

SUMMARY
Disturbances of the sleep/wake cycle in Alzheimer’s disease (AD) are common, frequently precede cognitive decline, and tend to worsen with disease progression. Sleep is critical to the maintenance of homeostatic and circadian function, and chronic sleep disturbances have significant cognitive and physical health consequences that likely exacerbate disease severity. Sleep-wake cycles are regulated by neuromodulatory centers located in the brainstem, the hypothalamus, and the basal forebrain, many of which are vulnerable to the accumulation of abnormal protein deposits associated with neurodegenerative conditions. In AD, while sleep disturbances are commonly attributed to the accumulation of amyloid beta, patients often first experience sleep issues prior to the appearance of amyloid beta plaques, on a timeline that more closely corresponds to the first appearance of abnormal tau neurofibrillary tangles in sleep/wake regulating areas of the brainstem. Sleep disturbances also occur in pure tauopathies, providing further support that tau is a major contributor. Here, we provide an overview of the neuroanatomy of sleep/wake centers discovered in animal models, and review the evidence that tau-driven neuropathology is a primary driver of sleep disturbance in AD.
Keywords: Alzheimer’s disease, Progressive supranuclear palsy, Tauopathies, Human, Neuropathology, Wake-promoting, Sleep-promoting
Introduction
Sleep is an essential physiological process governed by endogenous homeostatic and circadian mechanisms that arise via input from a distributed network of regulatory centers within the central nervous system as well as input from exogenous environmental stimuli. Chronic disruptionof sleep can have severe immunological, metabolic, and glymphatic consequences associated with failed clearance of pathogenic proteins [1]. Sleep affects synaptic and structural plasticity essential for memory formation and normal cognitive function [2]. Sleep disturbances are a common comorbidity of some neurodegenerative diseases, including Alzheimer’s disease (AD), for which severe decline in memory and other cognitive functions are major clinical hallmarks [3]. Pathologic changes in sleep/wake regulating neuronal populations likely underlie sleep disturbances in AD and other neurodegenerative diseases [4–7], and sleep disturbances may precede disease-defining symptoms, particularly in AD [8], potentially servingas anearly indicator of neurodegenerative disease. Whilesleep disturbances in AD have long been attributed to pathologic change caused by amyloid beta (Aβ) accumulation, little attention has been paid to the role of tau accumulation. However, many brain areas involved in sleep/wake control demonstrate tau-driven changes in AD early in disease, before the appearance of Aβ plaques. Furthermore, progressive supranuclear palsy (PSP), a pure tauopathy with tau-related changes in similar brain areas, displays sleep disturbances that are similar in pattern but more severe than AD. Characterizing tau-driven degeneration of sleep-wake promoting neuronal populations in AD and other tauopathies is critical for developing screening tools for early diagnosis and identifying potential therapeutic targets to treat debilitating symptoms of sleep disruption.
Sleep and wake brain states
The sleep/wake cycle is structurally organized by different cortical synchronization and de-synchronization states, representing the sleep architecture. Cortical de-synchronization defines the transition to wakefulness, and cortical synchronization marks the transition to sleep, with different sleep states featuring distinctive oscillatory activity [9]. Brain states include the sleep/ wake transition and light sleep (stages 1 and 2), non-rapid eye movement/slow-wave sleep (SWS; stage 3), and rapid eye movement sleep (REM, stage 4) [10].
Sleep architecture changes during life span. Older adults demonstrate a modest decrease in sleep quantity and quality, including decreases in total sleep time, proportionally more time spent in light sleep than in SWS and REM sleep, and increases in sleep stage shifts, leading to more awakenings and greater sleep fragmentation [11]. Sleep disruptions in individuals with neuro-degenerative diseases may range from an exacerbation of the pattern seen in normal aging, likely reflecting an early selective vulnerability in sleep-wake centers of the brain to the most common neurodegenerative conditions, such as AD [3,6,7], to complete and severe change in sleep architecture, as seen in PSP [12].
An interconnected network of nuclei in the brainstem, basal forebrain, and hypothalamus modulates cortical synchronization and oscillation in a hierarchical manner resulting in the sleep/wake cycle[13]. These neuronal populations are limited to discrete subcortical areas, but exert modulatory control over neuronal activity via vast projections throughout the brain [9]. Different circuits control and orchestrate wake, SWS and REM sleep, which will be discussed in more detail in subsequent sections. A prevailing hypothesis for the regulation of sleep is that specific cell populations containing wake- and sleep-promoting neurons interact via a “flip-flop switch” which, along with influences from circadian timekeepers in the superchiasmatic nucleus (SCN), compete through mutual inhibition to determine the arousal state [14].
Sleep disturbances in AD and PSP
Sleep disturbances are a common symptom of AD and other neurodegenerative diseases [3]. Although sleep disturbances are considered a non-cognitive symptom of neurodegenerative disease, disrupted sleep is associated with cognitive impairments in healthy subjects and has been shown to exacerbate cognitive decline in dementias [15].
Sleep disturbances are reported in around half of AD patients, with sleep changes frequently appearing in preclinical stages of disease one to two decades before first symptoms of cognitive decline, and increasing in severity as disease progresses [8,16]. Changes to sleep architecture in AD are characterized by a mild loss of total sleep time and REM sleep, a significant decrease in SWS time (a sleep stage critical to memory consolidation), and increased night-time awakenings resulting in fragmented sleep and reduced sleep efficiency [8,17]. AD patients and caregivers also report increased daytime propensity to sleep [17,18]. In AD patients with advanced dementia, circadian rhythms are significantly altered, demonstrating a decrease in amplitude and a phase delay [19].
The neuropathological hallmarks of AD include the presence of extracellular Aβ plaques, intraneuronal tau accumulation (neuro-fibrillary tangles, NFT) and neuron loss [20]. These neuropathological changes progress over several decades, with abnormal tau accumulation appearing before Aβ plaques and in several subcortical nuclei prior to changes in allocortex [21,22]. Sleep disturbances in AD have long been attributed to pathologic change caused by Aβ accumulation. Aβ clearance by the brain glymphatic system is dependent on sleep in mice, and transgenic mouse lines with mutations in APP and PSEN1 show a link between high concentrations of soluble Aβ in the extracellular matrix and vulnerability to early Aβ plaque accumulation, as well as increased sleep fragmentation [23,24]. In cognitively normal individuals with disrupted sleep, there appears to be a relationship between Aβ cerebrospinal fluid concentration and sleep loss, suggesting a similar relationship may be present in humans, although this is yet to be confirmed [8,23–25]. However, diurnal variation in Aβ CSF concentration has also been linked to changes in synaptic activity, and loss of SWS, the sleep stage characterized by reduced synaptic activity, is associated with increased Aβ (and tau) CSF concentration in humans, suggesting metabolic suppression during sleep may also be important in the clearance of pathogenic proteins [16].
While there is clear evidence of the relationship between Aβ and sleep, the role of tau has been comparatively under-explored. Although amyloid-driven mouse models of AD demonstrate sleep fragmentation, the pattern of sleep disruption lacks some of the features that are observed in AD in humans [24], for which both amyloid and tau pathologic change is present. Furthermore, human neuroimaging studies have found that alpha wave hyposynchrony is correlated with tau tracer uptake and tau deposits, while delta-theta hypersynchrony is correlated with Aβ tracer uptake, suggesting that Aβ and tau have differential contributions to alterations in brain state changes in AD [26]. Finally, evidence for the role of Aβ-driven neuropathologic change of sleep/wake centers in prodromal disease stages, when sleep disruptions first emerge in AD, is lacking. In contrast, the case for tau-driven neurodegeneration as the primary driver of sleep disruption in the early stages of AD is more robust. Sleep disturbances in AD often begin early in disease pathogenesis, overlapping with early stages of tau neuropathology in AD when abnormal tau begins to accumulate in multiple wake-promoting regions of the brainstem (Fig. 1), and notably, before the presence of Aβ plaques [4,21]. Additionally, sleep disturbances are even more severe in the pure tauopathy progressive supranuclear palsy.
Fig. 1.
