

Abstract
RNA helicases are responsible for virtually all of RNA metabolism. Viral and bacterial pathogens typically encode their own RNA helicases. Hence, this family of enzymes is increasingly recognized as potential targets for treatment of a variety of diseases. However, the conserved structural similarities amongst helicase families present an obstacle to the idea of developing specific inhibitors. In order to identify potential modulators of RNA helicase activity, rapid screening approaches are needed. This has been accomplished by optimizing and adapting standard helicase assays to function in high throughput modalities. These optimized assays have enabled the application of rapid screening approaches to be applied towards discovering helicase inhibitors. This chapter provides detailed protocols for utilizing a medium to high throughput approach for inhibitor discovery.
Keywords: NS3, helicase, nucleic acids, inhibitors
1. Introduction
Helicases participate in the myriad of molecular events associated with DNA and RNA metabolism, hence they have become more commonly considered targets for treatment of human disease (1). Transcription, RNA maturation, splicing, and translation all are dependent on multiple RNA helicases (8). Furthermore, pathogens encode their own RNA helicases, which are necessary for viral (5) and bacterial (11) replication. Newly identified mechanisms of RNA helicase activity in various processes reveals exciting new approaches for treating human disease including cancer (4). RNA helicases are divided into sub-families which share many of the helicase-motifs. Hence, development of inhibitors that are specific for a single member of this class of enzymes is a challenge due to shared structural features. However, similar challenges exist in other enzyme or protein families, for example, proteases, tyrosine kinases, and G-protein coupled receptors. Therefore, successful identification of highly specific inhibitors is well within the capability of existing approaches for drug development.
Search for specific inhibitors of a targeted RNA helicase requires a means to detect enzyme activity quickly and selectively. An outline of the steps involved in discovery of inhibitors of RNA helicases is shown in Figure 1. The process begins with identification of a suitable target helicase, for example, a viral helicase such as NS3 from the Hepatitis C virus. Once identified, a recombinant form of the enzyme is prepared. This process can take significant resources and time because some RNA helicases are difficult to express in large quantities.
Figure 1:
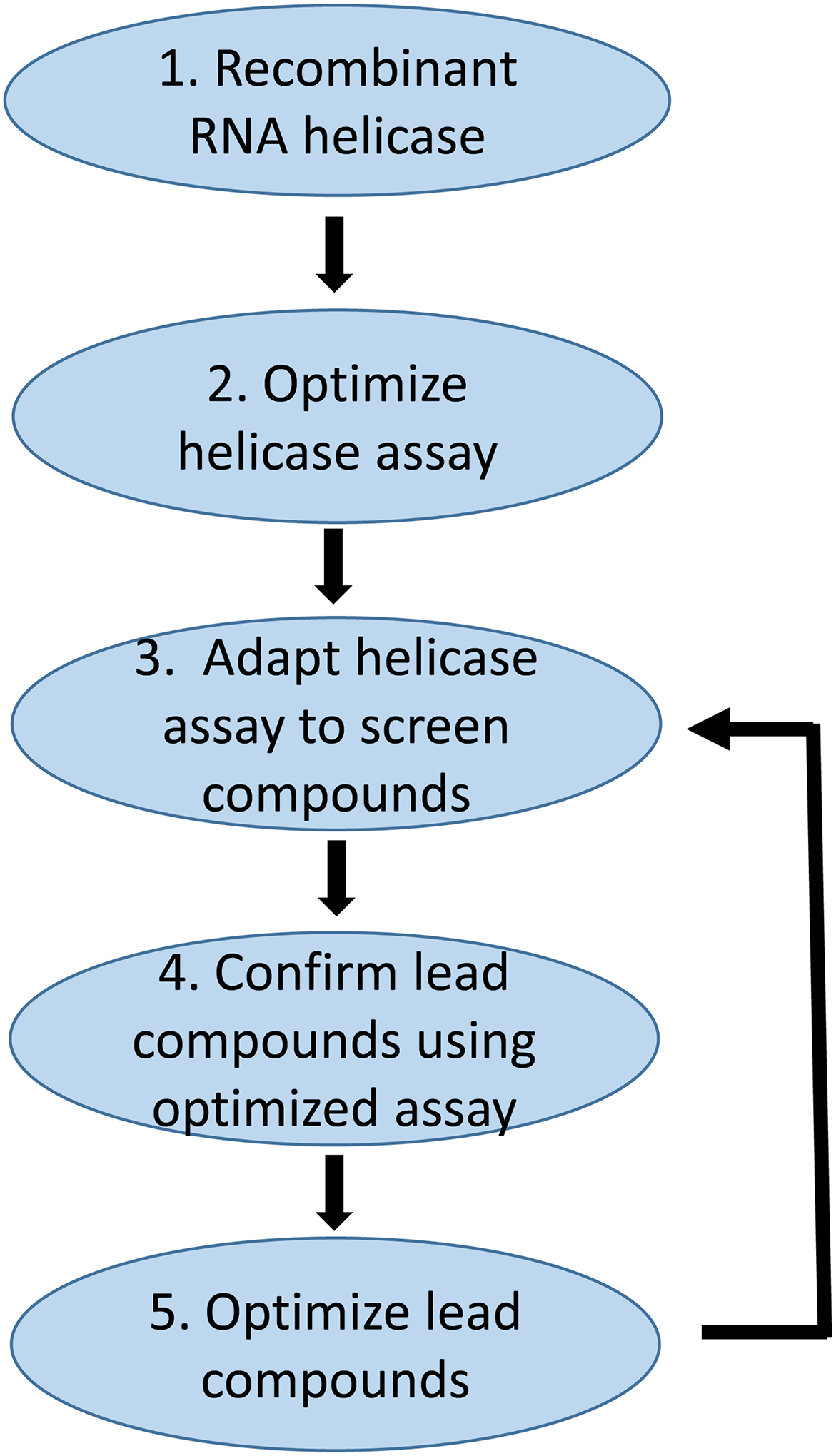
Simplified overview of steps involved in discovery of inhibitors of RNA helicases. Identifying and cloning the specific RNA helicase involves extensive effort but is summarized as step 1 in this figure. Step 2 is to develop an assay to characterize the RNA unwinding activity of the targeted helicase. Step 3 is to adapt the assay to a high throughput screen to test potential inhibitors. Step 4 is to confirm the results from the screening assay for selected inhibitors. Step 5 is to chemically modify potential inhibitors and then re-examine them in an iterative process using the helicase assays. This chapter focuses on steps 2, 3, and 4, to develop the unwinding assays to identify and confirm potential inhibitors.
A standard RNA unwinding assay serves as an initial screening tool. This assay utilizes commercially available oligonucleotides to create the helicase substrate. The first aspect in design of the substrate includes choosing the detection method. The helicase substrate can be radiolabeled on one of the strands, thereby allowing the substrate (duplex) to be separated from the product (single-stranded RNA) by gel electrophoresis (Figure 2). The radiolabeled strand can be detected by phosphorimager analysis. Using oligonucleotides allows for optimization of the substrate. Design of the substrate also takes into consideration the length of the duplex, the directionality of the helicase, the length of the single-strand overhang, and whether an RNA “fork” is preferred to a single-strand overhang. Some RNA helicases can utilize DNA as a substrate, so DNA substrates can sometimes be used to measure RNA helicase unwinding activity.
