

Abstract
Analysis of individual gametes has a number of applications in the study of the mechanism of repeat expansion in mouse models of the Fragile X-related disorders, as well as in mouse models of other Repeat Expansion Diseases. This chapter describes the techniques required to isolate oocytes and male gametes of different stages of maturity, along with the techniques required to accurately determine the repeat number in these gametes.
Keywords: oocyte, sperm, germ cell, fluorescence activated cell sorting, single cell analysis, small pool PCR
1. Introduction
The Fragile X-related disorders (FXDs) are members of the Repeat Expansion Diseases in which the expansion-prone repeat tract, located in exon 1 of the FMR1 gene, consists of >54 CGG-repeats (1). Expansion occurs primarily on intergenerational transmission with some additional somatic instability apparent in some individuals. We have made use of a mouse model of these disorders to try to understand the expansion mechanism responsible (2–6). The ability to isolate and accurately determine the repeat number present in the gametes in this mouse model has helped in our understanding of the timing of expansion and provided important clues as to the underlying mechanism responsible (7). In addition, the fact that we have found that expansion in male gametes is limited to the premeiotic stages of spermatogenesis (7), allows us to study the effect of various DNA repair/recombination mutations on intergenerational expansion even in animals that are sterile by isolating and analyzing the repeat size in immature gametes. Furthermore, the ability to analyze the repeats in individual gametes using single cell PCR greatly expedites the study of genetic factors important for intergenerational expansion without requiring the production of large numbers of offspring. Single cell or small pool PCR is also useful for the analysis of somatic instability in these animals since it gives a more realistic measurement of the extent of expansion and contraction than does the analysis of bulk DNA (8). This chapter will provide detailed protocols for the isolation of male and female gametes from mice and for the analysis of the repeat in these gametes in either bulk DNA or the DNA from single cells.
2. Materials
2.1. Oocyte collection
Female FX mice (see Note 1).
Needles (26-gauge), 1 mL sterile syringe.
Forceps, fine forceps, fine scissors.
35-mm petri dish
Oocyte handling pipette (consisting of hand-held pipette assembly and pulled capillary).
Stereomicroscope.
Pregnant Mare Serum Gonadotropin (PMSG), store the solution in aliquots at −20°C for up to 1 month (see Note 2).
Human chorionic gonadotropin (hCG), store the solution in aliquots at −20°C for up to 1 month (see Note 2).
M2 medium.
M2 with hyaluronidase.
Dulbecco’s Phosphate buffered saline (DPBS; without calcium or magnesium).
2.2. Male gamete collection
Male FX mice (see Note 3).
Forceps, fine forceps, fine scissors, needles (26-gauge).
35-mm petri dish
70-μm cell strainer
5-mL tube with cell strainer cap
Fluorescence activated cell sorter (FACS).
Dulbecco’s Phosphate buffered saline (DPBS; without calcium or magnesium).
DMEM Dulbecco’s modified Eagle medium (DMEM; 4.5 g/L glucose).
Collagenase type IV dissolved in DMEM to a final concentration of 10 mg/mL. Sterilize by passing through a 0.22-μm filter. Store the solution in aliquots at −20°C.
DNase I (10 U/μL).
Dispase (1 U/mL), store the solution in aliquots at −20°C.
Fetal Bovine Serum (FBS; heat inactivated), store the solution in aliquots at −20°C.
Penicillin-Streptomycin (Pen Strep; 10,000 U/mL).
Dimethyl sulfoxide (DMSO).
Hoechst 33342 (10 mg/mL).
Collagenase/DNase solution: Thaw collagenase IV stock solution and dilute with DMEM to a final concentration of 1 mg/mL. Add 10 U/μL DNase I to make a final concentration of 100 U/mL. Store at 4°C for up to a week.
Dispase/DNase solution: Thaw dispase stock solution and add 10 U/μL DNase I to a final concentration of 100 U/mL. Store at 4°C for up to a week.
MEF growth medium: Add FBS into DMEM to a final concentration of 10% (v/v) FBS. Add 10,000 U/mL Pen Strep to make a final concentration of 10 U/mL. Store at 4°C for up to 2 weeks.