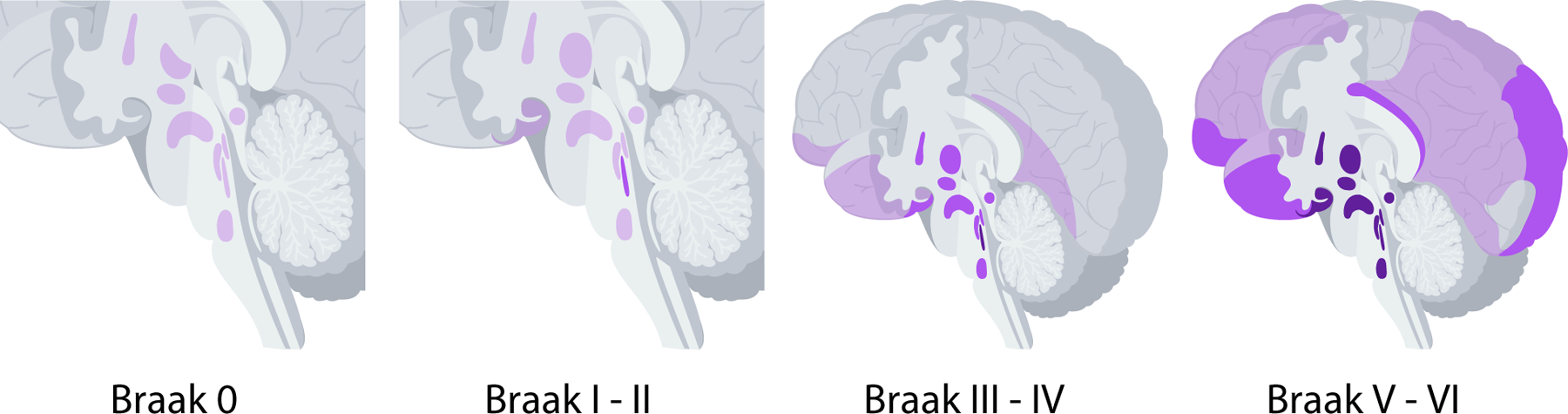
Brain areas containing tau neuropathology shown in purple in Braak stages 0 through VI in Alzheimer’s disease. Darker shades indicate increasing severity of tau pathology. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Progressive supranuclear palsy (PSP) is a rare neurodegenerative disease characterized by motor movement dysfunction and mild dementia [27]. Symptoms of PSP additionally include a profound disruption in sleep. Specifically, individuals with PSP have difficulty falling and staying asleep, resulting in significant sleep loss, a significant decrease in SWS and REM sleep time, and decreased ability to recover sleep loss with daytime napping, suggesting a disrupted homeostatic sleep drive [12]. In contrast to AD, which is a mixed proteinopathy, PSP is a pure tauopathy not associated with Aβ plaques, with neuropathological hallmarks that include both neuronal and glial tau inclusions [27,28]. As in AD, several brain regions selectively vulnerable to tau pathology in PSP include wake-promoting areas.
In summary, abnormal tau appears in wake-promoting regions of the brain earlier than Aβ plaques in AD pathogenesis, coinciding with the time course in which sleep disturbances first become apparent. Furthermore, sleep disturbances are also a feature of the pure tauopathy PSP. This suggests that abnormal tau is the primary driver of sleep disturbances, particularly in prodromal stages of AD, with Aβ contributing to sleep disturbances in concert with tau or through different neural pathways at later stages. Sleep-promoting areas, while scarcely examined in AD and PSP, also demonstrate tau neuropathologic change in the limited studies available. In the sections below, we review the evidence of tau neuropathologic change in wake- and sleep-promoting neurons in AD and PSP (see Supplementary Table 1 for summary of sleep/wake neurons in animal models and neuropathologic change in human postmortem brains).
Wake-promoting neurons
Wake-promoting neurons of the ascending reticular activating system (ARAS) are involved in arousal, and are spread across several nuclei in the reticular formation of the brainstem, basal forebrain, and hypothalamus. Wake-promoting neurons primarily include excitatory neurotransmitter-expressing populations [29].
Wake-promoting neurons with evidence of tau-driven neuropathologic change in AD
Located bilaterally in the pons near the wall of the fourth ventricle, the locus coeruleus (LC, Fig. 2) is a small nucleus composed of melanin-containing neurons. LC neurons provide the largest source of norepinephrine (NE) synthesis in the brain, with wide-ranging projections throughout the brain that modulate arousal and the sleep-wake cycle [30]. LC NE neurons have two distinct output modes, tonic and phasic, with distinct physiological properties [31]. LC NE neurons are wake-active, and are thought to contribute to the sleep-wake switch, as they stop firing shortly before cortical synchronization of the sleep state and begin firing shortly before cortical activation initiating the wake state [32].
Fig. 2.
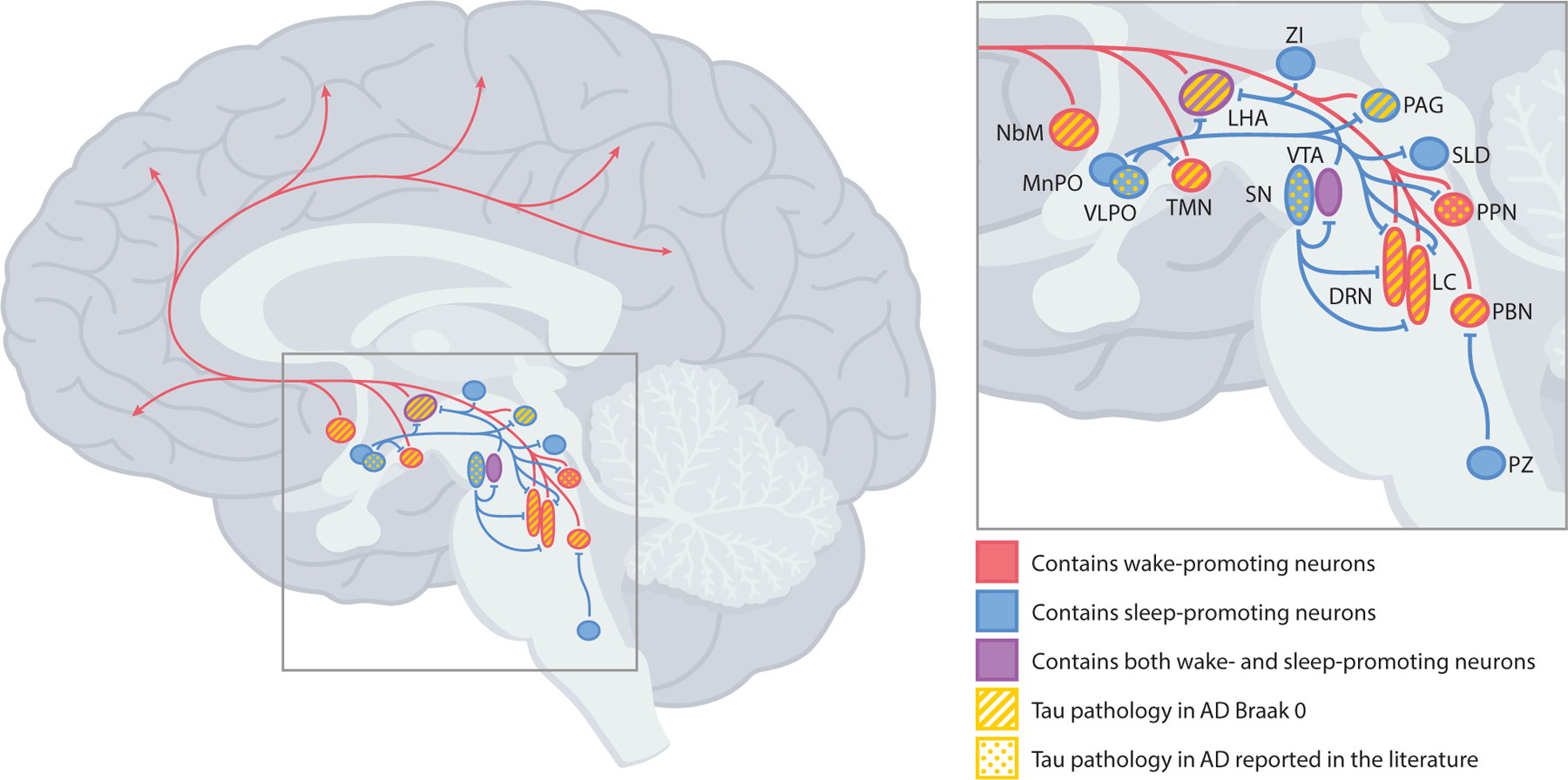
Brain regions in the brainstem, hypothalamus, and basal forebrain that contain sleep- and wake-promoting neurons, with areas containing abnormal tau and/or neurofibrillary tangles in AD noted. Red arrows indicate wake-promoting projections. Blue lines indicate sleep-promoting projections. DRN = dorsal raphe nucleus; LC = locus coeruleus; LHA = lateral hypothalamic area; MnPO = median preoptic nucleus; NbM = nucleus basalis of Meynert; PAG = periaqueductal gray; PBN = parabrachial nucleus; PPN = pedunculopontine nucleus; PZ = parafacial zone; SLD = sublaterodorsal nucleus; SN = substantia nigra; TMN = tuberomammillary nucleus; VLPO = ventrolateral preoptic nucleus; VTA = ventral tegmental area; ZI = zona incerta. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
LC NE neurons are some of the earliest affected in AD. A number of studies have observed NFTs and profound neuron loss in AD LC [33–35]. A study examining neuropathologic changes in individuals between the ages of four and 29 years found abnormal tau throughout the brainstem in 38 of 42 cases, most often affecting the LC, including a number of cases in which abnormal tau was confined to only to LC [21]. Later studies have found that LC is consistently affected early in AD, prior to changes in the entorhinal cortex, and furthermore, have found that NFT burden, neuron loss, and LC volumetric decreases positively correlate with disease progression [5,35]. LC is also greatly affected in PSP. Total neuron loss and loss of wake-promoting NE-positive neurons compared to controls is significant, but not as severe as in AD, while NFTs in the remaining neurons are greater in PSP compared to AD [6,7,36]. Early changes in the LC may underlie the symptoms of disrupted sleep that occur prior to cognitive and behavioral changes in AD [37,38].
