

EXECUTIVE SUMMARY
The past six decades have seen remarkable improvements in health outcomes for people with cystic fibrosis (CF), which was once a fatal disease of infants and young children. However, although life expectancy for people with CF has increased substantially, the disease continues to limit survival and quality of life, and results in a significant burden of care for people with CF and their families. Moreover, recent epidemiological studies in the past two decades have shown that CF occurs and is more frequent than was previously thought in non-European populations and in regions of the world where it had not previously been described. The Lancet Respiratory Medicine Commission was formed to assess the global health and economic costs of CF care over the next three decades and to identify opportunities for progress in the care of people with CF. challenges that need to be addressed, and to serve as a blueprint for the future of CF care. This Commission was established at a time of immense change in CF clinical care including enhanced survival, widespread genetic testing supporting the diagnosis of CF in regions of the world where it was previously thought to be or non-existent and the advent of a growing number of CFTR-directed therapies which are likely to further alter the natural trajectory of the disease. This document is intended to bring to the attention of patients, health-care professionals, researchers, funders, service providers, policy makers the variety of challenges associated with this changing landscape and to serve as a blueprint for the future of CF care. what was previously a universally fatal disease of children. In considering the future of CF care, the Commission focused on five key areas, which are discussed in this report: the changing epidemiology of CF (section 1); future challenges of clinical care and its delivery (section 2); the building of CF care globally (section 3); novel therapeutics (section 4); and patient engagement (section 5) (summarized in panel 1).
Since the discovery of the cystic fibrosis transmembrane regulator (CFTR) gene in the late 1980’s, triggered a surge of basic research which has enhanced understanding of the pathophysiology and the genotype/phenotype relationships of this clinically variable disease. Until recently, available treatments could be used to control symptoms and limit the complications of CF, but advances in CFTR modulator therapies to address the basic defect of CF have been phenomenal and the field is evolving rapidly.
Advances in clinical care have been multifaceted and include earlier diagnosis through the implementation of newborn screening programmes, formalised airway clearance therapy, and reduced malnutrition through the use of effective pancreatic enzyme replacement and a high-energy, high protein diet. Centre-based care has become the norm in high-income countries, allowing patients to benefit from the skills of expert members of multi-disciplinary teams. Pharmacological interventions to address pulmonary manifestations now include agents that target airway mucus and airway surface liquid hydration, as well as antimicrobial therapy, including antibiotic eradication treatment in early infection and protocols for maintenance therapy in chronic infection.
The challenges faced by all stakeholders in building and developing CF care globally are substantial, but many opportunities exist for improved care and better health outcomes for patients. Here, we discuss changes in diagnostic approaches including advances in genetic testing, advances in newborn screening and how it is changing landscape of the CF population, The occurrence and impact of CF in non-European populations and on different continents is explored as is. We highlight the current and future opportunities as well are the challenges for clinical care in both countries with established CF care programs and what care might look like for low- and middle-income countries where care programs more fragmented and less well funded. to the availability of integrated multi-disciplinary care and new disease-modifying treatments. The recent advances in CFTR modulator therapies to address the basic defect of CF have been phenomenal and current knowledge and future directions are appraised. The cost of new therapies is currently high and there are concerns about the affordability of those medications currently available and those under development especially when considered from a global perspective. As median age of CF has increased, there has been an associated rapid rise in the adult population, we evaluate opportunities for the use of new technologies to support engagement between patients and healthcare providers in novel ways to positively impact on the burden of care and support patient choice and yet to optimise healthcare outcomes. Challenges and opportunities in these key areas are being discussed to provide a conceptional framework for future CF care throughout the world.
Panel 1 Key Messages of the Lancet Commission for Cystic Fibrosis
Section 1 The changing epidemiology of cystic fibrosis
Newborn screening testing has been implemented in many parts of the world supporting an early diagnosis of CF.
Improved molecular genetic diagnostics have allowed the identification of CF in non-European populations and in individuals with nonclassical presentations of CF and related disorders.
CFTR related disease represents a spectrum ranging from single organ manifestations to a multi-system disease. Defining the threshold of CFTR function associated with disease manifestations is a priority to guide monitoring and treatment decisions.
Section 2 Clinical care and its delivery
Children with CF are healthier and the vast majority are living well into adulthood.
Diagnostics to allow earlier diagnosis of disease manifestations, deterioration of organ function and new airway infections are key priorities.
Models of care need to consider management approaches (including disease monitoring) to maintain health and delay lung transplantation and yet limit the burden of care.
Section 3 Cystic fibrosis care in developing nations
Information about genetic and clinical features CF in non-European populations have improved understanding of the disease in low- and middle-income countries.
Access to CF therapies which is sustained and affordable for people with CF living in LMIC is needed which requires partnerships between lay organisations, governments and the pharmaceutical industry.
Clinical registries in countries where CF is now recognised are developing and there is a need to harmonise data elements including established CF registries to support understanding of health care outcomes especially in LMIC.
Section 4 Novel therapeutics
CFTR modulator therapy targeting the basic molecular defect have been developed for specific CFTR mutations and is associated with improved health outcomes including better pulmonary function and nutritional status and enhanced quality of life.
New CFTR modulator drugs are showing promise in CFTR mutation where earlier modulators were ineffective. Early commencement of CFTR-directed therapies may prevent the establishment of irreversible airways complications and slow progression disease in older patients.
Drug development requires substantial investment and contributes to the current high-cost of approved CFTR modulators. This has in turn contributed to delays in funding for such therapies in many countries and current drug prices make them unaffordable for many LMICs.
Section 5 Patient experience, engagement and involvement
Complexity of care has increased for people with CF in parallel with increased life expectancy leading to significant burden of care and disease monitoring. Novel technologies have the potential to support self-monitoring and shared decision making between patient and health care team.
Mental health complications are more common in people with CF (including parents of children with CF) than the general community, impacting on quality of life. Adherence to complex therapeutic regimens is often sub-optimal and impacts of clinical outcomes.
Patients are highly engaged in the approaches to delivery of clinical care and their perspective of the important research priorities. CF patient organisations provide important roles in patient advocacy to delivery of clinical care, treatment access, and support and education for patients with CF and their families.
Keywords: Cystic fibrosis
INTRODUCTION
Since the original description of cystic fibrosis (CF)1, there has been very significant progress from what was a uniformly fatal disease in infancy to one which where the median survival approaches (and in some populations exceeds) 50 years of age. The recognition of the increased salt content of the sweat in people with CF by di Sant’Agnese in early 1950s2 led to the development of the pilocarpine iontophoresis stimulated sweat analysis as a practical diagnostic test3. In the 1980s, Quinton demonstrated chloride impermeability in sweat glands as the basis for the raised sweat electrolytes in patients with CF4. This work provided a crucial advance in the understanding the basic defect was a membrane electrolyte transport defect.
Focus then shifted to discover the genetic basis of CF and culminated in the discovery of the CF gene in 1989 by teams led by Lap-Chi Tsui, Francis Collins and Jack Riordan5–7 with the subsequent identification of its protein product which was termed the cystic fibrosis transmembrane conductance regulator (CFTR). CFTR is responsible for chloride ion transport across apical membranes of epithelial cells in tissues. CFTR is now known to also have additional functions such as bicarbonate secretion which regulates airway surface liquid pH and inhibition of the epithelial sodium channel (eNAC), which plays an important role in hydration of secretions and mucins (Figure 1)8.
Figure 1. Pathophysiology of Cystic Fibrosis.
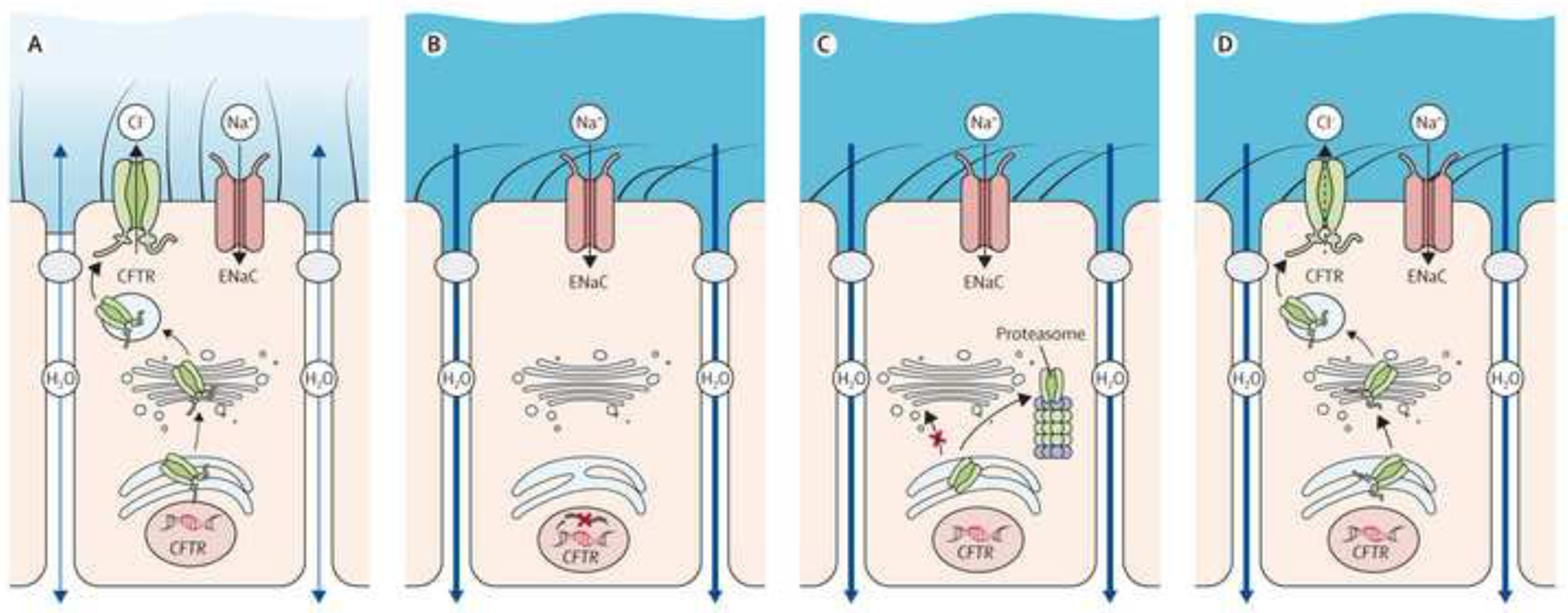
A-D, Role of CFTR in healthy airways and molecular mechanisms causing CFTR dysfunction in cystic fibrosis (CF). A, In healthy airways, CFTR is expressed at the apical surface of airway epithelial cells together with the epithelial sodium channel (ENaC). Coordinated regulation of CFTR and ENaC enables proper airway surface hydration and effective mucociliary clearance. B-D, In CF, different mutations in CFTR cause CFTR dysfunction via different molecular mechanisms. B, CFTR nonsense or splicing mutations abrogate CFTR production. C, Many missense mutations, including the common F508del mutation, impair proper folding of CFTR and lead to retention in the endoplasmic reticulum and degradation by the proteasome. D, Some missense and splicing mutations produce CFTR chloride channels that reach the cell surface but are not fully functional.
CFTR = cystic fibrosis transmembrane conductance regulator; ENaC = epithelial sodium channel.
Reproduced from Gentzsch and Mall, CHEST 2018; 154(2):383–393 by permission of Elsevier Ltd Copyright 2018 American College of Chest Physicians.
More than 2,000 variants in the cystic fibrosis transmembrane conductance regulator (CFTR) gene have been described to date9,10. To date the functional consequences have not been defined in all of these variants. Where the functional consequences are known variants can be divided into different function classes (Figure 2)11. Class I to III CF-causing variants (in legacy terminology ”mutations“) are associated with little or no CFTR function and therefore linked to a more severe phenotype including insufficiency of the exocrine pancreas, whereas Class IV and V variants have residual CFTR function which is often associated with preserved exocrine pancreatic function early in life. Overall, CFTR dysfunction causes a spectrum of disease with a range in the number of organs involved and a range in disease severity. For example, pancreas, liver and lung disease can be present in infancy and early childhood, or congenital bilateral absence of the vas deferens (CBAVD) in the male reproductive tract12 may be the only significant feature of CF in an adult. In addition, phenotypic manifestations vary widely even amongst people with severe CFTR variants underscoring the role of other factors such as environmental triggers and modifier genes in defining disease severity extensively reviewed by Cutting13.
Figure 2. Classes of CFTR Mutations.
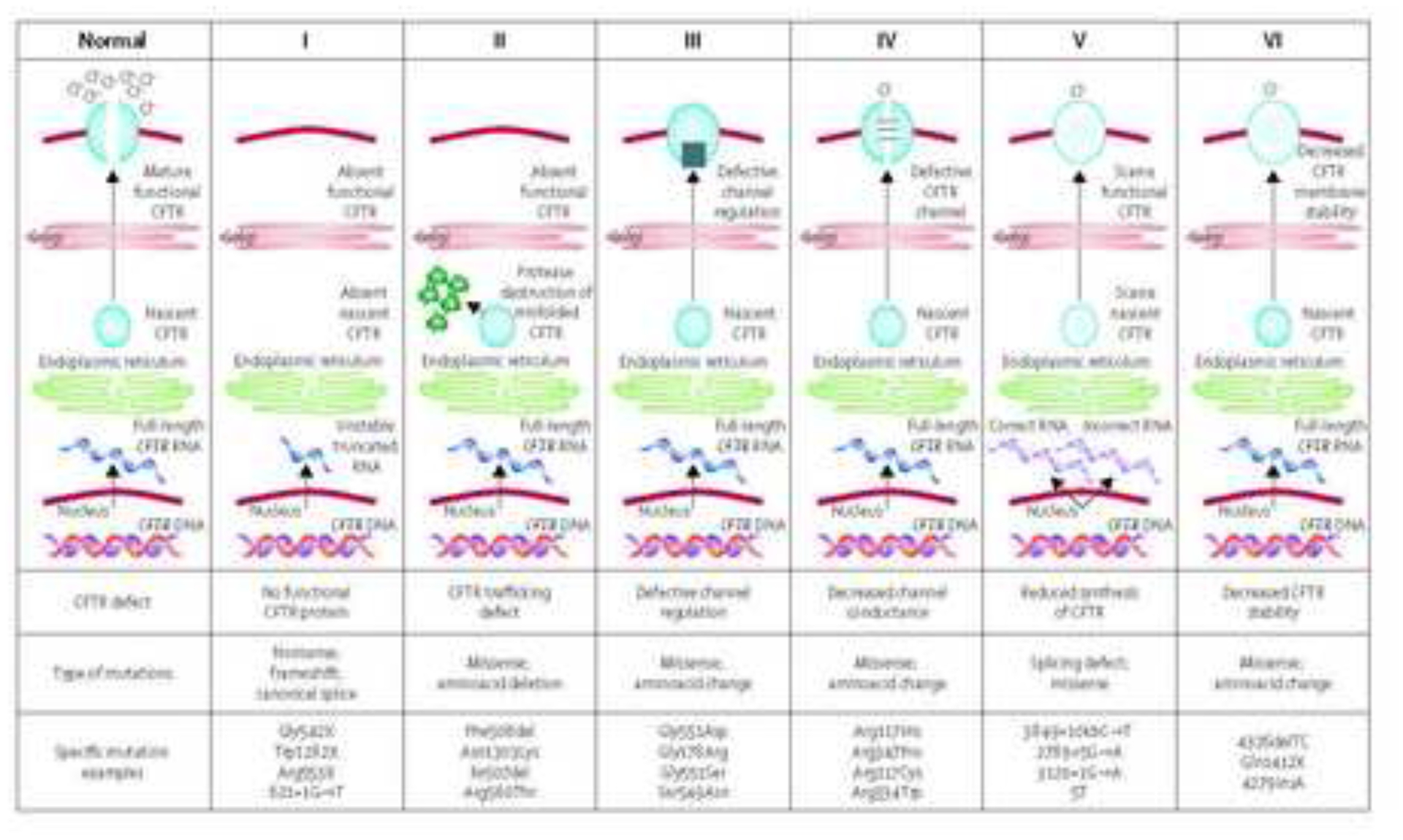
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene can be divided into six classes. Class I mutations result in no protein production. Class II mutations (including the most prevalent, Phe508del) cause retention of a misfolded protein at the endoplasmic reticulum, and subsequent degradation in the proteasome. Class III mutations affect channel regulation, impairing channel opening (eg, Gly551Asp). Class IV mutants show reduced conduction—ie, decreased flow of ions (eg, Arg117His). Class V mutations cause substantial reduction in mRNA or protein, or both, Class VI mutations cause substantial plasma membrane instability and include Phe508del when rescued by most correctors (rPhe508del).
Reproduced with permission Boyle MP, De Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med 2013; 1: 158–63.
The CF phenotype is characterised by lung disease (bronchiectasis with persistent airway-based infection and inflammation), exocrine pancreatic insufficiency associated with nutrient malabsorption contributing to under-nutrition, impaired growth, hepatobiliary manifestations and male infertility14 (Table 1). Evidence strongly supports the care of people with CF be coordinated by multi-disciplinary health-care teams and therapies for CF have developed rapidly over the past three decades (reviewed in detail elsewhere)15. The health of children with CF continues to improve with better lung function and growth by the time patients transition from pediatric to adult care with lower rates of chronic infection in particular P. aeruginosa (Figure 3). The progressive improvement of survival from CF over the past five decades16 has led to a dramatic increase in the number of adults surviving. Consequently there has been rapid expansion of adult CF care centres17 (Figure 4). Despite such improvements, mortality and morbidity from CF is still dominated by recurrent pulmonary infections (which are frequently multi-drug resistant), ultimately leading to lung destruction and respiratory failure. The emergence of complications of this disease including CF-related diabetes, metabolic bone disease, gastro-intestinal malignancy and co-morbidities including increased rates of mental health conditions (depression and anxiety) have required the development of specific expertise in providing CF clinical care18.
Table 1.
Phenotypic Features of Cystic Fibrosis
| Classic Features of CF |
| Respiratory |
| Bronchectasis with chronic infection |
| Pneumothorax |
| Haemoptysis |
| Respiratory failure |
| Chronic rhinosinusitis and nasal polyposis |
| Gastrointestinal |
| Luminal |
| Meconium ileus |
| Gastro-oespageal reflux syndrom |
| Distal intestinal obstruction syndrowm |
| Chronic constipation |
| Rectal Prolapse |
| Intersussception |
| Colorectal cancer and colonic polyposis |
| Other gastroinstestinal malignancies |
| Hepatobiliary |
| Pancreatic insufficiency |
| Recurrent acute pancreatitis (in patients with pancreatic sufficiency |
| Biliary sludge/cholelithiasis |
| Biliary cirrhosis |
| Metabolic complications |
| CF related diabetes |
| Microvascular complications (10+ years from diagnosis) |
| CF related bone disease/osteoporosis |
| Increased fracture risk |
| Ureteric calculi |
| Oligoammenorrhoea |
| Male infertility |
| Congential bilateral absence of the vas deferens |
| Common issues complicating life with CF or CF therapies |
| Mental health conditions |
| Depression |
| Anxiety |
| Vascular access complications |
| Thrombosis of vascular access devices |
| Drug complications |
| Antibiotic hypersensivity reactions and intolerance |
| Vestibulo-auditory disturbance including tinnitus |
| Chronic kidney disease |
| Metabolic complications |
| Overweight and obesity (especially in the older patient) |
| Post-tranplant complications (relevant to CF) |
| Chronic kidney disease and CFRD complications |
| Multiresistant organisms can complicate airways complications after transplantation |
| Cancer in long-term survivors (including gastrointestinal, skin and urogenital) |
Figure 3. Median FEV1 % predicted for people with Cystic Fibrosis in the United States by Age (1998 to 2018).
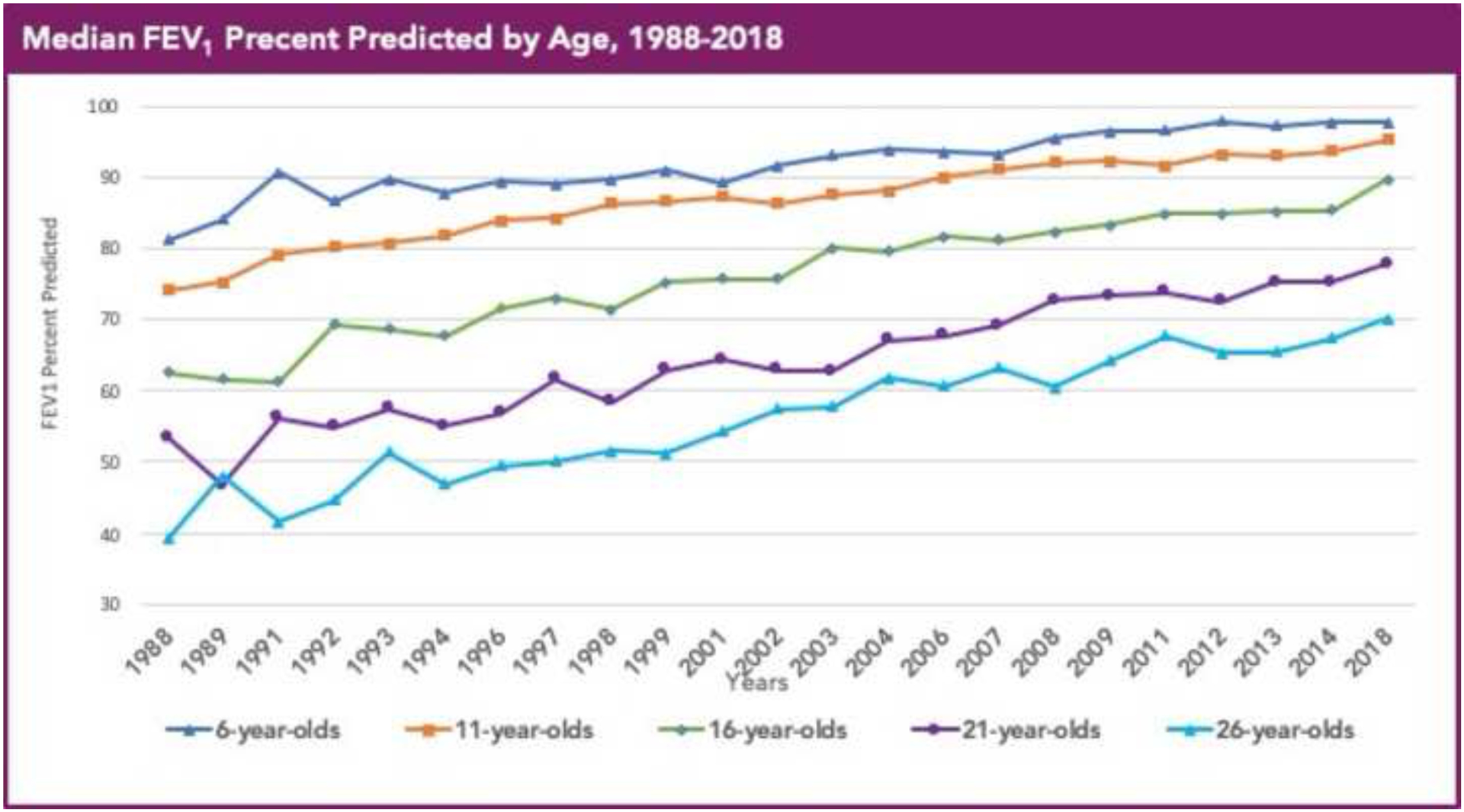
Cystic fibrosis patients under care at CF Foundation-accredited care center in the United States, who consented to have their data entered.
Reproduced with permission of the US Cystic Fibrosis Foundation, Bethesda Maryland.
Cystic Fibrosis Foundation Patient Registry
Bethesda, Maryland ©2019 Cystic Fibrosis Foundation.
Figure 4. The Number of Children and Adults with Cystic Fibrosis (USA),1987–2017.
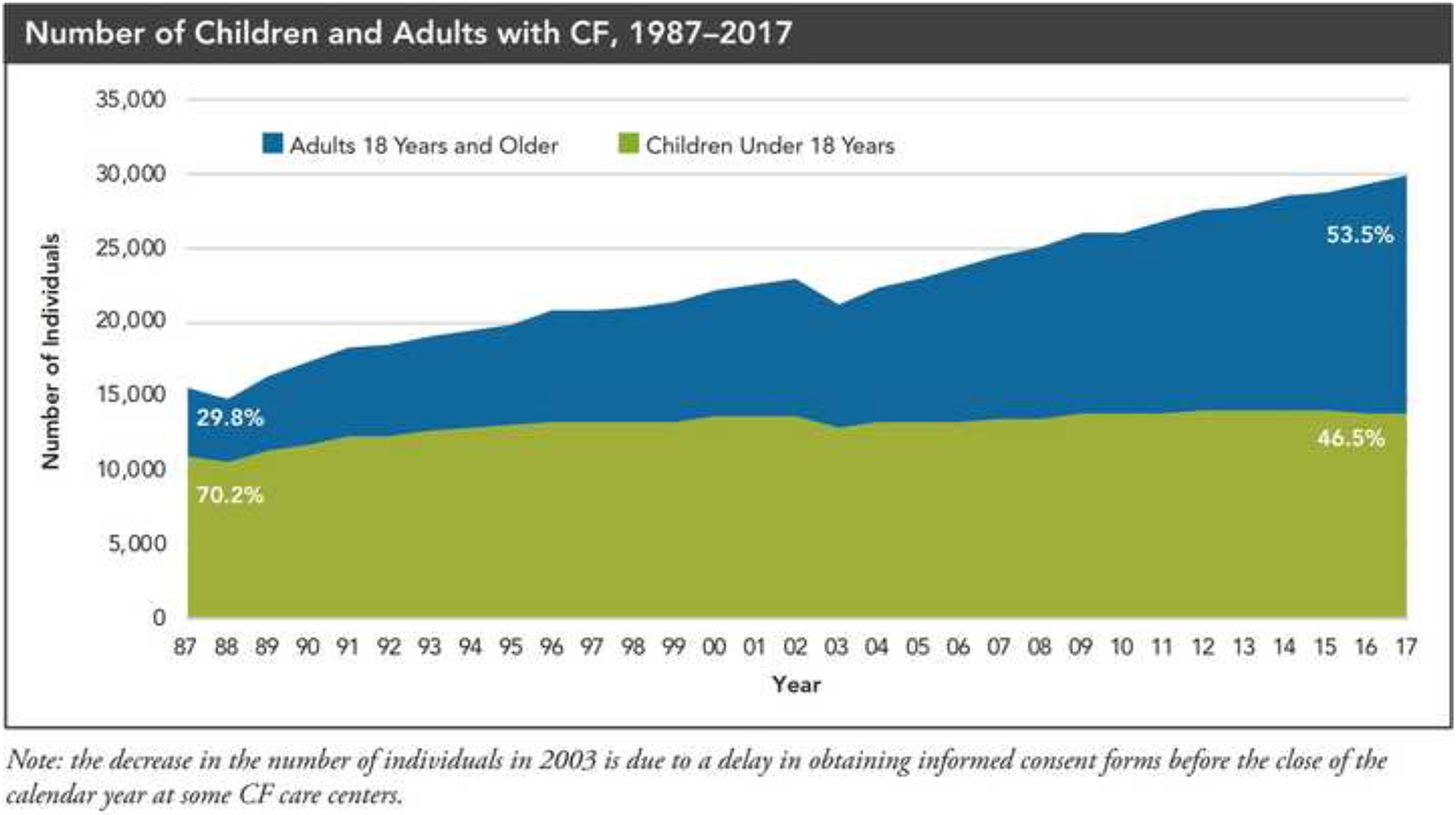
Cystic fibrosis patients under care at CF Foundation-accredited care centers in the United States, who consented to have their data entered.
Reproduced with permission of the US Cystic Fibrosis Foundation, Bethesda Maryland.
2017 Annual Data Report
Bethesda, Maryland ©2018 Cystic Fibrosis Foundation.
While structured care in specialised centres as well as improved strategies to treat disease manifestations have been the main drivers of better patients’ outcomes, over the past 15 years, a therapeutic “pipeline’ has been established with the aim to develop, safe and effective treatments for the basic defect of CF (correct dysfunctional CFTR protein)19–21. The first successful drug, a potentiator of CFTR function that increases the opening probability of the CFTR protein, was associated with remarkable clinical benefit (improved lung function, nutritional status, health-related quality of life (HRQoL) and lower rate of pulmonary exacerbations) in patients with residual expression of CFTR on the cell surface but a reduced open probability21,22. Subsequently, combinations of both CFTR potentiators and correctors that address the trafficking defect in the most common variant, p.Phe508del, have been shown to have positive impact on clinical outcomes23,24, and the field is rapidly evolving to a point where more than 90% of the current CF population could benefit from these therapies. However, access to these drugs varies widely amongst countries with could further enhance an already existing gap in outcomes for patients in different regions of the world.
At a time of very rapid therapeutic developments, advances in the genetic diagnostic testing and the explosion of the adult CF population associated with dramatic increases in the median survival for people with CF it is timely to review the advances and consider what CF care will look like into the future taking a global perspective. The Lancet Respiratory Medicine Commission on the Future of Cystic Fibrosis Care addresses the important health of CF care and economic costs including CFTR modulators over the next three decades.
SECTION 1 –. THE CHANGING EPIDEMIOLOGY OF CYSTIC FIBROSIS
1). The impact of genetic testing
Genetic testing has had a significant impact not only on the diagnosis of CF but also in defining a spectrum of disease manifestations for the different mutations described. Since the identification of the cystic fibrosis transmembrane regulator (CFTR) gene and initially the major pathogenic variant p.Phe508del in 1989, the number of sequence variants detected has increased to over 2,000; curated in the “CFTR1” locus-specific database9. The first genetic variants were described in clinically diagnosed cases of CF – implying that when present in trans, i.e. on both parental CFTR alleles, they are causative of CF. However, with the explosion in the number of gene variants discovered, the pattern of disease associated with each has become more difficult to confidently predict. Firstly, some CFTR variants are associated with a broader spectrum of CFTR-related disorders (CFTR-RD)25 and others do not appear to contribute to any disease state26. Secondly, the majority of CFTR variants are rare occurring in less than 0.5% of all patients with CF (or even present in a single family) making phenotypic predictions fragile. The “Clinical and Functional Translation of CFTR” project (“CFTR2”) was developed to try to address this by annotating variants with comprehensive clinical features when seen in at least four unrelated individuals worldwide27. Currently more than 340 variants are classified as “CF-causing” comprising over 89,000 cases from 43 countries. These “pathogenic variants” define the genetic mutations in more than 96% of all patients with CF of Northern European descent examined to date1. However, despite extensive international collaboration, the population variation in ‘non-European’ derived populations remains under-represented. This brings with it a challenge which impacts on newborn screening programme and carrier testing for individuals, as well as the accuracy of incidence and prevalence of CF for populations of ‘non-European’ background28 (see Newborn Screening in CF and Carrier Screening in CF below).
Nonetheless, despite some gene variant-clinical phenotype correlations being less robust, how the different CFTR variants pathogenically impair the amount and function of the CFTR protein has led to allocation into respective classes to better describe their disease-liability29,30,31. This stratification is important for the diagnosis of CF, neonatal screening, assuring reproductive choice in affected families, preconception carrier screening and clinical management. Finally this classification will enable precision medicine approaches in CF32.
2). Newborn screening for CF (and Cystic Fibrosis Screen Positive, Inconclusive Diagnosis or Cystic Fibrosis Transmembrane Conductance Regulator-Related Metabolic Syndrome (CFSPID/CRMS))
There is clear evidence to support newborn bloodspot screening (NBS) for CF and it fulfils the criteria set for a disease screening test33. Early diagnosis allows early institution of management to improve clinical outcomes. Specifically, early nutritional intervention after NBS leads to better growth and micronutrient status in the majority of children34. A direct link to early instigation of respiratory management and better respiratory outcomes in the first few years of life has been more difficult to demonstrate35. In part this is because respiratory outcome measures (lung function, airway inflammatory markers) are difficult to obtain in this age group, although these are being developed (see Monitoring lung disease and its progression below). Also risk factors such as P. aeruginosa acquisition can be unpredictable and impact outcome. However, we know that better nutritional status correlates with less severe lung disease during childhood. Survival has also increased in association with NBS suggesting mortality rates are improved as a result of early diagnosis36,37.
Less well defined benefits associated with the implementation of NBS for CF include the rationalisation and improvement of CF care provision across the area screened and the prevention of the well-documented diagnostic odyssey many families experience38. NBS for CF may not significantly reduce the gap in health outcomes recorded for infants with CF that relate to social inequalities as earlier recognition does not address the challenges that families from lower socio-economic circumstances face.
Most CF NBS programmes follow a two stage process: increased level of blood spot immunoreactive trypsinogen (IRT) measurement, which are caused by partial obstruction of pancreatic ducts leading to abnormal enzyme drainage into the intestine, followed by identification of causative CFTR variants (DNA)35. Some also employ an adjunctive biomarker (e.g. pancreatitis-associated protein, PAP) or a “safety net” repeat IRT at a later age if first one elevated (IRT/IRT). A recent study comparing strategies showed that the IRT/PAP combination was the most cost-effective in terms of cost-per-case detected and life-years gained39. However, it produces a large number of false-positive test results that necessitate a referral to a CF center for a diagnostic sweat test so the IRT/DNA sequence remains the most commonly used in developed countries40. The selection of CFTR variants varies worldwide, has a major influence on the screening-positive rate and reflects the aim of the screening programmes (detection of “severe” patients with CF versus “complete” detection). NBS programmes demand a high accountability of the CF centres to perform the confirmatory testing according to the highest standard. Despite extensive knowledge of the functional relevance of CFTR-mutations, a high quality sweat test still remains the most sensitive and specific diagnostic tool to diagnose CF, confirm the clinical relevance of CFTR variants and, more important, to make the diagnosis of CF unlikely. However, in some health systems including low and middle income countries (LMIC), the delivery of high quality sweat testing is technically challenging and availability is limited (detailed discussion following section 3).
NBS also identifies children are heterozygote carriers or infants with intermediate sweat chloride levels for whom the terms Cystic Fibrosis Screen Positive, Inconclusive Diagnosis or Cystic Fibrosis Transmembrane Conductance Regulator-Related Metabolic Syndrome (CFSPID/CRMS) are used. These infants are considered at risk for CFTR-related disorders such as Congenital Bilateral Absence of the Vas Deferens (CBAVD) or recurrent pancreatitis and recent studies have shown that 10–44% of those initially labeled CFSPID/CRMS may convert to a CF diagnosis— an observation that emphasises the importance of follow-up evaluations38,41.
Although the benefits are well established, implementation of NBS programmes for CF internationally faced some ethical queries and financial hurdles. Specifically, striking the right balance between sensitivity of NBS programmes to not miss affected individuals while minimising the detection of CFSPID infants; most of which will be healthy with no disease manifestations, is an ongoing challenge. Currently, it is estimated that approximately 10 million newborn infants are screened annually for CF worldwide. The future will be to get universal NBS programmes established, with population or ethnically appropriate derived gene mutation panels. The impact of NBS on the incidence of CF will be discussed in the section below.
3). Carrier screening in CF
The progress in laboratory and clinical annotation of the more common CF-causing variants allows screening of CFTR variants associated with the classical form of disease for a number of other key groups in the community. Preconception carrier screening42 in individuals without a family history of CF can be offered and is undertaken in some general populations including the US43, Israel44 and in North-eastern Italy45. Given the higher prevalence of CFTR variants in infertile males with congenital bilateral absence of vas deferens (CBAVD)46, genetic testing has been offered to infertile couples before assisted reproduction47,48. In addition, genetic testing for common population specific CF-causing variants is also often recommended by professional guidelines or even is mandatory in gamete donors49. Cascade screening of relatives of an index case can also be undertaken, as most CF babies are born to families with no known family history of CF.
Carrier screening is associated with various ethical questions including the high residual risk of not detecting a CFTR mutation depending on the selection of CFTR variant panels and inequitable access for minority populations. As providing screening is generally not mandated, but rather instituted by professional recommendations50 or position statements, the offer by health care professionals is therefore variable51 and its uptake depends on the caregivers and familes information on CF52 and awareness of CF53, reimbursement issues, stage of life when individuals are planning reproduction, as well as post-test information54and genetic counselling55,56. Often CF carrier screening is being implemented within the frame of extended carrier screening for multiple diseases57, thereby offering tested couples informed reproductive choice for other common rare Mendelian disorders. With decreasing family sizes in many countries there is increasing uptake of carrier screening already in first pregnancies58.
The main aim is to detect heterozygotes and offer couples reproductive choice before the birth of their child. Carrier couples have access to preimplantation genetic diagnosis within the context of assisted reproduction which assures transfer of an unaffected embryo and thus avoids elective termination of pregnancy59. At risk couples who conceive naturally can undergo prenatal diagnosis which utilises foetal DNA isolated by chorionic villus sampling or from amniotic fluid cells60 with well-defined alternative reproductive options61. In addition, routine prenatal ultrasound screening during pregnancy can identify foetal echogenic bowel between the 17th to 22nd week of gestation associated with CF62. Recently introduced non-invasive prenatal diagnosis utilises cell-free foetal DNA (cffDNA) to reliably assess the CFTR variant status of the foetus63. This approach offers a logistically and technically simpler, and more cost-effective, prenatal testing strategy that could be offered in future to a larger number of at-risk. However, in time, it would serve to increase equity in terms broader population coverage64 and assure broader ethnic access to such preventive approaches65
As indicated above (in The Impact of Genetic Testing), the gene variant panels developed for carrier testing are largely based on genetic testing of European derived populations which will disadvantage other ethnically-diverse individuals28. Population-based genomic variant frequencies from several major international genome66,67, projects could be used to extrapolate estimates of CFTR variant frequencies in European and non-European populations68. A UK 100,000 genomes project, for example, gathers data on representative cohorts of UK Indian and Pakistani immigrant populations69,70. The decreasing costs of parallel sequencing of the entire CFTR coding sequence and adjacent intronic regions71 could also overcome the challenge of lower CFTR variant detection rates and account for intra-CFTR structural variation. This could be advantageous for a changing population due to migration trends72 that disproportionately affect major multicultural cities worldwide73. Many newly described CFTR variants detected in this way may be classified as variants of unknown clinical significance given the fewer gene variant-clinical correlations of tested individuals of non-European descent to date74. Genome testing in individuals with no family history of CF may also identify pathogenic CFTR variants in trans in asymptomatic individuals which indicates their incomplete penetrance75. Thus, advanced DNA sequencing-based diagnostic strategy increases equity in terms broader population coverage64 and assures broader ethnic access to such preventive approaches65, yet needs to be balanced against the diagnostic uncertainty caused by an increased detection of variants of uncertain significance.
Finally, the rapid evolution in treatment, especially in the field of efficient CFTR-modulating therapies, may alter future responses to CF carrier screening or prenatal genetic testing. With the availability of CFTR modulator therapies, a higher proportion of parents may opt to continue the pregnancy than currently is the case. The commencement of modulator therapy may soon be licensed for use in early life, emphasising the importance of a prompt early pre- or post-natal diagnosis to institute prompt effective treatment. Indeed, carrier screening and prenatal genetic testing could be needed to render some forms of in utero treatments modalities in CF, as has been piloted in other rare diseases76. The possibility of effective in utero or infant treatment for CF may modify how carrier / prenatal / NBS genetic testing is utilised in the future. While research is needed to determine the reactions of prospective parents to medically actionable outcomes of preconception carrier screening programmes77 and the impact of religion and/or culture on reproductive choices78, there is a risk that affluent countries will be able to provide precision medicine therapy in CF to virtually all, while elsewhere these approaches might be economically out of reach79. In summary preconception carrier screening raises many complex and ethically challenging issues with respect to changing technologies and natural history of the condition. There is a risk of inequalities being widened through varied access and processing of screening programmes globally.