Cell freezing medium: Add FBS and DMSO into DMEM to a final concentration of 10% (v/v) FBS and 10% (v/v) DMSO. Store at 4°C for up to 2 weeks.
Cell sorting buffer: Add FBS into DPBS to a final concentration of 5% (v/v) FBS. Store at 4°C for up to 2 weeks.
2x cell staining buffer: Add 10 mg/mL Hoechst 33342 into cell sorting buffer to a final concentration of 10 μg/mL Hoechst 33342. Prepare fresh.
2.3. DNA isolation and determination of repeat number
Proteinase K (20 mg/mL, RNA grade). Add Proteinase K into equal volume of DPBS to make a final concentration of 10 mg/mL.
Dithiothreitol (DTT) dissolved in sterile distilled H2O to a final concentration of 1 M. Sterilize by passing through a 0.22-μm filter. Store the solution in aliquots at −20°C for up to 1 year.
Buffer ATL (Qiagen).
Sodium chloride (NaCl; 5 M).
Ethanol and 70% (v/v) ethanol.
Cell lysis buffer: Add 20 mg/mL Proteinase K and 1 M DTT into ATL buffer to a final concentration of 0.55 mg/mL Proteinase K and 30 mM DTT. Prepare fresh.
Tris-HCl (1 M, pH 9.0).
Magnesium chloride (MgCl2; 1 M).
Ammonium sulfate ((NH4)2SO4) dissolved in sterile double-distilled H2O to a final concentration of 2.2 M. Sterilize by passage through a 0.22-μm filter. Store the solution at room temperature.
Triton X-100.
Betaine dissolved in sterile distilled H2O to make a 5 M solution. Sterilize by passage through a 0.22-μm filter. Store the solution in aliquots at 4°C for up to 1 year. (see Note 4).
Dimethyl sulfoxide (DMSO).
dNTP set (50 mM each of dATP, dCTP, dGTP, and dTTP): Add the dNTP mix into 4 volumes of sterile double-distilled H2O to a final concentration of 10 mM each, store the solution in aliquots at −20°C (see Note 5).
dCTP (100 mM).
dGTP (100 mM).
Testicular cell lysis buffer: Add 20 mg/mL Proteinase K and 1 M DTT into ATL buffer to a final concentration of 0.55 mg/mL Proteinase K and 30 mM DTT.
10x Q5 CGG PCR buffer (9): Add 1 M Tris-HCl (pH 9.0), 1 M MgCl2, 2.2 M (NH4)2SO4, and Triton X-100 into sterile double-distilled H2O and make final concentration of 500 mM Tris-HCl, 15 mM MgCl2, 220 mM (NH4)2SO4, 2% Triton X-100. Store at room temperature.
2x Betaine-DMSO mix: Add 1 volume of DMSO into 25 volume of Betaine to make a final concentration of 4.8 M betaine, 4% DMSO.
dCTP-dGTP mix: Add equal volumes of dCTP and dGTP into 2 volumes of sterile double-distilled H2O to make a final concentration of 25 mM each. Store the solution in aliquots at −20°C (see Note 5).
Q5 Hot Start High-Fidelity DNA Polymerase (2,000 U/mL, New England Biolabs).
2x KAPA2G Fast HotStart Genotyping Mix (KAPA Biosystems).
3. Methods
Carry out all procedures in biosafety cabinets maintaining sterile working procedures. All biomedical/biohazard reagents and waste must be handled according to institutional lab safety guidelines. The methods described here suitable only for collecting cells for genomic DNA analysis, not for other purposes like in vitro fertilization.
3.1. Oocyte collection
This procedure is a slight modification of a previously published procedure (10).
Intraperitoneal injection of female mice with five international units (IU) PMSG (see Note 6).
Intraperitoneal injection of female mice with five IU hCG 47 hours after PMSG injection (see Note 6).
Thirteen hours after hCG injection, euthanize the females by CO2 inhalation. Place the animal(s) in the home cage and introduce 100% carbon dioxide by a fill rate of about 10%−30% of the cage volume per minute with carbon dioxide (11).