The dorsal raphe nucleus (DRN, Fig. 2) is composed of a linear midline region that begins just below the cerebral aqueduct, and two “wings” that extend laterally from the midline. The majority of DRN neurons are serotonin (5-HT) synthesizing, and serve as the origin of serotonergic pathways to the forebrain [39]. Most 5-HT neurons are wake-active, and appear to promote wakefulness and inhibit REM sleep [40,41]. Furthermore, projections of 5-HT neurons to wake-promoting glutamatergic neurons in the parabrachial nucleus (PBN) have been found to be critical for stimulating arousal in response to hypercapnia [42]. In AD, DRN 5-HT neurons are particularly vulnerable to NFTs [43]. Like LC, NFTs in the DRN appear in early Braak stages, before changes in the transentorhinal cortex [4]. DRN neurons project to allocortical areas affected in Braak stages I and II (Fig. 1) in AD and other wake-promoting brain areas that are vulnerable to tau pathology in AD[44,45]. In PSP, DRN tau burden is significant, but not as severe as AD [6].
The parabrachial nucleus (PBN, Fig. 2) surrounds the superior cerebellar peduncle in the dorsolateral pons. The PBN projects to the basal forebrain, intralaminar thalamus, lateral hypothalamus, amygdala, and cortex [46,47]. The PBN plays a critical role in arousal through glutamatergic signaling [48]. Contributions to arousal across the PBN differ by region. Lateral PBN neurons are involved in arousal due to hypercapnia, while medial PBN neurons are primarily involved in spontaneous arousal from sleep, possibly through SWS inhibition [48,49]. The PBN also has significant reciprocal connectivity with the sleep-promoting ventrolateral preoptic nucleus of the hypothalamus in rodents, and mutual inhibition between these two populations may contribute to the sleep-wake switch [50]. In AD, significant PBN tau pathology is found regardless of duration of disease or severity of dementia [51], including Braak stage 0 and I cases, which demonstrate tau pathology in the medial PBN [22]. In PSP, the medial PBN also demonstrates significant tau pathology[52].
The pedunculopontine nucleus (PPN, Fig. 2) consists of bilateral nuclei in the rostral pons that are anatomically identified by clusters of cholinergic neurons interspersed with excitatory glutamatergic and inhibitory GABAergic neuronal populations. The PPN projects to wake-promoting regions in the basal forebrain, lateral hypothalamus, reticular thalamic nuclei, and prefrontal cortex, and has significant reciprocal connectivity with midbrain dopaminergic centers [53]. While all three PPN neuronal populations are implicated in promoting cortical activation, only glutamatergic PPN neurons have been shown to strongly promote wakefulness [54]. Although cholinergic agonists injected into the PPN appear to induce REM sleep, leading some to suggest that cholinergic PPN neurons promote REM sleep, chemogenetic manipulations in mice found that while cholinergic PPN neurons inhibit slow thalamocortical rhythms associated with SWS, they do not drive REM states [54].
AD PPN contains a moderate amount of NFTs and fewer glutamatergic neurons [6]. PPN is the only region containing wake-promoting neurons found to have greater severity of neuropathologic change in PSP compared to AD is the PPN. PSP PPN demonstrates significant neuron loss, significantly fewer glutamatergic neurons, and significant NFT burden that is more severe than AD [6,55].
The nucleus basalis of Meynert (NbM, Fig. 2) comprises irregularly shaped clusters of magnocellular neurons within the basal forebrain, lying rostral and parallel to the optic nerve. The NbM is the primary source of cholinergic cortical projections [56]. NbM cholinergic neurons are wake- and REM-sleep active, and optogenetic stimulation of these neurons during SWS sleep elicits cortical activation and transition to wake state [57]. In AD, NbM NFTs are observed in all Braak stages, with cytoskeletal change severity paralleling Braak stage[45,58]. AD additionally NbM demonstrates substantial neuron loss, which is already apparent in pre-cortical stages of disease[59]. NbM tau pathology is also significant in PSP [60].
The tuberomammillary nucleus (TMN, Fig. 2) in the posterior hypothalamus contains the only source of histamine-synthesizing neurons in the brain. TMN histaminergic projections have wide-ranging targets, including wake-promoting regions in the brainstem and hypothalamus, and receive significant inhibitory projections from sleep-promoting neurons in the ventrolateral preoptic nucleus [61]. While histaminergic neurons alone are not sufficient for waking, they are wake-active, silent during sleep, and contribute to cortical arousal via interactions with orexinergic signaling [62]. In AD, TMN NFT burden and loss of histaminergic neurons and total number of neurons is significant [7]. Further-more, abnormal tau is present in Braak stage 0 and I, indicating that AD TMN pathological change occurs in prodromal disease stages [22]. In PSP, TMN neuronal loss is mild, and while NFT burden and loss of histaminergic neurons are significant, these changes are less severe than in AD [7].
The lateral hypothalamic area (LHA, Fig. 2) serves as an integrating center for several autonomic and endocrinologic regulatory and homeostatic systems. Orexin-expressing glutamatergic neurons in the LHA have excitatory projections to multiple wake-promoting targets within the ARAS, including dense projections to LC [63]. Orexinergic LHA neurons are exclusively wake-active, and inhibition or degeneration results in narcolepsy, demonstrating an essential role in the stability of wakefulness [64,65]. The LHA additionally contains a subpopulation of sleep-promoting neurons, which will be discussed in the next section. AD LHA contains significant NFT burden, significant loss in orexinergic and total neurons [66], and shows abnormal tau pathology in precortical stages of disease [22]. In PSP, loss of total neurons and orexinergic neurons is significant but not as severe as in AD, while NFT burden in surviving neurons is greater in PSP compared to AD [6,7].
Sleep-promoting neurons
In contrast to the primarily excitatory nature of wake-promoting neurons, sleep-promoting neurons are either GABAergic, or involved in the excitation of GABAergic neurons. Sleep-promoting neurons are thought to drive the “flip-flop switch” to sleep state via the inhibition of wake-promoting neurons and/or the coordination of brain state change [14].