Figure 2:
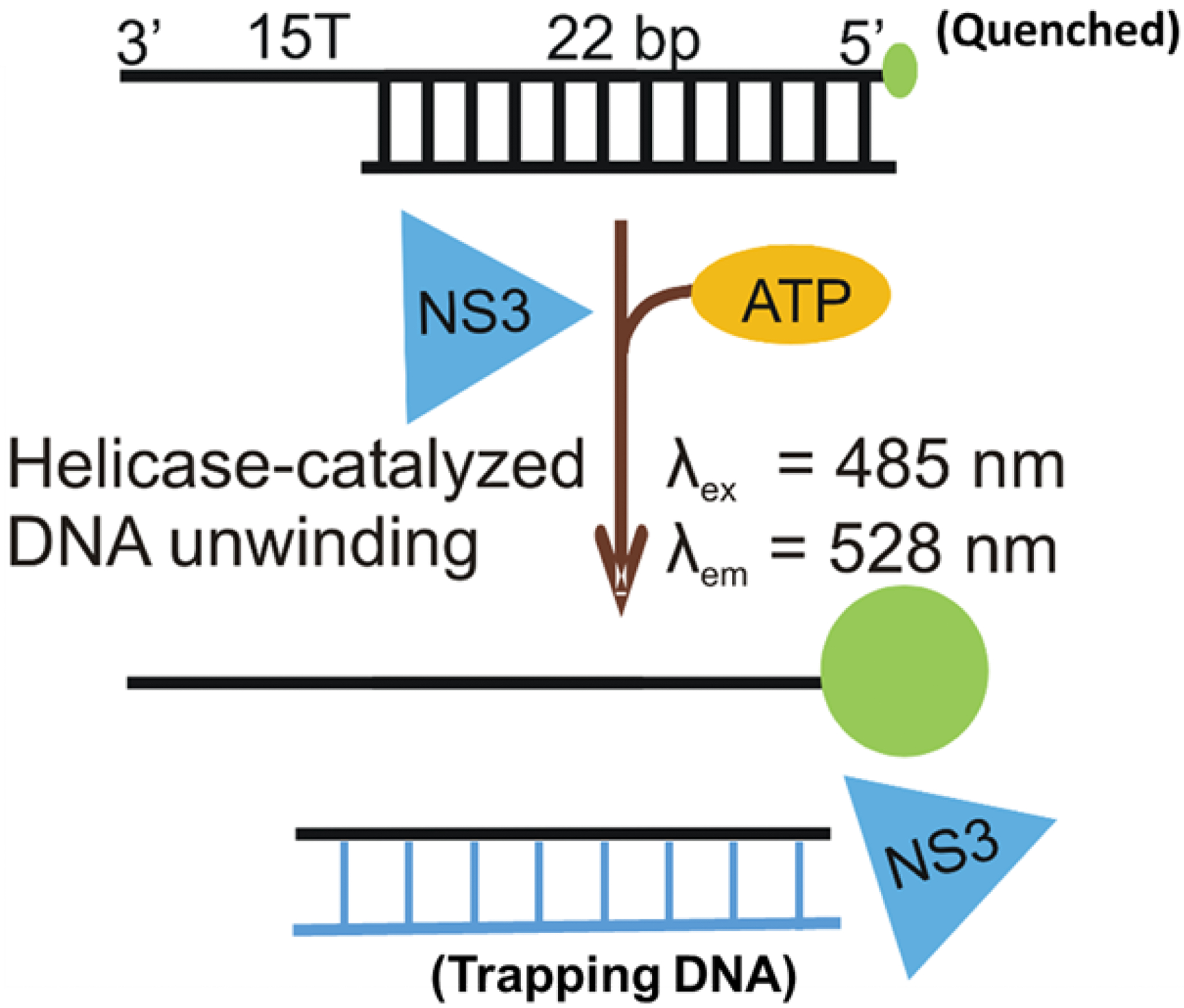
A simple helicase assay in which oligonucleotides are utilized as a substrate. Strands are designed to hybridize to form duplex RNA (or DNA) that contains a single stranded overhang for the helicase to bind. The directionality of the helicase, 5’-to-3’ or 3’-to-5’, determines the polarity of the single-stranded overhang. Duplex strands can be unwound by ATP-dependent helicase activity, and a trapping strand can be included to hybridize with the unlabeled substrate so that reannealing cannot occur. Native gel electrophoresis can separate single-stranded product from duplex substrate.
Once helicase activity is established, the standard helicase assay is revised to allow high throughput assays in order to identify lead compounds from a library of potential inhibitors. The assay shown in Figure 2 can be adapted to a fluorescence assay. Here, the end of one of the RNA (or DNA) strands is labeled with a fluorescence probe, often a fluorescein analog as first described in the Kodadek laboratory (3). The complementary strand is designed to hybridize to the labeled strand, but is shorter in order to leave a single-strand overhang in the substrate (Figure 2). This substrate can be altered slightly in order to make it useful for high throughput screening. The final three nucleotides on the non-labeled strand are guanine residues because the fluorescence of the fluorescein label is quenched when bound to the three guanines (2). Therefore, upon separation of the strands due to helicase activity, an increase in fluorescence occurs. A trapping strand can be included in this assay to prevent reannealing of the product strands. The fluorescence assay can be conducted in a plate assay format which enables a relatively high throughput for screening of thousands of compounds. As shown in Figure 3A, the fluorescence increase enables the identification of compounds that inhibit the helicase activity (6). Quantitation of the fluorescence change allows identification of potential inhibitors (Figure 3B). Once a potential inhibitor has been found, its ability to prevent unwinding activity can be confirmed by going back to the standard, gel-based assay as shown in Figure 4. If the compound inhibits the helicase, then further optimization of the compound can occur, and the process can continue in an iterative manner (step 5 in Figure 1). In the case of the NS3 helicase from the Hepatitis C virus, DNA substrates can replace RNA substrates which further simplifies the substrate design. Whether RNA or DNA is utilized, a general substrate that is likely to satisfy substrate requirements for many RNA helicases from superfamilies 1 and 2 is shown in Figure 2. Other approaches focused on the ATPase activity (9) or nucleic acid binding (7) are also effective in identifying RNA helicase inhibitors.