Quickly dissect out the oviducts with a small section of the uterus and transfer to a clean 35-mm petri dish with pre-warmed M2 media. The ampullae of the oviducts should be much enlarged.
Transfer one oviduct in a new 35-mm petri dish with M2 with hyaluronidase media. Under the stereomicroscope with 20x magnification, use fine forceps to grasp the ampullae, and use another fine forceps or 26-gauge needle to tear the ampullae to release the cumulus oocyte masses.
Incubate the oocytes in M2 with hyaluronidase medium for 2–5 min to allow the cumulus cells to dissociate.
Use an oocyte handling pipette to pick up the oocyte and transfer them to a new 35-mm petri dish containing several drops (100 μL/drop) of fresh M2 medium to rinse off the hyaluronidase solution, cumulus cells and debris.
Transfer individual oocytes into an 0.5 mL PCR tube containing 10 μL DPBS and freeze at −80°C.
3.2. Testicular cell collection
This procedure is a slight modification of a previously published procedure (7, 12).
Euthanize the male mice by CO2 inhalation as described above and quickly dissect out the testes.
Place the testes in a 60-mm petri dish containing DPBS.
Use two pairs of fine forceps to grasp and remove the tunica albuginea.
Cut the testes into small pieces and treat with 4 mL collagenase/DNase solution, incubate at 37°C for 20 min in a CO2 incubator.
Use a truncated 1000 μL tip to gently pipette and separate the seminiferous tubules.
Transfer the seminiferous tubules into 15 mL conical tube, centrifuge at 200 × g for 5 min at room temperature.
Carefully remove the supernatant, add 10 mL DPBS and gently pipette to wash the seminiferous tubules.
Centrifuge at 200 × g for 5 min at room temperature.
Repeat step 7 and 8 three times to remove cell debris.
Add 4 mL Dispase/DNase solution, incubate at 37°C for 20 min in a CO2 incubator, with gentle pipetting of the cells every 5 min.
Filter the mixture through a 70-μm cell strainer.
Centrifuge at 500 × g for 5 min at room temperature.
Carefully remove the supernatant. If possible, the cells should be processed immediately for staining and sorting as described in Subheading 3.3. If necessary, the cells can be frozen as described in steps 14 and 15 and recovered for later analysis.
Slowly resuspend cells in 6 mL prechilled (4°C) cell freezing media, transfer to six cryovials (1 mL/vial, about 1×107/mL, see Note 7).
Transfer the cryovials to the prechilled (4°C) StrataCooler Cryo preservation module. Place the module in a −80°C freezer overnight. Transfer the vials to liquid nitrogen for long-term storage.
3.3. Staining of testicular cells prior to isolation by fluorescence activated cell sorter (FACS)
Different testicular cell types were isolated by flow cytometry using a modification of a previously published procedure (13).
Warm the MEF growth media at 37°C.
If cells are fresh prepared, go to step 6.
Otherwise, remove the frozen vials of testicular cells from liquid nitrogen and thaw in a 37°C water bath until completely unfrozen, usually about 3–5 minutes (see Note 8).
Transfer the cells into a 15 mL conical tube with 10 mL prewarmed MEF growth media.
Centrifuge the cells at 200 × g for 5 min at room temperature. Carefully remove the supernatant.
Add 10 mL DPBS and pipette to gently wash the cells.
Centrifuge at 200 × g for 5 min at room temperature.
Carefully remove the supernatant and add 10 mL prewarmed cell sorting buffer (see Note 9).
Count the cells and adjust the cell number to 2 × 106/mL using cell sorting buffer (see Note 9).
Add equal volume of 2x cell staining buffer to stain cell at a final concentration of 5 μg/mL Hoechst 33342. Incubate at 37°C for 45 min, gently shaking the dish every 5 min.
Centrifuge at 200 × g for 5 min at room temperature.
Carefully remove the supernatant and resuspend cell in 3 mL testicular cell sorting buffer, cells are now ready for sorting (see Note 10).
3.4. Testicular cell sorting
This protocol describes the use of a FACSARIA™ (SORP) (BD) fluorescence activated cell sorter (FACS) system to isolate different murine male gametes. Note that other cell sorters may also be used, according to manufacturer recommendations.