Sleep-promoting neurons with evidence of tau-driven neuropathologic change in AD and PSP
The ventrolateral preoptic nucleus (VLPO, Fig. 2) of the mammalian brain, or the human brain homolog the intermediate nucleus (InH), is an oval-shaped group of neurons within the chiasmic region of the anterior hypothalamus. VLPO neurons are GABAergic and co-express galanin, a neuropeptide expressed in abundance in many regions of the hypothalamus, and project to numerous key wake-promoting centers [61]. Galanergic VLPO neurons are uniquely sleep-active [67], and strongly promote SWS via inhibitory projections to several wake-promoting areas [68]. The VLPO is sometimes called “sexually dimorphic nucleus” in the literature, as males demonstrate larger nuclei and greater numbers of neurons [69] (future studies are needed to determine if sex differences in the VLPO/InH contribute to sleep differences between sexes).
A human histologic study examining the relationship between neuron number in the InH and sleep actigraphy data in older adults, including some with AD, observed fewer galanergic neurons in individuals with AD and fewer galanergic neurons in individuals with more fragmented sleep [70]. While a future study is needed to examine the potential contributions of tau accumulation in degeneration of InH sleep-promoting neurons, these findings support the hypothesis that the InH plays a critical role in healthy sleep cycles and is selectively vulnerable to cell death in AD.
In addition to wake-promoting orexinergic neurons, the LHA is interspersed with a neuronal population expressing melanin-concentrating hormone (MCH), which promote sleep [71]. MCH neurons are sleep-active, with greatest activity during REM sleep, and project to neuronal populations involved in arousal, including local orexinergic neurons [71]. Orexinergic and MCH neuronal interactions appear to have a regulatory, and likely mutually inhibitory effect on mediating sleep and arousal [72]. Optogenetic stimulation of MCH neurons induces sleep during circadian wake periods, with greatest increases in REM sleep[73,74], suggesting MCH neurons may be involved in the sleep/wake switch. A study by Mladinov and colleagues quantified number and tau burden of MCH neurons in the hypothalamus in AD and PSP. In AD, neuron loss of non-MCH neurons and the proportion of MCH neurons with NFTs was significant, while the proportion of MCH neurons with NFTs in PSP was lower than in AD, suggesting that MCH neurons are more vulnerable to AD tau than to PSP tau [75].
The substantia nigra (SN, Fig. 2) is a sizeable bilateral nucleus in the midbrain located behind the cerebral peduncles. The SN is subdivided into two distinct regions: the more dorsal SN pars compacta (SNpc), which is primarily dopaminergic, densely packed and dark in appearance due to neuromelanin-containing pigmented neurons, and the more ventral SN pars reticulata (SNr), which primarily contains GABAergic neurons [76]. Optogenetic activation of SNr GABAergic neurons suppresses motor movement and enhances SWS, while inactivation enhances motor movement and suppresses sleep [77]. SNr GABAergic neurons are selectively active in states of low motor activity, and directly innervate brain-state-regulating monoaminergic cells in the DRN, LC, and VTA, suggesting that this cell population may be involved in the coordination of brain-state change and behavior to promote SWS [77]. Additionally, manipulations in mice have shown that dopaminergic SNpc neuronal activity acts on GABAergic SNr neurons to promote REM sleep [78].
While SN NFT burden is mild in AD, a number of studies have demonstrated significant neuron loss and NFT burden in PSP [6,79,80]. Although loss of dopaminergic neurons in the SNpc has been most studied and linked to motor deficits in PSP, one study found that GABAergic neurons in the SNr also demonstrate pro-found neuron loss in PSP [80]. The PPN and the SN share significant reciprocal projections [81,82], and severe degeneration of the two regions may be associated with the spread of tau through inter-connected networks. Degeneration and cell loss of these neuronal populations may contribute to the significant reduction in SWS and REM sleep time in PSP.
Examination of wake-promoting regions has demonstrated that pathological changes in PSP and AD, while present in both diseases, are more severe in AD. These findings suggest that while tau pathology may contribute to disruptions in sleep/wake regulation in both diseases, AD tau may be more toxic to wake-promoting neurons than PSP tau, contributing to greater deficiencies in wakefulness in AD. Given that sleep loss and disruptions to sleep architecture are more severe in PSP than in AD, in contrast to wake-promoting regions, we hypothesize that regions containing sleep-promoting neurons may demonstrate greater tau-driven degeneration in PSP. Future postmortem studies comparing PSP and AD in regions containing sleep-promoting neurons are needed to examine the potential contributions of neurodegeneration to sleep architecture differences between the two diseases.
Sleep-promoting neuronal populations not yet examined in AD and PSP
While AD neuropathology of wake-promoting regions is well-established, only three sleep-promoting regions, the InH, LHA, and SN, have been examined in AD postmortem studies. Work in animal models and some evidence from in-vivo studies have identified several other sleep-promoting neuronal populations that warrant future examination in AD and other tauopathies with disrupted sleep phenotypes.
The ventral tegmental area (VTA, Fig. 2) is a group of midbrain nuclei that lies between the SN and the superior cerebellar peduncle/red nucleus, and medial to these structures along the midline. Neuronal composition of the VTA is heterogeneous. The A10 dopaminergic cell group within the VTA serves as the origin of the mesocortical and mesolimbic dopaminergic pathways [83]. While dopaminergic neurons, which make up ~50e60% of the VTA, are wake-active, their contribution to wakefulness is unclear [84,85]. However, GABAergic neurons, which comprise 30e45% of the VTA neuronal population, strongly promote SWS [85,86]. Although these neurons promote SWS, they are primarily wake- and REM-sleep active, and are thought to restrain wakefulness via local inhibition of VTA dopaminergic and glutamatergic neurons, and via inhibitory projections to external wake-promoting targets [85,87]. While the VTA has not been extensively examined in postmortem AD, this region shows evidence of tau pathology as early as Braak stage 0 [22]. Other lines of evidence suggest this region warrants further study. A neuroimaging study examining the VTA in AD observed a significant positive correlation between VTA volume, hippocampal volume, and memory in healthy controls, but not in subjects with mild cognitive impairment or AD, suggesting that the VTA undergoes neuropathologic changes in AD that may occur early in disease pathogenesis [88]. Similarly, a mouse model of familial AD showed selective vulnerability of VTA dopaminergic neurons in pre-plaque stage mice that correlated with memory dysfunction [89]. Future studies of these brain areas in both human neuropathology and animal models of neurode-generative disease are needed to better understand the link between neurodegeneration and sleep disruption.
The periaqueductal gray (PAG, Fig. 2) is a midbrain structure composed of rostro-caudally oriented gray matter columns surrounding the cerebral aqueduct. PAG columns are functionally distinct and are involved in a broad range of functions including autonomic regulation, pain response, fear response, and sleep [90]. A group of sleep-active neurotensin-expressing (NTS) glutamatergic neurons in the ventrolateral PAG were found to promote slow-wave sleep via excitation of local GABAergic neurons and GABAergic neurons in the ventromedial medulla, which in turn send inhibitory projections to midbrain wake-promoting neurons [90]. Like the VTA, the PAG contains tau pathology in pre-cortical Braak stages [22].
The sublaterodorsal nucleus (SLD, Fig. 2) is a small nucleus in the dorsal pons containing a mix of glutamatergic and GABAergic neurons, which are thought to be involved in inducing REM sleep and suppressing wakefulness [91]. While glutamatergic SLD neurons project to regions in the brainstem that promote muscle atonia, GABAergic “REM-on” neurons in the SLD have reciprocal connections with “REM-off” GABAergic neurons in the ventrolateral periaqueductal gray and lateral pontine tegmentum regions, and these two cell populations are hypothesized to regulate REM sleep through mutual inhibition [92].