4). Trends in the changing incidence and prevalence of CF
Prior to the introduction of CF NBS, epidemiological CF incidence estimates suffered from an ascertainment bias due to the clinical underdiagnosis / misdiagnosis of CF, delayed clinical diagnosis of CF or underdevelopment and/or lack of standardised registry reporting80. Initial incidence estimates from the 1960s to late 1980s had been generally performed in more demographically stable populations of Western Europe and North America81 This would not have accounted for unregistered patients, unrecognised minorities with CF, or patients who died undiagnosed. With the advent of NBS which became increasing established in many countries national incidences of CF were more accurately determined. The epidemiological incidence estimates in non-European derived populations82 currently remain biased by the many issues noted above due to the insufficient awareness of CF, absence of registries and neonatal screening schemes.
Studies on the changing incidence of CF are confounded by the immigration rates from countries with a lower incidence of CF such as Asians, Africans, and groups from the Middle East that are also more challenging to detect with the traditional gene variant panels used in NBS. Additionally, distinguishing the impact of neonatal screening from that of prenatal screening in countries where both is offered may be impossible. Nevertheless, many, but not all, regions offering NBS for CF during the past decade have observed a decreasing incidence of the disease.
In the US a retrospective study 1994–2011 assessing the incidence of CF in NBS populations83 interestingly found the incidence of CF to be increasing (albeit not significantly) in Wisconsin, while decreasing in Massachusetts and remaining static in Minnesota and Colorado over the same period. The ethnic composition of a population and birth rates also figure in altering incidences. For example California demographic trends have predicted that 33% of all incident CF cases will be Hispanic and 2% non-Hispanic and 2% non-Hispanic people of African origin84. which significantly increase non-p.Phe508del genotypes.
Prenatal diagnosis in European-derived populations has been associated with decreased incidence of CF. In North-Western France an aggregate 22.6% reduction in CF births in families with a family history of CF was documented85. In Israel, a national-wide preconception carrier screening programme has been associated with a decrease of at birth prevalence (a term used when the incidence of a disease is influenced by preventive public health measures) of CF from 14.5 (in 1990) to 6.0 per 100,000 live births (in 2011)86. Similarly, in Northeast Italy, at birth prevalence of CF monitored by neonatal screening was 1 in 2,700 (in 1993) and 1 in 14,000 after 20 years of extensive population-based preconception carrier testing of couples87 from the general population.
Currently the US and European Union have a comparable prevalence of CF at 7.97 versus 7.37 per 100,000 in the general population, irrespective of different CFTR variant spectra of the population surveyed88 Improved standard therapies for CF and CFTR modulators impacting on survival are likely to increase the prevalence of CF. However, the complex interplay of migration, ethnic-specific birth rate, offer of preconception carrier screening, quality and quantity of registry data makes it very difficult to model the ultimate incidence and prevalence. Statistical modelling cannot fully account for the uptake of preventive genetic testing schemes, elective termination of pregnancies within prenatal diagnosis or even the rate spontaneous miscarriages. However, the Cystic Fibrosis Foundation Patient Registry has shown only a minor decrease in the prevalence of CF89,90. In the future the “negative” effect of carrier screening may be offset by the positive effect of improving care for patients with CF.
Comprehensive studies including large, well-defined populations are required to confirm that NBS leads to a decreased incidence of CF over time. In addition future studies on population responses with regard to reproductive behaviour would be informative as preconception testing becomes more widely available.
5). The changing face of the current CF population
a). What do CF Registries tell us about the current CF population?
National registries are powerful resources that enable understanding of population dynamics, disease progression, efficacy of clinical interventions and to allow benchmarking with other centres, countries and internationally91. It was the foresight of the CF community that created the first national CF registry in the 1960s to summarise data from Canadians and Americans living with CF. Since then, many countries (Australia, Belgium, Brazil, France, Germany, Ireland, Italy, the Netherlands, New Zealand, UK) now have well-established national CF registries capturing data on more than 90,000 individuals with the disease. However, the worldwide number of affected individuals will be an underestimate with a growing number of patients reported in continents or regions with less well-established registries (e.g. Africa, Asia, Middle East, most parts of South America) that are not included in current estimates. Registries reflect the disparity of health care systems including access to diagnosis and associated diagnostic procedures, clinical care and therapeutic options. It is critical to ensure a high proportion of the population is captured within the registry in order to minimise ascertainment bias and to draw accurate conclusions (Figure 5).
Figure 5. Maps of Countries with Cystic Fibrosis Registries and their Population Size.
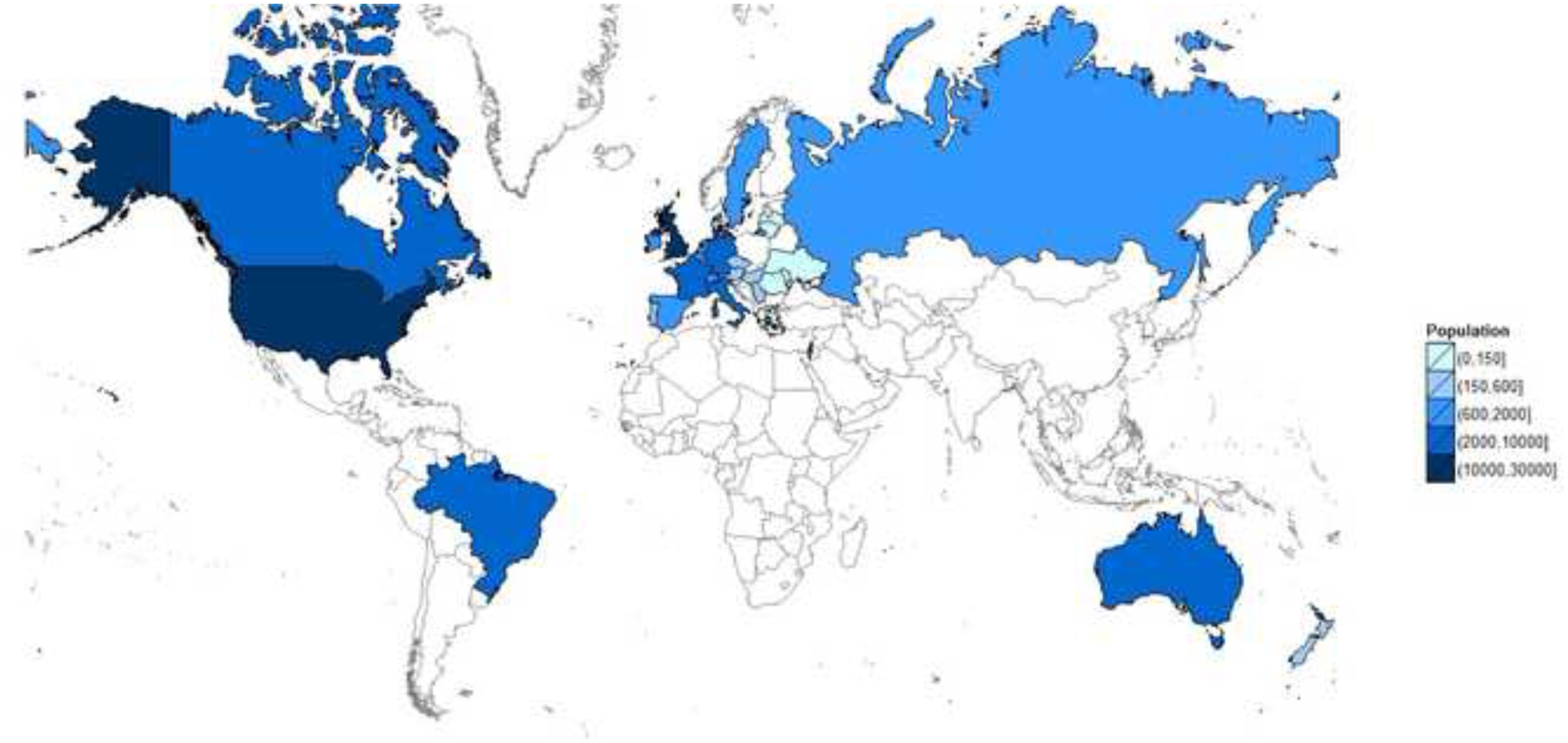
Countries with a cystic fibrosis registry
Reproduced with permission of the US Cystic Fibrosis Foundation, Bethesda Maryland.
One of the most dramatic changes observed has been the growing adult population, with the proportion of adults exceeding the proportion of children in many countries. This growth is largely driven by very low pediatric mortality and improved survival rather than an increasing number of adult diagnoses95,96. Registry data from several countries have shown that the median age of the population has increased over time, and the median age of survival has also significantly increased with time. A European CF Society Patient Registry study estimated that in 16 European countries the number of adults living with CF will increase by approximately 75% between 2010 and 202517. The median age of survival for patients born between 2012 and 2016 has been reported between 43 (US)90, 47 (UK, Germany)92,93 and 53 (Canada)94 years.
However, not all groups have enjoyed the same rate of improvement in survival. Despite improvements in outcomes, disease progression in adulthood and during the transition period from pediatric to adult care and the gender gap between males and females are still hallmarks of CF, even in countries with the best survival data. Early and severe infection, adherence to treatment in the case of high treatment burden and low socioeconomic status are the most important risk factors for poorer clinical outcomes independent of the specific health care system. Furthermore in US CF registry data in California from 1991 to 2010 mortality in Hispanic patients was 9.1% (44 deaths/485 subjects) compared to 3.3% (41 deaths/1234 subjects) in the non-Hispanic US population95. Worse survival persisted in the Hispanic group even after adjusting for clinical factors and socioeconomic status (HR 2.81; 95% CI 1.70–4.63). By highlighting cohorts at risk for higher mortality using registry data in this way can reveal such discrepancies and targeted strategies to improve health outcomes can be implemented.
Understanding the heterogenity of the CF population internationally through the use of CF registries is an important mechanism to identify risk factors for poor outcomes that can enable advancement of clinical care. However, before conducting any international comparison, one must be careful to ensure that registry data are harmonised in order make accurate comparisons and minimise bias.96 This includes careful review of variable definitions, data process and collection and assumptions made in calculations and statistical approach. Using a unified and harmonised approach, a recent study using Canadian and US CF registry data found that the median age of survival in Canadian CF patients from 2009–2013 was ten years longer than their American counterparts97. Although not specifically designed to investigate the reasons for this gap, the authors hypothesised that earlier nutritional support implemented in Canada, differential access to lung transplantation, or differences in healthcare delivery may be contributing factors. This has led to new targeted research to investigate these hypotheses in an effort to better understand the survival gap across countries.
Beyond survival, registry data has the ability to enhance our knowledge of the overall health of the CF population and possible growing complications inclusing those emerging such as gastro-intestinal malignancy and lifestyle diseases including cardiovascular conditions in the ageing CF population, which add to the burden of care98,99 With this increasing adult population comes increasing comorbidities.
Beyond survival, registry data has the ability to enhance our knowledge on the overall health of the CF population and the growing complications which add to the burden of care. Registry data has shown us that nutritional status and lung function is better for a given age than it was historically and that individuals with CF are living longer with lower lung function98,99. With an increasing adult population comes increasing comorbidities, including CF-related diabetes which is estimated to affect over 50% of the adult population and the ever-increasing number of people living post-lung transplantation.
The worldwide number of affected individuals is likely an underestimate as the growing number of patients reported in countries with less well-established registries for example, Argentina and Middle Eastern countries are not included in current estimates. The future for registry data will be to harmonise the registry capture worldwide in order make accurate international comparisons and minimise bias96.This includes careful review of definitions, data process and collection and assumptions made in calculations to harmonise baseline differences. International agreement on how to capture the CFSPID/CRMS population is ongoing.
b). The spectrum of CF is changing
CF has been defined as a diagnosis with a wide range of clinical symptoms, including positive newborn screening (NBS) for CF, in combination with well-defined measurement of well-defined spectrum of CFTR-dysfunction (sweat choride concentrations ≥60 mEq/L on two occasions and/or two CF-causing CFTR variants in trans and/or a CF-typical electrophysiology). However, the combination of the variable dysfunction related to the CFTR variant and the different organs involved leads to a wide spectrum of disease caused by dysfunction in the CFTR protein – from no disease to CF with pancreatic sufficiency to CF with pancreatic insufficiency (Figure 6).
Figure 6. CFTR Function and Clinical Phenotype.
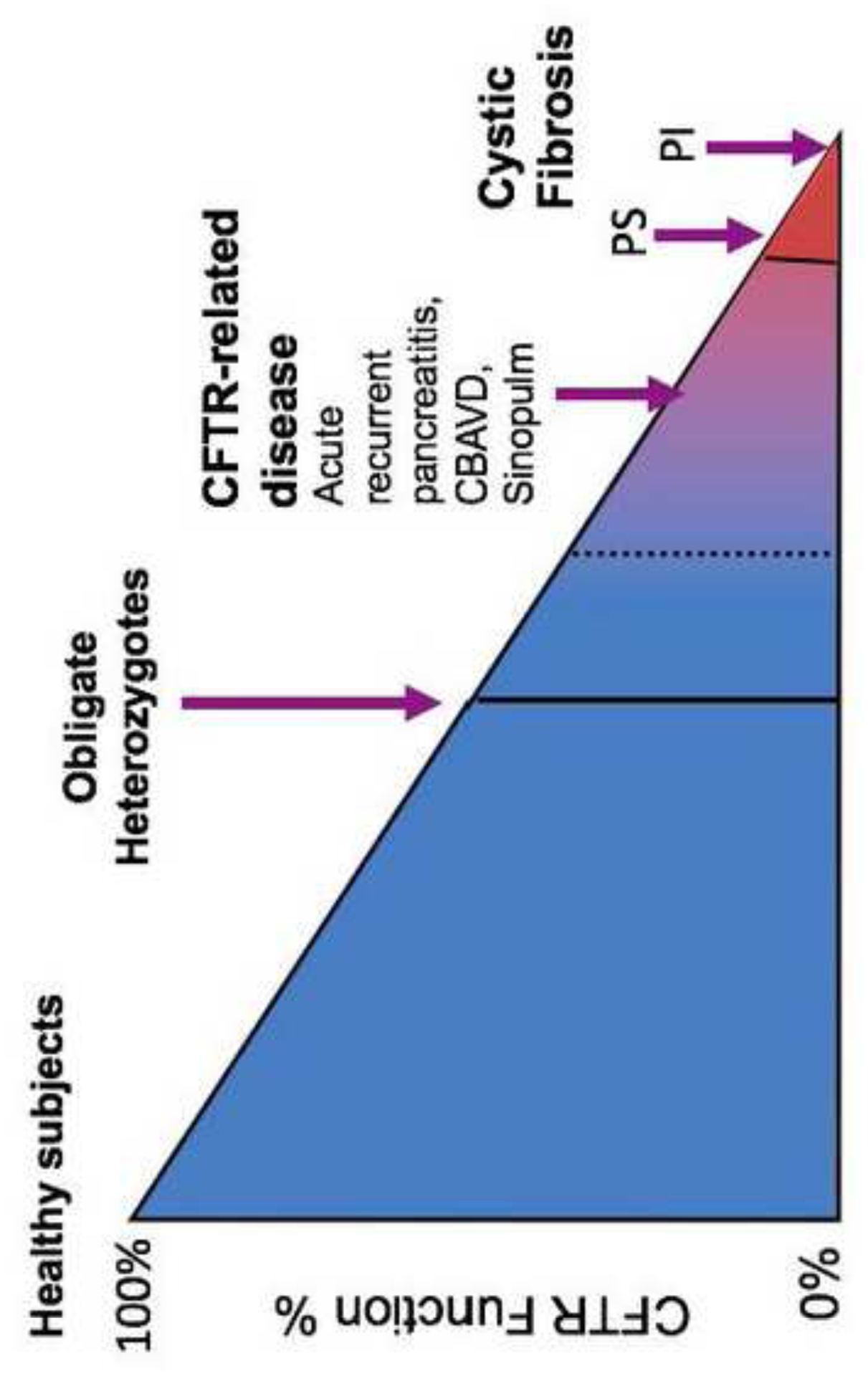
CBAVD – congenital bilateral absence of toe vas deferens.
Some individuals have clinical features of CF (such as CBAVD, or recurrent acute pancreatitis), and have CFTR variants but do not meet the diagnostic criteria for CF now, a group now coined CFTR related disease (CFTR-RD)100. NBS programmes for CF shift the diagnosis paradigm from symptomatic to (mainly) asymptomatic patients and a new group of infants with CFSPID/CRMS has been identified101,102,103,41.
CFSPID/CRMS is sometimes considered “the CFTR-RD of infants” where the “clinical phenotype” is hypertrysinogenemia but it is a biochemical abnormality with an uncertain prognostic significance. Whether it is of clinical benefit for individuals to screen for CFSPID has yet to be defined and the majority of infants in this category will kely not develop any phenotype. Longitudinal follow-up of the CFSPID/CRMS is needed to clarify what proportion of these infants will later meet diagnostic criteria for CF or CFTR-RD. Minimising the number of CFSPID is an important consideration in NBS programmes as referring and following these infants in centres creates a burden on both the families and the health care systems. The ratio of diagnosis of CFSPID/CRMS to the diagnosis of CF by NBS can vary greatly from one patient diagnosed with CFSPID/CRMS for every two to five patients diagnosed with CF to a reverse ratio of 3:126,104,105, likely based on the methodology, the use of gene sequencing in NBS and the ethnicity of the population studied. Of children referred to a single pediatric centre in USA, 80% met the criteria for CF, 15% for CFSPID/CRMS and 5% CFTR-RD41,102,105. What is not clear, and much more difficult to calculate, is the ratio of CF:CFTR-RD in a population that includes adults.
The answer to the question, “how much CFTR function is needed to prevent disease” is complex and depends on the organ involved (vas deferens vs lungs), the time frame under consideration (years vs decades) and the clinical phenotype (bronchiectasis vs risk for GI malignancy) (Figure 6). As has been shown in the CF population, variants in other genes may also modify the expression of disease. Thus CFTR-associated conditions (CF, CFSPID/CRMS and CFTR-RD) are now considered to be comprised of overlapping groups of patients along a spectrum from asymptomatic to live-shortening, severe multisystem disease. Engaging more representative populations worldwide both for genetic testing and registry capture are future goals to enhance understanding of the global distribution and clinical impact of rarer mutations.
6). CF Populations Beyond European Populations
In populations with a European background, there is rapid expansion of neonatal screening106 and publication of guidelines for the diagnosis of CF107,108, The most recent World Bank economic data109 demonstrate that the diagnosis of CF should not be discounted solely based on the ‘racial background’ of the individual. There is increasing evidence that CF is present in Asia, Africa, the Middle East and Latin America, albeit at a lower incidence than in European populations.
CF incidence in specific populations in non-European regionsis likely to be underestimated due to ascertainment bias resulting from limited and unrepresentative data. In the absence of comprehensive CF registries in these regions it is not possible to determine the extent of underdiagnosis, misdiagnosis, underreporting and or ethnic-specific variations in CFTR variant carrier rates or distribution or their ethnic-specific penetrance.
The use of diagnostic assays that are optimised for European populations110 and the reporting of CFTR variant frequencies by ‘countries’ rather than to individual ethnicities (which can be highly variable within countries) can contribute to underestimates of CF. The clinical course of people with CF living in these regions may be very different from that seen in European populations and relate to various factors involving late diagnosis, misdiagnosis, comorbidity with other recessive disorders due to consanguinity, different diet, exposure to different microorganisms, different genomic backgrounds and perhaps most importantly, socio-economic status and the lack of access to coordinated CF specialised care and treatments.
a). Asia
A recent meta-analysis described 160 variants in a total of 1,850 cases with CF of which 24 variants were found in broadly defined ‘South Asian’, 54 in the ‘Middle Eastern’ and 38 in ‘East Asian’ regions111. The p.Phe508del variant was most prevalent allele accounting for 12 to 31% in Asia patients with CF81. There are additional 10 variants with a frequency over 5% and ~ 50 variants were detected only once. Of note, p.Phe508del was not seen on CF alleles of East-Asian ancestry (e.g. China, Korea, Japan, Vietnam, Thailand).
Only 34 cases of CF patients of mainland China origin were diagnosed within the last four decades112. A retrospective analysis of 21 cases, originating from the Beijing area detected CFTR variants differed substantially from the European spectra although the novel ‘Asian’ variants require further characterization including intra-CFTR copy number analysis to capture the entire scope of variation and to study the likely impact on disease phenotype.
Clinical data of Asian CF patients suggests later diagnoses, a younger population, higher rates of pancreatic sufficiency, lower sweat chloride concentrations (albeit within the diagnostic range) and lower rates of classic CF respiratory pathogens. A recent study112 provided evidence that most CF patients in mainland China were diagnosed with a variety of more common respiratory conditions (such as bronchiectasis, pulmonary tuberculosis or diffuse panbronchiolitis). In Laos and Vietnam also infertile men with congenital bilateral absence of vas deferens (CBAVD) were reported. Many countries including Bhutan, Cambodia, Indonesia, Laos, Mongolia, Myanmar, Papua New Guinea, Philippines and all Pacific Islands have yet to reports CF in their indigenous populations in the literature.
b). Middle East, Transcaucasia and Central Asia
A recent literature review including studies over the past three decades described 5,481 CF patients derived from 22 Middle Eastern and North African (predominantly Arab)113. Many of the included studies were based on case series of people with CF living in Europe114. Thus, the reported distribution may not be representative. The p.Phe508del variant was found in 14 Arab countries, but notably not in Iraq, Qatar or North Sudan. Although most CFTR variants in this region were present in other regions such as Southern European, the Mediterranean rim and the Black Sea region, there were also 18 CFTR variants unique to the region.
Consanguinity is common in a proportion of Middle Eastern countries e.g. high rates of first cousin marriage115. In some parts of the region CF is commonly diagnosed in first- or second-cousin marriages as exemplified in Northern Sudan116 and Jordan in close to 70% of all incident cases117. In some countries given CFTR variants can even be attributed to “tribal-specific surnames”118, such as in the case of common CFTR variant p.Ser549Arg (T>G) in United Arab Emirates or Oman119.
The substantial population heterogeneity in individual Middle Eastern countries is further compounded by people with Sunni-, Shia and Ibadi religious affiliations120,121 leading to different CFTR variant distributions in groups of various religious denominations. Furthermore, analysis of 69 Iranian CF patients122 identified a broad spectrum of 37 CFTR variants all of which that had previously reported in European-derived populations (likely immigrant communities) with p.Phe508del being the most common (18%) followed by c.2051_2052delAAinsG, p.Gln637HisfsX26 and p.Ser466*. When Iranian CFTR variant distribution was divided by individual Iranian provinces substantial differences were observed123.
Many countries including Afghanistan, Tajikistan and Turkmenistan have yet to reports CF in their populations, although, there are ongoing studies on the distribution of CFTR variants in small cohorts of Afghan, Armenian, Azerbaijani, Georgian, Kazakh, Kyrgyz, Tadjik, Turkmen and Uzbek CF patients (Milan Macek, personal communication, 2019).
The CF phenotype is usually is more severe, with a common presenting symptom of CF in this region of electrolyte disbalance due to excessive salt loss124 and/or association with the Pseudo-Bartter syndrome125 which could be due to climatic (extreme heat). As an example, the earlier mortality reported in Saudi Arabia may be due to a combination of delayed diagnosis, the presence of multi-resistant strains of P. aeruginosa, more severe liver disease and poor nutritional status. However, given the high degree of consanguinity, CF may manifest simultaneously with other regionally prevalent inherited diseases, such as sickle cell disease in Saudi Arabia126 or G6PD deficiency127 or various forms of familial Mediterranean fever128, impacting on adverse health care outcomes and delayed diagnosis or misdiagnosis.
c). Africa
A recent retrospective review of published studies on the distribution of CFTR variants in classical forms of CF and CFTR-related disorders from 49 African countries129 reported 1,172 cases. Importantly, the majority of studies are drawn from Northern-Africa (667) and Southern Africa (380)130,131,132, and the majority of Sub-Saharan countries remain “unexplored”. Of 79 CFTR alleles reported, the p.Phe508del variant was the most common (48%) along with other variants reported in European, Middle Eastern/African American populations (e.g. p.Gly542*, p.Asn1303Lys, c.2988+1 G>A. Twenty-one CFTR alleles were unique to Africa and CFTR variant distribution in Africa is incomplete and significant challenges to undertake this work include the broad population and linguistic diversity, including the impact of intra-country / inter-country migration133.
In Africa, presenting symptoms of CF are accentuated by climate (excessive heat). More broadly, more prevalent diseases (e.g. malnutrition134, tuberculosis135, HIV infection136) which have phenotypic overlap with CF can lead to delayed or midiagnosis, especially in Sub-Saharan Africa.
d). Latin America, the Caribbean and African diaspora
Modern populations of Latin America established from the late 15th Century through gradually increasing genetic admixture of Europeans, West Africans, and indigenous Amerindian peoples. Exact proportions vary within and between countries137. In many countries the strong Amerindian heritage is mixed with European (predominantly Spanish or Portuguese populations), yet with a broad range of European ancestries. In other countries (such as Brazil, Colombia and Cuba), most of their populations is of African descent138. The impact of ethnicities on the incidence of CF is highlighted in Brazil (ranging from 1 in 1,600 in Euro-Brazilian to 1 in 14,000 in Afro-Brazilian infants).
The potential impact of Spanish ancestry139 on the distribution of CFTR variants in Latin America was studied by comparing distribution in CF populations from Argentina, Brazil, Colombia, Chile and Mexico with a CF patient cohort from Spain. The p.Phe508del variant was the most frequent CF causing mutation (51%), and reported 29 indigenous CFTR variants which were not seen in Spain. Another review included 2,177 unrelated CF patients140 from 10 Latin American countries described 89 CFTR variants. The p.Phe508del had a mean frequency of 47% (ranging from 23% in Costa Rica to 59% in Argentina) paralleling the proportion of European ancestry.
The impact of the African Diaspora141 is difficult to substantiate since there is a lack of data on the distribution of CFTR variants in African countries where an estimated total of 12 million West Africans involuntarily transferred to Latin America. Intra-CFTR haplotype analysis provided evidence that this “p.Phe508del of the Tropical Belt” is residing on the same haplotype in US African-Americans, thus far studied Africans, Saudi Arabians and Greeks suggesting its common origin and potential spread via Atlantic slave trade to Latin American (e.g. Brazil, Colombia or Cuba) and to Middle Eastern/North African/Mediterranean countries.
This section highlights the changing epidemiology of CF. Future opportunities and challenges are highlighted in Panel 2.
SECTION 2 –. CLINICAL CARE AND ITS DELIVERY
Note to Editors – Cases to be placed adjacent to text in this section – no specific place identified to date.
Early in the life of a child born with CF, gastrointestinal symptoms predominate. Pulmonary disease may become manifest during early childhood and most complications such as CF-related diabetes, later in life. The care required for both prevention and management of the multi-organ aspects of CF differ at these different disease stages, although the integrated involvement of a multidisciplinary team is key. With improved conventional therapy, this disease progression has, on average, occurred at a slower pace; further slowing may be evident in the medium term future with the widespread adoption of highly effective modulator therapies.
1). Care of the infant and child
The start point of clinical management and limitation of disease progression is a timely diagnosis. The changing epidemiology of diagnosis based on newborn screening (NBS) programmes has been outlined in the previous section. The diagnosis on NBS is not always clear cut, indeed, with the increasing implementation of protocols incorporating CFTR variant analysis, there are increasing numbers of babies identified as CF screen positive, determinate diagnosis (CF-SPID), the management of whom is discussed below.
Standards of care outline the skill mix essential for excellent clinical care, a team of sufficient size for the patient population comprising specialist nurses, physiotherapists, dieticians, psychologists and doctors142 143. One of the most useful developments of recent years has been training and integration of CF-specialised pharmacists into the multidisciplinary team of health care professionals; polypharmacy is common and as new drugs become increasingly adopted, this is a vital resource144. Although traditionally, medical care is delivered by respiratory clinicians, the group needs also to include ENT, gastroentero/ hepatologists and endocrinologists. Over the life course, this will need to expand further, requiring expertise in fertility, obstetrics, cardiothoracic/ general surgery, specialist anaesthesia, rheumatology, nephrology, psychiatry, intensive care, interventional radiology, transplantation and palliative care services145,146 146,147.
a). The importance of optimal nutrition
The benefits of early diagnosis are well-recognised particularly with regards to nutritional outcomes. Multiple studies confirm the growth benefits of early diagnosis with NBS. Later in life, lung health is closely related to weight; the early years are particularly important, faltering growth in the first two years of life being associated with impaired lung health out to adolescence148,149. For these reasons150, during the early months of life, focus of care will usually be on optimising nutrition through pancreatic enzyme replacement, hydration and electrolyte balance and dietary advice; frequency of hospital attendance will best be guided by progress in these aspects monitored by specialised CF dieticians.
b). Monitoring lung disease and its progression
There is good evidence that the lungs of a CF baby are normal at birth, but after some (variable and often ‘silent’) time, mucus accumulation, infection and inflammation will occur151–155. The sequence of these events is still being debated; specifically the role of mucus accumulation driving inflammation independent of infection. Such disease may initially be asymptomatic, although the majority will have ‘normal’ childhood respiratory infections, which are largely viral in nature156. Despite a lack of more specific symptoms, both BAL and CT studies demonstrate airway infection and inflammation, alongside structural airway wall changes during this silent period155. However, neither BAL nor CT are frequently repeatable. The challenge currently and into the future will be developing the tools to monitor this establishment of disease in a non-invasive and acceptable fashion.
i). Detection of lower airway infection
Most children are not sputum producers, a problem that will affect a larger proportion of patients as new drugs lead to a generally healthier population. Throat/ cough swabs lack specificity and in some studies, sensitivity157–159. A number of studies have confirmed the superior yield with induced sputum but160–163 this is time consuming (for both patient and staff) and is not universally popular with patients and feasibility is limited in infants and young children. Given the close relationship between frequency of sampling and detection of organisms, it essential for future care that methods are improved.
ii). Monitoring airway physiology
Most centres lack facilities for infant lung function testing; even those with this expertise consider it largely a research tool and164 indeed, the most recent studies report the majority of CF infants to have preserved lung health154,165. Into the pre-school years, this can be more informative, although spirometry is both challenging to achieve reproducibly and lacks sensitivity for distal airways disease, which predominates at this age. Sensitive, non-effort dependent techniques based on multibreath washout (MBW) show promise and guidelines for their performance in this age group have been published166. The resulting lung clearance index (LCI) is abnormal at an earlier age than spirometry167–170. There are several studies of this outcome measure as a clinical monitoring tool and with the increasing training of CF centres and central overreading of data for clinical trial purposes for both Europe and North America171,172, it is likely this measure will be in more widespread clinical use in the near future. Several devices and tracer gases are available, which are not interchangeable173. Alternative approaches such as impulse oscillometry require more of an evidence base before their utility can be judged174,175.
iii). Detecting and monitoring structural lung damage
A minority of paediatric centres advocate regular lung CT scanning, many others having concerns over the radiation involved and using them in a concern-driven fashion. With the advent of ultralow dose (ULD) CT protocols, this may become less of an issue; a recent publication176 described ULD CT with equivalent radiation to a plain chest radiograph, but yielding substantially more information regarding mucous plugging, bronchial wall thickening and atelectasis, all features of early CF lung disease. Lung MRI protocols are increasingly being developed177. This completely radiation-free technique could be used at frequent intervals to monitor longitudinal progression of lung disease or response to interventions, but generally requires sedation or anaesthesia in young children. Several techniques are described; those involving hyperpolarised gases have shown promise as an outcome measure178, but are currently limited by availability and cost. Less expensive alternatives based on oxygen-enhancement179 and gas-free systems180 are showing promise. MRI scanners are not yet widely used for pulmonary imaging but it could be envisaged that they may be more widely available in the near-mid term future.
iv). Stratification and optimised, person-centred management
Current monitoring for the majority of patients both in this age group and older is generally based around scheduled hospital out-patient visits (every 2–3 months once beyond the first year of life) and an annual full systems-based review. Many aspects of care are already personalised, a good example being airway clearance techniques181,182 which need adapting with growth/ maturity and exercise recommendations183. Similarly, patients will be prescribed specific antimicrobial therapies based on their culture results. Ideally, in the future, a more stratified approach could be envisaged, wherein a number of clinical features/biomarkers were sufficiently well understood that prognosis and level of intervention could be adjusted accordingly. Several features relating to outcome are well understood, for example early onset of infection and inflammation184,185, but others such as socioeconomic status and in some geographical factors, access to healthcare, have enormous impacts and may be more difficult to measure186,187. Currently, there are no biomarkers which are used in this fashion, although they are being frequently investigated188–192 and will hopefully emerge. The impact that such stratification could ultimately have on delivery of care will be discussed in a later section.
c). Emerging concepts of early intervention/preventive targeted therapies
Childhood is absolutely key as a time the scene is set for future health. The early initiation of airway clearance widely recommended and implemented; it has been the case for so long that providing evidence in support of this is challenging182. Adjuncts to airway clearance have a strong evidence base, with hypertonic saline (HS) leading to fewer pulmonary exacerbations in older children and adults193. Recent trials have examined the role of HS in younger children and infants; outcomes in the age group are inherently challenging, but improvements in LCI have been demonstrated194. The fact that this therapy is relatively cheap may make it an attractive intervention for areas with major economic challenges. The convincing relationship between neutrophilic inflammation (the cell of origin of the majority of extracellular DNA) and bronchiectasis lends support to the early use of rhDNase over and above the beneficial impact on lung function in older populations. The philosophy of prompt eradication protocols for first Pseudomonas infection is widely held, although various approaches are taken and the best of these not yet clear195. Such aggressive therapy may prevent or delay the onset of chronic infection, associated with worse disease outcomes.
Early interventions are less able to prevent disease in other organs. Pancreatic exocrine failure, for example, is bypassed with replacement therapy rather than treated. Until recently, it was probably considered that any ‘treatment’ was futile as pancreatic disease was irreversible. However, encouraging data from trials of the CFTR potentiator, ivacaftor, in preschool children have demonstrated this not neccessarily to be the case196,197. A proportion of children treated early regain sufficient pancreatic function to enable digestion. Anecdotal evidence is emerging that there may also be an impact on pancreatic endocrine function and the development of CF-related diabetes, although more study is required198–201.
This therefore raises the question of when to start modulators for maximal benefit. For most paediatricians, safety is paramount. To date, regulatory agencies have encouraged trial protocols which extrapolate clinical efficacy from adult studies as long as safety, pharmacokinetics and pharmacodynamics (sweat chloride) are addressed196. There are two possible reasons to advocate starting these drugs as early as possible, once safety is confirmed: 1) babies are born with CFTR dysfunction. The longer they spend with this, the more likely is any impact on longer term outcomes and 2) there may be a window of opportunity which could subsequently close; the impacts on fecal elastase in early childhood is perhaps an example of this. Conversely, there are those who consider a later introduction is more desirable, based on challenges quantifying efficacy in this age group, lack of data on long term safety and health economics. Over the next few years, the community will accumulate substantial data in this area and will, in our opinion, be convinced of the benefits of early intervention.
2). Management of the child with an unclear diagnosis: CF-SPID and CFTR-RD
As outlined above the detection of these babies poses a number of clinical challenges. Most paediatricians seem to agree a balance needs striking between over-medicalisation and losing an opportunity to prevent ill-health in later life and a number of published documents propose ways to do this41,102,103.
The incidence is reported to be one for every three to five cases of confirmed CF diagnosis cases in NBS programmes across the world and 10–20% may develop manifestations of CF or a CF-related disorder (CFTR-RD) over time202. Apart from parental anxiety and increased healthcare utilisation, appropriate follow-up and additional investigations to confirm or exclude CF diagnosis and surveillance of early CF disease manifestations are recommended in these infants202,203.
Ultimately, we need to develop tools to stratify these children into higher and lower risk groups. Waiting for evidence of disease in order to do this, for example with multibreath washout or time-point based imaging (available perhaps more widely as we emerge into an era of ultralow dose CT or radiation-free MRI) does not seem ideal. Biomarkers are available which correlate with CFTR function; repeated measurement of sweat chloride is already being used in this fashion in some clinics, although there are yet to be established long-term correlates with clinical outcomes. Assessment of CFTR function based on intestinal current measurement100 in tissue obtained form rectal biopsy (further details later) has been shown to help clarify the diagnosis and is probably more feasible than nasal potential difference in this age group. For any of these measures, long term clinical data is required. Currently these children are either entered into CF registries or not. We urgently need a separate interface on which we can map their clinical trajectory in order to fully understand their outcomes and hopefully stratify and rationally care for future cohorts.
3). Caring for the older child into adulthood
As the child grows up, all the issues discussed above remain important and newer ones may dominate.
a). The emergence of infections
The old concept that young children acquired organisms such as S. aureus early in life and later, Gram-negative bacteria, most commonly P. aeruginosa has evolved substantially with the use of molecular tools to diagnose infection. The lung microbiota is much richer than culture methods led us to believe204. As patients age, the natural diversity diminishes, whether due to antibiotic pressures alone, or other aspects of the disease205,206. The last few years have witnessed the burgeoning of this field of research, largely resulting in descriptive outputs. Looking forward, we urgently need research which will aid in the rational prescribing of antibiotics.
i). Cross-infection and strategies to prevent it
Cross-infection has long been recognised in patients with CF207. This included epidemics of infection with B. cepacia complex infection or specific, highly transmissible strains of P. aeruginosa and also MRSA has been reported. Such transmission was demonstrated to have occurred as the result of inpatient and/or outpatient visits to CF centres or at social events involving multiple CF patients. The more recent recognition that CF pathogens can be transmitted not only directly (e.g., from patient to patient) but also indirectly (airborne) a matter of great concern. For example, outbreaks of non-tuberculous mycobacteria due to M. abscessus complex have been described208,209 and genotyping of M. abscessus strains has raised the possibility of patient to patient transmission210,211. Recent data also suggest aerosol transmission of Aspergillus fumigatus in patients with CF212.
The ECFS consensus on standards of care for patients with CF already recommended segregation of outpatients with B. cepacia complex, MRSA and/or P. aeruginosa at the end of day, on separate days or in a different location213. However, the spectrum of bacteria that infect CF patients has evolved with the recognition of difficult to treat Gram-negative bacteria (e.g., Inquilinus spp., Ralstonia spp., Pandoraea spp., etc …) for which only limited data is available on possible cross infection risks and strategies to prevent or treat such infections. Recent recommendations emphasise roles for hand and environment hygiene and some recommend the wearing of masks when visiting CF centres214. Furthermore, patients are discouraged from mixing socially and guidelines limit attendance at events including education days and conferences to a single person with CF214. Many of these recommendations have been in use for several years in large CF centres, but a recent study indicated that they may be more complicated to adopt by smaller CF centres215. Most of these recommendations are based on low level of evidence and some of them may need to be tested in appropriate trials216. Suggestions have also been made to include negative-pressure inpatient and outpatient rooms to diminish the risk of airborne contamination of ward corridors and communal areas and to monitor air exchange rates in CF centres. No consensus exists on such hospital architectural recommendations.
ii). The adverse consequences of antibiotics including antimicrobial resistance (AMR)
The liberal use of antibiotics has undoubtedly contributed to the great improvements in respiratory health observed in today’s patient cohorts. However, this comes with something of a price. People with CF receive huge numbers of antibiotics over the course of their lives delivered via oral, inhaled or intravenous routes. In many countries, this starts from the time of diagnosis with anti-staphylococcal prophylaxis217. Although demonstrated as useful for reducing early S. aureus infections, some concern remains over whether such therapy increases the risk of acquiring other organisms, for example P. aeruginosa218. The UK-led CF-START trial is currently exploring this219.
Although most people tolerate antibiotics well, side-effects are common and include a) disturbance of natural host flora with diarrhea and/ or candidiasis; b) allergy and in some cases anaphylaxis; c) antimicrobial resistance (AMR). There is also some evidence to link the use of antibacterial agents with the increased incidence of fungal and mycobacterial infection.
