 1.81 – 1.68 (m, 2H), 1.60 (dt, J = 15.2, 7.5 Hz, 2H), 1.36 – 1.28 (m, 4H), 0.94 – 0.85 (m, 3H). 13C NMR (101 MHz, CDCl3) δ: 141.4, 140.7, 139.0, 132.2, 129.3, 128.7, 128.5, 128.4, 65.0, 61.8, 58.5, 54.4, 37.8, 37.6, 35.7, 31.7, 31.4, 27.7, 23.6, 22.7, 14.2, 7.3. HRMS (ESI + ): calcd for C25H36NO [M + H]+ 366.2791, found: 366.2792.
Supplementary Material
Acknowledgments
This work was supported by the NIH (R01 GM104366 and R01 GM121075).
Abbreviations:
- S1P
Sphingosine 1-phosphate
- Sph
Sphingosine
- SphK
Sphingosine kinase
- HCTU
O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
Footnotes
Declaration of Competing Interest
The authors declare the following competing financial interest(s): W. L.S. and K.R.L. are co-founders of Flux Therapeutics Inc, which was created to commercialize S1P-related discoveries, including SphK inhibitors, discovered and characterized in their laboratories.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2020.115941.
References
- 1.Spiegel S, Milstien S. Sphingosine 1-Phosphate, a Key Cell Signaling Molecule. J Biol Chem 2002;277(29):25851–25854. [DOI] [PubMed] [Google Scholar]
- 2.Van Brocklyn JR, Williams JB. The Control of the Balance between Ceramide and Sphingosine-1-Phosphate by Sphingosine Kinase: Oxidative Stress and the Seesaw of Cell Survival and Death. Comp Biochem Physiol B: Biochem Mol Biol 2012;163(1): 26–36. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez T, Hla T. Structural and Functional Characteristics of S1P Receptors. J Cell Biochem 2004;92(5):913–922. [DOI] [PubMed] [Google Scholar]
- 4.Pyne NJ, Pyne S. Sphingosine 1-Phosphate and Cancer. Nat Rev Cancer 2010;10(7): 489–503. [DOI] [PubMed] [Google Scholar]
- 5.Pyne NJ, El Buri A, Adams DR, Pyne S. Sphingosine 1-Phosphate and Cancer. Adv. Biol. Regul 2018;68:97–106. [DOI] [PubMed] [Google Scholar]
- 6.Singh SK, Spiegel S. Sphingosine-1-Phosphate Signaling: A Novel Target for Simultaneous Adjuvant Treatment of Triple Negative Breast Cancer and Chemotherapy-Induced Neuropathic Pain. Adv. Biol. Regul 2020;75, 100670. [DOI] [PubMed] [Google Scholar]
- 7.Li C, Kong Y, Wang H, et al. Homing of Bone Marrow Mesenchymal Stem Cells Mediated by Sphingosine 1-Phosphate Contributes to Liver Fibrosis. J Hepatol 2009; 50(6):1174–1183. [DOI] [PubMed] [Google Scholar]
- 8.Long DA, Price KL. Sphingosine Kinase-1: A Potential Mediator of Renal Fibrosis. Kidney Int 2009;76(8):815–817. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Berka V, Song A, et al. Elevated Sphingosine-1-Phosphate Promotes Sickling and Sickle Cell Disease Progression. J. Clin. Invest 2014;124(6):2750–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao S, Adebiyi MG, Zhang Y, et al. Sphingosine-1-Phosphate Receptor 1 Mediates Elevated IL-6 Signaling to Promote Chronic Inflammation and Multitissue Damage in Sickle Cell Disease. FASEB J 2018;32(5):2855–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patmanathan SN, Wang W, Yap LF, Herr DR, Paterson IC. Mechanisms of Sphingosine 1-Phosphate Receptor Signalling in Cancer. Cell Signal 2017;34:66–75. [DOI] [PubMed] [Google Scholar]
- 12.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-Phosphate Receptor Signaling. Annu Rev Biochem 2009;78:743–768. [DOI] [PubMed] [Google Scholar]
- 13.Pyne S, Adams DR, Pyne NJ. Sphingosine 1-Phosphate and Sphingosine Kinases in Health and Disease: Recent Advances. Prog Lipid Res 2016;62:93–106. [DOI] [PubMed] [Google Scholar]
- 14.Zemann B, Urtz N, Reuschel R, et al. Normal Neutrophil Functions in Sphingosine Kinase Type 1 and 2 Knockout Mice. Immunol Lett 2007;109(1):56–63. [DOI] [PubMed] [Google Scholar]
- 15.Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Essential Role for Sphingosine Kinases in Neural and Vascular Development. Mol Cell Biol 2005;25 (24):11113–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos WL, Lynch KR. Drugging Sphingosine Kinases. ACS Chem Biol 2015;10(1): 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams DR, Tawati S, Berretta G, et al. Topographical Mapping of Isoform-Selectivity Determinants for J-Channel-Binding Inhibitors of Sphingosine Kinases 1 and 2. J Med Chem 2019;62(7):3658–3676. [DOI] [PubMed] [Google Scholar]