The parafacial zone (PZ, Fig. 2) is a region in the caudal pons/ rostral medulla that lies dorsal and lateral to the facial nerve. While previously associated with respiration, recent studies have identified a population of GABAergic neurons in the PZ that are sleep-active and have been demonstrated to promote slow wave sleep via inhibitory projections to wake-promoting neurons in the medial PBN, which in turn results in a loss of excitatory drive to cortically-projecting basal forebrain neurons [93]. Furthermore, a rat model of sporadic AD observed a significant reduction in SWS and increased wakefulness that was linked to dysfunction of GABAergic neurons in the PZ, suggesting that selective vulnerability of this neuronal population may contribute to sleep disturbances in AD [94].
The median preoptic nucleus (MnPO, Fig. 2) lies anteromedial to the VLPO along the midline anterior wall of the third ventricle. The projections of the MnPO to the VLPO and wake-active centers in the LC and DRN suggest that this region is critically situated within sleep-wake circuitry [95]. Like the VLPO, the MnPO contains GABAergic neurons that are sleep-active. However, they also fire prior to sleep, with the most significant firing rate occurring in response to sleep deprivation, suggesting that they contribute to sleep pressure, an important homeostatic contributor to the sleep switch[96,97].
The zona incerta (ZI, Fig. 2) is a small region of gray matter extending ventrally from the thalamicreticular nucleus. A preliminary study found that a subgroup of GABAergic neurons in the ventral ZI become active with increasing sleep pressure, and promote both REM and SWS by inhibiting wake-promoting neurons in the lateral hypothalamus [98].
The SLD, PZ, MnPO, and ZI have not yet been examined in human AD postmortem studies, and warrant further examination for their potential role in sleep disruption in neurodegenerative disease.
Clinical diagnostic and therapeutic implications of tau-driven degeneration of sleep and wake nuclei in neurodegenerative disease.
Studies in animal models have identified several neuronal populations in the brainstem, hypothalamus, and basal forebrain that either promote wakefulness/inhibit sleep or promote sleep/inhibit wakefulness. Many regions containing wake-promoting neuronal populations in animal models show extensive tau burden and neuron loss in AD, with several containing neuropathologic change early in AD pathogenesis before cognitive changes, suggesting that tau, rather than Aβ, is the primary driver of sleep disturbance in the disease. PSP also shows neuropathologic changes in many of these same regions. However, changes in wake-promoting regions overall are less severe and without significant neuronal loss, suggesting that PSP tau is less fatal than AD tau in these regions, and may contribute to intact wake states observed in PSP relative to AD.
In contrast to differences in wakefulness, sleep disruption is more severe in PSP than in AD. PSP demonstrates a greater reduction in SWS than what is observed in AD and significant reductions in REM sleep and total sleep time, supporting the hypothesis that neuropathological changes in sleep-promoting areas may be more extensive in PSP compared to AD. Few studies have examined sleep-promoting regions in neurodegenerative disease. The significant disruption to sleep architecture observed in AD and PSP renders sleep-promoting regions as potentially high-yield areas for further exploration in human postmortem studies.
The postmortem studies reviewed here suggest that nuclei containing wake-promoting neurons, and possibly also nuclei containing sleep-promoting neurons, are selectively vulnerable to tau-driven degeneration in AD and PSP. While disease progression is not well understood in PSP, in AD, the LC and DRN midbrain nuclei are the sites of earliest tau neuropathologic change, and a number of other brainstem and hypothalamic nuclei also show tau abnormalities early in disease [4,21,35]. These affected regions are either within or closely associated with the isodendritic core of the brainstem and basal forebrain, and anatomically interconnected. The isodendritic core is a phylogenetically conserved neuronal network underlying several primary homeostatic functions and sharing a number of features, including poor myelination and volume transmission of neurotransmitters, which may make them more susceptible than other neurons to pathologic mechanisms [99]. Also, several hypotheses preconize that toxins or pathogens in CSF may trigger parenchymal tau changes that subsequently spread within the brain. All of the wake-promoting neurons which are very early affected by AD-tau are in close proximity with the ventricle system [100]. The pattern of tau spread in AD mirrors projection targets of these earliest affected regions, supporting the ““prion-like spread of tau”“ hypothesis, which states that misfolded tau proteins disseminate along neural processes, resulting in selective degeneration of interconnected functional systems, including arousal networks [99] (but see [101] for hypothesis regarding glymphatic spread of tau prions).
While the overlap of affected regions in AD and PSP may be due in part to selective vulnerability of specific functional systems due to properties of tau spread, the differences between diseases in severity of NFT burden and neuronal mortality in affected regions warrants further exploration. Current hypotheses for these differences between diseases include specificities of tau strains, which have been demonstrated in animal models to differ in rate and regional vulnerability of tau spread [102] and/or differences in genetic and specific disease-related physiological stressors [103]. An open question in the field is if AD-tau-related changes in sleep/wake nuclei exacerbate other pathological changes associated with AD. Disturbed sleep cycles cause an increase in Aβ levels in the brain of animal models [24,25]. It opens the possibility that sleep disruption caused by early tau-driven degeneration have downstream effects that exacerbate the accumulation of Aβ plaques suggesting that even symptomatic treatment of AD-tau-related sleep cycle disturbances could delay AD progression. However, tau-related neurotransmission unbalancing is unlikely the only mechanism for beta-amyloid exacerbation. Evidence from murine models suggests that LC dysfunction, an early event associated with AD-tau deposition, accelerates Aβ deposition in the cortex and norepinephrine replacement only is inferior to LC rescuing in reversing Aβ deposition [104]. Finally, tau-related degeneration of sleep nuclei or chronic sleep deprivation do not always result in Aβ deposition. PSP, a primary tauopathy featuring tau deposition in sleep centers and profound hyper-insomnia is devoid of Aβ accumulation [12]. More research examining the link between tau-related sleep disturbance and downstream Aβ accumulation is needed.
More extensive comparisons of neuropathological changes in AD and PSP in sleep- and wake-promoting neurons, as well as examination of the SCN and degradation of circadian function, will provide insight into differences and similarities in disease pathogenesis that can inform animal and cellular models aiming to understand underlying causes of differences in tau-driven selective vulnerability. Such comparative studies may additionally identify associations between neuropathological changes and sleep disturbances that will contribute to the development of screening tools used to detect prodromal stages of disease, prior to onset of cognitive decline, and may also identify targets useful in the development of potential therapeutic applications.
Supplementary Material
Practice points.
Disrupted sleep is a common feature of Alzheimer’s disease that frequently appears early in disease, and tends to worsen in severity as disease progresses, resulting in greater caregiver burden and earlier institutionalization.
While amyloid-beta deposition has long been the focus of sleep disruption in Alzheimer’s disease, pathologic tau is present in wake-promoting nuclei before Ab, suggesting that tau-driven neurodegeneration may be the first and primary driver of sleep disruption.
Pathologic tau is also present in wake-promoting nuclei in progressive supranuclear palsy, a pure tauopathy for which severe sleep disruption is a common disease feature.
While more research is needed, preliminary findings suggest that sleep-promoting nuclei may also be targeted by tau-driven neurodegeneration in Alzheimer’s disease and progressive supranuclear palsy.
Identifying the neuropathological basis of sleep disruption in Alzheimer’s disease is a critical step in diagnosing and treating associated debilitating sleep disorders.
Research agenda.
Quantitative studies in all wake- and sleep-promoting nuclei comparing neuropathology in Alzheimer’s disease and progressive supranuclear palsy in humans to better define tau-driven neurodegeneration in these regions.
Clinical-pathological studies in Alzheimer’s disease and progressive supranuclear palsy to examine relationships between sleep architecture and neuropathology.
Leverage targets identified in the above studies to develop clinical tools for diagnosing and treating sleep disruption in Alzheimer’s disease and other tauopathies.
Funding
This work was supported by Clinical features and neuropathological basis of sleep-wake behavior in Alzheimer’s Disease and PSP (NIA R01 AG060477), Linking sleep dysfunction to tau-related degeneration across AD progression (NIA R01 AG064314), NIH K24053435 and the Tau Consortium/Rainwater Charity Foundation.