Antimicrobial resistance (AMR) is increasing worldwide and is also a matter of concern in patients with CF, in whom bacteria, fungi and non-tuberculous mycobacteria (NTM) are increasingly tested as being resistant in vitro to multiple antimicrobial agents. Although some pathogens (e.g., M. abscessus) are intrinsically resistant to multiple antimicrobial agents, the widespread use of recurrent or prolonged systemic and/or inhaled antibiotic therapy220,221 has been directly linked with the emergence of resistant bacterial clones. Similarly, clinical and/or agricultural use of azole antifungal agents is associated with increase in isolation of A. fumigatus resistant strains222.
Furthermore, in people with CF who have chronic airways infection, the interpretation of microbial susceptibility is challenging. Many studies have shown the lack of association of in vitro susceptibility results to ceftazidime or tobramycin223 or to antibiotics tested in combination224 and clinical outcomes in patients with CF chronically infected with Gram-negative bacteria (e.g., P. aeruginosa). Multiple causes could account for this lack of association including compartmentalization of infection with multiple bacterial strains possessing different resistance characteristics within the lung225, modalities of in vitro testing (e.g., in planktonic vs. biofilm medium, sampling of various bacterial clones, interpretation of AMR results)226 and pharmacokinetics/pharmacodynamic characteristics of antibiotic use in patients with CF. Recent publications from the CFF/ECFS task force on AMR in patients with CF have proposed definitions of AMR in patients with CF and future research priorities227,228.
iii). Future strategies to improve anti-infective therapies and combat AMR
One of the drivers of AMR is the inappropriate use of antibiotics. CF Clinicians are often hindered by a lack of specific knowledge around lower airway pathogens when prescribing drugs. Although to date this has posed a practical challenge mostly in younger patients, it is likely to become more common as the health of older patients improves. Future directions may include point-of-care diagnostics229,230, allowing more frequent testing at home and novel detection methods, for example breath biomarkers231,232.
There are a number of groups developing adjuncts to conventional drugs which could allow the use of lower doses or shorter courses. Relatively few new antibiotic drugs are being developed, despite initiatives to encourage this. Novel approaches such as bacteriophage or immunotherapy remain experimental233.
b). Monitoring for complications
Most of the complications associated with CF become more common as the patient ages. Perhaps regarded as the most serious of these, CFRD is not preventable (there are early encouraging reports of patients on CFTR modulators, so this may change), but there are clear benefits to its early identification234. CFRD is well known to have clinical impacts on respiratory health, nutrition and mortality, with some studies reporting deterioration in the pre-diabetic stage235. Guidelines promote screening with glucose tolerance tests or continuous glucose monitoring and the early initiation of insulin. It is likely, with an expanding adult population, that the burden of CFRD will increase, at least in the medium term until populations have early access to highly effective modulators. Studies are needed to understand the benefits of early insulin therapy in the pre-diabetic stage. Alongside data collection on the impact of CFTR modulators, the role of new drugs such as incretin-based medications needs to be established236. New technologies are being developed such as ambulatory insulin pumps and continous glucose monitoring systems (CGM), which could improve both screening and management of CFRD. Close attention should be paid to prevention and monitoring of other complications such as bone disease with close attention to nutrition, vitamin supplementaton and bone mineral density scanning. Liver disease surveillance programmes, whilst not reducing the prevalence of this complication, can allow early initiation of treatment. Alternatives to ursodeoxycholic acid with greater efficacy are needed and237 research into modifier genes may ultimately help to define patients at risk for this complication238.
c). Recognising and treating pulmonary exacerbations (PEx)
The definitition of PEx is not straightforward nor universally accepted239. Features may include airway symptoms (increased cough and/or sputum production, change in sputum appearance, shortness of breath), systemic symptoms (fever, weight loss, fatigue), as well as new clinical findings (such as tachypnea, new crackles or wheeze, fall in oxygen saturation, decreased pulmonary function tests, new radiological findings). The constellations in which these present differ with age and disease stage and PEx are particularly challenging to diagnose in young children239. PEx are important both during after they are experienced: patients may require time off school or work, which if frequently repeated impacts on long-term educational/career potential; there are well-described adverse impacts on quality of life and mood240,241; there may be side-effects of treatment such as drug allergies and gastrointestinal disturbance. In the longer term, the importance of PEx relates to their association with prognosis. Each PEx episode carries a risk of permanent loss of lung function and it has been estimated that in ~25% of episodes, there is failure to regain 90% of previous baseline FEV1242. Thus, the frequency of PEx is closely related to chronic rates of decline in lung function243–245, in one study, it was estimated that approximately half the decline in lung function observed over a 10 year time period was related to PEx244.
There are multiple unmet needs in this area of CF care in addition to establishing standardised, age-appropriate definitions. In terms of treatment there are several problems: currently, antibiotics are used no matter what the trigger; many exacerbations are virally-associated but no effective antiviral therapies exist. The optimal selection of antimicrobial agents and the duration of treatment has not been determined246. Attempts to select antibiotics based on more advanced laboratory techniques such as biofilm sensitivities247 or synergy testing248 have to date failed to yield results. Whole sputum sensitivity testing249 rather than susceptibility of a single bacterial colony has been proposed as a superior technique but remains to be tested with regard to clinical impact. Another challenge is that PEx are identified once symptoms and signs have already appeared; earlier initiation of treatment, based perhaps on predictive biomarkers, would be more effective. Several biomarkers are being investigated190,250; if those showing promise could be feasibly used frequently at home, this could have clinical potential for the future. To date, there are no therapies that prevent PEx completely. Hypertonic saline251, azithromycin252, and CFTR modulators23,253 have been shown to significantly reduce the frequency of PEx. It is possible that with the introduction of new, highly effective therapies at an early age before lung disease is established, that the epidemiology and manifestation of PEx will be very different.
4). Transition into adult care
Adolescence is a time of great change for young people, physically and psychologically, and those with a chronic disease are no exception. Death in childhood is becoming less common that in previous decades, with most young people expected to transition to adult care, many of them in fact in good health. Numbers of children with CF are not expected to increase greatly in most geographies, although as diagnostic services develop in regions which previously had none, this will lead to increased numbers of young people requiring transition into an already over-stretched adult service. It is essential that transition is a) a process, not a single event and b) co-ordinated between paediatric and adult multidisciplinary team. Healthcare transition (HCT) is defined as a purposeful, planned movement of adolescents with chronic conditions from child-centred to adult-oriented health care that is supported by individualised planning in the paediatric setting, a coordinated transfer of care, and secure attachment to adult services. A framework for CF HCT should include transition strategies to prepare young people and their caregivers, and tools to document transition progress and measure readiness at set points throughout the HCT process254. A coordinated transfer of care should include a complete clinical transfer summary (contributed by all members of the CF multidisciplinary team), ideally an opportunity to meet and/or tour adult care team and CF center, and timely access and tracking of a first adult provider appointment255. In the adult setting, HCT practices which encourage refinement of skills and knowledge (begun in the paediatric setting) should continue until secure attachment within the adult-based setting and full healthcare autonomy are reached, likely around age 24 years256,257. The development of formalised adult-based HCT practices has lagged behind its paediatric counterpart and requires attention, but in both settings, there is a need for high quality research studies to guide evidence-based (and not just empirically-based) HCT practices.
5). Complexities of adult care
a). Diagnosis with CF in adulthood
While most CF diagnoses are at a young age, up to 10% are detected in adulthood (≥18 years of age). These individuals tend to have less severe or atypical phenotypes and often present with gastrointestinal (GI) issues such as pancreatitis or respiratory manifestations such as mild bronchiectasis initially labelled as asthma, chronic bronchitis or non-CF bronchiectasis258. Diagnosis by newborn screening has become more common over the last decades, however for older adults it may not have been available or may have been negative at the era of birth. Knowledge of CF genetic variants has increased and a late diagnosis may reveal rare variants not included in previous limited CF variants panels258,259. Confirmation of the diagnosis is often made at tertiary adult CF Centres where bioassays reflective of CFTR function, including nasal potential difference and intestinal current measurement, and/or CFTR sequencing have become increasingly available100.
Referrals to adult CF Centres from non-respiratory specialists commonly occur for patients with atypical CF or CFTR related disorders, often with single or multi-organ involvement100,260. Presentation may be with recurrent sinus infections (ENT specialists), male infertility and congenital bilateral absence of the vas deferens (CBAVD, fertility clinics), as well as acute or chronic pancreatitis and/or weight loss (gastroenterologists). Incidental diagnoses may also occur in young asymptomatic females or couples contemplating pregnancy who elect to perform genetic carrier screening tests.
b). Maintaining nutrition and lung health
The areas of focus outlined in children remain of primary importance in adults, in whom additional complications (of CF itself, of its treatment and, increasingly, of ageing) occur. Regular review of pulmonary health and infection remains key as does the rational use of drugs targeting bacteria, fungi and atypical mycobacteria. Anti-inflammatory therapies are not in widespread use outside of the US (ibuprofen), although this may well change in the near future as multiple entities are being passed through trials pipelines. Prevention of malnutrition in pancreatic insufficient patients and maintenance of a body mass index (BMI) of 22 kg/m2 in females and 23 kg/m2 in males is associated with better lung function261. The more recent issue of obesity and its impact on lung function and metabolic health is being further investigated in the CF population262. Early detection of inadequate bone health and CF-related diabetes (CFRD) are also key aspects of the annual review, with the latter being associated with increased mortality263.
c). Emerging diagnoses in the older patient
The CF population is currently ageing due to therapeutic progress leading to a survival benefit264. However, there may be a price to pay for this progress. Apart from the usual extrapulmonary manifestations observed in CF adults (e.g., malnutrition, bone disease, diabetes)(Table 1), novel comorbidities are emerging due to more prolonged survival, evolution of the disease and/or prolonged exposure to treatments (reviewed in18) (Table 2).
Table 2.
An example of day to day treatment in an adult with cystic fibrosis who has chronic infection with P. aeruginosa
| Objectives | Treatment |
|---|---|
| Day to day treatment | |
| Promote clearance of airway secretions | Airway clearance techniques (physiotherapist, PEP mask, oscillating devices, etc.) twice daily Increase physical activity level Nebulised dornase alfa once daily Nebulised hypertonic saline and or Inhaled mannitol twice daily |
| Prevent acute exacerbations | Nebulised antibiotics (tobramycin, colistin, aztreonam lysine) or Inhaled antibiotics (tobramycin inhalation powder, colistimethate sodium) two to three times daily# Azithromycin three times/week Influenza vaccination |
| Ensure appropriate nutritional intake | High-fat/high-calory diet Pancreatic enzyme replacement with every meal Liposoluble vitamins: A, D, E (± K) once daily |
| Correct underlying CFTR defect * | Ivacaftor twice daily Lumacaftor/Ivacaftor twice daily-Tezacaftor/Ivacaftor twice daily |
| Potential treatments of respiratory insufficiency | Oxygen therapy Non-invasive ventilation Pulmonary rehabilitation |
| Treatment of comorbidities | Diabetes: insulin (or repaglinide) Osteopenia/osteoporosis: ensure appropriate calcium and vitamin D uptake, consider bisphosphonates Drug therapy for anxiety and depression |
| Treatment of acute complications | |
| Treat pulmonary exacerbations | Oral and/or Inhaled and/or intravenous antibiotics aiming to return to “stable state” lung function |
| Treat other complications | Sinusitis and nasal polyposis: consider intranasal steroids and non-sedating antihistamines Hemoptysis: consider bronchial artery embolization Pneumothorax: consider chest tube and/or surgery Allergic bronchopulmonary aspergillosis: systemic steroids; consider antifungal azoles or anti-IgE Distal intestinal obstruction syndrome: oral laxatives and/or enemas |
depending on the drug
in patients with eligible CFTR mutations
i). Cancer
An increased risk of cancer is emerging in the adult CF population. Colorectal cancers were increased after lung transplantation, presumably related to the reduction of antitumoral immune control caused by immunosuppressive drugs. The risk was 15 times higher in CF patients receiving lung transplants than for other disease populations265. The risk of other digestive tract cancer (e.g., oesophageal) and non-hodgkin lymphoma was increased compared to non-CF lung transplant recipients266. Registry studies267,268 and a metanalysis269 have further highlighted the increased risk of digestive tract cancers including small bowel, colon, biliary tract, and pancreatic cancers in non-transplanted CF patients. Consensus recommendations for colorectal cancer screening in non-transplanted and transplanted patients with CF were recently published270. Screening is based on regular colonoscopy that should begin after 40 years in non-transplanted patients and after 30 years (or within 2 years after transplantation) in transplanted patients270. Evaluation of these expert recommendations will be necessary in future years. Mechanisms of increased risk of cancer in patients with CF are under investigation and may include dysbiosis induced by chronic bacterial infection and antibiotics271,272 and/or direct effects of CFTR dysfunction on carcinogenesis273,274. The number of cancers is predicted to increase in future years to ageing of the CF population and to the increased number of CF adults who have undergone lung transplantation. The effects of CFTR modulators on the risk of cancer are unknown and will require evaluation.
ii). Renal disease
Chronic kidney disease, sometimes leading chronic renal failure requiring hemodialysis and/or renal transplantation, is also an emerging complication in older adults with CF275,276. Causes include repeated use of nephrotoxic drugs (e.g., aminoglycosides, non-steroidal anti-inflammatory drugs), diabetic nephropathy, amyloidosis277, and oxalate nephropathy278. After lung transplantation, immunosuppressive drugs (e.g., anticalcineurin) also play a major role in deterioration of renal function. Preserving renal function throughout life should become an objective in all CF patients and the balance between increasing survival with advanced CF-lung disease and maintaining lung health with intensive antimicrobials, may impact on kidney health especially with longterm CFRD and in those undergoing transplantation where nephrotoxic therapies are common.
iii). Obesity and metabolic complications
CF is classically associated with malnutrition and low fat mass. However, with improvement of life expectancy, changes in nutritional habits (increase dietary intake with high calorie/high fat consumption) and pancreatic enzyme replacement therapy, the risk of overweight/obesity and metabolic syndrome is increasing in CF patients especially the older adult279,280. Treatment with CFTR modulators is also associated with weight gain and it is hypothesised that when next generation CFTR modulators are widely available, the usual recommendation for a high fat/high calorie diet may need to be reconsidered to avoid complications related to metabolic syndromes. Further, although CFRD is not usually associated with the macroangiopathy and increased macrovascular complications apparent in the general population with diabetes, they may emerge as patients continue ageing with longer exposure to CFRD.
The list of emerging comorbidities in older adults with CF will likely get longer in future years and objectives of CF care will shift progressively from the aims of preserving lung function and avoiding malnutrition to achieving these goals in parallel with preventative strategies for emerging comorbidities.
d). Transplantation
Since the first lung transplantation in the early 1980’s, significant progress has led to improved outcomes from transplantation281. Lung transplantation is now considered the standard of care for patients with severe respiratory disease refractory to usual therapy, as it has the potential to improve survival282. However, not all suitable patients gain such benefit, either because they are not referred in a timely fashion, because the fail to be transplanted once listed or because they have poor post-transplant outcomes. Tackling these issues for the future is imperative given the increasing adult CF population.
i). The need for improvement in lung transplant referral strategies
Recent studies have revealed that barriers exists to providing lung transplantation in patients with CF. Analyses of the US CFF patient registry have shown that patients who are economically poor and/or poorly educated are less likely to be referred283 and accepted for transplantation284, underscoring the need for improved funding for lung transplantation programmes224. Even in countries with fully funded transplant services, lung transplantation referral remains a problem. A study from France has revealed that at least 40% of patients who died without lung transplantation would have been eligible for transplantation, but were not transplanted due to late referral or no referral285.
Several barriers to timely referral for lung transplantation have been identified. The first is related to the identification of objective factors for deciding transplant referral. Percent predicted FEV1<30% has long been associated with high risk of death in patients with CF286 and has been considered a criteria for lung transplantation referral287. However, improvement in patient care and survival in CF adults has occurred even in patients with severe disease and median survival at FEV1<30% predicted has increased from a few months in the 1990’s to more than 6.5 years in the recent period99,288. This might in part explain that ~35% of patients with FEV1<30% were not referred for lung transplantation in the US283. These findings underscore the difficulties predicting prognosis based only on lung function and highlight many other prognostic factors (e.g., repeated exacerbations, hemoptysis, pneumothorax, malnutrition, infection with specific bacterial pathogens). Recent predictive models have incorporated several variables to predict 1 year289 or 3 year290,291 survival in CF patients and may help in optimising timing of transplant referral. Machine learning has also been recently applied to UK national registry data to develop a model of prediction of prognosis and need for tranplantation referral292. Scores developed based on such studies290,291 may also be used for improving physician knowledge on non-lung function based criteria. One study identified lack of physician knowledge as a possible limitation for timely transplant referral293.
Another barrier may be related to difficulties in discussing lung transplantation between physicians and patients. Patient decision aids may be helpful and programmes helping physicians to disseminate these aids to their patients have been suggested useful for patients considering lung transplantation293. Finally, patient-identified barriers and facilitators to lung transplant discussions can inform physicians as they discuss transplant in CF clinic294.
Recently, new recommendations for lung transplant referral have been produced by the CF Foundation (USA) aim to reduce the number of individuals with CF who die without the opportunity to be considered as a suitable lung transplant candidate295. The impact of these recommendations on promoting timely referral and improving survival in CF patients will require further studies.
ii). Ensuring that patients listed for transplantation get transplanted
Another aspect is ensuring that all patients listed for lung transplantation undergo lung transplantation and derive survival benefit from transplantation. In the US, the lung allocation score (LAS) has been developed to prioritise among patients on the waiting list296. The score is based on survival probability without transplantation and predicted post-transplantation survival. The implementation of LAS has decreased rates of death on waiting list for transplantation., However, some consider that CF patients are disadvantaged by the current system and question whether a lower threshold of LAS score should be applied297,298. In France, a high emergency lung transplantation programme has been implemented since 2007. This programme allows transplantation within 14 days in patients with imminent risk of death. In CF patients, eligible patients are those with severe respiratory failure and hypercapnia despite intensive non-invasive ventilation, invasive ventilation and/or ECMO. This programme has dramatically decreased mortality on the transplant waiting list with an acceptable post-transplant survival299,300. It remains to be established whether such programme can be implemented successfully in other healthcare systems.
iii). Post-transplant care in patients with CF
In most countries, post-transplant care is performed in transplant centres, which are often not co-located with CF centres. Although this model of care has proven efficacious with an improvement in post-transplant prognosis during the past three decades, novel models of care may be needed for CF patients are now requiring consideration for several reasons. First, transplanted patients live longer301, which is good news for the patients, but creates a significant burden for the ongoing care provision by transplant centres. They need to accommodate a large number of patients previously transplanted whilst also performing surgery on a growing number of candidates awaiting lung transplantation (and not only patients with CF). Second, patients with CF and their CF physicians often share a willingness to maintain their interaction developed before lung transplantation, especially when the transplant center is at a longer distance from the patient home. Third, the prognosis after lung transplantation in CF patients is driven by lung function (especially by chronic allograft dysfunction)285 but importantly also by extra-pulmonary comorbidities of CF (e.g., diabetes, renal failure, bowel cancers, etc…)281 that potentially may be managed optimally by multidisciplinary team in CF centres. One solution might be a system of alternating follow-up appointments between transplant centres and CF centres. Ultimately the aim should be to improve daily life, burden of follow-up and overall quality of life for the patient, without decreasing post-transplant survival. Developing such new systems could prove beneficial for patients and the centres but roles will need to be clearly defined.
6). Delivering care in the future
a). Training and sustaining the CF Multidisciplinary Team
Children diagnosed with CF from the year 2000 onwards can anticipate a median survival greater than 50 years302. Many will pursue higher education, hold full time jobs and build their own families. Some may even go one to obtain a pension.
In many developed countries, the number of adults is now greater than the number of children303, and the number of adults is predicted to increase markedly in the next two decades17. The increase in the number of patients will constitute a significant challenge to ensuring that enough highly qualified professional are present in CF centres, especially adult centres. Appropriate staffing (including physicians, specialist nursing and allied health professionals) for multidisciplinary team care has been defined according to the number of patients by the ECFS standards of care142. Training enough healthcare professionals and maintaining their level of expertise in a rapidly changing disease will require an important effort by academic institutions, learning societies, patient organizations and pharmaceutical industry. As CF physicians are often also provide primary health care to their patients, training should include knowledge on lung disease (management of airway infections, acute complications and respiratory insufficiency), comorbidities (e.g., diabetes, bone disease, liver disease, GI problems, malnutrition…), fertility and pregnancy, and emerging issues (screening for cancer, acute and chronic renal failure). As highlighted previously up to 20% adult patients live with lung transplants304, requiring specific and highly specialised care. Involving enough physicians from other specialties (e.g., endocrinologist, gastroenterologist, obstetrician) will also constitute an important aspect.
There is currently no clearly defined curriculum for becoming a CF physician305. Ideally training should involve acquisition of core competence (e.g., using a syllabus), post-graduate courses and/or workshops, E-learning modules, and other teaching materials (e.g., books, articles, videos…)305. The ERS/ECFS Taskforce has recommended trainees who aspire to practice as CF physicians should also spend a minimum of one year in a well-established CF centre to learn all the practical aspects of CF care. Training programmes will also be necessary for allied health and nursing professionals.
b). Models of care
Current follow-up of CF patients is based on recommendations defining the standards of care for CF145,306. Most care is hospital-based although geographical variations exist in whether a tertiary centre or through partnerships with general hospitals closer to the patients’ homes. Pros and cons to both of these models exist. Patients with stable disease usually have routine appointment every two to three months and an annual assessment to ensure that a full medical, dietetic, physiotherapy and psychosocial review is performed. Blood test, chest imaging and assessment for comorbidities are also recommended at these annual visits145,306.
Recommendations further emphasise the need to provide access for emergency consultations and inpatient care in case of acute exacerbations or other complications. These recommendations have proven useful and presumably contributed to the increase in life expectancy in CF patients over the past decades. However, demographical and clinical characteristics of patients have markedly changed during the same period, and patient’s needs and aspirations have evolved. In that context, an important question is to determine whether it is possible to provide evolution to the present model of care in the setting of rapidly growing numbers attending current adult CF centres and yet aiming to decrease the burden of care for the patient without impairing health outcomes. Some of these changes have already occurred: for example, many centres have outreach services comprising specialised nurses +/− physiotherapists who can visit the patients in their home, although these visits are likely limited by workforce capacity and should be considered complementary to the hospital-based service rather than seen as an alternative. Some paediatric centres provide a school or nursery visit system, although these would be infrequent at times of change for example. Most CF centres have access to treatment of exacerbations using intravenous antibiotics at home, which likely reduces the need for hospitalization307,308. Screening recommendation for diabetes is based on oral glucose tolerance test (OGTT) that should be performed annually for all CF patients aged ≥10 years. However, repetition of these tests is not well accepted by the patients as it adds to the burden of care; alternative approaches have recently been proposed using HbA1C as a screening tool to determine which patients should undergo more cumbersome testing (e.g., OGTT and/or CGM)309, although such screening has been uniformally been adopted, remains controversial and requires further study before it can be broadly recommended.
The growing population, alongside concerns around cross-infection, have led some clinics to explore the possibility of video-conferences to replace (at least some) direct clinic visits. These could provide tangible benefit to patients, who often experience visiting the CF centre as exhausting, although it is likely they will be more appropriate for some individuals than others. Systems are available to allow downloading and review of home-performed spirometry and sputum samples could be sent in by the patient; if this was done the week before the consultation, results could even be available in real time. Unscheduled contacts in between regular appointments are already current practice in many centres, where CF nurses and doctors provide information and care via emails an telephone calls310. These informal care practices are important aspect of CF care and are likely to evolve with evolving technology and further development and evaluation of the effectiveness and safety of telemedicine (reviewed in detail in later section).
This section highlights clinical care and its delivery for people with CF. Future opportunities and challenges are highlighted in Panel 3.
SECTION 3 –. CF CARE IN DEVELOPING NATIONS
On a global level, there is a glaring gap in CF median age of survival between countries based on gross domestic product or level of industrialization; while the life expectancy of individuals with CF in Canada, United States, and EU countries is in the mid-40s or above, it is half of that in Brazil311, and even less in El Salvador, India, the Middle East, and non-EU countries312. Disparities in CF mortality based on country-level income were most recently corroborated with data from the European Cystic Fibrosis Society Patient Registry (ECFPR).313 In addition, the EuroCareCF collaboration, utilised the 35-country European CF Registry at the time including 29,095 patients, reported that disparate country-specific prevalence of CF could not be explained by differential population frequency of CFTR variants or case under-ascertainment, but rather is indicative of excess premature childhood CF mortality in non-West European countries314. Estimates show that the CF population in non-West European countries would increase by 84% if individuals with CF there had a demographic profile comparable to that of patients in West European countries303. Inequities between developed and developing nations are observed not only in life expectancy but across all CF-specific outcome measures, from growth and nutrition to lung function and quality of life314. It also should be noted that inequality in CF care and outcomes is seen both between and within countries, typically as a reflection of existing social inequities146,187,315,316.
Over the past decade, initiatives to improve CF care have included: establishment of newborn screening and diagnostic confirmation via sweat testing results; populations specific genetic panels; refinement of patient registries for comparison of processes and outcomes; development of evidence- and consensus-based guidelines for care; training of care teams in a systems approach to quality improvement; data transparency; and encouragement of patient- and family-centered care147 This section outlines the limitations and challenges of CF care in developing countries, and proposes priorities for care delivery that address early diagnosis, laboratory support, medication availability, and multidisciplinary care. We also identify research directions to help improve CF health outcomes in developing nations. The main goal of the strategies and priorities proposed here is to close the gap in life expectancy and survival between individuals with CF in various parts of the developing world.
1). CF Care Delivery and Models of the Future
Individuals with CF have complex needs that require extensive medical expertise and a wide range of supportive health services. Despite existing guidelines and established standards for CF care in developed countries, individuals with CF in developing nations are diagnosed later and likely to receive substandard CF care due to organizational differences, limited healthcare infrastructure, and lack of human resources, medications, and adequate facilities (Figure 7)39,106,148,316.
Figure 7. Cystic fibrosis Standards of Care.
Green – comprehensive, Blue – partial, Orange – limited, Red – minimal or unknown.
2). Current diagnostic algorithms may not be appropriate in new populations
The adoption of current diagnostic algorithms in many LMIC according to the World Bank classiciation, where newborn screening programmes do often not exist, poses significant diagnostic challenges as they assume basic CF diagnostic tests are available. Furthermore, the recent revision of international consensus guidelines to lower the limit of intermediate range sweat chloride from 40mmol/L to 30mmol/L has created additional diagnostic challenges in LMIC where alternate measures of CFTR function e.g. nasal potential difference (NPD) or molecular CFTR testing are not available or limited to CFTR panel testing more suited to Caucasian people. Although the rationale for adopting the revised intermediate sweat chloride reference range is based on evidence of rare cases of confirmed CF with sweat chloride in this range, the suitability of automatically adopting these guidelines in new populations requires careful consideration with respect to new population CFTR profiles, cost-benefit and availability of testing modalities in the diagnostic algorithms.
Interpretation of sweat chloride concentrations in non-screened populations in LMIC must factor in the effect of malnutrition and acute illness on sweat chloride concentration or conductivity in these settings, as well as higher prevalence of CF-resembling diseases such as tuberculosis and non-CF bronchiectasis317,318. In a study from Rwanda of 57 children with CF-like illness (87% with protein energy malnutrition), 48 (84%) had a sweat chloride > 40 mmol/L, of which 37 (65%) were > 60 mmol/L. However, a single CF disease-causing CFTR variant was found in only four individuals319. Similar findings have been observed in Cape, Town, South Africa (Marco Zampoli, personal communication 2019). Whether these findings represent true false positives or the effect of other factors e.g. ENaC channel variants319 on sweat chloride concentration needs further investigation, but highlight the potential flaws of universal application of CF diagnosis algorithms in new populations or LMIC. Although not accepted as diagnostic for CF, sweat conductivity is more affordable, more widely available and less labour intensive thus sweat chloride measurement320,321. Inclusion of sweat conductivity and other CF screening investigations (e.g. fecal elastase) in CF diagnostic algorithms, as in the South African guidelines, may be useful in settings where access to standard CF diagnosis tests is limited322. The undeniable reality for many LMIC or new populations is that rapid molecular diagnosis of CF is becoming more accessible and affordable, especially for remote settings, thus providing an alternate pathway for CF diagnosis where sweat testing is not available or feasible. Suitable CFTR panels or screening strategies would need to be developed depending on the profile and prevalence of CFTR variants in the population.
3). Newborn Screening and Diagnostic Confirmation
Many challenges in the diagnosis and provision of CF care in LMIC have been highlighted above. NBS programmes in developing countries also face several challenges, and health officials need to balance the risk-benefit ratio of NBS for CF in their specific population and healthcare delivery structure. Most developing countries – particularly those outside of Europe - have a relatively low CF population incidence that could lower the priority of implementing NBS in these countries. Some countries lack the specialised laboratory testing resources and medical expertise needed for CF diagnosis and subsequent management.
Genetic testing in developing countries is challenging, as the frequency distributions of CF variants are heterogeneous depending on the genetic admixture of the population. Such is the case in Latin America, for example: panels that are commonly used in Europe or North America might detect only a fraction of alleles in the region. In Chile, the diagnostic panel of the 36 most common variants detects approximately 50% of all alleles, while for predominantly-Caucasian populations it is nearly 90%323. Thus, there is a need to increase the ability of genetic testing to detect CFTR variants in non-Caucasian populations, with full sequencing of all CFTR exons, untranslated regions, and copy number variants. The use of next-generation sequencing (NGS) will avoid the bias generated by use of commercial kits that limit their scope to a specific set of known variants. Once extensive NGS analysis has been performed to identify all variants across many populations, more sensitive panels would be able to be developed and utilised in NBS programmes, leaving NGS analysis for those cases for which the panel was not successful in identifying the two causative variants324.
Functional assessment of CFTR activity should be performed by sweat testing, particularly in cases with rare or novel variants for which clinical impact is unknown. Sweat testing should be performed in a center with significant experience (>150 sweat tests per year) and sweat chloride measured by a standard method (titration, potentiometry, or coulometric quantitative tests).146 Designated clinical laboratories with adequate geographic distribution are the most cost-effective step toward reliable diagnostic results from sweat testing.
4). Organization of CF Care
Among the organizational factors preventing optimal CF care delivery, the most prominent are paucity of specialised CF center networks and multidisciplinary teams with expertise in pulmonology, gastroenterology, endocrinology, respiratory care, physical therapy, nutrition, psychology, and social work, among others (four examples from LMIC are provided from different regions around the world in Panel 4). In many developing countries, CF is managed within multiple, disconnected health service subdivisions, resulting in a dispersion of patients and limiting providers from acquiring the expertise necessary for provision of specialised CF care325. The low volume of individuals with CF within each unit may discourage health service administrators from allocating sufficient resources for multidisciplinary CF care. Another organizational barrier is lack of integration or sharing of information within and amongst different health systems, allowing for individuals with CF to move between systems with limited follow-up. Finally, as salaries from public employment are meager, health professionals essential to the multidisciplinary CF team often supplement their income working as independent contractors to both public and private systems, limiting their availability or willingness to dedicate more time in CF care, which is mostly reimbursed from limited public sources and forcing families to pay out-of-pocket resulting in higher costs to insurance providers and families.324 This arrangement creates a two-tier system, lack of effective multidisciplinary work, and the false reassurance of receiving better care when paying the provider in private practice. While smaller countries, such as Uruguay, Paraguay or Serbia, tend to have centralised CF care with neonatal screening and centralised medication delivery, larger countries, such as Argentina or Brazil, have a more complex – and in some ways less well organised – CF care delivered in a wide variety of settings, from tertiary centres similar to those in the developed world to primary care facilities staffed by only one physician.
5). Training of Multidisciplinary Teams
It is well established that better survival for individuals with CF occurs when care is provided by multidisciplinary teams of experienced specialists that employ best practices, engage in quality improvement initiatives, share knowledge, and are organised in a care center network326. Unfortunately, this is not the case in the developing world. Most developing countries lack the resources to build such teams, and CF care is relegated to individuals with interest rather than expertise, often resulting in appropriate management of one aspect of CF while neglecting others. Building multidisciplinary teams through appropriate training, skill development, and provision of resources is critical in low-resource settings146,147,315. To establish such infrastructure in underserved parts of the world, it is not enough to disseminate information through traditional learning methods, such as care guidelines, lectures, and conferences. There is a critical need to support CF care providers in these regions with team training and ongoing professional development316. Even more useful is a sustained and tangible training partnership between CF teams with demonstrated excellence in the delivery of evidence-based CF care and the training of multidisciplinary CF care teams, on the one hand, and CF teams in developing countries, on the other327. These partnerships should work in three critical aspects of CF care: 1) delivery of standard, evidence-based CF care that is adapted to local healthcare settings; 2) establishment of a CF registry and data infrastructure to support such care; and 3) engagement and education of the local CF community and stakeholders.
6). CF Care Teams as a Clinical Microsystem
The significant changes in health care and medicine in the past several decades, including the provision of ongoing care, have weakened the autonomy of individual physicians, requiring that they work as part of medical teams either in a single institution or across institutional settings. The slow adaptation of individual clinicians to team-based medical care has been influenced by several factors, including a persistent culture of professional autonomy, a prominent cultural trait in most developing countries; the lack of formal training in teamwork techniques; and the absence of tools, infrastructure, and incentives to facilitate the change. The lack of team-based care has resulted in deviations from established standards for evidence-based, “best practice” CF protocols. Thus, in developing nations, it is essential to create effective CF teams and networks of teams that collaborate, even in the absence of a formal entity or accrediting body that requires or support team care, such as the CFF (USA) or the United Kingdom CF Trust.
One approach to solve the deficit of team-based CF care is the “clinical microsystem.” The CF care team is the basic building block of the clinical microsystem, which also includes the CF patient population, support staff, equipment, facilities, and information environment serving the needs of patients, family caregivers and healthcare professionals328. Significant improvement in clinical outcomes has been achieved by structuring CF care as a clinical microsystem, standardising care where possible, using best current evidence, stratifying patients based on medical need, and customising care to meet individual needs for patients and their families329,330. Structuring CF care as a microsystem should be a cost-effective strategy for CF care teams in developing countries that can improve clinical outcomes and quality of life of patients. Finally, problems in care management, such as lack of trained workforce, resources for sustaining CF care, lack of medications, and limited state support can be addressed through the organization of expertise at the regional level, under the clinical microsystem model.
7). Medication Access and Choice of Therapies
Medicines account for 20 to 60% of health spending in LMIC, compared with 18% in countries of the Organisation for Economic Co-operation and Development. Furthermore, many developed countries have programmes in place, either public or private, that subsidise the final cost of medications to consumers. Up to 90% of the population in developing countries purchase medicines through out-of-pocket payments, making medicines the largest family expenditure item after food. Although the absolute cost of a given medication may be lower in developing countries331 compared to the price in advanced economies, they are still unaffordable (as an arbitrary threshold, a treatment requiring more than one days’ wage will be considered “unaffordable”)24. Limited access to cost-effective supply of medical products – drugs, nutritional supplements, durable medical devices, and disposable supplies – impede the effective delivery of CF care according to established guidelines.332 In most developing countries, the current pattern of pricing of FDA-approved drugs, FDA-approved to market medical devices (so called 510k cleared devices), and nutritional supplements for CF care may be as high as 200–300% the U.S. costs, in relative terms. This pattern, seen across all CF care products, reflects large differences between the manufacturer selling price and the in-country distributor selling price. Pharmaceutical companies cite the relatively high cost of registration, regulatory framework, limited intellectual property rights, weak healthcare systems, as well as the lack of resources to pursue registration in every country, as a significant barrier to making their products available at an affordable price333. In addition, many companies do not sell directly to the government due to its lack of effective distribution systems in place, using instead a local distributor that has an unlimited freedom for price setting, resulting in prices several times higher than the manufacturer fair price.
Nutritional status has a prime impact on pulmonary disease progression and survival in CF334. Infancy and early childhood are periods of vulnerability to adverse exposures associated with poverty; therefore, an aggressive strategy should be employed to achieve good nutritional status as early as possible. Specific considerations in LMIC include some of the following: a) Breastfeeding, the most affordable and safest method of nutrition, should be encouraged for all infants along with proper enzyme supplementation; b) When there are no resources for gastrostomy, overnight nasogastric tube feeding can be administered; c) During hot, humid weather, intense physical activity, or fever, salt should be added along with adequate hydration335.
Ideally airway clearance treatment should begin at the time of diagnosis, even without symptoms and parents using techniques recommended by a physiotherapist including including autogenic drainage. Inexpensive mechanical devices that generate oscillating or non-oscillating positive expiratory pressure (PEP) should be favored over more expensive high-frequency chest wall oscillation (HFCWO) devices336.
Choice of antibiotics to treat pulmonary exacerbations is based on guidelines created in developed countries. Market availability and pricing may differ in the developing nations. For example, although in the US tobramycin is the aminoglycoside of choice for intravenous treatment of CF pulmonary exacerbations due to P. aeruginosa337, amikacin and gentamicin may be more affordable and easily available in the developing world. Similarly, use of injectable formulations of tobramycin and amikacin have been used instead of tobramycin solution for inhalation (TSI) to treat chronic P. aeruginosa infection. Although TSI might be advantageous, the use of the injectable preparations for nebulization is certainly better than no treatment338.
8). Patient Registry and Data Management
The development of national registries that collect patient data and illustrate population-based care processes and disease outcomes is an essential step to improved survival339. High-quality patient registries are vital for identifying epidemiologic trends in CF and monitoring the impact of interventions over time. Although advancements in CF care have led to dramatic improvements in survival on a population level, there are individual patient-level factors that can impact survival, and national differences in survival. It is important to understand these factors to ensure that each individual and overall population can achieve their full health potential. Established patient registries (PR), including the ECFS PR and the US CFF PR include comparisons between CF centres in terms of key clinical outcomes. Although such comparisons make no adjustments for differences in patient case mix among the different CF centres, they are very important, because describing and understanding the differences in CF survival is only possible through robust and transparent analysis of nationwide CF patient registries. Initiatives to improve CF care require a sophisticated patient registry that allows: a comparison of processes and outcomes between and within CF centres; development of evidence-based and consensus-based guidelines regarding standards of care; ability for the CF care teams to use a systems approach to quality improvement; data transparency; and encouragement of a patient- and family-centered care
9). Geographic, Economic, and Political Barriers
Studies in developing countries have shown that geographic proximity and physical accessibility of health services are strongly associated with healthcare utilization340. Geographical information systems (GIS) approaches and tools have been used to model accessibility to healthcare services341,342. GIS software could provide useful information for planning and delivery of health services and for the assessment of access to healthcare.343 Developing countries could consider using this GIS approach when designing and positioning CF care centres to optimise CF care access and delivery. At the governmental level, CF care can be improved by health systems strengthening interventions, such as the U.S.Agency for International Development’s “Acting on the Call -Ending Preventable Child and Maternal Deaths”37 which are increasingly being implemented to improve health services in IMIC344. Such initiatives can also be attractive to potential donors.
10). Influencing the Health System
Currently, there is little collaboration between practitioners and the health systems organizations where they practice, a reflection of the deeply ingrained culture of medical autonomy and the belief that the ultimate responsibility of a physician is to the individual patient. Unfortunately, this misalignment leads to suboptimal care to the very patient whom the physician treats. To support patient-centered care delivery by well-functioning clinical care teams, health organizations must find ways to bridge the divide between the CF team and the delivery system and allocate resources for improving its efficiency and impacting clinical outcomes.