Startup and QC the cytometer as per the manufacturer’s instructions.
Create a new template. The instrument must have a 488-nm blue laser to measure the scatter properties and a 355 nm UV laser (at 60 mW or higher) to excite the Hoechst dye. Select the instrument settings to include the height, area and width parameters of forward scatter (FSC), side scatter (SSC) and two detectors from the UV laser – one with a bandpass filter of 450/20 nm (Blue shift of HOECHST emission) and the other at 640/20 nm (Red shift of HOECHST emission), The fluorescence detectors should be on linear scale, not log (see Note 11).
Once the right parameters have been selected for collecting data for the HOECHST and FSC/SSC parameters, make a series of dot plots including (1) a plot showing FC vs SSC, (2) a plot showing FSC width vs FSC area, (3) a one parameter plot showing the 450/20 fluorescence, and (4) a dot plot showing 450/20 fluorescence vs the 640/20 fluorescence (Fig.1).
Immediately prior to running the samples on the sorter, transfer cell to a 5-mL tube with cell strainer cap to removes any residual clumps of cells.
Load the stained and filtered sample on sample port of sorter and start collecting data. Run the samples at a relatively slow acquisition rate (approximately 5000 cells per second to give tight coefficients of variations in the analysis of the samples). While instrument is acquiring data in real-time, adjust the voltages of the FSC and SSC detectors to place the cells on scale. Subsequently adjust the voltages to the 450/20 and 640/20 detectors to visualize 1C, 2C, and 4C populations on linear scales, after excluding doublets using width and area parameters. The populations can be viewed on either the one parameter plot of the 450/20 detector or on the dot plot of the 450/20 vs 640/20 detectors. The first peak (one parameter plot) or major cluster (on the bivariate dot plot) is the 1C population (population 1 and 2), the 2C peak (population 3) is at 1.8–2 times intensity of 1C signal and 4C (population 4) as 1.6–2 times of the 2C signal intensity. Initial gates were established signals using identically processed and stained samples obtained from FX mouse and control mouse lacking haploid population (Fig.1E, F).
The 1C population was further sub gated into two populations based on relative size using FSC parameter (1C Higher FSC and 1C Lower FSC) (Fig.1D).
After establishing 1C higher FSC, 1C lower FSC, 2C and 4C gates, those populations are sorted at a flow rate between 5000 to 10000 events per second and collected in DNAse, RNAse free sterile 1.8 mL microcentrifuge tubes containing 100 μL of cell sorting buffer.
Take 20 μL of sorted cells and place on a slide. Examine the purity of each subpopulation using fluorescence microscopy. The excitation and emission wavelength of Hoechst 33342 is 352 nm and 455 nm respectively. The blue fluorescence may be efficiently excited with a xenon or mercury-arc lamp or with a UV laser, and be detected using the common DAPI filter or blue GFP filters (see Note 12).
Centrifuge each of the cell subpopulations at 200 × g for 5 min at room temperature. Carefully remove the supernatant.
The cell pellet can be used directly for genomic DNA (gDNA) preparation or frozen at −80°C.
Fig. 1.