- 18.Sibley CD, Morris EA, Kharel Y, et al. Discovery of a Small Side Cavity in Sphingosine Kinase 2 That Enhances Inhibitor Potency and Selectivity. J Med Chem 2020;63(3): 1178–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Worrell BL, Brown AM, Santos WL, Bevan DR. In Silico Characterization of Structural Distinctions between Isoforms of Human and Mouse Sphingosine Kinases for Accelerating Drug Discovery. J Chem Inf Model 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allende ML, Sasaki T, Kawai H, et al. Mice Deficient in Sphingosine Kinase 1 Are Rendered Lymphopenic by FTY720. J Biol Chem 2004;279(50):52487–52492. [DOI] [PubMed] [Google Scholar]
- 21.Kharel Y, Raje M, Gao M, et al. Sphingosine Kinase Type 2 Inhibition Elevates Circulating Sphingosine 1-Phosphate. Biochem J 2012;447(1):149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kharel Y, Morris EA, Congdon MD, et al. Sphingosine Kinase 2 Inhibition and Blood Sphingosine 1-Phosphate Levels. J Pharmacol Exp Ther 2015;355(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kharel Y, Huang T, Salamon A, Harris TE, Santos WL, Lynch KR. Mechanism of Sphingosine 1-Phosphate Clearance from Blood. Biochem J 2020;477(5):925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maines LW, Fitzpatrick LR, Green CL, Zhuang Y, Smith CD. Efficacy of a Novel Sphingosine Kinase Inhibitor in Experimental Crohn’s Disease. Inflammopharmacology 2010;18(2):73–85. [DOI] [PubMed] [Google Scholar]
- 25.Pitman MR, Costabile M, Pitson SM. Recent Advances in the Development of Sphingosine Kinase Inhibitors. Cell Signal 2016;28(9):1349–1363. [DOI] [PubMed] [Google Scholar]
- 26.Schnute ME, McReynolds MD, Kasten T, et al. Modulation of Cellular S1P Levels with a Novel, Potent and Specific Inhibitor of Sphingosine Kinase-1. Biochem J 2012;444 (1):79–88. [DOI] [PubMed] [Google Scholar]
- 27.Schnute ME, McReynolds MD, Carroll J, et al. Discovery of a Potent and Selective Sphingosine Kinase 1 Inhibitor through the Molecular Combination of Chemotype-Distinct Screening Hits. J Med Chem 2017;60(6):2562–2572. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Knapp S, Pyne NJ, Pyne S, Elkins JM. Crystal Structure of Sphingosine Kinase 1 with PF-543. ACS Med Chem Lett 2014;5(12):1329–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Childress ES, Kharel Y, Brown AM, Bevan DR, Lynch KR, Santos WL. Transforming Sphingosine Kinase 1 Inhibitors into Dual and Sphingosine Kinase 2 Selective Inhibitors: Design, Synthesis, and in Vivo Activity. J Med Chem 2017;60(9): 3933–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kharel Y, Mathews TP, Kennedy AJ, Houck JD, Macdonald TL, Lynch KR. A Rapid Assay for Assessment of Sphingosine Kinase Inhibitors and Substrates. Anal Biochem 2011;411(2):230–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kharel Y, Morris EA, Congdon MD, et al. Sphingosine Kinase 2 Inhibition and Blood Sphingosine 1-Phosphate Levels. J Pharmacol Exp Ther 2015;355(1):23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Congdon MD, Kharel Y, Brown AM, et al. Structure-Activity Relationship Studies and Molecular Modeling of Naphthalene-Based Sphingosine Kinase 2 Inhibitors. ACS Med Chem Lett 2016;7(3):229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kharel Y, Agah S, Huang T, et al. Saccharomyces Cerevisiae as a Platform for Assessing Sphingolipid Lipid Kinase Inhibitors. PLoS ONE 2018;13(4), e0192179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, Min X, Xiao S-H, et al. Molecular Basis of Sphingosine Kinase 1 Substrate Recognition and Catalysis. Structure 2013;21(5):798–809. [DOI] [PubMed] [Google Scholar]
- 35.Patwardhan NN, Morris EA, Kharel Y, et al. Structure-Activity Relationship Studies and in Vivo Activity of Guanidine-Based Sphingosine Kinase Inhibitors: Discovery of SphK1- and SphK2-Selective Inhibitors. J Med Chem 2015;58(4):1879–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem 2009;30(16): 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trott O, Olson AJ. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J Comput Chem 2010;31(2):455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.