11). The Individual CF Patient/Family
Over the last decades, the role of the patient has changed from a passive recipient to an active participant in care. Fragmented healthcare systems, combined with the growing burden of chronic disease and the need for continuous care, have forced many individuals with CF to assume an active role in the design, coordination, and delivery of their care. Such patient activation is even more critical in developing countries. Information and communications systems can provide valuable information to the patient for disease self-management and enable ongoing interaction between patients and their care team outside of the clinic walls. Continuous, real-time communication of an individual’s physiologic data to CF care providers could accelerate the pace of diagnosis and treatment. Remote monitoring and care delivery could make care more convenient for patients, save them time, and conceivably improve adherence to treatments, although additional research is required to determine effectiveness and safety of these strategies. The use of apps such a Genia in Sweden or Folia in the US is an example of facilitating interactions between families and their care team and enhancing the quality of care. Apps also have the potential to change the nature of the relationship between patient and provider, encouraging patient activation, provider trust, and shared decision-making345,346. Modern-day CF care is technology-dependent and cost-intensive. In most developing countries, healthcare systems lack the resources (trained manpower, materials, and funds) for appropriate diagnosis, management, and regular monitoring of the disease with a focus on long-term outcomes, including CF-related metabolic disease, infertility, quality of life, etc. Options to overcome these challenges using available technology and resources needs to be explored.
12). Partnering with Stakeholders
Patient associations and advocacy organizations, which in most developing countries center their efforts on medication access, need to consider alternatives for reaching their objectives. Specifically, a change from “demanding” to “partnering” with local health systems and governing authorities, working collaboratively with CF care teams to develop strategies to improve care, and together reaching out to health care officials and policy makers can be a fruitful approach.
13). Meaningful CF Research for Developing Nations
Research is essential for delivering evidence-based and innovative CF care and improving the survival and quality of life of individuals with CF in developing nations where epigenetic, environmental, and socioeconomic factors, including access to health services and CF care, differ substantially compared to industrialised countries313. Lung function and nutritional status remain central indicators of CF health. Tracking such indicators through epidemiological and observational studies and identifying local determinants of these outcomes should be prioritised. An important first step toward meaningful CF research in developing countries is establishing of CF patient registries, which advance our understanding of CF epidemiology and help improve survival80,339. Countries in Latin America347 and South Africa348 have recently established registries for their CF patient population.
While developing patient registries, it is critical to capture not only clinical but also socioeconomic and environmental data. It is also essential to coordinate data fields and adopt definitions consistent with established CF registries to ensure harmonization, applicability, and meaningful comparisons of CF data sets within and between countries349,350.
Interventional studies addressing the unique needs of developing countries are equally important. Access to high-quality and affordable CF care and specialised CF drugs such as inhaled antibiotics, Dornase alfa, and CFTR modulators is often restricted or absent. Therefore, investigation of cost-effective alternatives achieving similar outcomes are critical, such as inexpensive nebulised hypertonic saline194,251, off-label use of nebulised gentamicin or amikacin for eradication of P. aeruginosa351, antibiotics delivered through non-standard inhalation devices, and non-inferiority studies of simple chest physiotherapy devices336. Other complex issues that need to be addressed through research include the varying genetic epidemiology of the disease, the acceptance of CF as a lifelong chronic condition, and the development of education platforms including adequate pathways for physician training and systems to disseminate research findings113.
An emphasis on local priorities must remain at the forefront of research investment in CF in developing nations. Global shifts from healthcare to health and from hospital to community care have relevance to CF, but in the challenging context of financial constraint, geographic variability, and environmental influences, cost-effectiveness is imperative. Prioritising research in areas of greatest need – from infection control, antimicrobial resistance, and the monitoring of colonization patterns to point-of-care alternatives for detecting infection in CF, lung function measurement, and clinical assessment – is crucial and requires a dialogue between healthcare sectors, providers, government entities, and CF patient organizations and stakeholders. For example, research to develop evidence-based CF care guidelines that account for local practices, culture, and diet could expand access to low-cost chest physiotherapy devices or identify unique local nutritional supplements.
Knowledge translation through improvement in clinical practice and monitoring change are important in all healthcare settings but have even greater relevance to CF care in developing nations. Engaging governments, healthcare teams, and individuals with CF and their families is key for performing robust clinical research to improve outcomes and demonstrate effectiveness. The pillars of successful research in developing nations are communication, cost, and community engagement. Communication is central to delivering high-quality CF care in low-resource settings and with widespread internet access (even in the remote locations) prompts work on connecting patients with remote CF care teams using digital health, telemedicine, and e-tools to facilitate care. Disparities between rural and urban CF populations352 as well as disparities based on distance to the CF center353 may be ameliorated through outreach care and the adoption of telehealth clinics.354,355 Telehealth approaches may help ensure more equitable access to CF care in geographically diverse areas. In terms of cost, quantitative economic modelling is particularly important, supplemented by qualitative, observational, and quasi-experimental studies addressing disease heterogeneity, geographic variation, and environmental influences on disease outcomes. Finally, community engagement is vital to successful CF research in developing countries. The delivery of CF care that is sensitive to cultural, ethnic, and religious beliefs and norms is fundamental for community engagement in CF research. Self-management approaches, including strategies to promote patient and family education and treatment adherence, activate and empower CF patients, families, and stakeholders to participate in research to improve outcomes.
Equitable access to emerging CF therapies, including therapeutic clinical trials, is an important issue for developing countries. Large, pharmaceutical-sponsored drug trials are often the only way for patients in developing countries to access novel and expensive therapies. However, significant and distinct challenges for the implementation of CF clinical trials exist. The unwillingness of the pharmaceutical industry to sponsor trials in low-resource settings or to assure continued post-trial access to drugs is often a stumbling block. Examples of this include trials of novel and costly oral CFTR potentiator-corrector combinations run by smaller biotech companies (e.g. Galapagos) with limited resources for larger studies in significant numbers of patients including lesser developed settings. The Tobramycin Inhaled Powder (TIP), a simple device reducing administration time and improving adherence is particularly important in lower resourced settings. Often long-term safety studies have been conducted excluding lesser developed nations where TIP benefits may arguably be even greater. In assessing where to conduct clinical trials, industry clearly considers future markets and drug affordability and with the high costs of newer agents and challenges with infrastructure to conduct clinical trials, this makes running clinical trials less feasible and desirable in lesser developed nations at an ethical cost to patient access.
Other challenges include geographic distance and terrain, language barriers requiring the use of medical translators, and cultural and religious beliefs that hinder the ‘acceptance’ of experimental therapies. Variability of the treatment response in a clinical trial setting may also be higher in lesser developed countries if patients are seen more frequently than the standard of routine clinical care making the ‘placebo’ effect potentially more pronounced. Tailored adjustments may be necessary to address these challenges as well as restrictions imposed by local healthcare structures. Simplified algorithms and condensed study protocols with focus on outcomes that do not require complex and expensive technology would improve trial quality and consistency. Innovative solutions to fund CF trials are needed because data derived from developing countries strengthens the local political case for drug access, providing evidence for national effectiveness.
Finally, improved survival of individuals with CF in some low-resource settings has led to detection of complications such as allergic bronchopulmonary aspergillosis, antibiotic-resistant organisms, side effects of prolonged/persistent antimicrobial therapy, infection with organisms not traditionally associated with CF, and more. The sheer numbers of CF patients surviving into adulthood has increased globally and this is likely to occur in lower-middle income countries over the next decades. Planning and resourcing this challenge is necessary and is limited at present. Clinical and health service research focusing on these issues is important to inform future CF clinical care in the developing world.
In summary, effective models for the improvement of CF care in developing countries should emphasise (1) early diagnosis, with a focus on greater awareness of the disease, expanding the use of quality sweat tests, and supporting NBS programmes in the countries where they already exist; (2) improvement of existing CF teams by regionalising care and providing training by experienced teams from developed countries; (3) intervention in medication costs to make treatments more accessible; (4) cooperation with governments and CF stakeholders to maximise effort; and (5) meaningful research that aims to address and improve the distinctive epigenetic, cultural, environmental, socioeconomic factors, and limitations to care access that makes CF such a challenging chronic disease to developing nations.
Effective models for the improvement of CF care in developing countries should emphasise (1) early diagnosis, with a focus on greater awareness of the disease, expanding the use of quality sweat tests, and supporting NBS programmes in the countries where they already exist; (2) improvement of existing CF teams by regionalising care and providing training by experienced teams from developed countries; (3) intervention in medication costs to make treatments more accessible; (4) cooperation with governments and CF stakeholders to maximise effort; and (5) meaningful research that aims to address and improve the distinctive epigenetic, cultural, environmental, socioeconomic factors, and limitations to care access that makes CF such a challenging chronic disease to developing nations.
Individuals with CF face increased challenges to successfully juggle complex treatment regimens and manage complications that come with aging. Registry data is a valuable resource that can be used to advocate for additional resources to care for the aging CF population so that we can best meet their current and future needs.
This section highlights clinical care and its delivery for people with CF. Future opportunities and challenges are highlighted in Panel 5.
SECTION 4 –. NOVEL THERAPEUTICS
1). The path to development of CFTR-directed therapeutics for restoration of CFTR function in all patients with CF: current status and remaining challenges
Based on the nature of the molecular defects in CFTR (Figure 1), it was conceived that small molecules could successfully restore activity to the mutant protein, thereby ameliorating disease manifestations8. These agents were discovered by high throughput screening, and optimised by rigorous medicinal chemistry that was driven by a remarkably faithful and predictive preclinical assay, the ion transport properties of primary human CF bronchial epithelial cells that can be maintained in cell culture. The first class of agents to be successfully developed were CFTR potentiators;small molecules that interact with the mutant channel to augment its open probability of the CFTR channel, enhancing anion flux through the plasma membrane (Table 3)356–358. Initial studies focused on the G551D gating mutation, as sufficient protein was already localised to the cell surface, but its function was almost complete absent356,359–362. In vitro, ivacaftor restored ~50% of CFTR activity to the channel, and improved impaired mucociliary clearance; a cornerstone of the CF pathophysiological defect356. Effects in patients were marked, rapid, dose-dependent, and reversible. Endpoints including lung function and effects on biomarkers of CFTR function, i.e. sweat chloride, nasal potential difference and intestinal current measurements were internally consistent, enhancing confidence in the findings21,363,364. This initial proof of concept was a harbinger of a relatively rapid transformation of the field as substantial clinical development accelerated to increase the number of mutations addressed by CFTR modulators, evaluate their effects in populations outside of the initial clinical trials (and in particular, young individuals), and understand the protean effects of these agents, including in vitro to in vivo correlates, which continue to evolve (Figure 8)365. One challenge for turning a CF concept product into an actual medicine for the benefit of patients resides in the fact that, although pre-clinical models have shown good predictability for certain CFTR mutations, a good correlation to clinical end-points or biomarkers, individual patient level correlation to clinical end-points or biomarkers (e.g. FEV1 and sweat chloride) has not yet been achieved. In this respect, the use of alternative end-points and innovative nonclinical models could be helpful for the understanding of those translational discrepancies. Collaborative endeavours to promote further research and development in these areas as well as early dialogue with the regulatory bodies are recommended366.
Table 3 │.
CFTR mutations by molecular class, functional abnormality and primary therapy type
| Molecular Classification | Molecular abnormality | Examples of potential therapy(ies) | Common mutations |
|---|---|---|---|
| Class I | Truncated protein undergoing nonsense medated decay | Translational readthrough | W1282X G542X |
| Class II | Trafficking defect | Corrector/Potentiator Combinations | 508del N1303K |
| Class III | Defective gating | Potentiator | G551D R117H, 508del |
| Class IV | Decreased conductance | Potentiator | R334W R117H |
| Class V | Abnormal splicing | Splice repair Potentiator* | 3849 + 10kbC>T |
| Class VI | Increased cell surface turnover | Cell surface stabiliser Potentiator | 508del S1455X |
For non-canonical (incomplete) splicing mutations
Figure 8. Schematic of approach to CFTR restoration with small molecules, by mutation class.
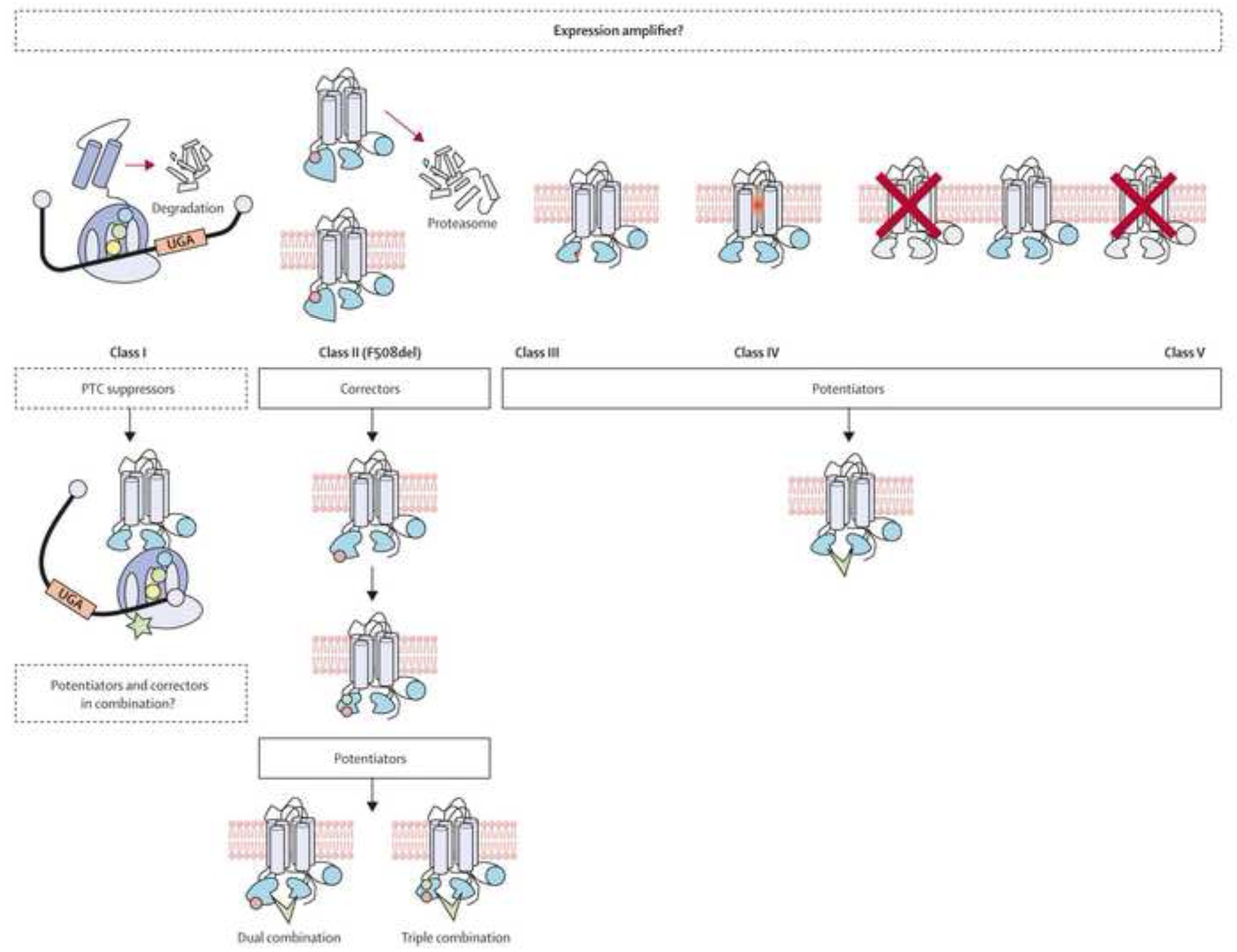
Adapted from Sloane PA, Rowe SM. Cystic fibrosis transmembrane conductance regulator protein repair as a therapeutic strategy in cystic fibrosis. Current Opinion in Pulmonary Medicine 2010;16(6):591–597.
Schematic of approach to CFTR restoration with small molecules, by mutation class. Therapeutic approaches with definitive studies in solid boxes; those under investigation with dotted boxes.
Regarding expansion beyond the original, rare G551D population, several developments have occurred. First, ivacaftor has been shown to be effective in up to 37 variants, based principally on in vitro findings but substantiated by clinical effects367,368. This includes gating mutations that have similar mechanism and efficacy of G551D gating mutations, missense alleles from mutations that reach the cell surface, but exhibit partial dysfunction from abnormal conductance or cell processing, or non-canonical splice variants that exhibit residual activity due to small but meaningful amounts of CFTR channels at the cell surface369. In general, ivacaftor responsiveness in vitro for these other alleles paralleled each other with a few important exceptions, and were proportionate to the amount of residual CFTR activity (e.g. those with milder phenotypes have a high likelihood of potentiator response as residual CFTR to potentiate is present). Due to confidence in the assay and the cell physiology that underlies it, cell model data has been considered sufficient to expand availability to rare variants by the FDA, and additional mutations are likely to follow as more comprehensive in vitro testing is accomplished with other rare CFTR variants370. Second, the underling population based on clinical severity has continued to expand, largely driven by the evaluation of patients younger in age. At present, ivacaftor is now approved in the U.S. for patients age 6 months and above, and even younger populations are presently under study in the U.S. and elsewhere22,196. Studies in progressively younger CF individuals have demonstrated the potential to preserve or restore pancreatic function371,372, and effective CFTR modulators such as Ivacaftor could slow or prevent lung disease progression if instituted prior to the onset of bronchiectasis373.
The roadmap provided by the development of ivacaftor has accelerated the pace of developing CFTR modulators for more common mutations (Figure 9). Correction of p.Phe508del, with the potential to apply to up to 90% of the CF population by genetic cause, was initially challenging due to the complexity of the p.Phe508del molecular defect that includes inefficient cell processing, abnormal gating, and shortened cell surface half-life (Table 3)374–376. Not surprisingly, CFTR potentiator or corrector therapy using a single drug was found to be ineffective, as augmenting processing of p.Phe508del without gating, or the converse, was inherently inefficient377,378. In contrast, combined two drug corrector-potentiator combination therapy was efficacious for patients homozygous for p.Phe508del, using either lumacaftor/ivacaftor23,379 or tezacaftor/ivacaftor380,381. However, a gene-dose effect made these regimen ineffective in patients heterozygous for p.Phe508del and an unresponsive ‘minimal function’ allele, where these corrector-potentiator combination therapies were not active enough to confer clinical benefit. This stood in contrast to the beneficial effect of tezacaftor/ivacaftor in patients with p.Phe508del and an ivacaftor responsive allele, in which the combined effect of tezacaftor/ivacaftor on the p.Phe508del allele and ivacaftor on the ivacaftor-responsive residual function allele was sufficient to confer clinical benefit that exceeded ivacaftor alone382.
Figure 9. Cystic fibrosis drug development pipeline of the US Cystic Fibrosis Foundation.
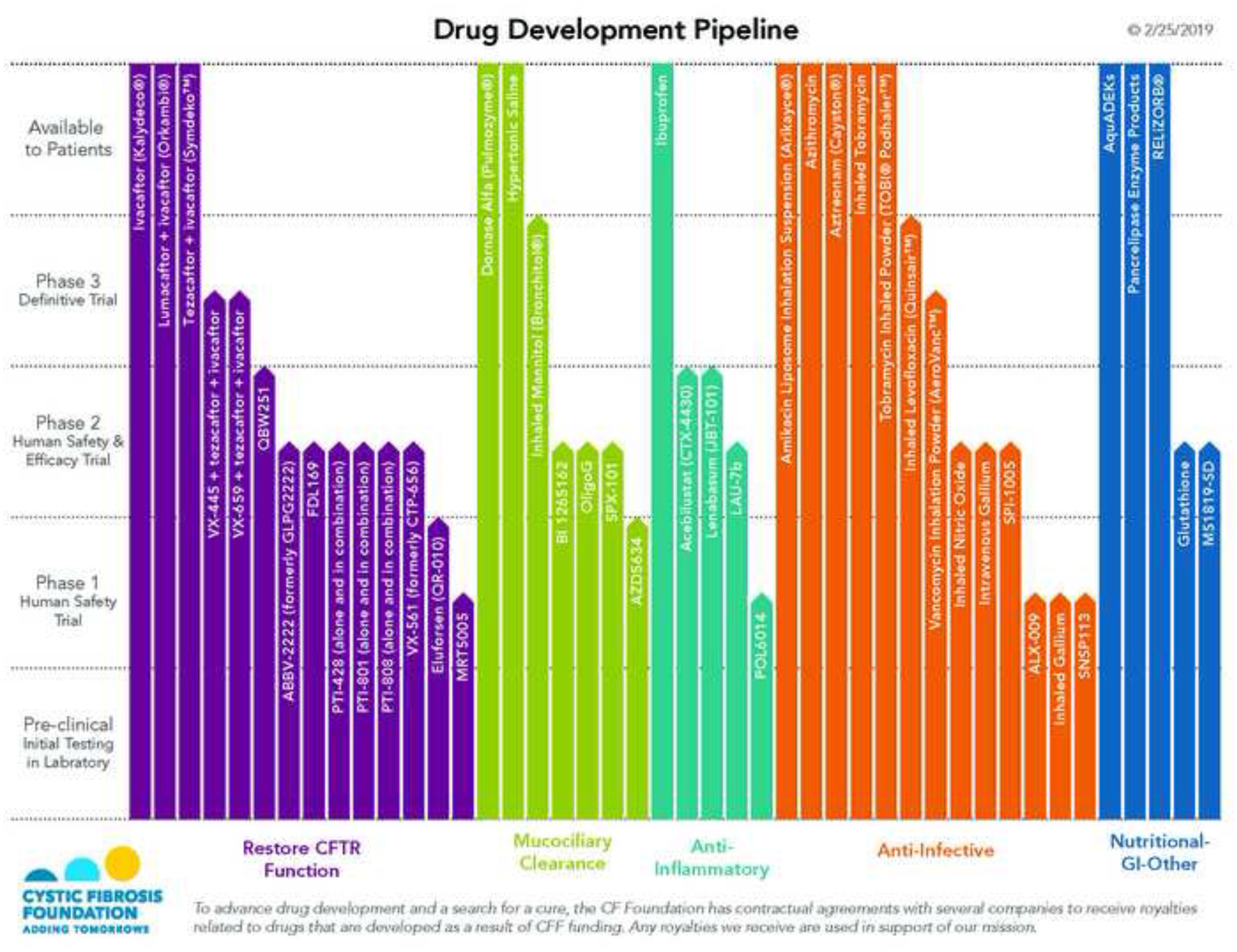
Reproduced with permission of the US Cystic Fibrosis Foundation, Bethesda Maryland.
Applying the concept that treatment with multiple CFTR correctors, in addition to ivacaftor as a potentiator, could restore greater function to p.Phe508del and provide benefit even in p.Phe508del heterozygous individuals, has accelerated progress, with recent results suggesting efficacy similar to potentiation of G551D by ivacaftor383,384. The potential for multi-drug therapy consisting of two correctors acting at different sites of the p.Phe508del protein to correct distinct folding defects and a potentiator is likely to extend to multiple corrector-potentiator regimen, or even in combination with CFTR amplifiers to increase CFTR mRNA substrate pools applicable to pharmacological restoration by correctors385.
Adoption of highly effective CFTR modulator regimens could entirely alter the landscape of CF treatment if availability of the regimen is assured to all patients with the potential to benefit. Several issues still have to be solved to assure maximal impact of CFTR-directed therapeutics and each of them requires distinct research efforts. First, patient-specific drug testing strategies may be needed, particularly given that ultra-rare mutations in which CFTR modulator regimen have not been clinically tested have the potential to respond to these therapies. Individualised testing regimen could assist this effort, including N of 1 trial designs that incorporate in vivo assessments of CFTR biomarkers coupled with cell-based assays derived from trial participants386–388. Agreement among regulatory bodies regarding the approach to patients with CF who harbor ultra-rare mutations that are insufficiently common to be addressed with typical clinical trial designs is needed to help secure access to effective medications for this patient population. Second, mutations in which current CFTR corrector-potentiator combination regimen are unlikely to provide benefit need further drug development. Premature termination codons are particularly prevalent in this group and have the potential to be addressed by the pharmacologic suppression via translational readthrough (Table 3)389–392. This concept has translated to proof-of-principle in vivo studies with aminoglycosides such as gentamicin, but not with sufficient efficacy or safety to envision chronic therapy as of yet393–395. Several strategies are in development, including both rational drug design396 and new high throughput screening approaches.
In addition to CFTR modulators, several novel small-molecule therapeutic strategies are presently in development to expand pharmacological rescue to CFTR mutations that are currently not covered, or to provide durable long-term benefit via gene replacement therapy397–399. A relatively approachable strategy would be mRNA based repair via anti-sense oligonucleotides of individual and severe splice mutations in CFTR, as has been conceptually demonstrated for Duchenne’s muscular dystrophy400,401 and spinal muscular atrophy402. Codon optimised CFTR mRNA replacement therapy could provide an additional alternative in a mutation agnostic fashion, noting that intermittent recurrent delivery would be required with mRNA based therapies. The ultimate goal of providing a life-long cure for CF may require the emergence of highly efficient gene therapy, either in the form of full-length functional CFTR delivery via viral and non-viral vectors, or gene editing of the genome. Substantial barriers remain to these genetic approaches, including efficacy of delivered genes and assuring off target events do not occur, particularly if gene editing enzymes are delivered403. Moreover, all nucleotide-based treatments rely on the emergence of efficient nucleotide delivery strategies to the lung, in which innate defense mechanisms need to be circumvented. In addition, a better understanding of where CFTR must be delivered, both anatomically and at the level of respiratory cell type, to impart durable improvemens to the lung is needed, particularly given the undefined role of the recently discovered ionocyte (a rare subtype of airway epithelial cells that express high levels of CFTR) in airway physiology404. Finally, how gene repair might be feasible in other affected organs and improve the health of CF patient in the absence of CFTR restoration beyond the lung is also unknown.
2). Patient-derived model systems for advancement of personalised medicine in CF
In parallel to these therapeutic strategies designed to restore mutant CFTR function, tremendous progress has been made in the development of patient-derived cell and tissue models that are now available for advancement of personalised medicine for patients with CF405–407. Planar cultures of primary bronchial epithelial cells obtained from lung explants were shown to be a suitable model to predict response to CFTR modulator therapy in vitro356,361, but their availability remains limited especially for rare CFTR genotypes. Therefore, the development of techniques such as conditional reprogramming of airway epithelial that enables the use of brushed nasal cells that can be easily obtained from patients at any age408–411, as well as the possibility to generate airway epithelial-like cells from skin or blood cells using induced pluripotent stem cell technology412, are major advances for in vitro testing of individual responses to CFTR-directed therapeutics. In addition to planar cultures for electrophysiological studies of CFTR chloride channel function, three-dimensional organoid cultures of intestinal and nasal epithelial cells have become available for studies of CFTR-mediated fluid transport and have been used increasingly for in vitro testing of CFTR modulator effects on specific CFTR mutations387,413–415. While these cell based models provide unique opportunities for preclinical in vitro testing, recent studies showed that intestinal current measurements (ICM) for assessment of CFTR function in native rectal tissue biopsies is sensitive to detect in vivo responses to treatment with ivacaftor in G551D patients and lumacaftor/ivacaftor combination therapy p.Phe508del homozygous patients364,388,416,417. These data suggest ICM as a sensitive biomarker of CFTR function that may be used to determine in vivo response of rare CFTR mutations and compare responsiveness to different approved CFTR-directed drugs in individual CF patients.These patient-derived models are important expansions of the precision medicin toolbox for CF that can be employed to predict and assess in vivo responses of individual CF patients to multiple emerging CFTR-directed therapeutics and are therefore expected to be instrumental for realising the full potential of personalised medicine for CF.
3). Clinical trial designs that led to the approval of current CFTR modulators
As outlined above, to date, three CFTR modulator drugs have been advanced through the whole process of clinical drug development, regulatory review and approval: the potentiator ivacaftor for patients bearing responsive CFTR gating or residual function mutations, and two corrector/potentiator combinations for patients homozygous for the p.Phe508del mutation (lumacaftor/ivacaftor and tezacaftor/ivacaftor), or compound heterozygous for the p.Phe508del mutation and select residual function mutations (tezacaftor/ivacaftor)8. For all of these three CFTR modulator treatments, clinical trial designs have been very similar. Whether phase 2 or phase 3 trials, they were all conducted in genotype-specific populations21–24,363,379,380. Even though CF is a rare disease, the restriction of eligibility to patients with specific genotypes has not been a major limitation for the clinical development of these compounds, as they were the first in their class to be tested. Moreover, enthusiasm from clinicians and eagerness from patients to participate enabled fast enrollment in all studies. Nearly all phase 2 and phase 3 trials were randomised, placebo-controlled, parallel-group, multicentre studies)21–24,363,380. Phase 2 trials were dose-ranging studies of 4 to 8 weeks conducted in adults with CF that evaluated safety, functional rescue of CFTR activity and early clinical benefit363,379,381. In addition to sweat chloride, nasal potential difference measurements were used as a biomarker of CFTR activity in the airway epithelium in the early phase 2 trial evaluating ivacaftor363. However, due to its high variability despite standardization418, later trials only relied on change in sweat chloride concentration as a biomarker of CFTR function. Clinical benefit was assessed through measurement of effects on forced expiratory volume in 1 s (FEV1) and quality of life questionnaires. Pivotal phase 3 trials were 24 to 48 week-long studies conducted in children over 6 years of age and adults21 22 23,24 380. Their aim was to study safety and long-term clinical benefit with the primary endpoint being change in FEV1 percent predicted. Other clinical endpoints were the time to the first pulmonary exacerbation and the rate of exacerbations, change in weight and quality of life questionnaires. Studies conducted in children younger than 6 years of age were limited to open label trials and focused on pharmacokinetics, safety, sweat chloride and fecal elastase as key trial endpoints196,372.
A slightly different approach was used in trials of the novel potentiator GLP1837. A phase 2 study of the potentiator GLP1837 was based on a 4-week open-label study with a 7 day-withdrawal of ivacaftor before the start of dosing the new potentiator419. A placebo-controlled phase 2 study on an amplifier designed to increase the amount of immature CFTR protein in the cell and thus provide additional substrate for CFTR correctors and potentiators to act upon was conducted in patients on stable lumacaftor/ivacaftor treatment (NCT02718495). Some phase 3 trials that included patients with less common mutations had a crossover design with two 8-week intervention periods separated by a 4 to 8-week washout period382,420. In the randomized superiority phase 2 and phase 3 studies of triple combination therapies consisting of a next generation CFTR corrector (either VX-445 or VX-659) in combination with the first generation corrector tezacaftor and the potentiator ivacaftor in p.Phe508del homozygous patients, tezacaftor/ivacaftor was used as the active control because dual-combination CFTR modulators were considered the standard of care for these patients383,384 (VX17-445-103, VX17-659-103). This approach of testing added benefit of a triple combination CFTR modulator therapy on top of current combination therapy with tezacaftor/ivacaftor was more straight forward because all the drugs tested were developed by the same sponsor. For the first time, two different compounds with the same mode of action, i.e. the next generation correctors VX-445 and VX-659, were tested simultaneously in parallel phase 2 trials and both were advanced to phase 3 trials to mitigate the risk of unforeseen adverse effects that is associated with new chemical entities. Finally, because previous studies have shown that the efficacy of CFTR modulator therapies on lung function could be reliably evaluated after 4 weeks of treatment21–24,363,380, the phase 3 trials of triple combination CFTR modulator therapies in p.Phe508del homozygous patient had a trial primary endpoint at 4 weeks, although the duration of the randomized phase was longer to evaluate efficacy and safety. This was then followed by an open-label study (NCT03224351, NCT03224351). The rapid increase of people with CF treated with approved CFTR modulators may impede future trials by other sponsors to follow this path.
4). Current challenges in trial design
The advances of the last few years, while providing much promise for patients with CF, pose some challenges in the approach to testing further and emerging novel drugs in clinical trials. There are two specific issues, both related to the emergence of effective mutation-specific CFTR modulators as standard care, namely i) how new CFTR corrector/potentiator combinations can be tested; and ii) how trials with compounds targeting more ‘conventional’ or down-stream targets in CF lung disease such as airways mucus plugging, inflammation and infection should be designed.
Three currently licensed CFTR modulator therapies have been mentioned above and phase 3 trials of triple combinations including a next generation CFTR corrector (VX-659 or VX-445) added to tezacaftor/ivacaftor have been completed in patients with CF homozygous for p.Phe508del and compound heterozygous for p.Phe508del and a minimal function mutation (VX17-445-102, VX17-445-103, VX17-659-102, VX17-659-103). If licensed, a triple combination CFTR modulator therapy may be suitable for more than 85% of the CF population that carries at least one allele of the common CFTR mutation, p.Phe508del. In regions where these drugs are reimbursed, any new investigational compound will therefore be trialled in patients already receiving drug, from which they may be experiencing clinical benefit. Such new agents may seek to compete with, or replace the current drug(s) or may be specifically designed as an ‘add-on’. An example of the latter are CFTR amplifiers, designed to increase the amount of p.Phe508del CFTR protein upon which correctors can work385,421.
New CFTR modulator compounds developed to further enhance efficacy of functional rescue of p.Phe508del and other CFTR mutations could be tested in head-to-head active comparison trials against existing CFTR modulator drugs or with a placebo-controlled design422,423. The former can be powered either for superiority or non-inferiority, potentially requiring large number of patients depending on the hypothesis and the trial endpoint(s)424. For an active comparator trial, there will be substantial costs for the trial sponsor in supplying and blinding the control drug if manufactured by another company. Placebo-controlled trials, while often considered gold-standard and requiring smaller, possibly shorter studies, will not only require patients to ‘wash-out’ their current therapy, but also potentially to remain off drug if randomised to placebo, noting that the effcacy of the new therapy is not established. A small study has surveyed opinions on this, reporting reasonable support from patients for short-term placebo periods425. However, this data may already be out of date. As the eligible population grows and compound efficacy of modulators in the pipeline is greater, it is essential that those involved in design and implementation of CF trials continue to appreciate how such issues influence patient acceptability. Ultimately, this decision will rest with regulatory bodies, but they may be prepared to take into account patient opinions426. Agents designed as ‘add-ons’ perhaps have fewer challenges; the original therapy can continue to be supplied through the clinic with the add-on molecule being administered in a standard placebo-controlled fashion (eg. clinicaltrials.gov NCT03258424).
Despite the recent breakthrough with CFTR modulators, the development of novel mucolytic, anti-inflammatory and anti-infective therapies will likely remain important, especially for patient populations with more advanced stages of CF lung disease. There are multiple current trials of these agents running through the CF clinical trial networks, some of which may be more challenging to recruit to if CFTR modulator trials are also on offer for the same patient group. Casting the eye forward to an environment within which the majority of our patients are receiving effective CFTR modulators, this particular challenge may have lessened, but it is clear there may be new ones; i) patients may feel so well they do not wish to enrol in future trials and ii) the clinical impact of any intervention may be more difficult to discern on a background of better health, leading to the need for larger group sizes or longer trial duration. Specific challenges of anti-infective and anti-inflammatory trials in the era of highly effective CFTR modulators have been reviewed recently427,428. In the case of the latter, one aim may be creative adaptation of protocols in later stage trials to reduce the burden on participants. Home monitoring with blue-tooth enabled devices for lung function, weight and symptoms is being explored for clinical care429,430 and could also be considered by sponsors in a trial setting. Low volume blood samples obtained by finger prick that could be sent into the laboratory by patients or community research nurses are being explored in some other diseases431,432. In terms of signal detection, we may need more sensitive outcome measures than the conventional FEV1. Multi-breath washout derived lung clearance index has proved sensitive and extremely useful in young children and adults with mild disease and has been accepted as efficacy endpoint in pediatric regulatory trials24,167,433,434. In addition, lung imaging by CT and MRI hold promise as quantitative outcome measures in this patient population178,435–440. As the population hopefully moves towards this ‘milder’ state, LCI and new imaging modalities may prove useful. It is encouraging that consideration of these outcome measures is increasingly acceptable not only to sponsors but also to regulatory bodies, the EMA in fact convening a workshop around this issue several years ago441.
5). The role of different stakeholders in CF drug development
The development of novel drugs for CF relies on successful collaboration between the different stakeholders including academia, industry, regulatory bodies, patient organizations and learned societies, and the development of CFTR modulators probably exemplifies the best case scenario in the CF field so far. Researchers from different fields including genetics, biochemistry and physiology applied basic science to understand the molecular and functional consequences of mutations in the CFTR gene, and developed tools to screen for compounds improving mutant CFTR function442. Patient organisations have often participated in funding such research. The CF Foundation (CFF) in particular has funded and collaborated with pharmaceutical companies to develop drugs emerging from that research. Patient organisations and learned societies like the European Cystic Fibrosis Society (ECFS) have established clinical trial networks such as the CFF Therapeutics Development Network (TDN) and the ECFS Clinical Trials Network (CTN) to optimize the delivery of clinical trials to test candidate drugs443–445. Throughout this process, clinician scientists, care givers and patients were actively involved in supporting basic research, building and studying large patients cohorts, developing/validating outcome measures and by giving their time to clinical trial visits.
Orphan drug designation and its incentives put in place by regulatory bodies including the expedited review of applications has indeed attracted the interest of pharma companies to develop drugs for CF as a rare disease. A central pillar to protect public health is the strengthening of collaboration at international level to promote harmonisation of regulatory requirements, sharing of information and addressing common challenges. International cooperation is moving from harmonisation of technical requirements towards a more convergence-based approach, emphasising information and work-sharing through multilateral cooperation and coalitions. Regular meetings by phone or videoconference are held with other non-EU regulators in so-called ‘clusters’ initially set up by EMA and United States (US) Food and Drug Adminstration (FDA). Other global regulatory bodies, including Health Canada, the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) and the Australian Therapeutic Goods Administration (TGA), now participate in some of the clusters446. Recognising the rapid pace of innovation and its own role in catalysing and enabling regulatory science and innovation, the European Medicines Agency (EMA) has recently launched for public consultation its new regulatory science strategy (RSS) (EMA Regulatory Science to 2025 – Strategic reflection447. One of the key goals identified in the RSS is “Driving collaborative evidence generation”, fostering innovation in clinical trials (with a focus on novel trial designs, endpoints, or techniques for gathering data) and developing methodologies to incorporate clinical care data sources in regulatory decision-making. A portfolio of regulatory tools and procedures are in place to implement this strategy including multi-stakeholder meetings, such as the workshop on endpoints for CF clinical trials in collaboration with the ECFS441, or the Enpr-EMA workshop on gastrointestinal (GI) outcome measures to evaluate CFTR modulators for the treatment of cystic fibrosis448. The most important tool is scientific advice provided by regulatory bodies during all phases of product development. In addition to product-specific advice, the EMA is also offering a scientific advice procedure to support the qualification of novel methodologies for medicine development447. During an initial consultation and advice phase repeat interactions between the innovators and regulators define what studies or other activities will be required to make a new methodology “fit for purpose”. Before final adoption of qualification opinion on the acceptability of the specific use of a method, the evaluations are made available for public consultation by the scientific community. This ensures that all relevant information is open to scientific scrutiny and discussion.
Recently, a qualification opinion on the ECFS Patient Registry and CF Pharmacoepidemiology studies has been adopted (Qualification Opinion on The European Cystic Fibrosis Society Patient Registry (ECFSPR) and CF Pharmacoepidemiology Studies449. Several methodology advice procedures have been concluded successfully in collaboration with EU HTA bodies and patient groups (Parallel consultation with regulators and health technology assessment bodies450). This was done to ensure the widest possible input from and acceptance of useful methodologies by key healthcare decision makers, beyond regulators only. It is hoped that researchers from academia, industry and/or public private consortia will choose this pathway to open up their methodology developments to external scrutiny and, in the process, familiarise decision makers with their concepts and enhance their acceptability, where justified. The development and validation of novel clinical study methods and data sources will require the long-term, collaborative efforts of all stakeholders in the healthcare ecosystem. However, the potential benefits are high: to bring important information on drugs’ effects to patients faster and thus enable the translation of scientific progress into timely access to much-needed treatments for patients with CF.
Despite these attempts, this encouraging collaborative work between industry, academia, patient organizations and regulatory bodies is however countered by failures in the system at the final stage, i.e. general access to safe and effective drugs. This has proved difficult or currently impossible even in a number of ‘high income’ countries around the world, largely due to the ultra-high price of novel break-through therapies for people with CF (this issue is discussed further below)451.