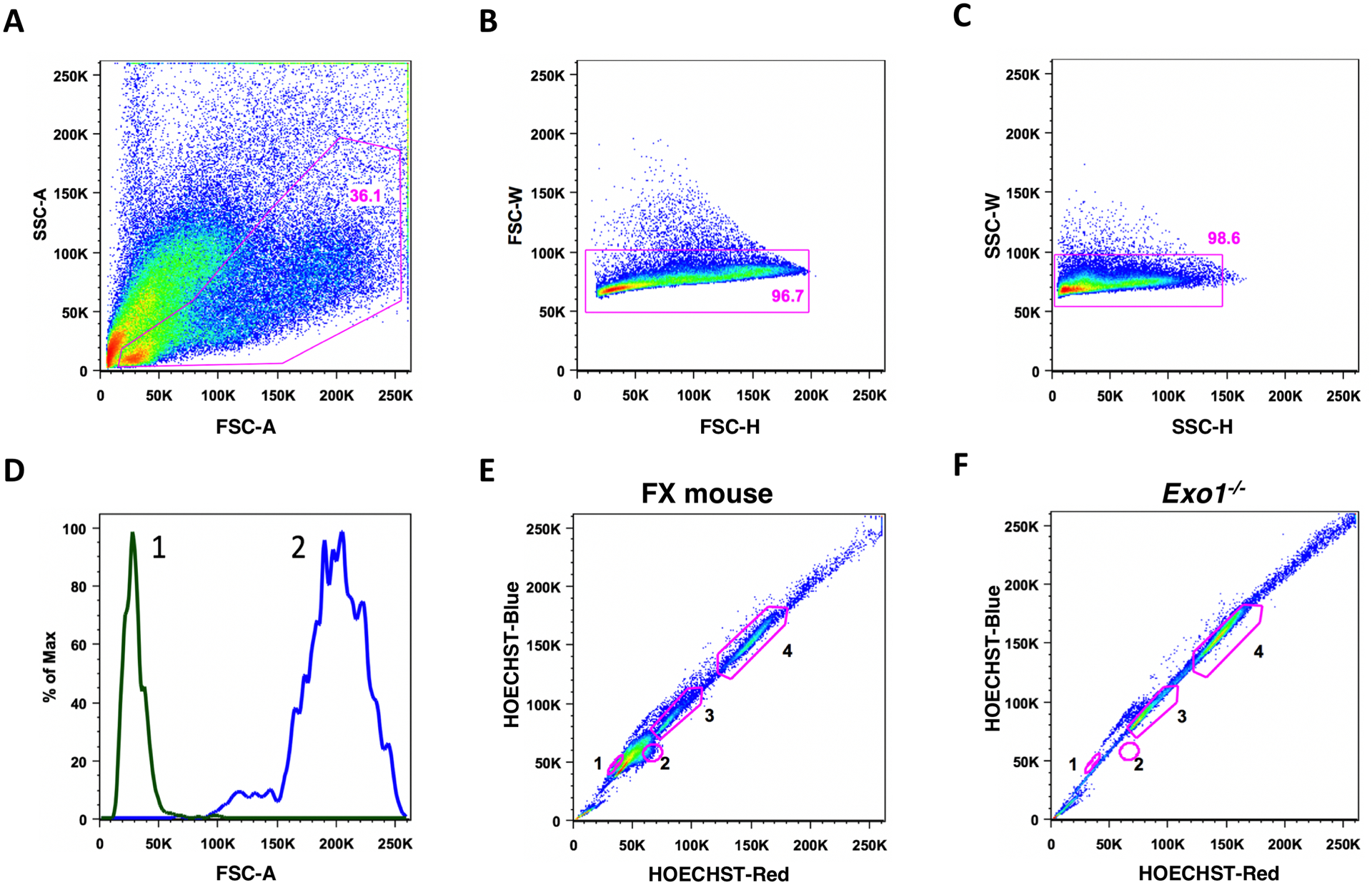
Gating scheme to purify different testicular cells types. Live cells were differentiated from dead cells and debris based on scatter properties (A). Further doublets were removed on the basis of width parameters of the FSC detector (B) and then the SSC detector (C). The 450/20 (Blue shift) and 640/20 (Red shift) signals were then used to identify the 1C (population 1 and 2), 2C (population 3) and 4C (population 4) cell clusters (E and F). Among the 1C population (first cluster population 1 and 2) gates were made to distinguish the smaller elongating spermatids from the larger round spermatids (D).
3.5. Sperm collection
Warm DPBS at 37°C.
Euthanize the male mice by CO2 inhalation as described in Subheading 3.2 and quickly dissect out the cauda epididymis with part of the vas deferens.
Place the epididymis with the vas deferens in a 35-mm petri dish with 1 mL prewarmed DPBS, remove as much fat and blood vessels as possible and rinse with prewarmed DPBS (see Note 13).
Place the epididymis with vas deferens in a new 35-mm petri dish with 2 mL prewarmed DPBS.
Use forceps to gently squeeze out the sperm from the vas deferens.