Figure 3:
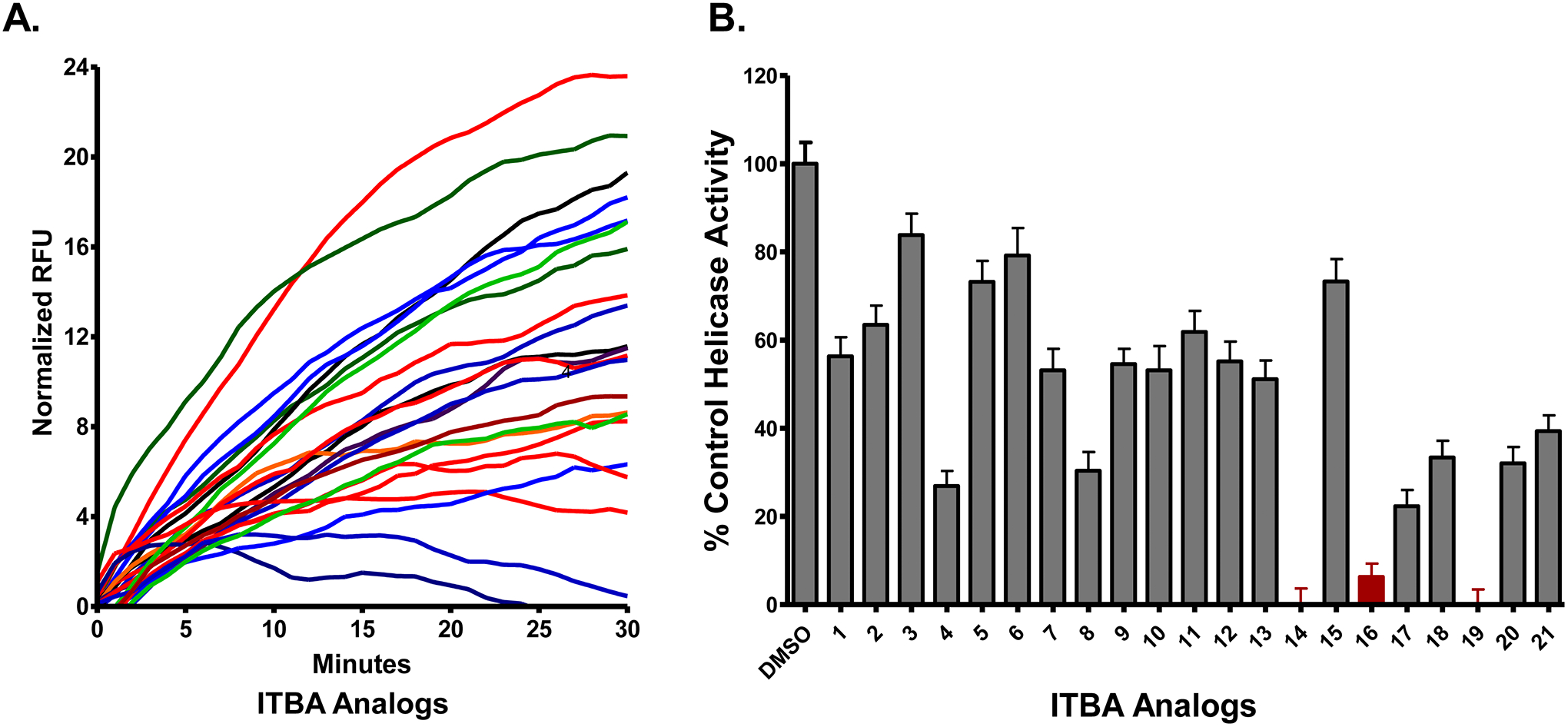
The RNA unwinding assay using a plate-reader format can measure helicase activity in the presence of possible inhibitors. One of the strands of the substrate can be synthesized with a fluorescein label. The complementary strand is synthesized with three guanine residues opposite of the fluorescein label, which results in quenching of the fluorescein probe upon hybridization (2) or a fluorescence quencher can be placed opposite the probe. Thus, helicase-catalyzed unwinding results in an increase in fluorescence (panel A). The inhibition of helicase activity is quantified by the decrease in the amount of substrate unwound as measured by the change in fluorescence due to “unquenching” of fluorescein. B) In this example, the Hepatitis C virus NS3 helicase was targeted. Unwinding rates from the plate–based assay in the presence of the inhibitors were normalized to the DMSO-containing control samples (Reproduced from reference (6) with permission from Elsevier).The ITBA analogs used in this assay resulted in three “hits” (compounds 14, 16, and 19 in the chemical library).
Figure 4:
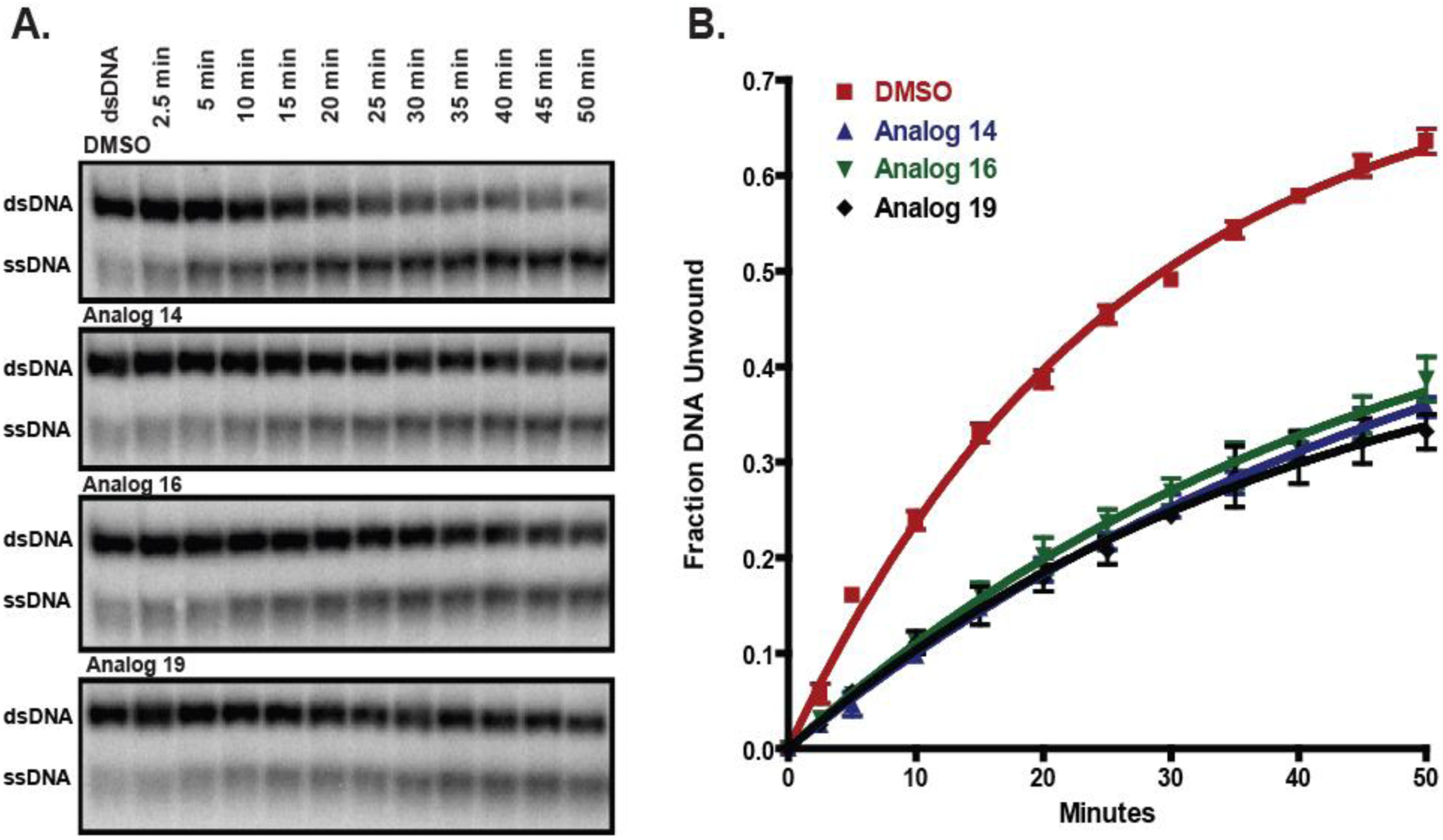
Compounds that are identified using the plate-reader assay can be confirmed as inhibitors by using the standard, gel-based helicase assay (Benchtop Unwinding Assay). A) Unwinding of radiolabeled duplex DNA by HCV NS3 in the presence of 25 μM of the indicated inhibitors. B) Densitometric measurements from each time point were mathematically fit to determine the following DNA unwinding rate constants: DMSO control (red squares): 0.039 ± 0.002 min−1, Analog 14 (blue triangles): 0.018 ± 0.001 min−1, Analog 16 (green inverted triangles): 0.021 ± 0.002 min−1, and Analog 19 (black diamonds): 0.024 ± 0.002 min−1 (Reproduced from reference (6) with permission from Elsevier).