Abbreviations:
- 5-HT
serotonin
- AD
Alzheimer’s Disease
- ARAS
ascending reticular activating system
- Aβ
amyloid beta
- DRN
dorsal raphe nucleus
- EEG
electroencephalogram
- InH
intermediate nucleus
- LC
locus coeruleus
- LHA
lateral hypothalamic area
- MCH
melanin-concentrating hormone
- MnPO
median preoptic nucleus
- NbM
nucleus basalis of Meynert
- NE
norepinephrine
- NFT
neurofibrillary tau tangle
- NTS
neurotensin-expressing
- PAG
periaqueductal gray
- PBN
parabrachial nucleus
- PPN
pedunculopontine nucleus
- PSG
polysomnography
- PSP
progressive supranuclear palsy
- PZ
parafacial zone
- REM
rapid eye movement sleep
- SNpc
substantia nigra pars compacta
- SCN
superchiasmatic nucleus
- SLD
sublaterodorsal nucleus
- SN
substantia nigra
- SNr
substantia nigra par reticulate
- SWS
slow-wave sleep
- TMN
tuberomammillary nucleus
- VLPO
ventrolateral preoptic nucleus
- VTA
ventral tegmental area
- ZI
zona incerta
Footnotes
Conflicts of interest
The authors do not have any conflicts of interest to disclose.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.smrv.2021.101541.
References
* The most important references are denoted by an asterisk.
- [1].Krueger JM, Frank MG, Wisor JP, Roy S. Sleep function: toward elucidating an enigma. Sleep Med Rev 2016;28:46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Raven F, Van der Zee EA, Meerlo P, Havekes R. The role of sleep in regulating structural plasticity and synaptic strength: implications for memory and cognitive function. Sleep Med Rev 2018;39:3–11. [DOI] [PubMed] [Google Scholar]
- [3].Raggi A, Ferri R. Sleep disorders in neurodegenerative diseases. Eur J Neurol 2010;17(11):1326–38. [DOI] [PubMed] [Google Scholar]
- [4].Grinberg LT, Rüb U, Ferretti REL, Nitrini R, Farfel JM, Polichiso L, et al. The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimers disease. A precocious onset? Neuropathol Appl Neurobiol 2009;35(4):405–16. [DOI] [PubMed] [Google Scholar]
- *[5].Theofilas P, Ehrenberg AJ, Dunlop S, Di Lorenzo Alho AT, Nguy A, Leite REP, et al. Locus coeruleus volume and cell population changes during Alzheimer’s disease progression: a stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimer’s Dementia 2017;13(3):236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *[6].Eser RA, Ehrenberg AJ, Petersen C, Dunlop S, Mejia MB, Suemoto CK, et al. Selective vulnerability of brainstem nuclei in distinct tauopathies: a postmortem study. J Neuropathol Exp Neurol 2018;77(2):149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *[7].Oh J, Eser RA, Ehrenberg AJ, Morales D, Petersen C, Kudlacek J, et al. Profound degeneration of wake-promoting neurons in Alzheimer’s disease. Alzheimer’s Dementia 2019. July:1–11 [Internet]Available from: https://linkinghub.elsevier.com/retrieve/pii/S1552526019340816. [DOI] [PMC free article] [PubMed]
- [8].Ju YES, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurol 2013;70(5): 587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. J Neuropsychiatry Clin Neurosci 1949;7(2):251–67. [DOI] [PubMed] [Google Scholar]
- [10].Šušmáková K, Krakovská A. Discrimination ability of individual measures used in sleep stages classification. Artif Intell Med 2008;44(3):261–77. [DOI] [PubMed] [Google Scholar]
- [11].Peter-Derex L, Yammine P, Bastuji H, Croisile B. Sleep and Alzheimer’s disease. Sleep Med Rev 2015;19:29–38. 10.1016/j.smrv.2014.03.007 [Internet]Available from:. [DOI] [PubMed] [Google Scholar]
- [12].Walsh CM, Ruoff L, Walker K, Emery A, Varbel J, Karageorgiou E, et al. Sleepless night and day, the plight of progressive supranuclear palsy. Sleep 2017;40(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Saper CB, Fuller PM. Wakeesleep circuitry: an overview. Curr Opin Neurobiol 2017;44:186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci 2001;24(12):726–31. [DOI] [PubMed] [Google Scholar]
- [15].Xu L, Jiang CQ, Lam TH, Liu B, Jin YL, Zhu T, et al. Short or long sleep duration is associated with memory impairment in older Chinese: the Guangzhou Biobank Cohort Study. Sleep 2011;34(5):575–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang C, Holtzman DM. Bidirectional relationship between sleep and Alzheimer’s disease: role of amyloid, tau, and other factors. Neuropsychopharmacology 2020;45(1):104–20. 10.1038/s41386-019-0478-5 [Internet]Available from:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Westerberg CE, Mander BA, Florczak SM, Weintraub S, Mesulam MM, Zee PC, et al. Concurrent impairments in sleep and memory in amnestic mild cognitive impairment. J Int Neuropsychol Soc 2012;18(3):490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Little JT, Satlin A, Sunderland T, Volicer L. Sundown syndrome in severely demented patients with probable alzheimer’s disease. J Geriatr Psychiatr Neurol 1995;8(2):103–6. [DOI] [PubMed] [Google Scholar]
- [19].Ancoli-Israel S, Klauber MR, Jones DW, Kripke DF, Martin J, Mason W, et al. Variations in circadian rhythms of activity, sleep, and light exposure related to dementia in nursing-home patients. Sleep 1997;20(1):18–23. [PubMed] [Google Scholar]
- [20].Stelzmann RA, Norman Schnitzlein H, Reed Murtagh F. An English translation of alzheimer’s 1907 paper, “über eine eigenartige erkankung der hirnrinde. Clin Anat 1995;8(6):429–31. [DOI] [PubMed] [Google Scholar]
- [21].Braak H, Del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol 2011;121(2):171–81. [DOI] [PubMed] [Google Scholar]
- *[22].Stratmann K, Heinsen H, Korf HW, Del Turco D, Ghebremedhin E, Seidel K, et al. Precortical phase of alzheimer’s disease (AD)-Related tau cytoskeletal pathology. Brain Pathol 2016;26(3):371–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bero AQ, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, et al. Neuronal activity regulates the regional vulnerability to amyloidb deposition. Nat Neurosci 2011;14(6):750–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sethi M, Joshi SS, Webb RL, Beckett TL, Donohue KD, Murphy MP, et al. Increased fragmentation of sleep-wake cycles in the 5XFAD mouse model of Alzheimer’s disease. Neuroscience 2015;290:80–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science 2013;342(6156):373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *[26].Ranasinghe KG, Petersen C, Kudo K, Srivatsan S, Beagle AJ, Mizuiri D, et al. Alpha-frequency synchronization deficits during life predict postmortem neurofibrillary tangle burden in Alzheimer’s disease. Alzheimer’s Dementia 2020;16(S4):1–3. [Google Scholar]
- [27].Steele J, Richardson J, Olszewski J. Progressive supranuclear palsy. JAMA Neurol 1964;10(4):333–59. [DOI] [PubMed] [Google Scholar]
- [28].Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 2010;23(4):394–400. [DOI] [PubMed] [Google Scholar]
- [29].Chokroverty S, Montagna P. Sleep, breathing, and neurologic disorders. Sleep Disord Med 2009:436–98.