6). The anticipated future impact of CFTR-directed therapeutics for people with CF
The unprecedented possibility to treat CF patients with drugs that restore the function of mutant CFTR raises the questions of how these novel CFTR-directed therapeutics will be used in future CF care, how they will impact on the clinical phenotype and to what extent they can replace the current ‘gold standard’ of symptomatic therapies for CF? The mode of action of CFTR modulators, i.e. direct targeting of CFTR dysfunction in all affected organs including the lungs, pancreas and gastrointestinal track, forms a strong rationale for employing this class of drugs as the new backbone of the standard of care of patients with responsive CF genotypes. The clinical benefit of these CFTR-directed drugs will likely depend on i) efficacy of restoration of CFTR function; ii) initiation of therapy with regard to the stage of disease; and iii) long-term safety and tolerability.
The notion that the clinical benefit of CFTR modulators is related to their efficacy in the rescue of mutant CFTR function is based on observational studies on the relationship between CF genotype, the level of residual CFTR chloride channel function and clinical phenotype. These studies demonstrated that mutations that confer residual CFTR chloride channel function are associated with a milder disease phenotype including better pulmonary and nutritional outcomes, and reduced mortality compared to CF patients carrying minimal function mutations416,452,453. This concept has been corroborated by the pivotal pase 3 trials of current CFTR modulators where restoration of CFTR function to a level of ~50% of normal in patients with the G551D mutation treated with ivacaftor resulted in a substantially higher (~3-fold) improvement in lung function (FEV1 percent predicted) compared to the restoration of ~10 to 20% of CFTR function in p.Phe508del homozygous patients treated with lumacaftor/ivacaftor or tezacaftor/ivacaftor21,23,363,364,380,388,417. This relationship between functional rescue and clinical response, and recent preclinical/early phase clinical data of the next generation CFTR correctors VX-659 and VX-459 in combination with tezacaftor/ivacaftor, predicts that triple combination therapy has the potential to extend the level of functional correction and clinical benefit that has so far only been achieved with ivacaftor in the subpopulation of ~4% of CF patients carrying the rare G551D mutation to up to 90 % of the CF population that carries at least one p.Phe508del allele8,383,384. Collectively, these data indicate that the level of functional correction is an important determinant of clinical efficacy and that full restoration of CFTR function should remain the ultimate goal to achieve maximal clinical benefit.
In addition to the magnitude of restoration of CFTR function, the timing of initiation of therapy with respect to the stage of disease is another critical determinant of the efficacy of CFTR modulator therapy. In the context of chronic lung disease with irreversible structural damage of the airways and lung parenchyma, restoration of CFTR function is likely limited to anatomically preserved regions of the lung. This effect has been shown to improve overall mucus clearance and lung function in adult patients with a spectrum of lung function impairment21,23,365. However, it cannot be expected that CFTR modulators will revert bronchiectasis and it remains uncertain to what extent these drugs can break the viscious cycle of chronic polymicrobial infection, inflammation and tissue damage in structually abnormal regions of the lung454,455. The main therapeutic effect of CFTR modulators in this scenario is probably the protection of structually preserved airways thereby reducing the number of pulmonary exacerbations and lung function decline, lung transplantation and mortality in people with CF21,23,373,380,456. In patients with irreversible structural lung damage it is therefore expected that adjunct therapeutics targeting impaired mucociliary clearance, chronic airways infection and inflammation continue to be important components of multimodal CF care. Similar to the lung, CFTR-targeting therapies are not expected to revert irreversible structural damage of other organs such as the pancreas and current symptomatic therapies such as pancreatic enzyme replacement and insulin therapy will remain mainstay therapies of CF-related exocrine and endocrine pancreatic insufficiency365.
Although first tested and approved for the treatment of adolescent and adult patients with chronic disease manifestations, CFTR modulators may be most effective when started early prior to the onset of irreversible organ damage. The lungs of CF patients are structurally normal at birth, but initial abnormalities such as airway mucus plugging, inflammation and transient infection with CF pathogens are observed in the first months of life even in the absence of respiratory symptoms457. In contrast to irreversible bronchiectasis that develop over time in the muco-inflammatory milieu of CF airways, these early changes are potentially reversible. Therefore, early initiation of CFTR-directed therapies in CF infants has great potential to delay or even prevent the onset of irreversible lung damage. Such a preventive approach has become feasible by the interplay of two recent developments. First, widespread implementation of CF newborn screening has enabled establishment of the diagnosis of CF in first weeks of life in a growing number of countries worldwide458. Second, the development of non-invasive quantitative outcome measures of lung function and structure such as the lung clearance index (LCI) derived from the multiple breath washout (MBW) technique or radiation-free cross-sectional chest imaging by MRI have enabled sensitive detection of the onset and progression of early abnormalities, as well as response to therapeutic interventions in infants and preschool children with CF168,194,433,437. The concept that preventive CFTR modulator therapy may indeed be most effective is supported by recent studies in a CF ferret model with the G551D mutation where in utero initiation and postnatal continuation of ivacaftor therapy led to improved pancreatic exocrine and endocrine function, growth and survival and prevented mucus accumulation and bacterial infections in the lungs371. Furthermore, studies in this CF model showed that withdrawal of CFTR modulator therapy at any age rapidly reestablished CF-like multiorgan disease. Of note, a similar withdrawal syndrome was recently reported in CF patients with the G551D mutation in whom discontinuation of ivacaftor therapy led to a rapid deterioration of lung function and clinical symptoms459. Besides the development of more efficacious CFTR modulators, these data persuasively argue for accelerated clinical development of preventive therapy, ideally starting in early infancy (potentlally in utero) and point to the importance of life-long adherence to avoid reestablishment and progression of CF disease manifestations upon withdrawal of CFTR-directed therapeutics.
Another important determinant of the future clinical use of CFTR modulators will be their long-term safety and tolerability. While current CFTR modulator drugs have so far shown good safety and tolerability profiles across independent clinical studies and in real life for almost one decade for ivacaftor monotherapy and approximately 5 years for lumacaftor/ivacaftor combination therapy respectively, potential side effects caused by the addition of next generation CFTR corrector compounds and/or other new chemical entities such as amplifiers remain unknown. While it has been established that current CFTR modulators directly bind to CFTR and thereby improve the folding, trafficking and gating of mutant CFTR chloride channels, their potential to bind to other (wild-type and mutant) proteins and thereby interfere with other cellular functions has not been studied in great detail460. Therefore, establishment of long-term safety by pharmacovigilance programmes remains an important task during the implementation of increasingly complex combination therapies based on new chemical entities in patients of different age groups.
With the improvements of health and life expectancy anticipated with the implementation of efficacious CFTR modulators as standard of care, it will also be important to determine if these medicines can be administered safely durining pregnancy in increasingly healthy female patients with a desire to start their own family. In addition, establishment of safety of in utero exposure of the embryo and fetus would also be a crucial prerequisite for in utero treatment of patients that have been diagnosed by prenatal diagnostics to prevent damage of the pancreas and other gastro-intestinal disease manifestations as recently demonstrated for the CF ferret model459. Furthermore, it remains to be seen what impact CFTR modulators will have on the prevalence of common extrapulmonary disease manifestations including CFRD, liver fibrosis or osteoporosis, as well as emerging complications, such as increased risk of gastrointestinal cancers that has been associated with CFTR mutations269,461, or difficult to treat infections with NTM that have emerged in adult patients with CF with increased life expectancy462.
7). The cost and access to novel therapeutics
From a clinical perspective, it is understandable that healthcare professionals as well as CF patients and their families expect timely access to new therapies once they successfully pass all phases of their clinical trials. However, as healthcare budgets are limited, it is necessary for reasons of equity and efficiency that regulatory authorities and third party payers consider the impact on all patients of a decision to reimburse therapies for any given patient group. Third party payers must in other words compare alternative uses of scarce resources in terms of their opportunity cost - the benefits that could have been enjoyed had those resources been put to their next best use - as well as their benefits. In CF, the emergence of CFTR modulator therapy, currently extremely high cost therapies, has thrown into sharp focus the issues of equity, drug pricing and even how we fund research and development.
Ivacaftor with its approximate cost of 180,000 GBP per annum highlighted these issues in CF463 and became a precedent for other CF orphan drug newcomers464. Ivacaftor was a groundbreaking clinical research achievement and obtained rapid approvals by FDA and EMA regulatory agencies21. However, the staggering price of ivacaftor not only raised the question of its relative value for money, but re-ignited complex arguments about what constitutes “value for money”, how we incorporate equity into decision making and what are the longer terms implications for research and development of decisions about whether or not to reimburse new expensive therapies; issues health economists have debated for many years465–471 When conventional cost effectiveness analyses based on calculating incremental cost per quality-adjusted life years (QALY) gained – the incremental cost effectiveness ratio (ICER) - were applied to determine the value of the new treatment, the ICER varied considerably between 334,000 GBP and 1,270,000 GBP per QALY gained472. These figures crossed the usual benchmark for the maximum allowable cost to be paid (e.g., in England and Wales where National Health Systems set the conventional threshold around 20,000 to 30,000 GBP per year, although recently they have excluded ultra-rare disorders and life-limiting conditions from the requirement to fit this threshold). As a result, economic arguments were used in several European countries to delay or deny cover for ivacaftor for some of its age- and mutation-specific indications (e.g., Irish National Centre for Pharmacoeconomics did not recommend the reimbursement for CF patients aged 2 to 5 years473, Scottish Medicines Consortium did not recommend ivacaftor for use in patients with R117H mutation474). Ultimately, other arguments not entirely based on value-for-money methodology, but emphasising clinical effectiveness and unmet medical needs prevailed: as of end 2018 the drug has been accepted for reimbursement in United States, Canada, Australia, Brazil, England, Wales and Scotland, Ireland, Denmark, Sweden, Norway, Iceland, Germany, Austria, Czech Republic, Switzerland, Slovenia, France, Italy, Spain, Portugal, Belgium, Netherlands, Luxembourg and Greece.
Ivacaftor happened to be a classic example of the best possible therapeutic agent available, yet not accessible to suitable patients living outside the countries listed above where it was reimbursed (Figure 10 panels a-c). This inevitably means different standards of care will apply in regions where ivacaftor or similar therapies remain inaccessible compared those where it is.
Figure 10. CFTR Modulators – countries where approval and funded.
Panel A Countries with significant population of European Origin
Panel B Ivacaftor (inset Europe)
Panel C Lumacaftor/Ivacaftor (inset Europe)
Panel D Tezacaftor/Ivacaftor (inset Europe)
Phase III triple combination therapy studies (Vertex Pharmaceuticals Inc) have been completed but as yet approval applications have not been submitted to Regulatory Authorities.
This is seen in the even more complicated situation regarding reimbursement for another CFTR modulator drug that combines molecules of ivacaftor and lumacaftor. This drug, very similar in price to ivacaftor, also showed a clinical effect that reached statistical significance23, yet in a much more moderate way than ivacaftor and therefore raised concerns about its true long-term benefits. The high cost accompanied by debatable clinical effectiveness created a less favourable reimbursement context. So far, public funding or insurance coverage for lumacaftor/ivacaftor combination therapy has been secured in the United States, Canada, Australia, Scotland, Ireland, Denmark, Sweden, Norway, Iceland, Germany, Austria, Italy, Spain, Netherlands, Luxembourg and Greece, but not for instance in England or France (Figure 10 panels a-c).
The marketing experience with CFTR modulators indicates that price relative to the value placed on benefit of innovative orphan drugs is a key to the determination of whether or not the drug becomes accessible. Other things being equal, high prices certainly hamper, if not prevent completely, the adoption of new therapies by regulatory authorities and third party payers, unless compelling arguments can be presented to balance those related to cost per QALY gain (Panel 6)475.
Finding solutions to global access will be critical to ensure all people with treatable CFTR mutations can benefit from innovative and transformative medicines, including those in LMIC. This will reduce the likelihood of individuals facing the dire consequences of denied access and of socities facing “two-classes” of medicine for those who can and those who can’t afford access to novel therapies. More broadly, it may help avoid the unintended consequences of negative reimbursement decisions such as counterfeit drug production and distribution476, or of uncontrolled use by patients who are denied access to novel therapies in their own health care system.
The proportion of potentially eligible patients suitable for existing CFTR modulators varies and likely to be significantly lower than the >50% in some countries e.g. USA due to lower rates of the mutations suitable for modulators therapy (e.g. patients with the G551D mutation and those who have p.Phe508del/p.Phe508del). In many regions globally, it’s not possible to make such estimations due to the very limited CFTR variant analysis. Furthermore, diagnosis is often missed or delayed and the availability of conventional standards of care limited. There is an imperative to get these issues better coordinated in LMIC so accurate diagnosis can be made. Of course, on the other hand the options for CFTR modulators to manage other mutations (e.g. those with non- p.Phe508del minimal function mutations will increase the numbers of the CF population globally who will potentially benefit from combinations of CFTR modulators. The international approaches to ensure access of anti-retroviral agents for HIV in LMIC is laudable but perhaps less likely to gain the political traction due to the relatively small numbers of patients with CF compared with those with HIV disease477,478.
This section highlights issues related to novel therapeutics in CF. Future opportunities and challenges are highlighted in Panel 7.
SECTION 5 –. PATIENT EXPERIENCE, ENGAGEMENT AND INVOLVEMENT
Living with CF means living with a life-long, chronic illness. For some, relentless daily recurrent symptoms requiring treatment at home or in hospital and a reliance on lifelong center care dominates life481. For others there are fewer reminders of the disease with less symptom and treatment burden, and a limited reliance on healthcare systems. The challenge however, can be accepting or even believing that there is a need for CF therapies. Regardless of symptoms, all people with CF (and their families) live with the knowledge of the life-limiting nature of the disease.
Managing a life with CF has been associated with a significant psychological and practical burden. The literature describes elevated symptoms of anxiety and depression and impacts on quality of life482. Well-being fluctuates over time and is determined by the interaction between illness parameters, burden of treatment, availability of expertise and care, individual characteristics including resilience and positive coping, and available social support systems.
Support from and involvement with CF multidisciplinary teams and Patient Organisations can assist to mitigate the day to day burden of treatment and encourage participation in daily life activities.
1). Living With Cystic Fibrosis
a). The early years
NBS programmes can have specific challenges of a diagnosis including the acceptance of the disease when the infant appears completely well and the need to learn to maintain a treatment regimen. A diagnosis of CF can be overwhelming and not only impacts on the infant and parents, but also siblings and the extended family483. Coping with a life-limiting disease causes emotional turmoil in parents including anxiety, disbelief and concern484. Learning about CF and the requirements of treatment is an immense responsibility and accepting the diagnosis takes time. Families need expert information, education and support from the CF multidisciplinary team, family and friends. Within a short period of time, CF treatment must become integrated into normal family routine with demanding day to day adaptions impacting on family relationships and decision making, particularly around schooling, employment and social activities485.
b). Toddler and pre- school years
During the pre-school years children develop rapidly and these changes affect families living with CF486. For example, language acquisition gives children the opportunity to verbally demonstrate their feelings about their treatment demands, while increased mobility and independent play may challenge the routine of CF therapies. New cognitive and emotional skills require age appropriate information and support about CF, especially as children take CF to school and learn to communicate with friends about CF. During these years education and support are also essential as children slowly begin to take some responsibility and learn to understand and manage aspects of their treatment487.
c). Adolescence and early adulthood
Adolescence and early adulthood are the years in which a person searches for social, emotional, educational, and economic resources to maintain health and wellbeing throughout their life. The processes of identification and independence are central to psychosocial development however, living with CF can make this challenging. Being different from peers requires social and emotional adaptation488. Independence from parents is a crucial developmental task for young people with CF however, as well as these normal tasks, young people also need to start taking responsibility for and managing treatment.
During these years the impact of CF on social, romantic and intimate relationships grows, and education and career choices are often made. Unfortunately, CF often interferes with or at least influences such decision making due to clinical status, level of education or poor support. A well-established transition programme which provides information and counselling on these themes is crucial for present and future lives489.
d). Adulthood
Many of issues important in adolescence and early adulthood will continue during adult life, adding new themes including planning for a family and becoming parents, finding accommodation, career decisions and financial independence. Parenthood is as important to people with CF as it is to their peers, however becoming a parent often changes their perspective of CF and CF treatment490. Although most adults with CF were diagnosed in infancy or early childhood, for about 10% of the population the diagnosis comes much later in life491. For this group, the literature describes disbelief, anger or anxiety, but also relief of finally having a diagnosis. These (often) older people must learn to adapt to needs of CF therapy which will depend on the severity of symptoms. In addition, a late diagnosis will impact family members, employment, relationships, and future plans.
CF is changing, thanks to changes in clinical management, early and aggressive treatment, novel therapeutics and options around medication delivery systems. The development of new medicines and the potential they unlock brings hope across the ages. Older people with CF are recognising a significant slowing down of their symptoms, while parents anticipate that their children will delay experiencing or even avoid signs of CF.
CF may be relatively stable for many years however, there will inevitably be a deterioration in health leading to a change in treatment requirements. In the later stages as health declines, treatment demands will increase, lung transplant options will be discussed, end-of-life decisions will be discussed and documented, and individuals will have to cope with the unpredictable character of CF492,493.
2). Adherence, Burden of Treatment and Well-Being (Patients and Carers)
Whilst early studies suggested the mental health of people with CF was not different from the general population, a recent large international study reported a high prevalence of depression and anxiety people with CF494–496. This study included children and adults with CF (n=6088) and parents of children (n=4102) from nine countries (154 CF centres) in Europe and the USA. The key findings were two to three-fold increases in depression and anxiety when compared with community samples. As a consequence, the International Committee on Mental Health in CF was created to develop a consensus for screening and treatment of mental health in CF497. Annual mental health screening with reliable, valid measures allows early detection and a stepped care approach to intervention498. Research shows that adherence is related to knowledge and information, age, family and social support, treatment burden and illness severity499. Non-adherence is related to a worsening prognosis, increased health care needs, lack of support and work or school absence498. In CF, adherence is reported to be sub-optimal although there is no clear definition of optimal adherence. Adherence is difficult to measure, often overestimated by people with CF yet underestimated by health care professionals495,500. For many however, perfect adherence is an unhelpful ambition unless it results in a improvement in health (actual or perceived). Healthcare professionals must be aware that avoidance is a common coping mechanism and that distraction and escapism is understandable as treatment is a constant reminder of the disease501. Many individuals find that adherence to all prescribed treatments is not an option as it interferes with their aim to lead a normal life and some just misunderstand treatment recommendations502. People with CF describe time related barriers as one of the main reasons for not managing to keep to treatment requests (social/family life, work/education commitments and childcare).
For the adolescent with CF, emerging therapeutic independence can challenge the balancing of health maintenace and aiming for normalcy amonst their peers Poor adherence is a potential factor contributing to clinical deterioration as the adolescent approachs adulthood and adult care. Poorer adherence to prescribed treatments is likely to relate to challenges with time management, complexity of regimens, decreased parental oversight, individual concerns of necessity of treatments, social pressure and reluctance to disclose their condition, and depression503–507. Optimal approaches to support the adolescent is still not well established.
Adults with CF therefore often make day to day decisions about the level of treatment they can carry out508. Healthcare professionals should be aware of this decision-making process and work together with people with CF to achieve an optimal treatment regimen while maintaining an optimal life balance.
CF teams should be promoting good mental and physical health from childhood, including sleep hygiene, healthy eating, exercise and learning to balance CF with pleasurable activities509. Focusing on building coping strategies and resilience in young people with CF to reduce the risk of depression and anxiety will play a large part in paediatric and adult care of the future498. This shift from active management of deterioration in both physical and mental health, to the promotion of health and resilience has started in some areas however, this must be highlighted in future models of care. Promoting health and wellbeing is a challenge for society and specialist CF services need to engage with wider public health initiatives. Rather than attempting to design CF specific initiatives to encourage exercise and physical activity, good nutrition and managing mental health, CF teams should utilise the excellent work already being completed for the general population. Finally, in this technology driven age, utilising technology and apps to encourage self-management and control of health will also lead to patients feeling empowered and more in control of their disease.
As CFTR modulators become more readily available consideration of the impact of these therepies on adherence to current therapies which are part of the standard of care will be required. It is tempting to consider reduced the burden of treatments is the patient whose health has improved dramatically following the initiation of modulator therapy. It is important to recognise that all pivotal studies have included standard medications and whilst the medium-term impact of modulators appears to reduce rate of lung function decline – decline continues510. Pragmatic studies are required to assess the impact of CFTR modulators on long-term adherence to therapies in general and in turn, the impact on health over time.
3). Communication, Telehealth and Self Monitoring
a). Communicating with people with CF and their families
Communication about chronic illness and treatment expectation is demanding and important however, the heterogeneous character of the population makes communication even more challenging511,512. Children, their families and adults with CF deserve appropriate information and education about the disease, treatment, prognosis and new developments to be able to make choices in their lives513. Even when there are cognitive or emotional challenges, it is the task of healthcare professionals to provide information adapted to individual needs. Randomly providing information is irresponsible without assessing each person’s ability to process and cope with such information. The CF multidisciplinary team should never withhold any information because it is considered to be too difficult, demotivating or frightening. The ability to explain, listen, reflect and adapt is crucial and should be an integral part of any training.
The loss of patient contact with each other at lay conferences, camps. etc may also contribute isolation from “peers“ with similar lived experience supports the concept of remote contact (utilising social media e.g chat rooms, remote partcipation in conferences, etc).
Parents and carers, the growing child, adolescent and adults are exposed to the internet and social media where information, comment, opinion and advice are constantly available. Trust, honesty, openness and support from CF healthcare professionals can mitigate dubious or untrue messaging and support useful and helpful suggestions. However, navigating this constantly changing media is challenging and CF multidisciplinary teams need to stay up to date with the latest beliefs and opinions. More positively, the internet can be a useful medium for CF healthcare professionals to communicate sharing experiences and methods of providing care, including clinical pathways, protocols, guidelines and shared decision-making tools514–516.
b). Telehealth and self-monitoring
Telemedicine is a remote, patient-centred, clinical service including the use of video and audio connections, telemonitoring and mobile applications relating to health and/or well-being and connected health-related wearable devices517. A factor which has supported the move to remote assessment of patients has been the need to progressively limit contact between patients due to increasing eveidence of cross-infection with patients potentially sharing infections518,519. In CF, studies of telemedicine have predominantly consisted of small studies including patients with limited disease variability, short feasibility trials using a wide age range of patients, and pilot studies with often poorly defined outcome measures520. Nevertheless, the availability and use of new technologies for people with chronic illness is increasing and has led to an immense growth of web-based and mobile information and communication technologies521.
The use of these developing technologies is appealing and links into the social world of today’s patient population. The notion of telemedicine is familiar to many with remote healthcare services using telecommunication technologies (spirometers, pulse oximeters, activity tracking, text reminders, e-diaries and smartphone applications) to identify intervention points, potentially preventing the need for aggressive treatment, including hospitalisation430,522. There is also interest in and development of physical activity or exercise programmes and pharmacy video training523.
However, several problems remain. Cost and availability will be limiting factors as the move towards telehealth goes forward. Many people do not have regular access to the internet, mobile phones or personal computers, although this is changing rapidly with the advent of smartphone devices524. In addition, little is known about the cost benefit of using new technologies and little is known about costs or the problem of invoicing for the use of e-healthcare. Common technological issues need to be addressed such as the time lag in audio-feed and disrupted connections. There is also a need for clear criteria (e.g. what constitutes an exacerbation) in order to use new technologies more reliably and further insight is needed into the daily variation in CF symptoms. In addition, although telemonitoring appears feasible and readily accepted, to date few studies have demonstrated a significant impact on health outcomes such as exacerbation rates, lung function, HRQoL and health care utilisation525. To establish and prove the long-term effectiveness and cost-effectiveness, more well-designed clinical studies, including psychological and behavioural issues, are needed526.
For people with CF and their families, the main advantage of telehealth is cost reduction (travel, days off work/education etc), convenience and efficiency (e.g. electronic documents, appointments via e-mail, psychological support via e-mail, video conferencing). The literature to date does not provide any evidence of negative effects for people with CF, however this does not mean they don’t exist. Potentially, self-monitoring can cause anxiety due to the lack of immediate feedback or obsessive over-monitoring (e.g. lung function, oxygen saturation). Conversely, several studies have shown low adherence to self-monitoring with an association with increased burden527,528.
Ideally, telemedicine should not be an all or nothing option, more likely into the future it will be used periodically throughout the patient journey, particularly for those living in regional and remote environments and during changes such as trying a new therapy to monitor success, or maybe at times of clinical deterioration and frequent exacerbations. In the near future most people will engage with new technologies and home monitoring as they are already using e-mail, mobile phones, appliances, websites etc on a day-to-day basis. Consideration needs to be given to e-health research and in setting up new systems and people with CF need to be involved in the development of such programmes. Cost–benefit issues need to be addressed and patient reported outcomes are essential. However, the most important aim of telemedicine is to monitor and improve clinical outcome for individuals with CF.
As a step further studies are underway on the feasibility and utility of more continuous remote monitoring of patients. Examples include lung function performed on a regular basis and uploaded via bluetooth devices into a shared system from which the clinical team can access it; machine learning approaches could ultimately allow ‘flagging’ of a result outside the subject’s normal range for rapid review. Point of care technologies to diagnose airway infection are being explored and outside the CF field, remote monitoring of diabetic control is already becoming a reality. Such approaches will require rigorous assessment as adherence to monitoring can wane with time and provides a constant reminder of CF which is not always embraced by the patient and may have additional adverse impacts on HRQoL529,530. It is quite likely they will enhance convenience, but there may not be the expected cost savings. An approach of this nature could be particularly attractive to populations dispersed over a very large geography or for whom transport links are limited.
4). The Role of Lay Organisations and Foundations
Patient Organisations (PO) support and advocate for the needs of people with CF and their families with the aim of improving quality and length of life. Mission statements include funding research, collecting and publishing information, providing emotional and welfare support and lobbying for access to high quality care. The role of POs in lobbying activities is an area of tangible success with recent focus on new and frequently high-cost therapies becoming licensed for use in healthcare systems where affordability is a point of contention. A list of Patinet Organisations is provided in Panel 8.
Patient Organisations can empower and unite the community directly affected by CF under a harmonised campaigning banner, maximising a professional approach and access to expertise. They can utilise their reach within the press, social media, and governments, to penetrate decision making processes and influencers in ways that would not be achievable for individuals or unofficial groups. Patient Organisations also have more freedom to respond with ingenuity and emotion to non-clinical issues that strike a chord with decision-makers and are not beholden to impartiality requirements. That is not to say that this role is straightforward; balancing influencing tactics and high-level strategies against perceived responsiveness and representativeness is a challenge. This is not without its challenges especially in advocating for funding of high-cost CFTR modulator therpes over the past decade531.
Patient Organisations have both opportunity and obligation to galvanise key stakeholders to anticipate and accommodate the future needs of people with CF. Existing outside of, and less constrained by systems, POs can evolve with agility ensuring that the needs of people with CF and their families continue to be met.
a). Patient engagement through Registries
As outlined above patient Registries have tracked the evolution of CF since the 1960s. They have demonstrated the real-world ups and downs of infection rates, nutritional status, access to and outcomes of lung transplantation and survival across the globe. As we consider the future of CF, observations about the course of the disease over the last 50 years are a vital tool to drive the adaptations in care needed to optimise life chances for people with CF around the world.
A recent review describes the utilisation of Registries to advance our understanding of the CF population, including an international overview of Registries80. As the global effort to understand and advance outcomes for people with CF continues, we will increasingly see support for Registry establishment and evolution outside these geographical areas – where genotypic characteristics and prevalence may differ from our current understanding. Documentation will open doors for those populations allowing them to benefit from benchmarking, safety monitoring and clinical trial participation.
Compared to more sophisticated and well-established Registries, the impact of collecting even a minimal dataset is easy to underestimate. The capability of the CF Community to report CF population numbers in different areas around the world sets us apart as a data-rich exemplar for other diseases. A good example of this is the European CF Registry (ECFSPR) which has supported countries with lower incomes to estimate their CF population. From 2008 to 2017, the registry has increased patient coverage by over 150%, from 18,999 patients in 19 countries, to >48,000 patients in 35 countries.
Supporting research is another important role of CF Registries, with recent examples including identifying trends in NTM transmission and cross-country comparisons.
This is however reliant on one key characteristic - high quality, systematically reported longitudinal data with minimal loss to follow up underpinned by transparency. Data request processes mean that pseudonymised, anonymised or aggregate data can be made available for epidemiological research. Sharing data with the clinical and research community is a key way to maximise the impact of Registries. Providing Registry data encourages researchers to cultivate an interest in CF and sharing Registry information leverages the skills of people outside the nuclear Registry teams532. Use of data by researchers also establishes a useful feedback loop to improve the quality and format of Registry data over time and instills within the research community buy-in to Registries and their impact.
Moving forward, patient groups are asking to be part of the Registry process and more personally involved in data entry. Registry teams in a number of countries are now exploring, in partnership with patient representatives, the potential for individuals to contribute either aspects of their own clinical data or data from Patient Reported Outcome Measures using portals designed for data entry. Work is also being done around the way that Registry data can be fed back to people with CF using patient accessed dashboards533,534. When taking this step, registries need to ensure they are acting on behalf of pateints and ask them which measures are important and how data should be be presented, rather than make assumptions based on reports designed for clinical teams.
This section highlights issues related to patient experience, engagement and involvement for people with CF. Future opportunities and challenges are highlighted in Panel 9.
CONCLUSION
Over the last decades we have seen major advances in how to diagnose and treat people with CF early and efficiently. These include, but are not limited to, a more widespread adoption of newborn screening, an increased understanding of the importance of implementing interdisciplinary care teams and in defining better ways of treating its main manifestations of the disease. All of these aspects have led to a changes in the population of people with CF and those with CFTR dysfunction.
With a changing spectrum of CF new challenges have emerged. Many patients have a milder phenotype and striking the right balance between implementing necessary therapies and treatment burden has become a growing priority in clinical care. Advances in genetic technologies facilitates the annotation of variants as potentially disease-causing with their number rapidly increasing over time, but genetic testing also identifies individuals with limited phenotypic manifestations for whom the risk of disease progression and thus the benefit of making the diagnosis is not yet well defined. Despite genome sequencing technologies now becoming more widely available, functional tests defining the extent of CFTR dysfunction and critical assessment of the clinical disease manifestation continue to be essential and their role requires further assessment. How we utilise these different methodologies into the future is not only relevant to countries where CF has traditionally been diagnosed, but also for LMIC where access to facilities offering functional testing such as the sweat testing is limited.
New drugs targeting the basic defect of CF have provided hope for people with CF and the progress has been substantial over the last decade. However, these drugs come at a high cost which is mainly driven by the costs associated with drug development rather than drug production costs. It will be interesting to see, how “patient” people with CF will be to wait for health care systems to fund the costs of these new therapies with availability of current compounds already varying widely – even between Western countries. New forms of partnerships between pharmaceutical companies and reimbursement bodies may be needed and innovative solutions may be required for low and middle income countries to ensure that patients throughout the world have access to effective therapies and that the gap between “have and have nots” will not wider even further.
With continuous progress the shift from CF as mainly a paediatric disease to one where many people with CF will go through early adulthood maintaining a good level of health albeit at the cost of requiring continuous ongoing and often increasingly intense treatments. With early mortality becoming less common, care of the ageing person with CF will not be a rare oddity anymore and this may require a new generation to health care providers with specific training with broad skills to be able to manage the emerging complications of CF. It may also require us to reconsider which components of the care will need to be conducted directly by the multidisciplinary team at CF centres and which can be undertaken remotely utilising new technologies to monitor patients’ wellbeing
These are exciting times for CF, a disease that is often considered a model of care for other chronic disease, but also for the progress of targeted therapies for genetic disorders. With these advances come new opportunities challenges and the community should now prepare for these so we can continue to provide exceptional care to individuals with this multi-facet disease in the future.
Panel 2 The Changing Epidemiology of Cystic Fibrosis – the Future.
Challenges
To appreciate the socioeconomic impact of the changing demography and epidemiology of CF worldwide.
To standardise diagnostic testing for CF globally considering accuracy and accessibilty.
To improve specificity of NBS programmes and limit false positive and inconclusive case recalls while maintaining the high sensitivity of testing.
To adjust diagnostic algorithms for CF, currently optimised for European-derived populations, to support accurate diagnosis of the “new” populations with different CF phenotypes.
To improve knowledge on the correlation between CFTR genotype and clinical phenotype for rare CFTR mutations.
Opportunities
To support a more balanced understanding on diagnosis and clinical course of cystic fibrosis in European and non-European populations, globally.
To provide equal diagnostic opportunity to non-European populations and assure equal access to early and stratified care globally.
To initiate effective therapies from the first weeks’ of life by early diagnosis through NBS.
To better understand the long-term natural history of individuals with inconclusive diagnosis of CF.
To improve general awareness of CF in non-European populations, overcome its misdiagnosis and under-diagnosis to avoid delays in the diagnosis.
Panel 3 Clinical Care and it’s Delivery – the Future.
Challenges
To clarify the role of conventional evidenced-based therapies in CF, following the implementation of effective CFTR modulator therapies.
To develop accurate biomarkers suitable for the assessment of infection and inflammatory status in children and healthier adults with CF who rarely produce sputum.
Funding and the provision of services for screening and treatment of CF complications occuring through improved paediatric outcomes and the increasing adult CF population.
To develop therapeutic approaches to reduce the risk of metabolic complications and obesity the CF population.
Training and sustaining the CF workforce for the expanding adult patient population especially in regions where the adult population is predicted to grow most rapidly.
Opportunities
To develop models of care which limit the burden of care and of disease monitoring.
To develop novel infection diagnostics to improve and support earlier diagnosis and clinical outcomes and in parallel limit the emergence of antimicrobial resistance.
To enhance the experience and expertise of the CF multidisciplinary team and the recruitment of new specialities to support emerging long-term complications of CF in the older person with CF.
To reduce the burden of maintenance therapies such as antibiotics and mucolytics and the need to treat pulmonary exacerbations by the early introduction of highly effective CFTR modulators.
To determine the best approach to maintain health to delay lung transplantation, yet protecting end-organs so that when transplantation is required these organs are not adversely impacted.
Panel 4 Description of health care systems, funding models and resources for cystic fibrosis.
These countries were selected as examples of LMIC from Africa, South America, Eurasia and Eastern Europe to highlight the models of CF care and health system structures.
Panel 4a South Africa
There are approximately 500 people with CF under medical care, a conservative estimate as significant numbers of infants and young children succumb to CF-related illness before diagnosis.
The South African CF patient registry was launched in 2018. However, there is no newborn screening. However, South Africa is the only country in sub-Saharan Africa with the capacity to diagnose and treat people with CF, and undertake CF-related research.
CF care is fragmented and separated along socioeconomic lines between a well-resourced private health sector and an under-resourced public health sector.
Private sector:
Individual practitioners and health professionals with expertise in CF. Provision of multidisciplinary care in private clinics is difficult. Access to care and medications is determined by levels of health insurance coverage. CF is not recognised as a prescribed minimum benefit illness; therefore, health insurance coverage is inadequate and variable, without regulation.
Public sector:
High-quality CF care and laboratory services are concentrated at university CF clinics and hospitals, where coordinated and multidisciplinary care model is adopted. The public health sector is under resource constraints and serves predominantly those who cannot afford private health insurance. With the overwhelming public health priorities (e.g. HIV and tuberculosis), rare diseases like CF receive limited attention. Poverty, lack of awareness about CF, and poor infection control policies are additional barriers to optimal CF care.

Access to expensive medications such as inhaled tobramycin and dornase alpha is very restricted. Pancreatic enzymes, azithromycin, and suitable antibiotics are widely and available without charge.
Laboratory services are maintained with high standards in line with international norms.
The South African CF Association - www.sacfa.org.za – is an active and well-organised lay CF community that aims to create CF awareness, support CF scientific meetings, and provide food and equipment such as nebulisers to CF families with limited financial resources and no health insurance.
Panel 4b Argentina
There are approximately 1,300 people with CF, a conservative number due to under reporting to the National Registry.
A patient registry was established in 2011, and falls under the responsibility of The National Institute of Epidemiology and the Argentinian Pediatric Society. Data entry is voluntary, with significant under-reporting by private practitioners.
Newborn screening is available. Method used is IRT/IRT in most of the country (city of Buenos Aires uses IRT/PAP). A panel of 50 mutations is used with a sensitivity of around 70%. Expanded genetic sequencing testing is not widely available.
CF care is fragmented and separated along socioeconomic lines. There is no specific recognition of the centers by the Ministry of Health.
Private sector:
Care is provided by individual practitioners and health professionals with expertise in CF. Most patients are seen by pulmonologists or gastroenterologists and do not receive multidisciplinary care.
Public sector:
Care is provided at specialised CF centres. Most of these centres have reasonable resources, including multidisciplinary team care and laboratory services. They are part of large hospitals and universities. There are at least nine centres with more than 50 patients, and three with >150 patients. A few other centres report caring for 20+ patients, but their resources are limited.
Around 20% of the patients are older than 18 years, and are cared for one of the three adult CF centres.
All medications are available, including ivacaftor and ivacaftor/lumacaftor, produced by Argentinian manufacturers. There is no national coverage for all patients. Cost of medications and medical care coverage varies depending of payers (government, employer-supported, or fully private), and provincial governmental resources. Advocacy organizations: Asociación Argentina de Lucha contra la Enfermedad Fibroquística del Páncreas (FIPAN); fipan.org.ar.
Panel 4c Turkey
There are approximately 3,000 people with CF. CF Centres are supported by the Ministry of Health (MoH). There are two major CF Centres, in Ankara (Hacettepe University CF Centre) which follows ~600 patients and in Istanbul (Marmara University CF Cente) which follows ~300 patients. Both centres have reasonable infrastructure support, including multidisciplinary teams and laboratory support (sweat testing, microbiology, and genetic testing). There are 25 other smaller CF centres across the country. Smaller centres in consult with the centers in Ankara and Istanbul periodically. Most patients in Istanbul that are followed in smaller centres are evaluated once a year by the multidisciplinary team at Marmara CF centre.
CF newborn screening programme using a immunoreactive trypsinogen – IRT/IRT/Sweat Test/DNA sequencing protocol is available. Sweat testing labs are certified by the government and the method has been compared to the standard one (1). CF centers are either part of the European CF Registry (Marmara CF Cenrer) or have developed their own (Hacettepe CF Centre). Most CF medications are available, covered by the government, and are free for patients. There is a strong CF patient/family organization (MECFA; mecfa.org) that is connected to the European CF patient/family organizations. This group works closely with the government and is very active in lobbying, advocacy and education in collaboration with the CF centres.
Panel 4d Serbia
There are approximately 210 reported patients with CF, 70% of them under age 18 (median age = 13 years). The country has one CF centre, located in Belgrade (Mother and Child Health Care Institute “Dr. Vukan Cupic”), but the European CF Society standards of care including a multidisciplinary team approach for CF care are not fullfilled. There is no dedicated adult CF centre in Serbia.
NBS screening is available in some parts of the country, with plans for a national programme to begin in the near future. The mean age at diagnosis is currently 3 years, (~50% of patients diagnosed <8 months of age). Although genotyping is performed on 98% of patients, 17.5% have at least one mutation unknown.
Availability of CF medications is limited. The only CF drugs available to patients are Pulmozyme and inhaled tobramycin, but their coverage for those older than 18 years of age is problematic. CFTR modulator drugs are not available. Medical equipment, including nebulisers and compressors, is not covered. Similarly, vitamins and nutritional supplements are neither available nor covered. CF care in the country doesn’t include psychosocial care or physical therapy/respiratory therapy. Transplantation services are not offered.
An active patient advocacy organization, the Serbian Cystic Fibrosis Association (www.cfsrbija.rs), works to foster implementation of European standards of CF care, facilitate CF education and research, create awareness of the disease, and provide legal, material, and other types of support to CF patients and families.
Panel 5 CF Care In Developing Nations – the Future.