2. Materials
All materials should be of high quality and purity. All solutions should be prepared with ultrapure water, and solutions used with RNA oligonucleotides and duplexes should be treated for RNase contamination with DEPC, or purchased as RNase free reagents.
2.1. Plate-Based Fluorescent Detection of the Inhibition of Helicase Unwinding of DNA or RNA Duplexes
HE Buffer: 10 mM Hepes, pH 7.5, 1 mM EDTA. Store at room temperature.
Loading Strand Oligomer: 5’-FAM-labelled 37-mer that has been HPLC or PAGE purified [See NOTE 1]. For screening inhibitors of the HCV NS3 helicase, the DNA oligonucleotide is dissolved in HE buffer and diluted to 5 μM.
Displaced Strand DNA Oligomer: An unlabeled HPLC or PAGE-purified 25-mer complimentary to the first 22 nucleotides of the loading strand with three additional guanines that are positioned near the FAM in the duplex substrate. The oligonucleotide is dissolved in HE buffer and diluted to 5 μM.
- Trapping Oligomer: An unlabeled HPLC or PAGE-purified 22-mer containing the first 22 nucleotides of the loading strand dissolved in HE buffer and diluted to 5 μM.
Loading Strand 5’ FAM-CATCATGCAGGACAGTCGGATCTTTTTTTTTTTTTTT-3’ Displaced Strand 5’ GATCCGACTGTCCTGCATGATG-GGG-3’ Trapping Strand 5’-CATCATGCAGGACAGTCGGATC-3’ Duplex DNA, 200 nM annealed as described in the “Annealing Reaction for the FAM-Labelled, Quenched DNA Substrate”.
5X Reaction Buffer: 125 mM MOPS, pH 7.0, 250 mM NaCl, 10 mM β-mercaptoethanol, 500 μg/ml bovine serum albumin, 0.5 mM EDTA stored in aliquots at −20°C [See NOTE 2].
250 mM ATP: dissolve in water and adjust pH to 7.0 using NaOH. Store in aliquots at −20°C.
1 M MgCl2 in water. Store at room temperature.
Black, 96-well flat bottom low-binding assay plates (such as those available from Corning).
Inhibitor compounds dissolved in DMSO at 15–20 mM [See NOTE 3].
Purified helicase protein in storage buffer (25 mM HEPES pH 7.5, 100 mM NaCl, 1mM EDTA, 5 mM β-mercaptoethanol, 20% Glycerol) [See NOTE 4].
Temperature-controlled plate reader capable of kinetic fluorescent measurements (Ex = 485 nm, Em = 528 nm).
2.2. Inhibition of the Benchtop Duplex Nucleic Acid Substrate Unwinding
The benchtop duplex unwinding assay utilizes either DNA or RNA substrates, depending on the helicase being studies. HCV NS3 is capable of unwinding either DNA or RNA duplex, but with differing kinetics (10). Thus, the enzyme concentration, pre-incubation time and time course must be carefully optimized. Solutions used with RNA and RNA duplexes should be prepared with ultrapure water treated for RNase contamination with DEPC.
2.2.1. Oligonucleotide labelling with 32P and Annealing Reaction
Loading Strand Oligomer: Synthetic 37-mer that has been HPLC or PAGE purified and dissolved in HE buffer and diluted to 5 μM.
- Displace Strand oligonucleotide: An unlabeled HPLC or PAGE-purified 22-mer complimentary to the first 22 nucleotides of the loading strand. The oligo is dissolved in HE buffer and diluted to 5 μM [See NOTE 5].
Loading Strand (DNA) 5’-CATCATGCAGGACAGTCGGATCTTTTTTTTTTTTTTT-3’ Displaced Strand (DNA) 5’-GATCCGACTGTCCTGCATGATG-3’ Loading Strand (RNA) 5’-CAUCAUGCAGGACAGUCGGAUGUUUUUUUUUUUUUUU-3’ Displaced Strand (RNA) 5’-GAUCCGACUGUCCUGCAUGAUG-3’ 10X Polynucleotide kinase (PNK) buffer.
T4 polynucleotide kinase (PNK) (10U/μl)
Nucleotide removal spin columns [See NOTE 6].
ATP, [γ−32P] - 3000Ci/mmol 10mCi/ml.
(For RNA substrates) RNase inhibitor RNasin (40 U/μl, Promega).
(For RNA Substrates) DEPC-Treated ultrapure water [See NOTE 7]
Heating block set to 37°C.
Benchtop centrifuge.
2.2.2. Non-Denaturing Polyacrylamide Gel
Large glass electrophoresis plates (Model S2 Sequencing Plates) and S2 Sequencing gel stand.
Power supply capable of operating at high voltage (~1000 V).
10% dichlorodimethylsilane in hexane: Store tightly capped at 4°C. [See NOTE 8]
(For RNA substrates) RNase inhibitor RNasin (40 U/μl, Promega).
(For RNA Substrates) DEPC-Treated ultrapure water.
10X TBE, pH 8.0 [See NOTE 9].
40% Acrylamide: N,N-methylene-bis-Acrylamide Solution (19:1) [See NOTE 10].
Ammonium persulfate: 10% solution prepared fresh in water.
N,N,N,N’-tetramethyl-ethylenediamine (TEMED).
2.2.3. Benchtop Duplex Unwinding Reaction
Labeled duplex DNA or RNA: 200 nM annealed duplexes as described in the “Oligonucleotide labelling with 32P and Annealing Reaction” section.
- Trapping Oligomer: A 22-mer containing the first 22 nucleotides of the loading strand dissolved in HE buffer and diluted to 5 μM.
Trapping Strand 5’-CATCATGCAGGACAGTCGGATC-3’ 5X Reaction Buffer: 125 mM MOPS, pH 7.0, 250 mM NaCl, 10 mM β-mercaptoethanol, 500 μg/ml bovine serum albumin, 0.5 mM EDTA stored in aliquots at −20°C [See NOTE 2].