Use a 30-gauge needle on a syringe to make 5–10 slit on the cauda epididymis (see Note 14).
Incubate the dish at 37°C for 30 min. Gentle shake the dish every 5 min. Sperm will swim out from the slit and the solution will become cloudy.
Gently shake the dish and collect 1 mL suspension into a 1.5 mL microcentrifuge tube (see Note 15).
Centrifuge at 300 × g for 5 min at room temperature.
Carefully remove the supernatant. The pellet should be white and without visible red blood cells at the bottom (see Note 16).
Gently tap the tube one or two times to loosen the pellet.
Slowly add 1 mL prewarm DPBS along the tube wall.
Incubate the tube at 37°C for 20 min and allow the sperm to swim out.
Collect 500 μL supernatant into a new 1.5 mL microcentrifuge tube.
Centrifuge at 300 × g for 5 min at room temperature.
Carefully remove the supernatant. If there is visible red blood cell at the bottom, repeat the swim out step 11–16 until there is no visible red blood cell at the bottom.
The sperm pellet can be used directly for gDNA preparation or frozen at −80°C.
3.6. DNA isolation from oocytes
Take out the tubes with individual oocytes from −80°C and incubate at 37°C for 10 min.
Place the tube on dry ice for 10 min.
Repeat step 1 and 2 three times.
Add 2.5 μL 10 mg/mL Proteinase K to the tube to make a final concentration of 2 mg/mL Proteinase K.
Incubate at 55°C for 2 h.
Incubate at 95°C for 10 min.
The oocyte DNA in a total volume of 12.5 μL can be used directly for PCR or frozen at −20°C.
3.7. DNA isolation from testicular cells and sperm
Add 300 μL cell lysis buffer to each tube of the sorted testicular cells and the sperm sample. Either fresh prepared or frozen samples can be used.
Vortex for 10 s to mix the sample.
Incubate at 55°C overnight.
Spin down the liquid that has accumulated in the lid at 300 × g for 1 min.
Add 90 μL 5 M NaCl, gently invert the tube to mix.
Centrifuge at 13,000 × g for 10 min at room temperature.
Transfer as much of supernatant as possible to a new 1.5 mL microcentrifuge tube, taking care not to transfer the debris.
Add an equal volume of ethanol and invert gently to mix.
Precipitate the gDNA at −20°C for 1 h (see Note 17).
Centrifuge at 13,000 × g for 10 min at room temperature.
Carefully remove the supernatant and add 800 μL 70% ethanol to wash the pellet.
Centrifuge at 13,000 × g for 10 min at room temperature.
Carefully remove the supernatant and leave the lid open for 5 min to dry the pellet.
Add 5–100 μL PCR grade H2O to dissolve the DNA pellet. Incubate at 55°C for 15 min (see Note 18).
Genomic DNA samples can be used directly for PCR or frozen at −20°C.
3.8. Analysis of the repeat number in male gametes
To analyze the repeats in individual gametes, determine the DNA concentration and dilute to 3 pg/μL. Use 1 μL as the template to carry out nested PCR using the small pool PCR/Nested PCR strategy described in Subheading 3.10 below.
To analyze the repeats from bulk gDNA from sperm or different testicular cell samples, dilute the gDNA to a concentration of 100 ng/μL. Use 1 μL to do a single round of PCR as described in steps 3–6 of Subheading 3.10.
3.9. Analysis of the repeat number in oocytes
Use the small pool PCR/nested PCR strategy described in Subheading 3.10 to amplify the repeats from individual oocyte samples using 4 μL of the oocyte DNA solution for each reaction.
3.10. Small pool PCR/nested PCR analysis of repeat number
Carried out the first round of PCR using the Not_mFraxC and Not_FraxR4 primer pair (Table 1) in a 20 μL reaction. The PCR mix contains 1x Q5 CGG PCR buffer, 1x Betaine-DMSO mix (2.4 M Betaine, 2% DMSO), 0.2 mM dNTPs plus an additional 0.1 mM dCTP-dGTP, 0.5 μM of each primer and 0.02 U/μL of the DNA Q5 polymerase. The first round PCR parameters should be 98°C for 10 min, 33 cycles of (98°C for 30 s, 59°C for 30 s, and 72°C for 2.5 min), followed by 72°C for 10 min (Table 1).