- [30].Berridge CW. Noradrenergic modulation of arousal. Brain Res Rev 2008;58(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Devilbiss DM, Waterhouse BD. Phasic and tonic patterns of locus coeruleus output differentially modulate sensory network function in the awake rat. J Neurophysiol 2011;105(1):69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Takahashi K, Kayama Y, Lin JS, Sakai K. Locus coeruleus neuronal activity during the sleep-waking cycle in mice [Internet] Neuroscience 2010;169(3):1115–26. 10.1016/j.neuroscience.2010.06.009. Available from:. [DOI] [PubMed] [Google Scholar]
- [33].Ishii T Distribution of Alzheimer’s neurofibrillary changes in the brain stem and hypothalamus of senile dementia. Acta Neuropathol 1966;6(2):181–7. [DOI] [PubMed] [Google Scholar]
- [34].Tomlinson BE, Irving D, Blessed G. Cell loss in the locus coeruleus in senile dementia of Alzheimer type. J Neurol Sci 1981;49(3):419–28. [DOI] [PubMed] [Google Scholar]
- *[35].Ehrenberg AJ, Nguy AK, Theofilas P, Dunlop S, Suemoto CK, Di Lorenzo Alho AT, et al. Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: the pathological building blocks of early Alzheimer’s disease. Neuropathol Appl Neurobiol 2017;43(5):393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kaalund SS, Passamonti L, Allinson KSJ, Murley AG, Robbins TW, Spillantini MG, et al. Locus coeruleus pathology in progressive supranuclear palsy, and its relation to disease severity. Acta Neuropathol Commun 2020;8(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ehrenberg AJ, Suemoto CK, França Resende E de P, Petersen C, Leite REP, Rodriguez RD, et al. Neuropathologic correlates of psychiatric symptoms in alzheimer’s disease. J Alzheimers Dis 2018;66(1):115–26.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lucey BP, McCullough A, Landsness EC, Toedebusch CD, McLeland JS, Zaza AM, et al. Reduced nonerapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci Transl Med 2019;11(474). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jacobs BL, Azmitia EC. Structure and function of the brain serotonin system. Physiol Rev 1992;72(1):165–230. [DOI] [PubMed] [Google Scholar]
- [40].Sakai K, Crochet S. Differentiation of presumed serotonergic dorsal raphe neurons in relation to behavior and wake-sleep states. Neuroscience 2001;104(4):1141–55. [DOI] [PubMed] [Google Scholar]
- [41].Monti JM. The role of dorsal raphe nucleus serotonergic and non-serotonergic neurons, and of their receptors, in regulating waking and rapid eye movement (REM) sleep. Sleep Med Rev 2010;14(5):319–27. [DOI] [PubMed] [Google Scholar]
- [42].Kaur S, De Luca R, Khanday MA, Bandaru SS, Thomas RC, Broadhurst RY, et al. Role of serotonergic dorsal raphe neurons in hypercapnia-induced arousals. Nat Commun 2020;11(1):1–15. 10.1038/s41467-020-16518-9 [Internet]Available from:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Halliday GM, Mccann HL, Pamphlett R, Brooks WS, Creasey H, Mccusker E, et al. Brainstem serotonin-synthesizing neurons in alzheimer’s disease: a clinicopathological correlation. Acta Neuropathol 1992;(84):638–50. [DOI] [PubMed] [Google Scholar]
- [44].Parvizi J, Van Hoesen GW, Damasio A. The selective vulnerability of brainstem nuclei to Alzheimer’s disease. Ann Neurol 2001;49(1):53–66.* [DOI] [PubMed] [Google Scholar]
- [45].Sassin I, Schultz C, Thal DR, Rüb U, Arai K, Braak E, et al. Evolution of Alzheimer’s disease-related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathol 2000;100(3):259–69. [DOI] [PubMed] [Google Scholar]
- [46].Saper CB, Loewy AD. Efferent connections of the parabrachial nucleus in the rat. Brain Res 1980;197(2):291–317. [DOI] [PubMed] [Google Scholar]
- [47].Fulwiler CE, Saper CB. Subnuclear organization of the efferent connections of the parabrachial nucleus in the rat. Brain Res Rev 1984;7(3):229–59. [DOI] [PubMed] [Google Scholar]
- [48].Kaur S, Pedersen NP, Yokota S, Hur EE, Fuller PM, Lazarus M, et al. Glutamatergic signaling from the parabrachial nucleus plays a critical role in hypercapnic arousal. J Neurosci 2013;33(18):7627–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kaur S, Wang JL, Ferrari L, Thankachan S, Kroeger D, Venner A, et al. A genetically defined circuit for arousal from sleep during hypercapnia. Neuron 2017;96(5):1153–67. 10.1016/j.neuron.2017.10.009 [Internet]e5. Available from:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fuller P, Sherman D, Pedersen NP, Saper CB, Lu J. Reassessment of the structural basis of the ascending arousal system. J Comp Neurol 2011;519. 3817–3817.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Parvizi J, Van Hoesen GW, Damasio A. Severe pathological changes of parabrachial nucleus in Alzheimer’s disease. Neuroreport 1998;9(18): 4151–4. [DOI] [PubMed] [Google Scholar]
- [52].Rüb U, Del Tredici K, Schultz C, De Vos RAI, Jansen Steur ENH, Arai K, et al. Progressive supranuclear palsy: neuronal and glial cytoskeletal pathology in the higher order processing autonomic nuclei of the lower brainstem. Neuropathol Appl Neurobiol 2002;28(1):12–22. [DOI] [PubMed] [Google Scholar]
- [53].Oakman SA, Faris PL, Kerr PE, Cozzari C, Hartman BK. Distribution of pontomesencephalic cholinergic neurons projecting to substantia nigra differs significantly from those projecting to ventral tegmental area. J Neurosci 1995;15(9):5859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kroeger D, Ferrari LL, Petit G, Mahoney CE, Fuller PM, Arrigoni E, et al. Cholinergic, glutamatergic, and GABAergic neurons of the pedunculopontine tegmental nucleus have distinct effects on sleep/wake behavior in mice. J Neurosci 2017;37(5):1352–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jellinger K The pedunculopontine nucleus in Parkinson’s disease, progressive supranuclear palsy and Alzheimer’s disease. J Neurol Neurosurg Psychiatry 1988;51(4):540–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Grinberg LT, Heinsen H. Computer-assisted 3D reconstruction of the human basal forebrain complex. Dement Neuropsychol 2007;1(2):140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ozen Irmak S, De Lecea L. Basal forebrain cholinergic modulation of sleep transitions. Sleep 2014;37(12):1941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hanna Al-Shaikh FS, Duara R, Crook JE, Lesser ER, Schaeverbeke J, Hinkle KM, et al. Selective vulnerability of the nucleus basalis of meynert among neuropathologic subtypes of alzheimer disease. JAMA Neurol 2020;77(2):225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Arendt T, Brückner MK, Morawski M, Jäger C, Gertz HJ. Early neurone loss in Alzheimer’s disease: cortical or subcortical? Acta Neuropathol Commun 2015;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tagliavini F, Pilleri G, Gemignani F, Lechi A. Neuronal loss in the basal nucleus of meynert in progressive supranuclear palsy. Acta Neuropathol 1983;61(2):157–60. [DOI] [PubMed] [Google Scholar]
- [61].Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci 1998;18(12):4705–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fujita A, Bonnavion P, Wilson MH, Mickelsen LE, Bloit J, de Lecea L, et al. Hypothalamic tuberomammillary nucleus neurons: electrophysiological diversity and essential role in arousal stability. J Neurosci 2017;37(39): 9574–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Peyron C, Tighe DK, Van Den Pol AN, De Lecea L, Heller HC, Sutcliffe JG, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 1998;18(23):9996–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Takahashi K, Lin JS, Sakai K. Neuronal activity of orexin and non-orexin waking-active neurons during wake-sleep states in the mouse. Neuroscience 2008;153(3):860–70. [DOI] [PubMed] [Google Scholar]
- [65].Branch AF, Navidi W, Tabuchi S, Terao A, Yamanaka A, Scammell TE, et al. Progressive Loss of the orexin neurons reveals dual effects on wakefulness. Sleep 2016;39(2):369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Oh J, Petersen C, Walsh CM, Bittencourt JC, Neylan TC, Grinberg LT. The role of co-neurotransmitters in sleep and wake regulation. Mol Psychiatr 2019;24(9):1284–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Szymusiak R, Alam N, Steininger TL, McGinty D. Sleep-waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats. Brain Res 1998;803(1–2):178–88. [DOI] [PubMed] [Google Scholar]
- [68].Kroeger D, Absi G, Gagliardi C, Bandaru SS, Madara JC, Ferrari LL, et al. Galanin neurons in the ventrolateral preoptic area promote sleep and heat loss in mice. Nat Commun 2018;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Garcia-falgueras A, Ligtenberg L, Kruijver FPM, Swaab DF. Galanin neurons in the intermediate nucleus ( InM ) of the human hypothalamus in relation to sex, age, and gender identity 2011;3084:3061–84. [DOI] [PubMed] [Google Scholar]
- [70].Lim ASP, Ellison BA, Wang JL, Yu L, Schneider JA, Buchman AS, et al. Ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer ‘ s disease 2014.* [DOI] [PMC free article] [PubMed]
- [71].Hassani OK, Lee MG, Jones BE. Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle. Proc Natl Acad Sci U S A 2009;106(7):2418–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Apergis-Schoute J, Iordanidou P, Faure C, Jego S, Schöne C, Aitta-Aho T, et al. Optogenetic evidence for inhibitory signaling from orexin to MCH neurons via local microcircuits. J Neurosci 2015;35(14):5435–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Konadhode RR, Pelluru D, Blanco-Centurion C, Zayachkivsky A, Liu M, Uhde T, et al. Optogenetic stimulation of MCH neurons increases sleep. J Neurosci 2013;33(25):10257–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Jego S, Glasgow SD, Herrera CG, Ekstrand M, Reed SJ, Boyce R, et al. Optogenetic identification of a rapid eye movement sleep modulatory circuit in the hypothalamus. Nat Neurosci 2013;16(11):1637–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Mladinov M, Oh JY, Petersen C, Eser R, Li SH, Theofilas P, et al. Specific pattern of melanin-concentrating hormone (MCH) neuron degeneration in Alzheimer’s disease and possible clinical implications. medRxiv 2021. Jan 1 [Internet]2021.01.27.21250608. Available from: http://medrxiv.org/content/early/2021/01/29/2021.01.27.21250608.abstract.