250 mM ATP: dissolve in water and adjust pH to 7.0 using NaOH. Store in aliquots at −20°C.
1 M MgCl2 in water. Store at room temperature.
(For RNA substrates) RNase inhibitor RNasin (40 U/μl, Promega).
(For RNA Substrates) DEPC-Treated ultrapure water.
(For RNA Substrates) RNaseZap or RNase AWAY surface treatment solutions.
Inhibitor compounds dissolved in DMSO at 15–20 mM [See NOTE 3].
Purified helicase protein in storage buffer (Our storage buffer for HCV NS3 is 25 mM HEPES pH 7.5, 100 mM NaCl, 1mM EDTA, 5 mM β-mercaptoethanol, 20% glycerol) [See NOTE 4].
Quenching Buffer: 200 mM EDTA, 0.7% SDS made fresh daily.
6X loading buffer (0.25% bromophenol blue, 0.25% xylene cyanol FF, 30% glycerol) stored at room temperature [See NOTE 11].
Heating block set to 37°C and a heating block set to 95°C [See NOTE 12].
Large cassette for exposing the radioactive gel to a phosphor screen or film [See NOTE 13].
3. Methods
3.1. Plate-Based Fluorescent Detection of the Inhibition of Helicase Unwinding of DNA Duplexes
Real-time duplex DNA or RNA unwinding helicase activity is measured by the appearance of the fluorescein fluorescence following the displacement of a strand of DNA or RNA containing a FAM-quenching tri-guanosine (2). Trapping this displaced oligonucleotide with an excess of a complementary oligonucleotide prevents re-annealing of the substrate duplex and the loss of signal (Figure 2). The binding of small molecule inhibitors slows the unwinding reactions sufficiently to observe changes in the rate of increase in fluorescence. Instrumental temperature control ensures homogeneous conditions and optimal activity across the plate. Screening the potential inhibitors involves assembly of a mix containing helicase, substrate and inhibitor in the wells followed by the initiation of the reactions with the rapid addition of ATP. Fluorescence changes are monitored until the reaction is completed, and the initial linear portion of the fluorescence change is used as a measure of helicase’s DNA unwinding activity. Alterations in the rate of fluorescence appearance in the presence of inhibitor compounds measures the degree of helicase activity inhibition.
3.1.1. Annealing Reaction for the FAM-Labelled, Quenched DNA Substrate.
Mix 20 pmol of the FAM-labelled Loading Strand oligomer, and 24 pmol of Displaced Strand Oligomer (1:1.2 molar ratio, See NOTE 14). Bring to 100 μl with HE buffer.
Heat to 95° C for 5 minutes, and allow the strands to anneal with slow cooling to room temperature by removing the heating block from the unit [See NOTE 15].
Briefly vortex and spin in a tabletop centrifuge to collect all the condensate, and store on ice until used. The stock FAM-labelled substrate concentration will be 200 nM in HE.
3.1.2. Microtiter Plate-based Fluorescence Helicase Unwinding Reaction
Equilibrate the plate reader to 37° for at least 20 minutes prior to the start of the assay. Stock reagents are thawed at room temperature and maintained on ice prior to the assembly of the reaction mixture. Aliquots of helicase preparation are slowly thawed on ice and gently mixed to reduce the potential for denaturation and precipitation [See NOTE 16].
Assemble 2X master mixtures for the positive control (No Inhibitor), a negative control (Heated Reaction), as well as one mix for each potential helicase inhibitor compound. If the compound is dissolved in a solvent other than water, be sure to include a mixture containing the solvent. Calculate the amount of master mix needed for 100 μl/well containing 500 nM helicase, 4 nM fluorescently labeled DNA or RNA substrate (See “Annealing Reaction” above), 200 nM Trapping Strand, 1X Reaction buffer and the desired amount of inhibitor. All concentrations are 2X the final concentration since samples will be mixed to initiate the reaction However, the assay buffer concentration is 1X since it is included at 1X in both half reactions.
Assemble a 2X mixture of Mg2+-ATP solution. Calculate the amount of master mix needed for 100 μl/well for all of the samples. The solution contains 10 mM ATP, 20 mM MgCl2 AND 1X Reaction buffer. This solution is used to initiate the reaction. Pre-warm in a water bath for 20 minutes prior to use.
Place the Negative Control (Heated Reaction) at 95°C for 10 minutes [See NOTE 16]. Allow to cool and briefly spin in a tabletop centrifuge to collect the condensate.
Add 100 μl of the Negative Control (Heated Reaction) to individual wells of the plate in triplicate.
Add 100 μl of the Positive Control (No Inhibitor) to individual wells of the plate in triplicate.
Add 100 μl of each inhibitor reaction to individual wells of the plate in triplicate. Place the plate into the thermostatted plate reader for 5–30 minutes to equilibrate.
Measure the FAM fluorescence of each well of the plate at an interval 20–30 seconds. This rate is the background fluorescence in each of the wells.
Rapidly add 100 μl of the 2X Mg2+-ATP mixture to each of the wells using a multi-channel pipette and begin monitoring the fluorescence.
Continue monitoring the fluorescence of each well every 20–30 seconds for a total of 20–30 minutes, or until the fluorescence of the wells no longer changes.
Subtract the background fluorescence obtained during the equilibration period from the values obtained for the Negative Control, the Positive Control, and the inhibitor reactions.
Calculate the rate of fluorescence change for the linear portion of the curve in units of Relative Fluorescence Units (RFU)/min for the Negative Control, the Positive Control, and the inhibitor reactions [See NOTE 17]. The fluorescence changes in each well should be examined for linearity, and the window of measurements (with corresponding time interval) are used to ensure the best fit with linear regression analysis.
The rate from the Negative Control (Background Rate) should be less than 5% of the Positive Control (No Inhibitor) rate.
Compare the Positive Control (No Inhibitor) rate with samples that include the solvent for the inhibitors (if other than water) to determine the effect (if any) of the solvent on the control helicase unwinding activity.
- The percent of the nominal enzyme activity of helicase for each well is calculated as:
where ki is the inhibitor rate and kc is the rate of the positive control (Figure 3). Comparison of the unwinding kinetics in the absence and presence of the inhibitors provides a semi-quantitative measure of the effect. Additional experiments varying the amount of inhibitor under the standard conditions established for the helicase allow determination of the IC50 for select molecules.
3.2. Inhibition of the Benchtop Duplex Nucleic Acid Substrate Unwinding
The benchtop unwinding reaction involves combining a mixture of the protein-substrate complex and the potential helicase inhibitors with a solution of Mg2+-ATP and the Trapping Oligonucleotide to initiate the reaction. Serial samples are removed from the mixture at defined times, the helicase activity quenched with EDTA and SDS, mixed with loading buffer, and the DNA or RNA complexes separated with polyacrylamide gel electrophoresis. An additional sample representing the “0” time point is also collected, as well as a sample that is heated to 95°C and rapidly cooled to test the trapping efficiency. The radioactive substrate and product are visualized and quantified. The effect of the potential inhibitors on the unwinding activity of the helicase is also readily quantified (Figure 4).