Challenges
To improve general awareness of CF and its diagnosis in LMIC countries
Addressing the global disparities in access to healthcare (both established and emerging).
The delivery of basic accurate and reliable CF diagnostic testing platforms including sweat testing and appropriate CFTR gene panels.
More prevalent health priorities in LIMC limit the practical application of CF-specific resources to enhance clinical care.
Challenges in delivering state of the art education for families affected by CF where the health care community is restricted.
The ability to support the initial training and then ongoing mentoring of enthusiastic clinicians from LMIC to participate in international conferences is limited by support and finances.
Opportunities
Improved understanding of the genetic mix of CFTR variants in non-European populations which support the develop of better and more targeted diagnostic panels to allow earlier diagnosis of CF in LMIC.
To understand variant phenotypes of CF in non-European populations which may improve our current understanding of the disease and thus improve its diagnosis and targeted therapies.
Enhanced access to CF therapies in a sustainable and affordable method working with governments and industry.
Partnering of CF health care providers in LMIC settings with those with clinical, educational and research expertise for settings with well-established and highly functional multidisciplinary teams.
To create registries for countries where CF is increasingly being recognised and ensure that they are harmonised with longstanding CF registries to support understanding of health care outcomes and as a mode of advocacy for enhanced resourcing to allow multidisciplinary team care in LMIC.
Develop partnerships with established Patient Organisations and fledgling parallel organisations in LMIC.
Panel 6 Proposed method for future Health Technology Assessments (HTA) that involve CF orphan therapies.
A proposed method for future Health Technology Assessments (HTA) that involve CF orphan therapies could be considered more appropriate, where they include the following inputs:
Robust evidence for clinical effectiveness measured on a long-term basis that include disease specific measures such as annual rate of pulmonary exacerbations or time to first exacerbation as well as measures of mortality and health-related quality of life. The parameters should ideally be monitored over the longer term, for instance by means of post authorisation efficacy studies.
As drug prices may be set before the availability of complete data on effectiveness and on a basis that can therefore lack transparency479, there should be greater transparency as to how prices are determined and the opportunity to revise assessments in light of new information. For example, what savings are associated with use of the new drug in terms of other procedures that would otherwise be consumed; in markets where there exist more than one payer, what discounts, rebates and co-pays have been agreed so that users can more easily compare actual prices; details of how long the drug will remain on patent and; what is the projected rate of return based on current price and actual expenditures on research and development for the drug. Such transparency would not only provide a better understanding of the total drug costs, but more importantly provide solid evidence for the comparison of the value of the drug’s benefits relative to its costs, its accuracy and how long before it should be re-appraised when the drug comes off patent for example. While such information may not address issues such as potential spill-over effects from innovation in one area for other areas, it will facilitate price adjustments, corrections or discounts upon fair negotiations between healthcare policy stakeholders and pharma companies, on an ongoing basis as information changes.
An integral part of the HTA could be the budget impact analysis, with the aim to quantify the exposure of a given healthcare system to adoption of a new therapy. Studies which took into account disease prevalence, patent exclusivity (of 10 years) and the fact that only a limited number of new drugs enter the market have shown that annual budget impact of drugs for orphan diseases (one of them being lumacaftor/ivacaftor) and ultra-orphan diseases (ivacaftor) can in fact be modest with increases by 5 to 10% per year only480 480
Panel 7 Novel Therapeutics – the Future.
Challenges
To develop medicines that restore CFTR function in all people with CF irrespective of CFTR genotype.
To increase efficacy of current CFTR-directed therapeutics with the ultimate goal to achieve full restoration of CFTR function for optimal clinical benefits.
To develop preventive strategies for the use of CFTR-directed therapeutics in infants and young children with CF that may prevent or delay irreversible damage of the lungs and other affected organs.
To determine to what extent current symptomatic therapies can be discontinued in patients treated with medicines that restore CFTR function.
To provide global access to these transformative therapies to all people with CF.
Opportunities
To treat people with CF with medicines that target the basic molecular defect of a growing spectrum of CFTR mutations in all affected organ systems.
To leverage early diagnosis by CF NBS for early treatment with CFTR-directed therapeutics that may have the greatest long-term benefits.
To slow progression of CF multiorgan disease and thereby reduce disease burden and improve quality of life by initiating these transformative therapies in people with CF.
To transform CF from a fatal disease to a treatable chronic disease through highly efficacious CFTR-directed medicines and specialized multidisciplinary care.
To provide a model for successful development of transformative medicines for other severe genetic diseases.
Panel 8 Professional and Patient Organisations.
Europe
https://www.ecfs.eu/ (European Cystic Fibrosis Society)
https://www.cf-europe.eu/ (CF Europe)
USA
Middle East
Australia
Argentina*
Austria*
Belgium*
Brazil
Bulgaria
Canada
https://www.cysticfibrosis.ca/ (English or French)
Czech Republic
Denmark*
Ecuador
Estonia*
Finland
France
https://www.vaincrelamuco.org/ English option
Germany
https://www.muko.info/ English option
Greece
Iran*
Italy*
Ireland
Israel
https://cff.org.il/ English option
Lithuania*
Luxemburg*
Macedonia
http://www.cf.mk/ English option
Moldova*
Netherlands
New Zealand
Norway
https://www.cfnorge.no/ English option
Poland*
Portugal*
Romania*
Russia*
Slovakia*
Slovenia
https://www.drustvocf.si/ English option
South Africa
Spain*
Sweden
https://www.rfcf.se/ English option
Switzerland*
http://www.cfch.ch/ Italian, French and German options
Turkey
UK
https://www.cysticfibrosis.org.uk/
Where countries are not listed, points of contact can be accessed through the Cystic Fibrosis Worldwide website https://www.cfww.org/global-associations
Panel 9 Patient Experience, Engagement and Involvement – the Future.
Challenges
Growing population, not only of adults but children in some developing countries where currently they are not being diagnosed or effectively treated.
Develop individual patient contribution and access to national CF registries.
Personalised models of care and its delivery versus a ‘one size fits all’ approach in the setting of enhanced longevity.
Opportunities
Utilize technologies allowing for self-monitoring and shared decision making between patient and health care team that is evidence-based.
Develop pathways for patients and families to actively participate in shaping models of clinical care and the research priorities.
Enhance communication between patients and their families about therapeutic advances.
ACKNOWLEDGEMENTS
The authors acknowledge the expertise and support of Hajany Jeyakumar (Hospital for Sick Kids,Toronto), Chris Parker (Medical Librarian, TPCH) (reference management), Mary Phillips and Dr Luke Knibbs (Illustrations and Map Development), Dr Hector Hernan Ruiz (UMAE Pediatric Hospital of the Medical Center of the West, Mexico), Emily Bateman (European Cystic Fibrosis Society) and Dr Bruce Marshall and Dr Alexander Elbert (CF Foundation (USA)).
FUNDING SUPPORT
SCB has been supported by Queensland Health (Health Research Fellowship) and Cystic Fibrosis Foundation. MAM has been supported by grants from the German Ministry for Education and Research (82DZL004A1), the German Research Foundation (SFB-TR84TP B08), and the Einstein Foundation Berlin (EP-2017-393). HG has been supported by CF Foundation (GUTIER18QI1). MM has been supported by IP00064203/6003; NF-CZ11-PDP-3-003-2014, CZ.02.1.01/0.0/0.0/16_013/0001634, and LM2015091. PMF has been supported by Cystic Fibrosis Foundation (LAI17A0), NIH/NIDDK (R01 DK109692) and The Legacy of Angels Foundation (133-AAC7732). SMR has been supported by the NIH (R35HL135816 and 2P30DK072482) and the CF Foundation (RDP Award). FR has been supported by Genome Canada, Cystic Fibrosis Canada, Cystic Fibrosis Foundation.
DECLARATIONS
No funding was provided specifically for this commission paper.
SCB has received institutional support from Vertex Pharmaceuticals, Galapagos NV and Abbvie and travel support to attend investigator meetings from Vertex Pharmaceuticals, Galapagos NV, Flatley Discovery Lab and Abbvie. MAM has received institutional support from Vertex Pharmaceuticals Limited, and personal fees from Bayer, Boehringer Ingelheim, Polyphor, Arrowhead Pharmaceuticals, ProQR Therapeutics, Spyryx Biosciences, Vertex Pharmaceuticals Limited, Santhera, Galapagos NV, Sterna Biologicals, Enterprise Therapeutics and Celtaxys. MM has received institutional support from Vertex Pharmaceuticals for CFTR genotyping of in non-European cystic fibrosis populations. JCD has received institutional support from Abbvie, AlgiPharma AS, Bayer AG, Boehringer Ingelheim, Chiesi, Eloxx, Enterprise Therapeutics, Flatley Discovery Lab, Galapagos NV, GSK, ImevaX GmbH, Ionis, Nivalis Therapeutics Inc, Novartis, Proteostasis, PTC Therapeutics, Pulmocide, Roche; personal fees from Proteostasis, Teva Pharmaceuticals, Vertex and grants from CF Trust. PRB has received grants from Boeringher Ingelheim and personal fees from Astra-Zeneca, Chiesi CZ, GSK, Novartis, Pfizer, Teva Pharmaceutical Industries, Vertex Pharmaceuticals Limited and Zambon. ET has received funding from the Cystic Fibrosis Canada and personal fees from Vertex Pharmaceuticals Limited, Proteostasis Therapeutics Inc and Raptor Pharmaceuticals Inc. CAB has received grant support from the National Research Council of New Zealand, National Health and Medical Research Council of Australia, Auckland Medical Research Fund. CC has received institutional support Vertex Pharmaceuticals Inc, Teva Pharmaceuticals and Actelion Pharmaceuticals and personal fees from Vertex Pharmaceuticals Inc. RC has received institutional support from Vertex Pharmaceuticals Limited, Chiesi, Teva Pharmaceutical Industries and Pharmaxis. IF has received personal and institutional fees from Vertex Pharmaceuticals Limited and Proteostasis Therapeutics and personal fees from Boehringer Ingelheim. CHG has received grant support from The Cystic Fibrosis Foundation US, European Commission, NIH (NHLBI) NIH (NIDDK and NCRR) and the FDA, and personal fees from Gilead Sciences, Novartis and institutional support from Boehringer Ingelheim. PD has received personal fees from Vertex Pharmaceuticals Limited, Galapagos NV, Flatley Discovery Lab, Proteostasis Therapeutics, Actelion Pharmaceuticals, Chiesi CZ, Corbus Pharmaceuticals. NH has received personal fees from Calithera. LN has received institutional fees for site particpataion in clinical trials from Vertex Pharmaceuticals Limited. EM has received grant support from Vertex Pharmaceuticals Limited and personal fees from Vertex Pharmaceuticals Limited, Proteostasis and Novartis. KR has received grant support from the Cystic Fibrosis Foundation US. SR has received grant support from Bayer Pharmaceuticals, Forest Research Institute, Astra Zeneca, N30/Nivalis, Novartis, Galapagos NV, AbbVie, Proteostasis, Eloxx, Celtaxsys, PTC Therapeutics and Vertex Pharmaceuticals Inc. Personal fees received from Celtaxsys, Vertex Pharmaceuticals Inc, Bayer, Novartis and Renovion. AS has received grant support from Cystic Fibrosis Foundation US and personal fees from Cystic Fibrosis Canada and Vertex Pharmaceuticals Inc. FR has received grant support from Vertex Pharmaceuticals Inc. Personal feeds received from Novartis, Bayer, Roche, and Genetech.
HG, SM, ClC, CaC, FC, SC, IE, PMF, AG, TH, CO, NK, EK, JM, SN, GO, UP, KWS, SS, MZ all make no declarations.
Footnotes
The views expressed in this article are the personal views of the author(s) and may not be understood or quoted as being made on behalf of or reflecting the position of the regulatory agency/agencies or organisations with which the author(s) is/are employed/affiliated.
REFERENCES
- 1.Andersen DH, Hodges RG. Celiac syndrome; genetics of cystic fibrosis of the pancreas, with a consideration of etiology. Am J Dis Child 1946; 72: 62–80. [DOI] [PubMed] [Google Scholar]
- 2.Di Sant’Agnese PA, Darling RC, Perera GA, Shea E. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease. Pediatrics 1953; 12(5): 549–63. [PubMed] [Google Scholar]
- 3.Gibson LE, Cooke RE. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics 1959; 23(3): 545–9. [PubMed] [Google Scholar]
- 4.Quinton PM. Chloride impermeability in cystic fibrosis. Nature 1983; 301(5899): 421–2. [DOI] [PubMed] [Google Scholar]
- 5.Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245(4922): 1073–80. [DOI] [PubMed] [Google Scholar]
- 6.Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245(4922): 1066–73. [DOI] [PubMed] [Google Scholar]
- 7.Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245(4922): 1059–65. [DOI] [PubMed] [Google Scholar]
- 8.Gentzsch M, Mall MA. Ion Channel Modulators in Cystic Fibrosis. Chest 2018; 154(2): 383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. http://www.genet.sickkids.on.ca/
- 10.http://www.cftr2.org/.
- 11.Boyle MP, De Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med 2013; 1(2): 158–63. [DOI] [PubMed] [Google Scholar]
- 12.de Souza DAS, Faucz FR, Pereira-Ferrari L, Sotomaior VS, Raskin S. Congenital bilateral absence of the vas deferens as an atypical form of cystic fibrosis: reproductive implications and genetic counseling. Andrology 2018; 6(1): 127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cutting GR. Modifier genes in Mendelian disorders: the example of cystic fibrosis. Ann N Y Acad Sci 2010; 1214: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ratjen F, Bell SC, Rowe SM, Goss CH, Quittner AL, Bush A. Cystic fibrosis. Nat Rev Dis Primers 2015; 1: 15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elborn JS. Cystic fibrosis. Lancet 2016; 388(10059): 2519–31. [DOI] [PubMed] [Google Scholar]
- 16.Stephenson AL, Stanojevic S, Sykes J, Burgel PR. The changing epidemiology and demography of cystic fibrosis. Presse Med 2017; 46(6 Pt 2): e87–e95. [DOI] [PubMed] [Google Scholar]
- 17.Burgel PR, Bellis G, Olesen HV, et al. Future trends in cystic fibrosis demography in 34 European countries. Eur Respir J 2015; 46(1): 133–41. [DOI] [PubMed] [Google Scholar]
- 18.Plant BJ, Goss CH, Plant WD, Bell SC. Management of comorbidities in older patients with cystic fibrosis. Lancet Respir Med 2013; 1(2): 164–74. [DOI] [PubMed] [Google Scholar]
- 19.Bell SC, De Boeck K, Amaral MD. New pharmacological approaches for cystic fibrosis: promises, progress, pitfalls. Pharmacol Ther 2015; 145: 19–34. [DOI] [PubMed] [Google Scholar]
- 20.De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med 2016; 4(8): 662–74. [DOI] [PubMed] [Google Scholar]
- 21.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365(18): 1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med 2013; 187(11): 1219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med 2015; 373(3): 220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ratjen F, Hug C, Marigowda G, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med 2017; 5(7): 557–67. [DOI] [PubMed] [Google Scholar]
- 25.Simmonds NJ. Is it cystic fibrosis? The challenges of diagnosing cystic fibrosis. Paediatr Respir Rev 2019. [DOI] [PubMed] [Google Scholar]
- 26.Kharrazi M, Yang J, Bishop T, et al. Newborn Screening for Cystic Fibrosis in California. Pediatrics 2015; 136(6): 1062–72. [DOI] [PubMed] [Google Scholar]
- 27.Mishra A, Greaves R, Massie J. The relevance of sweat testing for the diagnosis of cystic fibrosis in the genomic era. Clin Biochem Rev 2005; 26(4): 135–53. [PMC free article] [PubMed] [Google Scholar]
- 28.Kabra SK, Kabra M, Shastri S, Lodha R. Diagnosing and managing cystic fibrosis in the developing world. Paediatr Respir Rev 2006; 7 Suppl 1: S147–50. [DOI] [PubMed] [Google Scholar]
- 29.Ferec C, Cutting GR. Assessing the Disease-Liability of Mutations in CFTR. Cold Spring Harb Perspect Med 2012; 2(12): a009480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marson FAL, Bertuzzo CS, Ribeiro JD. Classification of CFTR mutation classes. Lancet Respir Med 2016; 4(8): e37–e8. [DOI] [PubMed] [Google Scholar]
- 31.Amaral MD, Balch WE. Hallmarks of therapeutic management of the cystic fibrosis functional landscape. J Cyst Fibros 2015; 14(6): 687–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martiniano SL, Sagel SD, Zemanick ET. Cystic fibrosis: a model system for precision medicine. Curr Opin Pediatr 2016; 28(3): 312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson JMG JG. Principles and practice of screening for disease. Geneva: WHO. http://wwwwhoint/bulletin/volumes/86/4/07-050112BPpdf 1968. [Google Scholar]
- 34.Farrell PM, Kosorok MR, Rock MJ, et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics 2001; 107(1): 1–13. [DOI] [PubMed] [Google Scholar]
- 35.Gonska T, Ratjen F. Newborn screening for cystic fibrosis. Expert Rev Respir Med 2015; 9(5): 619–31. [DOI] [PubMed] [Google Scholar]
- 36.Coffey MJ, Whitaker V, Gentin N, et al. Differences in Outcomes between Early and Late Diagnosis of Cystic Fibrosis in the Newborn Screening Era. J Pediatr 2017; 181: 137–45 e1. [DOI] [PubMed] [Google Scholar]
- 37.Tridello G, Castellani C, Meneghelli I, Tamanini A, Assael BM. Early diagnosis from newborn screening maximises survival in severe cystic fibrosis. ERJ Open Res 2018; 4(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayeems RZ, Miller FA, Barg CJ, et al. Psychosocial Response to Uncertain Newborn Screening Results for Cystic Fibrosis. J Pediatr 2017; 184: 165–71 e1. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt M, Werbrouck A, Verhaeghe N, et al. Strategies for newborn screening for cystic fibrosis: A systematic review of health economic evaluations. J Cyst Fibros 2018; 17(3): 306–15. [DOI] [PubMed] [Google Scholar]
- 40.Sommerburg O, Stahl M, Hammermann J, et al. [Newborn Screening on Cystic Fibrosis in Germany: Comparison of the new Screening Protocol with an Alternative Protocol]. Klin Padiatr 2017; 229(2): 59–66. [DOI] [PubMed] [Google Scholar]
- 41.Ren CL, Borowitz DS, Gonska T, et al. Cystic Fibrosis Transmembrane Conductance Regulator-Related Metabolic Syndrome and Cystic Fibrosis Screen Positive, Inconclusive Diagnosis. J Pediatr 2017; 181S: S45–S51 e1. [DOI] [PubMed] [Google Scholar]
- 42.Girodon-Boulandet E, Cazeneuve C, Goossens M. Screening practices for mutations in the CFTR gene ABCC7. Hum Mutat 2000; 15(2): 135–49. [DOI] [PubMed] [Google Scholar]
- 43.Committee Opinion No. 691: Carrier Screening for Genetic Conditions. Obstet Gynecol 2017; 129(3): e41–e55. [DOI] [PubMed] [Google Scholar]
- 44.Zlotogora J, Israeli A. A comprehensive screening program for cystic fibrosis. Isr Med Assoc J 2009; 11(9): 555–7. [PubMed] [Google Scholar]
- 45.Janssens S, Kalokairinou L, Chokoshvilli D, et al. Attitudes of cystic fibrosis patients and their parents towards direct-to-consumer genetic testing for carrier status. Per Med 2015; 12(2): 99–107. [DOI] [PubMed] [Google Scholar]
- 46.Thauvin-Robinet C, Munck A, Huet F, et al. CFTR p.Arg117His associated with CBAVD and other CFTR-related disorders. J Med Genet 2013; 50(4): 220–7. [DOI] [PubMed] [Google Scholar]
- 47.Harper JC, Aittomaki K, Borry P, et al. Recent developments in genetics and medically assisted reproduction: from research to clinical applications. Eur J Hum Genet 2018; 26(1): 12–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dequeker E, Stuhrmann M, Morris MA, et al. Best practice guidelines for molecular genetic diagnosis of cystic fibrosis and CFTR-related disorders--updated European recommendations. Eur J Hum Genet 2009; 17(1): 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landaburu I, Gonzalvo MC, Clavero A, et al. Genetic testing of sperm donors for cystic fibrosis and spinal muscular atrophy: evaluation of clinical utility. Eur J Obstet Gynecol Reprod Biol 2013; 170(1): 183–7. [DOI] [PubMed] [Google Scholar]
- 50.ACOG Committee Opinion No. 486: Update on carrier screening for cystic fibrosis. Obstet Gynecol 2011; 117(4): 1028–31. [DOI] [PubMed] [Google Scholar]
- 51.Morgan MA, Driscoll DA, Mennuti MT, Schulkin J. Practice patterns of obstetrician-gynecologists regarding preconception and prenatal screening for cystic fibrosis. Genet Med 2004; 6(5): 450–5. [DOI] [PubMed] [Google Scholar]
- 52.Castellani C, Perobelli S, Bianchi V, et al. An interactive computer program can effectively educate potential users of cystic fibrosis carrier tests. Am J Med Genet A 2011; 155a(4): 778–85. [DOI] [PubMed] [Google Scholar]
- 53.Braido F, Baiardini I, Sumberesi M, Canonica GW, Blasi F, Castellani C. Public awareness on cystic fibrosis: results from a national pragmatic survey. Eur Respir J 2015; 46(1): 264–7. [DOI] [PubMed] [Google Scholar]
- 54.Metcalfe SA. Genetic counselling, patient education, and informed decision-making in the genomic era. Semin Fetal Neonatal Med 2018; 23(2): 142–9. [DOI] [PubMed] [Google Scholar]
- 55.Castellani C, Massie J. Newborn screening and carrier screening for cystic fibrosis: alternative or complementary? Eur Respir J 2014; 43(1): 20–3. [DOI] [PubMed] [Google Scholar]
- 56.Foil KE, Powers A, Raraigh KS, Wallis K, Southern KW, Salinas D. The increasing challenge of genetic counseling for cystic fibrosis. J Cyst Fibros 2019; 18(2): 167–74. [DOI] [PubMed] [Google Scholar]
- 57.King JR, Klugman S. Ethnicity-Based Carrier Screening. Obstet Gynecol Clin North Am 2018; 45(1): 83–101. [DOI] [PubMed] [Google Scholar]
- 58.Henneman L, Borry P, Chokoshvili D, et al. Responsible implementation of expanded carrier screening. Eur J Hum Genet 2016; 24(6): e1–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Girardet A, Viart V, Plaza S, et al. The improvement of the best practice guidelines for preimplantation genetic diagnosis of cystic fibrosis: toward an international consensus. Eur J Hum Genet 2016; 24(4): 469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Levy B, Stosic M. Traditional Prenatal Diagnosis: Past to Present. Methods Mol Biol 2019; 1885: 3–22. [DOI] [PubMed] [Google Scholar]
- 61.Cunningham S, Marshall T. Influence of five years of antenatal screening on the paediatric cystic fibrosis population in one region. Arch Dis Child 1998; 78(4): 345–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scotet V, Dugueperoux I, Audrezet MP, et al. Focus on cystic fibrosis and other disorders evidenced in fetuses with sonographic finding of echogenic bowel: 16-year report from Brittany, France. Am J Obstet Gynecol 2010; 203(6): 592.e1–6. [DOI] [PubMed] [Google Scholar]
- 63.Guissart C, Dubucs C, Raynal C, et al. Non-invasive prenatal diagnosis (NIPD) of cystic fibrosis: an optimized protocol using MEMO fluorescent PCR to detect the p.Phe508del mutation. J Cyst Fibros 2017; 16(2): 198–206. [DOI] [PubMed] [Google Scholar]
- 64.Sosnay PR, Castellani C, Penland CM, et al. Bias in CFTR screening panels. Genet Med 2016; 18(2): 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holtkamp KCA, Mathijssen IB, Lakeman P, et al. Factors for successful implementation of population-based expanded carrier screening: learning from existing initiatives. Eur J Public Health 2017; 27(2): 372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hetu M, Koutouki K, Joly Y. Genomics for All: International Open Science Genomics Projects and Capacity Building in the Developing World. Front Genet 2019; 10: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.An JY. National human genome projects: an update and an agenda. Epidemiol Health 2017; 39: e2017045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.http://www.genomeasia100k.com.
- 69.https://www.genomicsengland.co.uk/ (Accessed April 18, 2019).
- 70.https://www.genomicsengland.co.uk/wp-content/uploads/2018/07/black-african-black-caribbean-communities-participation-100kgp.pdf.
- 71.Loukas YL, Thodi G, Molou E, Georgiou V, Dotsikas Y, Schulpis KH. Clinical diagnostic Next-Generation sequencing: the case of CFTR carrier screening. Scand J Clin Lab Invest 2015; 75(5): 374–81. [DOI] [PubMed] [Google Scholar]
- 72.Davis KF, D’Odorico P, Laio F, Ridolfi L. Global spatio-temporal patterns in human migration: a complex network perspective. PLoS One 2013; 8(1): e53723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lamanna F, Lenormand M, Salas-Olmedo MH, Romanillos G, Goncalves B, Ramasco JJ. Immigrant community integration in world cities. PLoS One 2018; 13(3): e0191612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Steward CA, Parker APJ, Minassian BA, Sisodiya SM, Frankish A, Harrow J. Genome annotation for clinical genomic diagnostics: strengths and weaknesses. Genome Med 2017; 9(1): 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen R, Shi L, Hakenberg J, et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol 2016; 34(5): 531–8. [DOI] [PubMed] [Google Scholar]
- 76.Hui L, Bianchi DW. Prenatal pharmacotherapy for fetal anomalies: a 2011 update. Prenat Diagn 2011; 31(7): 735–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kauffman TL, Wilfond BS, Jarvik GP, et al. Design of a randomized controlled trial for genomic carrier screening in healthy patients seeking preconception genetic testing. Contemp Clin Trials 2017; 53: 100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Khan MAZ, Konje JC. Ethical and religious dilemmas of modern reproductive choices and the Islamic perspective. Eur J Obstet Gynecol Reprod Biol 2019; 232: 5–9. [DOI] [PubMed] [Google Scholar]
- 79.Dunne CP, Dunne SS. Personalized medicine should not be restricted to the wealthy. Nature 2018; 559(7712): 32. [DOI] [PubMed] [Google Scholar]
- 80.Jackson AD, Goss CH. Epidemiology of CF: How registries can be used to advance our understanding of the CF population. J Cyst Fibros 2018; 17(3): 297–305. [DOI] [PubMed] [Google Scholar]
- 81.Bobadilla JL, Macek M Jr., Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations--correlation with incidence data and application to screening. Hum Mutat 2002; 19(6): 575–606. [DOI] [PubMed] [Google Scholar]
- 82.Banjar H, Angyalosi G. The road for survival improvement of cystic fibrosis patients in Arab countries. Int J Pediatr Adolesc Med 2015; 2(2): 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Parker-Mc Gill K, Nugent M, Bersie R, et al. Changing incidence of cystic fibrosis in Wisconsin, USA. Pediatr Pulmonol 2015; 50(11): 1065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alper OM, Wong LJ, Young S, et al. Identification of novel and rare mutations in California Hispanic and African American cystic fibrosis patients. Hum Mutat 2004; 24(4): 353. [DOI] [PubMed] [Google Scholar]
- 85.Scotet V, Dugueperoux I, Saliou P, et al. Evidence for decline in the incidence of cystic fibrosis: a 35-year observational study in Brittany, France. Orphanet J Rare Dis 2012; 7: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stafler P, Mei-Zahav M, Wilschanski M, et al. The impact of a national population carrier screening program on cystic fibrosis birth rate and age at diagnosis: Implications for newborn screening. J Cyst Fibros 2016; 15(4): 460–6. [DOI] [PubMed] [Google Scholar]
- 87.Castellani C, Picci L, Tridello G, et al. Cystic fibrosis carrier screening effects on birth prevalence and newborn screening. Genet Med 2016; 18(2): 145–51. [DOI] [PubMed] [Google Scholar]
- 88.Farrell PM. The prevalence of cystic fibrosis in the European Union. J Cyst Fibros 2008; 7(5): 450–3. [DOI] [PubMed] [Google Scholar]
- 89.Grody WW, Cutting GR, Klinger KW, Richards CS, Watson MS, Desnick RJ. Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet Med 2001; 3(2): 149–54. [DOI] [PubMed] [Google Scholar]
- 90.https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Patient-Registry-Annual-Data-Report.pdf.
- 91.Jackson AD, Goss CH. Epidemiology of CF: How registries can be used to advance our understanding of the CF population. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2017. [DOI] [PubMed] [Google Scholar]
- 92.registry GC. https://www.muko.info/fileadmin/user_upload/angebote/qualitaetsmanagement/register/berichtsband_2017.pdf 2017.
- 93.registry UT. https://www.cysticfibrosis.org.uk/news/registry-report-2017. 2017.
- 94.registry CC. https://www.cysticfibrosis.ca/our-programs/cf-registry. 2017.
- 95.Buu MC, Sanders LM, Mayo JA, Milla CE, Wise PH. Assessing Differences in Mortality Rates and Risk Factors Between Hispanic and Non-Hispanic Patients With Cystic Fibrosis in California. Chest 2016; 149(2): 380–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sykes J, Stanojevic S, Goss CH, et al. A standardized approach to estimating survival statistics for population-based cystic fibrosis registry cohorts. Journal of clinical epidemiology 2016; 70: 206–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stephenson AL, Sykes J, Stanojevic S, et al. Survival Comparison of Patients With Cystic Fibrosis in Canada and the United States: A Population-Based Cohort Study. Annals of internal medicine 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Goss CH, Sykes J, Stanojevic S, et al. Comparison of Nutrition and Lung Function Outcomes in Patients with Cystic Fibrosis Living in Canada and the United States. American journal of respiratory and critical care medicine 2018; 197(6): 768–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.George PM, Banya W, Pareek N, et al. Improved survival at low lung function in cystic fibrosis: cohort study from 1990 to 2007. BMJ 2011; 342: d1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bombieri C, Claustres M, De Boeck K, et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros 2011; 10 Suppl 2: S86–102. [DOI] [PubMed] [Google Scholar]
- 101.Mayell SJ, Munck A, Craig JV, et al. A European consensus for the evaluation and management of infants with an equivocal diagnosis following newborn screening for cystic fibrosis. J Cyst Fibros 2009; 8(1): 71–8. [DOI] [PubMed] [Google Scholar]
- 102.Munck A, Mayell SJ, Winters V, et al. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J Cyst Fibros 2015; 14(6): 706–13. [DOI] [PubMed] [Google Scholar]
- 103.Cystic Fibrosis F, Borowitz D, Parad RB, et al. Cystic Fibrosis Foundation practice guidelines for the management of infants with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome during the first two years of life and beyond. J Pediatr 2009; 155(6 Suppl): S106–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ren CL, Desai H, Platt M, Dixon M. Clinical outcomes in infants with cystic fibrosis transmembrane conductance regulator (CFTR) related metabolic syndrome. Pediatr Pulmonol 2011; 46(11): 1079–84. [DOI] [PubMed] [Google Scholar]
- 105.Ooi CY, Castellani C, Keenan K, et al. Inconclusive diagnosis of cystic fibrosis after newborn screening. Pediatrics 2015; 135(6): e1377–85. [DOI] [PubMed] [Google Scholar]
- 106.Barben J, Castellani C, Dankert-Roelse J, et al. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J Cyst Fibros 2017; 16(2): 207–13. [DOI] [PubMed] [Google Scholar]
- 107.Farrell PM, White TB, Howenstine MS, et al. Diagnosis of Cystic Fibrosis in Screened Populations. J Pediatr 2017; 181s: S33–S44.e2. [DOI] [PubMed] [Google Scholar]
- 108.Sosnay PR, White TB, Farrell PM, et al. Diagnosis of Cystic Fibrosis in Nonscreened Populations. J Pediatr 2017; 181S: S52–S7 e2. [DOI] [PubMed] [Google Scholar]
- 109.Worldbank. http://datatopics.worldbank.org/world-development-indicators/the-world-by-income-and-region.html (Accessed May 8, 2019).
- 110.Camajova J, Berwouts S, Matthijs G, Macek M Jr., Dequeker E. Variability in the use of CE-marked assays for in vitro diagnostics of CFTR gene mutations in European genetic testing laboratories. Eur J Hum Genet 2009; 17(4): 537–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Singh M, Rebordosa C, Bernholz J, Sharma N. Epidemiology and genetics of cystic fibrosis in Asia: In preparation for the next-generation treatments. Respirology 2015; 20(8): 1172–81. [DOI] [PubMed] [Google Scholar]
- 112.Shen Y, Liu J, Zhong L, et al. Clinical Phenotypes and Genotypic Spectrum of Cystic Fibrosis in Chinese Children. J Pediatr 2016; 171: 269–76.e1. [DOI] [PubMed] [Google Scholar]
- 113.Al-Sadeq D, Abunada T, Dalloul R, et al. Spectrum of mutations of cystic fibrosis in the 22 Arab countries: A systematic review. Respirology 2019; 24(2): 127–36. [DOI] [PubMed] [Google Scholar]
- 114.Biomed. www.bmbtrj.org/temp/BiomedBiotechnolResJ12105-5448768_150807.pdf
- 115.Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and reproductive health among Arabs. Reprod Health 2009; 6: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ibrahim SA, Fadl Elmola MA, Karrar ZA, et al. Cystic fibrosis in Sudanese children: First report of 35 cases. Sudan J Paediatr 2014; 14(1): 39–44. [PMC free article] [PubMed] [Google Scholar]
- 117.Bahrain. Bahrain Medical Bulletin 201; (23): 157–9. [Google Scholar]
- 118.Al-Gazali LI, Alwash R, Abdulrazzaq YM. United Arab Emirates: communities and community genetics. Community Genet 2005; 8(3): 186–96. [DOI] [PubMed] [Google Scholar]
- 119.Dawson KP, Frossard PM. Cystic fibrosis in the Middle East: the historical perspective. Ann Saudi Med 2000; 20(1): 20–3. [DOI] [PubMed] [Google Scholar]
- 120.Maps G. http://gulf2000.columbia.edu/maps.shtml.
- 121.Desgeorges M, Megarbane A, Guittard C, et al. Cystic fibrosis in Lebanon: distribution of CFTR mutations among Arab communities. Hum Genet 1997; 100(2): 279–83. [DOI] [PubMed] [Google Scholar]
- 122.Alibakhshi R, Kianishirazi R, Cassiman JJ, Zamani M, Cuppens H. Analysis of the CFTR gene in Iranian cystic fibrosis patients: identification of eight novel mutations. J Cyst Fibros 2008; 7(2): 102–9. [DOI] [PubMed] [Google Scholar]
- 123.Koomesh. http://koomeshjournal.semums.ac.ir/browse.php?a_code=A-10-2167-1&slc_lang=fa&sid=1
- 124.Issa AR, Teebi AS, Issa MA, Shaabani IS, Ramadan DJ. Metabolic alkalosis in cystic fibrosis: atypical presentation in Kuwait. Ann Trop Paediatr 1988; 8(4): 271–2. [DOI] [PubMed] [Google Scholar]
- 125.Dahabreh MM, Najada AS. Pseudo-bartter syndrome, pattern and correlation with other cystic fibrosis features. Saudi J Kidney Dis Transpl 2013; 24(2): 292–6. [DOI] [PubMed] [Google Scholar]
- 126.Banjar HH. Cystic fibrosis: presentation with other diseases, the experience in Saudi Arabia. J Cyst Fibros 2003; 2(3): 155–9. [DOI] [PubMed] [Google Scholar]
- 127.Al-Gazali L, Ali BR. Mutations of a country: a mutation review of single gene disorders in the United Arab Emirates (UAE). Hum Mutat 2010; 31(5): 505–20. [DOI] [PubMed] [Google Scholar]
- 128.Koshy R, Sivadas A, Scaria V. Genetic epidemiology of familial Mediterranean fever through integrative analysis of whole genome and exome sequences from Middle East and North Africa. Clin Genet 2018; 93(1): 92–102. [DOI] [PubMed] [Google Scholar]
- 129.Stewart C, Pepper MS. Cystic fibrosis on the African continent. Genet Med 2016; 18(7): 653–62. [DOI] [PubMed] [Google Scholar]
- 130.Van Rensburg J, Alessandrini M, Stewart C, Pepper MS. Cystic fibrosis in South Africa: A changing diagnostic paradigm. S Afr Med J 2018; 108(8): 624–8. [DOI] [PubMed] [Google Scholar]
- 131.Zampoli On Behalf Of The Msac M. Cystic fibrosis: What’s new in South Africa in 2019. S Afr Med J 2018; 109(1): 16–9. [DOI] [PubMed] [Google Scholar]
- 132.Wang SS, O’Leary LA, Fitzsimmons SC, Khoury MJ. The impact of early cystic fibrosis diagnosis on pulmonary function in children. J Pediatr 2002; 141(6): 804–10. [DOI] [PubMed] [Google Scholar]
- 133.Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet 2008; 9: 403–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Akombi BJ, Agho KE, Merom D, Renzaho AM, Hall JJ. Child malnutrition in sub-Saharan Africa: A meta-analysis of demographic and health surveys (2006–2016). PLoS One 2017; 12(5): e0177338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Slagle T, Ben Youssef M, Calonge G, Ben Amor Y. Lessons from Africa: developing a global human rights framework for tuberculosis control and prevention. BMC Int Health Hum Rights 2014; 14: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Reid SR. Injection drug use, unsafe medical injections, and HIV in Africa: a systematic review. Harm Reduct J 2009; 6: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Adhikari K, Mendoza-Revilla J, Chacon-Duque JC, Fuentes-Guajardo M, Ruiz-Linares A. Admixture in Latin America. Curr Opin Genet Dev 2016; 41: 106–14. [DOI] [PubMed] [Google Scholar]
- 138.Norris ET, Wang L, Conley AB, et al. Genetic ancestry, admixture and health determinants in Latin America. BMC Genomics 2018; 19(Suppl 8): 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Alonso MJ, Heine-Suner D, Calvo M, et al. Spectrum of mutations in the CFTR gene in cystic fibrosis patients of Spanish ancestry. Ann Hum Genet 2007; 71(Pt 2): 194–201. [DOI] [PubMed] [Google Scholar]
- 140.Perez MM, Luna MC, Pivetta OH, Keyeux G. CFTR gene analysis in Latin American CF patients: heterogeneous origin and distribution of mutations across the continent. J Cyst Fibros 2007; 6(3): 194–208. [DOI] [PubMed] [Google Scholar]
- 141.Stewart C, Pepper MS. Cystic Fibrosis in the African Diaspora. Ann Am Thorac Soc 2017; 14(1): 1–7. [DOI] [PubMed] [Google Scholar]
- 142.Conway S, Balfour-Lynn IM, De Rijcke K, et al. European Cystic Fibrosis Society Standards of Care: Framework for the Cystic Fibrosis Centre. J Cyst Fibros 2014; 13 Suppl 1: S3–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Boyle MP, Sabadosa KA, Quinton HB, Marshall BC, Schechter MS. Key findings of the US Cystic Fibrosis Foundation’s clinical practice benchmarking project. BMJ Qual Saf 2014; 23 Suppl 1: i15–i22. [DOI] [PubMed] [Google Scholar]
- 144.Young DC, Autry E, Zobell JT, et al. Patients and families experience with pharmacist care at cystic fibrosis foundation accredited clinics. Pediatr Pulmonol 2019. [DOI] [PubMed] [Google Scholar]
- 145.Smyth AR, Bell SC, Bojcin S, et al. European Cystic Fibrosis Society Standards of Care: Best Practice guidelines. J Cyst Fibros 2014; 13 Suppl 1: S23–42. [DOI] [PubMed] [Google Scholar]
- 146.Castellani C, Duff AJA, Bell SC, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros 2018; 17(2): 153–78. [DOI] [PubMed] [Google Scholar]
- 147.Elborn JS, Bell SC, Madge SL, et al. Report of the European Respiratory Society/European Cystic Fibrosis Society task force on the care of adults with cystic fibrosis. Eur Respir J 2016; 47(2): 420–8. [DOI] [PubMed] [Google Scholar]
- 148.Farrell PM, Lai HJ, Li Z, et al. Evidence on improved outcomes with early diagnosis of cystic fibrosis through neonatal screening: enough is enough! J Pediatr 2005; 147(3 Suppl): S30–6. [DOI] [PubMed] [Google Scholar]
- 149.Sanders DB, Zhang Z, Farrell PM, Lai HJ, Wisconsin CFNSG. Early life growth patterns persist for 12years and impact pulmonary outcomes in cystic fibrosis. J Cyst Fibros 2018; 17(4): 528–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cystic Fibrosis F, Borowitz D, Robinson KA, et al. Cystic Fibrosis Foundation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr 2009; 155(6 Suppl): S73–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Esther CR Jr., Muhlebach MS, Ehre C, et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci Transl Med 2019; 11(486). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ranganathan SC, Hall GL, Sly PD, Stick SM, Douglas TA, Australian Respiratory Early Surveillance Team for Cystic F. Early Lung Disease in Infants and Preschool Children with Cystic Fibrosis. What Have We Learned and What Should We Do about It? Am J Respir Crit Care Med 2017; 195(12): 1567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nguyen TT, Thia LP, Hoo AF, et al. Evolution of lung function during the first year of life in newborn screened cystic fibrosis infants. Thorax 2014; 69(10): 910–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bush A, Sly PD. Evolution of cystic fibrosis lung function in the early years. Curr Opin Pulm Med 2015; 21(6): 602–8. [DOI] [PubMed] [Google Scholar]
- 155.Rosenow T, Mok LC, Turkovic L, et al. The cumulative effect of inflammation and infection on structural lung disease in early CF. Eur Respir J 2019. [DOI] [PubMed] [Google Scholar]
- 156.Deschamp AR, Hatch JE, Slaven JE, et al. Early respiratory viral infections in infants with cystic fibrosis. J Cyst Fibros 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Eyns H, Pierard D, De Wachter E, Eeckhout L, Vaes P, Malfroot A. Respiratory Bacterial Culture Sampling in Expectorating and Non-expectorating Patients With Cystic Fibrosis. Front Pediatr 2018; 6: 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Doumit M, Belessis Y, Stelzer-Braid S, Mallitt KA, Rawlinson W, Jaffe A. Diagnostic accuracy and distress associated with oropharyngeal suction in cystic fibrosis. J Cyst Fibros 2016; 15(4): 473–8. [DOI] [PubMed] [Google Scholar]
- 159.Equi AC, Pike SE, Davies J, Bush A. Use of cough swabs in a cystic fibrosis clinic. Arch Dis Child 2001; 85(5): 438–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ferreira ACM, Marson FAL, Cohen MA, et al. Hypertonic Saline as a Useful Tool for Sputum Induction and Pathogen Detection in Cystic Fibrosis. Lung 2017; 195(4): 431–9. [DOI] [PubMed] [Google Scholar]
- 161.Ahmed MI, Kulkarni H, Shajpal S, et al. Early detection of non-tuberculous mycobacteria in children with cystic fibrosis using induced sputum at annual review. Pediatr Pulmonol 2019; 54(3): 257–63. [DOI] [PubMed] [Google Scholar]
- 162.Ronchetti K, Tame JD, Paisey C, et al. The CF-Sputum Induction Trial (CF-SpIT) to assess lower airway bacterial sampling in young children with cystic fibrosis: a prospective internally controlled interventional trial. Lancet Respir Med 2018; 6(6): 461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zampoli M, Pillay K, Carrara H, Zar HJ, Morrow B. Microbiological yield from induced sputum compared to oropharyngeal swab in young children with cystic fibrosis. J Cyst Fibros 2016; 15(5): 605–10. [DOI] [PubMed] [Google Scholar]
- 164.Rosenfeld M, Allen J, Arets BH, et al. An official American Thoracic Society workshop report: optimal lung function tests for monitoring cystic fibrosis, bronchopulmonary dysplasia, and recurrent wheezing in children less than 6 years of age. Ann Am Thorac Soc 2013; 10(2): S1–S11. [DOI] [PubMed] [Google Scholar]
- 165.Davies G, Stocks J, Thia LP, et al. Pulmonary function deficits in newborn screened infants with cystic fibrosis managed with standard UK care are mild and transient. Eur Respir J 2017; 50(5). [DOI] [PubMed] [Google Scholar]
- 166.Robinson PD, Latzin P, Ramsey KA, et al. Preschool Multiple-Breath Washout Testing. An Official American Thoracic Society Technical Statement. Am J Respir Crit Care Med 2018; 197(5): e1–e19. [DOI] [PubMed] [Google Scholar]
- 167.Saunders C, Bayfield K, Irving S, Short C, Bush A, Davies JC. Developments in multiple breath washout testing in children with cystic fibrosis. Curr Med Res Opin 2017; 33(4): 613–20. [DOI] [PubMed] [Google Scholar]
- 168.Stanojevic S, Davis SD, Retsch-Bogart G, et al. Progression of Lung Disease in Preschool Patients with Cystic Fibrosis. Am J Respir Crit Care Med 2017; 195(9): 1216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Owens CM, Aurora P, Stanojevic S, et al. Lung Clearance Index and HRCT are complementary markers of lung abnormalities in young children with CF. Thorax 2011; 66(6): 481–8. [DOI] [PubMed] [Google Scholar]
- 170.Lum S, Gustafsson P, Ljungberg H, et al. Early detection of cystic fibrosis lung disease: multiple-breath washout versus raised volume tests. Thorax 2007; 62(4): 341–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.https://lab.research.sickkids.ca/ratjen/.