Amplify 1 μL of the first round PCR reaction or in the case of bulk DNA analysis, 1 μL of a 100 ng/μL gDNA solution, using the FAM-FraxM4 and FraxM5 primer pair (Table 1) in a 21 μL reaction (see Note 19). The PCR mix contains 1x KAPA2G Fast HotStart Genotyping Mix, 1x Betaine-DMSO mix (2.4 M Betaine, 2% DMSO), 0.5 μM each of the primers FraxM4 and FraxM5. Subject the PCR mix to amplification using the following cycling parameters: 95°C for 10 min, 35 cycle of (95°C for 30 s, 65°C for 30 s, and 72°C for 70 s), followed by incubation 72°C for 10 min. The 70 s annealing step can be extended to 1.5 min for amplification of alleles with >300 repeats.
Resolve the PCR products by capillary electrophoresis on an ABI Genetic Analyzer. The resultant fsa files can be analyzed using GeneMapper or equivalent software. An R script that can be used for this purpose is available on request (9). Repeat sizes are calculated using the formula: repeat number= (size of PCR products – 49 bp)/3 (see Note 20).
Table 1.
Primers used in this study
| Name | Sequence |
|---|---|
| Not_mFraxC | AGTTCAGCGGCCGCGCTGGGGAGCGTTTCGGTTTCACTTCCGGT |
| Not_FraxR4 | CAAGTCGCGGCCGCCTTGTAGAAAGCGCCATTGGAGCCCCGCA |
| FraxM5 | CGGGGGGCGTGCGGTAACGGCCCAA |
| FAM-FraxM4 | FAM-CTTGAGGCCCAGCCGCCGTCGGCC |
Dissolve the primers in PCR grade H2O at a concentration of 100 μM. Store the solution in aliquots at −20°C.
4. Notes
All experiments involving live rodents must conform to national and institutional regulations.
Store PMSG and hCG in a single use aliquot, avoid freeze-thaw cycles.
In addition to the FX male mice, male mice, for example Exo1−/− mice (4, 7), which do not complete meiosis and therefore lack haploid sperm can be used as a sorting control to confirm the specificity of the haploid population isolated.
To make 20 mL 5 M betaine weigh 13.5 g betaine and add 8 mL sterile double-distilled H2O. Once most of the betaine has dissolved, make the volume up to 20 mL and wait until the betaine has dissolved completely.
Store the dNTPs and dCTP-dGTP mix in small aliquots at −20°C and discard after 3 freeze-thaw cycles.
The dose needed of each gonadotropin is strain-dependent. Generally, doses of 2.0–5.0 IU per mouse of each gonadotropin and ages of 21–35 days (body weights of 12–14 g) will give the best yield of oocytes.
Normally a total number of 5–7×107 testicular cells can be collected from a 6-month old FX male mouse by this method. The Exo1−/− males have smaller testes and fewer testicular cells. The total testicular cell number is ~3–5×106.
Thawing procedure is time and temperature-sensitive. For better recovery of cryopreserved cells, this step should be performed at 37°C as gently and quickly as possible.
Reduce the volume of cell sorting buffer if total testicular cell number is low. If the cell number is lower than 2 × 106/mL, use 1 mL for staining.
A good concentration of cells for sorting is about 1–2×107/mL. If the cell number is less than this, use at least 500 μL for sorting.
It is advisable to shut down the 407 nm LASER in order to avoid any partial excitation of HOECHST dye by the violet LASER line.
Subpopulation 1 included elongated spermatids and spermatozoa with a 1C DNA content, and subpopulation 2 included post-meiotic haploid round spermatids with a 1C DNA content. Subpopulation 3 included secondary spermatocytes, germinal stem cells and some somatic cells with a 2C content. Subpopulation 4 contains prophase spermatocytes with a 4C DNA content. The purity of subpopulations 1, 2, and 4 should be over 90%.