3.2.1. Oligonucleotide labelling with 32P and Annealing Reaction
Assemble the 32P-labelling reaction for the Loading Strand Oligonucleotide as follows: Add 5 μl of the 10X PNK buffer, 4 μl (20 pmol) of the Loading Strand Oligo, 3–6 μl of 32P-ATP (depending on the number of half-lives that have passed since the 32P was assayed, 2 μl of PNK and H2O to 50 μl [See NOTE 18].
Place the reaction in a heat block set to 37°C for 1 hour.
Inactivate the PNK by heating the tube to 70°C for 10 minutes. Briefly centrifuge the sample to collect the condensate.
Add 4.8 μl (24 pmoles) of the Displaced Strand Oligonucleotide to the reaction, and dilute the sample to 100 μl with HE.
Prepare a Sephadex G-25 nucleotide removal column with a fresh 1.5 ml collection tube and apply the entire reaction to the top of the column. Immediately centrifuge the sample at 3,500 rpm for 7 minutes. Repeat with an additional G-25 column [See NOTE 6].
Check the resulting solution for radioactivity using a Geiger counter. To anneal the substrate DNA, heat the sample to 95°C for 5 minutes and allow the tube to slowly cool to room temperature.
The concentration of the labelled substrate is 200 nM in HE. Store at −20°C for DNA or 4 °C for RNA. We have found that RNA substrates can be stored at 4 °C for a few weeks with no loss of quality. Freeze-thaw cycles from storage at 20 °C result in RNA degradation. The half-life of 32P is 14 days so the substrate will need to be remade with fresh 32P every week or two.
3.2.2. Non-Denaturing Polyacrylamide Gel
Clean both glass electrophoresis plates (Model S2 Sequencing Plates) with warm soap and water and allow to dry. In a fume hood, apply a thin coating of 10% silane using Kimwipes. Allow the hexane to evaporate and clean the excess silane with a small amount of ultrapure water (or DEPC treated water if using RNA) and wipe dry. If using RNA substrates treat the glass plates, spacers and comb with RNaseZap for 10–15 minutes and wipe away the excess before silanizing the plates [See NOTE 19].
Place the 0.4 mm spacers on each side of the large plate, and cover with the small plate so that the silanized surfaces are facing each other.
Seal the bottom and the side edges of the glass sandwich with sealing tape or a rubber boot, starting at a bottom corner and working across and up the sides. Make sure the spacers and plates are properly aligned to prevent leakage of the acrylamide solution. Prop the plates at a slight angle so that liquid will run down toward the bottom of the plates.
Prepare 20% acrylamide in 1X TBE by adding 37.5 ml of 40% acrylamide, 7.5 ml 10X TBE, and 30 ml H20 in a beaker with slow stirring. Add 75 μl of TEMED and 750 μl of 10% APS.
Immediately following the addition of the TEMED/APS, pour the solution between the glass sandwich beginning at one corner while holding the plates angled toward that side. After the acrylamide reaches the bottom of the plates, the acrylamide should be allowed to fill the space from the bottom to the top to prevent accumulation of air bubbles. As needed, rotate the plate sandwich to eliminate air bubbles. Fill to the top of the smaller plate [See NOTE 20].
Prop the glass plate sandwich at a slight angle and insert a 32 well, 0.4 mm comb, making sure to position the top of the wells even with the top of the gel.
Place 6 large binder clamps across the top of the glass sandwich and allow the acrylamide to polymerize for at least 20 minutes at room temperature.
Once the gel is fully polymerized, carefully remove the clamps and use a new razor blade to remove excess gel from around the comb.
Slip the razor blade behind the comb, and gently slide it across while adding water under the comb to separate it from the large glass plate.
Gently remove the comb and inspect the gel to ensure that the wells are not distorted or broken. Remove the sealing tape or boot.
Place the glass sandwich onto the S2 Sequencing gel stand with the large plate facing out and clamp it into place.
Fill the top and bottom reservoirs with 1X TBE (1L total).
Using a 30-mL syringe filled with 1X TBE, gently flush the wells to remove any unpolymerized acrylamide or air bubbles.
Connect the electrodes to the gel stand and pre-run the gel at 22 mA for 30–45 minutes.
Disconnect the electrodes and using a 30-mL syringe filled with 1X TBE, gently flush the wells again.
3.2.3. Benchtop Nucleic Acid Duplex Unwinding Reaction
Label microfuge tubes for each reaction. The reactions components will be split between two half reactions (helicase-substrate, and ATP). A tube will be needed for each of these half reactions and a third tube is needed for the Reaction mixture. An additional tube containing Quench will be needed. The Quench will be the same for all reactions. For each set of reactions, include a Control reaction (No Inhibitor) and reactions containing the desired inhibitors [See NOTE 21].
Label microfuge tubes for each time course: a tube for Time 0, a tube for the Heated sample, and tubes for the time course for unwinding, typically 6 samples. The specific helicase used and length of the duplex will determine the appropriate time frame for the reaction [See NOTE 22].
Mix 1 ml of Quenching buffer by adding 197 μl of H2O, 400 μl of 0.5 M EDTA and 70 μl of 10% SDS and mix with inversion. Add 333 μl of the 6X Loading Buffer and mix by inversion.
Add 10 μl of the Quenching/Loading Buffer to each of the tubes for the time course including the Time 0, Heated, and each additional time point.
- Calculate the volumes and concentrations for each Reaction for a final volume of 50 μl for each reaction half. The concentration are 2-times the final reaction concentrations as the reaction components will be diluted 2-fold when the reaction is initiated:
- DNA/RNA Substrate: 10 nM [See NOTE 23]
- Helicase: 200–1000 nM
- Inhibitor Diluent (Control Reaction): Same as the inhibitor
- Candidate Inhibitor: At or near the IC50
- (For RNA substrates) RNasin (40 U)
- 5X Buffer: 1X
- Calculate the volumes and concentrations for the ATP solution for each reaction (Control and Inhibitor) for a final volume of 50 μl. The ATP Solutions components will be diluted 2-fold when the reaction is initiated so these are 2X the final concentration:
- ATP: 10 mM
- MgCl2: 20 mM
- Trapping Oligo: 300 nM (30X greater than the substrate)
- Assay Buffer: 1X
Assemble the components for all of the Reactions and pre-incubate the tubes at 37°C for 5–30 minutes [See NOTE 24].
Remove 5 μl from the ATP solution tube and mix with the Quenching/Loading Buffer in the designated “Time 0” tubes and Heated tubes for each time course.
Add 5 μl of helicase-substrate mixture to the Time 0 and Heated tubes and mix well.
Place the heated tube in a heat block at 95°C for 5 minutes and allow the sample to cool on ice.
Transfer 40 μl of the ATP solution to the Reaction tube.
Initiate the first Reaction by adding 40 μl of the helicase-substrate solution to the Reaction tube while simultaneously beginning a timer. At each designated time point, rapidly remove 10 μl from the Reaction tube and mix with the Quenching/Loading buffer in the designated tube. Place on ice [See NOTE 25].
Initiate the Reactions containing the potential Inhibitors in the same manner until all time points for all of the Reactions have been collected. Briefly centrifuge all samples to collect any condensate.