- 172.https://www.ecfs.eu/ctn/lci/lci-core-facility-definition;.
- 173.Stahl M, Joachim C, Wielputz MO, Mall MA. Comparison of lung clearance index determined by washout of N2 and SF6 in infants and preschool children with cystic fibrosis. J Cyst Fibros 2018. [DOI] [PubMed] [Google Scholar]
- 174.Sakarya A, Uyan ZS, Baydemir C, et al. Evaluation of children with cystic fibrosis by impulse oscillometry when stable and at exacerbation. Pediatr Pulmonol 2016; 51(11): 1151–8. [DOI] [PubMed] [Google Scholar]
- 175.Buchs C, Coutier L, Vrielynck S, Jubin V, Mainguy C, Reix P. An impulse oscillometry system is less efficient than spirometry in tracking lung function improvements after intravenous antibiotic therapy in pediatric patients with cystic fibrosis. Pediatr Pulmonol 2015; 50(11): 1073–81. [DOI] [PubMed] [Google Scholar]
- 176.Lin S, Lin M, Lau KK. Efficacy of model-based iterative reconstruction in cystic fibrosis assessment using CT. Clin Radiol 2019. [DOI] [PubMed] [Google Scholar]
- 177.Mall MA, Stahl M, Graeber SY, Sommerburg O, Kauczor HU, Wielputz MO. Early detection and sensitive monitoring of CF lung disease: Prospects of improved and safer imaging. Pediatr Pulmonol 2016; 51(S44): S49–S60. [DOI] [PubMed] [Google Scholar]
- 178.Rayment JH, Couch MJ, McDonald N, et al. Hyperpolarised (129)Xe magnetic resonance imaging to monitor treatment response in children with cystic fibrosis. Eur Respir J 2019; 53(5). [DOI] [PubMed] [Google Scholar]
- 179.Martini K, Gygax CM, Benden C, Morgan AR, Parker GJM, Frauenfelder T. Volumetric dynamic oxygen-enhanced MRI (OE-MRI): comparison with CT Brody score and lung function in cystic fibrosis patients. Eur Radiol 2018; 28(10): 4037–47. [DOI] [PubMed] [Google Scholar]
- 180.Wielputz MO, Eichinger M, Wege S, et al. Mid-Term Reproducibility of Chest MRI in Adults with Clinically Stable Cystic Fibrosis and Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 2019. [DOI] [PubMed] [Google Scholar]
- 181.Rand S, Hill L, Prasad SA. Physiotherapy in cystic fibrosis: optimising techniques to improve outcomes. Paediatr Respir Rev 2013; 14(4): 263–9. [DOI] [PubMed] [Google Scholar]
- 182.Flume PA, Robinson KA, O’Sullivan BP, et al. Cystic fibrosis pulmonary guidelines: airway clearance therapies. Respir Care 2009; 54(4): 522–37. [PubMed] [Google Scholar]
- 183.Ward N, Stiller K, Holland AE. Exercise as a therapeutic intervention for people with cystic fibrosis. Expert Rev Respir Med 2019; 13(5): 449–58. [DOI] [PubMed] [Google Scholar]
- 184.Acosta N, Heirali A, Somayaji R, et al. Sputum microbiota is predictive of long-term clinical outcomes in young adults with cystic fibrosis. Thorax 2018; 73(11): 1016–25. [DOI] [PubMed] [Google Scholar]
- 185.Khan MA, Ali ZS, Sweezey N, Grasemann H, Palaniyar N. Progression of Cystic Fibrosis Lung Disease from Childhood to Adulthood: Neutrophils, Neutrophil Extracellular Trap (NET) Formation, and NET Degradation. Genes (Basel) 2019; 10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sanders DB, Emerson J, Ren CL, et al. Early Childhood Risk Factors for Decreased FEV1 at Age Six to Seven Years in Young Children with Cystic Fibrosis. Ann Am Thorac Soc 2015; 12(8): 1170–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Oates GR, Schechter MS. Socioeconomic status and health outcomes: cystic fibrosis as a model. Expert Rev Respir Med 2016; 10(9): 967–77. [DOI] [PubMed] [Google Scholar]
- 188.Breuer O, Caudri D, Stick S, Turkovic L. Predicting disease progression in cystic fibrosis. Expert Rev Respir Med 2018; 12(11): 905–17. [DOI] [PubMed] [Google Scholar]
- 189.Sherrard LJ, Bell SC. Lower airway microbiota for ‘biomarker’ measurements of cystic fibrosis disease progression? Thorax 2018; 73(11): 1001–3. [DOI] [PubMed] [Google Scholar]
- 190.Scott LK, Toner R. Clinically Promising Biomarkers in Cystic Fibrosis Pulmonary Exacerbations. Lung 2017; 195(4): 397–401. [DOI] [PubMed] [Google Scholar]
- 191.Nick JA, Sanders LA, Ickes B, et al. Blood mRNA biomarkers for detection of treatment response in acute pulmonary exacerbations of cystic fibrosis. Thorax 2013; 68(10): 929–37. [DOI] [PubMed] [Google Scholar]
- 192.Liou TG, Adler FR, Argel N, et al. Prospective multicenter randomized patient recruitment and sample collection to enable future measurements of sputum biomarkers of inflammation in an observational study of cystic fibrosis. BMC Med Res Methodol 2019; 19(1): 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 2006; 354(3): 229–40. [DOI] [PubMed] [Google Scholar]
- 194.Stahl M, Wielputz MO, Ricklefs I, et al. Preventive Inhalation of Hypertonic Saline in Infants with Cystic Fibrosis (PRESIS). A Randomized, Double-Blind, Controlled Study. Am J Respir Crit Care Med 2019; 199(10): 1238–48. [DOI] [PubMed] [Google Scholar]
- 195.Mogayzel PJ Jr., Naureckas ET, Robinson KA, et al. Cystic Fibrosis Foundation pulmonary guideline. pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann Am Thorac Soc 2014; 11(10): 1640–50. [DOI] [PubMed] [Google Scholar]
- 196.Davies JC, Cunningham S, Harris WT, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med 2016; 4(2): 107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rosenfeld M, Cunningham S, Harris WT, et al. An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2–5years (KLIMB). J Cyst Fibros 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Christian F, Thierman A, Shirley E, Allen K, Cross C, Jones K. Sustained Glycemic Control With Ivacaftor in Cystic Fibrosis-Related Diabetes. J Investig Med High Impact Case Rep 2019; 7: 2324709619842898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kelly A, De Leon DD, Sheikh S, et al. Islet Hormone and Incretin Secretion in Cystic Fibrosis after Four Months of Ivacaftor Therapy. Am J Respir Crit Care Med 2019; 199(3): 342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hayes D Jr., McCoy KS, Sheikh SI. Resolution of cystic fibrosis-related diabetes with ivacaftor therapy. Am J Respir Crit Care Med 2014; 190(5): 590–1. [DOI] [PubMed] [Google Scholar]
- 201.Bellin MD, Laguna T, Leschyshyn J, et al. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediatr Diabetes 2013; 14(6): 417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ren CL, Borowitz DS, Gonska T, et al. Cystic Fibrosis Transmembrane Conductance Regulator-Related Metabolic Syndrome and Cystic Fibrosis Screen Positive, Inconclusive Diagnosis. J Pediatr 2017; 181s: S45–S51.e1. [DOI] [PubMed] [Google Scholar]
- 203.Hayeems RZ, Miller FA, Vermeulen M, et al. False-Positive Newborn Screening for Cystic Fibrosis and Health Care Use. Pediatrics 2017; 140(5). [DOI] [PubMed] [Google Scholar]
- 204.Caverly LJ, LiPuma JJ. Cystic fibrosis respiratory microbiota: unraveling complexity to inform clinical practice. Expert Rev Respir Med 2018; 12(10): 857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Carmody LA, Caverly LJ, Foster BK, et al. Fluctuations in airway bacterial communities associated with clinical states and disease stages in cystic fibrosis. PLoS One 2018; 13(3): e0194060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zemanick ET, Wagner BD, Robertson CE, et al. Airway microbiota across age and disease spectrum in cystic fibrosis. Eur Respir J 2017; 50(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Schaffer K. Epidemiology of infection and current guidelines for infection prevention in cystic fibrosis patients. J Hosp Infect 2015; 89(4): 309–13. [DOI] [PubMed] [Google Scholar]
- 208.Aitken ML, Limaye A, Pottinger P, et al. Respiratory outbreak of Mycobacterium abscessus subspecies massiliense in a lung transplant and cystic fibrosis center. American journal of respiratory and critical care medicine 2012; 185(2): 231–2. [DOI] [PubMed] [Google Scholar]
- 209.Johnston DI, Chisty Z, Gross JE, Park SY. Investigation of Mycobacterium abscessus outbreak among cystic fibrosis patients, Hawaii 2012. The Journal of hospital infection 2016; 94(2): 198–200. [DOI] [PubMed] [Google Scholar]
- 210.Bryant JM, Grogono DM, Greaves D, et al. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet (London, England) 2013; 381(9877): 1551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bryant JM, Grogono DM, Rodriguez-Rincon D, et al. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science (New York, NY) 2016; 354(6313): 751–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Engel TGP, Erren E, Vanden Driessche KSJ, et al. Aerosol Transmission of Aspergillus fumigatus in Cystic Fibrosis Patients in the Netherlands. Emerging infectious diseases 2019; 25(4): 797–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kerem E, Conway S, Elborn S, Heijerman H, Consensus C. Standards of care for patients with cystic fibrosis: a European consensus. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2005; 4(1): 7–26. [DOI] [PubMed] [Google Scholar]
- 214.Saiman L, Siegel JD, LiPuma JJ, et al. Infection prevention and control guideline for cystic fibrosis: 2013 update. Infection control and hospital epidemiology 2014; 35 Suppl 1: S1–S67. [DOI] [PubMed] [Google Scholar]
- 215.Stoudemire W, Jiang X, Zhou JJ, et al. Cystic fibrosis program characteristics associated with adoption of 2013 infection prevention and control recommendations. American journal of infection control 2019. [DOI] [PubMed] [Google Scholar]
- 216.Rowbotham NJ, Palser SC, Smith SJ, Smyth AR. Infection prevention and control in cystic fibrosis: a systematic review of interventions. Expert review of respiratory medicine 2019; 13(5): 425–34. [DOI] [PubMed] [Google Scholar]
- 217.Smyth AR, Rosenfeld M. Prophylactic anti-staphylococcal antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2017; 4: CD001912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ratjen F, Comes G, Paul K, et al. Effect of continuous antistaphylococcal therapy on the rate of P. aeruginosa acquisition in patients with cystic fibrosis. Pediatr Pulmonol 2001; 31(1): 13–6. [DOI] [PubMed] [Google Scholar]
- 219.http://www.cfstart.org.uk/.
- 220.Gramegna A, Moore JE, McCaughan J, et al. Increasing Burden of Antimicrobial Resistance in Pseudomonas Aeruginosa from Adult Patients with Cystic Fibrosis (Cf) in Northern Ireland: Then and Now. The Ulster medical journal 2018; 87(2): 129–30. [PMC free article] [PubMed] [Google Scholar]
- 221.Sherrard LJ, Tunney MM, Elborn JS. Antimicrobial resistance in the respiratory microbiota of people with cystic fibrosis. Lancet 2014; 384(9944): 703–13. [DOI] [PubMed] [Google Scholar]
- 222.Burgel PR, Baixench MT, Amsellem M, et al. High prevalence of azole-resistant Aspergillus fumigatus in adults with cystic fibrosis exposed to itraconazole. Antimicrobial agents and chemotherapy 2012; 56(2): 869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Smith AL, Fiel SB, Mayer-Hamblett N, Ramsey B, Burns JL. Susceptibility testing of Pseudomonas aeruginosa isolates and clinical response to parenteral antibiotic administration: lack of association in cystic fibrosis. Chest 2003; 123(5): 1495–502. [DOI] [PubMed] [Google Scholar]
- 224.Aaron SD, Chaparro C. Referral to lung transplantation--too little, too late. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2016; 15(2): 143–4. [DOI] [PubMed] [Google Scholar]
- 225.Jorth P, Staudinger BJ, Wu X, et al. Regional Isolation Drives Bacterial Diversification within Cystic Fibrosis Lungs. Cell host & microbe 2015; 18(3): 307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Somayaji R, Parkins MD, Shah A, et al. Antimicrobial susceptibility testing (AST) and associated clinical outcomes in individuals with cystic fibrosis: A systematic review. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2019; 18(2): 236–43. [DOI] [PubMed] [Google Scholar]
- 227.Kidd TJ, Canton R, Ekkelenkamp M, et al. Defining antimicrobial resistance in cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2018; 17(6): 696–704. [DOI] [PubMed] [Google Scholar]
- 228.Waters VJ, Kidd TJ, Canton R, et al. Reconciling Antimicrobial Susceptibility Testing and Clinical Response in Antimicrobial Treatment of Chronic Cystic Fibrosis Lung Infections. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2019. [DOI] [PubMed] [Google Scholar]
- 229.Qvist T, Johansen IS, Pressler T, et al. Urine lipoarabinomannan point-of-care testing in patients affected by pulmonary nontuberculous mycobacteria--experiences from the Danish Cystic Fibrosis cohort study. BMC Infect Dis 2014; 14: 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Songkhla MN, Tantipong H, Tongsai S, Angkasekwinai N. Lateral Flow Urine Lipoarabinomannan Assay for Diagnosis of Active Tuberculosis in Adults With Human Immunodeficiency Virus Infection: A Prospective Cohort Study. Open Forum Infect Dis 2019; 6(4): ofz132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Sanchez C, Santos JP, Lozano J. Use of Electronic Noses for Diagnosis of Digestive and Respiratory Diseases through the Breath. Biosensors (Basel) 2019; 9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Christiansen A, Davidsen JR, Titlestad I, Vestbo J, Baumbach J. A systematic review of breath analysis and detection of volatile organic compounds in COPD. J Breath Res 2016; 10(3): 034002. [DOI] [PubMed] [Google Scholar]
- 233.Smith WD, Bardin E, Cameron L, et al. Current and future therapies for Pseudomonas aeruginosa infection in patients with cystic fibrosis. FEMS Microbiol Lett 2017; 364(14). [DOI] [PubMed] [Google Scholar]
- 234.Moheet A, Moran A. CF-related diabetes: Containing the metabolic miscreant of cystic fibrosis. Pediatr Pulmonol 2017; 52(S48): S37–S43. [DOI] [PubMed] [Google Scholar]
- 235.Bridges N, Rowe R, Holt RIG. Unique challenges of cystic fibrosis-related diabetes. Diabet Med 2018. [DOI] [PubMed] [Google Scholar]
- 236.Moheet A, Moran A. Pharmacological management of cystic fibrosis related diabetes. Expert Rev Clin Pharmacol 2018; 11(2): 185–91. [DOI] [PubMed] [Google Scholar]
- 237.West NE, Flume PA. Unmet needs in cystic fibrosis: the next steps in improving outcomes. Expert Rev Respir Med 2018; 12(7): 585–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Cutting GR. Modifier genetics: cystic fibrosis. Annu Rev Genomics Hum Genet 2005; 6: 237–60. [DOI] [PubMed] [Google Scholar]
- 239.Waters V, Ratjen F. Pulmonary Exacerbations in Children with Cystic Fibrosis. Ann Am Thorac Soc 2015; 12 Suppl 2: S200–6. [DOI] [PubMed] [Google Scholar]
- 240.Solem CT, Vera-Llonch M, Liu S, Botteman M, Castiglione B. Impact of pulmonary exacerbations and lung function on generic health-related quality of life in patients with cystic fibrosis. Health Qual Life Outcomes 2016; 14: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Habib AR, Manji J, Wilcox PG, Javer AR, Buxton JA, Quon BS. A systematic review of factors associated with health-related quality of life in adolescents and adults with cystic fibrosis. Ann Am Thorac Soc 2015; 12(3): 420–8. [DOI] [PubMed] [Google Scholar]
- 242.Sanders DB, Bittner RC, Rosenfeld M, Hoffman LR, Redding GJ, Goss CH. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am J Respir Crit Care Med 2010; 182(5): 627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Cogen J, Emerson J, Sanders DB, et al. Risk factors for lung function decline in a large cohort of young cystic fibrosis patients. Pediatr Pulmonol 2015; 50(8): 763–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Waters V, Stanojevic S, Atenafu EG, et al. Effect of pulmonary exacerbations on long-term lung function decline in cystic fibrosis. Eur Respir J 2012; 40(1): 61–6. [DOI] [PubMed] [Google Scholar]
- 245.Sanders DB, Bittner RC, Rosenfeld M, Redding GJ, Goss CH. Pulmonary exacerbations are associated with subsequent FEV1 decline in both adults and children with cystic fibrosis. Pediatr Pulmonol 2011; 46(4): 393–400. [DOI] [PubMed] [Google Scholar]
- 246.Plummer A, Wildman M, Gleeson T. Duration of intravenous antibiotic therapy in people with cystic fibrosis. Cochrane Database Syst Rev 2016; 9: CD006682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Waters V, Ratjen F. Standard versus biofilm antimicrobial susceptibility testing to guide antibiotic therapy in cystic fibrosis. Cochrane Database Syst Rev 2017; 10: CD009528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Aaron SD. Antibiotic synergy testing should not be routine for patients with cystic fibrosis who are infected with multiresistant bacterial organisms. Paediatr Respir Rev 2007; 8(3): 256–61. [DOI] [PubMed] [Google Scholar]
- 249.Serisier DJ, Tuck A, Matley D, Carroll MP, Jones G. Antimicrobial susceptibility and synergy studies of cystic fibrosis sputum by direct sputum sensitivity testing. Eur J Clin Microbiol Infect Dis 2012; 31(11): 3211–6. [DOI] [PubMed] [Google Scholar]
- 250.Shoki AH, Mayer-Hamblett N, Wilcox PG, Sin DD, Quon BS. Systematic review of blood biomarkers in cystic fibrosis pulmonary exacerbations. Chest 2013; 144(5): 1659–70. [DOI] [PubMed] [Google Scholar]
- 251.Wark P, McDonald VM. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev 2018; 9: CD001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Southern KW, Barker PM, Solis-Moya A, Patel L. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2012; 11: CD002203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Patel S, Sinha IP, Dwan K, Echevarria C, Schechter M, Southern KW. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database Syst Rev 2015; (3): CD009841. [DOI] [PubMed] [Google Scholar]
- 254.Goralski JL, Nasr SZ, Uluer A. Overcoming barriers to a successful transition from pediatric to adult care. Pediatr Pulmonol 2017; 52(S48): S52–S60. [DOI] [PubMed] [Google Scholar]
- 255.Six Core Elements of Health Care Transition.
- 256.Gleeson H, McCartney S, Lidstone V. ‘Everybody’s business’: transition and the role of adult physicians. Clin Med (Lond) 2012; 12(6): 561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.ON TRAC TRANSITION CLINICAL PATHWAY.
- 258.Keating CL, Liu X, Dimango EA. Classic respiratory disease but atypical diagnostic testing distinguishes adult presentation of cystic fibrosis. Chest 2010; 137(5): 1157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Desai S, Wong H, Sykes J, Stephenson AL, Singer J, Quon BS. Clinical Characteristics and Predictors of Reduced Survival for Adult-diagnosed Cystic Fibrosis. Analysis of the Canadian CF Registry. Ann Am Thorac Soc 2018; 15(10): 1177–85. [DOI] [PubMed] [Google Scholar]
- 260.De Boeck K, Wilschanski M, Castellani C, et al. Cystic fibrosis: terminology and diagnostic algorithms. Thorax 2006; 61(7): 627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Turck D, Braegger CP, Colombo C, et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clin Nutr 2016; 35(3): 557–77. [DOI] [PubMed] [Google Scholar]
- 262.Panagopoulou P, Fotoulaki M, Nikolaou A, Nousia-Arvanitakis S. Prevalence of malnutrition and obesity among cystic fibrosis patients. Pediatr Int 2014; 56(1): 89–94. [DOI] [PubMed] [Google Scholar]
- 263.Lewis C, Blackman SM, Nelson A, et al. Diabetes-related mortality in adults with cystic fibrosis. Role of genotype and sex. Am J Respir Crit Care Med 2015; 191(2): 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Regard L, Martin C, Chassagnon G, Burgel PR. Acute and chronic non-pulmonary complications in adults with cystic fibrosis. Expert Rev Respir Med 2019; 13(1): 23–38. [DOI] [PubMed] [Google Scholar]
- 265.Safaeian M, Robbins HA, Berndt SI, Lynch CF, Fraumeni JF Jr., Engels EA. Risk of Colorectal Cancer After Solid Organ Transplantation in the United States. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2016; 16(3): 960–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Fink AK, Yanik EL, Marshall BC, et al. Cancer risk among lung transplant recipients with cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2017; 16(1): 91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Maisonneuve P, FitzSimmons SC, Neglia JP, Campbell PW 3rd, Lowenfels AB. Cancer risk in nontransplanted and transplanted cystic fibrosis patients: a 10-year study. Journal of the National Cancer Institute 2003; 95(5): 381–7. [DOI] [PubMed] [Google Scholar]
- 268.Maisonneuve P, Marshall BC, Knapp EA, Lowenfels AB. Cancer risk in cystic fibrosis: a 20-year nationwide study from the United States. Journal of the National Cancer Institute 2013; 105(2): 122–9. [DOI] [PubMed] [Google Scholar]
- 269.Yamada A, Komaki Y, Komaki F, Micic D, Zullow S, Sakuraba A. Risk of gastrointestinal cancers in patients with cystic fibrosis: a systematic review and meta-analysis. Lancet Oncol 2018; 19(6): 758–67. [DOI] [PubMed] [Google Scholar]
- 270.Hadjiliadis D, Khoruts A, Zauber AG, et al. Cystic Fibrosis Colorectal Cancer Screening Consensus Recommendations. Gastroenterology 2018; 154(3): 736–45 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Dai Z, Coker OO, Nakatsu G, et al. Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome 2018; 6(1): 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Dorsey J, Gonska T. Bacterial overgrowth, dysbiosis, inflammation, and dysmotility in the Cystic Fibrosis intestine. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2017; 16 Suppl 2: S14–S23. [DOI] [PubMed] [Google Scholar]
- 273.Zhang J, Wang Y, Jiang X, Chan HC. Cystic fibrosis transmembrane conductance regulator-emerging regulator of cancer. Cellular and molecular life sciences : CMLS 2018; 75(10): 1737–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Than BLN, Linnekamp JF, Starr TK, et al. CFTR is a tumor suppressor gene in murine and human intestinal cancer. Oncogene 2017; 36(24): 3504. [DOI] [PubMed] [Google Scholar]
- 275.Berg KH, Ryom L, Faurholt-Jepsen D, Pressler T, Katzenstein TL. Prevalence and characteristics of chronic kidney disease among Danish adults with cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2018; 17(4): 478–83. [DOI] [PubMed] [Google Scholar]
- 276.Quon BS, Mayer-Hamblett N, Aitken ML, Smyth AR, Goss CH. Risk factors for chronic kidney disease in adults with cystic fibrosis. American journal of respiratory and critical care medicine 2011; 184(10): 1147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Yahiaoui Y, Jablonski M, Hubert D, et al. Renal involvement in cystic fibrosis: diseases spectrum and clinical relevance. Clinical journal of the American Society of Nephrology : CJASN 2009; 4(5): 921–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Lefaucheur C, Nochy D, Amrein C, et al. Renal histopathological lesions after lung transplantation in patients with cystic fibrosis. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2008; 8(9): 1901–10. [DOI] [PubMed] [Google Scholar]
- 279.Gonzalez Jimenez D, Munoz-Codoceo R, Garriga-Garcia M, et al. [Excess weight in patients with cystic fibrosis: is it always beneficial?]. Nutricion hospitalaria 2017; 34(3): 578–83. [DOI] [PubMed] [Google Scholar]
- 280.Stephenson AL, Mannik LA, Walsh S, et al. Longitudinal trends in nutritional status and the relation between lung function and BMI in cystic fibrosis: a population-based cohort study. The American journal of clinical nutrition 2013; 97(4): 872–7. [DOI] [PubMed] [Google Scholar]
- 281.Snell G, Reed A, Stern M, Hadjiliadis D. The evolution of lung transplantation for cystic fibrosis: A 2017 update. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2017; 16(5): 553–64. [DOI] [PubMed] [Google Scholar]
- 282.Thabut G, Christie JD, Mal H, et al. Survival benefit of lung transplant for cystic fibrosis since lung allocation score implementation. American journal of respiratory and critical care medicine 2013; 187(12): 1335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Ramos KJ, Quon BS, Psoter KJ, et al. Predictors of non-referral of patients with cystic fibrosis for lung transplant evaluation in the United States. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2016; 15(2): 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Quon BS, Psoter K, Mayer-Hamblett N, Aitken ML, Li CI, Goss CH. Disparities in access to lung transplantation for patients with cystic fibrosis by socioeconomic status. American journal of respiratory and critical care medicine 2012; 186(10): 1008–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Martin C, Hamard C, Kanaan R, et al. Causes of death in French cystic fibrosis patients: The need for improvement in transplantation referral strategies! J Cyst Fibros 2016; 15(2): 204–12. [DOI] [PubMed] [Google Scholar]
- 286.Kerem E, Reisman J, Corey M, Canny GJ, Levison H. Prediction of mortality in patients with cystic fibrosis. N Engl J Med 1992; 326(18): 1187–91. [DOI] [PubMed] [Google Scholar]
- 287.Yankaskas JR, Mallory GB Jr., Lung transplantation in cystic fibrosis: consensus conference statement. Chest 1998; 113(1): 217–26. [DOI] [PubMed] [Google Scholar]
- 288.Ramos KJ, Quon BS, Heltshe SL, et al. Heterogeneity in Survival in Adult Patients With Cystic Fibrosis With FEV1 < 30% of Predicted in the United States. Chest 2017; 151(6): 1320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Aaron SD, Stephenson AL, Cameron DW, Whitmore GA. A statistical model to predict one-year risk of death in patients with cystic fibrosis. Journal of clinical epidemiology 2015; 68(11): 1336–45. [DOI] [PubMed] [Google Scholar]
- 290.Nkam L, Lambert J, Latouche A, Bellis G, Burgel PR, Hocine MN. A 3-year prognostic score for adults with cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2017; 16(6): 702–8. [DOI] [PubMed] [Google Scholar]
- 291.Coriati A, Sykes J, Nkam L, Hocine MN, Burgel PR, Stephenson AL. Validation of the French 3- year prognostic score using the Canadian Cystic Fibrosis registry. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2018. [DOI] [PubMed] [Google Scholar]
- 292.Alaa AM, van der Schaar M. Prognostication and Risk Factors for Cystic Fibrosis via Automated Machine Learning. Sci Rep 2018; 8(1): 11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Ramos KJ, Somayaji R, Lease ED, Goss CH, Aitken ML. Cystic fibrosis physicians’ perspectives on the timing of referral for lung transplant evaluation: a survey of physicians in the United States. BMC pulmonary medicine 2017; 17(1): 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Ramos KJ, Hobler MR, Engelberg RA, et al. Addressing lung transplant with adults with cystic fibrosis: A qualitative analysis of patients’ perspectives and experiences. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Ramos KJ, Smith PJ, McKone EF, et al. Lung transplant referral for individuals with cystic fibrosis: Cystic Fibrosis Foundation consensus guidelines. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Egan TM, Murray S, Bustami RT, et al. Development of the new lung allocation system in the United States. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2006; 6(5 Pt 2): 1212–27. [DOI] [PubMed] [Google Scholar]
- 297.Li SS, Miller R, Tumin D, Stewart WCL, Tobias JD, Hayes D Jr., Lung Allocation Score Thresholds Prioritize Survival After Lung Transplantation. Chest 2019. [DOI] [PubMed] [Google Scholar]
- 298.Braun AT, Dasenbrook EC, Shah AS, Orens JB, Merlo CA. Impact of lung allocation score on survival in cystic fibrosis lung transplant recipients. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation 2015; 34(11): 1436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Roux A, Beaumont-Azuar L, Hamid AM, et al. High Emergency Lung Transplantation: dramatic decrease of waiting list death rate without relevant higher post-transplant mortality. Transplant international : official journal of the European Society for Organ Transplantation 2015; 28(9): 1092–101. [DOI] [PubMed] [Google Scholar]
- 300.Boussaud V, Mal H, Trinquart L, et al. One-year experience with high-emergency lung transplantation in France. Transplantation 2012; 93(10): 1058–63. [DOI] [PubMed] [Google Scholar]
- 301.Chaparro C, Keshavjee S. Lung transplantation for cystic fibrosis: an update. Expert review of respiratory medicine 2016; 10(12): 1269–80. [DOI] [PubMed] [Google Scholar]
- 302.Urquhart DS, Thia LP, Francis J, et al. Deaths in childhood from cystic fibrosis: 10-year analysis from two London specialist centres. Arch Dis Child 2013; 98(2): 123–7. [DOI] [PubMed] [Google Scholar]
- 303.McCormick J, Mehta G, Olesen HV, et al. Comparative demographics of the European cystic fibrosis population: a cross-sectional database analysis. Lancet 2010; 375(9719): 1007–13. [DOI] [PubMed] [Google Scholar]
- 304.ECFS Patient Registry: Annual Data Report 2016.
- 305.Blasi F, Elborn JS, Palange P. Adults with cystic fibrosis and pulmonologists: new training needed to recruit future specialists. Eur Respir J 2019; 53(1). [DOI] [PubMed] [Google Scholar]
- 306.Kerem E, Conway S, Elborn S, Heijerman H. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros 2005; 4: 7–26. [DOI] [PubMed] [Google Scholar]
- 307.Balaguer A, Gonzalez de Dios J. Home versus hospital intravenous antibiotic therapy for cystic fibrosis. The Cochrane database of systematic reviews 2015; (12): CD001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Collaco JM, Green DM, Cutting GR, Naughton KM, Mogayzel PJ Jr., Location and duration of treatment of cystic fibrosis respiratory exacerbations do not affect outcomes. Am J Respir Crit Care Med 2010; 182(9): 1137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Burgess JC, Bridges N, Banya W, et al. HbA1c as a screening tool for cystic fibrosis related diabetes. J Cyst Fibros 2016; 15(2): 251–7. [DOI] [PubMed] [Google Scholar]
- 310.Burnet E, Hubert D, Champreux J, et al. A prospective analysis of unplanned patient-initiated contacts in an adult cystic fibrosis centre. J Cyst Fibros 2018; 17(5): 636–42. [DOI] [PubMed] [Google Scholar]
- 311.registry B. Registro Brasileiro de Fibrose Cistica 2016.
- 312.www.cfww.org.