- [76].Preston RJ, McCrea RA, Chang HT, Kitai ST. Anatomy and physiology of substantia nigra and retrorubral neurons studied by extra- and intracellular recording and by horseradish peroxidase labeling. Neuroscience 1981;6(3):331–44. [DOI] [PubMed] [Google Scholar]
- [77].Liu D, Li W, Ma C, Zheng W, Yao Y, Tso CF, et al. A common hub for sleep and motor control in the substantia nigra- supplementary materials. Science 2020;367(6476):440–5. [DOI] [PubMed] [Google Scholar]
- [78].Yadav RK, Khanday MA, Mallick BN. Interplay of dopamine and GABA in substantia nigra for the regulation of rapid eye movement sleep in rats. Behav Brain Res 2019;376(May). [DOI] [PubMed] [Google Scholar]
- [79].Inoue M, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of progressive supranuclear palsy. Neuropathology 1996;16:257–61. [Google Scholar]
- [80].Hardman CD, Halliday GM, McRitchie DA, Cartwright HR, Morris JGL. Progressive supranuclear palsy affects both the substantia nigra pars compacta and reticulata. Exp Neurol 1997;144(1):183–92. [DOI] [PubMed] [Google Scholar]
- [81].Kang Y, Kitai ST. Electrophysiological properties of pedunculopontine neurons and their postsynaptic responses following stimulation of substantia nigra reticulata. Brain Res 1990;535(1):79–95. [DOI] [PubMed] [Google Scholar]
- [82].Lavoie B, Parent A. Pedunculopontine nucleus in the squirrel monkey: cholinergic and glutamatergic projections to the substantia nigra. J Comp Neurol 1994;344(2):232–41. [DOI] [PubMed] [Google Scholar]
- [83].Luo SX, Huang EJ. Dopaminergic neurons and brain reward pathways: from neurogenesis to circuit assembly. Am J Pathol 2016;186(3):478–88. 10.1016/j.ajpath.2015.09.023 [Internet]Available from:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Eban-Rothschild A, Rothschild G, Giardino WJ, Jones JR, De Lecea L. VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nat Neurosci 2016;19(10):1356–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Takata Y, Oishi Y, Zhou XZ, Hasegawa E, Takahashi K, Cherasse Y, et al. Sleep and wakefulness are controlled by ventral medial midbrain/pons GABAergic neurons in mice. J Neurosci 2018;38(47):10080–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Nair-Roberts RG, Chatelain-Badie SD, Benson E, White-Cooper H, Bolam JP, Ungless MA. Stereological estimates of dopaminergic, GABAergic and glutamatergic neurons in the ventral tegmental area, substantia nigra and retrorubral field in the rat. Neuroscience 2008;152(4):1024–31. 10.1016/j.neuroscience.2008.01.046 [Internet]Available from:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Yu X, Li W, Ma Y, Tossell K, Harris JJ, Harding EC, et al. GABA and glutamate neurons in the VTA regulate sleep and wakefulness. Nat Neurosci 2019;22(1):106–19. 10.1038/s41593-018-0288-9 [Internet] Available from:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].De Marco M, Venneri A. Volume and connectivity of the ventral tegmental area are linked to neurocognitive signatures of Alzheimer’s disease in humans. J Alzheim Dis 2018;63(1):167–80. [DOI] [PubMed] [Google Scholar]
- [89].Nobili A, Latagliata EC, Viscomi MT, Cavallucci V, Cutuli D, Giacovazzo G, et al. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat Commun 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zhong P, Zhang Z, Barger Z, Ma C, Liu D, Ding X, et al. Control of non-REM sleep by midbrain neurotensinergic neurons. Neuron 2019;104(4): 795–809. 10.1016/j.neuron.2019.08.026 [Internet]e6. Available from:. [DOI] [PubMed] [Google Scholar]
- [91].Clément O, Sapin E, Bérod A, Fort P, Luppi PH. Evidence that neurons of the sublaterodorsal tegmental nucleus triggering paradoxical (REM) sleep are glutamatergic. Sleep 2011;34(4). 0–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature 2006;441(7093):589–94. [DOI] [PubMed] [Google Scholar]
- [93].Alam MA, Kostin A, Siegel J, McGinty D, Szymusiak R, Alam MN. Characteristics of sleep-active neurons in the medullary parafacial zone in rats. Sleep 2018;41(10):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Song JZ, Cui SY, Cui XY, Hu X, Ma YN, Ding H, et al. Dysfunction of GABAergic neurons in the parafacial zone mediates sleep disturbances in a streptozotocin-induced rat model of sporadic Alzheimer’s disease. Metab Brain Dis 2018;33(1):127–37. [DOI] [PubMed] [Google Scholar]
- [95].Uschakov A, Gong H, McGinty D, Szymusiak R. Efferent projections from the median preoptic nucleus to sleep- and arousal-regulatory nuclei in the rat brain. Neuroscience 2007;150(1):104–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Gong H, McGinty D, Guzman-Marin R, Chew KT, Stewart D, Szymusiak R. Activation of c-fos in GABAergic neurones in the preoptic area during sleep and in response to sleep deprivation. J Physiol 2004;556(3): 935–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Gvilia I, Xu F, McGinty D, Szymusiak R. Homeostatic regulation of sleep: a role for preoptic area neurons. J Neurosci 2006;26(37):9426–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Liu K, Kim J, Kim DW, Zhang YS, Bao H, Denaxa M, et al. Lhx6-positive GABA-releasing neurons of the zona incerta promote sleep. Nature 2017;548(7669):582–7. 10.1038/nature23663 [Internet] Available from:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Guest WC, Silverman JM, Pokrishevsky E, O’Neill MA, Grad LI, Cashman NR. Generalization of the prion hypothesis to other neurodegenerative diseases: an imperfect fit. J Toxicol Environ Health Part A Curr Issues 2011;74(22–24):1433–59. [DOI] [PubMed] [Google Scholar]
- [100].Rüb U, Stratmann K, Heinsen H, Seidel K, Bouzrou M, Korf H-W. Alzheimer’s disease: characterization of the brain sites of the initial tau cytoskeletal pathology will improve the success of novel immunological anti-tau treatment approaches. J Alzheimers Dis 2017;57(3):683–96. [DOI] [PubMed] [Google Scholar]
- [101].Nedergaard M, Goldman SA. Glymphatic failure as a final common pathway to dementia. Science 2020;370(6512):50–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol 2018;136(1):57–67. 10.1007/s00401-018-1855-6 [Internet]. Available from:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: fromstressor thresholds todegeneration. Neuron 2011;71(1):35–48. 10.1016/j.neuron.2011.06.031 [Internet]Available from:. [DOI] [PubMed] [Google Scholar]
- [104].Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, et al. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci 2006;26(5):1343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.