3.2.4. Separation and Detection of Unwound Products with Non-Denaturing PAGE
Flush the wells of the gel using a pipettor or a syringe with 1X TBE. Load 5 μl of the time points from each reaction starting with Time 0 through the time points and ending with the Heated sample. Store the remaining samples at −20°C for DNA or 4°C for RNA in case there is a need to re-run the samples.
Run the samples at 22 mA for at least 2 hours, or until the bromophenol blue has migrated a distance of at least 50% of the length of the gel. If a shorter duplex is used, the gel may need to run longer than 2 hours to separate the duplex substrate and single-stranded products.
Once the electrophoresis is complete, disconnect the power and remove the glass sandwich from the gel stand. Carefully remove the spacers and separate the glass plates, ensuring that the gel remains adhered to one of the plates.
Place a large piece of plastic wrap or clear plastic transparency over the gel, and flip the gel so that the plate is on top. Carefully peel the gel away from the glass plate, and cover the second side of the gel with plastic wrap or a clear plastic transparency. Check the gel for radioactivity using a Geiger counter. Seal the edges of the gel with plastic wrap to prevent damage to the phosphorimager screen. [See NOTE 26]
Place the gel in the phosphorimager (or film) cassette overnight [See NOTE 27].
Remove the gel from the cassette and image the screen according to the manufacturer’s instructions for the imager used. Scan the entire gel.
3.2.5. Analysis of Helicase Activity from the Non-Denaturing PAGE
Open the scanned image in the image software provided by the scanner, or other analysis software such as ImageJ. If using ImageJ, import the image in the TIFF or BMP format to prevent data loss through compression.
Identify the substrate and product using the Time 0 lane (duplex substrate) and the Heated lane (single-stranded product).
Measure the density of the substrate and the product. Most programs have methods of automatically quantifying the density of each lane. We prefer to draw a rectangle around the first band and copy and paste rectangles of the same size for each band (substrate and product in each lane) making sure that the regions of interests do not overlap. Three rectangles in regions of the gel image with no signal are included to determine the background.
Average the background density and subtract from the values obtained from the complexes in each lane.
Calculate the “Fraction Product” by dividing the density of the ”Product” by the sum of the densities of the ”Product” and the “Substrate”.
- A correction to the “Fraction Product” is made to account for the amount of substrate that is unwound during the pre-incubation period:
where Pt is the fraction product at a time point and P0 is the fraction product at Time 0. - Plot the Fraction Unwound (y) versus time (x), and analyze the goodness of fit to a single exponential function:
where A is the amplitude, k is the rate constant, and t is the time. - Calculate the percent effect of the inhibitor on the rate constant(s) by dividing the inhibitor values by the control values and multiplying by 100%:
where ki is the inhibitor rate and kc is the rate of the positive control.
4. Notes
Companies offer to HPLC or PAGE purify oligonucleotides for a cost. We prefer to PAGE purify all oligonucleotide in-house to ensure that n-1 and smaller products are not present in the oligonucleotide sample. Depending on the activity of the helicase, the length of the duplex may need to be adjusted. The ideal length is one which the helicase can completely unwinding in 15–60 min. If the reaction is too fast, it will be completed before sufficient data has been collected for each well in the plate. If the helicase does not have sufficient processivity to unwind the entire duplex, no product will be observed. The reaction time can also be adjusted by varying the concentration of the helicase [See NOTE 18].
Purified RNA helicases for in vitro assays have an optimal pH and salt concentration for maximal activity. Our HCV NS3 helicase has optimal activity at pH 7.0 with 50 mM NaCl. Different buffer compositions, pH ranges and salt concentrations may need to be examined for other helicases.
The NS3 inhibitors we screen readily dissolve in DMSO, and we have found that the addition of DMSO up to 5% of the reaction does not interfere with the unwinding activity. If other solvents are used to dissolve the inhibitors, or other helicases are being used, additional controls are necessary to ensure that the solvent does not interfere with its ATPase or unwinding activities.
We purify the HCV NS3 from Escherichia coli as described in (10). The purified protein should be stored at −80°C in small aliquots to prevent repeated freeze-thaw cycles. Ensure that the protein has ATPase activity and the molar concentration of protein has been confirmed with UV-visible spectrophotometry using a known extinction coefficient, as well as an additional colorometric assay such as the Bradford or BCA assays. The two measurements should agree to within 5%. Stock concentrations of protein should be 5 μM or greater.
We find that oligonucleotides labelled with the fluorophore FAM nicely substitute for 32P-labelled oligonucleotides in the benchtop unwinding assay. Detecting the fluorescently labelled complexes when using sequencing gels requires a scanner with a 488 nm laser and 520 nm emission filter such as the GE Typhoon Trio.
We use in-house developed G-25-Sephadex columns to remove unincorporated nucleotides from our DNA labelling reaction. We purchase prepared RNase free G-25 Sephadex columns for use with RNA. To prepare columns, a 1 ml syringe barrel is packed with a small amount of glass wool, and a 50% slurry of G-25-Sephadex/10 mM Hepes, 1 mM EDTA, pH 7.5 is added and allowed to settle until the packed bed volume is approximately 1 ml. The syringe is placed in a 15 ml Falcon tube containing a 1.5 ml microfuge collection tube in the bottom. Centrifuge for 7 minutes at 3,500 RPM in a tabletop centrifuge to remove excess buffer. Remove the microfuge tube with forceps and discard. Replace with a new tube, apply labeled substrate to the top of the column, and centrifuge again for 7 minutes at 3,500 RPM. Carefully remove tube containing labeled substrate with forceps and repeat the process with a second column.
For work with RNA substrates all buffers must be made with DEPC-treated water or purchased as RNase free reagents. Add 1 ml of diethylpyrocarbamate to 1 liter of ultrapure water, incubate overnight at room temperature, and autoclave for 30 minutes. Allow the bottle to cool to room temperature and cap tightly. We prefer to purchase all components of the reactions and oligonucleotide storage solutions as RNase free reagents but use DEPC-treated reagents for the gel.
Silane is highly toxic and should be handled in a fume hood with gloves.
RNase and DNase-free 10X TBE is commercially available and reduces the potential for contamination of solutions with RNase. For in-house preparation of 10X TBE: Dissolve 108 g of Tris-Base in approximately 800 ml of ultrapure water. Add 55 grams of boric acid and 40 ml of 0.5 M EDTA, pH 8.0. Bring to 1 L with water. Filter through a 0.44 μm pore size (or smaller) filter to remove particulates, and store at room temperature.
Commercially available DNase and RNase-free acrylamide solutions may be purchased. If making in-house, use DEPC-treated water for solutions that will be used with RNA. Acrylamide is a neurotoxin. Gloves must be worn when working with acrylamide solutions and any spills should be cleaned immediately.
If using FAM-labeled oligonucleotides, omit the xylene cyanol FF from the Loading Buffer, as it will be visible in the fluorescent scan.
Different helicases may have a different optimal reaction temperature.