- 313.Zolin A, Bossi A, Cirilli N, Kashirskaya N, Padoan R. Cystic Fibrosis Mortality in Childhood. Data from European Cystic Fibrosis Society Patient Registry. Int J Environ Res Public Health 2018; 15(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Mehta G, Macek M Jr., Mehta A, European Registry Working G. Cystic fibrosis across Europe: EuroCareCF analysis of demographic data from 35 countries. J Cyst Fibros 2010; 9 Suppl 2: S5–S21. [DOI] [PubMed] [Google Scholar]
- 315.Madge S, Bell SC, Burgel PR, et al. Limitations to providing adult cystic fibrosis care in Europe: Results of a care centre survey. J Cyst Fibros 2017; 16(1): 85–8. [DOI] [PubMed] [Google Scholar]
- 316.Walicka-Serzysko K, Peckova M, Noordhoek JJ, Sands D, Drevinek P. Insights into the cystic fibrosis care in Eastern Europe: Results of survey. J Cyst Fibros 2018; 17(4): 475–7. [DOI] [PubMed] [Google Scholar]
- 317.Rodrigues ME, Melo MC, Reis FJ, Penna FJ. Concentration of electrolytes in the sweat of malnourished children. Arch Dis Child 1994; 71(2): 141–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Yigit H, Selimoglu MA, Altinkaynak S. Sweat test results in children with primary protein energy malnutrition. J Pediatr Gastroenterol Nutr 2003; 37(3): 242–5. [DOI] [PubMed] [Google Scholar]
- 319.Mutesa L, Azad AK, Verhaeghe C, et al. Genetic analysis of Rwandan patients with cystic fibrosis-like symptoms: identification of novel cystic fibrosis transmembrane conductance regulator and epithelial sodium channel gene variants. Chest 2009; 135(5): 1233–42. [DOI] [PubMed] [Google Scholar]
- 320.Mattar AC, Leone C, Rodrigues JC, Adde FV. Sweat conductivity: an accurate diagnostic test for cystic fibrosis? Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2014; 13(5): 528–33. [DOI] [PubMed] [Google Scholar]
- 321.Lezana JL, Vargas MH, Karam-Bechara J, Aldana RS, Furuya ME. Sweat conductivity and chloride titration for cystic fibrosis diagnosis in 3834 subjects. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2003; 2(1): 1–7. [DOI] [PubMed] [Google Scholar]
- 322.Zampoli M. Cystic fibrosis: What’s new in South Africa in 2019. SAMJ: South African Medical Journal 2019; 109(1): 16–9. [DOI] [PubMed] [Google Scholar]
- 323.Liu J, Lewinger JP, Gilliland FD, Gauderman WJ, Conti DV. Confounding and heterogeneity in genetic association studies with admixed populations. Am J Epidemiol 2013; 177(4): 351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Ruiz-Cabezas JC, Barros F, Sobrino B, et al. Mutational analysis of CFTR in the Ecuadorian population using next-generation sequencing. Gene 2019; 696: 28–32. [DOI] [PubMed] [Google Scholar]
- 325.Gutierrez HH, Sanchez I, Schidlow DV. Cystic fibrosis care in Chile. Curr Opin Pulm Med 2009; 15(6): 632–7. [DOI] [PubMed] [Google Scholar]
- 326.Mogayzel PJ Jr., Dunitz J, Marrow LC, Hazle LA. Improving chronic care delivery and outcomes: the impact of the cystic fibrosis Care Center Network. BMJ Qual Saf 2014; 23 Suppl 1: i3–8. [DOI] [PubMed] [Google Scholar]
- 327.Sabadosa KA, Godfrey MM, Marshall BC. Trans-Atlantic collaboration: applying lessons learned from the US CF Foundation quality improvement initiative. Orphanet J Rare Dis 2018; 13(Suppl 1): 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Nelson EC, Batalden PB, Mohr JJ, Plume SK. Building a quality future. Front Health Serv Manage 1998; 15(1): 3–32. [PubMed] [Google Scholar]
- 329.Godfrey MM, Oliver BJ. Accelerating the rate of improvement in cystic fibrosis care: contributions and insights of the learning and leadership collaborative. BMJ Qual Saf 2014; 23 Suppl 1: i23–i32. [DOI] [PubMed] [Google Scholar]
- 330.Marshall BC, Nelson EC. Accelerating implementation of biomedical research advances: critical elements of a successful 10 year Cystic Fibrosis Foundation healthcare delivery improvement initiative. BMJ Qual Saf 2014; 23 Suppl 1: i95–i103. [DOI] [PubMed] [Google Scholar]
- 331.Schweitzer SO, Comanor WS. Prices of pharmaceuticals in poor countries are much lower than in wealthy countries. Health Aff (Millwood) 2011; 30(8): 1553–61. [DOI] [PubMed] [Google Scholar]
- 332.Patel MR, Press VG, Gerald LB, et al. Improving the Affordability of Prescription Medications for People with Chronic Respiratory Disease. An Official American Thoracic Society Policy Statement. Am J Respir Crit Care Med 2018; 198(11): 1367–74. [DOI] [PubMed] [Google Scholar]
- 333.Kremer M. Pharmaceuticals and the developing world. J Econ Perspect 2002; 16(4): 67–90. [DOI] [PubMed] [Google Scholar]
- 334.Matel JL, Milla CE. Nutrition in cystic fibrosis. Semin Respir Crit Care Med 2009; 30(5): 579–86. [DOI] [PubMed] [Google Scholar]
- 335.Cohen-Cymberknoh M, Shoseyov D, Breuer O, Shamali M, Wilschanski M, Kerem E. Treatment of cystic fibrosis in low-income countries. Lancet Respir Med 2016; 4(2): 91–2. [DOI] [PubMed] [Google Scholar]
- 336.McIlwaine M, Button B, Dwan K. Positive expiratory pressure physiotherapy for airway clearance in people with cystic fibrosis. Cochrane Database Syst Rev 2015; (6): CD003147. [DOI] [PubMed] [Google Scholar]
- 337.Prescott WA Jr., A survey of extended-interval aminoglycoside dosing practices in United States adult cystic fibrosis programs. Respir Care 2014; 59(9): 1353–9. [DOI] [PubMed] [Google Scholar]
- 338.Nikolaizik WH, Vietzke D, Ratjen F. A pilot study to compare tobramycin 80 mg injectable preparation with 300 mg solution for inhalation in cystic fibrosis patients. Can Respir J 2008; 15(5): 259–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Fink AK, Loeffler DR, Marshall BC, Goss CH, Morgan WJ. Data that empower: The success and promise of CF patient registries. Pediatr Pulmonol 2017; 52(S48): S44–S51. [DOI] [PubMed] [Google Scholar]
- 340.Muller I, Smith T, Mellor S, Rare L, Genton B. The effect of distance from home on attendance at a small rural health centre in Papua New Guinea. Int J Epidemiol 1998; 27(5): 878–84. [DOI] [PubMed] [Google Scholar]
- 341.Okwaraji YB, Edmond KM. Proximity to health services and child survival in low- and middle-income countries: a systematic review and meta-analysis. BMJ Open 2012; 2(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Clark RA, Coffee N, Turner D, et al. Application of geographic modeling techniques to quantify spatial access to health services before and after an acute cardiac event: the Cardiac Accessibility and Remoteness Index for Australia (ARIA) project. Circulation 2012; 125(16): 2006–14. [DOI] [PubMed] [Google Scholar]
- 343.Kapwata T, Manda S. Geographic assessment of access to health care in patients with cardiovascular disease in South Africa. BMC Health Serv Res 2018; 18(1): 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Zeng W, Li G, Ahn H, Nguyen HTH, Shepard DS, Nair D. Cost-effectiveness of health systems strengthening interventions in improving maternal and child health in low- and middle-income countries: a systematic review. Health Policy Plan 2018; 33(2): 283–97. [DOI] [PubMed] [Google Scholar]
- 345.Martinez-Millana A, Zettl A, Floch J, et al. The Potential of Self-Management mHealth for Pediatric Cystic Fibrosis: Mixed-Methods Study for Health Care and App Assessment. JMIR Mhealth Uhealth 2019; 7(4): e13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Hilliard ME, Hahn A, Ridge AK, Eakin MN, Riekert KA. User Preferences and Design Recommendations for an mHealth App to Promote Cystic Fibrosis Self-Management. JMIR Mhealth Uhealth 2014; 2(4): e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.http://portalgbefc.org.br/.
- 348.http://www.sacfa.org.za/.
- 349.Taylor-Robinson D, Schechter MS. Health inequalities and cystic fibrosis. BMJ 2011; 343: d4818. [DOI] [PubMed] [Google Scholar]
- 350.Taylor-Robinson DC, Smyth RL, Diggle PJ, Whitehead M. The effect of social deprivation on clinical outcomes and the use of treatments in the UK cystic fibrosis population: a longitudinal study. Lancet Respir Med 2013; 1(2): 121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Van Stormbroek B, Zampoli M, Morrow BM. Nebulized gentamicin in combination with systemic antibiotics for eradicating early Pseudomonas aeruginosa infection in children with cystic fibrosis. Pediatr Pulmonol 2019; 54(4): 393–8. [DOI] [PubMed] [Google Scholar]
- 352.Vahedi L, Jabarpoor-Bonyadi M, Ghojazadeh M, Hazrati H, Rafeey M. Association Between Outcomes and Demographic Factors in an Azeri Turkish Population With Cystic Fibrosis: A Cross-Sectional Study in Iran From 2001 Through 2014. Iran Red Crescent Med J 2016; 18(4): e29615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Roberts JM, Wilcox PG, Quon BS. Evaluating Adult Cystic Fibrosis Care in BC: Disparities in Access to a Multidisciplinary Treatment Centre. Can Respir J 2016; 2016: 8901756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Thomas CL, O’Rourke PK, Wainwright CE. Clinical outcomes of Queensland children with cystic fibrosis: a comparison between tertiary centre and outreach services. Med J Aust 2008; 188(3): 135–9. [DOI] [PubMed] [Google Scholar]
- 355.Wood J, Mulrennan S, Hill K, Cecins N, Morey S, Jenkins S. Telehealth clinics increase access to care for adults with cystic fibrosis living in rural and remote Western Australia. J Telemed Telecare 2017; 23(7): 673–9. [DOI] [PubMed] [Google Scholar]
- 356.Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 2009; 106(44): 18825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Jih KY, Hwang TC. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc Natl Acad Sci U S A 2013; 110(11): 4404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Eckford PD, Li C, Ramjeesingh M, Bear CE. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem 2012; 287(44): 36639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Galietta LV, Jayaraman S, Verkman AS. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am J Physiol Cell Physiol 2001; 281(5): C1734–42. [DOI] [PubMed] [Google Scholar]
- 360.Pedemonte N, Lukacs GL, Du K, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest 2005; 115(9): 2564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 2011; 108(46): 18843–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Van Goor F, Straley KS, Cao D, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol 2006; 290(6): L1117–30. [DOI] [PubMed] [Google Scholar]
- 363.Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 2010; 363(21): 1991–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Graeber SY, Hug MJ, Sommerburg O, et al. Intestinal Current Measurements Detect Activation of Mutant CFTR in Patients with Cystic Fibrosis with the G551D Mutation Treated with Ivacaftor. Am J Respir Crit Care Med 2015; 192(10): 1252–5. [DOI] [PubMed] [Google Scholar]
- 365.Rowe SM, Heltshe SL, Gonska T, et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med 2014; 190(2): 175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Ponzano S, Nigrelli G, Fregonese L, et al. A European regulatory perspective on cystic fibrosis: current treatments, trends in drug development and translational challenges for CFTR modulators. Eur Respir Rev 2018; 27(148). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros 2014; 13(1): 29–36. [DOI] [PubMed] [Google Scholar]
- 368.Yu H, Burton B, Huang CJ, et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros 2012; 11(3): 237–45. [DOI] [PubMed] [Google Scholar]
- 369.Sosnay PR, Siklosi KR, Van Goor F, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet 2013; 45(10): 1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Ratner M. FDA deems in vitro data on mutations sufficient to expand cystic fibrosis drug label. Nat Biotechnol 2017; 35(7): 606. [DOI] [PubMed] [Google Scholar]
- 371.Sun X, Yi Y, Yan Z, et al. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci Transl Med 2019; 11(485). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Rosenfeld M, Wainwright CE, Higgins M, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir Med 2018; 6(7): 545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Bessonova L, Volkova N, Higgins M, et al. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax 2018; 73(8): 731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Rabeh WM, Bossard F, Xu H, et al. Correction of both NBD1 energetics and domain interface is required to restore DeltaF508 CFTR folding and function. Cell 2012; 148(1–2): 150–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Mendoza JL, Schmidt A, Li Q, et al. Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell 2012; 148(1–2): 164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Wang C, Protasevich I, Yang Z, et al. Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein Sci 2010; 19(10): 1932–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Clancy JP, Rowe SM, Accurso FJ, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012; 67(1): 12–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Flume PA, Liou TG, Borowitz DS, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012; 142(3): 718–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Boyle MP, Bell SC, Konstan MW, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med 2014; 2(7): 527–38. [DOI] [PubMed] [Google Scholar]
- 380.Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med 2017; 377(21): 2013–23. [DOI] [PubMed] [Google Scholar]
- 381.Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med 2018; 197(2): 214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Rowe SM, Daines C, Ringshausen FC, et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med 2017; 377(21): 2024–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Davies JC, Moskowitz SM, Brown C, et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med 2018; 379(17): 1599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Keating D, Marigowda G, Burr L, et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med 2018; 379(17): 1612–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Molinski SV, Ahmadi S, Ip W, et al. Orkambi(R) and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol Med 2017; 9(9): 1224–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Mutyam V, Libby EF, Peng N, et al. Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J Cyst Fibros 2017; 16(1): 24–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Berkers G, van Mourik P, Vonk AM, et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep 2019; 26(7): 1701–8 e3. [DOI] [PubMed] [Google Scholar]
- 388.Graeber SY, Dopfer C, Naehrlich L, et al. Effects of Lumacaftor-Ivacaftor Therapy on Cystic Fibrosis Transmembrane Conductance Regulator Function in Phe508del Homozygous Patients with Cystic Fibrosis. Am J Respir Crit Care Med 2018; 197(11): 1433–42. [DOI] [PubMed] [Google Scholar]
- 389.Bedwell DM, Kaenjak A, Benos DJ, et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med 1997; 3(11): 1280–4. [DOI] [PubMed] [Google Scholar]
- 390.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med 1996; 2(4): 467–9. [DOI] [PubMed] [Google Scholar]
- 391.Wilschanski M, Yahav Y, Yaacov Y, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med 2003; 349(15): 1433–41. [DOI] [PubMed] [Google Scholar]
- 392.Clancy JP, Bebok Z, Ruiz F, et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J Respir Crit Care Med 2001; 163(7): 1683–92. [DOI] [PubMed] [Google Scholar]
- 393.Wilschanski M, Miller LL, Shoseyov D, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J 2011; 38(1): 59–69. [DOI] [PubMed] [Google Scholar]
- 394.Kerem E, Konstan MW, De Boeck K, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med 2014; 2(7): 539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Sermet-Gaudelus I, Boeck KD, Casimir GJ, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med 2010; 182(10): 1262–72. [DOI] [PubMed] [Google Scholar]
- 396.Xue X, Mutyam V, Tang L, et al. Synthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftor. Am J Respir Cell Mol Biol 2014; 50(4): 805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Mutyam V, Du M, Xue X, et al. Discovery of Clinically Approved Agents That Promote Suppression of Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations. Am J Respir Crit Care Med 2016; 194(9): 1092–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Roy B, Friesen WJ, Tomizawa Y, et al. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc Natl Acad Sci U S A 2016; 113(44): 12508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Xue X, Mutyam V, Thakerar A, et al. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Hum Mol Genet 2017; 26(16): 3116–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.van Deutekom JC, Janson AA, Ginjaar IB, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med 2007; 357(26): 2677–86. [DOI] [PubMed] [Google Scholar]
- 401.Anthony K, Arechavala-Gomeza V, Ricotti V, et al. Biochemical characterization of patients with in-frame or out-of-frame DMD deletions pertinent to exon 44 or 45 skipping. JAMA Neurol 2014; 71(1): 32–40. [DOI] [PubMed] [Google Scholar]
- 402.Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med 2017; 377(18): 1723–32. [DOI] [PubMed] [Google Scholar]
- 403.Pranke I, Golec A, Hinzpeter A, Edelman A, Sermet-Gaudelus I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front Pharmacol 2019; 10: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Montoro DT, Haber AL, Biton M, et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018; 560(7718): 319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Cholon DM, Gentzsch M. Recent progress in translational cystic fibrosis research using precision medicine strategies. J Cyst Fibros 2018; 17(2S): S52–S60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Clancy JP, Cotton CU, Donaldson SH, et al. CFTR modulator theratyping: Current status, gaps and future directions. J Cyst Fibros 2019; 18(1): 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Eckford PDW, McCormack J, Munsie L, et al. The CF Canada-Sick Kids Program in individual CF therapy: A resource for the advancement of personalized medicine in CF. J Cyst Fibros 2019; 18(1): 35–43. [DOI] [PubMed] [Google Scholar]
- 408.Gentzsch M, Boyles SE, Cheluvaraju C, et al. Pharmacological Rescue of Conditionally Reprogrammed Cystic Fibrosis Bronchial Epithelial Cells. Am J Respir Cell Mol Biol 2017; 56(5): 568–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Veit G, Xu H, Dreano E, et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat Med 2018; 24(11): 1732–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Brewington JJ, Filbrandt ET, LaRosa FJ 3rd, et al. Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight 2018; 3(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Pranke I, Hatton A, Masson A, et al. Might Brushed Nasal Cells Be a Surrogate for CFTR Modulator Clinical Response? Am J Respir Crit Care Med 2019; 199(1): 123–6. [DOI] [PubMed] [Google Scholar]
- 412.Wong AP, Bear CE, Chin S, et al. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat Biotechnol 2012; 30(9): 876–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Dekkers JF, Wiegerinck CL, de Jonge HR, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med 2013; 19(7): 939–45. [DOI] [PubMed] [Google Scholar]
- 414.van Mourik P, Beekman JM, van der Ent CK. Intestinal organoids to model Cystic Fibrosis. Eur Respir J 2019. [DOI] [PubMed] [Google Scholar]
- 415.Guimbellot JS, Leach JM, Chaudhry IG, et al. Nasospheroids permit measurements of CFTR-dependent fluid transport. JCI Insight 2017; 2(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Hirtz S, Gonska T, Seydewitz HH, et al. CFTR Cl-channel function in native human colon correlates with the genotype and phenotype in cystic fibrosis. Gastroenterology 2004; 127(4): 1085–95. [DOI] [PubMed] [Google Scholar]
- 417.Masson A, Schneider-Futschik EK, Baatallah N, et al. Predictive factors for lumacaftor/ivacaftor clinical response. J Cyst Fibros 2018. [DOI] [PubMed] [Google Scholar]
- 418.De Boeck K, Kent L, Davies J, et al. CFTR biomarkers: time for promotion to surrogate end-point. Eur Respir J 2013; 41(1): 203–16. [DOI] [PubMed] [Google Scholar]
- 419.JC D. https://www.sciencedirect.com/science/article/pii/S1569199319301110?via%3Dihub. 2019.
- 420.De Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros 2014; 13(6): 674–80. [DOI] [PubMed] [Google Scholar]
- 421.Giuliano KA, Wachi S, Drew L, et al. Use of a High-Throughput Phenotypic Screening Strategy to Identify Amplifiers, a Novel Pharmacological Class of Small Molecules That Exhibit Functional Synergy with Potentiators and Correctors. SLAS Discov 2018; 23(2): 111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Mayer-Hamblett N, Boyle M, VanDevanter D. Advancing clinical development pathways for new CFTR modulators in cystic fibrosis. Thorax 2016; 71(5): 454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.VanDevanter DR, Mayer-Hamblett N. Innovating cystic fibrosis clinical trial designs in an era of successful standard of care therapies. Curr Opin Pulm Med 2017; 23(6): 530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Goss CH, Bell SC. Aztreonam for inhalation solution, challenges to drug approval and integration into CF care. J Cyst Fibros 2013; 12(2): 99–101. [DOI] [PubMed] [Google Scholar]
- 425.VanDevanter DR, Mayer-Hamblett N, Boyle M. Feasibility of placebo-controlled trial designs for new CFTR modulator evaluation. J Cyst Fibros 2017; 16(4): 496–8. [DOI] [PubMed] [Google Scholar]
- 426.https://www.ema.europa.eu/en/documents/report/report-workshop-patients-voice-evaluation-medicines_en.pdf.
- 427.Nichols DP, Durmowicz AG, Field A, Flume PA, VanDevanter DR, Mayer-Hamblett N. Developing Inhaled Antibiotics in Cystic Fibrosis: Current Challenges and Opportunities. Ann Am Thorac Soc 2019; 16(5): 534–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Perrem LRF. Anti-inflammatories and mucociliary clearance therapies in the age of CFTR modulators. Pediatr Pulmonol 2019; in press. [DOI] [PubMed] [Google Scholar]
- 429.van Horck M, Winkens B, Wesseling G, et al. Early detection of pulmonary exacerbations in children with Cystic Fibrosis by electronic home monitoring of symptoms and lung function. Sci Rep 2017; 7(1): 12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Lechtzin N, Mayer-Hamblett N, West NE, et al. Home Monitoring of Patients with Cystic Fibrosis to Identify and Treat Acute Pulmonary Exacerbations. eICE Study Results. Am J Respir Crit Care Med 2017; 196(9): 1144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Zwart TC, Gokoel SRM, van der Boog PJM, et al. Therapeutic drug monitoring of tacrolimus and mycophenolic acid in outpatient renal transplant recipients using a volumetric dried blood spot sampling device. Br J Clin Pharmacol 2018; 84(12): 2889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Elliott TG, Dooley KC, Zhang M, Campbell HSD, Thompson DJS. Comparison of Glycated Hemoglobin Results Based on At-Home and In-Lab Dried Blood Spot Sampling to Routine Venous Blood Sampling In-Lab in Adult Patients With Type 1 or Type 2 Diabetes. Can J Diabetes 2018; 42(4): 426–32 e1. [DOI] [PubMed] [Google Scholar]
- 433.Subbarao P, Stanojevic S, Brown M, et al. Lung clearance index as an outcome measure for clinical trials in young children with cystic fibrosis. A pilot study using inhaled hypertonic saline. Am J Respir Crit Care Med 2013; 188(4): 456–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Stahl M, Graeber SY, Joachim C, et al. Three-center feasibility of lung clearance index in infants and preschool children with cystic fibrosis and other lung diseases. J Cyst Fibros 2018; 17(2): 249–55. [DOI] [PubMed] [Google Scholar]
- 435.Mott LS, Park J, Murray CP, et al. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax 2012; 67(6): 509–16. [DOI] [PubMed] [Google Scholar]
- 436.Sly PD, Gangell CL, Chen L, et al. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med 2013; 368(21): 1963–70. [DOI] [PubMed] [Google Scholar]
- 437.Wielputz MO, Puderbach M, Kopp-Schneider A, et al. Magnetic resonance imaging detects changes in structure and perfusion, and response to therapy in early cystic fibrosis lung disease. Am J Respir Crit Care Med 2014; 189(8): 956–65. [DOI] [PubMed] [Google Scholar]
- 438.Stahl M, Wielputz MO, Graeber SY, et al. Comparison of Lung Clearance Index and Magnetic Resonance Imaging for Assessment of Lung Disease in Children with Cystic Fibrosis. Am J Respir Crit Care Med 2017; 195(3): 349–59. [DOI] [PubMed] [Google Scholar]
- 439.Pennati F, Salito C, Borzani I, et al. Quantitative multivolume proton-magnetic resonance imaging in patients with cystic fibrosis lung disease: comparison with clinical indicators. Eur Respir J 2019; 53(5). [DOI] [PubMed] [Google Scholar]
- 440.Wielputz MO, von Stackelberg O, Stahl M, et al. Multicentre standardisation of chest MRI as radiation-free outcome measure of lung disease in young children with cystic fibrosis. J Cyst Fibros 2018; 17(4): 518–27. [DOI] [PubMed] [Google Scholar]
- 441.https://www.ema.europa.eu/en/documents/report/report-workshop-endpoints-cystic-fibrosis-clinical-trials_en.pdf.
- 442.Mall MA, Hartl D. CFTR: cystic fibrosis and beyond. Eur Respir J 2014; 44(4): 1042–54. [DOI] [PubMed] [Google Scholar]
- 443.Dasenbrook EC, Sawicki GS. Cystic fibrosis patient registries: A valuable source for clinical research. J Cyst Fibros 2018; 17(4): 433–40. [DOI] [PubMed] [Google Scholar]
- 444.Goss CH, Mayer-Hamblett N, Kronmal RA, Ramsey BW. The cystic fibrosis therapeutics development network (CF TDN): a paradigm of a clinical trials network for genetic and orphan diseases. Adv Drug Deliv Rev 2002; 54(11): 1505–28. [DOI] [PubMed] [Google Scholar]
- 445.De Boeck K, Bulteel V, Tiddens H, et al. Guideline on the design and conduct of cystic fibrosis clinical trials: the European Cystic Fibrosis Society-Clinical Trials Network (ECFS-CTN). J Cyst Fibros 2011; 10 Suppl 2: S67–74. [DOI] [PubMed] [Google Scholar]
- 446.https://www.ema.europa.eu/en/partners-networks/international-activities.
- 447.https://www.ema.europa.eu/documents/regulatory-procedural-guideline/ema-regulatory-science-2025-strategic-reflection_en.pdf.
- 448.https://www.ema.europa.eu/en/documents/report/report-european-network-paediatric-research-european-medicines-agency-workshop-gastrointestinal-gi_en.pdf.
- 449.https://www.ema.europa.eu/documents/regulatory-procedural-guideline/qualification-opinion-european-cystic-fibrosis-society-patient-registry-ecfspr-cf-pharmaco_en.pdf.
- 450.https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-advice-protocol-assistance/parallel-consultation-regulators-health-technology-assessment-bodies.
- 451.Wise J. NHS and Vertex remain deadlocked over price of cystic fibrosis drug. BMJ 2019; 364: l1094. [DOI] [PubMed] [Google Scholar]
- 452.McKone EF, Emerson SS, Edwards KL, Aitken ML. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet 2003; 361(9370): 1671–6. [DOI] [PubMed] [Google Scholar]
- 453.Wilschanski M, Dupuis A, Ellis L, et al. Mutations in the cystic fibrosis transmembrane regulator gene and in vivo transepithelial potentials. Am J Respir Crit Care Med 2006; 174(7): 787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Ronan NJ, Einarsson GG, Twomey M, et al. CORK Study in Cystic Fibrosis: Sustained Improvements in Ultra-Low-Dose Chest CT Scores After CFTR Modulation With Ivacaftor. Chest 2018; 153(2): 395–403. [DOI] [PubMed] [Google Scholar]
- 455.Hisert KB, Heltshe SL, Pope C, et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am J Respir Crit Care Med 2017; 195(12): 1617–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Konstan MW, McKone EF, Moss RB, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med 2017; 5(2): 107–18. [DOI] [PubMed] [Google Scholar]
- 457.Sly PD, Brennan S, Gangell C, et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med 2009; 180(2): 146–52. [DOI] [PubMed] [Google Scholar]
- 458.Castellani C, Massie J, Sontag M, Southern KW. Newborn screening for cystic fibrosis. Lancet Respir Med 2016; 4(8): 653–61. [DOI] [PubMed] [Google Scholar]
- 459.Trimble AT, Donaldson SH. Ivacaftor withdrawal syndrome in cystic fibrosis patients with the G551D mutation. J Cyst Fibros 2018; 17(2): e13–e6. [DOI] [PubMed] [Google Scholar]
- 460.Savitski MM, Reinhard FB, Franken H, et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014; 346(6205): 1255784. [DOI] [PubMed] [Google Scholar]
- 461.Hou Y, Guan X, Yang Z, Li C. Emerging role of cystic fibrosis transmembrane conductance regulator - an epithelial chloride channel in gastrointestinal cancers. World J Gastrointest Oncol 2016; 8(3): 282–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Floto RA, Olivier KN, Saiman L, et al. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis. Thorax 2016; 71 Suppl 1: i1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Kaiser J. Personalized medicine. New cystic fibrosis drug offers hope, at a price. Science 2012; 335(6069): 645. [DOI] [PubMed] [Google Scholar]
- 464.Jones AM, Barry PJ. Lumacaftor/ivacaftor for patients homozygous for Phe508del-CFTR: should we curb our enthusiasm? Thorax 2015; 70(7): 615–6. [DOI] [PubMed] [Google Scholar]
- 465.Claxton K, Martin S, Soares M, et al. Methods for the estimation of the National Institute for Health and Care Excellence cost-effectiveness threshold. Health Technol Assess 2015; 19(14): 1–503, v–vi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Claxton K, Sculpher M, Palmer S, Culyer AJ. Causes for concern: is NICE failing to uphold its responsibilities to all NHS patients? Health Econ 2015; 24(1): 1–7. [DOI] [PubMed] [Google Scholar]
- 467.Appleby J. Crossing the line: NICE’s value for money threshold. BMJ 2016; 352: i1336. [DOI] [PubMed] [Google Scholar]
- 468.Collins M, Latimer N. NICE’s end of life decision making scheme: impact on population health. BMJ 2013; 346: f1363. [DOI] [PubMed] [Google Scholar]
- 469.Paulden M. Recent amendments to NICE’s value-based assessment of health technologies: implicitly inequitable? Expert Rev Pharmacoecon Outcomes Res 2017; 17(3): 239–42. [DOI] [PubMed] [Google Scholar]
- 470.Vernon JA, Goldberg R, Golec J. Economic evaluation and cost-effectiveness thresholds: signals to firms and implications for R & D investment and innovation. Pharmacoeconomics 2009; 27(10): 797–806. [DOI] [PubMed] [Google Scholar]
- 471.Williams A. Intergenerational equity: an exploration of the ‘fair innings’ argument. Health Econ 1997; 6(2): 117–32. [DOI] [PubMed] [Google Scholar]
- 472.Whiting P, Al M, Burgers L, et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health Technol Assess 2014; 18(18): 1–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.http://www.ncpe.ie/wp-content/uploads/2015/12/Ivacaftor-2-5-years-web-summary.pdf.
- 474. https://www.scottishmedicines.org.uk/medicines-advice/ivacaftor-kalydeco-fullsubmission-119316/
- 475.Schlander M, Garattini S, Holm S, et al. Incremental cost per quality-adjusted life year gained? The need for alternative methods to evaluate medical interventions for ultra-rare disorders. J Comp Eff Res 2014; 3(4): 399–422. [DOI] [PubMed] [Google Scholar]
- 476.https://www.who.int/en/news-room/fact-sheets/detail/substandard-and-falsified-medical-products.
- 477.Hoen E, Berger J, Calmy A, Moon S. Driving a decade of change: HIV/AIDS, patents and access to medicines for all. J Int AIDS Soc 2011; 14: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Global Burden of Disease Health Financing Collaborator N. Past, present, and future of global health financing: a review of development assistance, government, out-of-pocket, and other private spending on health for 195 countries, 1995–2050. Lancet 2019; 393(10187): 2233–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Picavet E, Morel T, Cassiman D, Simoens S. Shining a light in the black box of orphan drug pricing. Orphanet J Rare Dis 2014; 9: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Schey C, Milanova T, Hutchings A. Estimating the budget impact of orphan medicines in Europe: 2010 – 2020. Orphanet J Rare Dis 2011; 6: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Muther EF, Polineni D, Sawicki GS. Overcoming psychosocial challenges in cystic fibrosis: Promoting resilience. Pediatr Pulmonol 2018; 53(S3): S86–S92. [DOI] [PubMed] [Google Scholar]
- 482.Abbott J, Havermans T, Jarvholm S, et al. Mental Health screening in cystic fibrosis centres across Europe. J Cyst Fibros 2019; 18(2): 299–303. [DOI] [PubMed] [Google Scholar]
- 483.Havermans T, Tack J, Vertommen A, Proesmans M, de Boeck K. Breaking bad news, the diagnosis of cystic fibrosis in childhood. J Cyst Fibros 2015; 14(4): 540–6. [DOI] [PubMed] [Google Scholar]
- 484.Simms R, Cole FS. The many roles of family members in “family-centered care”--part II. Interview by Deborah Dokken. Pediatr Nurs 2007; 33(1): 51–2, 70. [PubMed] [Google Scholar]
- 485.Tluczek A, Clark R, McKechnie AC, Brown RL. Factors affecting parent-child relationships one year after positive newborn screening for cystic fibrosis or congenital hypothyroidism. J Dev Behav Pediatr 2015; 36(1): 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Anna Elderton KV, Trudy Havermans, Doris Staab. Understanding and working with challenging behaviours and fears in pre-school children with Cystic Fibrosis. ECFS publication 2018; The early Cystic Fibrosis Years. [Google Scholar]
- 487.Jamieson N, Fitzgerald D, Singh-Grewal D, Hanson CS, Craig JC, Tong A. Children’s experiences of cystic fibrosis: a systematic review of qualitative studies. Pediatrics 2014; 133(6): e1683–97. [DOI] [PubMed] [Google Scholar]
- 488.Coyne I, Sheehan AM, Heery E, While AE. Improving transition to adult healthcare for young people with cystic fibrosis: A systematic review. J Child Health Care 2017; 21(3): 312–30. [DOI] [PubMed] [Google Scholar]
- 489.Haarbauer-Krupa J, Alexander NM, Mee L, et al. Readiness for transition and health-care satisfaction in adolescents with complex medical conditions. Child Care Health Dev 2019; 45(3): 463–71. [DOI] [PubMed] [Google Scholar]
- 490.Barker H, Moses J, O’Leary C. ‘I’ve got to prioritise’: being a parent with cystic fibrosis. Psychol Health Med 2017; 22(6): 744–52. [DOI] [PubMed] [Google Scholar]
- 491.Nick JA, Nichols DP. Diagnosis of Adult Patients with Cystic Fibrosis. Clin Chest Med 2016; 37(1): 47–57. [DOI] [PubMed] [Google Scholar]
- 492.Sawicki GS, Sellers DE, McGuffie K, Robinson W. Adults with cystic fibrosis report important and unmet needs for disease information. J Cyst Fibros 2007; 6(6): 411–6. [DOI] [PubMed] [Google Scholar]
- 493.Braithwaite M, Philip J, Tranberg H, et al. End of life care in CF: patients, families and staff experiences and unmet needs. J Cyst Fibros 2011; 10(4): 253–7. [DOI] [PubMed] [Google Scholar]
- 494.Quittner AL, Goldbeck L, Abbott J, et al. Prevalence of depression and anxiety in patients with cystic fibrosis and parent caregivers: results of The International Depression Epidemiological Study across nine countries. Thorax 2014; 69(12): 1090–7. [DOI] [PubMed] [Google Scholar]
- 495.Butcher JL, Nasr SZ. Direct observation of respiratory treatments in cystic fibrosis: parent-child interactions relate to medical regimen adherence. J Pediatr Psychol 2015; 40(1): 8–17. [DOI] [PubMed] [Google Scholar]
- 496.Goldbeck L, Fidika A, Herle M, Quittner AL. Psychological interventions for individuals with cystic fibrosis and their families. Cochrane Database Syst Rev 2014; (6): CD003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Quittner AL, Abbott J, Georgiopoulos AM, et al. International Committee on Mental Health in Cystic Fibrosis: Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus statements for screening and treating depression and anxiety. Thorax 2016; 71(1): 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Narayanan S, Mainz JG, Gala S, Tabori H, Grossoehme D. Adherence to therapies in cystic fibrosis: a targeted literature review. Expert Rev Respir Med 2017; 11(2): 129–45. [DOI] [PubMed] [Google Scholar]
- 499.Faint NR, Staton JM, Stick SM, Foster JM, Schultz A. Investigating self-efficacy, disease knowledge and adherence to treatment in adolescents with cystic fibrosis. J Paediatr Child Health 2017; 53(5): 488–93. [DOI] [PubMed] [Google Scholar]
- 500.Oates GR, Stepanikova I, Rowe SM, Gamble S, Gutierrez HH, Harris WT. Objective Versus Self-Reported Adherence to Airway Clearance Therapy in Cystic Fibrosis. Respir Care 2019; 64(2): 176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Velez-Velez E, Bosch RJ. Illness perception, coping and adherence to treatment among patients with chronic kidney disease. J Adv Nurs 2016; 72(4): 849–63. [DOI] [PubMed] [Google Scholar]
- 502.Pakhale S, Baron J, Armstrong M, et al. Lost in translation? How adults living with Cystic Fibrosis understand treatment recommendations from their healthcare providers, and the impact on adherence to therapy. Patient Educ Couns 2016; 99(8): 1319–24. [DOI] [PubMed] [Google Scholar]
- 503.Shakkottai A, Kidwell KM, Townsend M, Nasr SZ. A five-year retrospective analysis of adherence in cystic fibrosis. Pediatr Pulmonol 2015; 50(12): 1224–9. [DOI] [PubMed] [Google Scholar]
- 504.Modi AC, Lim CS, Yu N, Geller D, Wagner MH, Quittner AL. A multi-method assessment of treatment adherence for children with cystic fibrosis. J Cyst Fibros 2006; 5(3): 177–85. [DOI] [PubMed] [Google Scholar]
- 505.Bucks RS, Hawkins K, Skinner TC, Horn S, Seddon P, Horne R. Adherence to treatment in adolescents with cystic fibrosis: the role of illness perceptions and treatment beliefs. J Pediatr Psychol 2009; 34(8): 893–902. [DOI] [PubMed] [Google Scholar]
- 506.Zindani GN, Streetman DD, Streetman DS, Nasr SZ. Adherence to treatment in children and adolescent patients with cystic fibrosis. J Adolesc Health 2006; 38(1): 13–7. [DOI] [PubMed] [Google Scholar]
- 507.Goodfellow NA, Hawwa AF, Reid AJ, Horne R, Shields MD, McElnay JC. Adherence to treatment in children and adolescents with cystic fibrosis: a cross-sectional, multi-method study investigating the influence of beliefs about treatment and parental depressive symptoms. BMC Pulm Med 2015; 15: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Sawicki GS, Tiddens H. Managing treatment complexity in cystic fibrosis: challenges and opportunities. Pediatr Pulmonol 2012; 47(6): 523–33. [DOI] [PubMed] [Google Scholar]
- 509.Flewelling KD, Sellers DE, Sawicki GS, Robinson WM, Dill EJ. Social support is associated with fewer reported symptoms and decreased treatment burden in adults with cystic fibrosis. J Cyst Fibros 2019. [DOI] [PubMed] [Google Scholar]
- 510.Volkova N, Moy K, Evans J, et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J Cyst Fibros 2019. [DOI] [PubMed] [Google Scholar]
- 511.Nakagawa S. Communication--The Most Challenging Procedure. JAMA Intern Med 2015; 175(8): 1268–9. [DOI] [PubMed] [Google Scholar]
- 512.Nakagawa S. Communicating With Patients--The Other Side of the Conversation--Reply. JAMA Intern Med 2015; 175(12): 1997. [DOI] [PubMed] [Google Scholar]
- 513.Takaki H, Abe T, Hagihara A. Physicians’ and pharmacists’ information provision and patients’ psychological distress. J Interprof Care 2017; 31(5): 575–82. [DOI] [PubMed] [Google Scholar]
- 514.Transition to adult care, Ready Steady, Go. www.uhs.nhs.uk/OurServices/Childhealth/TransitiontoadultcareReadySteadyGo/Transitiontoadultcare.aspx
- 515.Cystic Fibrosis Trust: Factsheets, information packs and publications. [Google Scholar]
- 516.Age-Specific Care Guidelines. [Google Scholar]
- 517.Ketchell RI. Telemedicine is the way forward for the management of cystic fibrosis - the case in favour. Paediatr Respir Rev 2018; 26: 19–21. [DOI] [PubMed] [Google Scholar]
- 518.Govan JR, Brown AR, Jones AM. Evolving epidemiology of Pseudomonas aeruginosa and the Burkholderia cepacia complex in cystic fibrosis lung infection. Future Microbiol 2007; 2(2): 153–64. [DOI] [PubMed] [Google Scholar]
- 519.Kidd TJ, Soares Magalhaes RJ, Paynter S, Bell SC, Group ACI. The social network of cystic fibrosis centre care and shared Pseudomonas aeruginosa strain infection: a cross-sectional analysis. Lancet Respir Med 2015; 3(8): 640–50. [DOI] [PubMed] [Google Scholar]
- 520.Cox NS, Alison JA, Rasekaba T, Holland AE. Telehealth in cystic fibrosis: a systematic review. J Telemed Telecare 2012; 18(2): 72–8. [DOI] [PubMed] [Google Scholar]
- 521.Hardisty AR, Peirce SC, Preece A, et al. Bridging two translation gaps: a new informatics research agenda for telemonitoring of chronic disease. Int J Med Inform 2011; 80(10): 734–44. [DOI] [PubMed] [Google Scholar]
- 522.Sarfaraz S, Sund Z, Jarad N. Real-time, once-daily monitoring of symptoms and FEV in cystic fibrosis patients--a feasibility study using a novel device. Clin Respir J 2010; 4(2): 74–82. [DOI] [PubMed] [Google Scholar]
- 523.Chen JJ, Cooper DM, Haddad F, Sladkey A, Nussbaum E, Radom-Aizik S. Tele-Exercise as a Promising Tool to Promote Exercise in Children With Cystic Fibrosis. Front Public Health 2018; 6: 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Garner SL, Sudia T, Rachaprolu S. Smart phone accessibility and mHealth use in a limited resource setting. Int J Nurs Pract 2018; 24(1). [DOI] [PubMed] [Google Scholar]
- 525.Van Allen J, Davis AM, Lassen S. The use of telemedicine in pediatric psychology: research review and current applications. Child Adolesc Psychiatr Clin N Am 2011; 20(1): 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Coleman MT, Newton KS. Supporting self-management in patients with chronic illness. Am Fam Physician 2005; 72(8): 1503–10. [PubMed] [Google Scholar]
- 527.Polonsky WH, Fisher L, Hessler D, Edelman SV. What is so tough about self-monitoring of blood glucose? Perceived obstacles among patients with Type 2 diabetes. Diabet Med 2014; 31(1): 40–6. [DOI] [PubMed] [Google Scholar]
- 528.Daniels T, Goodacre L, Sutton C, Pollard K, Conway S, Peckham D. Accurate assessment of adherence: self-report and clinician report vs electronic monitoring of nebulizers. Chest 2011; 140(2): 425–32. [DOI] [PubMed] [Google Scholar]
- 529.Roehrer E, Cummings E, Beggs S, et al. Pilot evaluation of web enabled symptom monitoring in cystic fibrosis. Inform Health Soc Care 2013; 38(4): 354–65. [DOI] [PubMed] [Google Scholar]
- 530.Roehrer E, Cummings E, Turner P, et al. Supporting cystic fibrosis with ICT. Stud Health Technol Inform 2013; 183: 137–41. [PubMed] [Google Scholar]
- 531.Community unites to call for #OrkambiNow!
- 532.Stephenson AL, Sykes J, Stanojevic S, et al. Survival Comparison of Patients With Cystic Fibrosis in Canada and the United States: A Population-Based Cohort Study. Ann Intern Med 2017; 166(8): 537–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Keogh RH, Bilton D, Cosgriff R, Kavanagh D, Rayner O, Sedgwick PM. Results from an online survey of adults with cystic fibrosis: Accessing and using life expectancy information. PLoS One 2019; 14(4): e0213639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.https://www.cysticfibrosis.org.uk/news/darzi-fellowship-brings-new-insights-into-registry-development. https://www.cysticfibrosis.org.uk/news/darzi-fellowship-brings-new-insights-into-registry-development.