Instead of removing the blood vessels attached to the cauda epididymis, forceps can be used to gently squeeze out the blood from blood vessels. This step can remove most of the blood, but at the risk of some sperm loss.
Do not mince the cauda epididymis. Minimizing trauma will avoid contamination with somatic cells.
Gently shake the dish and tilt the dish a bit to allow the epididymis and vas deferens to slip to the bottom of the dish. Avoid the tissue debris while collecting the suspension. Up to 1.8 mL can be collected.
Visible red blood cells may appear at the bottom of the pellet if the blood vessels were not removed properly. Repeating the next swim out step (11–16) will improve the purity of sperm sample.
The DNA pellet can be very small and hard to see in the tube if total cell number is less than 100 thousand. Addition of 1 μL of pellet paint (EMD Millipore) helps in the visualization of the DNA.
Adjust the volume of water base on the size of the pellet. Normally add 100 μL for sperm sample and add 5 μL water for samples with less than 100,000 cells.
These primers are specific for the FXD mouse generated in our laboratory (14). Primers specific for other mouse models should aim to have a similar Tm.
The number 49 is the number of bases that flank the repeats in amplicons generated by the FraxM5 and FAM-FraxM4 primer pair. This number may change if other primers are used.
Acknowledgements:
The authors would like to thank Dr John Philip McCoy, Jr (NHLBI) for his careful reading of this manuscript. This work was made possible with funding to KU from the Intramural Program of NIDDK, NIH (DK057808).
References
- (1).Lozano R, Rosero CA, Hagerman RJ (2014) Fragile X spectrum disorders, Intractable Rare Dis Res 3(4) 134–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lokanga RA, Senejani AG, Sweasy JB et al. (2015) Heterozygosity for a hypomorphic Polbeta mutation reduces the expansion frequency in a mouse model of the Fragile X-related disorders, PLoS Genet 11(4) e1005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lokanga RA, Zhao XN, Usdin K (2014) The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model, Hum. Mutat 35(1) 129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhao X-N, Zhang Y, Wilkins K et al. (2018) Mlh3 promotes while Exo1 protects against repeat expansion in mouse model for the Fragile X-related disorders, PLoS Genet. 14(10) e1007719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhao XN, Usdin K (2018) FAN1 protects against repeat expansions in a Fragile X mouse model, DNA Repair (Amst) 69 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhao XN, Lokanga R, Allette K et al. (2016) A MutSbeta-Dependent Contribution of MutSalpha to Repeat Expansions in Fragile X Premutation Mice?, PLoS Genet 12(7) e1006190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhao XN, Usdin K (2018) Timing of Expansion of Fragile X Premutation Alleles During Intergenerational Transmission in a Mouse Model of the Fragile X-Related Disorders, Front Genet 9 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhao X-N, Gazy I, Hayward BE et al. Repeat Instability in the Fragile X-related disorders: lessons from a mouse model. Brain Sci 8:E52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hayward BE, Zhou Y, Kumari D et al. (2016) A Set of Assays for the Comprehensive Analysis of FMR1 Alleles in the Fragile X-Related Disorders, J. Mol. Diagn 18(5) 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Nagy A, Gertsenstein M, Vintersten K et al. , Manipulating the Mouse Embryo: A laboratory manual, CSH Press2003. [Google Scholar]
- (11).Danneman PJ, Stein S, Walshaw SO (1997) Humane and practical implications of using carbon dioxide mixed with oxygen for anesthesia or euthanasia of rats, Lab. Anim. Sci 47(4) 376–85. [PubMed] [Google Scholar]
- (12).Guan K, Wolf F, Becker A et al. (2009) Isolation and cultivation of stem cells from adult mouse testes, Nat. Protoc 4(2) 143–54. [DOI] [PubMed] [Google Scholar]
- (13).Hayama T, Yamaguchi T, Kato-Itoh M et al. (2016) Practical selection methods for rat and mouse round spermatids without DNA staining by flow cytometric cell sorting, Mol. Reprod. Dev 83(6) 488–96. [DOI] [PubMed] [Google Scholar]
- (14).Entezam A, Biacsi R, Orrison B et al. (2007) Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model, Gene 395(1–2) 125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]