Detection of the radioactive RNA/DNA in sequencing format gels using film is adequate, though the dynamic range is much smaller and accurate quantitation will require scintillation counting of gel pieces corresponding to each substrate and product band. We use the GE Typhoon Trio and phosphorimaging cassettes to detect radioactive oligonucleotides with a greater linear range of sensitivity.
To ensure complete duplex formation, we add an excess of the FAM-labelled Loading Strand to our annealing reaction.
Removing the heating block from the unit allows for a slow cooling and annealing of the oligomers. This process requires 2–3 hours for the block to reach room temperature.
Purified protein preparations stored at −80°C should be slowly thawed on ice prior to use. If excess protein is thawed, it should not be refrozen, but most helicases may be kept at 4°C for a few days with no loss of activity. NS3 can be stored at 4°C for up to a week. Heating of the sample to 95°C for 10 minutes ensures that the proteins have denatured. The sample can be placed on ice to cool before use.
The concentration of HCV NS3, or other model helicase that produces a measureable and linear change in fluorescence will need to be optimized. The concentration in this protocol with the described substrate results in a linear change in fluorescence over the first 8–10 minutes of the reaction, and all substrate is exhausted within 1 hour. Depending on the activity of the helicase used and the length of duplex substrate used, the amount of helicase will need to be altered accordingly. The rate of fluorescence change should be robust to visualize the helicase activity and the inhibition with added compounds. The rate of acquisition of data points will also depend on the rate of unwinding, and we do not recommend single end-point measurements.
32P is radioactive and must be used and disposed of following the safety rules of your institution.
RNaseZap and RNase AWAY remove RNase contamination of hard surfaces. They should be subsequently removed with DEPC-treated water. Ensure that all work surfaces and electrophoresis components are treated for RNase contamination.
If bubble form on the edges or in a region of the gel that will not be used for separating the complexes, the gel may still be used. If the bubbles persist, the glass sandwich must be disassembled, the plates cleaned thoroughly, dried and reassembled.
As with the plate assay, the effect of the solvent on the helicase activity must be determined, thus including control reactions without and with the solvent is critical.
Six data points appropriately spread out are required to adequately define a single exponential unwinding curve. In our studies of NS3 inhibitors, we measure the extent of unwinding of the DNA substrates at 1, 2, 5, 10 15 and 30 minutes. When measuring the rate unwinding for the RNA duplex substrates, we must condense the time course to a total of 5 minutes. It is critical to optimize the helicase concentration and time course to sufficiently measure the inhibition by small molecules.
For visualization using a GE Typhoon Trio, we have found that when using FAM-labeled substrates instead of 32P, the minimum final concentration of substrate in the reaction is 20 nM so the helicase-substrate half reaction concentration must be at least 40 nM. If the concentration of the substrate is increased, the concentration of the trapping strand must be increased proportionally.
The pre-incubation time is dependent on the helicase being used in the studies. HCV NS3 requires 30 minutes of pre-incubation with the RNA substrate but only 5 minutes of pre-incubation with the DNA substrate in vitro for full unwinding activity.
It is critical that the time point sample is mixed with the quench at the appropriate time. If a time point is missed, note the new time point for proper graphing and curve-fitting results.
For FAM-labeled substrates, after removing the first glass plate, cover the gel carefully with plastic wrap as any bubbles in the wrap will be visible in the scan. Leave the gel attached to the second plate and scan with the plastic side down on a scanner with a 488 nm laser and 520 nm emission filter. We use a GE Typhoon Trio.
The time required to adequately visualize the substrate and product with the phosphorimager can range from 4 hours to overnight. Due to the wide dynamic range of the phosphor screens, longer exposures should not saturate the screen. The time needed also varies based on the efficiency of oligonucleotide labelling. For experiments that utilize fluorescent oligonucleotides, the gel can be directly imaged without the need for the cassette.
References
- 1.Datta A and Brosh RM Jr. 2018. New Insights Into DNA Helicases as Druggable Targets for Cancer Therapy. Front Mol.Biosci 5:59. doi: 10.3389/fmolb.2018.00059 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donmez I and Patel SS. 2008. Coupling of DNA unwinding to nucleotide hydrolysis in a ring-shaped helicase. EMBO J. 27:1718–1726. doi:emboj2008100 [pii]; 10.1038/emboj.2008.100 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Houston P and Kodadek T. 1994. Spectrophotometric assay for enzyme-mediated unwinding of double-stranded DNA. Proc.Natl.Acad.Sci.U.S.A 91:5471–5474. doi: 10.1073/pnas.91.12.5471 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim DS, Camacho CV, Nagari A, Malladi VS, Challa S, and Kraus WL. 2019. Activation of PARP-1 by snoRNAs Controls Ribosome Biogenesis and Cell Growth via the RNA Helicase DDX21. Mol.Cell 75:1270–1285. doi:S1097–2765(19)30476–9 [pii]; 10.1016/j.molcel.2019.06.020 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leitao AL, Costa MC, and Enguita FJ. 2015. Unzippers, resolvers and sensors: a structural and functional biochemistry tale of RNA helicases. Int.J.Mol.Sci 16:2269–2293. doi:ijms16022269 [pii]; 10.3390/ijms16022269 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marecki JC, Aarattuthodiyil S, Byrd AK, Penthala NR, Crooks PA, and Raney KD. 2019. N-Naphthoyl-substituted indole thio-barbituric acid analogs inhibit the helicase activity of the hepatitis C virus NS3. Bioorg.Med.Chem.Lett 29:430–434. doi:S0960–894X(18)30972–7 [pii]; 10.1016/j.bmcl.2018.12.026 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukherjee S, Hanson AM, Shadrick WR, Ndjomou J, Sweeney NL, Hernandez JJ, Bartczak D, Li K, Frankowski KJ, Heck JA, Arnold LA, Schoenen FJ, and Frick DN. 2012. Identification and analysis of hepatitis C virus NS3 helicase inhibitors using nucleic acid binding assays. Nucleic Acids Res. 40:8607–8621. doi:gks623 [pii]; 10.1093/nar/gks623 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sloan KE and Bohnsack MT. 2018. Unravelling the Mechanisms of RNA Helicase Regulation. Trends Biochem.Sci. 43:237–250. doi:S0968–0004(18)30022–7 [pii]; 10.1016/j.tibs.2018.02.001 [doi]. [DOI] [PubMed] [Google Scholar]
- 9.Sweeney NL, Hanson AM, Mukherjee S, Ndjomou J, Geiss BJ, Steel JJ, Frankowski KJ, Li K, Schoenen FJ, and Frick DN. 2015. Benzothiazole and Pyrrolone Flavivirus Inhibitors Targeting the Viral Helicase. ACS Infect.Dis 1:140–148. doi: 10.1021/id5000458 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tackett AJ, Wei L, Cameron CE, and Raney KD. 2001. Unwinding of nucleic acids by HCV NS3 helicase is sensitive to the structure of the duplex. Nucleic Acids Res. 29:565–572. doi: 10.1093/nar/29.2.565 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu ZQ and Dixon NE. 2018. Bacterial replisomes. Curr.Opin.Struct.Biol 53:159–168. doi:S0959–440X(18)30095–2 [pii]; 10.1016/j.sbi.2018.09.006 [doi]. [DOI] [PubMed] [Google Scholar]